

Abstract
Here we present a new series of hydrazide-bearing class I selective HDAC inhibitors designed based on panobinostat. The cap, linker, and zinc-binding group were derivatized to improve HDAC affinity and anti-leukemia efficacy. Lead inhibitor 13a shows picomolar or low nanomolar IC50 against HDAC1 and HDAC3 and exhibits differential toxicity profiles toward multiple cancer cells with different FLT3 and p53 status. 13a indirectly inhibits the FLT3 signaling pathway and down-regulates the master anti-apoptotic proteins, resulting in the activation of pro-caspase3 in in wt-p53 FLT3-ITD MV4–11 cells. While in the wt-FLT3 and p53-null cells, 13a is incapable of causing apoptosis at therapeutic concentration. The MDM2 antagonist and the proteasome inhibitor promote 13a-triggered apoptosis by preventing p53 degradation. Furthermore, we demonstrate that apoptosis rather than autophagy is the key contributing factor for 13a-triggered cell death. When compared to panobinostat, 13a is not mutagenic and displays superior in vivo bioavailability and higher AUC0-inf.
Graphical Abstract
Introduction
Acute myelogenous leukemia (AML) is characterized by the uncontrolled proliferation and survival of immature malignant myeloid cells in parallel with the concurrent loss of normal hematopoiesis.1–2 The standard anti-AML therapies since 1973 are based on cytotoxic chemotherapy using antimetabolites such as cytarabine (ara-C), and the DNA intercalating anthracyclines such as daunorubicin or idarubicin.3 Although a series of targeted drugs including FLT3 inhibitors, IDH2 inhibitors, and Bcl-2 inhibitors have been approved for the treatment of AML, their uses limit specific patient population and undergo a high possibility of clonal resistance.4–5 Three-quarters of all AML patients are > 60 years of age, only less than 10% of them achieve disease-free survival greater than 5 years.5 With an increase in life expectancy in the U.S., AML cases are expected to become more prevalent, and there is a need for more effective and better-tolerated therapies.6
Unlike chronic myelogenous leukemia (CML), which is characterized by a more uniform genetic abnormality and a reciprocal translocation of the BCR and ABL genes,7 AML has various cytogenetic abnormalities and mutations, such as FLT3, NPM1, c-kit tyrosine kinase and Ras mutations.8–15 These constitutively active kinases initiate multiple pro-growth and pro-survival signaling through the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), signal transducer and activator of transcription 5 (STAT5), and PI3K/Akt kinase family mediated pathways and confer poor prognosis in AML.8–9, 16–19 These genetic aberrations are not mutually exclusive and commonly coexist in AML cells.20 Thus, the biggest challenge is to develop pharmacologic agents that possess significant specificity, yet are capable of attenuating multiple oncogenic signals in AML.
Class I HDACs play a crucial role in the transformation and survival of myeloid and lymphoid malignancies.21–23 Inhibition or co-depletion of HDACs 1 and 2 elicits pro-apoptotic responses in leukemia.22 HDAC3 activity is required for the initiation of leukemogenesis in acute leukemia.24 Relevant to cancer therapy, HDAC3 depletion or inhibition significantly reduces proliferation and promotes differentiation in leukemia.22 In our previous study, we demonstrated that our HDAC1, 2, and 3 selective inhibitors cause apoptosis in the AML cell line MV4–11, and displayed low nano-molar EC50, which suggests that class I HDACs 1, 2, and 3 are potential molecular targets for the treatment of AML.25–26
The mechanisms of HDACIs lethality against leukemia and other cancer types can be elucidated as follows: 1) HDACIs activate the endogenous cyclin-dependent kinase (CDK) inhibitor p2127 and disrupt cell cycle (especially mitotic spindle assembly) checkpoints;28–29 2) HDACIs activate both the intrinsic (mitochondrial) and extrinsic (death receptor-mediated) pathways of apoptosis by down-regulating the anti-apoptotic proteins such as X-linked inhibitor of apoptosis (XIAP) and cellular FLICE-like inhibitory protein (c-FLIP),30–32 while up-regulating the pro-apoptotic proteins (Bim, Bmf and Noxa) through acetylation of p5333–34 and inducing Bid cleavage;35 3) induction of autophagy by HDACIs through acetylation of the autophagy signaling component including Atg336 and regulation of mammalian target of rapamycin (mTOR) pathway.37
The actions of HDACIs in cancer cells reveal that in addition to epigenetic modifications, HDACs also control cell proliferation, differentiation, migration, and death by modification of non-histone proteins.38 The tumor suppressor p53 is the first characterized example of non-histone protein acetylation.39 It plays an important role in cellular signaling and stress responses and can either positively or negatively regulate apoptosis, cell cycle arrest, and autophagy.40 P53 regulates apoptosis through control of transcription of pro-apoptotic members of the Bcl-2 family, including Bax, Puma, Noxa, and Bid.41 P53 transcriptionally activates the endogenous CDK inhibitor p21, which can in turn inhibit cyclin E(A)/CDK2 and preserve the association of the tumor suppressor retinoblastoma protein.42 Additionally, damage-regulated autophagy modulator (DRAM) that modulates autophagosome formation is also activated by p53.37, 43
Four HDACIs have been approved by the FDA: vorinostat,44 romidepsin,45 belinostat46 and panobinostat,47 among these, panobinostat is the most potent HDACI in vitro and in vivo. Structural regions and pharmacophore models of HDACIs contain three critical parts: a cap group (interacts with the surface of the enzyme), a linker (occupies the long hydrophobic tunnel), and a zinc-binding group (ZBG, functioning within the catalytic site).48–49 Although hydroxamic acid is the most commonly used ZBG, it has disadvantages in its structural instability, lack of isoform selectivity, and the hydroxamic acid ZBG is mutagenic. Hydrazide-based ZBG-HDACIs have emerged in recent years in exploring new ZBGs of HDACIs that confirms to the physiological substrate geometric.25–26, 50–51 As a new motif for HDACIs, its potency, structural stability, and off-target toxicity all need to be addressed. In our previous studies, we have demonstrated HDAC inhibitory activity and superior stability of the hydrazide ZBG.25–26 However, the potential genotoxicity is a remains an important question. In fact, the hydrazine/hydrazide motif was extensively used in the past in anti-depressants as well as first line antituberculosis agents today best characterized by isocarboxazid and isoniazid for chronic diseases.52–54 Therefore, the hydrazide motif is expected to be better tolerated in vivo. Here we will describe and discuss the discovery of hydrazide-bearing HDACIs based on the structure of panobinostat with the comparative chemical structural studies, detailed p53 and FLT3 dependent anti-tumor mechanisms of action studies in AML cells, and preliminary mutagenic and pharmacokinetic studies.
Results and discussion
Chemistry
Scheme 1 showed the synthesis of intermediates 6a-6c, 10a, and 10b. Phenylhydrazine (1) and 5-chloropentan-2-one (2) afford 2-(2-methyl-1H-indol-3-yl)ethanamine (3). 3 was mixed with aldehyde 4a-4c, NaBH3CN and CH3COOH in methanol to give 5a-5c, respectively. Boc-protection of 5a-5c got 6a-6c. Similarly, compound 3, 8a, and 8b were reacted with benzaldehyde, then NaBH3CN was added to obtain 9a and 9b, respectively, which also converted to Boc-protected product 10a and 10b.
Scheme 1.
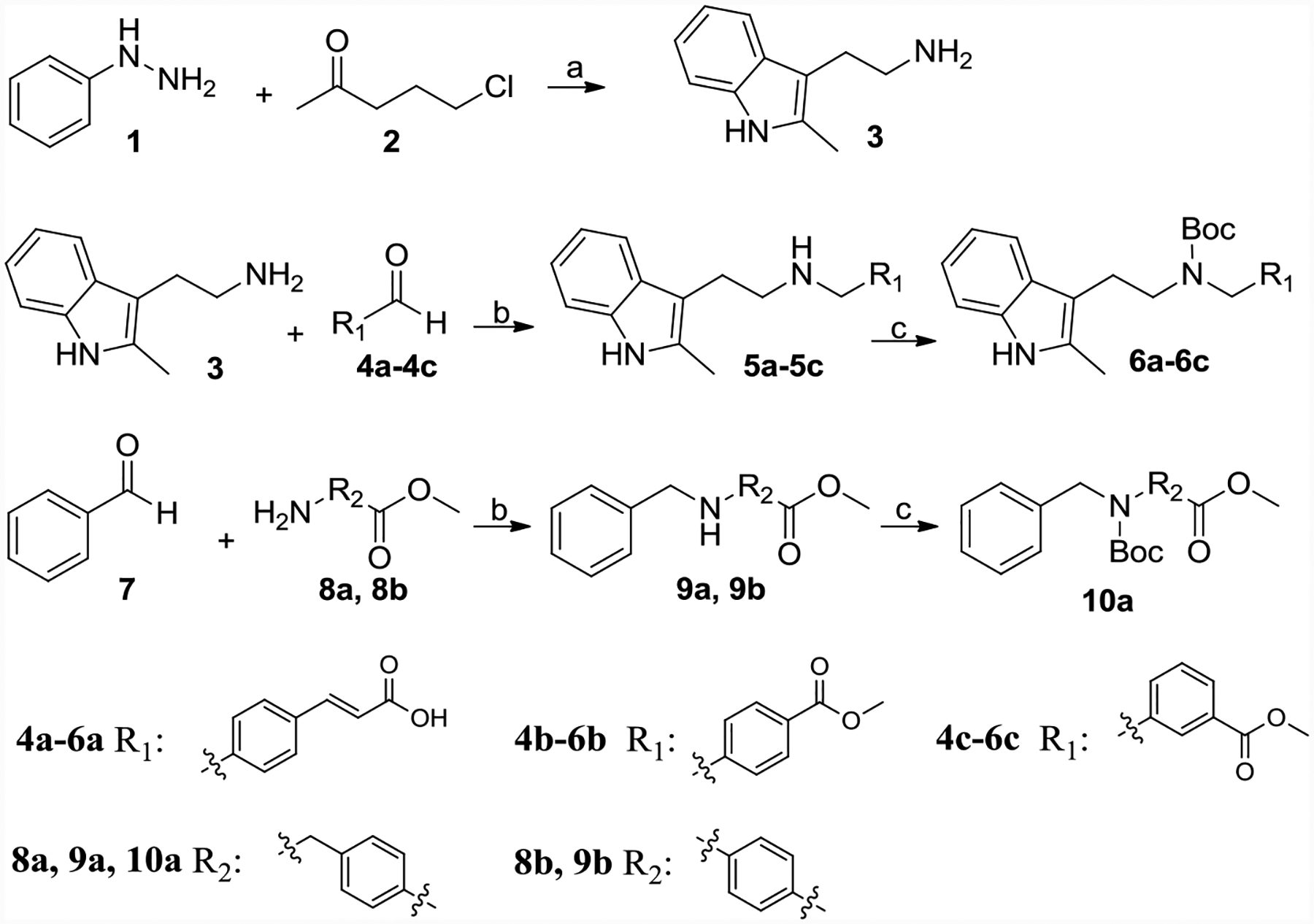
Synthesis of 3, 6a-6c, 10a and 10b.
Reagents and conditions: (a) Ethanol, NaOH, reflux, 47%; (b) NaBH3CN, CH3COOH, methanol, 70–80% yield; (c) Boc2O, TEA, DCM, 80–85% yield; (d) Propionaldehyde or benzaldehyde, NaBH3CN, HCl, Ethanol, 60–70%.
Synthesis of product 13a-13d was shown in Scheme 2. 6a connected with hydrazine monohydrate by TBTU-mediated amide formation afforded 11. 11 was reacted with propionaldehyde or N-Boc-2-aminoacetaldehyde to get Schiff base, which was suffered by NaBH3CN gave reductant product 12a or 12b, respectively. 12c and 12d were achieved by reaction of 11 with acrylonitrile or methyl acrylate in ethanol. Boc de-protection of 12a-12d leading end products 13a-13d, respectively.
Scheme 2.
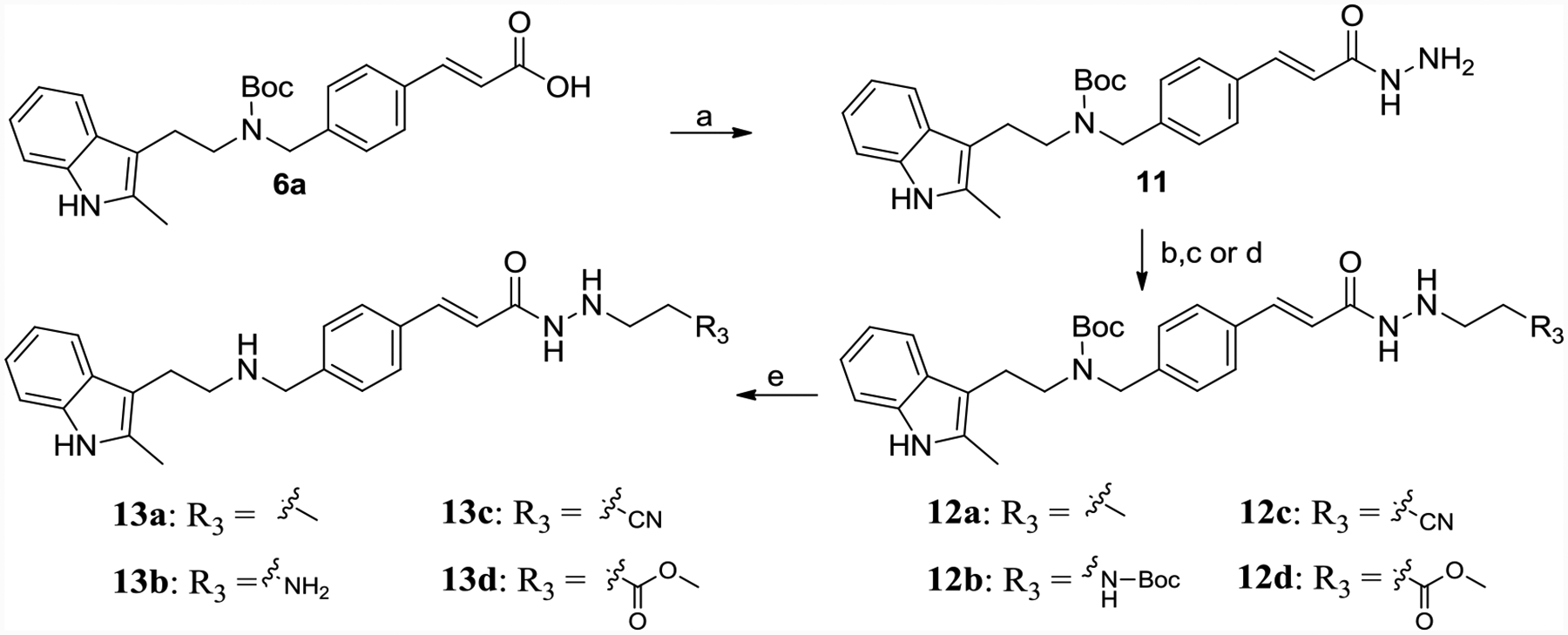
Synthesis of 13a-13d.
Reagents and conditions: (a) hydrazine monohydrate, TBTU, TEA, DMF, yield 50%; (b) propionaldehyde or N-Boc-2-aminoacetaldehyde, MgSO4, ethanol, yield 60–70%; (c) NaBH3CN, HCl, methanol, H2O, methyl orange, yield 60–70%; (d) acrylonitrile or methyl acrylate, ethanol, refluxed, yield 40–50%; (e) TFA, DCM, yield 60%.
6b, 6c, and 10a were refluxed with hydrazine monohydrate to get 14a-14c, which was reacted with propionaldehyde in the presence of magnesium sulfate followed by a reduction with sodium cyanoborohydride in acidified methanol to give the desired 15a-15c (Scheme 3). Boc de-protection by TFA afforded 16a-16c.
Scheme 3.
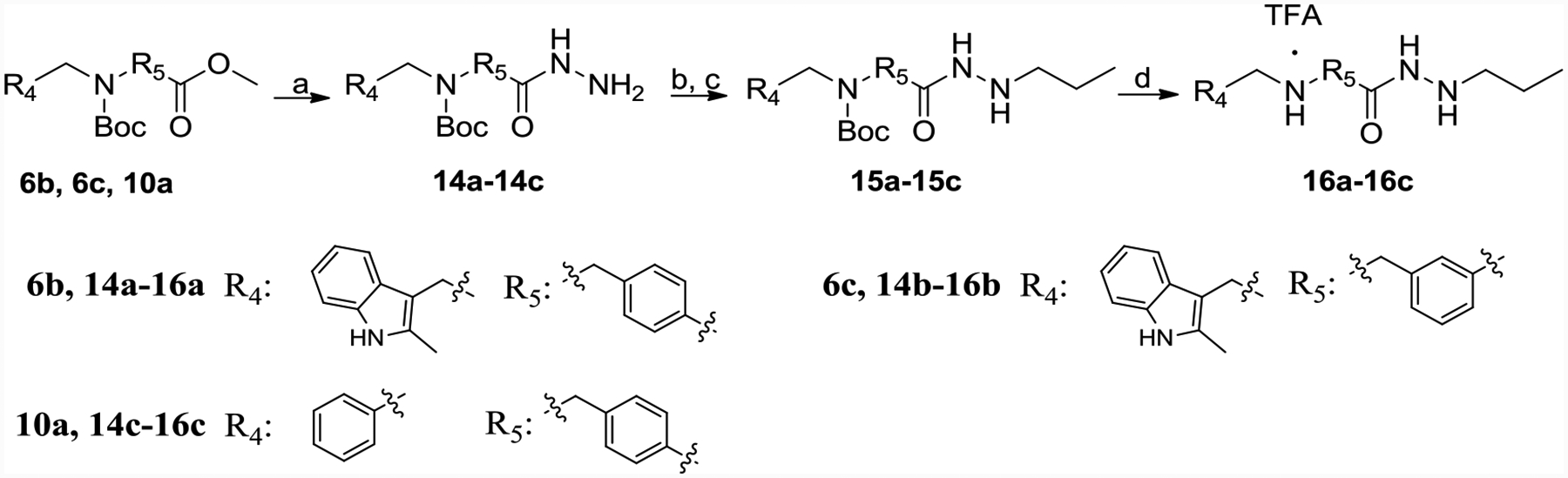
Synthesis of 16a-16c.
Reagents and conditions: (a) hydrazine monohydrate, methanol, reflux, yield 90–95%; (b) propionaldehyde, MgSO4, ethanol, yield 60–70%; (c) NaBH3CN, HCl, methanol, H2O, methyl orange, yield 60–70%; (d) TFA, DCM, yield 60–70%.
Scheme 4 described the synthesis of 24a-24c. 17 connected with 18 got amide 19, 20 connected with 21 by Mitsunobu reaction afforded ether 22. 23a, 23b, and 23c, the hydrazine product of 9b, 19 and 22 respectively, were treated with propionaldehyde followed by NaBH3CN to get end product 24a, 24b, and 24c.
Scheme 4.
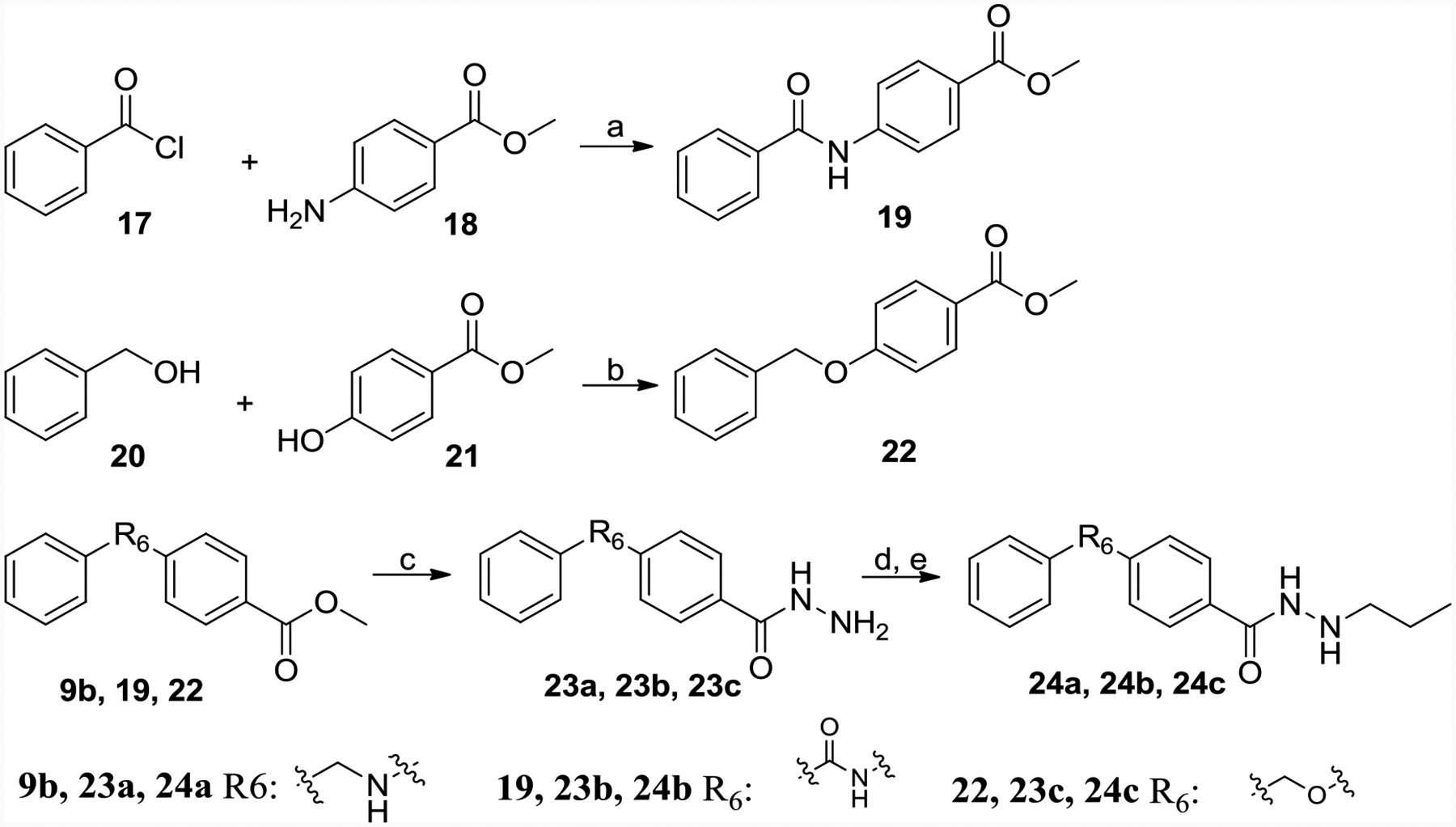
Synthesis of 24a-24c.
Reagents and conditions: (a) DCM, TEA, DMF, yield 80%; (b) PPh3, DEAD, anhydrous THF, yield 53%; (c) hydrazine monohydrate, methanol, reflux, yield 95%; (d) propionaldehyde, MgSO4, ethanol, yield 60–70%; (e) NaBH3CN, HCl, methanol, H2O, methyl orange, yield 60–70%.
6a connected with 1,1-dimethylhydrazine, pyrrolidin-1-amine, piperidin-1-amine or pyrazolidine by TBTU-mediated amide formation to get intermediates 25a-25c, respectively, which suffered TFA to get rid of the Boc group achieved 26a-26c (Scheme 5). 12a reacted with ethanal, propanal, n-butanal, n-pentanal or n-hexanal, followed by the addition of NaBH3CN provided tertiary amine 27a-27e. The end products 28a-28e were achieved by Boc deprotection (Scheme 5).
Scheme 5.
