

Abstract
Over the past four decades, the number of people with Type 1 Diabetes (T1D) has increased by 4% per year, making it an important public health challenge. Currently, no curative therapy exists for T1D and the only available treatment is insulin replacement. HLA-DQ8 has been shown to present antigenic islet peptides driving the activation of CD4+ T-cells in T1D patients. Specifically, the insulin peptide InsB:9-23 activates self-reactive CD4+ T-cells, causing pancreatic beta cell destruction. The aim of the current study was to identify retro-inverso-D-amino acid based peptides (RI-D-peptides) that can suppress T-cell activation by blocking the presentation of InsB:9-23 peptide within HLA-DQ8 pocket. We identified a RI-D-peptide (RI-EXT) that inhibited InsB:9-23 binding to recombinant HLA-DQ8 molecule, as well as its binding to DQ8 expressed on human B-cells. RI-EXT prevented T-cell activation in a cellular antigen presentation assay containing human DQ8 cells loaded with InsB:9-23 peptide and murine T-cells expressing a human T-cell receptor specific for the InsB:9-23–DQ8 complex. Moreover, RI-EXT blocked T-cell activation by InsB:9-23 in a humanized DQ8 mice both ex vivo and in vivo, as shown by decreased production of IL-2 and IFN-γ and reduced lymphocyte proliferation. Interestingly, RI-EXT also blocked lymphocyte activation and proliferation by InsB:9-23 in PBMCs isolated from recent onset DQ8-T1D patients. In summary, we discovered a RI-D-peptide that blocks InsB:9-23 binding to HLA-DQ8 and its presentation to T-cells in T1D. These findings set the stage for using our approach as a novel therapy for patients with T1D and potentially other autoimmune diseases.
Keywords: Type 1 Diabetes, RI-D-peptides, HLA-DQ8, InsB:9-23
1. Introduction
Type 1 diabetes mellitus (T1D) is a common autoimmune disorder, and is associated with significant morbidity and mortality in children and adults [1, 2]. According to recent studies the worldwide estimates of numbers of people with T1D continue to increase [3] especially among children under 5 years of age [1, 4]. These rapid trends are alarming and underscore the urgent need for newer therapies for T1D.
T1D is caused by the interaction of environmental triggers and susceptibility genes and is characterized by the generation of islet-specific T-cells and autoantibodies, inflammatory infiltration of the islets, and beta cell destruction [5-7]. Beta cell death characteristic of T1D leads to insulin deficiency and hyperglycemia which result in potential long-term complications, including cardiovascular disease, neuropathy, retinopathy, and nephropathy [8-11]. Currently no curative therapeutic or prevention modalities exist for T1D to reverse or prevent the autoimmune destruction of the islets, and the disease can only be managed with insulin replacement therapy [12]. While patients can achieve good glucose control on insulin therapy and a normal life expectancy if they maintain near normoglycemia, the challenges of insulin treatment include multiple injections or the use of an insulin pump, multiple blood glucose measurements per day, and risk of hypoglycemia [13]. Therefore, many patients have sub-optimal glycemic control resulting in complications, especially in teenage T1D patients, who often have uncontrolled diabetes due to non-adherence [14, 15]. Even when fully adherent to the treatment regimen, studies have shown that T1D patients often have sub-optimal glucose control, due to the fact that even the best insulin regimens cannot mimic endogenous secretion of insulin by pancreatic beta cells [14]. Therefore, novel therapies and prevention strategies are needed for T1D. Ideally, such therapeutic approaches should target the autoimmune attack against beta cells without causing systemic immunosuppression.
There is substantial evidence indicating that the destruction of beta cells by CD8+ cytotoxic T-cells and other mechanisms is triggered by CD4+ T-cells activated against diabetogenic islet peptides (such as InsB:9-23) [16, 17], which are activated by the presentation of islet peptides within HLA class II molecules expressed on antigen presenting cells (APCs). Interestingly, HLA class II-restricted T-cell responses to InsB:9–23 peptide are highly associated with T1D in humans [18]. Furthermore, one of the most important HLA class II molecules associated with T1D is HLA-DQ8, which is present in >50% of T1D patients [16, 19]. HLA-DQ8 has been shown to present diabetogenic peptides to CD4+ T-cells, and CD4+ T-cell clones isolated from inflamed islets of patients with T1D were found to recognize InsB:9–23 [20]. Strikingly, adoptive transfer of HLA-DQ8–restricted InsB:9–23-specific human CD4+ T-cells is capable of inducing diabetes in HLA-DQ8–transgenic humanized-mice, consistent with the potential of T-cell responses to the InsB:9–23 epitope to initiate T1D in humans [21]. Thus, preventing the binding of diabetogenic peptides to HLA-DQ8 and blocking their presentation to CD4+ T-cells seems an ideal target for precision immune therapies [22, 23].
The goal of the present study was to test an innovative peptide therapeutics approach to prevent beta cell death in T1D by blocking the interaction between HLA-DQ8 positive APCs and CD4+ T-cells. We believe that peptide therapeutics provide greater efficacy, selectivity, specificity, and safety compared to small molecules and antibodies [24]. To overcome the peptides’ intrinsic disadvantage of sensitivity to proteases we harnessed D-amino acid based peptides (RI-D-peptides) harboring the reverse sequence (retro-inverso peptides) of the corresponding L-amino acid peptides. The RI-D-peptides able to block the binding of pathogenic peptides (e.g. InsB:9-23) to HLA-DQ8, occupy the same pockets in the HLA groove but are unable to activate the T-cell receptor (TCR) since it cannot recognize D-amino acids. This novel approach is precise (only T-cells recognizing diabetogenic peptides are targeted) and if successful it will be used to treat T1D patients carrying the HLA-DQ8 at early stages of the disease, corresponding to at least 50% of patients. Moreover, if proven useful this therapeutic strategy can be expanded to patients with other autoimmune diseases associated with HLA class II alleles
2. Materials and methods
2.1. Designing the RI-D-peptides
The design of RI-D-peptides in this work was based on the original sequence of InsB:9-23, which has been shown to bind in the groove of the HLA-DQ8 [16]. The specificity of this binding resides in part from the insertion of their side chains into specific binding pockets. Indeed, the P1 pocket in the HLA-DQ8 protein is very polar and comfortably accommodates a Glutamic Acid from the InsB:9-23 peptide that interacts with the HLA-Q8 Arginine-52α; in addition, HLA-DQ8 pocket P4 is very deep and holds in it the side chain of InsB:9-23 Tyrosine [16]. With the goal of maintaining the positions of these side chains in their respective pockets, we inverted the stereochemistry of the L- to D-amino acid switching the positions of the N-H and C=O backbone groups. Such a transformation inverts the stereochemistry of Cα while maintaining the positions of the side chains in their respective pockets, but reverses the direction of the chain. Hence, we identified our peptides as retro-inverso (RI). Using the program Simulaid [25] we converted the original InsB:9-23 into the RI form. The RI-CT variant was designed by replacing the C-term end from Leucine-Histidine to Glycine-Glycine to reduce potential repulsion with the HLA-DQ8 protein (note that the C-term of the Rl-peptide resides in the same position as the N-term of the original InsB:9-23). Variants #3-#5 and variants #6-#9 were designed respectively by replacing the original Valine (P6) and the original Leucine (P3) with the residues listed in Fig. 1A. The RI-EXT series (#10-#14) was designed by extending the N- and C-termini by two residues on each side to enhance the effect of flanking residues in order to improve the affinity of the peptides to HLA-DQ8.
Fig. 1. In vitro testing of RI-D-peptides designed in silico.
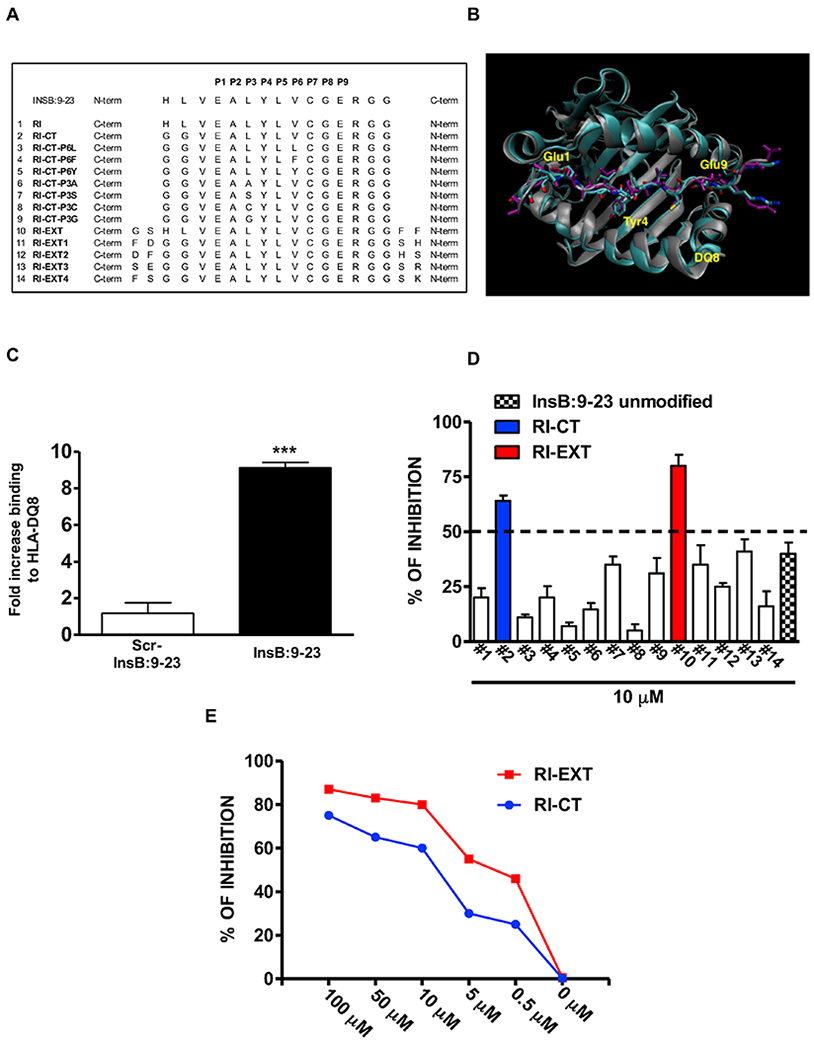
(A) 14 RI-D-peptides were predicted in silico to block the HLA-DQ8 pocket. (B) Representative structure of InsB:9-23-DQ8 (purple/grey) complex superimposed on the RI-DQ8 complex. Highlighted are three important residues Glu1, Tyr4 and Glu9, which are superimposed in both complexes. (C) testing of recombinant HLA-DQ8 protein used for the screening using DELFIA immunoassay. (D) RI-D-peptides RI-CT and RI-EXT inhibited InsB:9-23 binding to HLA-DQ8 more than 50% and (E) both compounds showed a significant dose-dependent inhibition of the InsB:9-23 binding to HLA-DQ8. Data represent the mean ± SEM and are representative of 3 independent experiments. ***p < 0.001, by Student’s t-test for comparison of InsB:9-23 versus scrambled InsB:9-23 binding to recombinant HLA-DQ8.