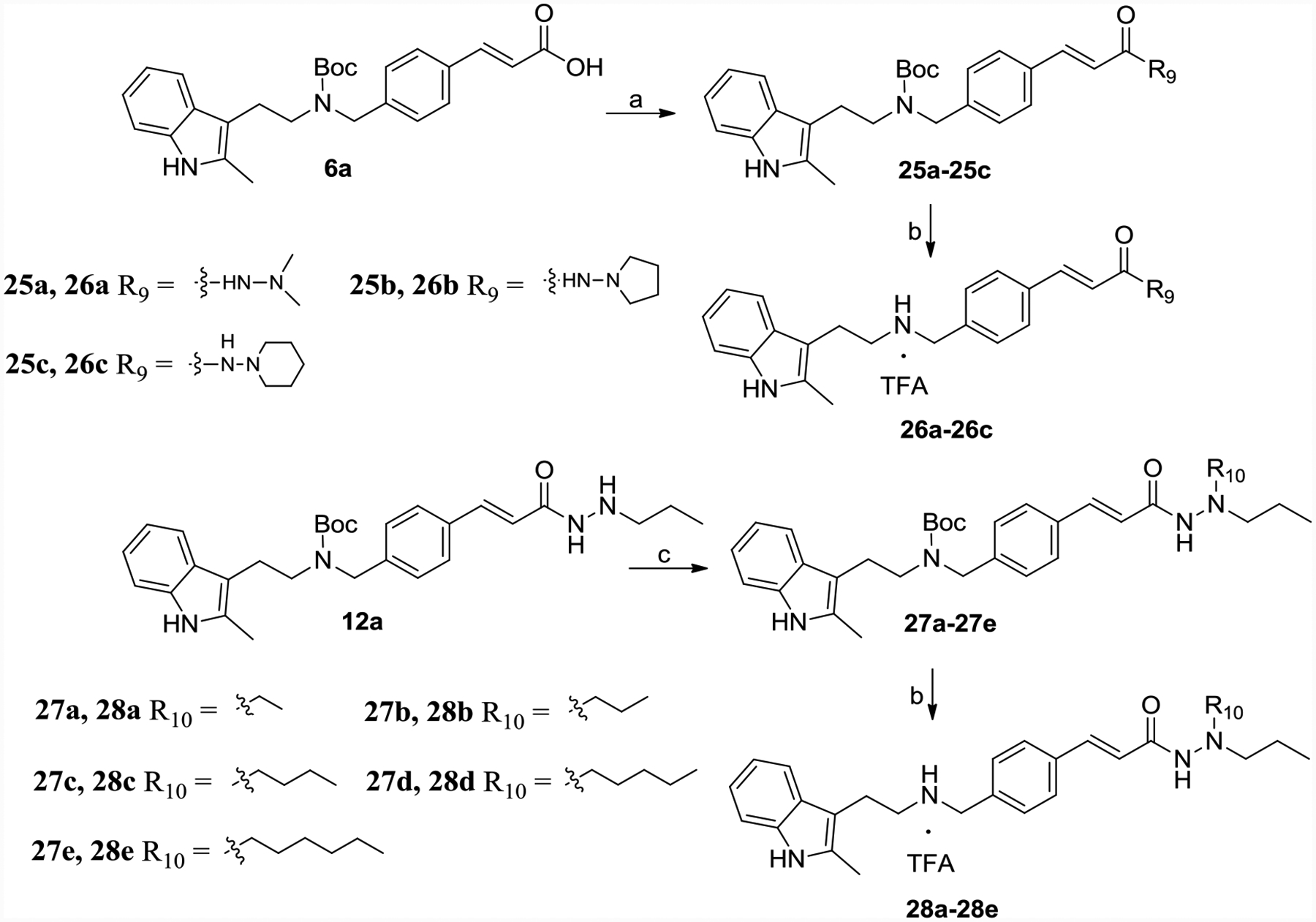
Synthesis of 26a-26c, 28a-28e.
Reagents and conditions: (a) 1,1-dimethylhydrazine, pyrrolidin-1-amine, piperidin-1-amine or pyrazolidine, TBTU, TEA, DMF, yield 50–60%; (b) TFA, DCM, yield 60–65%. (c) ethanal, propanal, n-butanal, n-pentanal, n-hexanal, NaBH3CN, CH3COOH, methanol, yield 50–60%.
Intermediate 11 reacted with 3-Bromo-1-propanol to get the propanol di-substituent product 29, which was treated with TFA to afford 30 (Scheme 6).
Scheme 6.
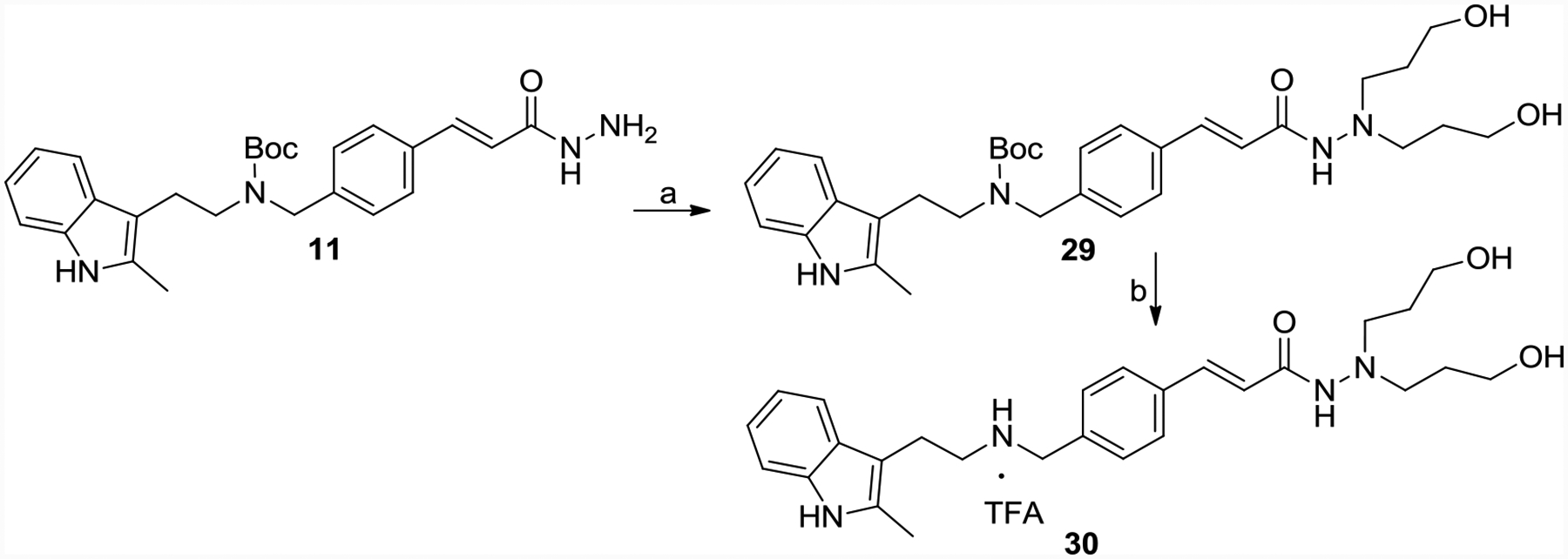
Synthesis of 30.
Reagents and conditions: (a) 3-Bromo-1-propanol, K2CO3, ethanol, reflux, yield 70%;(b) TFA, DCM, yield 60%.
Compounds design
Panobinostat is a highly potent hydroxamic acid-bearing pan-HDACI among all of the clinical HDACIs with low nano-molar IC50s against HDACs and cancer cells but with limited selectivity. Although hydroxamic acid is the most commonly used ZBG for HDACIs, it shows limitations in isoform-selectivity, which is hypothesized to cause greater side effects than more selective HDACIs. Further, hydroxamic acid also suffers from reduction, hydrolysis, and glucuronidation in vivo, leading to the rapid inactivation of HDACIs.55 This structural instability is the main reason for the poor pharmacokinetic profiles (short t1/2 and low Cmax and bioavailability) of clinical hydroxamate HDACIs.56–59 Therefore, the exploration of new ZBG motifs for HDACIs holds great interest. Initially, Wang and colleagues demonstrated a novel hydrazide-based HDACI with class I selectivity.51 We have shown further that the hydrazide motif as a potential ZBG displayed allosteric inhibition kinetics and was impervious to glucuronidation,25–26 indicating that hydrazide could be a preferable ZBG fragment with improved selectivity and in vivo stability. Most recently, Son et al. also demonstrated hydrazide with long chain can be used for the design of HDAC11 selective inhibitor.50
Therefore, we modify the hydroxamic acid group of panobinostat using hydrazide motifs. It should be noted that among all of the hydrazide-based HDAC inhibitor studies, only one single N-substituent at the hydrazide tail was explored.25–26, 50–51 Here in this study, we first design compounds with di-N- substituent at the hydrazide tail (Figure 1, R4 group). As modifications in the cap and linker groups can also generate changes in enzymatic activity and selectivity, we also incorporate different fragments in the cap and linker (Figure 1). These newly designed compounds are expected to have the following improvements: 1) selectivity against class I HDACs; 2) similar potency toward class I HDACs and anti-leukemia activity compared to the parent molecule panobinostat; 3) less toxicity and improved in vivo PK profiles.
Figure 1.
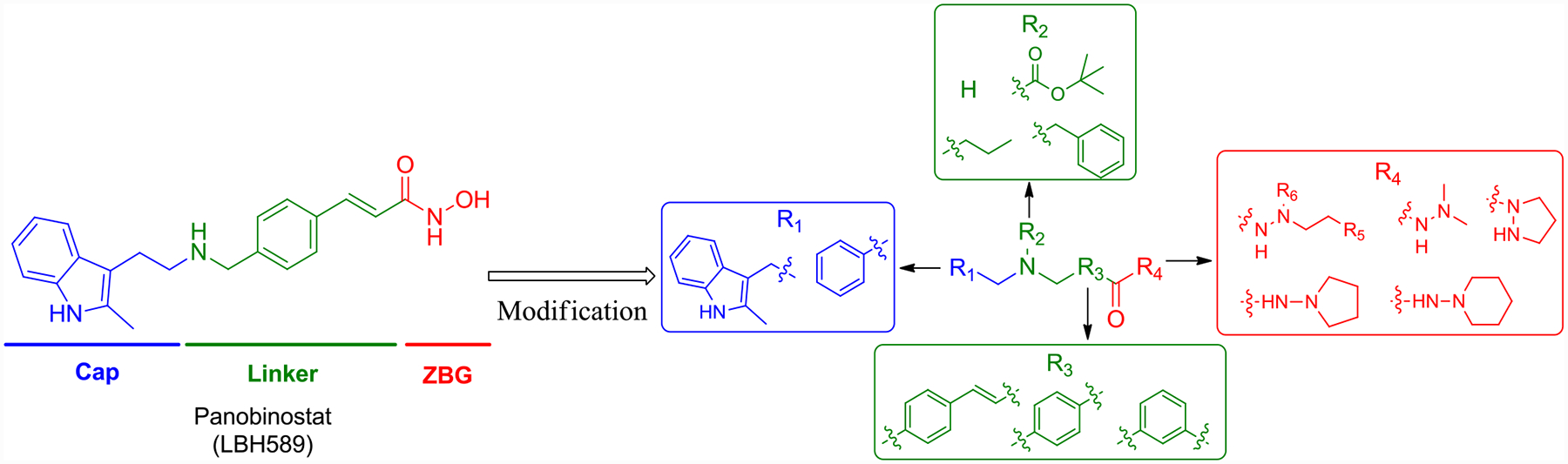
Design of hydrazide-bearing HDACIs based on the structure of panobinostat.
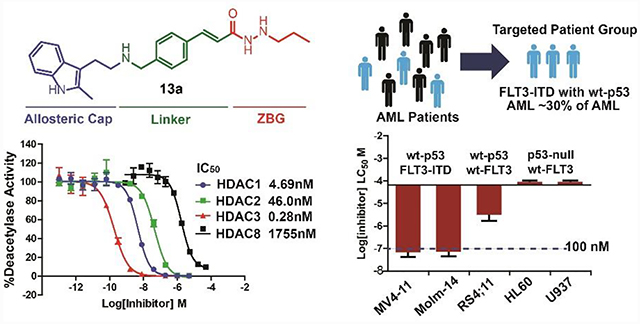
In our previous studies, hydrazide compounds with a 3 carbon on the hydrazide motif exhibited excellent HDAC inhibitory activity.25–26 Therefore, we initiated this study by designing compounds in series 1 (Table 1) with N’-propylacetohydrazide as their ZBG and with modifications of panobinostat in the cap and linker groups. In detail, direct modification of hydroximic acid of panobinostat with N’-propylacetohydrazide affords 13a. While keeping the cap group and ZBG unchanged, substituting the linker group of 13a with 1-ethyl-4-methylbenzene and 1-ethyl-3-methylbenzene results in 16a and 16b, respectively; modifying both cap and linker of 13a affords 16c, 24a, 24b, and 24c. Compounds 12a, 15a, 15b, and 15c are the Boc-containing structures of 13a, 16a, 16b, and 16c, respectively. Recombinant HDACs 1, 2, 3, and 6 are used to screen the newly designed compounds. IC50 of 13a for HDAC1, 2, and 3 are 5.17, 49.5 and 0.28 nM, respectively, while they do not show inhibition against HDAC6 up to 100,000 nM. This indicates that modification of panobinostat by replacing hydroxamic acid with N’-propylacetohydrazide maintains its inhibitory activity toward class I HDACs, as well as acquired selectivity between class I HDACs and HDAC6. The IC50 of 13a is much lower than that of 16a and 16b; therefore, (E)-1-ethyl-4-(prop-1-en-1-yl)benzene of 13a is the optimized linker. Compound 16a with 2-methyl-3-propyl-1H-indole as cap group is much more potent than 16c with an ethylbenzene cap. Compounds 24a, 24b, and 24c with modifications in both cap and linker groups all show weaker activity than 13a. Compounds 12a, 15a, 15b, and 15c with a Boc group exhibit reduced (12a, 15b, 15c) or equivalent (15a) activity when compared with their relative compounds; therefore, a branched aliphatic cap group may not improve the HDAC inhibitory activity of compounds.
Table 1.
Structure IC50 of compounds in series 1 against HDAC1, 2, 3 and 6.
| Compound No. | R1 | IC50 (nM)a | |||
|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | ||
| 12a | 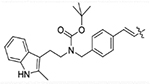 |
180.9 | 1298 | 16.76 | >50,000 |
| 13a | 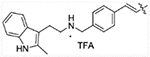 |
5.17 | 49.5 | 0.28 | >50,000 |
| 15a | 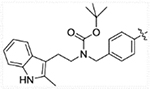 |
28.84 | 278.6 | 12.59 | >50,000 |
| 16a | 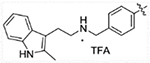 |
29.43 | 107.5 | 19.94 | >50,000 |
| 15b | 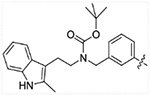 |
2524 | > 5000 | > 5000 | >50,000 |
| 16b | 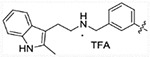 |
1674 | 4942 | 673.9 | >50,000 |
| 15c | 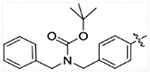 |
216.5 | 579.3 | 78.14 | >50,000 |
| 16c | 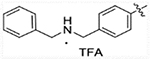 |
87.98 | 441.3 | 96.71 | >50,000 |
| 24a | 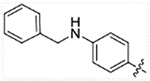 |
15.8 | 67.48 | 6.12 | >50,000 |
| 24b | 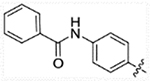 |
93.05 | 474.1 | 48.75 | >50,000 |
| 24c | 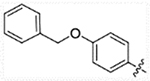 |
56.32 | 174.6 | 24.82 | >50,000 |
| Panobinostat | 2.5 | 13.2 | 2.1 | 10.5 | |
| SAHA | 43.76 | 89.23 | 144.7 | 78.3 | |
Values are average of three independent determinations, the SD values are < 20% of the mean.
As HDACs 1, 2, and 3 also have significant differences in surface charges and anchor interactions with the binding site, we further modified the hydrazide anchor (R3) group of 13a with motifs of varying electronegativity (13c and 13d) and ionization 13b (Table 2). HDAC inhibitory activity shows that the newly designed 13b-13d all display less potency and selectivity when compare with 13a. We then designed compounds with di-N-substituted and cyclic hydrazide groups (26a-26c, 28a-28e, 30) based on the ZBG space size difference between HDACs 1, 2, and 3 which are expected to achieve improved activity and selectivity these compounds. IC50s of compounds with cyclic hydrazide (26b-26c) are all > 2,000 nM, which indicates cyclized hydrazide is a weak ZBG. Although some of the di-N-substituted compounds (28b and 28e) display selectivity for HDAC3 to some extent, their activity is at least 100 times lower than N’-propylacetohydrazide 13a. Therefore, HDACIs with di-N-substituted and cyclized hydrazide groups all have much lower potency than those with a 3-carbon linker chain mono-substitution hydrazide. To the best of our knowledge, it is the first time we discuss the structure-activity relationship (SAR) study of di-N-substituted hydrazide based HDACIs. These conclusions are important in the design of hydrazide HDACIs.
Table 2.
Inhibition of HDAC1, 2, 3, and 6 for Compounds in series 2.
| R2 | R3 | IC50 (nM)a | ||||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC6 | |||
| 13a | H | 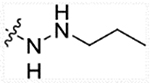 |
5.17 | 49.5 | 0.28 | >50,000 |
| 13b | H | 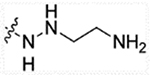 |
9842 | >10000 | 5241 | >50,000 |
| 13c | H | 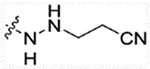 |
58.12 | 172 | 34.82 | >50,000 |
| 13d | H | 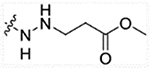 |
1089 | 4214 | 309.7 | >50,000 |
| 26a | H | 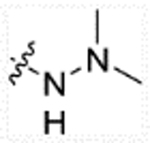 |
11000 | 12930 | 996.4 | >50,000 |
| 26b | H | 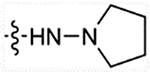 |
8599 | >10000 | 3091 | >50,000 |
| 26c | H | 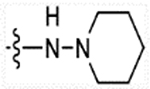 |
>10000 | >10000 | >10000 | >50,000 |
| 28a | H | 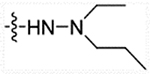 |
2925 | 8071 | 550.1 | >50,000 |
| 28b | H | 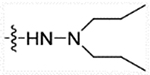 |
412.8 | 1366 | 25.19 | >50,000 |
| 28c | H | 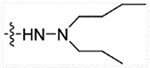 |
209.7 | 3222 | 229.4 | >50,000 |
| 28d | H | 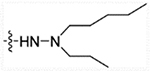 |
381.5 | 7316 | 321.1 | >50,000 |
| 28e | H | 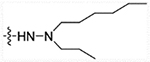 |
2880 | 9835 | 143.2 | >50,000 |
| 30 | H | 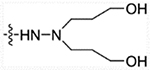 |
>10000 | >10000 | >10000 | >50,000 |
Values are average of three independent determinations, the SD values are < 20% of the mean.
MV4–11 is widely used for the biological evaluation of drugs targeting AML.60–62 What’s more, in our previous study, MV4–11 cells showed high sensitivity to our hydrazide HDACIs.26 Therefore, we chose the MV4–11 cell line to screen the antiproliferative activity of representative compounds 13a, 16a, 16b, and 16c with different linker- and cap-structures, plus the HDAC3 selective compound 28b (Table 3). Class I selective inhibitors entinostat, as well as pan-inhibitors vorinostat and panobinostat, were chosen as positive controls. Results show that compounds 13a, 16a, 16b, and 16c have similar selective profiles. 13a has the strongest enzyme inhibitory activity and antiproliferative activity with an EC50 of 15.35 nM, which is much lower than that of vorinostat and entinostat and comparable to that of panobinostat. HDACIs 26a, 26b, 26c, and 30, with little HDAC inhibitory activity, also do not affect cell proliferation at 50 μM.
Table 3.
EC50 of representative compounds.
| Cpd No. | EC50a (nM) of MV4–11 |
|---|---|
| 13a | 15.35±2.22 |
| 16a | 81.64±5.29 |
| 16b | 7696±1002 |
| 16c | 172.6±38.75 |
| 28b | 1054±240.6 |
| Vorinostat | 418.1±72.27 |
| Entinostat | 806.2±68.72 |
| Panobinostat | 5.2±1.87 |
EC50 values were shown as the mean of three experiments ± the standard error of the mean.
13a is slow-on/slow-off class I inhibitor
Enzymatic and cell-based assays indicate that 13a is the most potent HDACI among all of the compounds we designed. Thus, we chose 13a for further biological study. To assess the HDAC isozymes selectivity of 13a, we determined its IC50 for the other Zn2+-dependent HDACs (HDAC4, 5, 7, 8, and 9), and compared it with pan-HDAC inhibitor panobinostat, as well as class I selective inhibitor entinostat (Table 4). As there are no appropriate substrates for HDAC10 and HDAC11, inhibitory activity toward these two isotypes was not determined.63 IC50s of 13a for class I HDAC1, 2, and 3 are 4.69, 46.0, and 0.28 nM, respectively, which is comparable to those of panobinostat. Compound 13a displays micro-molar IC50 (1.75 μM) for HDAC8 and does not inhibit (up to 10 μM) HDAC4, 5, 6, 7, or 9; therefore, 13a is an HDAC1, 2, and 3 selective inhibitor. Although panobinostat shows nanomolar IC50 for all types of HDACs, its EC50 for MV4–11 cells (5.2 nM) is close to that of class I HDAC selective HDACI 13a (15.35 nM). This indicates that 13a achieves HDAC isoform selectivity while maintaining potent anti-leukemia activity. We also find that 13a does not show any inhibition to two metalloproteases, APN, and MMP (supporting information Figure S4), and trypsin used for the HDAC assay. Therefore, the anti-tumor activity of 13a comes from its inhibition toward HDACs rather than non-specific metalloprotease inhibition.
Table 4.
IC50 of 13a against HDAC1–9.
| HDAC Classes | HDAC isotypes | IC50a of 13a (nM) | IC50 of Panobinostat (nM)65 | IC50a of Entinostat (nM) |
|---|---|---|---|---|
| Class I | HDAC1 | 4.69±1.28 | 2.5 | 53.89±5.29 |
| HDAC2 | 46.0±5.39 | 13.2 | 108.2±8.28 | |
| HDAC3 | 0.28±0.12 | 2.1 | 77.18±6.30 | |
| HDAC8 | 1755±110.2 | 277 | >10,000 | |
| Class IIa | HDAC4 | >10,000 | 203 | >10,000 |
| HDAC5 | >10,000 | 7.8 | >10,000 | |
| HDAC7 | >10,000 | 531 | >10,000 | |
| HDAC9 | >10,000 | 5.7 | >10,000 | |
| Class IIb | HDAC6 | >10,000 | 10.5 | >10,000 |
IC50 values were shown as the mean of at least three experiments ± the standard error of the mean.
Enzyme kinetic studies were also performed for HDACs 1 and 3 (Figure 2A and B). It is interesting to note that 13a displays a non-competitive enzyme inhibition for HDAC1 and a mixed mode of inhibition for HDAC3. To better understand the potential binding modes and patterns between HDAC1 and HDAC3, we tested 13a in four conditions each by probing the competitive site (CS) and the allosteric site (AS) of HDAC1 and HDAC3 using computational docking. As shown using the free-energy calculation in the docking simulations (Figure 2C), there is very large discrimination between the high affinity AS for 13a and the very low-affinity CS of 13a in HDAC1, in silico binding mode also shows 13a fails to chelate due to the geometric constraint of the indole cap group preventing ZBG chelation with the Zn metal ion at the CS (supporting information Figure S6). This might be the reason that 13a exhibits the non-competitive inhibition against HDAC1. For HDAC3, 13a has a high binding affinity for both AS and CS sites based on free-energy calculation (Figure 2C). Potential binding mode of 13a with HDAC3 shows in the catalytic site, the top five docking poses all display a reasonable distance between ZBG and Zn metal ion (Figure 2D). These results are consistent with the kinetic studies that 13a inhibits HDAC1 non-competitively, and 13a has a mixed inhibition mode due to its interaction with both AS and CS of HDAC3.