2.2. Molecular Dynamics Simulations and Energy Decomposition Analysis
Molecular Dynamics (MD) simulations were conducted with AMBER v.16 [26]. InsB:9-23 peptide in complex with HLA-DQ8 (constructed from the crystal structure 1JK8.pdb) was converted to a RI-peptide by using the program Simulaid [25]; the original peptide was replaced by the RI-peptide and the initial structure served to construct the complete system with TLEAP in AMBER. Water was added to create a truncated octahedron and ions were added to neutralize the system. The entire system was contained within a truncated octahedron of ~100 Å with approximately 70,000 atoms. The system was minimized, heated and equilibrated with positional restraints on the solute that were reduced gradually. The MD simulations were carried out at normal temperature and pressure conditions. Analysis was conducted with CPPTRAJ and the energy decomposition was calculated with the MMGBSA module.
2.3. Production of recombinant HLA-DQ8 protein
HLA-DQ8 protein was produced using the baculovirus system. We designed 2 constructs for both the α and the β chains of HLA-DQ8. The β chain construct contained the extracellular portion of the DQB1*0302 β chain fused to the coiled-coil region of the basic leucine zipper domain of JunB, and the α chain construct contained the extracellular portion of the DQA1*0301 α chain fused to the coiled-coil region of the basic leucine zipper domain of Fos. The β chain had an histidine tag at the C-terminus in order to enable binding to nickel coated plates. A tobacco etch virus protease cutting site was introduced in each chain to allow removal of the dimerization motif. The JunB and Fos dimerization motifs were added in order to allow the α and β chains to dimerize and form the final HLA-DQ8 protein. These constructs were used to produce the HLA-DQ8 protein in a baculovirus system using the Life Technologies Baculovirus protein production custom services (Carlsbad, CA).
2.4. In vitro testing of RI-D-peptides designed by MD simulations
RI-D-peptides were produced by Fisher Scientific (Hanover Park, IL) in dry form and dissolved in PBS. All RI-D-peptides were screened at a concentration of 10 μM for their ability to bind to the HLA-DQ8 pocket and block it from binding the InsB:9-23 peptide using a specific DELFIA (dissociation-enhanced lanthanide fluorescence immunoassay) assay. In this assay 0.012 mg/ml of recombinant HLA-DQ8 protein were incubated with 10 μM biotinylated (or unmodified) InsB:9-23 peptide, GAD65, gliadin or their scrambled versions (Genscript, Piscataway, NJ), with or without the tested RI-D-peptide (10 μM), for 48 hours, at 37 °C, in binding buffer (0.1% BSA/PBS with 0.05% Triton (PBST), (Sigma). We used GAD65 and gliadin as controls for their known ability to bind to HLA-DQ8 [27, 28]. Following the same protocol, biotinylated RI-CT and RI-EXT or their scrambled version were incubated alone with HLA-DQ8 protein in order to check their binding to HLA-DQ8. After 48 hours incubation, a 100 μl solution containing either HLA-DQ8 protein and InsB:9-23, with or without RI-D-peptides, or HLA-DQ8 protein in the presence of RI-D-peptides or scrambled RI-D-peptides was added to nickel coated plates (Sigma) and shaken at slow speed for 2 h at room temperature. Since the β chain of the HLA-DQ8 protein had a His tag the DQ8 protein complexed with the InsB:9-23 peptide bound to the plate. After washing 4 times, DELFIA Eu-labeled streptavidin (Perkin Elmer) diluted in DELFIA assay buffer (Perkin Elmer) was added for 30 min and shaken at slow speed at room temperature. Since the InsB:9-23 peptide was biotinylated the complex HLA-DQ8-InsB:9-23 could be detected using the Eu-streptavidin. After washing for 6 times, DELFIA Enhancement Solution was added for 1 h or until the optimal signal was reached. Time-resolved fluorescence was measured using the BMG reader (BMG Labtech, Cary, NC). As negative control we used scrambled biotinylated InsB:9-23 peptide that was preincubated with HLA-DQ8. Percent inhibition of InsB:9-23 binding to HLA-DQ8 by the RI-D-peptides or by unmodified InsB:9-23 was calculated by the following formula: 100 – 100 x [HLA-DQ8-InsB:9-23 (with RI-D-peptide or unmodified InsB:9-23) / HLA-DQ8-InsB:9-23 (no RI-D-peptide or unmodified InsB:9-23)].
2.5. Determining potency of identified RI-D-peptides
To determine the potency of the RI-D-peptides that showed inhibition of InsB:9-23 binding to HLA-DQ8 greater than 50%, we tested the % inhibition at decreasing concentrations of RI-D-peptides. Two of the RI-D-peptide hits, RI-CT and RI-EXT, were serially diluted to 0.5 μM final concentration and incubated with the HLA-DQ8-InsB:9-23 complex for 48 hours at 37 °C for assessing the percentage of inhibition. Immunoassay was performed as described above.
2.6. Cell culture
BSM cells, that are homozygous for HLA-DQ8 (European Collection of Authenticated Cell Cultures), were cultured in RPMI (ATCC, Manassas, VA) supplied with 10% FBS (Sigma), 1% penicillin-streptomycin (Corning, NY), and 2mM glutamine (Corning). Cells were grown at 37°C, 5% CO2, and passaged 1–2 times a week. T-cell hybridoma cells (5KC cells, generously provided by Dr. Maki Nakayama, University of Colorado, Denver), transduced with the human InsB:9-23 responding TCR, were produced and cultured as described [29]. Cell viability before and after RI-D-peptides treatment was performed by MTT (Sigma-Aldrich) as previously described [30, 31].
2.7. Flow cytometric analysis of inhibition of InsB:9-23 peptide binding to antigen presenting cells by hit RI-D-peptides
N-terminal biotinylated peptide InsB:9-23 or gliadin were used to test for binding to BSM cells that express HLA-DQ8 on their surface. APC-streptavidin (BD Biosciences, Franklin Lakes, NJ) was used to detect the biotinylated peptides. BSM cells were seeded at 2.5 x 106 cells/ml in a 24-well plate (BD Biosciences) and preincubated overnight with 10 μM RI-CT or RI-EXT RI-D-peptides. After 24 hours incubation, cells were stained with PE-mouse anti-human HLA-DQ8 (BD Biosciences) and APC-streptavidin to detect the percent binding of the peptide to BSM cells with or without RI-D-peptides. Percent binding was analyzed by flow cytometry gated on live BSM cells. ReadiDrop Propidium Iodide Cell Viability Assay (Bio-Rad, USA), was used to exclude dead cells.
2.8. Cellular antigen presentation assay to test in vitro functional inhibition of T-cell activation by hit RI-D-peptides
To functionally test in vitro RI-D-peptides ability to block T-cell activation, 106 BSM cells were incubated overnight with 200 μg/ml InsB:9-23 (or with scrambled InsB:9-23 or gliadin as negative peptides); on the following day InsB:9-23 loaded BSM cells were washed twice with PBS to remove excess peptide, and RI-CT or RI-EXT peptides (or scrambled RI-CT/RI-EXT, as negative controls) were added to the cells. Following 2 hours of incubation at 37 °C 5KC cells were added to the mixture BSM-InsB:9-23-RI-D-peptides (1:1 proportion) and incubated overnight. (5KC cells are mouse T-cells transfected with human T-cell receptor that is activated only upon engagement with HLA-DQ8-InsB:9-23 complex. Upon activation by HLA-DQ8-InsB:9-23 complex 5KC cells produce and secrete IL-2). After overnight incubation of 5KC cells with BSM cells loaded with InsB:9-23 with or without different RI-D-peptides, IL-2 production was measured in the supernatants by Luminex assay as previously described [32, 33]. 5KC cells incubated with anti CD3-CD28 beads (which activate IL-2 production in non-antigen dependent fashion) were used as positive control.
2.9. Mice
Mice transgenic for HLA-DQ8 (DQA1*0301 / DQB1*0302) were originally generated by Dr. C.S. David and co-workers as previously described [34]. These mice do not develop autoimmune responses to InsB:9-23 peptide (data not shown), likely because their background is mostly C57Bl/6 which is autoimmune resistant. We therefore crossed these mice into the SJL background (which is highly autoimmune prone) [35] to produce the SJL-DQ8 mice. Mice were bred in a pathogen-free facility (Albert Einstein College of Medicine, Bronx, NY). The expression of HLA-DQ8 was tested by PCR using DQ8-specific primers: forward primer, 5′- AGGG ATC CCC GCA GAG GAT TTC GTG-3′ and reverse primer 5′- CACC TGC AGT GCG GAG CTC CAA CTG GTA-3′ and by flow using anti human PE-DQ8 antibody (Abcam, Cambridge, UK). Anti-mouse biotin-CD3 antibody (BioLegend, San Diego, CA), anti-mouse biotin-CD2 antibody, anti-mouse biotin-B220 antibody (both eBioscience, Hanover Park, IL) and anti-mouse biotin-CD11c antibody were used by flow to check the subpopulation of cells expressing DQ8 in lymphocytes isolated from DQ8 transgenic mice. APC-conjugated avidin was used as secondary antibody. (Both CD11c antibody and APC-conjugated avidin are from BD, Franklin Lakes, NJ).
2.10. Induction of autoimmune T-cell responses to InsB:9-23 in SJL-DQ8 humanized mice
20 SJL-DQ8 mice (12 females and 8 males), 6–8 weeks old, were injected subcutaneously with InsB:9-23 (150 μg/mouse) in Complete Freund’s Adjuvant (CFA, from Sigma) and boosted on day 7 in Incomplete Freund’s Adjuvant (IFA, from Sigma) to induce T-cell activation. Mice were sacrificed on day 14 and their spleens and draining lymph nodes removed. 10 wild type SJL mice (4 females and 6 males) were used as controls. Experimental and control animals were co-housed.