Figure 2A and B:
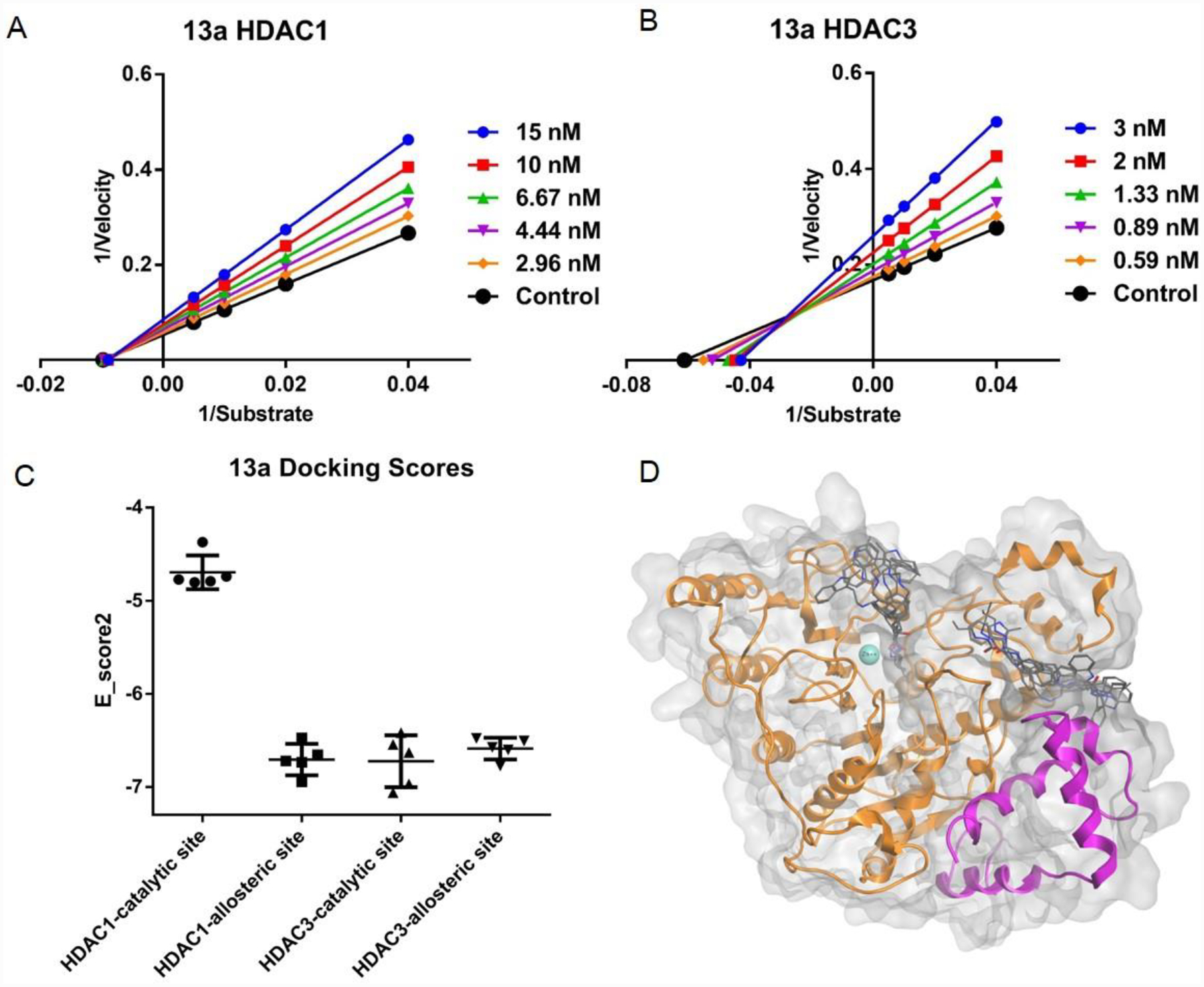
Lineweaver-Burke plots of enzyme kinetics data in the presence of inhibitors. Y-axes units: (pmoles acetylated substrate cleaved/min)−1, Xaxes units: (μmoles)−1. Compound 13a for HDAC1, and 3, respectively. Intersection on Xaxes are indicative of non-competative inhibition (HDAC1, 3A), while intersections in 2nd quadrant are indicative of mixed and competitive inhibition (HDAC3, 3B). Representative plots of n ≥ 3 experiments. 2C. Molecular docking probe of the catalytic and allosteric binding pockets of HDAC1 (PDB: 5ICN) and HDAC3 (PDB: 4A69). Data is represented as whisker plot using the top 5 docking poses for each condition. The catalytic site was defined as the catalytic pocket that incorporated the Zn metal iron, and the allosteric site was defined as the allosteric pocket created by the interface between the HDAC heterodimer. 2D. Potential binding mode of 13a with catalytic site and allosteric site of HDAC3 in silico. Top 5 docking poses for each site are displayed.
Inhibition of HDACs by hydrazide compound 13a is time-dependent (slow-on) as concentration-response curves shift and IC50 values change with enzyme-inhibitor pre-incubation time (Figure 3A). For example, the IC50 of 13a for HDAC3 following 90 min pre-incubation is 0.17 nM, which is 3 times lower than IC50 measured after 10 min pre-incubation. However, panobinostat reaches a steady-state within 10 min pre-incubation, indicating classical fast-on binding (Figure 3A). We also studied the release profile of 13a and panobinostat by western blot. MV4–11 cells were treated with 13a or panobinostat for 3 h. The drugs were then washed out and the cells cultured for 30 min, 1 h, 3 h, and 6 h without drugs. The t = 0 min time point was collected immediately after 3 h incubation with drugs. Histone acetylation caused by 13a does not decrease within 6 h (Figure 3B). Acetylation of histone H3 caused by panobinostat begins to decrease after 1 h, but persisted through 6 h (Figure 3B). Therefore, 13a and panobinostat both display slow-off release kinetics for class I HDACs. Interestingly, the acetylation of tubulin caused by panobinostat completely disappears after 30 min (Figure 3B). Inhibition of HDAC6 leads to tubulin acetylation, so panobinostat exhibits fast-off release kinetics against HDAC6, which is different from the class I HDACs. 13a does not increase the acetylation of tubulin (Figure 3B), which is in agreement with its isoform selectivity. Although our HDAC inhibitors display slow-off and likely irreversible inhibition, the potent inhibition of HDACs are likely required because non-substituted hydrazine and hydrazides with bulky, branched, and long-chain acyl-groups are significantly less effective at inhibiting HDACs and cancer cell proliferation.25–26, 64
Figure 3A:
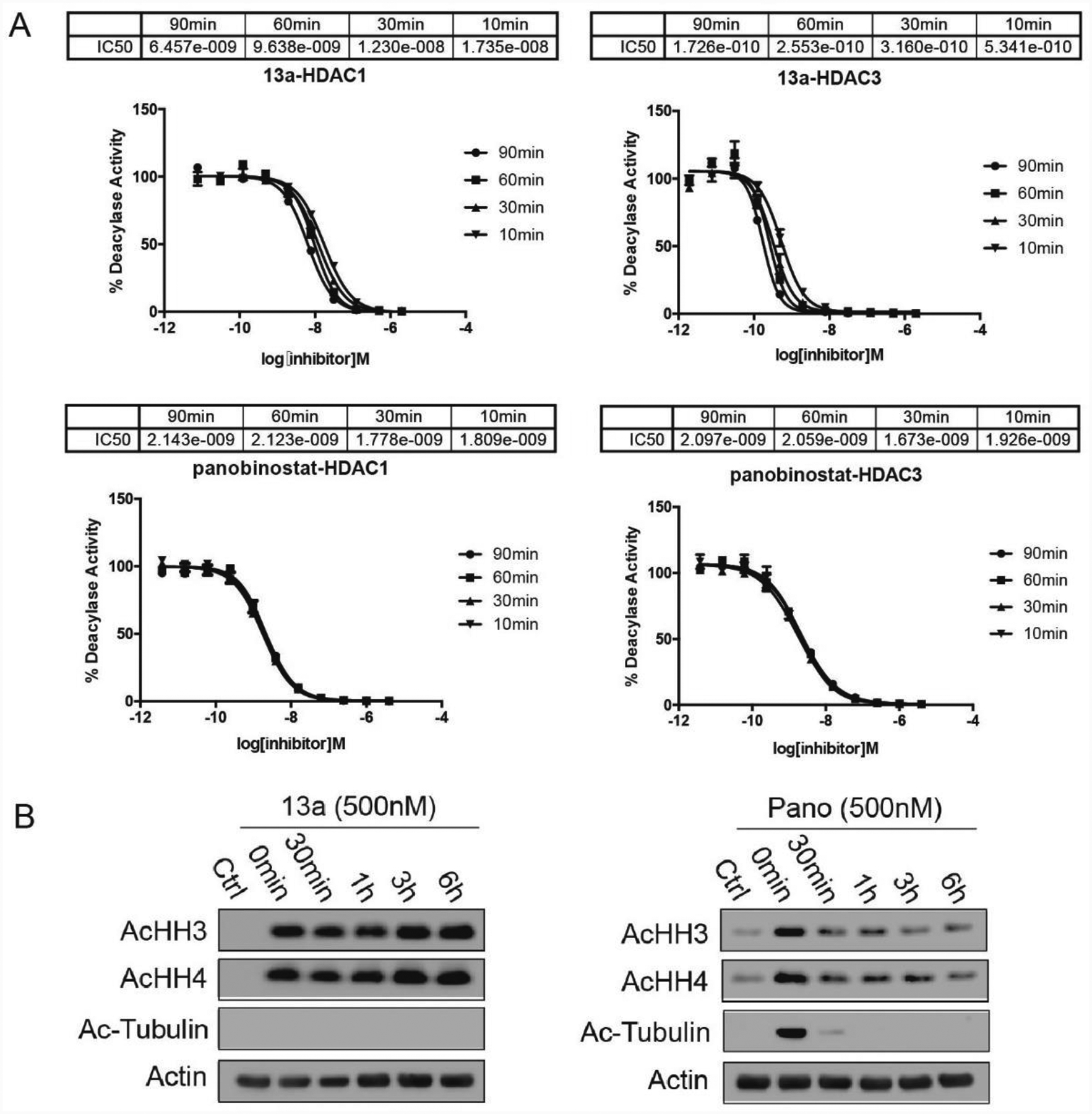
IC50 curves of 13a and panobinostat for HDACs 1, 2, and 3 after pre-incubation of 10 min, 30 min, 60 min, and 90 min, respectively. 13a shows time-dependent inhibition toward HDAC1 and 3, which indicates slow-on inhibition; while panobinostat reaches a steady-state with 10 min pre-incubation, indicating fast-on kinetics. Dose response curves for 13a and panobinostat performed in triplicates were generated using GraphPad Prism software. 3B: Histone acetylation caused by 13a does not reduce within 6 h of drug removal, while panobinostat reduces acetylation within 30 min after removal of drug. MV4–11 cells are treated with 13a or panobinostat (Pano) for 3 h, control cells are collected for the 0 min point, the drugs are washed out and the cells are cultured for various lengths of time.
Anti-cancer activity of 13a is p53 and FLT3 status-dependent
In our previous study, we concluded hydrazide-based HDACI 13e displayed different mechanisms in MV4–11 and PC-3 cells depending on their p53 status.26 However, only two cell lines were used and we only showed a simple mechanism with the phenomenon of apoptosis and cell cycle arrest.26 Therefore, in this study, we further verify this hypothesis by conducting experiments to determine 13a’s GI50 (average growth inhibition concentration) and LC50 (average lethal concentration) in the NCI60 tumor cell lines via the National Cancer Institute therapeutics program (Table 5). GI50s of 13a in 60 cell lines range from 17.3–338 nM; however, 13a does not induce cell death at these concentrations. 13a only induces lethality in 10 of the 60 cell lines at concentrations under 10 μM, and LC50 values of 13a towards 7 of the 10 lethality-sensitive cell lines are below 1 μM. Unlike 13a, panobinostat induces death in 18 of the 60 cell lines under the concentration of 10 μM (Figure 4A), which indicates that 13a has a more selective profile in the NCI 60 cell screen. Deeper analysis of the NCI 60 cell line screen revealed that 4 (NCI-H226, HCT116, LOX IMVI, and UACC-62) of the 7 most sensitive cell lines to 13a are p53 wild type (wt-p53); the other three sensitive cell lines are p53 mutants. Notably, none of the p53-null cell lines display sensitivity to 13a induced lethality. This result coincides with our previous hypothesis that the response of cancer cell lines to hydrazide inhibitor is related to the p53 status.
Table 5.
National Cancer Institute Developmental Therapeutics Program In Vitro Testing Results of 13a.
| Cancer Types | Cell Line | GI50 (nM) | LC50 (nM) |
|---|---|---|---|
| Leukemia | CCRF-CEM | 40.6 | >10,000 |
| HL-60 | 34.2 | >10,000 | |
| K-562 | 35.2 | >10,000 | |
| MOLT-4 | 47.7 | >10,000 | |
| RPMI-8226 | 26.8 | >10,000 | |
| SR | 94.7 | >10,000 | |
| Non-Small Cell Lung Cancer | A549/ATCC | 176 | >10,000 |
| EKVX | 285 | >10,000 | |
| HOP-62 | 102 | >10,000 | |
| HOP-92 | 23.0 | >10,000 | |
| NCI-H226 | 52.3 | 460 | |
| NCI-H23 | 182 | >10,000 | |
| NCI-H322M | 173 | >10,000 | |
| NCI-H460 | 163 | >10,000 | |
| NCI-H522 | 96.9 | >10,000 | |
| Colon Cancer | COLO 205 | 29.8 | 660 |
| HCC-2998 | 143 | 1760 | |
| HCT-116 | 22.9 | 879 | |
| HCT-15 | 313 | >10,000 | |
| HT29 | 32.4 | >10,000 | |
| KM12 | 116 | >10,000 | |
| SW-620 | 53.0 | >10,000 | |
| CNS Cancer | SF-268 | 198 | >10,000 |
| SF-295 | 131 | >10,000 | |
| SF-539 | 98.9 | >10,000 | |
| SNB-19 | 231 | >10,000 | |
| SNB-75 | 312 | >10,000 | |
| U251 | 80.1 | 4280 | |
| Melanoma | LOX IMVI | 118 | 537 |
| MALME-3M | 48.2 | >10,000 | |
| M14 | 72.5 | >10,000 | |
| MDA-MB-435 | 97.5 | >10,000 | |
| SK-MEL-2 | 73.9 | >10,000 | |
| SK-MEL-28 | 68.7 | 893 | |
| SK-MEL-5 | 192 | >10,000 | |
| UACC-257 | 79.6 | >10,000 | |
| UACC-62 | 72.6 | 589 | |
| Ovarian Cancer | IGROV1 | 115 | >10,000 |
| OVCAR-3 | 135 | >10,000 | |
| OVCAR-4 | 105 | >10,000 | |
| OVCAR-5 | 31.6 | >10,000 | |
| OVCAR-8 | 41.3 | >10,000 | |
| NCI/ADR-RES | 516 | >10,000 | |
| SK-OV-3 | 98.3 | >10,000 | |
| Renal Cancer | 786–0 | 161 | >10,000 |
| ACHN | 85.5 | >10,000 | |
| CAKI-1 | 101 | 4300 | |
| RXF 393 | 23.4 | 666 | |
| SN12C | 158 | >10,000 | |
| TK-10 | 17.3 | >10,000 | |
| UO-31 | 173 | >10,000 | |
| Prostate Cancer | PC-3 | 54.2 | >10,000 |
| DU-145 | 90.8 | >10,000 | |
| Breast Cancer | MCF7 | 106 | >10,000 |
| MDA-MB-231/ATCC | 203 | >10,000 | |
| HS 578T | 121 | >10,000 | |
| BT-549 | 338 | >10,000 | |
| T-47D | 23.9 | >10,000 | |
| MDA-MB-468 | 50.4 | >10,000 |
Figure 4A.
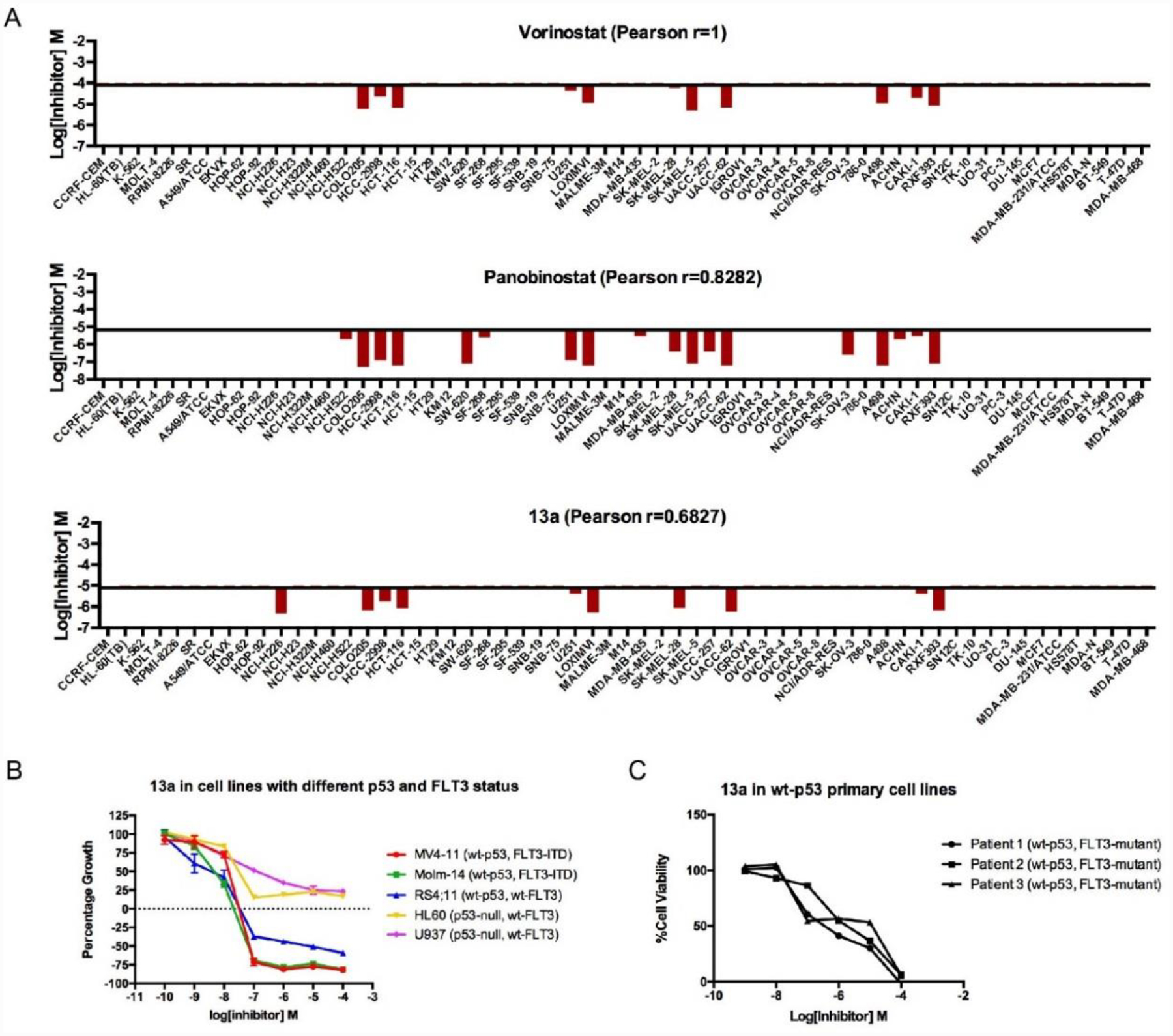
NCI 60 cells panel screen of 13a, panobinostat and SAHA. Compared to panobinostat, 13a shows better selectivity among all cell lines, and most of the cell lines sensitive to 13a is p53 wild type. 4B. In leukemia cells, 13a is altered due to different p53 and FLT3 status. 3C. 13a causes wt-p53 primary cell death. Concentration response curves performed in triplicates were generated using GraphPad Prism software.
In AML, p53 mutations occur in only ~8% of de novo AML.66–67 Interestingly, over-expression of wt-p53 is associated with FLT3-ITD mutations suggesting a potential therapeutic niche.68 FLT3 is important for the development of the hematopoietic and activating mutations of FLT3, which is now recognized as the most common molecular abnormality in AML and other hematologic malignancies as well. Patients with this activating mutation were found to have an increased incidence of leukocytosis and a decreased overall survival (OS).69 Therefore, we also checked the dependency of 13a with FLT3 status in addition to p53.
We chose 6 different leukemia and multiple myeloma cell lines with different p53 and FLT3 status, including MV4–11 and Molm-14 cell lines with wt-p53 and FLT3-ITD, the RS4;11 cell line with wt-p53 and wt-FLT3, as well as U937 and HL60 cell lines with p53-null and wt-FLT3 to test our compounds using the NCI-60 screening methodology. Results show that the wt-p53 leukemia cell lines are more sensitive to 13a than p53-null cell lines. Furthermore, 13a can cause cell death in wt-p53 MV4–11, Molm-14, and RS4;11, but only inhibits proliferation of p53-null HL-60 and U937 (Figure 4B). Within the wt-p53 cell lines, FLT3-ITD MV4–11 and Molm-14 cell lines are highly sensitive to HDACI treatment with significantly lower LC50 than wt-FLT3 cell line RS4;11. Therefore, wt-p53 and FLT3-ITD cell lines have the highest sensitivity of the 6 cell lines tested. We also tested the effect of 13a in primary wt-p53 AML cells (Figure 4C). 13a is toxic in all three patients’ cells with < 1 μM EC50.
We then explored the anti-tumor mechanism of 13a in cell lines with different p53 and FLT3 status, including wt-p53 FLT3-ITD cell line MV4–11 (Figure 5A), wt-p53 wt-FLT3 cell line RS4;11 (Figure 5C), p53-null wt-FLT3 cell line HL60 (Figure 5D) and wt-p53 FLT3-non-expression SR cell line (supporting information Figure S7). LP411 (50, 100, and 200 nM), panobinostat (100 nM), vorinostat (5000 nM) and entinostat (1000 nM and 5000 nM) were used as controls. LP411 (3b25) is the most potent hydrazide HDACI in our previous study. As p53 can function as a “master regulator” of the apoptotic program and FLT3-ITD potently activates the STAT5 pathway in contrast to wild-type FLT3 signaling,70–72 13a may have different mechanisms in cell lines with different p53 and FLT3 status.
Figure 5A.