2.11. T-cell stimulation analysis using CFSE
Spleens and draining lymph nodes were collected from mice upon sacrifice. Cells from spleens and draining lymph nodes were harvested in complete RPMI (Corning) supplemented with 10% FBS (Sigma) and 1 mM sodium pyruvate (Sigma). The harvested cells were resuspended at 2 x 106 cells/ml in 0.1% BSA/PBS. 106 cells were labeled with 1.5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Life Technologies). After incubating for 10 min at 37 °C, the staining was terminated by the addition of 4 volumes of ice-cold RPMI, 10% FBS. After 5 min of incubation on ice, the cells were washed 3 times with fresh RPMI and resuspended in fresh medium for counting. The CFSE-labeled cells were plated at 2 x 105 cells/well in 100 μl of medium (RPMI, 10% FBS). The cells were treated with medium or with InsB:9-23 (with or without RI-D-peptides). All peptides were used at 20 μg/ml . The cells were collected after 5 days for flow cytometry analysis. Cells were gated on live splenocytes in singlets. All experiments were performed at least in triplicates. The percent of proliferation inhibition in DQ8-lymphocytes by the RI-D-peptides was calculated using the following formula: 100 – 100 x (proliferation with RI-D-peptide / proliferation without RI-D-peptide).
2.12. T-cell activation analysis using cytokine assays
Lymphocytes from immunized mice were plated at 2 x 105 cells/well in 100 μl of medium (RPMI, 10% FBS). The cells were treated with medium, InsB:9-23 (with or without RI-D-peptides or scrambled RI-D-peptides), or scrambled InsB:9-23, (all peptides at 20 μg/ml). Supernatants from stimulated lymphocytes were collected 48 hours after stimulation and stored at −80 °C until the Luminex assay was performed. The Milliplex mouse cytokines/chemokines magnetic panel (EMD Millipore Corporation, Billerica, MA) and a Luminex 200 with xPONENT software (Luminex, Austin, Texas) were used to assay the cytokines following the manufacturer’s instructions.
2.13. ELISPOT assay
ELISPOT were performed as previously described [36]. A total of 5X105 lymphocytes isolated from SJL-DQ8 mice immunized with InsB:9-23 as described above were plated onto 96-well flat-bottom nitrocellulose ELISPOT plates (Millipore) coated with 4 μg/ml IL-2 or 10 μg/ml anti-IFN-γ (both BD Pharmigenin, San Diego, CA). 6 SJL-DQ8 mice were used in this ELISPOT assay (3 females and 3 males). Lymphocytes were incubated for 48 hours in the presence of InsB:9-23, InsB:9-23+RI-CT and InsB:9-23+RI-EXT. Anti CD3-CD28 beads were used as positive control. After 48 hours of incubation at 37 °C, plates were washed with PBS containing 0.05% Tween 20. Wells were incubated with 4 μg/ml biotinylated anti-IL-2, or 1 μg/ml biotinylated anti- IFN-γ (both BD Pharmigenin, San Diego, CA) for 2 h at 37 °C. Plates were washed as described above and then incubated with streptavidin-alkaline phosphatase (Life Technologies) for 1 hour at 37 °C. Plates were washed as described above and developed by the addition of 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium chloride (NBT) tablets (Sigma). Plates were counted with the aid of computer-assisted image analysis by using an AID ELISPOT reader (Autoimmun Diagnostika GmbH, Strassberg, Germany).
2.14. Validation of RI-D-peptides’ ability to block T-cell stimulation in PBMCs from T1D donors
Subjects with a diagnosis of T1D, according to American Diabetes Association criteria, within the previous 2 years, were recruited from the Pediatric Endocrine Clinic at Children Hospital at Montefiore and from the adult Endocrine Clinics at Montefiore and Mount Sinai Hospitals (New York). Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood using Ficoll-Paque (GE Healthcare); freshly isolated PBMCs were used for T-cell activation studies and for genotyping. DNA purified from the blood of the patients was typed for HLA-DQ as previously described [37]. PBMCs isolated from patients positive for at least one DQ8 allele (DQB*03:02) were seeded in 96 well plates with 200 μl medium (RPMI, 10% FBS). The cells were stimulated with InsB:9-23 or scrambled InsB:9-23, with or without the RI-D-peptides RI-CT or scrambled RI-CT, RI-EXT or scrambled RI-EXT for 48 hours (all peptides at 20 μg/ml). The stimulated PBMCs were assessed for cytokine production and lymphocytes proliferation as described above for mouse lymphocytes. PBMCs were gated on live T-cells for flow cytometry analysis. DQ8 typed patients with long standing T1D (> 10 years) were used as negative controls in our experiments (no T-cell responses to islet peptides). The percent of proliferation inhibition in DQ8-lymphocytes by the RI-D-peptides was calculated using the following formula: 100 – 100 x (proliferation with RI-D-peptide / proliferation without RI-D-peptide).
2.15. Confirmation of RI-D-peptide’ activity in vivo using SJL-DQ8 humanized mice
SJL-DQ8 mice were immunized with InsB:9-23 in CFA on day 1, and then injected with 100 μg of the RI-D-peptides RI-EXT (10 females and 10 males) or scrambled RI-EXT as negative control (8 females and 6 males) intraperitoneally (IP) on days 4 and 7. The mice were boosted with InsB:9-23 in IFA on day 8, and were injected with 100 μg of the RI-D-peptides again on days 11 and 14. Mice were sacrificed on day 17.
2.16. Statistical analysis
Prism 5 software was used to perform statistical analysis. Student’s t-tests (paired t and unpaired t-tests, one tailed) were used for the comparison of normally distributed values between groups of samples. A p value < 0.05 was considered statistically significant. For ELISPOT assays, responses were considered positive if the net number of spot-forming cells per million cells was > 20 and the P value was < 0.05 (as determined by Student’s t test for the means of triplicate values for the response against treatment lymphocytes versus control lymphocytes).
2.17. Study approval
The study was approved by the Albert Einstein College of Medicine and Icahn School of Medicine at Mount Sinai institutional review boards. Written informed consent was obtained from all study participants. All animal studies were approved by the Institutional Animal Care and Use Committees (IACUC) of the Albert Einstein College of Medicine and carried out according to its guidelines.
3. Results
3.1. Designing RI-D-peptides that block InsB:9-23 binding to the HLA-DQ8 pocket
An in silico approach was used to rank RI-D-peptides predicted to bind structural pockets in the HLA-DQ8 peptide-binding groove. First, we designed a retro-inverso InsB:9-23 (RI-InsB:9-23), i.e. a RI-D-peptide in which the sequence of InsB:9-23 was reversed (Fig. 1A and Supplementary Table 1). Of note, all residues in Fig. 1A are D-amino acids except for glycine (G). An example of the structural alignment of the RI-InsB:9-23 peptide and the original InsB:9-23 peptide is depicted in Fig. 1B. The resulting side chains of the peptide resided in nearly identical pockets but the backbone direction was reversed thereby creating different H-bonds with the HLA-DQ8 protein. We then used molecular dynamics (MD) simulations to generate RI-peptides that can block InsB:9-23 binding to HLA-DQ8 pockets and inhibit T-cell stimulation.
3.2. In vitro screening of lead-candidate RI-D-peptides
Using MD simulations we designed 14 RI-D-peptides able to block in silico the HLA-DQ8 pocket (Fig. 1A and Supplementary Table 1); the RI-D-peptides were tested in vitro using a recombinant HLA-DQ8 protein in a unique immunoassay depicted in Supplemental Fig. 1A and 1B. Briefly, to first establish that our immunoassay detects significant binding of InsB:9-23 to HLA-DQ8 we incubated the recombinant HLA-DQ8 protein with biotinylated InsB:9-23 or biotinylated scrambled InsB:9-23 as a negative control. Using this immunoassay we detected significant binding of InsB:9-23 to the recombinant HLA-DQ8 protein compared to scrambled InsB:9-23 (Fig. 1C). Next, the in silico designed 14 RI-D-peptides and the unmodified/unlabelled InsB:9-23 (used to check the binding inhibition by L-InsB:9-23) were incubated with the HLADQ8-InsB:9-23 complex to evaluate whether they could block InsB:9-23 binding to HLA-DQ8; two peptides, GGREGCVLYLAEVGG, (designated RI-CT) and FFGGREGCVLYLAEVLHSG (designated RI-EXT) showed significant inhibition of InsB:9-23 binding to HLA-DQ8 (Fig. 1D). Furthermore, we tested the binding of RI-CT, RI-EXT, scrambled RI-CT and scrambled RI-EXT peptides alone to HLA-DQ8 recombinant protein by DELFIA showing a significant stronger binding of RI-CT and RI-EXT to HLA-DQ8 compared to scrambled peptides and compared to InsB:9-23 (Supplementary Fig. 1C). This is explained in part by the fact that RI-D-peptides have additional hydrogen bonds than corresponding L-peptides, and therefore they have higher affinity to HLA-DQ8 (changes in hydrogen bonds of RI, RI-CT, and RI-EXT peptides are shown in Supplementary Fig. 1D). In addition, for both RI-CT and RI-EXT peptides the inhibition of the InsB:9-23 binding to HLA-DQ8 was dose-dependent (Fig. 1E). Next, we tested the effect of RI-CT and RI-EXT in inhibiting other HLA-DQ8-binders such as GAD65 or gliadin. First we tested the binding of biotinylated GAD65 peptide or biotinylated gliadin peptide (and their scrambled versions) to our recombinant HLA-DQ8 molecule (Supplementary Table 1 and Supplementary Fig. 1E) and then the same binding in the presence of RI-CT and RI-EXT. Interestingly, the HLA-DQ8 blocking was specific to the selected corresponding L-peptide, indeed, RI-CT and RI-EXT competed only with InsB:9-23 and not with other HLA-DQ8-binders (Supplementary Fig. 1F).
3.3. RI-CT and RI-EXT block antigen presentation in vitro
To validate that the two RI-D-peptide hits (RI-CT and RI-EXT) could also block InsB:9-23 binding to HLA-DQ8 expressed on antigen presenting cells we used a cell-based in vitro assay. Briefly, we incubated BSM cells, a B-cell line homozygous for HLA-DQ8 [38], with biotinylated InsB:9-23 with or without unlabeled RI-CT and RI-EXT (Fig. 2A-D). To confirm HLA-DQ8 expression, BSM cells were incubated with PE-labeled anti-DQ8 antibody or PE-labeled IgG antibody as negative control; in addition, BSM cells were incubated with APC-labeled streptavidin for detection of biotinylated InsB:9-23 binding. Binding of InsB:9-23 peptide to HLA-DQ8 and inhibition by RI-D-peptides were evaluated by flow cytometry. The binding of InsB:9-23 to HLA-DQ8 positive BSM cells went from 2.5% to 84.8% without RI-D-peptides (Fig. 2A and B), whereas in the presence of RI-CT or RI-EXT the binding was reduced to 39.4 % and 25%, respectively (Fig. 2C and D). InsB:9-23 bound to HLA-DQ8 expressed on BSM cells in a dose-dependent manner (Fig. 2E). These results indicate that both RI-D-peptides can block the binding and presentation of InsB:9-23 within HLA-DQ8 to T-cells.
Fig. 2. RI-CT and RI-EXT inhibit the binding of InsB:9-23 to HLA-DQ8 expressed on BSM cells.
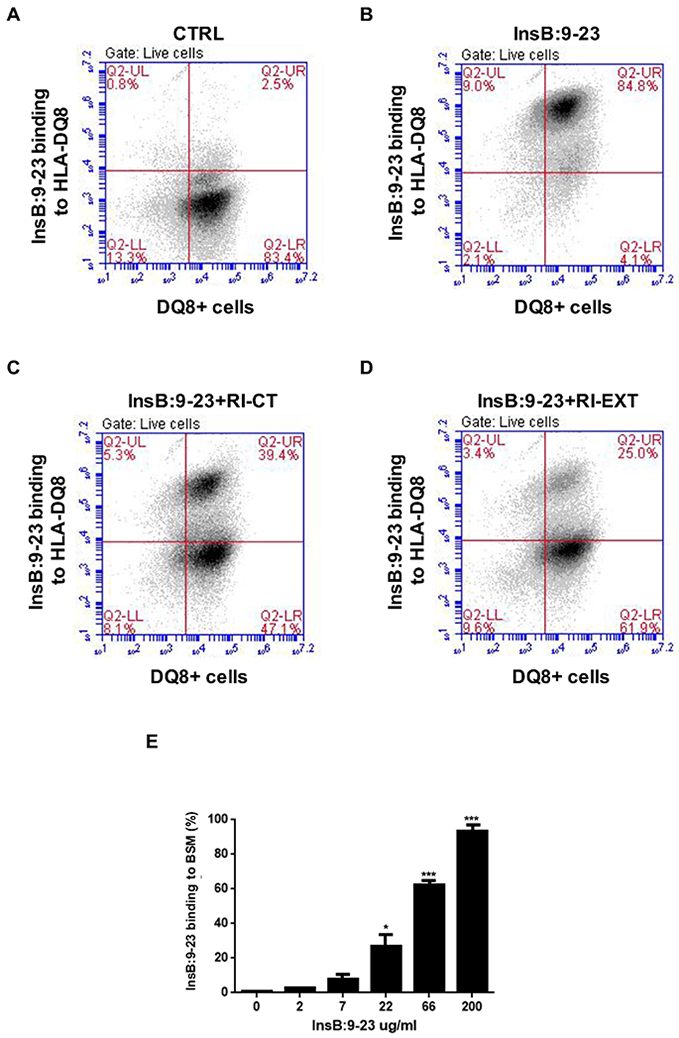
(A-E) HLA-DQ8 expression on BSM cells was evaluated using a PE-DQ8 antibody (PE-labeled IgG antibody was used as negative control), and APC-streptavidin was used to detect the peptide binding (cells double positive for PE and APC are shown in the upper-right quadrant). (A) and (B) BSM cells were loaded or not with InsB:9-23 in the presence of (C) and (D) RI-CT and RI-EXT. Inhibition of binding was analyzed by flow cytometry; cells were gated on live BSM cells. Both RI-D-peptides significantly blocked the binding between InsB:9-23 and HLA-DQ8 expressed on BSM cells. (E) Dose-dependent InsB:9-23 binding to HLA-DQ8 molecule. Bars represent mean ± SEM from 3 independent experiments. *p < 0.001; ***p < 0.001, by Student’s t-test.
3.4. RI-CT and RI-EXT block T-cell activation in vitro
RI-CT and RI-EXT were tested for their functional activity in inhibiting T-cell activation using an in vitro cellular antigen presentation assay. In this assay as T-cells we used a murine T-cell clone expressing human co-receptors and a human TCR specific for the InsB:9-23–DQ8 complex (5KC cells, kindly provided by Dr. Maki Nakayama, University of Colorado School of Medicine, Denver, CO), and as APCs cells we used BSM cells (Fig. 3A). When co-incubated with InsB:9-23 loaded BSM cells, the 5KC T-cell clone secreted IL-2 upon engaging the InsB:9-23–DQ8 complex (Fig. 3A and B); however, no IL-2 production was detected when BSM cells or 5KC cells were incubated alone with InsB:9-23 (Fig. 3B). As control peptides we used scrambled InsB:9-23 and gliadin peptides (sequence listed in Supplementary Table 1), and neither induced IL-2 secretion in 5KC cells (Supplementary Fig. 2A). Of note, although we showed that gliadin was able to bind the DQ8 expressed on BSM cells by flow (Supplementary Fig. 2B), gliadin did not activate 5KC cells to produce IL-2 (Supplementary Fig. 2A), confirming the rigorosity of our in vitro model. Furthermore, we observed that InsB:9-23 was able to activate 5KC cells in a dose-dependent manner (Supplementary Fig. 2C). Both RI-CT and RI-EXT significantly inhibited IL-2 production in our system, at concentrations of ≥100 μg/ml and ≥50 μg/ml, respectively, confirming their ability to block TCR engagement to the InsB:9-23–DQ8 complex (Fig. 3C). Moreover, we demonstrated that InsB:9-23-specific 5KC T-cells were not activated by RI-D-peptides alone demonstrating the specificity of our system (Fig. 3D). Therefore, both peptides were further tested ex vivo using murine models.
Fig. 3. RI-CT and RI-EXT inhibit the production of IL-2 in a cellular antigen presentation assay with BSM cells and 5KC cells.
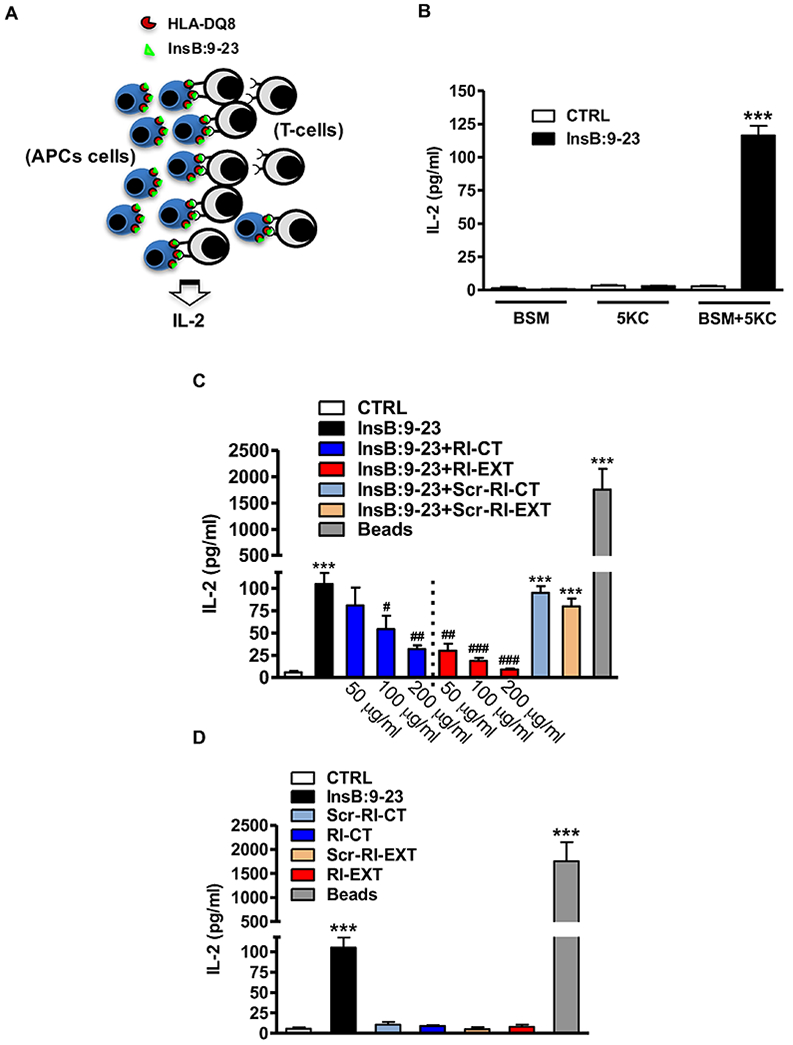
(A) BSM cell line (APCs) loaded with InsB:9-23 peptide and a murine T-cell clone (T-cells) expressing a human TCR specific for the InsB:9-23–DQ8 complex (5KC cells) were used to test functionally in vitro RI-CT and RI-EXT. (B) In our system, IL-2 was detected only when BSM cells and 5KC cells were incubated together with InsB:9-23. (C) Both RI-CT and RI-EXT inhibited significantly IL-2 production compared to untreated cells, starting at a concentration of 100 μg/ml and 50 μg/ml respectively. (D) RI-D-peptides alone or their scrambled versions did not induce IL-2 production. CD3/CD28 mouse beads were used as positive control; supernatants were analyzed by Luminex for IL-2. Bars represent mean ± SEM from 4 to 5 independent experiments. ***p < 0.001, by Student’s t-test for comparison of treated cells versus control cells. ###p < 0.001; ##p < 0.01; #p < 0.001 by Student’s t-test for comparison of RI-D-peptides treated cells versus InsB:9-23 treated cells.
3.5. RI-CT and RI-EXT block T-cell activation ex vivo
To test the effects of the RI-CT and RI-EXT RI-D-peptides ex vivo, we induced T-cell activation to the insulin peptide in SJL-DQ8 mice by immunizing them with InsB:9-23. The presence of the HLA-DQ8 transgene in SJL-DQ8 mice was verified by PCR as well as flow cytometry analysis (Fig. 4A and B). Moreover, we confirmed by flow cytometry that in lymphocytes isolated from SJL-DQ8 mice the HLA-DQ8 molecule was expressed mostly on APCs (Fig. 4C). Lymphocytes from immunized SJL-DQ8 mice were incubated with InsB:9-23 with or without RI-CT or RI-EXT. As negative control for lymphocytes stimulation by InsB:9-23 we stimulated the cells with scrambled InsB:9-23, whereas as negative control for the RI-D-peptides we used scrambled RI-CT and RI-EXT. Both RI-D-peptides significantly blocked IL-2 and IFN-γ production by lymphocytes isolated from InsB:9-23-immunized mice (Fig. 4D and 4E); scrambled RI-D-peptides had no significant effect in inhibiting cytokine production (Fig. 4D and 4E). In addition, lymphocyte proliferation (analyzed by the CFSE assay) was significantly suppressed by both RI-CT and RI-EXT (Fig. 4F and Supplementary Fig. 3A). Of note, using wild type SJL mice as a control mouse line, we demonstrated that InsB:9-23 is specifically presented to T-cells by the human HLA-DQ8; indeed, in WT SJL mice immunized with InsB:9-23 we did not detect significant T-cell activation when compared to SJL-DQ8 mice (Supplementary Fig. 3B and C). We thus set out to determine whether RI-CT and RI-EXT were able to inhibit specific T-cell populations making IL-2 and INF-γ. To do this, we used IL-2 and INF-γ ELISPOT plates and we incubated lymphocytes isolated from SJL-DQ8 mice immunized with InsB:9-23 with insulin peptide with or without RI-D-peptides. Importantly, the ELISPOT data agreed with the Luminex results: SJL-DQ8 mice immunized with InsB:9-23 elicited a strong ELISPOT response to the InsB:9-23 peptide and the response was significantly decreased with the addition of RI-CT or RI-EXT in the assay corroborating our previous ex vivo results (Fig. 5A and B).