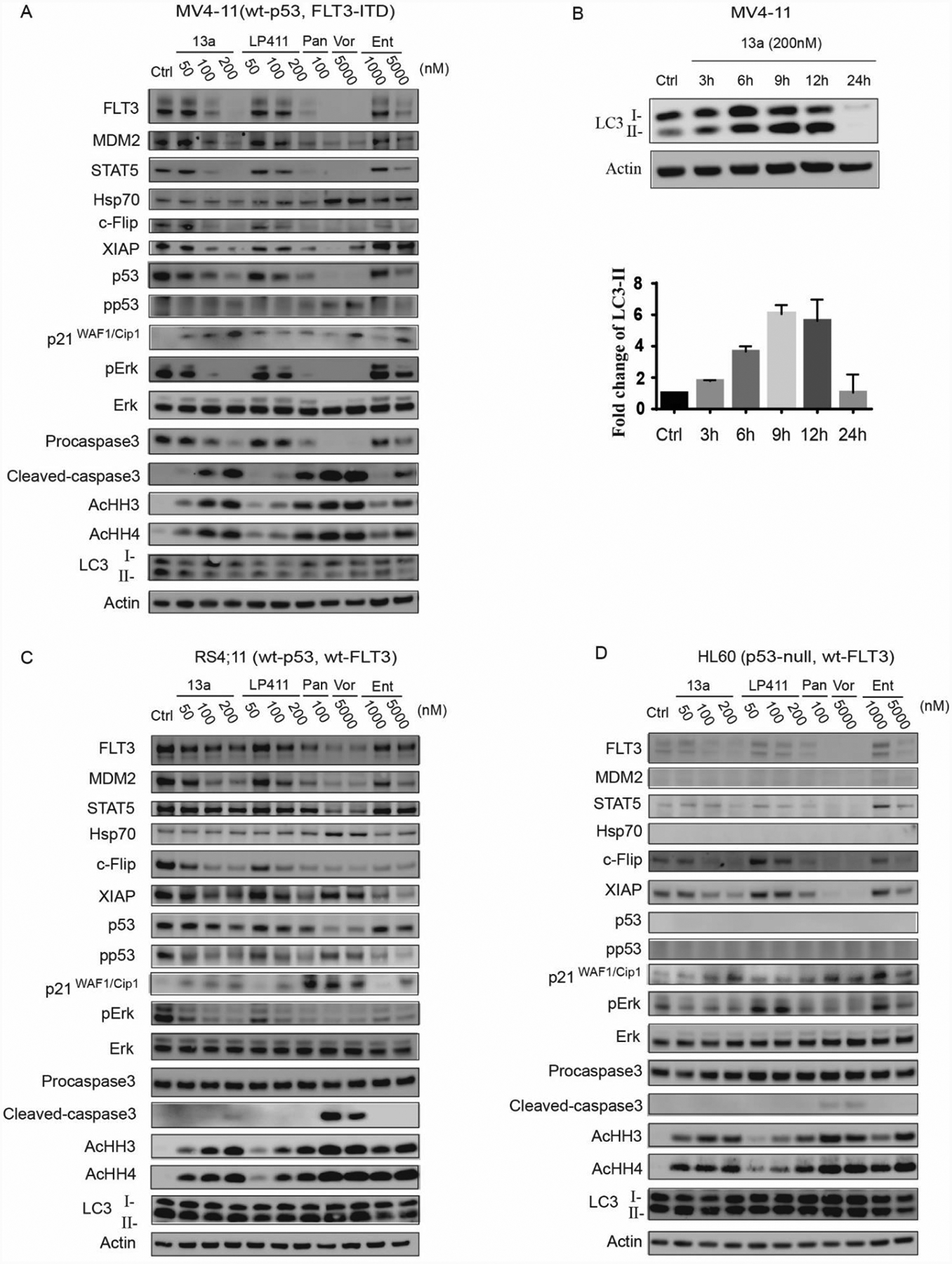
Treatment of 13a, LP411, panobinostat (Pan), vorinostat (Vor) and entinostat (Ent) in wt-p53, FLT3-ITD MV4–11 cell line for 24 h. 5B. (top) Treatment of 200 nM 13a with 3 h, 6 h, 9 h, 12 h, and 24 h, respectively. LC3-II accumulated within 12 h and is fully degraded in 24 h; (bottom) Quantification of LC3-II values were normalized to actin levels. 5C. Treatment of 13a, LP411, panobinostat (Pan), vorinostat (Vor) and entinostat (Ent) in wt-p53, wt-FLT3 RS4;11 cell line for 24 h. 5D. Treatment of 13a, LP411, panobinostat (Pan), vorinostat (Vor) and entinostat (Ent) in p53-null, wt-FLT3 HL60 cell line for 24 h.
In wt-p53 FLT3-ITD cell line MV4–11, pro-caspase3 cleavage is observed for the treatments of 13a, LP411, panobinostat, vorinostat, and entinostat (Figure 5A). As caspase3 is the apoptotic effector, apoptosis occurs after the treatment of all the compounds. In addition, 13a and LP411 down-regulate FLT3 and STAT5. Overexpression of FLT3 leads to the stimulation of proliferation and inhibition of apoptosis in many primary samples of leukemia.73–75 Activation of FLT3 potently activates the STAT5 pathway, which constitutively induces cyclin D1, c-myc, and serine/threonine kinases Pim-1 and Pim-2, leading to aberrant cell growth of leukemia cells.71, 76–78 We observe that pERK is down-regulated by 13a. Activation of FLT3 kinase and its downstream proliferative signaling Ras/MEK/ERK pathway can also accelerate cell growth.79 Therefore, our HDAC inhibitors can indirectly inhibit the FLT3 pathway via the down-regulation of FLT3, STAT5, and pERK. Due to the important role played by FLT3 in the survival and proliferation of AML blasts, and the fact that FLT3 inhibitor midostaurin (Rydapt) is an FDA-approved targeted small molecule for the treatment of AML with FLT3-ITD mutation, our compounds are likely to have a promising therapeutic effect in the treatment of AML with FLT3 mutations. 13a also causes degradation of master anti-apoptotic proteins c-Flip and XIAP, eventually leading to cell apoptosis (cleavage of pro-caspase3) (Figure 5A). c-FLIP isoforms are major anti-apoptotic proteins that suppress caspase 8 and 10 activations, and therefore prevent the downstream apoptosis cascade,80 while XIAP can directly bind to and inhibit caspase3 and 781–82 as well as prevent caspase 9 from activating.83 P21WAF1/Cip1, a CDK inhibitor, is markedly up-regulated by 13a, which may contribute 13a’s anti-proliferative activity. Furthermore, LC3 degradation is also observed in wt-p53 MV4–11 cells (Figure 5A). A recent study showed that LC3-II (an LC3 phospholipid conjugate) accumulation is related to autophagy, and intra-autophagosomal LC3-II may be degraded by lysosomal hydrolase.84 Therefore, we hypothesize that the degradation of LC3 after 24h treatment with 13a might indicate activation of autophagy. In a time-chased western blot experiment (Figure 5B), LC3-II fluxes and accumulates within 12 h and is later degraded within 24 h. Based on the western results, both apoptosis and autophagy occur in wt-p53 MV4–11 cells.
In the wt-p53 and wt-FLT3 RS4;11 cell line, cleavage of pro-caspase3 is only observed at high dose 13a (200 nM), panobinostat (100 nM) and vorinostat (5000 nM), which means that wt-FLT3 RS4;11 is less sensitive than FLT3-ITD MV4–11 (Figure 5C). Another obvious difference between these two cell lines with different FLT3 status is that STAT5 does not down-regulate with the treatment of 13a. It has been described that the signaling mechanisms of FL3-ITD differed from those conferred by wt-FLT3. STAT5 activation was part of the FLT3 signaling chain, and that STAT5 was constitutively active in FLT3-ITD cells, whereas FLT3 ligand (FL) did not induce STAT5 phosphorylation in wt-FLT3 cells.85 These might be the reason that our compounds behave differently in the FLT3-ITD MV4–11 cells and the wt-FLT3 RS4;11 cells. LC3 degradation is also not observed in wt-FLT3 RS4;11 cells (Figure 5C). However, p21WAF1/Cip1 is up-regulated by 13a in RS4;11 cells. In p53-null wt-FLT3 HL60 cells, 13a can result in the degradation of anti-apoptotic protein c-Flip and XIAP but is unable to trigger apoptosis as pro-caspase3 is not cleaved to its active form (Figure 5D). 13a also induces p21 up-regulation in HL60 cells consistent with previous reports that the p21 up-regulation by HDACIs is independent of p53 status resulting in the cell-cycle arrest but not cell death.26 Moreover, LC3 is not degraded in HL60 cells; therefore, 13a is incapable of causing apoptosis and autophagy in p53-null HL60 cells. It must be noted that 13a does not cause wt-p53 SR cell line death (Table 5); the western blot result of 13a in SR cells is shown in supporting information Figure S7. Pro-caspase3 does not cleave after treatment of 13a for 24h. Interestingly, 13a can up-regulate pERK, which is down-regulated in the other 3 cell lines. Additionally, FLT3 is undetectable in the SR cell line. In summary, the effect of 13a depends on the cellular p53 status, which controls the levels of anti-apoptotic proteins c-FLT and XIAP, and caspase-3 cleavage and activation. 13a also controls the expression of FLT-3 affecting cellular pro-survival and proliferation signaling pERK and STAT5 pathways, especially in AML with oncogenic FLT3 mutations. In combination, AML cells with wt-p53 and FLT3-ITD are the most sensitive to 13a treatment.
It is also interesting that an increase in Hsp70 levels is not observed for 13a but was observed for SAHA and panobinostat treatments (Figures 5A and C). We determined that 17-AAG, an Hsp90 inhibitor that can up-regulate Hsp70,86 antagonized with 13a in anti-proliferation activity when used at lower concentrations (supporting information Figure S8). Hsp70 up-regulation contributes to chemo-resistance in cancer therapeutic development,87–89 which suggests that 13a can be a more sustainable therapy compared to the classical HDACIs.
Preventing p53 degradation promotes 13a-triggered apoptosis
We also observe that 13a leads to a down-regulation of p53 in MV4–11 and RS4;11 cells. As MDM2 is also down-regulated (via p53 down-regulation) by 13a (Figure 5A), we hypothesize that the p53 down-regulation is mainly due to proteasome degradation instead of acetylation or phosphorylation. To study the mechanism of p53 down-regulation, we treated MV4–11 cells with 13a plus the MDM2-p53 antagonist RG7388 and the p53 reactivator PRIMA-1met for 24 h. RG7388 prevented the binding of p53 and MDM2 and inhibited ubiquitin-mediated proteasome degradation of p53. PRIMA-1met can reactivate p53 by binding to and modifying thiol groups in the central domain of the mutated protein, so we used it as a negative control, as it will not restore p53 in the wt-p53 MV4–11 cell line. RG7388 blocked p53 degradation caused by 13a, thus promoting the cleavage of pro-caspase3 (Figure 6A). However, degradation of MDM2 caused by 13a is not fully restored by RG7388. We speculate that this is because of the significant cell death after the 24 h treatment (Figure 6A). Therefore, we treated with 13a and RG7388 for 3 h, 6 h, 9 h, 12 h, and 24 h, respectively, and then detected the level of p53 and MDM2. As expected, RG7388 fully blocks the degradation of p53 and MDM2 caused by 13a within 12 h (Figure 6B). Similar results are also shown in T-PLL cells treated with panobinostat and MDM2-p53 antagonist or p53 reactivator.90–91 Furthermore, 13a and RG7388 synergistically inhibit cell proliferation (Figure 6C, CI < 0.9). Collectively, we conclude that the mechanism of p53 down-regulation is due to MDM2 activation and proteasome degradation, which is further confirmed via proteasome inhibitor. In Figure 7A, proteasome inhibitor bortezomib blocks the 13a-induced p53 degradation. Treatment with 13a in combination with bortezomib facilitates the cleavage of pro-caspase3 (Figure 7B), indicating that bortezomib can promote 13a-triggered apoptosis. 13a and bortezomib also synergistically inhibit cell proliferation (Figure 7C, CI < 0.9), which is consistent with the previous results in Figure 5.
Figure 6A.
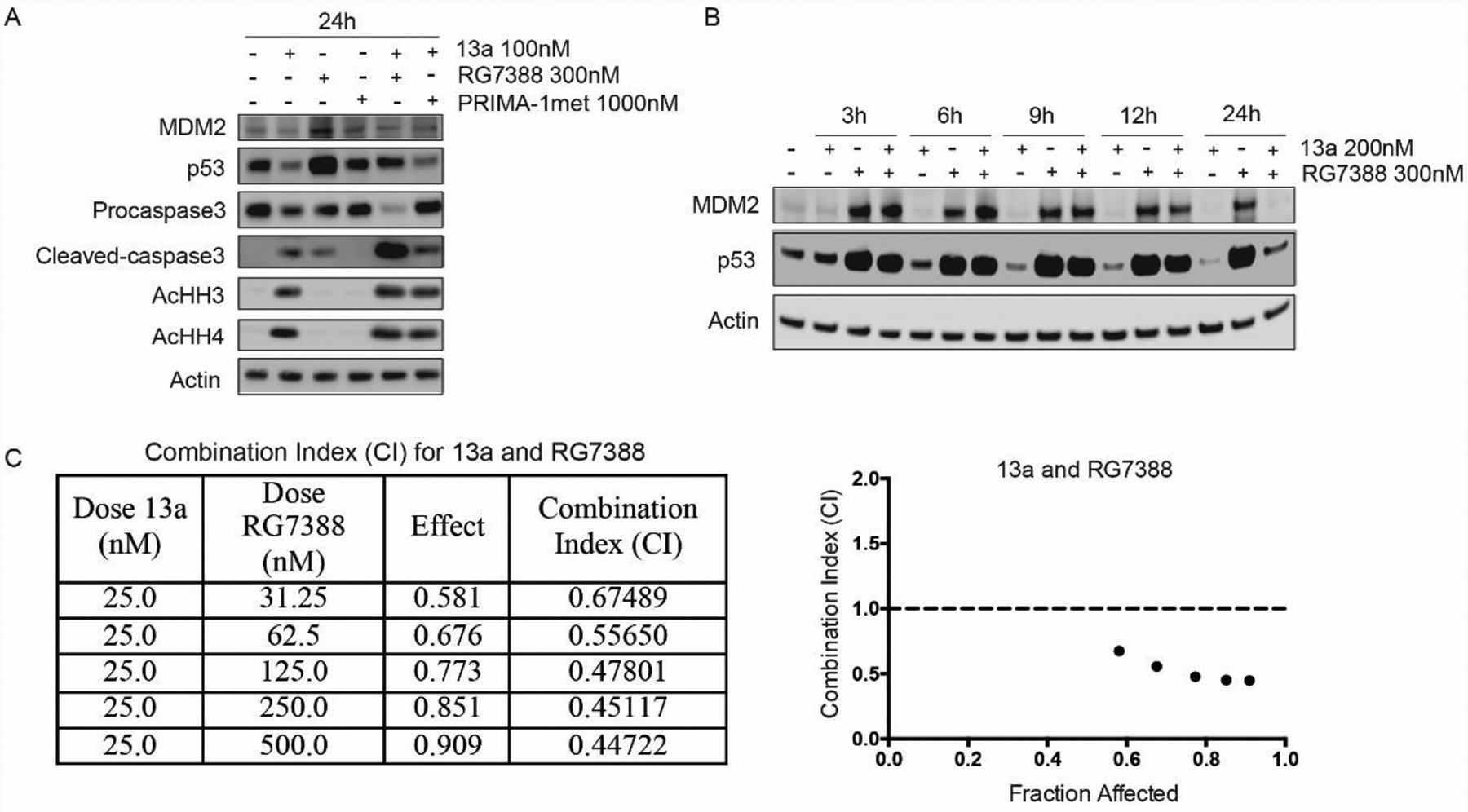
Treatment of 13a (100 nM) or 13a (100 nM) in combination with p53-MDM2 inhibitor RG7388 (300 nM) and p53 activator Prima-1met (1000 nM) for 24h, respectively. RG7388 can prevent p53 degradation and promote 13a-triggered apoptosis obviously; 6B. Treatment of 13a (200 nM) and 13a (200 nM) in combination with RG7388 (300 nM) for 3h, 6h, 9h, 12h and 24h, respectively. p53 begins to degrade after treatment by 13a for 6h-9h, and the degradation of p53 can be fully recovered by RG7388 within 12h; 6C. Combination Index (CI) for 13a and RG7388 after treatment 0f 24 h. Data was analyzed using CompuSyn Software. CI < 1, = 1, and > 1 indicate synergism, additive effect, and antagonism, respectively.
Figure 7A.
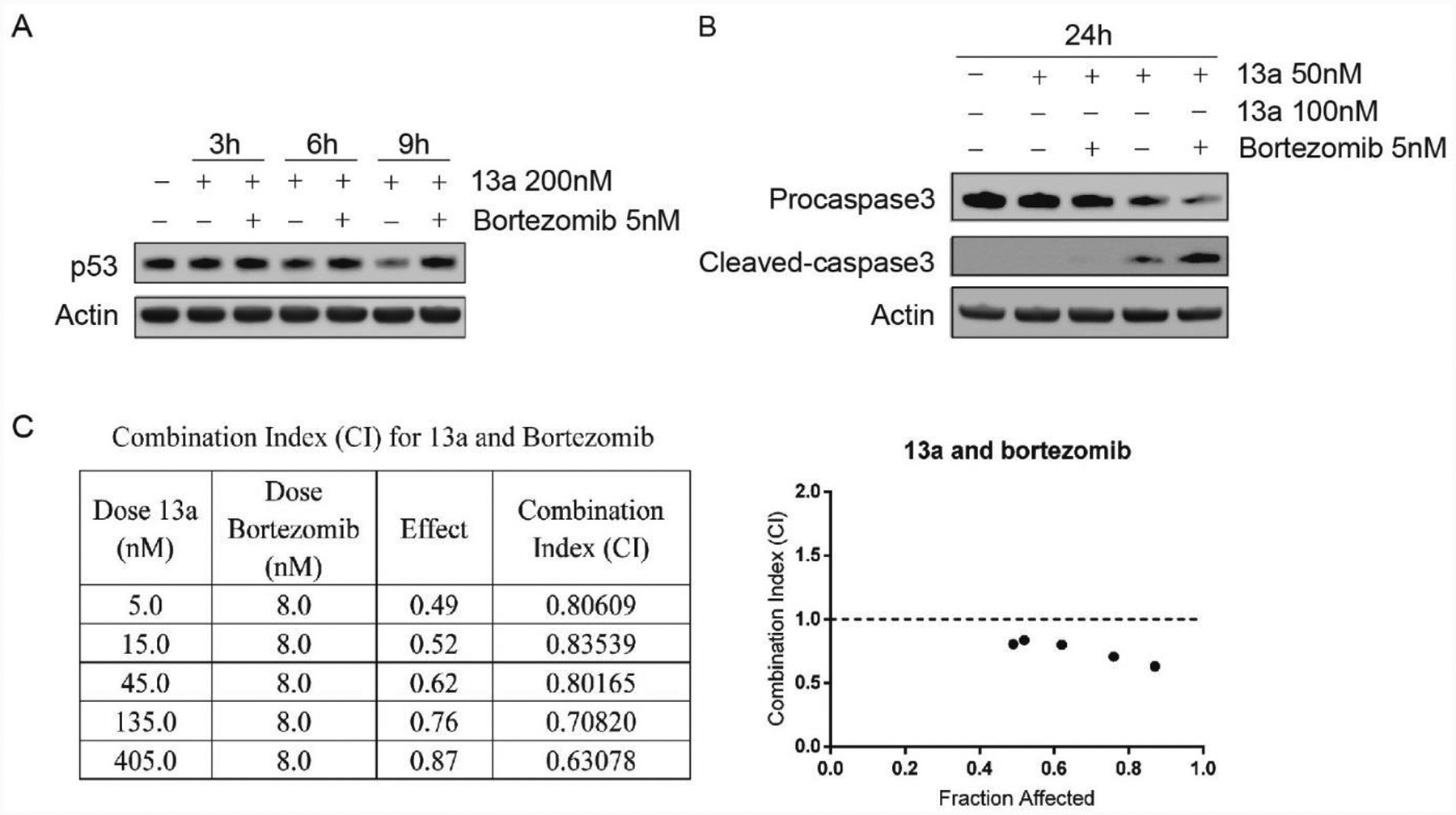
Treatment of 13a (200 nM) or 13a (200 nM) in combination with bortezomib (5 nM) for 3 h, 6 h and 9 h, respectively. p53 begins to degrade in treatment of 13a at 6 h, and the degradation can be rescued by bortezomib. 7B. Treatment of 13a 50 nM, 100 nM or in combination with bortezomib (5 nM). Bortezomib can promote 13a-triggered apoptosis. 7C. Combination Index (CI) for 13a and Bortezomib after treatment 0f 24 h. Data was analyzed using CompuSyn Software. CI < 1, = 1, and > 1 indicate synergism, additive effect, and antagonism, respectively.
Caspase inhibitor z-VAD inhibits MV4–11 death caused by 13a
As shown in Figure 5A and B, 13a results in cell apoptosis and also triggers autophagy in wt-p53 MV4–11 cells. Apoptosis and autophagy) are two forms of programmed cell death, which is in contrast with the non-physiological necrotic process that occurs as a result of infection or injury.92 To explore the mechanism of MV4–11 cell death, we treated MV4–11 cells with 13a plus the pan-caspase inhibitor z-VAD and the autophagy inhibitors chloroquine or wortmannin for 24 h, and then measured cell viability (Figure 8A). Autophagy inhibitors chloroquine and wortmannin are incapable of attenuating cell death, while in the presence of pan-caspase inhibitor z-VAD, 13a-triggered cell death was rescued. Western blots also indicated that among the three co-treatment agents with 13a, z-VAD can prevent the cleavage of pro-caspase3, suggesting z-VAD can rescue cell death by inhibiting apoptosis (Figure 8B). The autophagy inhibitor chloroquine did block the degradation of LC3-II and prevent the progression of autophagy but it does not prevent 13a-induced cell death (Figure 8B). Therefore, apoptosis is likely the main mechanism leading to 13a-triggered cell death rather than lethal autophagy.
Figure 8A.
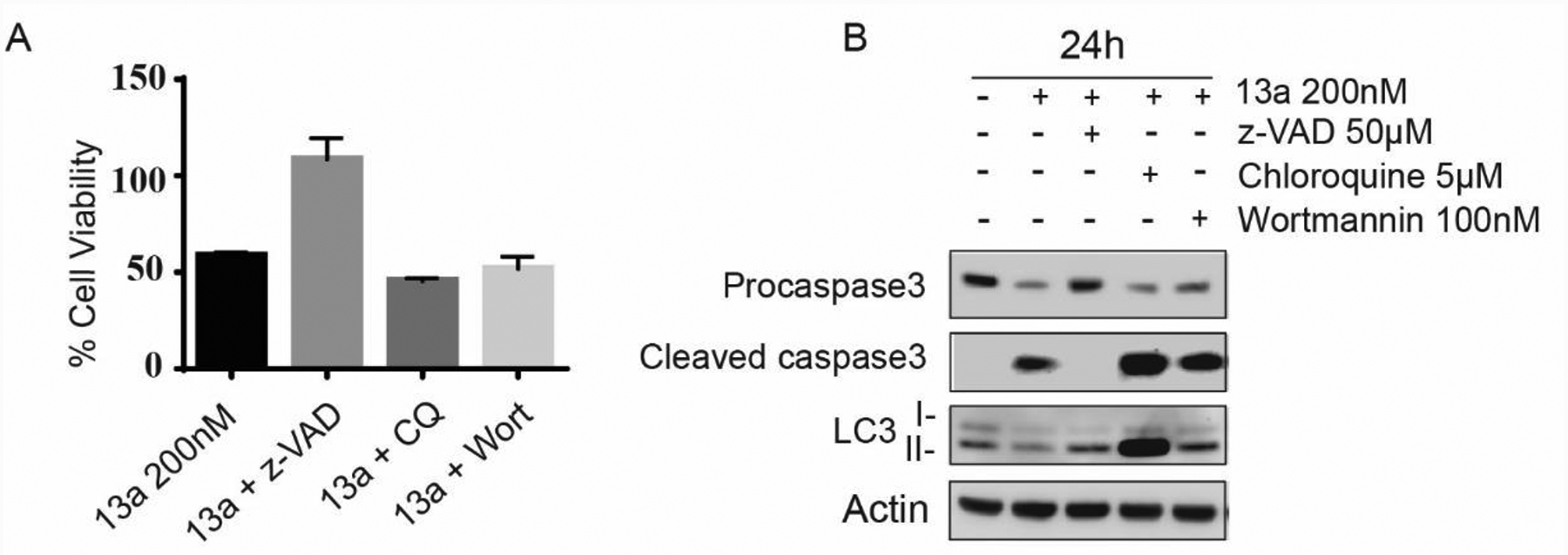
Treatment of 13a (200 nM) with z-VAD (50 μM), chloroquine (CQ 5 μM) or wortmannin (Wort 100 nM) for 24 h incubation (*p < 0.001, n = 3). The pan-caspase inhibitor z-VAD is capable of attenuating cell death rather than the autophagy inhibitors wortmannin and chloroquine, suggesting although both of apoptosis and autophagy occur after treatment of 13a, apoptosis is the key factor leading to cell death. 8B. Treatment of 13a (200 nM) with z-VAD (50 μM), chloroquine (CQ 5 μM) or wortmannin (Wort 100 nM) for 24 h incubation (*p < 0.001, n = 3). z-VAD rescues procaspase activation, and chloroquine blocks the degradation of LC3-II in 24 h and prevent the progression of autophagy.