Fig. 4. Ex vivo effect of RI-CT and RI-EXT.
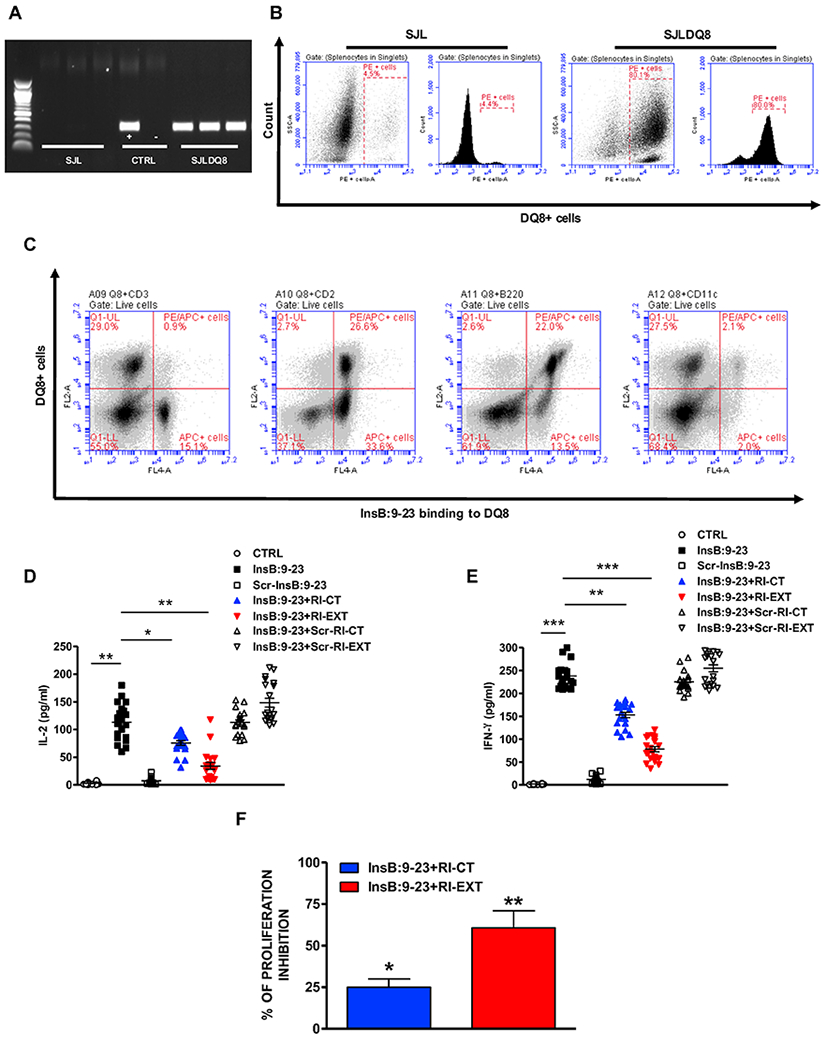
(A) and (B) PCR and flow analysis confirming the presence of the transgene in SJL-DQ8 mice compared to WT SJL mice. Cells were gated on live splenocytes in singlets. (C) Flow assay showing the expression of HLA-DQ8 molecule in lymphocytes isolated from SJL-DQ8 mice. HLA-DQ8 molecule is expressed mostly on APCs. Cells were gated on live splenocytes in singlets. (D-F) 20 SJL-DQ8 mice were immunized subcutaneously with InsB:9-23 in CFA on day 1 and in IFA on day 8. 9 days after the second immunization (day 17) mice were sacrificed. (D) and (E) Lymphocytes isolated from SJL-DQ8 mice were stimulated with InsB:9-23 or with scrambled InsB:9-23 as negative peptide and incubated with RI-CT or RI-EXT (scrambled RI-CT or RI-EXT were used as negative peptides). Supernatants were analyzed by Luminex for IL-2 and IFN-γ. (F) Inhibition of T-cell proliferation by RI-CT or RI-EXT was analyzed by the CFSE assay after stimulation of lymphocytes with InsB:9-23 with or without addition of RI-CT or RI-EXT. Cells were gated on live splenocytes in singlets. Both RI-D-peptides significantly decreased secretion of cytokines and proliferation induced by InsB:9-23. *p < 0.001; **p < 0.01; ***p < 0.001, by Student’s t-test.
Fig. 5. Effect of RI-CT and RI-EXT on InsB:9-23 specific T-cell responses measured by ELISPOT.
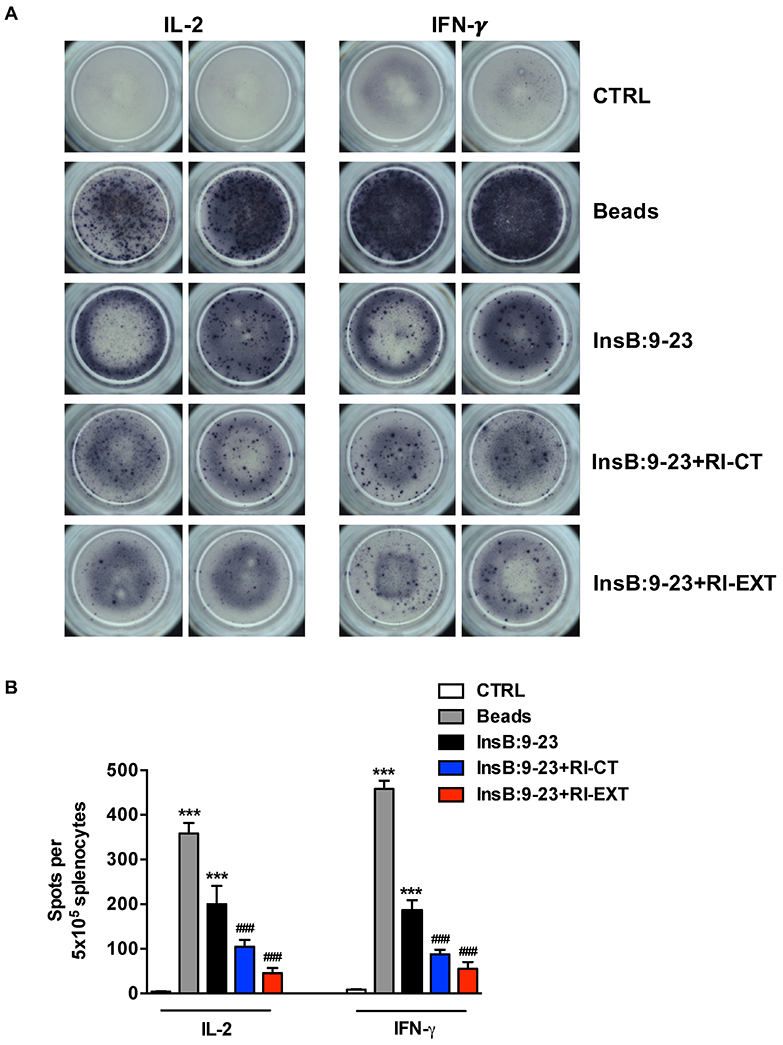
(A) and (B) ELISPOT assay of IL-2 and IFN-γ secretion by SJL-DQ8 mice immunized with 150 μg InsB:9-23. 5X105 lymphocytes from spleen and draining lymph node were restimulated without antigen (CTRL) or with InsB:9-23 peptide in the presence or absence of RI-D-peptides (RI-CT or RI-EXT), all at a dose of 20 μg/ml. DQ8-lymphocytes incubated with anti CD3-CD28 beads were used as positive control. Data shown in panel B depict the number of spots per 500,000 lymphocytes and represent the mean values and SEM for at least triplicate samples. ***p < 0.001, by Student’s t-test for comparison of beads or InsB:9-23 treated lymphocytes versus control lymphocytes. ###p < 0.001 by Student’s t-test for comparison of RI-D-peptides treated lymphocytes versus InsB:9-23 treated lymphocytes.
3.6. RI-CT and RI-EXT block T-cell activation in PBMCs isolated from recent-onset T1D patients.
To test the effects of RI-CT and RI-EXT on human T-cells from T1D patients we used an ex vivo functional mixed lymphocytes assay. Briefly, we isolated PBMCs from HLA-DQ8 positive T1D patients with no longer than 2-year duration (in order to maximize the likelihood that they retain strong T-cell responses to InsB:9-23) [39]; as controls we recruited T1D patients with longstanding disease that lost their T-cell responses to islet antigens [39]. Among 97 T1D patients recruited at the Pediatric Endocrine Clinic at Children Hospital at Montefiore and the Einstein-Mount Sinai Diabetes Research Center in NY, 52 were found to be HLA-DQ8 positive; the stimulation of PBMCs with InsB:9-23 induced a significant T-cell activation in 31 HLA-DQ8 positive T1D patients. As controls we used 17 HLA-DQ8 T1D patients with a disease duration longer than 10 years. PBMCs were isolated and stimulated with InsB:9-23 with or without RI-CT or RI-EXT; scrambled InsB:9-23, scrambled RI-CT, and scrambled RI-EXT were used as control peptides. Lymphocyte activation was assessed by their proliferative responses (using the CFSE assay) and by their cytokine production (using the Luminex assay). Both RI-CT and RI-EXT significantly suppressed pro-inflammatory cytokine production (IL-2 and IFN-γ) and lymphocytes proliferation, while scrambled RI-D-peptides had no effect (Fig. 6A-C and Supplementary Fig. 4). Taken together, our results indicate that these RI-D-peptides specifically inhibit InsB:9-23 presentation by HLA-DQ8 to T-cells in PBMCs cells from new-onset T1D patients.
Fig. 6. Effect of RI-CT and RI-EXT in PBMCs from new onset T1D patients.
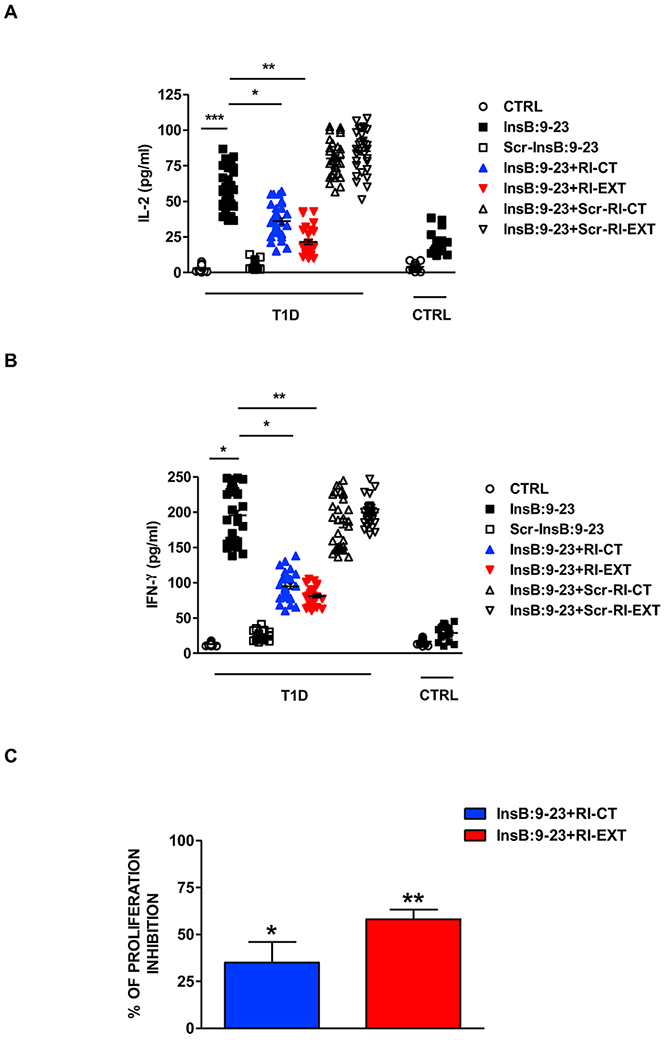
(A) and (B) PBMCs were isolated from 31 new onset DQ8 positive T1D patients and 17 DQ8 positive controls and were stimulated for 48h with InsB:9-23, with or without RI-CT and RI-EXT (scrambled InsB:9-23, scrambled RI-CT and scrambled RI-EXT were used as negative peptides). The production of IL-2 and IFN-γ were assessed by Luminex. RI-CT and RI-EXT significantly decreased T-cell activation induced by InsB:9-23, whereas scrambled RI-CT or RI-EXT had no effect. (C) Inhibition of T-cell proliferation by RI-CT or RI-EXT was analyzed by the CFSE assay after stimulation of human PBMCs with InsB:9-23 with or without addition of RI-CT or RI-EXT. PBMCs were gated on live T-cells for flow cytometry analysis. Both RI-D-peptides significantly decreased proliferation induced by InsB:9-23 in PBMCs isolated from DQ8-T1D patients. *p < 0.001; **p < 0.01; ***p < 0.001, by Student’s t-test.
3.7. RI-EXT blocks T-cell activation by InsB:9-23 in vivo
We tested in vivo the inhibitory effects of our most active RI-D-peptide, RI-EXT, using the SJL-DQ8 humanized mouse model of islet autoimmunity. We injected mice with RI-EXT (or scrambled RI-EXT as negative control peptide) IP at days 4, 7, 11, and 14 after immunizing them twice with InsB:9-23 on days 1 and 8 (Fig. 7A). Mice were sacrificed on day 17, spleens and draining lymph nodes were collected from mice upon sacrifice and lymphocyte were isolated and tested for their responses to InsB:9-23 or scrambled InsB:9-23. Treating mice IP with RI-EXT after immunizations significantly blocked the activation of T-cells by InsB:9-23. Instead, IP injections of scrambled RI-EXT peptide did not block T-cell activation in vivo (Fig. 7B and 7C). A representative structure of RI-EXT bound to the groove of HLA-DQ8 is shown in Fig. 7D.
Fig. 7. RI-EXT blocks activation of T-cells to InsB:9-23 in vivo.
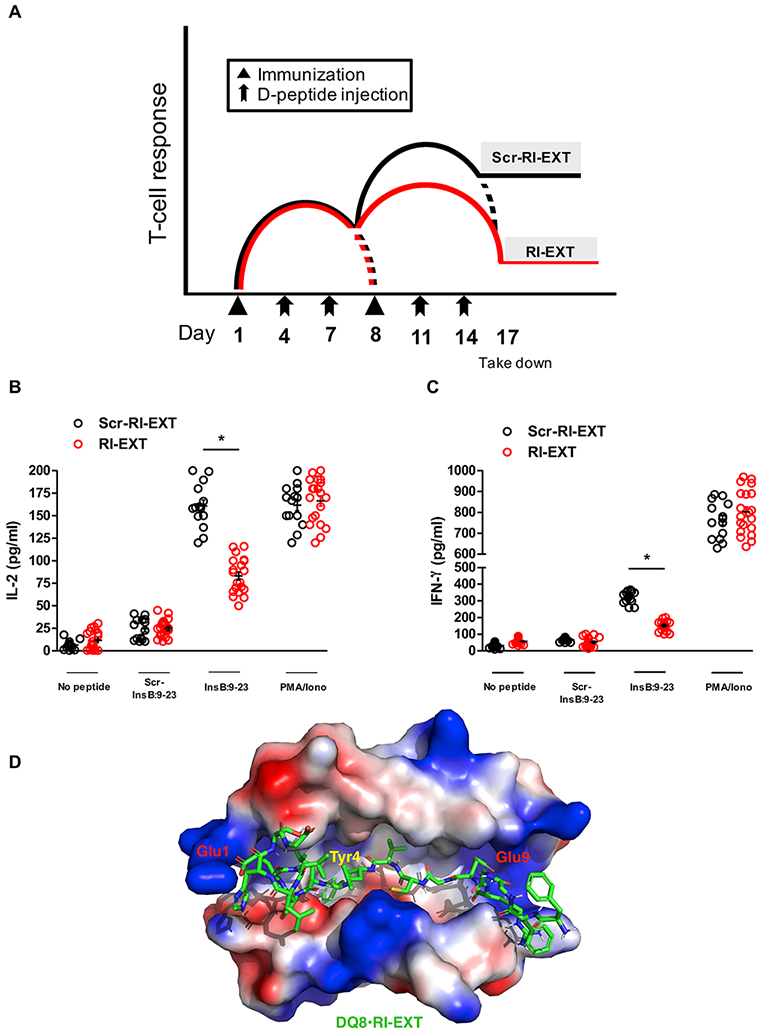
(A-C) SJL-DQ8 mice were immunized subcutaneously with InsB:9-23 in CFA on day 1 and with InsB:9-23 in IFA on day 8. At days 4, 7, 11 and 14 mice were injected with RI-EXT (20 mice) or scrambled RI-EXT as negative control peptide (14 mice). 9 days after the second immunization (day 17) mice were sacrificed. Lymphocytes isolated from SJL-DQ8 mice were stimulated with InsB:9-23 or with scrambled InsB:9-23 as negative peptide. PMA/Ionomycin was used as positive control. (B) and (C) Supernatants were analyzed by Luminex for IL-2 and IFN-γ. RI-EXT significantly blocked the InsB:9-23 induced activation of T-cells in InsB:9-23-immunized mice. There was no significant decrease of T-cell activation in response to scrambled RI-EXT. *p < 0.001, by Student’s t-test for comparison of RI-EXT versus scrambled RI-EXT treated mice. (D) Molecular model of the HLA-DQ8 binding cleft and RI-EXT; the critical residues Glu1, Tyr4 and Glu9 are highlighted.
4. Discussion
T1D is caused by autoimmune responses targeting pancreatic beta cells that lead to the killing and/or silencing of beta cells by various immune effectors mechanisms, including CD8+ cytotoxic T-cells, macrophages, and cytokines [40]. Therefore, the best approach to cure and/or prevent T1D will have to be based on immune modulation to suppress the immune effector mechanisms that target the beta cells. Several non-targeted immune therapies have been examined in pre-clinical and clinical testing, such as anti-CD3 MAb [41] and Rituximab [42], but while showing promise (especially anti-cD3 [43]) they cause generalized immunosuppression [44]. Importantly, in autoimmune diseases such as multiple sclerosis, some non-targeted immune therapies have caused very serious complications [45]. Therefore, new targeted or precision approaches to block the immune mediated beta cell destruction are needed. For the first time, we demonstrate that uniquely designed RI-D-peptides can block pathogenic TCR activation by self-antigen–MHC class II complexes providing a novel tool for preventing and potentially reversing T1D.
Other investigators have attempted to block antigen presentation in T1D using small compounds [23], but our study is the first to test antigen presentation blockade using RI-D-peptides. We used RI-D-peptides to block HLA-DQ8 for the following reasons: (1) their unique chemical properties enable them to bind to the HLA-DQ8 pocket with higher affinity compared to L-peptides because their backbone forms alternate hydrogen bonding with the HLA pocket (as depicted in Supplementary Fig. 1C and 1D); (2) RI-D-peptides are resistant to degradation by proteases and have a much longer half-life than L-peptides [46]; (3) the contact residues of RI-D-peptides with the TCR are not expected to activate it (Fig. 3D) but to block T-cell activation (as we have demonstrated in Figs. 3-7); (4) RI-D-peptides are much less likely to be antigenic in vivo compared to L-peptides and therapeutic antibodies [47]; (5) RI-D-peptides are manufactured chemically and their sequences can be easily engineered to improve stability, affinity, and pharmacokinetic profiles in order to maximize their effects [48]; and (6) if one RI-D-peptide is not sufficiently effective it is possible to produce (using the same approach we described in our study) a panel of RI-D-peptides that can be delivered together to target multiple epitopes. Thus, RI-D-peptides may have advantages over small molecules or therapeutic monoclonal antibodies [49]. Indeed, both small molecule drugs and antibodies targeting specific HLA molecules or peptide-HLA complexes have been used to inhibit HLA-specific antigen presentation but while these approaches showed promise they were unable to fully prevent the disease and had off-target effects [22, 23]. Of note, our RI-D-peptides are completely different than altered peptide ligands (APL) which are composed of L-amino acids and which have not been successful in previous studies [50].