13a has favorable toleration and in vivo PK properties
As a potential clinical therapy, the inhibitor’s efficacy and ability to influence targeted biomarkers must be validated in vivo. The chemical agent should be well tolerated with no overbearing toxicity and with optimized PK properties in addition to its pharmacological efficacy. We have already demonstrated the in vitro anti-tumor activity of 13a. Our next step is to test its mutagenic toxicity and pharmacokinetic properties, two major limiting factors for HDACIs in the clinic. Mini-Ames tests were conducted to determine the mutagenic toxicity of 13a, and mutagen 2-aminoanthracene and panobinostat were used as positive controls. 13a is not mutagenic compared to the known mutagen 2-aminoanthracene and panobinostat (Figure 9), which suggests that 13a bear less genotoxicity than panobinostat. We then conducted a pharmacokinetic study for 13a by iv as well as oral administration and compared with panobinostat. The in vivo t1/2 of 13a iv and oral are 15.2 h and 7.45 h, which are significantly longer than panobinostat in mice (2.9 h oral). AUC0-inf (ng·h/mL) of 13a is 265 ng/mL at 20 mg/kg oral which is 2 times that of panobinostat 50 mg/kg oral (126 ng/mL) (Table 6).93 What’s more, the bioavailability of 13a (19.8%) is much superior than that of panobinostat (4.62%).93 Therefore, 13a displays much more favorable PK profiles in mice. The slow-on/slow-off kinetics profile reveals that 13a binds tightly and acts much longer than the classical inhibitors, regardless of its actual t1/2. This provides the possibility of using lower doses or longer intervals between dosing, which should minimize any potential adverse effects and make this a more effective and durable therapy than vorinostat and panobinostat.
Figure 9.
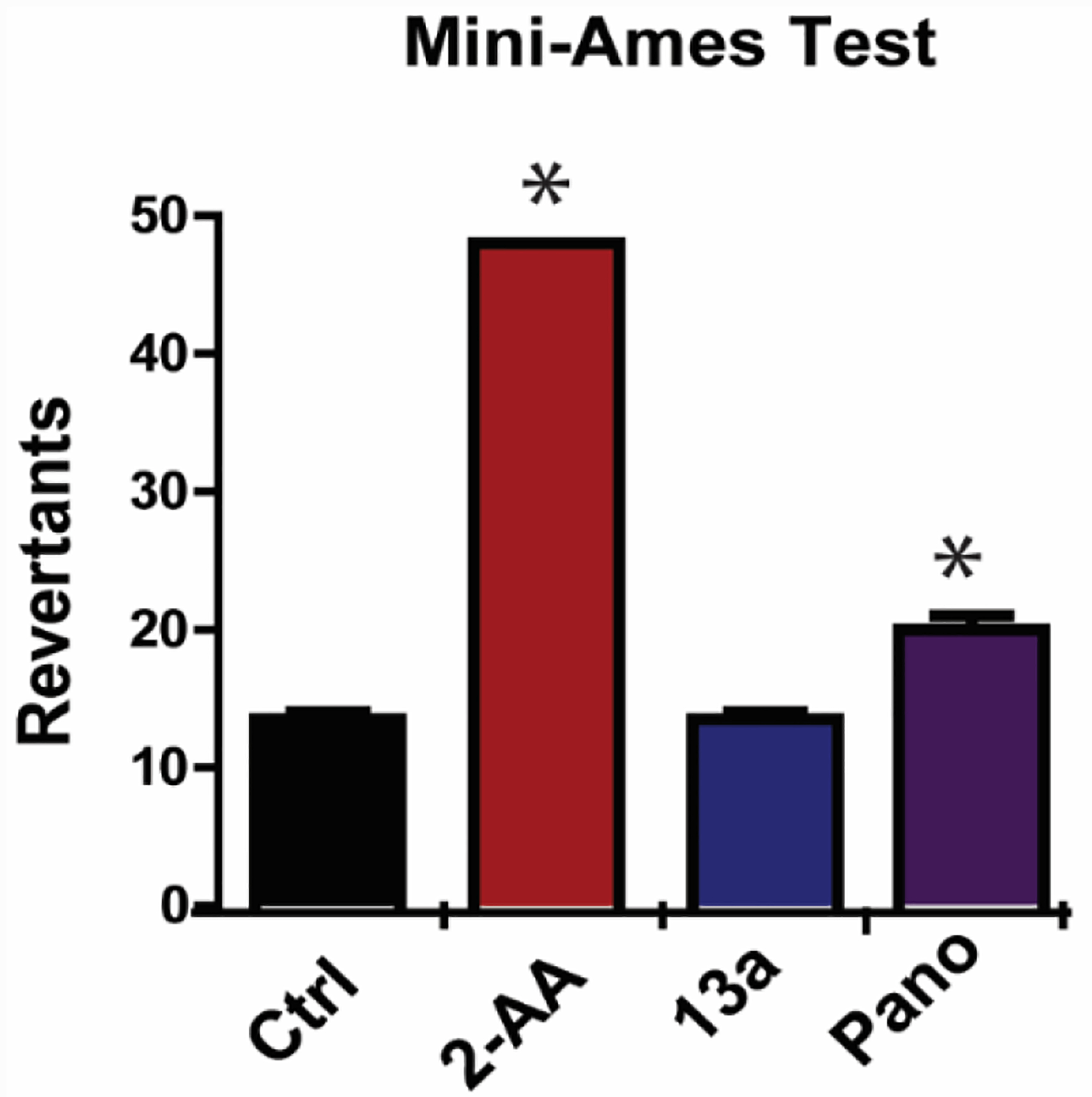
Mini-Ames tests with liver S9 fraction activation (EBPI, Canada). Mutagen controls 2-aminoanthracene (2-AA) and HDAC inhibitor panobinostat (*p < 0.01 versus Ctrl) are Ames positive. 13a is not mutagenic as compared to panobinostat and the known mutagen 2-aminoanthracene.
Table 6.
Pharmacokinetic characterization of 13a with iv and oral administration.
| 13a | 13a | Panobinostat93 | |
|---|---|---|---|
| Administered dose (mg/kg) | iv at 5 mg/kg | oral at 20 mg/kg | oral at 50 mg/kg |
| Cmax (ng/mL) | -- | 45.1 | 116 |
| t1/2 (h) | 15.2 | 7.45 | 2.9 |
| AUC0-inf (ng·h/mL) | 334 | 265 | 126 |
| F% | -- | 19.8 | 4.62 |
13a was injected via iv and oral (n = 3), blood was sampled at different time-points after dosing for 24 h, and the inhibitor plasma concentration is determined via LC-MS/MS. The area under the plasma concentration versus time curve (AUC) is calculated using the linear trapezoidal method. The PK data are fitted to obtain PK parameters using the non-compartmental method.
Conclusion
We have designed and synthesized a new series of hydrazine-bearing class I selective HDACIs based on the modification of the three domains (cap, linker, and ZBG) of panobinostat. For the first time, we design di-N-substituents hydrazide based inhibitor and discuss their SAR. The representative compound 13a exhibits potent activity against class I HDAC 1, 2, and 3, with IC50 values of 5.17 nM, 49.5 nM, and 0.28 nM, respectively. However, 13a displays micromolar IC50 for HDAC8 and does not show inhibition up to 10 μM for HDACs 4, 5, 6, 7, and 9. Therefore, 13a not only maintains panobinostat’s potency toward class I HDACs and has HDACs 1, 2, and 3 selectivity. Interestingly, 13a displays mixed binding mode to HDAC3, but a non-competitive binding to HDAC1, which could potentially be used to identify the allosteric site these hydrazide inhibitors targets. The EC50 of 13a against wt-p53 MV4–11 cells is 15.35 nM, which is comparable to that of panobinostat (5.2 nM). As 13a has differential activities in cell lines with different p53 and FLT3 status, we conducted a multitude of molecular biological experiments to reveal the anti-tumor mechanism in wt-p53 FLT3-ITD MV4–11 cell line, wt-p53 wt-FLT3 RS4;11 cell line, p53-null wt-FLT3 HL-60 cell line and wt-p53 FLT3-undetectable SR cell line together with LP41l (a comparable lead hydrazide HDACI), vorinostat, and panobinostat. In the wt-p53 FLT3-ITD MV4–11 cell line, 13a indirectly inhibits the FLT3 signaling pathway via down-regulation of FLT3, STAT5, and pERK, making this compound a promising therapeutic strategy for the treatment of AML with FLT3 mutation. It also down-regulates master anti-apoptotic proteins c-Flip and XIAP as well as leads to cleavage of pro-caspase3. Also, in the wt-p53 and wt-FLT3 cell line RS4;11, 13a cannot down-regulate activation of the STAT5 pathway, and this might be the main contributing factor for RS4;11 being less sensitive to the HDACI treatment. In addition, 13a is incapable of causing apoptosis in p53-null HL60 cells as expected, indicating that a functional p53 is required for the selective toxicity. This hypothesis is further strengthened when p53-MDM2 antagonist RG7388 and proteasome inhibitor bortezomib are used to prevent the degradation of p53, resulting in effects with 13a to induce cell death (CI from 0.5–0.9). All of these results reveal that 13a displays selective class I HDAC inhibition, inducing targeted lethality against leukemia cell lines depending on their p53 status and FLT3 status. We conclude that FLT3-ITD cell lines are more sensitive than wt-FLT3 cell lines among cells with wt-p53, and 13a displays wt-p53 and FLT3-ITD dependent anti-leukemia activity, which can be a potential therapeutic niche for AML and a useful diagnostic indication for patient selection in the clinic.
MV4–11 cell death caused by 13a can be rescued by the pan-caspase inhibitor z-VAD, unlike the autophagy inhibitors chloroquine and wortmannin. This indicates that the mechanism of 13a’s lethality against wt-p53 cells is apoptosis, rather than autophagy. Although compound 13a shows slightly lower potency than panobinostat in vitro, it displays an HDAC class I selective profile. In addition, an increase in Hsp70 level is not observed for 13a but is observed for panobinostat and vorinostat (Figure 5A). As Hsp70 up-regulation has contributed to chemoresistance in cancer therapeutic development,87–89 13a could be a more effective and durable therapy compared to panobinostat in vivo. 13a is not mutagenic and showed superior bioavailability and half-life with a higher AUC exposure, and a longer t1/2 in vivo, which suggests that it is likely to be a better therapeutic agent than panobinostat in the clinic. Furthermore, the slow-on/slow-off inhibition profile reveals that 13a can tightly bind to HDACs, which makes them act much longer than classical inhibitors, despite the t1/2, if its Cmax reaches effective concentration. This provides the possibility of using lower doses or longer intervals between dosing.
Experimental Section
General chemistry
All solvents, reagents, and compound precursors were purchased from Sigma-Aldrich or other chemical vendors and used as received unless otherwise noted. NMR data were collected in deuterated solvents using a Bruker Nanobay 400 MHz instrument with TMS as an internal standard. Chemical shifts (δ) are given in parts per million, and coupling constants (J) are reported in hertz (Hz). Mass spectral data were gathered on a Thermo LCQ Fleet mass spectrometer using electrospray ionization. Purification was performed using a Teledyne Isco Combiflash 200 on prepacked C18-Aq columns. All target compounds were at least 95% pure as confirmed via UV detection of ESI-LCMS, performed on an Waters e2695 HPLC instrument using an XBridge C18 column (5 μm, 4.6 mm × 150 mm) using a gradient of water/methanol plus 0.1% formic acid (0–1 mins from 0–50% methanol, 1–12 mins from 50% to 100% methanol, 12–14 min to 0% methanol, and maintained at 0% for 1 minute).
Procedure for preparation of target compounds
2-(2-methyl-1H-indol-3-yl)ethanamine (3)
Phenyl hydrazine (1, 1.08 g, 10.0 mmol) was dissolved in absolute ethanol (50 mL), to this solution was added 5-chloropentan-2-one (2, 1.80 g, 15.0 mmol) dropwise, the mixed solution was refluxed at 80 °C for 4 h. After the reaction finished, volatiles were removed under vacuum and resulting residue was dissolved in DCM. The organic layer was washed with brine, dried over anhydrous MgSO4 and evaporated under vacuum. The crude product was purified on reverse phase columns eluted with acetonitrile and water to yield pure product 3 (1.36 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 7.39 (d, J = 7.7 Hz, 1H), 7.21 (d, J = 7.9 Hz, 1H), 6.98 (t, J = 7.2 Hz, 1H), 6.92 (t, J = 7.2 Hz, 1H), 2.69 (s, 4H), 2.31 (s, 3H).
(E)-3-(4-(((2-(2-methyl-1H-indol-3-yl)ethyl)amino)methyl)phenyl)acrylic acid (5a)
Compound 3 (0.87 g, 5 mmol) was dissolved in 100 mL methanol, to this solution was added (E)-3-(4-formylphenyl)acrylic acid (4a, 0.88g, 5mmol), sodium cyanoborohydride (1.24 g, 20 mmol) and 2 drops of acetic acid. The mixture was allowed to stir overnight and volatiles were removed under vacuum. The crude product was purified on reverse phase columns eluted with acetonitrile and water to yield pure product 5a (1.17g, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 7.57 (d, J = 8.1 Hz, 2H), 7.47 (d, J = 16.0 Hz, 1H), 7.38–7.35 (m, 3H), 7.21 (d, J = 7.9 Hz, 1H), 6.98–6.93 (m, 1H), 6.92–6.86 (m, 1H), 6.50 (d, J = 16.0 Hz, 1H), 3.78 (s, 2H), 2.82 (t, J = 7.3 Hz, 2H), 2.70 (t, J = 7.4 Hz, 2H), 2.31 (s, 3H). ESI-MS m/z: 335.17 [M + H]+.
Methyl 4-(((2-(2-methyl-1H-indol-3-yl)ethyl)amino)methyl)benzoate (5b)
Using the synthetic method of 5a, compound 3 and 4b gave 5b as a white solid, 72% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 7.99 (d, J = 8.2 Hz, 2H), 7.59 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.9 Hz, 1H), 7.01–6.91 (m, 2H), 4.12 (s, 2H), 3.86 (s, 3H), 2.92–2.88 (m, 4H), 2.32 (s, 3H). ESI-MS m/z: 323.08 [M + H]+.
Methyl 3-(((2-(2-methyl-1H-indol-3-yl)ethyl)amino)methyl)benzoate (5c)
Using the synthetic method of 5a, compound 3 and 4c gave 5c as a white solid, 70% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 7.97 (s, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.48 (t, J = 8.1 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 6.98 (t, J = 7.3 Hz, 1H), 6.91 (t, J = 7.4 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 2H), 2.83 (t, J = 7.5 Hz, 2H), 2.70 (t, J = 7.5 Hz, 2H), 2.31 (s, 3H). ESI-MS m/z: 323.17 [M + H]+.
(E)-3-(4-(((tert-butoxycarbonyl)(2-(2-methyl-1H-indol-3 yl)ethyl)amino)methyl)phenyl)acrylic acid (6a)
Compound 5a (0.67 g, 2 mmol) was dissolved in 15 mL 1 mol/L NaOH aqueous solution, to which was added 0.48 g (Boc)2O and 2 mL THF. The mixture was stirred overnight and THF condensed under vacuum. The left NaOH aqueous solution was acidification by diluted hydrochloric acid and then was extracted by 20 mL ethyl estate for three times. The organic layer was washed by brine and dried over anhydrous MgSO4. The solvent was evaporated under vacuum, the product was used for next step without purification. 1H NMR (400 MHz, DMSO-d6) δ 12.52 (s, 1H), 10.78 (s, 1H), 7.88–7.55 (m, 5H), 7.31–7.16 (m, 2H), 6.99–6.87 (m, 2H), 6.53 (d, J = 16.0 Hz, 1H), 4.49–4.31 (m, 2H), 3.27–3.20 (m, 2H), 2.79–2.73 (m, 2H), 2.27 (s, 3H), 1.38 (s, 9H). ESI-MS m/z: 435.17 [M + H]+.
methyl 4-(((tert-butoxycarbonyl)(2-(2-methyl-1H-indol-3-yl)ethyl)amino)methyl)benzoate (6b)
Using the synthetic method of 6a, compound 5b gave 6b as a white solid, 72% yield. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.1 Hz, 2H), 7.77 (s, 1H), 7.42–7.35 (m, 1H), 7.23 (t, J = 1.0 Hz, 1H), 7.16–7.14 (m, 1H), 7.11–7.02 (m, 2H), 4.56 (s, 1H), 4.23 (s, 1H), 3.89 (s, 3H), 3.42–3.30 (m, 2H), 2.94–2.84 (m, 2H), 2.37 (s, 3H), 1.48 (s, 9H). ESI-MS m/z: 423.17 [M + H]+.
methyl 3-(((tert-butoxycarbonyl)(2-(2-methyl-1H-indol-3-yl)ethyl)amino)methyl)benzoate (6c)
Using the synthetic method of 6a, compound 5c gave 6c as a white solid, 65% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 7.88–7.86 (m, 2H), 7.52–7.50 (m, 2H), 7.40–7.30 (m, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.98 (t, J = 7.2 Hz, 1H), 6.91 (t, J = 7.5 Hz, 1H), 4.49–4.37 (m, 2H), 3.86 (s, 3H), 3.25 (t, J = 7.2 Hz, 2H), 2.78–2.74 (m, 2H), 2.27 (s, 3H), 1.37 (s, 9H). ESI-MS m/z: 423.17 [M + H]+.
methyl 4-((benzylamino)methyl)benzoate (9a)
Using the synthetic method of 5a, compound 7 and 8a gave 9a as a white solid, 72% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.02 (d, J = 7.9 Hz, 2H), 7.63 (d, J = 7.9 Hz, 2H), 7.48–7.38 (m, 5H), 4.17 (s, 2H), 4.10 (s, 2H), 3.87 (s, 3H). ESI-MS m/z: 256.17 [M + H]+.
methyl 4-(benzylamino)benzoate (9b)
Using the synthetic method of 5a, compound 7 and 8b gave 9b as a white solid, 72% yield. 1H NMR (400 MHz, DMSO-d6) δ 7.67 (d, J = 8.8 Hz, 2H), 7.35–7.32 (m, 4H), 7.16 (t, J = 5.6 Hz, 1H), 6.62 (d, J = 8.8 Hz, 2H), 4.34 (d, J = 6.0 Hz, 2H), 3.73 (s, 3H). ESI-MS m/z: 323.08 [M + H]+. ESI-MS m/z: 242.08 [M + H]+.
methyl 4-((benzyl(tert-butoxycarbonyl)amino)methyl)benzoate (10a)
Using the synthetic method of 6a, compound 9a gave 10a as a white solid, 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 7.9 Hz, 2H), 7.61 (d, J = 7.9 Hz, 2H), 7.48–7.37 (m, 5H), 4.48–4.32 (m, 4H), 3.84 (s, 3H), 1.40 (s, 9H). ESI-MS m/z: 356.17 [M + H]+.
(E)-tert-butyl 4-(3-hydrazinyl-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (11)
To a solution of compound 6a (0.86 g, 2 mmol) in DCM was added 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU, 0.71 g, 2.4 mmol), followed by TEA (0.4 mL, 3 mmol). 30 mins later, hydrazine monohydrate (0.2 g, 4 mmol) was added. The reaction was allowed to stir overnight, and monitored by TLC. After the reaction finished, the solution of DCM was washed by brine and dried over anhydrous MgSO4. Volatile was removed under vacuum, the residue was recrystallized by ethyl estate and hexane to yield a white solid 11 (0.44 g, 50% yield) was obtained on reverse phase columns eluted with acetonitrile and water. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 9.35 (s, 1H), 7.54 (d, J = 7.8 Hz, 2H), 7.45 (d, J = 16.0 Hz, 1H), 7.32–7.21 (m, 4H), 6.99 (t, J = 7.3 Hz, 1H), 6.92 (t, J = 7.3 Hz, 1H), 6.55 (d, J = 16.0 Hz, 1H), 4.47–4.31 (m, 4H), 3.25–3.21 (m, 2H), 2.79–2.76 (m, 2H), 2.27 (s, 3H), 1.37 (s, 9H). ESI-MS m/z: 449.17 [M + H]+.
(E)-tert-butyl (2-(2-methyl-1H-indol-3-yl)ethyl)(4-(3-oxo-3-(2-propylhydrazinyl)prop-1-en-1-yl)benzyl)carbamate (12a)
Compound 11 (0.45 g, 1 mmol) was dissolved in 20 mL ethanol followed by the addition of 1.2 g anhydrous MgSO4 and propionaldehyde (0.07 g, 1.2 mmol). The reaction was monitored by TLC, after finished, MgSO4 was filtered and ethanol was removed under vacuum. The resulting residue was dissolved in 20 mL methanol, to this solution was added 5 mg methyl orange and 0.31 g (5 mmol) sodium cyanoborohydride (NaBH3CN). Mixture of methanol and concentrated hydrochloric acid (1:1) was added dropwise until the solution turned and stayed red. Six h later, volatiles were removed under vacuum and purified on reverse phase columns eluted with acetonitrile and water to yield pure product 12a (0.29 g, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 9.67 (s, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.41 (d, J = 15.8 Hz, 1H), 7.29–7.24 (m, 2H), 7.21 (d, J = 7.9 Hz, 2H), 6.96 (t, J = 7.4 Hz, 1H), 6.89 (t, J = 7.0 Hz, 1H), 6.51 (d, J = 15.8 Hz, 1H), 5.03 (s, 1H), 4.39 (s, 1H), 4.27 (s, 1H), 3.21 (s, 2H), 2.76 (s, 2H), 2.68 (t, J = 7.0 Hz, 2H), 2.25 (s, 3H), 1.46–1.41 (m, 2H), 1.34 (s, 9H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.28, 155.53, 140.65, 138.63, 135.63, 134.25, 132.50, 128.65, 128.51, 128.09, 120.44, 120.39, 118.52, 117.48, 110.83, 107.47, 79.20, 53.55, 49.59, 47.39, 28.43, 23.30, 21.24, 12.01, 11.49. ESI-MS m/z: 491.25 [M + H]+. HRMS (AP-ESI) m/z calcd for C29H38N4O3 [M + H]+ 491.30167, found 491.30157.
(E)-2-(2-methyl-1H-indol-3-yl)-N-(4-(3-oxo-3-(2-propylhydrazinyl)prop-1-en-1-yl)benzyl)ethanaminium 2,2,2-trifluoroacetate (13a)
Compound 12a (0.24 g, 0.5 mmol) was dissolved in 10 mL mixed solution of DCM and TFA (1:1), the solution was stirred at room temperature for 1 hour. The reaction was monitored by TLC, after finished, volatiles were removed under vacuum. Resulting residues were purified by C18Aq reverse phase column eluted with acetonitrile and water to achieve pure product 13a (0.13 g, 50% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.12 (s, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.61 (d, J = 16.0 Hz, 1H), 7.57 (d, J = 8.2 Hz, 2H), 7.41 (d, J = 7.7 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.04–6.98 (m, 1H), 6.97 – 6.91 (m, 1H), 6.71 (d, J = 15.9 Hz, 1H), 4.26 (s, 2H), 3.06 (s, 2H), 3.04–2.98 (m, 2H), 2.98–2.91 (m, 2H), 2.33 (s, 3H), 1.62–1.50 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.31, 140.56, 135.70, 135.44, 134.28, 133.16, 130.93, 128.56, 128.21, 120.72, 119.85, 118.82, 117.52, 111.07, 105.41, 52.52, 50.02, 47.48, 21.14, 19.27, 11.57, 11.53. HRMS (AP-ESI) m/z calcd for C23H29N5O [M + H]+ 391.24923, found 391.24972.