We focused on blocking HLA-DQ8 in view of the well-known association of HLA class II, and particularly HLA-DQ8 with T1D [51-53]. These genetic associations reflect the key role that HLA class II proteins play in the etiology of T1D, specifically in the presentation of pathogenic islet peptides to CD4+ T-cells. Furthermore, we focused on blocking the InsB:9-23 peptide since it selectively binds to HLA-DQ8 and has been shown to be a major T-cell epitope in T1D [16, 54, 55]. Studies performed by Eisenbarth and colleagues [54, 56] and confirmed by others [57], have shown that the insulin peptide InsB:9-23 is a major peptide activating diabetogenic CD4+ T-cells that escaped tolerance, and triggering the autoimmune response against beta cells. Interestingly, while several GAD65 peptides have also been implicated in the etiology of T1D, transfer of anti-GAD65 T-cells does not transfer disease in mouse models suggesting that insulin peptides are the primary pathogenic epitopes [58]. Indeed, insulin antibodies are significantly associated with young age of onset of T1D, with the highest frequency seen in patients < 6-year-old [59], further supporting the notion that insulin-producing cells are the earliest target of immune response in T1D. Also, epidemiological studies have shown a significant association between the presence of insulin antibodies and HLA-DQ8, corroborating the key role played by HLA-DQ8 in the presentation of insulin peptides to CD4+ T-cells [59]. Furthermore, our results in hPBMCs highlight the functional importance of the InsB:9-23 – HLA-DQ8 complex in triggering T1D in DQ8 carrying individuals, and demonstrate that blocking this complex by RI-D-peptides may be an effective strategy to block the activation of diabetogenic T-cells in early onset T1D. Indeed, RI-EXT significantly inhibited T-cell islet peptide recall responses when incubated with PBMCs from recent-onset T1D patients with DQ8 haplotype (Fig. 6). It is important to consider the timing of therapeutic intervention with RI-D-peptides, we believe that patients at earlier stages of the disease would likely have greater clinical benefit due to a larger remaining beta cell reserve than patients at later stage of the disease.
The most potent RI-D-peptide we identified is RI-EXT: according to our evaluation of the interaction energies, the affinity to HLA-DQ8 of RI-EXT compared to RI-CT is better by about 0.8 kcal/mol. Analysis of the H-bonds in RI-EXT revealed that the flanking residues at both termini added stability through H-bonds (Supplementary Fig. 1D). In addition, the Phe at the C-terminus of the RI-EXT adds stabilization by Van der Waals interactions with the hydrophobic portions of HLA-DQ8 Lys75α. Notably, RI-EXT showed promising results both ex vivo and in vivo in our humanized mouse model carrying HLA-DQ8 (SJL-DQ8 mice). In this model, RI-EXT blocked InsB:9-23 peptide presentation by HLA-DQ8 and the consequent T-cell activation/proliferation (Fig. 4, 5 and 7). Treatment of DQ8-splenocytes and DQ8-hPBMCs isolated from T1D patients with RI-EXT significantly reduced lymphocytes activation and proliferation at a concentration of 20 μg/ml (or 10 μM) consistent with other studies using RI-D-peptide-based therapies [60].
Importantly, RI-D-peptides have shown promise as a novel approach to target different proteins in several diseases. For example, Uppalapati and colleagues have designed an effective RI-D-peptide antagonist of VEGF-A [61]. Similarly, RI-D-peptides have been designed to target: p53 as a potential therapy for certain cancers [62], amyloid-β peptide as a therapy for Alzheimer’s disease [63], c-Myc as inhibitors of proliferation [64], and α-synuclein as a therapeutic approach for Parkinson’s disease [65]. Moreover, RI-D-peptides have also been developed to block the expansion of encephalitogenic T-cells in vitro, corroborating the potential usefulness of these peptides for the treatment of autoimmune disease conditions via down-regulation of T-cell responses [66]. However, to the best of our knowledge our study is the first to use RI-D-peptides to target HLA class II molecules as a potential therapy for autoimmune diseases and in particular, to treat the underlying autoimmunity in T1D.
Our study has some limitations, for example, there are several potential concerns to using RI-D-peptides to block HLA class II in order to treat/prevent autoimmunity. First, the RI-D-peptides themselves might potentially amplify the autoimmune response. While this is a theoretical concern, it has been recently shown that retro inverso RI-D-peptides are significantly less immunogenic than the corresponding L-peptides [61]. This aspect is also supported by our results showing that RI-CT and RI-EXT did not activate TCR. Another concern might be that blocking only the HLA-DQ8-InsB:9-23 complex may not be effective since T-cells targeting other islet antigens and epitopes will not be blocked. Indeed, autoimmune responses (as well as normal immune responses) propagate by epitope spreading, and in T1D this would result in T-cell targeting of additional islet antigens and epitopes [67]. However, epitope spreading is the result of tissue damage and does not by itself trigger or perpetuate the autoimmune attack on the islets. In fact, Prasad and colleagues have shown that blocking InsB:9-23 can reverse the autoimmune response in NOD mice even after epitope spreading has begun [68]. In humans this concept is best exemplified in celiac disease where, as epitope spreading develops, the autoimmune response propagates from gliadin to tissue trans-glutaminase; yet removal of gliadin from the diet (equivalent to blocking T-cells recognizing gliadin) reverses the autoimmune responses against all antigens and epitopes, and results in complete remission even in late stages of the disease [69]. Similarly, blocking the dominant epitope in multiple sclerosis (MOG35-55 peptide) has been shown to reverse experimental autoimmune encephalitis even after epitope spreading has occurred and the disease established [70]. Taken together, these evidence strongly support our hypothesis that targeting the dominant epitope in T1D, i.e. InsB:9-23 peptide, and blocking its presentation to T-cells will halt the autoimmune responses even when other antigens such as GAD65, IA-2, and ZnT8 become targeted by epitope spreading [68]. Although our data and evidence summarized above suggest that blocking InsB:9-23 may be sufficient to prevent beta cell death in early stages of T1D, it is possible that targeting a single peptide may not be sufficient to revert T1D permanently, especially considering the heterogeneity of T1D in humans. Therefore, we are now expanding our work to test multiple RI-D-peptide antagonist in order to target several T-cell epitopes including those recognized by human islet-infiltrating T-cells, neo-epitopes, and insulin hybrid peptides [71].
5. Conclusions
In summary, we have developed a novel therapeutic approach to block the autoimmune response in T1D using RI-D-peptides targeting the HLA-DQ8–InsB:9-23 interaction. RI-D-peptides have been used in other contexts but this is the first study to actually show their efficacy in blocking autoantigen - HLA interaction. The main advantage of our therapeutic approach is its selectivity, since only T-cells recognizing pancreatic-specific peptides in T1D are targeted; moreover, RI-D-peptides could represent an unexplored antigen-specific tolerization approach in the context of T1D. Finally, our methodology can be easily expanded to patients with other autoimmune diseases, representing, therefore, a new strategy to treat autoimmunity in general.
Supplementary Material
Highlights.
There is no cure or prevention treatment for Type 1 Diabetes (T1D)
Insulin peptide InsB:9-23 and HLA-DQ8 molecule are key elements in T1D development
Our D-amino acid peptides block the binding between InsB:9-23 and HLA-DQ8
Our D-amino acid peptides block the presentation of InsB:9-23 to autoreactive T-cells
D-amino acid peptides could lead to innovative therapies for the treatment of T1D
Acknowledgements
We thank Dr. Maki Nakayama at the University of Colorado for generously providing us with the 5KC cells and Dr. Chella David from the Mayo Clinic for generously providing us with the humanized B6-DQ8 mice. We also thank Dr. Teresa DiLorenzo at Albert Einstein College of Medicine for helpful comments and advice and Dr. Tony Ng and Dr. Steven Porcelli (Albert Einstein College of Medicine) for helping us with ELISPOT assays.
Funding
This work was supported by the National Institute of Health (KL2TR002558 and part of 2P30DK020541-45 to A.L., and DK067555 to Y.T.), as well as by the Juvenile Diabetes Research Foundation (1-INO-2020-924-A-N to A.L.). None of the sources of funding have an interest in the subject matter or materials discussed in the submitted manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
Angela Lombardi, Yaron Tomer, and Roman Osman are listed as inventors on a patent disclosure relating to the compounds described in this manuscript. The other authors have declared that there is no duality of interest associated with their contribution to this manuscript.
References
- [1].Norris JM, Johnson RK, Stene LC. Type 1 diabetes-early life origins and changing epidemiology. Lancet Diabetes Endocrinol, 2020;8:226–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Herold KC, Vignali DA, Cooke A, Bluestone JA. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nature reviews Immunology, 2013;13:243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinology and metabolism clinics of North America, 2010;39:481–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. Jama, 2014;311:1778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barnett R. Type 1 diabetes. Lancet, 2018;391:195. [DOI] [PubMed] [Google Scholar]
- [6].Lombardi A, Tsomos E, Hammerstad SS, Tomer Y. Interferon alpha: The key trigger of type 1 diabetes. Journal of autoimmunity, 2018;94:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ahmed R, Omidian Z, Giwa A, Cornwell B, Majety N, Bell DR et al. A Public BCR Present in a Unique Dual-Receptor-Expressing Lymphocyte from Type 1 Diabetes Patients Encodes a Potent T Cell Autoantigen. Cell, 2019;177:1583–99 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sousa GR, Pober D, Galderisi A, Lv H, Yu L, Pereira AC et al. Glycemic Control, Cardiac Autoimmunity, and Long-Term Risk of Cardiovascular Disease in Type 1 Diabetes Mellitus. Circulation, 2019;139:730–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vinik AI, Nevoret ML, Casellini C, Parson H. Diabetic neuropathy. Endocrinology and metabolism clinics of North America, 2013;42:747–87. [DOI] [PubMed] [Google Scholar]
- [10].Wong TY, Cheung CM, Larsen M, Sharma S, Simo R. Diabetic retinopathy. Nature reviews Disease primers, 2016;2:16012. [DOI] [PubMed] [Google Scholar]
- [11].Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clinical journal of the American Society of Nephrology : CJASN, 2017;12:2032–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barbetti F, Taylor SI. Insulin: still a miracle after all these years. The Journal of clinical investigation, 2019;130:3045–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes, 2008;57:3169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Juvenile G Diabetes Research Foundation Continuous Glucose Monitoring Study. Prolonged nocturnal hypoglycemia is common during 12 months of continuous glucose monitoring in children and adults with type 1 diabetes. Diabetes care, 2010;33:1004–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Borus JS, Laffel L. Adherence challenges in the management of type 1 diabetes in adolescents: prevention and intervention. Curr Opin Pediatr, 2010;22:405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nature immunology, 2001;2:501–7. [DOI] [PubMed] [Google Scholar]
- [17].Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature, 2005;435:220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alleva DG, Crowe PD, Jin L, Kwok WW, Ling N, Gottschalk M et al. A disease-associated cellular immune response in type 1 diabetics to an immunodominant epitope of insulin. The Journal of clinical investigation, 2001;107:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yoshida K, Corper AL, Herro R, Jabri B, Wilson IA, Teyton L. The diabetogenic mouse MHC class II molecule I-Ag7 is endowed with a switch that modulates TCR affinity. The Journal of clinical investigation, 2010;120:1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes, 2017;66:722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan S, Li Y, Xia J, Jin CH, Hu Z, Duinkerken G et al. Type 1 diabetes induction in humanized mice. Proceedings of the National Academy of Sciences of the United States of America, 2017;114:10954–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW et al. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proceedings of the National Academy of Sciences of the United States of America, 2014;111:2656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ostrov DA, Alkanani A, McDaniel KA, Case S, Baschal EE, Pyle L et al. Methyldopa blocks MHC class II binding to disease-specific antigens in autoimmune diabetes. The Journal of clinical investigation, 2018;128:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug discovery today, 2015;20:122–8. [DOI] [PubMed] [Google Scholar]
- [25].Mezei M. Simulaid: a simulation facilitator and analysis program. Journal of computational chemistry, 2010;31:2658–68. [DOI] [PubMed] [Google Scholar]
- [26].Case DSCDA, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, Izadi S, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, Merz KM, Monard G, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM and Kollman AMBER PA 2017, University of California, San Francisco: . 2017. [Google Scholar]
- [27].Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol, 2007;148:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Broughton SE, Petersen J, Theodossis A, Scally SW, Loh KL, Thompson A et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity, 2012;37:611–21. [DOI] [PubMed] [Google Scholar]
- [29].White J, Pullen A, Choi K, Marrack P, Kappler JW. Antigen recognition properties of mutant V beta 3+ T cell receptors are consistent with an immunoglobulin-like structure for the receptor. J Exp Med, 1993;177:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].De Vitis S, Sonia Treglia A, Ulianich L, Turco S, Terrazzano G, Lombardi A et al. Tyr phosphatase-mediated P-ERK inhibition suppresses senescence in EIA + v-raf transformed cells, which, paradoxically, are apoptosis-protected in a MEK-dependent manner. Neoplasia, 2011;13:120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lombardi A, Gambardella J, Du XL, Sorriento D, Mauro M, Iaccarino G et al. Sirolimus induces depletion of intracellular calcium stores and mitochondrial dysfunction in pancreatic beta cells. Scientific reports, 2017;7:15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee HJ, Lombardi A, Stefan M, Li CW, Inabnet WB 3rd, Owen RP et al. CD40 Signaling in Graves Disease Is Mediated Through Canonical and Noncanonical Thyroidal Nuclear Factor kappaB Activation. Endocrinology, 2017;158:410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Blackard JT, Kong L, Lombardi A, Homann D, Hammerstad SS, Tomer Y. A preliminary analysis of hepatitis C virus in pancreatic islet cells. Virology journal, 2017;14:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kudva YC, Rajagopalan G, Raju R, Abraham RS, Smart M, Hanson J et al. Modulation of insulitis and type 1 diabetes by transgenic HLA-DR3 and DQ8 in NOD mice lacking endogenous MHC class II. Hum Immunol, 2002;63:987–99. [DOI] [PubMed] [Google Scholar]
- [35].Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H et al. Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology, 2008;149:3461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Johnson AJ, Kennedy SC, Ng TW, Porcelli SA. Identification of Novel Mycobacterial Targets for Murine CD4(+) T-Cells by IFNgamma ELISPOT. Methods Mol Biol, 2018;1808:143–50. [DOI] [PubMed] [Google Scholar]
- [37].Sengar DP, Goldstein R. Comprehensive typing of DQB1 alleles by PCR-RFLP. Tissue Antigens, 1994;43:242–8. [DOI] [PubMed] [Google Scholar]
- [38].Turner TR, Hayhurst JD, Hayward DR, Bultitude WP, Barker DJ, Robinson J et al. Single molecule real-time DNA sequencing of HLA genes at ultra-high resolution from 126 International HLA and Immunogenetics Workshop cell lines. Hla, 2018;91:88–101. [DOI] [PubMed] [Google Scholar]
- [39].Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med, 2012;209:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet, 2014;383:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. The New England journal of medicine, 2002;346:1692–8. [DOI] [PubMed] [Google Scholar]
- [42].Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. The New England journal of medicine, 2009;361:2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. The New England journal of medicine, 2019;381:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Michels AW, Eisenbarth GS. Immune intervention in type 1 diabetes. Seminars in immunology, 2011;23:214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. The New England journal of medicine, 2012;366:1870–80. [DOI] [PubMed] [Google Scholar]
- [46].Liu M, Li X, Xie Z, Xie C, Zhan C, Hu X et al. D-Peptides as Recognition Molecules and Therapeutic Agents. Chemical record, 2016;16:1772–86. [DOI] [PubMed] [Google Scholar]
- [47].Teyton L. The saga of MHC-bound peptides: a renaissance for antigen presentation? The Journal of clinical investigation, 2007;117:3164–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug discovery today, 2010;15:40–56. [DOI] [PubMed] [Google Scholar]
- [49].Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chemical biology & drug design, 2013;81:136–47. [DOI] [PubMed] [Google Scholar]
- [50].Spear TT, Wang Y, Smith TW Jr., Simms PE, Garrett-Mayer E, Hellman LM et al. Altered Peptide Ligands Impact the Diversity of Polyfunctional Phenotypes in T Cell Receptor Gene-Modified T Cells. Mol Ther, 2018;26:996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Onengut-Gumuscu S, Concannon P. The genetics of type 1 diabetes: lessons learned and future challenges. Journal of autoimmunity, 2005;25 Suppl:34–9. [DOI] [PubMed] [Google Scholar]
- [52].Jahromi MM, Eisenbarth GS. Genetic determinants of type 1 diabetes across populations. Ann N Y Acad Sci, 2006;1079:289–99. [DOI] [PubMed] [Google Scholar]
- [53].Cucca F, Dudbridge F, Loddo M, Mulargia AP, Lampis R, Angius E et al. The HLA-DPB1--associated component of the IDDM1 and its relationship to the major loci HLA-DQB1, -DQA1, and -DRB1. Diabetes, 2001;50:1200–5. [DOI] [PubMed] [Google Scholar]
- [54].Nakayama M, Beilke JN, Jasinski JM, Kobayashi M, Miao D, Li M et al. Priming and effector dependence on insulin B:9-23 peptide in NOD islet autoimmunity. The Journal of clinical investigation, 2007;117:1835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wucherpfennig KW. Insights into autoimmunity gained from structural analysis of MHC-peptide complexes. Curr Opin Immunol, 2001;13:650–6. [DOI] [PubMed] [Google Scholar]
- [56].Eisenbarth GS, Moriyama H, Robles DT, Liu E, Yu L, Babu S et al. Insulin autoimmunity: prediction/precipitation/prevention type 1A diabetes. Autoimmun Rev, 2002;1:139–45. [DOI] [PubMed] [Google Scholar]
- [57].Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. The Journal of clinical investigation, 2004;114:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yu B, Gauthier L, Hausmann DH, Wucherpfennig KW. Binding of conserved islet peptides by human and murine MHC class II molecules associated with susceptibility to type I diabetes. Eur J Immunol, 2000;30:2497–506. [DOI] [PubMed] [Google Scholar]
- [59].Graham J, Hagopian WA, Kockum I, Li LS, Sanjeevi CB, Lowe RM et al. Genetic effects on age-dependent onset and islet cell autoantibody markers in type 1 diabetes. Diabetes, 2002;51:1346–55. [DOI] [PubMed] [Google Scholar]
- [60].Rosener NS, Gremer L, Reinartz E, Konig A, Brener O, Heise H et al. A d-enantiomeric peptide interferes with heteroassociation of amyloid-beta oligomers and prion protein. The Journal of biological chemistry, 2018;293:15748–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Uppalapati M, Lee DJ, Mandal K, Li H, Miranda LP, Lowitz J et al. A Potent d-Protein Antagonist of VEGF-A is Nonimmunogenic, Metabolically Stable, and Longer-Circulating in Vivo. ACS chemical biology, 2016;11:1058–65. [DOI] [PubMed] [Google Scholar]
- [62].Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proceedings of the National Academy of Sciences of the United States of America, 2010;107:14321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gregori M, Taylor M, Salvati E, Re F, Mancini S, Balducci C et al. Retro-inverso peptide inhibitor nanoparticles as potent inhibitors of aggregation of the Alzheimer's Abeta peptide. Nanomedicine : nanotechnology, biology, and medicine, 2017;13:723–32. [DOI] [PubMed] [Google Scholar]
- [64].Pescarolo MP, Bagnasco L, Malacarne D, Melchiori A, Valente P, Millo E et al. A retro-inverso peptide homologous to helix 1 of c-Myc is a potent and specific inhibitor of proliferation in different cellular systems. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 2001;15:31–3. [DOI] [PubMed] [Google Scholar]
- [65].Shaltiel-Karyo R, Frenkel-Pinter M, Egoz-Matia N, Frydman-Marom A, Shalev DE, Segal D et al. Inhibiting alpha-synuclein oligomerization by stable cell-penetrating beta-synuclein fragments recovers phenotype of Parkinson's disease model flies. PloS one, 2010;5:e13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Srinivasan M, Wardrop RM, Gienapp IE, Stuckman SS, Whitacre CC, Kaumaya PT. A retro-inverso peptide mimic of CD28 encompassing the MYPPPY motif adopts a polyproline type II helix and inhibits encephalitogenic T cells in vitro. Journal of immunology, 2001;167:578–85. [DOI] [PubMed] [Google Scholar]
- [67].Sohnlein P, Muller M, Syren K, Hartmann U, Bohm BO, Meinck HM et al. Epitope spreading and a varying but not disease-specific GAD65 antibody response in Type I diabetes. The Childhood Diabetes in Finland Study Group. Diabetologia, 2000;43:210–7. [DOI] [PubMed] [Google Scholar]
- [68].Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. Journal of autoimmunity, 2012;39:347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Green PH, Cellier C. Celiac disease. The New England journal of medicine, 2007;357:1731–43. [DOI] [PubMed] [Google Scholar]
- [70].Ji N, Somanaboeina A, Dixit A, Kawamura K, Hayward NJ, Self C et al. Small molecule inhibitor of antigen binding and presentation by HLA-DR2b as a therapeutic strategy for the treatment of multiple sclerosis. Journal of immunology, 2013;191:5074–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nakayama M, Michels AW. Determining Antigen Specificity of Human Islet Infiltrating T Cells in Type 1 Diabetes. Frontiers in immunology, 2019;10:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.