(E)-N-(4-(3-(2-(2-ammonioethyl)hydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (13b)
Using the synthetic method of 12a and 13a, 11 gave 13b as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 9.76 (s, 1H), 9.08 (s, 2H), 7.82 (s, 3H), 7.67 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 15.9 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.04–6.98 (m, 1H), 6.98–6.92 (m, 1H), 6.65 (d, J = 15.8 Hz, 1H), 4.25 (d, J = 5.4 Hz, 2H), 3.06–3.00 (m, 4H), 2.95 (t, J = 5.8 Hz, 2H), 2.86 (s, 2H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.00, 139.26, 135.75, 134.04, 133.22, 131.00, 128.42, 128.26, 121.15, 120.79, 118.89, 117.59, 111.14, 105.49, 50.05, 48.53, 47.50, 37.37, 21.19, 11.62. ESI-MS m/z: 392.17 [M + H]+. HRMS (AP-ESI) m/z calcd for C23H29N5O [M + H]+ 392.24449, found 392.24384.
(E)-N-(4-(3-(2-(2-cyanoethyl)hydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (13c)
Compound 11 (0.45 g, 1 mmol) was dissolved in ethanol followed by the addition of Acrylonitrile (0.066 g, 1.25 mmol), the mixed solution was refluxed at 80 °C for 48 h. Volatiles were evaporated under vacuum, the crude product was purified by C18Aq column on reverse phase columns eluted with acetonitrile and water to achieve pure product 12c (0.25 g, 50% yield). ESI-MS m/z: 446.17 [M + H]+. Using the synthetic method of 13a, 12c gave 13c as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.76 (s, 1H), 9.00 (s, 2H), 7.66 (d, J = 8.2 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 15.9 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.05–6.98 (m, 1H), 6.98–6.92 (m, 1H), 6.59 (d, J = 15.9 Hz, 1H), 4.25 (t, J = 5.5 Hz, 2H), 3.06 (s, 2H), 3.01–2.97 (m, 4H), 2.62 (t, J = 6.4 Hz, 2H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.95, 138.81, 135.88, 135.75, 133.92, 133.22, 131.01, 128.33, 128.28, 121.38, 120.78, 120.41, 118.88, 117.62, 111.13, 105.52, 50.05, 47.51, 47.22, 21.17, 16.84, 11.62. ESI-MS m/z: 402.25 [M + H]+. HRMS (AP-ESI) m/z calcd for C24H27N5O [M + H]+ 402.22884, found 402.22818.
(E)-N-(4-(3-(2-(3-methoxy-3-oxopropyl)hydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (13d)
Using the synthetic method of 12c, compound 11 and methyl acrylate gave compound 12d. ESI-MS m/z: 535.25 [M + H]+. Using the synthetic method of 13a, 12d gave 13d as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 10.02 (s, 1H), 9.15 (s, 2H), 7.66 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 15.8 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.04–6.98 (m, 1H), 6.98–6.92 (m, 1H), 6.63 (d, J = 15.9 Hz, 1H), 4.25 (t, J = 5.3 Hz, 2H), 3.61 (s, 3H), 3.08–3.03 (m, 6H), 2.54–2.52 (m, 2H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.54, 164.53, 139.12, 135.84, 135.75, 133.98, 133.23, 131.00, 128.41, 128.27, 121.03, 120.80, 118.89, 117.60, 111.14, 105.48, 51.94, 50.09, 47.53, 47.11, 32.43, 21.20, 11.63. ESI-MS m/z: 435.25 [M + H]+. HRMS (AP-ESI) m/z calcd for C25H30N4O3 [M + H]+ 435.23907, found 435.23871
tert-butyl 4-(hydrazinecarbonyl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (14a)
Compound 6a (0.42 g, 1 mmol) was dissolved in 20 mL methanol followed by the addition of 0.25 g (5 mmol) hydrazide monohydrate. The mixed solution was refluxed at 80 °C for 48 hours, after the reaction finished, volatiles were removed to afford product 14a (0.34 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 9.72 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.36–7.18 (m, 4H), 7.01–6.86 (m, 2H), 4.47 (d, J = 18.9 Hz, 2H), 3.29–3.19 (m, 2H), 2.82–2.72 (m, 2H), 2.27 (s, 3H), 1.36 (s, 9H). ESI-MS m/z: 423.17 [M + H]+.
tert-butyl 3-(hydrazinecarbonyl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (14b)
Using the synthetic method of 14a, 6c and hydrazide monohydrate gave 14b. 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 9.77 (s, 1H), 7.78–7.64 (m, 2H), 7.47–7.26 (m, 3H), 7.21 (d, J = 7.9 Hz, 1H), 7.00–6.87 (m, 2H), 4.45 (s, 2H), 3.24 (s, 2H), 2.84–2.66 (m, 2H), 2.27 (s, 3H), 1.38 (s, 9H). ESI-MS m/z: 423.17 [M + H]+.
tert-butyl benzyl(4-(hydrazinecarbonyl)benzyl)carbamate (14c)
Using the synthetic method of 14a, 10a and hydrazide monohydrate gave 14c. 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.35 (t, J = 7.2 Hz, 2H), 7.31–7.20 (m, 5H), 4.55–4.29 (m, 6H), 1.39 (s, 9H). ESI-MS m/z: 356.17 [M + H]+.
tert-butyl (2-(2-methyl-1H-indol-3-yl)ethyl)(4-(2-propylhydrazinecarbonyl)benzyl)carbamate (15a)
Using the synthetic method of 12a, compound 14a and propionaldehyde gave compound 15a. 1H NMR (400 MHz, DMSO-d6) δ 10.72 (s, 1H), 9.98 (d, J = 6.0 Hz, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 7.21 (d, J = 7.9 Hz, 1H), 6.96 (t, J = 7.3 Hz, 1H), 6.90 (t, J = 7.1 Hz, 1H), 5.08 (d, J = 6.0 Hz, 1H), 4.45 (s, 1H), 4.33 (s, 1H), 3.28–3.21(m, 2H), 2.78–2.73 (m, 4H), 2.27 (s, 3H), 1.51–1.41 (m, 2H), 1.36 (s, 9H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.50, 155.53, 142.52, 135.64, 132.52, 128.65, 127.75, 127.70, 127.36, 120.39, 118.52, 117.49, 110.83, 107.44, 79.24, 53.55, 49.58, 47.47, 28.41, 23.31, 21.30, 12.10, 11.50. ESI-MS m/z: 465.00 [M + H]+. HRMS (AP-ESI) m/z calcd for C27H36N4O3 [M + H]+ 465.28602, found 465.28555.
tert-butyl (2-(2-methyl-1H-indol-3-yl)ethyl)(3-(2-propylhydrazinecarbonyl)benzyl)carbamate (15b)
Using the synthetic method of 12a, compound 14b and propionaldehyde gave compound 15b. 1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 1H), 10.04 (s, 1H), 7.68 (d, J = 5.9 Hz, 2H), 7.43–7.33 (m, 3H), 7.21 (d, J = 7.9 Hz, 1H), 6.95 (t, J = 7.4 Hz, 1H), 6.88 (t, J = 6.8 Hz, 1H), 4.43 (s, 2H), 3.23 (s, 2H), 2.73 (t, J = 7.1 Hz, 4H), 2.25 (s, 3H), 1.49–1.40 (m, 2H), 1.35 (s, 9H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.77, 155.63, 135.58, 132.52, 128.60, 120.45, 118.58, 110.85, 107.45, 79.37, 53.50, 28.39, 21.21, 12.05, 11.42. HRMS (AP-ESI) m/z calcd for C27H36N4O3 [M + H]+ 465.28602, found 465.28622.
tert-butyl benzyl(4-(2-propylhydrazinecarbonyl)benzyl)carbamate (15c)
Using the synthetic method of 12a, compound 14c and propionaldehyde gave compound 15c. 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.37–7.34 (m, 2H), 7.31–7.20 (m, 5H), 5.10 (s, 1H), 4.38 (d, J = 21.7 Hz, 4H), 2.75 (t, J = 7.1 Hz, 2H), 1.50–1.42 (m, 2H), 1.39 (s, 9H), 0.91 (t, J = 7.4 Hz, 3H). ESI-MS m/z: 398.25 [M + H]+. 13C NMR (101 MHz, DMSO-d6) δ 165.47, 155.53, 142.15, 138.62, 132.46, 128.95, 127.96, 127.68, 79.84, 53.55, 31.16, 28.43, 21.31, 12.14. HRMS (AP-ESI) m/z calcd for C23H31N3O3 [M + H]+ 398.24382, found 398.24393.
2-(2-methyl-1H-indol-3-yl)-N-(4-(2-propylhydrazinecarbonyl)benzyl)ethanaminium 2,2,2-trifluoroacetate (16a)
Using the synthetic method of 13a, 15a gave 16a as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 10.12 (s, 1H), 9.01 (s, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.7 Hz, 1H), 7.04–6.99 (m, 1H), 6.96 (td, J = 7.5, 1.1 Hz, 1H), 5.24 (s, 1H), 4.28 (s, 2H), 3.15–2.95 (m, 4H), 2.77 (t, J = 7.1 Hz, 2H), 2.33 (s, 3H), 1.56–1.40 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.14, 159.16, 158.85, 135.67, 133.98, 133.14, 130.22, 128.19, 127.84, 120.70, 119.15, 118.80, 117.51, 116.17, 111.05, 105.41, 53.45, 49.90, 47.51, 21.22, 21.11, 12.10, 11.53. HRMS (AP-ESI) m/z calcd for C22H28N4O [M + H]+ 365.23359, found 365.23303.
2-(2-methyl-1H-indol-3-yl)-N-(3-(2-propylhydrazinecarbonyl)benzyl)ethanaminium 2,2,2-trifluoroacetate (16b)
Using the synthetic method of 13a, 15b gave 16b as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 10.21 (s, 1H), 8.97 (s, 2H), 7.98 (s, 1H), 7.88–7.78 (m, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.51 (t, J = 7.7 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.02–6.83 (m, 2H), 4.24 (s, 2H), 3.09–2.88 (m, 4H), 2.76 (t, J = 7.2 Hz, 2H), 2.29 (s, 3H), 1.51–1.36 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.41, 135.75, 133.99, 133.36, 133.22, 133.04, 129.82, 129.32, 128.27, 127.74, 120.81, 118.90, 117.60, 111.15, 105.46, 53.41, 50.25, 47.63, 21.21, 21.08, 12.13, 11.66. ESI-MS m/z: 365.17 [M + H]+. HRMS (AP-ESI) m/z calcd for C22H28N4O [M + H]+ 365.23359, found 363.23306.
N-benzyl-1-(4-(2-propylhydrazinecarbonyl)phenyl)methanaminium 2,2,2-trifluoroacetate (16c)
Using the synthetic method of 13a, 15c gave 16c as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (d, J = 3.9 Hz, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.42 (d, J = 8.3 Hz, 2H), 7.38–7.29 (m, 4H), 7.25–7.21 (m, 1H), 5.08 (d, J = 4.8 Hz, 1H), 3.72 (s, 2H), 3.67 (s, 2H), 2.75 (d, J = 2.7 Hz, 3H), 1.52–1.42 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.67, 144.78, 141.16, 131.94, 128.57, 128.38, 128.14, 127.38, 127.01, 53.58, 52.59, 52.22, 21.33, 12.15. HRMS (AP-ESI) m/z calcd for C18H23N3O [M + H]+ 298.19139, found 298.19150.
methyl 4-benzamidobenzoate (19)
methyl 4-aminobenzoate (18, 0.30 g, 2 mmol) was dissolved in 20 mL DCM followed by the addition of benzoyl chloride (17, 0.28 g, 2 mmol) and TEA (0.4 mL, 3 mmol) at 0 °C, the mixture was allowed to react at room temperature overnight. After the reaction finished, organic solution was washed by saturated NaHCO3 solution (2 × 30 mL), 1 M aqueous citric acid (2 × 30 mL), and brine (2 × 30 mL) and dried over MgSO4 overnight. The solvent was evaporated, resulting residues were recrystallized by ethyl estate and hexane to yield 19, a white solid (0.41 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 7.99–7.93 (m, 6H), 7.65–7.47 (m, 3H), 3.85 (s, 3H).
methyl 4-(benzyloxy)benzoate (22)
Phenylmethanol (20, 0.22 g, 2 mmol), methyl 4-hydroxybenzoate (21, 0.33 g, 2.2 mmol) and triphenylphosphine (0.79 g, 3 mmol) were dissolved in 40 mL anhydrous THF, to this solution was added Diethylazodicarboxylate (DEAD, 0.52 g, 3 mmol) at 0 °C. The reaction solution was allowed to stir at room temperature for 2 hours and monitored via TLC. After the reaction finished, volatiles were removed under vacuum, the crude product was purified by flash chromatography with hexanes and ethyl acetate to get pure product 22 (0.22 g, 45% yield). ESI-MS m/z: 243.17 [M + H]+.
4-(benzylamino)benzohydrazide (23a)
Using the synthetic method of 14a, 9b and hydrazide monohydrate gave 23a. 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 7.59 (d, J = 8.8 Hz, 1H), 7.37–7.31 (m, 4H), 7.26–7.22 (m, 1H), 6.80 (t, J = 6.0 Hz, 1H), 6.57 (d, J = 8.8 Hz, 2H), 4.36–4.30 (m, 4H).
N-(4-(hydrazinecarbonyl)phenyl)benzamide (23b)
Using the synthetic method of 14a, 19 and hydrazide monohydrate gave 23b. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 9.70 (s, 1H), 7.99–7.96 (m, 2H), 7.88–7.83 (m, 4H), 7.65–7.54 (m, 3H), 4.47 (s, 2H). ESI-MS m/z: 256.17 [M + H]+.
4-(benzyloxy)benzohydrazide (23c)
Using the synthetic method of 14a, 22 and hydrazide monohydrate gave 23c. ESI-MS m/z: 243.17 [M + H]+.
N-(4-(2-propylhydrazinecarbonyl)benzyl)benzenaminium 2,2,2-trifluoroacetate (24a)
Using the synthetic method of 12a, 23a gave 24a as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H), 7.56–7.48 (m, 2H), 7.34–7.24 (m, 4H), 7.21–7.16 (m, 1H), 6.78 (t, J = 6.1 Hz, 1H), 6.55–6.48 (m, 2H), 4.28 (d, J = 6.0 Hz, 2H), 2.65 (t, J = 7.1 Hz, 2H), 1.44–1.35 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.11, 151.66, 140.25, 128.95, 128.88, 127.69, 127.28, 120.45, 111.68, 53.88, 46.48, 21.39, 12.24. HRMS (AP-ESI) m/z calcd for C17H21N3O [M + H]+ 284.17574, found 284.17574.
N-(4-(2-propylhydrazinecarbonyl)phenyl)benzamide (24b)
Using the synthetic method of 12a, compound 23b and propionaldehyde gave compound 24b. 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.95 (s, 1H), 8.00–7.95 (m, 2H), 7.90–7.81 (m, 4H), 7.65–7.59 (m, 1H), 7.58–7.52 (m, 2H), 5.09 (s, 1H), 2.75 (t, J = 7.1 Hz, 2H), 1.48–1.36 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.37, 165.40, 142.37, 135.24, 135.19, 132.34, 130.77, 128.99, 128.58, 128.29, 119.97, 53.69, 21.41, 12.23. HRMS (AP-ESI) m/z calcd for C17H19N3O2 [M + H]+ 298.15500, found 298.15427.
4-(benzyloxy)-N’-propylbenzohydrazide (24c)
Using the synthetic method of 12a, compound 23c and propionaldehyde gave compound 24c. 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 7.84–7.75 (m, 2H), 7.49–7.44 (m, 2H), 7.43–7.38 (m, 2H), 7.37–7.32 (m, 1H), 7.10–7.03 (m, 2H), 5.20–5.12 (m, 2H), 5.04 (s, 1H), 2.73 (t, J = 7.0 Hz, 2H), 1.53–1.37 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.31, 161.05, 137.15, 129.27, 128.92, 128.40, 128.22, 126.04, 114.82, 69.76, 53.64, 21.33, 12.15. HRMS (AP-ESI) m/z calcd for C17H20N2O2 [M + H]+ 285.15975, found 285.15966.
(E)-N-(4-(3-(2,2-dimethylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (26a)
Using the synthetic method of 11, compound 6a and 1,1-dimethylhydrazine gave compound 25a. ESI-MS m/z: 477.33 [M + H]+. Using the synthetic method of 13a, 25a gave 26a as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.14 (s, 2H), 7.73–7.62 (m, 2H), 7.56–7.52 (m, 2H), 7.48–7.41 (m, 2H), 7.35 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.04–6.98 (m, 1H), 6.98–6.92 (m, 1H), 6.58 (d, J = 15.8 Hz, 1H), 4.25 (s, 2H), 3.06–3.01 (m, 4H), 2.54 (s, 6H), 2.33 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.94, 162.74, 158.93, 138.50, 135.94, 135.67, 133.68, 133.14, 130.90, 128.57, 128.19, 122.32, 120.71, 119.26, 118.80, 117.51, 116.17, 111.05, 105.41, 50.02, 48.52, 47.46, 46.87, 21.13, 11.57. HRMS (AP-ESI) m/z calcd for C23H28N4O [M + H]+ 377.23359, found 377.23434.
(E)-2-(2-methyl-1H-indol-3-yl)-N-(4-(3-oxo-3-(pyrrolidin-1-ylamino)prop-1-en-1-yl)benzyl)ethanaminium 2,2,2-trifluoroacetate (26b)
Using the synthetic method of 11, compound 6a and pyrrolidin-1-amine gave compound 25b. ESI-MS m/z: 503.33 [M + H]+. Using the synthetic method of 13a, 25b gave 26b as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 9.06 (s, 2H), 7.75–7.61 (m, 2H), 7.58–7.51 (m, 2H), 7.47 (d, J = 15.8 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.36 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.04–6.98 (m, 1H), 6.98–6.93 (m, 1H), 6.60 (d, J = 15.8 Hz, 1H), 4.24 (s, 2H), 3.06–3.00 (m, 4H), 2.88 (s, 4H), 2.33 (s, 3H), 1.72–1.82 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 167.50, 163.29, 159.01, 138.62, 136.15, 135.93, 135.67, 133.78, 133.72, 133.14, 130.90, 130.83, 128.21, 128.19, 122.08, 120.71, 118.81, 117.51, 111.05, 105.40, 55.89, 54.61, 50.03, 47.47, 22.33, 21.13, 11.56. HRMS (AP-ESI) m/z calcd for C25H30N4O [M + H]+ 403.24924, found 403.24993.
(E)-2-(2-methyl-1H-indol-3-yl)-N-(4-(3-oxo-3-(piperidin-1-ylamino)prop-1-en-1-yl)benzyl)ethanaminium 2,2,2-trifluoroacetate (26c)
Using the synthetic method of 11, compound 6a and piperidin-1-amine gave compound 25c. ESI-MS m/z: 517.42 [M + H]+. Using the synthetic method of 13a, 25c gave 26c as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.14 (s, 2H), 7.72–7.64 (m, 2H), 7.59–7.53 (m, 2H), 7.49 (d, J = 15.8 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.36 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.05–6.98 (m, 1H), 6.98–6.92 (m, 1H), 6.64 (d, J = 15.8 Hz, 1H), 4.31–4.17 (m, 2H), 3.06–2.98 (m, 4H), 2.89–2.76 (m, 4H), 2.32 (s, 3H), 1.70–1.51 (m, 4H), 1.39 (s, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.62, 139.20, 135.83, 135.69, 133.82, 133.15, 130.90, 128.30, 128.19, 121.80, 120.73, 118.81, 117.51, 111.06, 105.37, 57.52, 50.06, 47.49, 24.94, 23.04, 21.15, 11.58. HRMS (AP-ESI) m/z calcd for C26H32N4O [M + H]+ 417.26489, found 417.26410.
(E)-tert-butyl 4-(3-(2-ethyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (27a)
12a (0.24 g, 0.5 mmol) was dissolved in methanol, then acetaldehyde (0.044 g, 1 mmol) was added, 2 hours later, NaBH3CN (0.31 g, 5 mmol) was added followed by 2 drop of acetic acid. The mixed solution was allowed to stir overnight, after the reaction finished, volatiles were removed under vacuum. Resulting residues were purified by C18Aq reverse phase column eluted with acetonitrile and water to achieve pure product 27a (0.12 g, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 7.55–7.50 (m, 2H), 7.40 (d, J = 15.8 Hz, 1H), 7.34–7.18 (m, 4H), 7.00–6.85 (m, 2H), 6.56 (d, J = 15.8 Hz, 1H), 4.36 (s, 2H), 3.23 (s, 2H), 2.84–2.71 (m, 4H), 2.70–2.60 (m, 2H), 2.27 (s, 3H), 1.44–1.40 (m, 2H), 1.37 (s, 9H), 0.97 (td, J = 7.1, 3.0 Hz, 3H), 0.86 (td, J = 7.4, 2.1 Hz, 3H). ESI-MS m/z: 519.33 [M + H]+.
(E)-tert-butyl 4-(3-(2,2-dipropylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (27b)
Using the synthetic method of 27a, compounds 12a and propanal gave 27b. ESI-MS m/z: 533.42 [M + H]+.
(E)-tert-butyl 4-(3-(2-butyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (27c)
Using the synthetic method of 27a, compounds 12a and n-butanal gave 27c. ESI-MS m/z: 547.42 [M + H]+.
(E)-tert-butyl (2-(2-methyl-1H-indol-3-yl)ethyl)(4-(3-oxo-3-(2-pentyl-2-propylhydrazinyl)prop-1-en-1-yl)benzyl)carbamate (27d)
Using the synthetic method of 27a, compounds 12a and n-pentanal gave 27d. 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 8.16 (s, 1H), 7.61–7.48 (m, 2H), 7.39 (d, J = 15.7 Hz, 1H), 7.34–7.15 (m, 4H), 6.98–6,.89 (m, 2H), 6.54 (d, J = 15.8 Hz, 1H), 4.42 (s, 2H), 3.31–3.23 (m, 2H), 2.77 (s, 2H), 2.72–2.53 (m, 4H), 2.27 (s, 3H), 1.42–1.33 (m, 13H), 1.31–1.18 (m, 4H), 0.90–0.75 (m, 6H). ESI-MS m/z: 561.42 [M + H]+.
(E)-tert-butyl 4-(3-(2-hexyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (27e)
Using the synthetic method of 27a, compounds 12a and n-hexanal gave 27e. ESI-MS m/z: 575.42 [M + H]+.
(E)-N-(4-(3-(2-ethyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (28a)
Using the synthetic method of 13a, compound 27a gave 28a as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 9.11 (s, 2H), 7.67 (d, J = 7.3 Hz, 2H), 7.60–7.48 (m, 3H), 7.42–7.33 (m, 2H), 7.26 (d, J = 7.7 Hz, 1H), 7.00 (t, J = 7.2 Hz, 1H), 6.99–6.94 (m, 1H), 6.68 (d, J = 15.8 Hz, 1H), 4.23 (s, 2H), 3.05–2.95 (m, 6H), 2.90–2.78 (m, 2H), 2.32 (s, 3H), 1.49–1.38 (m, 2H), 1.01 (dt, J = 26.6, 6.7 Hz, 3H), 0.87 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.61, 164.29, 158.81, 136.16, 135.64, 133.97, 133.19, 130.91, 128.46, 128.15, 120.76, 118.86, 117.50, 111.07, 105.34, 60.21, 59.09, 50.06, 47.49, 21.09, 19.31,12.50, 12.09, 11.51. HRMS (AP-ESI) m/z calcd for C26H34N4O [M + H]+ 419.28053, found 419.28153.
(E)-N-(4-(3-(2,2-dipropylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (28b)
Using the synthetic method of 13a, compound 27b gave 28b as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.12 (s, 2H), 7.66 (t, J = 8.3 Hz, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.48 (d, J = 16.1 Hz, 1H), 7.43–7.40 (m, 1H), 7.36 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.98–6.91 (m, 1H), 6.66 (d, J = 15.8 Hz, 1H), 4.25 (t, J = 5.4 Hz, 2H), 3.13–2.92 (m, 4H), 2.76 (t, J = 7.2 Hz, 2H), 2.71–2.55 (m, 2H), 2.33 (s, 3H), 1.46–1.39 (m, 4H), 0.96–0.82 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.25, 164.11, 159.19, 139.91, 136.20, 135.81, 135.68, 133.85, 133.14, 130.96, 128.29, 128.19, 120.69, 119.84, 118.80, 117.52, 111.05, 105.42, 60.62, 59.35, 50.01, 47.46, 21.12, 20.24, 19.92, 12.12, 11.90, 11.54. HRMS (AP-ESI) m/z calcd for C27H36N4O [M + H]+ 433.29619, found 433.29717.
(E)-N-(4-(3-(2-butyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (28c)
Using the synthetic method of 13a, compound 27c gave 28c as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.15 (s, 2H), 7.67–7.64 (m, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 14.5 Hz, 1H), 7.41 (dd, J = 7.5, 3.7 Hz, 1H), 7.34 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.98–6.91 (m, 1H), 6.67 (d, J = 15.8 Hz, 1H), 4.25 (t, J = 5.2 Hz, 2H), 3.15–2.92 (m, 4H), 2.82–2.75 (m, 2H), 2.73–2.55 (m, 2H), 2.33 (s, 3H), 1.50–1.24 (m, 6H), 0.93–0.77 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.24, 164.05, 158.68, 136.20, 135.85, 135.67, 133.78, 133.14, 130.96, 130.89, 128.30, 128.27, 128.19, 120.70, 118.80, 118.58, 117.51, 111.05, 105.40, 60.61, 59.30, 50.01, 47.43, 29.12, 21.13, 20.24, 20.15, 14.35, 12.13, 11.56. HRMS (AP-ESI) m/z calcd for C28H38N4O [M + H]+ 447.31183, found 447.31278.
(E)-2-(2-methyl-1H-indol-3-yl)-N-(4-(3-oxo-3-(2-pentyl-2-propylhydrazinyl)prop-1-en-1-yl)benzyl)ethanaminium 2,2,2-trifluoroacetate (28d)
Using the synthetic method of 13a, compound 27d gave 28d as a pale solid 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 8.99 (s, 2H), 7.68–7.63 (m, 2H), 7.58–7.52 (m, 2H), 7.52–7.38 (m, 2H), 7.35 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.98–6.92 (m, 1H), 6.63 (d, J = 15.8 Hz, 1H), 4.24 (d, J = 5.1 Hz, 2H), 3.04 (d, J = 16.6 Hz, 4H), 2.78–2.53 (m, 4H), 2.33 (s, 3H), 1.45–1.23 (m, 8H), 0.90–0.77 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.32, 164.12, 159.03, 139.92, 136.29, 136.00, 135.75, 133.87, 133.80, 133.23, 131.03, 130.98, 128.38, 128.33, 128.26, 120.80, 118.89, 117.60, 111.14, 105.47, 60.73, 59.29, 50.08, 47.53, 29.64, 26.71, 26.63, 22.59, 22.55, 21.21, 20.33, 20.28, 14.49, 12.21, 11.65. HRMS (AP-ESI) m/z calcd for C29H40N4O [M + H]+ 461.32749, found 461.32721.
(E)-N-(4-(3-(2-hexyl-2-propylhydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (28e)
Using the synthetic method of 13a, compound 27e gave 28e as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.00 (s, 2H), 7.67–7.63 (m, 2H), 7.59–7.52 (m, 2H), 7.48 (d, J = 13.8 Hz, 1H), 7.43–7.39 (m, 1H), 7.34 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 7.04–6.98 (m, 1H), 6.98–6.91 (m, 1H), 6.63 (d, J = 15.8 Hz, 1H), 4.25 (s, 2H), 3.05–3.02 (m, 4H), 2.74–2.53 (m, 4H), 2.33 (s, 3H), 1.44–1.35 (m, 4H), 1.32–1.19 (m, 6H), 0.92–0.74 (m, 6H). 13C NMR (101 MHz, DMSO-d6) δ 168.33, 164.14, 158.95, 139.14, 136.29, 135.90, 135.75, 133.86, 133.22, 131.00, 128.38, 128.26, 120.79, 118.88, 117.59, 111.14, 105.47, 60.74, 59.39, 50.08, 47.54, 47.47, 31.67, 27.06, 26.99, 26.65, 22.59, 21.20, 20.32, 20.00, 14.38, 12.21, 11.99, 11.64. HRMS (AP-ESI) m/z calcd for C30H42N4O [M + H]+ 475.34314, found 475.34277.
(E)-tert-butyl 4-(3-(2,2-bis(3-hydroxypropyl)hydrazinyl)-3-oxoprop-1-en-1-yl)benzyl(2-(2-methyl-1H-indol-3-yl)ethyl)carbamate (29)
Compound 11 (0.22 g, 0.5 mmol) was dissolved in 10mL ethanol, to this solution was added 3-Bromo-1-propanol (0.14 g, 1 mmol) followed by the addition of K2CO3 (0.27 g, 2 mmol). The mixture was refluxed at 90 °C for 1 hour, K2CO3 was filtered, the volatiles were removed under vacuum. Resulting residues were purified by C18Aq reverse phase column eluted with acetonitrile and water to achieve pure product 29 (0.20 g, 70% yield). 1H NMR (400 MHz, MeOD) δ 8.34 (s, 1H), 7.67 (d, J = 15.2 Hz, 1H), 7.42–7.21 (m, 4H), 7.15–6.99 (m, 4H), 6.36 (d, J = 15.6 Hz, 1H), 4.31–4.16 (m, 2H), 3.74–3.62 (m, 4H), 3.36–3.27 (m, 2H), 2.91–2.60 (m, 6H), 2.24 (s, 3H), 1.71–1.63 (m, 4H), 1.47 (s, 9H). ESI-MS m/z: 565.42 [M + H]+.
(E)-N-(4-(3-(2,2-bis(3-hydroxypropyl)hydrazinyl)-3-oxoprop-1-en-1-yl)benzyl)-2-(2-methyl-1H-indol-3-yl)ethanaminium 2,2,2-trifluoroacetate (30)
Using the synthetic method of 13a, 29 gave 30 as a pale solid. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1H), 9.15 (s, 2H), 7.69–7.63 (m, 2H), 7.56 (dd, J = 8.3, 2.0 Hz, 2H), 7.52–7.38 (m, 2H), 7.33 (d, J = 16.1 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.00 (dd, J = 11.0, 4.0 Hz, 1H), 6.95 (dd, J = 10.7, 3.9 Hz, 1H), 6.65 (d, J = 15.8 Hz, 1H), 4.25 (s, 2H), 3.49–3.44 (m, 4H), 3.08–3.00 (m, 4H), 2.86–2.56 (m, 4H), 2.33 (s, 3H), 1.64–1.47 (m, 4H). 13C NMR (101 MHz, DMSO-D6) δ 164.38, 138.88, 135.96, 135.75, 133.21, 130.99, 128.46, 128.27, 120.77, 118.87, 117.61, 111.12, 105.53, 59.23, 55.01, 50.06, 49.13, 21.18, 11.59. HRMS (AP-ESI) m/z calcd for C27H36N4O3 [M + H]+ 465.28602, found 465.28540.
Cell culture
All of the cell lines were purchased from ATCC. The MV4–11 cell line was cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) Anti-Anti in 5% CO2 at 37 °C. The HL-60 cell line was cultured in IMDM medium supplemented with 20% FBS and 1% (v/v) Anti-Anti in 5% CO2 at 37 °C. Molm-14, RS4;11, and U937 were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) Anti-Anti in 5% CO2 at 37 °C. Primary AML cells were cultured in RPMI-1640 medium supplemented with human hematopoietic stem cell expansion cytokine package (IL-3) from Peprotech. All human studies were approved by the Yale University Human Investigation Committee.
In vitro HDACs inhibition fluorescence assay
All of the HDAC enzymes were purchased from BPS Bioscience. In vitro HDAC inhibition assays were conducted as previously described.26 In brief, 20 μL of recombinant HDAC enzyme solution (HDAC1–9) was mixed with various concentrations of tested compound (20 μL). The mixture was incubated at 30 °C for 1 h (for the time-dependent assay, the mixture was incubated for 15 min, 30 min, 60 min and 90 min, respectively), then 10 μL fluorogenic substrate (Boc-Lys (acetyl)-AMC for HDAC1, 2, 3, and 6, Boc-Lys (triflouroacetyl)-AMC for HDAC 4, 5, 7, 8, and 9) was added to a final concentration of 50 μM. After incubation at 30 °C for 2 h, the catalysis was stopped by addition of 10 μL of developer containing 30 mg/mL trypsin and 6 μM trichostatin A (TSA). 30 mins later, fluorescence intensity was measured using a microplate reader at excitation and emission wavelengths of 360 and 460 nm, respectively. The inhibition ratios were calculated from the fluorescence intensity readings of tested wells relative to those of control wells, and the IC50 curves and values were determined by GraphPad Prism 6.0, using the “log(inhibitor) vs. normalized response-variable slope” function.
HDACs 1 and 3 kinetics study
All of the HDAC enzymes were bought from BPS Bioscience. 40 μL of recombinant human HDAC enzyme (HDAC1 and 3) solution was added in 96-well format to black U-bottom plates. 40 μL of 13a in HDAC buffer was added to a final concentration of 15 nM, 10 nM, 6.67 nM, 2.44 nM, and 2.96 nM for HDAC1 and 3, 2 nM, 1.33 nM, 0.89 nM, 0.59 nM for HDAC3, respectively. The mixture was allowed to incubate for 2 h at 30 °C before the addition of 20 μL of fluorogenic substrate Boc-Lys(acetyl)-AMC serially diluted from 200 μM to 25 μM, 2 times dilution. Two hours later, 20 μL of 5 mg/mL trypsin and 1 μM TSA solution was added to quench the reaction. Fluorescence was read at 360 nm (ex.)/460 nm (em.) using a Tecan M200 Pro. Vmax and Km were determined using GraphPad Prism’s Michaelis-Menten function. Corresponding Lineweaver-Burke double reciprocal plots were derived from these values and plotted accordingly.
Molecular Docking Against HDAC1 and HDAC3
Docking sites were defined using sitefinder, and simulations used a rigid receptor and triangle matcher mythology for ligand site placement with London dG scoring. Thirty poses were calculated per binding site and the top 5 poses refined using GBVI/WSA rescoring (E_Score2, implemented in MOE 2019 from CCG). Structural files were PDB: 5ICN for HDAC1 and PDB: 4A69 for HDAC3.
MMP-2/MMP-9 inhibitory activity
The InnoZyme™ Gelatinase (MMP-2/MMP-9) Activity Assay Kit was purchased from EMD Chemicals, Inc. 13a (45 μL), MMP inhibitor (positive control from Kit, 45 μL) and activation buffer (45 μL) were added to designated wells, the final comcentrations of 13a were 50 μM and 10 μM. 45 μL of MMP-2/MMP-9 diluted in the activation buffer was then added to each well. After 10–15 min incubation, 10 μL substrate working solution was added. The mixture was incubated at 37 °C for 2 h, and fluorescence was read at 320 nm (ex.)/405 nm (em.) using a Tecan M200 Pro.
CD13 inhibition assay
40 μL of 13a (final comcentrations were 50 μM and 10 μM) and CD13 inhibitor bestatin (final comcentrations was 10 μM) were added into 96-well clear plate followed by the addition of CD13 enzyme (microsomal aminopeptidase from porcine kidney microsomes, Sigma) solution. 5 min later, the substrate (L-leucine-p-nitroanilide) was added and the mixture was incubated at 37 °C for 30 min. The absorbance was measured at 405 nm with a microplate reader.
EC50 analysis.
MV4–11 (20,000 per well) or primary leukemia (100,000 per well) cells were plated in 96-well clear plates and pre-incubated overnight, then serially diluted inhibitors (3 times dilution) with 12 concentrations from 5 μM were added. Cells were incubated with compounds for 48 h. CellTiter-Blue was added to a final concentration of 0.125 mg/mL. The mixture was allowed to incubate for 2–4 h until sufficient color changed occurred. Cell viability was measured as a function of resorufin fluorescence intensity using a Tecan M200 Pro spectrophotometer, 560 nm/590 nm (excitation/emission). Data were normalized to control wells and background was removed. EC50s were determined using GraphPad Prism’s “log(inhibitor) vs. normalized response-variable slope” function.
NCI-60 screening methodology
All cell lines were maintained in RPMI1640 medium containing 10% FBS at 37°C in a 5% CO2 humidified incubator. 5,000–10,000 cells were plated per well in 96-well clear plates and pre-incubated overnight. CellTiter-Blue was added to selected wells to a final concentration of 0.125 mg/mL to present a measurement of the cell population for each cell line at the time of drug addition (TZ). At the same time, serially 10 times diluted inhibitor 13a (6 concentrations) and medium at the same volume (100% control) was added to the rest wells, the maximal final concentration was 10 μM. Following drug addition, the plates were incubated for an additional 48 h at 37°C in a 5% CO2, then CellTiter-Blue was added to each well to a final concentration of 0.125 mg/mL. The mixture was allowed to incubate for 2–4 h until sufficient color change occurred. Cell viability was measured as a function of resorufin fluorescence intensity using a Tecan M200 Pro spectrophotometer, 560 nm/590 nm (excitation/emission). The absorbance of time zero (Tz), 100% control with no drugs (C), and test growth in the presence of the drug at 6 concentration levels (Ti) were used for calculation of percentage growth inhibition.
Dose-response parameters were calculated for each experimental agent. Growth inhibition of 50% (GI50) was calculated from [(Ti-Tz)/(C-Tz)] ×100 = 50, which is the drug concentration resulting in a 50% reduction in control cells during the drug incubation. LC50 (concentration of drug resulting in a 50% reduction as compared to that at the beginning) was calculated from [(Ti-Tz)/Tz] ×100 = −50.
Western blot analysis
MV4–11 or HL-60 cells were plated (1,500,000 per well) in flat bottom 6-well plates and allowed to grow for 12 h, and then treated with desired drugs. Final concentrations of tested drugs were shown in the figure legends of Figure 3, Figure 5, Figure 6, Figure 7, and Figure 8. After treatment, cells were harvested, washed by PBS, and lysed with RIPA buffer, which was comprised of 50 mM Tris Base,150 mM NaCl, 5 mM EDTA, 0.1% (v/v) SDS, 0.5% (v/v) Sodium Deoxycholate, and 1% (v/v) Triton-x-100. After lysing, the suspension was ultra-sonicated and centrifuged at 14000 RPM for 15 min at 4°C. The mixture of 75 mL of supernatant and 25 mL of β-mercaptoethanol: NuPAGE® Lithium dodecyl sulfate (LDS) Sample Buffer (4X) (15:85) was heated at 90 °C for 15 min and normalized according to a BCA test before loading. Lysates were run on Invitrogen NuPAGE 4–12% Bis-Tris 15 well gels at 170 V for approximately 60 min in MES buffer. Gels were transferred to the methylcellulose membrane and ran at 30 V for 3 h. Membranes were incubated at 4 °C overnight with 1/1000 primary antibodies, which were diluted in 2.5% (w/v) milk or 5% (w/v) bovine serum albumin. The membrane was washed twice with Tris-buffered saline and with Tween (TBST buffer) before incubation with secondary antibodies. Images were acquired using a GE ImageQuant LAS 4000.
Antibodies
Ac-Histone H3 (Lys9/14)-R, Ac-Histone H4 (Ser 1/Lys 5/Lys 8/Lys 12), acetylated α-tubulin (6–11B-1), Flt-3/Flk-2 (C-20), STAT5 (A-9), p-ERK 1/2 (Thr202/Tyr204)-R, HSP70 (K-20), p53 (DO-1), XIAP (E-2), caspase-3 (31A1067), ERK 1 (K-23), p-p53 (59. Ser315), MDM2 (SMP14) and FlipS/L (G-11) antibodies were purchased from Santa Cruz Biotechnology. Actin and LC3B antibodies were purchased from Abcam.
Combination index (CI) test
20,000 MV4–11 cells were plated per well in 96-well clear plates and pre-incubated overnight, then serially diluted 13a and RG7388, bortezomib, or 17-AAG were added alone and combined. For the experiment for 13a and RG7388, concentration of 13a was 25 nM, RG7388 was 2 times dilution from 500 nM. For the experiment of 13a and bortezomib, concentrations of 13a were 405 nM, 135 nM, 45 nM, 15 nM, and 5 nM, respectively, while concentration of bortezomib was 8 nM. In the experiment of 13a in combination with 17-AAG, final comcentration of 13a was 9.87 nM, concentrations of 17-AAG were 300 nM, 200 nM, 133 nM, 88 nM, and 59 nM, respectively. Then cells were incubated for 24 h (RG7388 and bortezomib) and 48 h (17-AAG), CellTiter-Blue was added to each well to a final concentration of 0.125 mg/mL. The mixture was allowed to incubate for 2–4 h until sufficient color changed occurred. Cell viability was measured as a function of resorufin fluorescence intensity using a Tecan M200 Pro spectrophotometer, 560 nm/590 nm (excitation/emission). The combination index (CI) was calculated using CompuSyn software.
MV4–11 cells rescue study
MV4–11 cells were maintained in IMDM medium containing 10% FBS at 37 °C in a 5% CO2 humidified incubator. Cells were plated (50,000 per well) in flat-bottom 24-well plates and allowed to grow for 12 h, then CellTiter-Blue was added to selected wells to a final concentration of 0.125 mg/mL to measure the cell viability at the time of inhibitor addition (t = 0), and was incubated for 2–4 h until sufficient color changed occurred. At the same time, the remaining wells were treated with 13a (200 nM), 13a (200 nM) with z-VAD (20 μM), chloroquine (5 μM) or wortmannin (100 nM) for 24 h. CellTiter-Blue was added to each well to a final concentration of 0.125 mg/mL. The mixture was allowed to incubate for the same time as the t = 0 wells. Cell viability was measured as a function of resorufin fluorescence intensity using a Tecan M200 Pro spectrophotometer, 560 nm/590 nm (excitation/emission). Data were normalized to control wells and background was removed. Cell viability with each treatment was compared with t = 0.
Mini-Ames tests
Compound mutagenic properties were tested using EBPI’s Modified Ames ISO kit. Bacterial strains used in this test carry a specific mutation in the histidine biosynthesis pathway. As a result, Ames strains are histidine dependent and will not survive in histidine-deficient media. However, specific mutations caused by toxicant interactions at sites in the bacterial DNA can produce reversions back to the wild type genome, a state in which the bacteria regain the ability to synthesize histidine. Lyophilized Salmonella typhimurium strain (with modified TA100) was rehydrated in growth media, covered with a rubber stopper, and incubated at 37 °C with shaking for 16 h. Overnight TA100 cultures were diluted to OD600 = 0.05 into 1× Exposure media. To a 24 well plate, 1600 μL of water was added into negative control and sterility control wells while 1550 μL of water with 50 μL of 2-aminoanthracene (2-AA) was added into positive control wells. 1600 μL sterilized inhibitor solution was added into “No dilution” wells to a final concentration of 10 μM. Four sets of serial dilutions of compounds were performed in a separate sterilized container. 1.6 mL of diluted sample was transferred into the appropriate wells of the 24-well exposure plate. As 2-AA is a pro-mutagen that needs metabolic bioactivation through the use of S9 liver homogenate, 68 μL of S9 mix was added followed by the addition of 200 μL exposure solution containing D-glucose, D-biotin and bit of L-histidine. Then 200 μL of the overnight culture was added to all wells, except the sterility control well, and the mixture was allowed to incubate at 37 °C for 100 min. 1600 μL mixture was transferred from each well of the exposure plate to conical tubes and 8 mL reversion media was added. Each tube was vortex-mixed and emptied onto a loading boat. Using a multichannel pipette, 200 μL of the reversion mix from the reagent boats were immediately aliquoted into a 96 well plate (one 15 mL conical tube should be aliquoted in 48 wells from the 96 well plate). Plates were sealed into the included zip-top bags and the 96 well plates were incubated for 2–3 days at 37 °C (no shaking). Plates were scored visually; yellow and partial yellow wells are scored as positive while purple wells are scored as negative.
Pharmacokinetics study
All animal experiments were conducted under the guidelines of the “Guide for the care and use of laboratory animals” published by the National Institutes of Health (revised 2011) and under the approval of the Institutional Animal Care and Use Committee (IACUC# IACUC-2017–00079) of the Medical University of South Carolina (MUSC). Male CD‐1 Mice (Charles River Laboratories) were fed a standard laboratory rodent diet and housed in individual cages on a 12 h light and 12 h dark cycle with room temperature maintained at 22 ± 3 °C and relative humidity at 50 ± 20%. Animals were typically fasted overnight before dosing, with food returned after the 6 h blood samples were obtained. Compound 13a was dissolved in PBS containing 5% DMSO. Intravenous bolus injection (iv) and oral gavage (po) were performed at a dose of 5 mg/kg and 20 mg/kg with a volume of 10 mL/kg, respectively. 3 mice were used for iv and po administration. All blood samples (100–300 μL per sample) were taken via appropriate vein (saphenous, jugular, or submandibular vein) at 5, 15, and 30 min and 1, 2, 4, 6, 8, and 24 h after dosing. Fluid replacement (1.5 mL of 0.9% sodium chloride injection, USP) was administered subcutaneously once after the 2 h blood sampling. Blood samples were collected in BD Microtainer tubes coated with anticoagulant, placed on ice, and within 30 min, centrifuged at 15,000 × g for 5 min to obtain plasma samples. All plasma samples were stored at −70 °C until analysis.
Plasma samples were prepared as follows. Two or three volumes of acetonitrile containing internal standard were added to one volume of plasma to precipitate proteins. Samples were centrifuged (3000 × g for 10 min) and the supernatant removed for analysis by LC-MS/MS. Calibration standards and quality controls were made by preparation of a 1 mg/mL stock solution and subsequently a series of working solutions in methanol: water (1/:1, v/v) which were spiked into the blank plasma to yield a series of calibration standard samples in the range of 1 ng/mL to 10 μg/mL and quality control samples at three concentration levels (low, middle and high). All incurred PK/PD plasma samples were treated identically to the calibration standards and quality control samples. LC-MS-MS analysis was performed utilizing multiple reaction monitoring for detection of characteristic ions for each drug candidate, additional related analytes and internal standard.
Plasma concentrations were measured as described above to determine a concentration vs. time profile. The area under the plasma concentration vs. time curve (AUC) was calculated using the linear trapezoidal method. Fitting of the data to obtain pharmacokinetic parameters was carried out using non-compartmental analysis. Key PK parameters reported following intravenous administration are as follows: terminal half-life t1/2, initial plasma concentration C0, area under the plasma concentration vs. time curve AUC, the volume of distribution at steady-state Vss, total plasma clearance CLp, and mean residence time MRT. Key PK parameters reported following extravascular administration are as follows: terminal half-life t1/2, maximum plasma concentration Cmax, time to reach maximum plasma concentration tmax, area under the plasma concentration vs. time curve AUC, mean residence time MRT, and bioavailability F. All parameters are expressed for individual animals as well as mean, standard deviation, and coefficient of variation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health/National Cancer Institute (CA163452 to C.J.C.) and the National Institutes of Health/National Institute of General Medical Sciences (P20GM103542). We’d like to thank the National Cancer Institute for the NCI60 screening. We also like to thank Joshua A. Wilhide from Molecular Characterization and Analysis Complex of the University of Maryland, Baltimore County for high-resolution mass spectrometry support.
Abbreviations used
- Ara-C
cytarabine
- FLT3
FMS-like tyrosine kinase 3
- IDH
isocitrate dehydrogenase
- Bcl-2
B-cell lymphoma-2
- NPM1
Nucleophosmin 1
- STAT5
signal transducer and activator of transcription 5
- XIAP
X-linked inhibitor of apoptosis
- c-FLIP
cellular FLICE-like inhibitory protein
- mTOR
mammalian target of rapamycin
- DRAM
damage-regulated autophagy modulator
- ZBG
zinc-binding group
- HDACI
HDAC inhibitor
- NaBH3CN
sodium cyanoborohydride
- Boc2O
di-tert-butyl decarbonate
- TEA
triethylamine
- TBTU
O-(Benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate
- MgSO4
magnesium sulfate anhydrous
- PPh3
triphenylphosphine
- K2CO3
potassium carbonate
- CS
catalytic site
- AS
allosteric site
- OS
overall survival
- FLT3-ITD
FLT3 internal tandem duplication
- GI50
concentration for 50% of maximal inhibition of cell proliferation
- LC50
Median lethal concentration
- Cmax
mean peak plasma concentration
- FL
FLT3 ligand
- MDM2
murine double minute 2
- CI
combination index
Footnotes
Supporting Information:
IC50 curves of representative compounds 12a, 16a-16c, 24a, 24b, 28b against HDAC1, 2, and 3. EC50 curves of SAHA, MS275, panobinostat, 13a, 16a, 16b, 16c and 28b for MV4–11 cells. Inhibitory activity of 13a against APN/CD13 and MMP2/9. IC50 curves of 13a against HDAC1–9. Western blot study of 13a, CZ411, panobinostat (Pan), vorinostat (Vor) and entinostat (Ent) in wt-p53 SR cell line. Combination Index (CI) for 13a and 17-AAG after treatment for 48 h. 1H-NMR and 13C-NMR spectrum and HPLC trance of representative target compounds (PDF).
Molecular Formula Strings (CSV).
Competing interests
C.J.C., S.S.L.C., and R.A.H. are the co-founders of Lydex Pharmaceuticals. The other authors declare no competing financial interest.
Accession Code
Atomic coordinates include HDAC1 (PDB: 5ICN) in complex with 13a and HDAC3 (PDB: 4A69) in complex with 13a. Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Lowenberg B; Downing JR; Burnett A, Acute myeloid leukemia. N. Engl. J. Med 1999, 341, 1051–1062. [DOI] [PubMed] [Google Scholar]
- (2).Redaelli A; Lee JM; Stephens JM; Pashos CL, Epidemiology and clinical burden of acute myeloid leukemia. Expert Rev. Anticancer Ther 2003, 3, 695–710. [DOI] [PubMed] [Google Scholar]
- (3).Tallman MS; Wang ES; Altman JK; Appelbaum FR; Bhatt VR; Bixby D; Coutre SE; De Lima M; Fathi AT; Fiorella M, Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Network 2019, 17, 721–749. [DOI] [PubMed] [Google Scholar]
- (4).Wei AH; Tiong S, Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood. 2017, 130, 2469–2474. [DOI] [PubMed] [Google Scholar]
- (5).Miller KD; Nogueira L; Mariotto AB; Rowland JH; Yabroff KR; Alfano CM; Jemal A; Kramer JL; Siegel RL, Cancer treatment and survivorship statistics, 2019. Ca-Cancer J. Clin 2019, 69, 363–385. [DOI] [PubMed] [Google Scholar]
- (6).Deschler B; Lubbert M, Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006, 107, 2099–2107. [DOI] [PubMed] [Google Scholar]
- (7).Druker BJ, Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008, 112, 4808–4817. [DOI] [PubMed] [Google Scholar]
- (8).Kindler T; Lipka DB; Fischer T, FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010, 116, 5089–5102. [DOI] [PubMed] [Google Scholar]
- (9).Stirewalt DL; Radich JP, The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [DOI] [PubMed] [Google Scholar]
- (10).Kelly LM; Liu Q; Kutok JL; Williams IR; Boulton CL; Gilliland DG, FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002, 99, 310–318. [DOI] [PubMed] [Google Scholar]
- (11).Verhaak RG; Goudswaard CS; van Putten W; Bijl MA; Sanders MA; Hugens W; Uitterlinden AG; Erpelinck CA; Delwel R; Lowenberg B; Valk PJ, Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005, 106, 3747–3754. [DOI] [PubMed] [Google Scholar]
- (12).Ikeda H; Kanakura Y; Tamaki T; Kuriu A; Kitayama H; Ishikawa J; Kanayama Y; Yonezawa T; Tarui S; Griffin JD, Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood. 1991, 78, 2962–2968. [PubMed] [Google Scholar]
- (13).Neubauer A; Dodge RK; George SL; Davey FR; Silver RT; Schiffer CA; Mayer RJ; Ball ED; Wurster-Hill D; Bloomfield CD; et al. , Prognostic importance of mutations in the ras proto-oncogenes in de novo acute myeloid leukemia. Blood. 1994, 83, 1603–1611. [PubMed] [Google Scholar]
- (14).Radich JP; Kopecky KJ; Willman CL; Weick J; Head D; Appelbaum F; Collins SJ, N-ras mutations in adult de novo acute myelogenous leukemia: prevalence and clinical significance. Blood. 1990, 76, 801–807. [PubMed] [Google Scholar]
- (15).Coghlan DW; Morley AA; Matthews JP; Bishop JF, The incidence and prognostic significance of mutations in codon 13 of the N-ras gene in acute myeloid leukemia. Leukemia. 1994, 8, 1682–1687. [PubMed] [Google Scholar]
- (16).Min YH; Eom JI; Cheong JW; Maeng HO; Kim JY; Jeung HK; Lee ST; Lee MH; Hahn JS; Ko YW, Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia. 2003, 17, 995–997. [DOI] [PubMed] [Google Scholar]
- (17).Birkenkamp KU; Geugien M; Lemmink HH; Kruijer W; Vellenga E, Regulation of constitutive STAT5 phosphorylation in acute myeloid leukemia blasts. Leukemia. 2001, 15, 1923–1931. [DOI] [PubMed] [Google Scholar]
- (18).Milella M; Kornblau SM; Estrov Z; Carter BZ; Lapillonne H; Harris D; Konopleva M; Zhao S; Estey E; Andreeff M, Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J. Clin. Invest 2001, 108, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Grandage VL; Gale RE; Linch DC; Khwaja A, PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 2005, 19, 586–594. [DOI] [PubMed] [Google Scholar]
- (20).Patel JP; Gonen M; Figueroa ME; Fernandez H; Sun Z; Racevskis J; Van Vlierberghe P; Dolgalev I; Thomas S; Aminova O; Huberman K; Cheng J; Viale A; Socci ND; Heguy A; Cherry A; Vance G; Higgins RR; Ketterling RP; Gallagher RE; Litzow M; van den Brink MR; Lazarus HM; Rowe JM; Luger S; Ferrando A; Paietta E; Tallman MS; Melnick A; Abdel-Wahab O; Levine RL, Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med 2012, 366, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fredly H; Gjertsen BT; Bruserud Ø, Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. epigenet 2013, 5, 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Matthews GM; Mehdipour P; Cluse LA; Falkenberg KJ; Wang E; Roth M; Santoro F; Vidacs E; Stanley K; House CM, Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood. 2015, 126, 2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wada T; Kikuchi J; Nishimura N; Shimizu R; Kitamura T; Furukawa Y, Expression levels of histone deacetylases determine the cell fate of hematopoietic progenitors. J. Biol. Chem 2009, 284, 30673–30683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mehdipour P; Santoro F; Botrugno O; Romanenghi M; Pagliuca C; Matthews G; Johnstone R; Minucci S, HDAC3 activity is required for initiation of leukemogenesis in acute promyelocytic leukemia. Leukemia. 2017, 31, 995–997. [DOI] [PubMed] [Google Scholar]
- (25).McClure JJ; Zhang C; Inks ES; Peterson YK; Li J; Chou CJ, Development of Allosteric Hydrazide-Containing Class I Histone Deacetylase Inhibitors for Use in Acute Myeloid Leukemia. J. Med. Chem 2016, 59, 9942–9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li X; Peterson YK; Inks ES; Himes RA; Li J; Zhang Y; Kong X; Chou CJ, Class I HDAC inhibitors display different antitumor mechanism in leukemia and prostatic cancer cells depending on their p53 status. J. Med. Chem 2018, 61, 2589–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang H; Zhou W; Zheng Z; Zhang P; Tu B; He Q; Zhu W-G, The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA repair. 2012, 11, 146–156. [DOI] [PubMed] [Google Scholar]
- (28).Ishii S; Kurasawa Y; Wong J; Yu-Lee L. y., Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore–microtubule attachment. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 4179–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stevens F; Beamish H; Warrener R; Gabrielli B, Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008, 27, 1345–1354. [DOI] [PubMed] [Google Scholar]
- (30).Aron JL; Parthun MR; Marcucci G; Kitada S; Mone AP; Davis ME; Shen T; Murphy T; Wickham J; Kanakry C, Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8–mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003, 102, 652–658. [DOI] [PubMed] [Google Scholar]
- (31).Mitsiades CS; Mitsiades NS; McMullan CJ; Poulaki V; Shringarpure R; Hideshima T; Akiyama M; Chauhan D; Munshi N; Gu X, Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rosato RR; Almenara JA; Kolla SS; Maggio SC; Coe S; Giménez MS; Dent P; Grant S, Mechanism and functional role of XIAP and Mcl-1 down-regulation in flavopiridol/vorinostat antileukemic interactions. Mol. Cancer Ther 2007, 6, 692–702. [DOI] [PubMed] [Google Scholar]
- (33).Chen S; Dai Y; Pei X-Y; Grant S, Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol. Cell. Biol 2009, 29, 6149–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xargay-Torrent S; López-Guerra M; Saborit-Villarroya I; Rosich L; Campo E; Roué G; Colomer D, Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin. Cancer Res 2011, 17, 3956–3968. [DOI] [PubMed] [Google Scholar]
- (35).Lindemann R; Newbold A; Whitecross K; Cluse L; Frew A; Ellis L; Williams S; Wiegmans A; Dear A; Scott C, Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 8071–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yi C; Ma M; Ran L; Zheng J; Tong J; Zhu J; Ma C; Sun Y; Zhang S; Feng W, Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012, 336, 474–477. [DOI] [PubMed] [Google Scholar]
- (37).Mrakovcic M; Kleinheinz J; Fröhlich LF, Histone deacetylase inhibitor-induced autophagy in tumor cells: implications for p53. Int. J. Mol. Sci. 2017, 18, 1883–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Grant S; Dai Y, Histone deacetylase inhibitors and rational combination therapies In Advances in cancer research, Elsevier: 2012; Vol. 116, pp 199–237. [DOI] [PubMed] [Google Scholar]
- (39).Gu W; Roeder RG, Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- (40).Vousden KH; Lane DP, p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [DOI] [PubMed] [Google Scholar]
- (41).Fridman JS; Lowe SW, Control of apoptosis by p53. Oncogene. 2003, 22, 9030–9040. [DOI] [PubMed] [Google Scholar]
- (42).Bucher N; Britten C, G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Crighton D; Wilkinson S; O’Prey J; Syed N; Smith P; Harrison PR; Gasco M; Garrone O; Crook T; Ryan KM, DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006, 126, 121–134. [DOI] [PubMed] [Google Scholar]
- (44).Duvic M; Vu J, Update on the treatment of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biol.: Targets Ther 2007, 1, 377–392. [PMC free article] [PubMed] [Google Scholar]
- (45).Bertino EM; Otterson GA, Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin. Invest. Drugs 2011, 20, 1151–1158. [DOI] [PubMed] [Google Scholar]
- (46).Rashidi A; Cashen AF, Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Future Oncol. 2015, 11, 1659–1664. [DOI] [PubMed] [Google Scholar]
- (47).Greig SL, Panobinostat: a review in relapsed or refractory multiple myeloma. Target oncol. 2016, 11, 107–114. [DOI] [PubMed] [Google Scholar]
- (48).Traoré MD; Zwick V; Simões-Pires CA; Nurisso A; Issa M; Cuendet M; Maynadier M; Wein S; Vial H; Jamet H, Hydroxyl ketone-based histone deacetylase inhibitors to gain insight into class I HDAC selectivity versus that of HDAC6. ACS Omega. 2017, 2, 1550–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Micelli C; Rastelli G, Histone deacetylases: structural determinants of inhibitor selectivity. Drug discovery today. 2015, 20, 718–735. [DOI] [PubMed] [Google Scholar]
- (50).Son SI; Cao J; Zhu CL; Miller SP; Lin H, Activity-guided design of HDAC11-specific inhibitors. ACS Chem. Biol 2019, 14, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang Y; Stowe RL; Pinello CE; Tian G; Madoux F; Li D; Zhao LY; Li JL; Wang Y; Wang Y; Ma H; Hodder P; Roush WR; Liao D, Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol 2015, 22, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Koechlin BA; Schwartz MA; Oberhaensli WE, Metabolism of C14-iproniazid and C14-isocarboxazid in man. J. Pharmacol. Exp. Ther 1962, 138, 11–20. [PubMed] [Google Scholar]
- (53).Nelson SD; Mitchell JR; Snodgrass W; Timbrell JA, Hepatotoxicity and metabolism of iproniazid and isopropylhydrazine. J. Pharmacol. Exp. Ther 1978, 206, 574–585. [PubMed] [Google Scholar]
- (54).Girling D, The hepatic toxicity of antituberculosis regimens containing isoniazid, rifampicin and pyrazinamide. Tubercle. 1977, 59, 13–32. [DOI] [PubMed] [Google Scholar]
- (55).Clive S; Woo MM; Nydam T; Kelly L; Squier M; Kagan M, Characterizing the disposition, metabolism, and excretion of an orally active pan-deacetylase inhibitor, panobinostat, via trace radiolabeled 14 C material in advanced cancer patients. Cancer Chemother. Pharmacol 2012, 70, 513–522. [DOI] [PubMed] [Google Scholar]
- (56).McClure JJ; Li X; Chou CJ Advances and challenges of HDAC inhibitors in cancer therapeutics In Advances in cancer research, Elsevier: 2018; Vol. 138, pp 183–211. [DOI] [PubMed] [Google Scholar]
- (57).Steele NL; Plumb JA; Vidal L; Tjørnelund J; Knoblauch P; Rasmussen A; Ooi CE; Buhl-Jensen P; Brown R; Evans TJ, A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res 2008, 14, 804–810. [DOI] [PubMed] [Google Scholar]
- (58).Kelly WK; Richon VM; O’Connor O; Curley T; MacGregor-Curtelli B; Tong W; Klang M; Schwartz L; Richardson S; Rosa E, Phase I clinical trial of histone deacetylase inhibitor. Clin. Cancer Res 2003, 9, 3578–3588. [PubMed] [Google Scholar]
- (59).Giles F; Fischer T; Cortes J; Garcia-Manero G; Beck J; Ravandi F; Masson E; Rae P; Laird G; Sharma S, A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res 2006, 12, 4628–4635. [DOI] [PubMed] [Google Scholar]
- (60).Peschel I; Podmirseg SR; Taschler M; Duyster J; Götze KS; Sill H; Nachbaur D; Jäkel H; Hengst L; Tirol ÖK, FLT3 and FLT3-ITD phosphorylate and inactivate the cyclin dependent kinase inhibitor p27Kip1 in acute myeloid leukemia. haematologica. 2017,102, 1378–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mali RS; Lasater EA; Doyle K; Malla R; Boghaert E; Souers A; Leverson JD; Sampath D, Abstract B052: FLT3-ITD activation mediates resistance to the BCL-2 selective antagonist, venetoclax, in FLT3-ITD mutant AML models. Mol. Cancer Ther 2018, 17, B052–B052. [Google Scholar]
- (62).Ranganathan P; Yu X; Santhanam R; Hofstetter J; Walker A; Walsh K; Bhatnagar B; Klisovic R; Vasu S; Phelps MA, Decitabine priming enhances the anti-leukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood. 2015,125, 2689–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bradner JE; West N; Grachan ML; Greenberg EF; Haggarty SJ; Warnow T; Mazitschek R, Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol 2010, 6, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Mohamed MFA; Youssif BGM; Shaykoon MSA; Abdelrahman MH; Elsadek BEM; Aboraia AS; Abuo-Rahma GEA, Utilization of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidinone as a cap moiety in design of novel histone deacetylase inhibitors. Bioorg. Chem 2019, 91, 103127. [DOI] [PubMed] [Google Scholar]
- (65).Atadja P, Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer lett. 2009, 280, 233–241. [DOI] [PubMed] [Google Scholar]
- (66).Malkin D; Nichols KE; Zelley K; Schiffman JD, Predisposition to pediatric and hematologic cancers: a moving target. Am Soc Clin Oncol Educ Book. 2014, 34, e44–55. [DOI] [PubMed] [Google Scholar]
- (67).Network CGAR, Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med 2013, 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Assi R; Gur HD; Loghavi S; Konoplev SN; Konopleva M; Daver N; Tashakori M; Kadia T; Routbort M; Salem A, P53 protein overexpression in de novo acute myeloid leukemia patients with normal diploid karyotype correlates with FLT3 internal tandem duplication and worse relaps-free survival. Am. J. Hematol 2018, 93, 1376–1383. [DOI] [PubMed] [Google Scholar]
- (69).Levis M; Small D, FLT3: ITDoes matter in leukemia. Leukemia. 2003, 17, 1738–1752. [DOI] [PubMed] [Google Scholar]
- (70).Choudhary C; Schwäble J; Brandts C; Tickenbrock L; Sargin B; Kindler T; Fischer T; Berdel WE; Müller-Tidow C; Serve H, AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005, 106, 265–273. [DOI] [PubMed] [Google Scholar]
- (71).Mizuki M; Schwäble J; Steur C; Choudhary C; Agrawal S; Sargin B; Steffen B; Matsumura I; Kanakura Y; Böhmer FD, Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood. 2003, 101, 3164–3173. [DOI] [PubMed] [Google Scholar]
- (72).Grundler R; Miething C; Thiede C; Peschel C; Duyster J, FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005, 105, 4792–4799. [DOI] [PubMed] [Google Scholar]
- (73).Drexler H, Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia. 1996, 10, 588–599. [PubMed] [Google Scholar]
- (74).Piacibello W; Fubini L; Sanavio F; Brizzi MF; Severino A; Garetto L; Stacchini A; Pegoraro L; Aglietta M, Effects of human FLT3 ligand on myeloid leukemia cell growth: heterogeneity in response and synergy with other hematopoietic growth factors. Blood. 1995, 86, 4105–4114. [PubMed] [Google Scholar]
- (75).Stacchini A; Fubini L; Severino A; Sanavio F; Aglietta M; Piacibello W, Expression of type III receptor tyrosine kinases FLT3 and KIT and responses to their ligands by acute myeloid leukemia blasts. Leukemia. 1996, 10, 1584–1591. [PubMed] [Google Scholar]
- (76).Calò V; Migliavacca M; Bazan V; Macaluso M; Buscemi M; Gebbia N; Russo A, STAT proteins: from normal control of cellular events to tumorigenesis. J. Cell. Physiol 2003, 197, 157–168. [DOI] [PubMed] [Google Scholar]
- (77).Takahashi S; Harigae H; Kaku M; Sasaki T; Licht JD, Flt3 mutation activates p21 WAF1/CIP1 gene expression through the action of STAT5. Biochem. Biophys. Res. Commun 2004, 316, 85–92. [DOI] [PubMed] [Google Scholar]
- (78).Kim K-T; Baird K; Ahn J-Y; Meltzer P; Lilly M; Levis M; Small D, Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005, 105, 1759–1767. [DOI] [PubMed] [Google Scholar]
- (79).Gilliland DG; Griffin JD, The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002, 100, 1532–1542. [DOI] [PubMed] [Google Scholar]
- (80).Safa A, c-FLIP, a master anti-apoptotic regulator. Exp. Oncol 2012, 34, 176–184. [PMC free article] [PubMed] [Google Scholar]
- (81).Riedl SJ; Renatus M; Schwarzenbacher R; Zhou Q; Sun C; Fesik SW; Liddington RC; Salvesen GS, Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001, 104, 791–800. [DOI] [PubMed] [Google Scholar]
- (82).Scott FL; Denault JB; Riedl SJ; Shin H; Renatus M; Salvesen GS, XIAP inhibits caspase-3 and −7 using two binding sites: evolutionarily conserved mechanism of IAPs. EMBO J. 2005, 24, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Shiozaki EN; Chai J; Rigotti DJ; Riedl SJ; Li P; Srinivasula SM; Alnemri ES; Fairman R; Shi Y, Mechanism of XIAP-mediated inhibition of caspase-9. Mol cell. 2003, 11, 519–527. [DOI] [PubMed] [Google Scholar]
- (84).Tanida I; Minematsu-Ikeguchi N; Ueno T; Kominami E, Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005, 1, 84–91. [DOI] [PubMed] [Google Scholar]
- (85).Quentmeier H; Reinhardt J; Zaborski M; Drexler H, FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003, 17, 120–124. [DOI] [PubMed] [Google Scholar]
- (86).Ma L; Sato F; Sato R; Matsubara T; Hirai K; Yamasaki M; Shin T; Shimada T; Nomura T; Mori K, Dual targeting of heat shock proteins 90 and 70 promotes cell death and enhances the anticancer effect of chemotherapeutic agents in bladder cancer. Oncol. Rep 2014, 31, 2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Beere HM; Wolf BB; Cain K; Mosser DD; Mahboubi A; Kuwana T; Tailor P; Morimoto RI; Cohen GM; Green DR, Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol 2000, 2, 469–475. [DOI] [PubMed] [Google Scholar]
- (88).Garrido C; Schmitt E; Candé C; Vahsen N; Parcellier A; Kroemer G, HSP27 and HSP70: potentially oncogenic apoptosis inhibitors. Cell cycle. 2003, 2, 578–583. [PubMed] [Google Scholar]
- (89).Ravagnan L; Gurbuxani S; Susin SA; Maisse C; Daugas E; Zamzami N; Mak T; Jäättelä M; Penninger JM; Garrido C, Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol 2001, 3, 839–843. [DOI] [PubMed] [Google Scholar]
- (90).Schrader A; Crispatzu G; Oberbeck S; Mayer P; Pützer S; Jan J; Vasyutina E; Warner K; Weit N; Pflug N, Actionable perturbations of damage responses by TCL1/ATM and epigenetic lesions form the basis of T-PLL. Nat. Commun 2018, 9, 697–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Andersson E; Pützer S; Yadav B; Dufva O; Khan S; He L; Sellner L; Schrader A; Crispatzu G; Oleś M, Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018, 32, 774–787. [DOI] [PubMed] [Google Scholar]
- (92).D’Arcy MS, Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019, 43, 582–592. [DOI] [PubMed] [Google Scholar]
- (93).Yeo P; Xin L; Goh E; New LS; Zeng P; Wu X; Venkatesh P; Kantharaj E, Development and validation of high-performance liquid chromatography-tandem mass spectrometry assay for 6-(3-benzoyl-ureido)-hexanoic acid hydroxyamide, a novel HDAC inhibitor, in mouse plasma for pharmacokinetic studies. Biomed. Chromatogr. 2007, 21, 184–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.