

Abstract
The inflammatory response to periodontal pathogens is dynamically controlled by the chromatin state on inflammatory gene promoters. In the present study, we have focused on the effect of the methyltransferase SETD1B on histone H3 lysine K4 (H3K4) histone trimethylation on inflammatory gene promoters. Experiments were based on 3 model systems: 1) an in vitro periodontal ligament (PDL) cell culture model for the study of SETD1 function as it relates to histone methylation and inflammatory gene expression using Porphyromonas gingivalis lipopolysaccharide (LPS) as a pathogen, 2) a subcutaneous implantation model to determine the relationship between SETD1 and nuclear factor κB (NF-κB) through its activation inhibitor BOT-64, and 3) a mouse periodontitis model to test whether the NF-κB activation inhibitor BOT-64 reverses the inflammatory tissue destruction associated with periodontal disease. In our PDL progenitor cell culture model, P. gingivalis LPS increased H3K4me3 histone methylation on IL-1β, IL-6, and MMP2 gene promoters, while SETD1B inhibition decreased H3K4me3 enrichment and inflammatory gene expression in LPS-treated PDL cells. LPS also increased SETD1 nuclear localization in a p65-dependent fashion and the nuclear translocation of p65 as mediated through SETD1, suggestive of a synergistic effect between SETD1 and p65 in the modulation of inflammation. Confirming the role of SETD1 in p65-mediated periodontal inflammation, BOT-64 reduced the number of SETD1-positive cells in inflamed periodontal tissues, restored periodontal tissue integrity, and enhanced osteogenesis in a periodontal inflammation model in vivo. Together, these results have established the histone lysine methyltransferase SETD1 as a key factor in the opening of the chromatin on inflammatory gene promoters through histone H3K4 trimethylation. Our studies also confirmed the role of BOT-64 as a potent molecular therapeutic for the restoration of periodontal health through the inhibition of NF-κB activity and the amelioration of SETD1-induced chromatin relaxation.
Keywords: periodontal disease, chromatin dynamics, histone methylation, small molecule, homeostasis, methyltransferase
Introduction
Periodontal inflammation is a meticulously controlled immune response to bacterial pathogens that avoids the tissue injury of prolonged infection while at the same time preventing septic exacerbation of excessive inflammation (Nicodeme et al. 2010; Bartold and Van Dyke 2013). The fine-tuning of the periodontal inflammatory response is accomplished through Toll-like receptor 2- and 4 (TLR2- and TLR4-)–mediated nuclear factor κB (NF-κB) signaling in response to lipopolysaccharide (LPS) from the cell walls of mostly gram-negative bacteria (Li et al. 2014). NF-κB nuclear translocation initiates the expression of proinflammatory cytokines, chemokines, and matrix metalloproteinases in the periodontium (Parker et al. 2007), which in turn are controlled by changes in chromatin accessibility (Foster et al. 2007; Foster and Medzhitov 2009; Ghisletti et al. 2010; Schmidt et al. 2016; Luan et al. 2018). There is a reciprocal relationship between chromatin and NF-κB; not only is transcription factor binding controlled by chromatin, but NF-κB also affects chromatin dynamics through the recruitment of chromatin complexes, competitive eviction of negative chromatin modifications, and the recruitment of components of the general transcriptional machinery (Natoli et al. 2005; Bhatt and Ghosh 2014).
Chromatin remodeling occurs either as a result of the effect of chromatin remodeling complexes on nucleosome structure or through the action of enzymatically mediated histone modifications, including phosphorylation, acetylation, ubiquitination, and methylation (Greer and Shi 2012). Among the posttranslational histone modifications, histone methylation dynamics have been associated with important roles in many biological processes, including cell-cycle regulation, stress response, and immunity (Busslinger and Tarakhovsky 2014), and histone H3 lysine K4 (H3K4) methylation is generally associated with active transcription or with genes that are poised for activation (Dangaria et al. 2011; Greer and Shi 2012; Gopinathan et al. 2013). H3K4 methylation is mediated by several SET domain-containing methyltransferases, including mixed-lineage leukemia (MLL) (Miller et al. 2001), SETD1A/B (Bledau et al. 2014), SET7/9 (Keating and El-Osta 2013), and SET and Mynd domain-containing protein 1–3 families (Hamamoto et al. 2004).
Previously, we have demonstrated that inflammation triggers repressive histone mark enrichment on extracellular matrix gene promoters and active marks on defensins and chemokines (Francis et al. 2019). In the present study, we have focused on SETD1B as a histone methyltransferase that specifically methylates the lysine 4 position of histone H3. Consequently, we have hypothesized that inflammatory conditions and NF-κB activation alter the histone methylation state of the periodontal connective tissues, resulting in open chromatin on inflammatory gene promoters and a diminished ability to function physiologically, to heal, and to regenerate. To test our hypothesis, we have assessed SETD1-mediated chromatin dynamics on IL-1β, IL-6, and MMP2 proinflammatory and matrix-degrading gene promoters. To elucidate the relationship between SETD1 and NF-κB, we have asked whether SETD1 and NF-κB act in concert and whether the NF-κB activation inhibitor BOT-64 reverses the inflammatory tissue destruction that occurs during periodontal disease. BOT-64 blocks the inhibitory κB (IκB) kinase beta (IKKbeta). The results of our study provide insights into the intimate relationship between chromatin methyltransferases and the inflammatory master regulator NF-κB, revealing how NF-κB avails itself of chromatin dynamics to facilitate inflammatory processes.
Materials and Methods
Animals and Periodontitis Model
Six-week-old athymic mice were obtained from Charles River and C57/BL6 mice were bred at the University of Illinois at Chicago. All studies were conducted in strict accordance with the recommendations on the Guide for Care and Use of Laboratory Animals of the National Institutes of Health. The C57 mice (6 wk old) underwent anesthesia with an induction of 5% isoflurane and maintenance of 1% to 2% isoflurane. The periodontitis model in this study was generated by intragingival injections of 50 µL Porphyromonas gingivalis LPS (Pg-LPS,1 µg/mL in phosphate-buffered saline [PBS]; Invivogen) on the palatal and buccal aspects of the first maxillary molars 3 times/wk for 15 wk.
Tissue Processing and Histology
Maxillae and collagen implants were dissected, fixed, embedded in paraffin, and then sectioned. For hematoxylin and eosin (H&E) staining, sections were dipped in hematoxylin and then in eosin; for Alizarin red staining, sections were stained with 1% Alizarin red solution. The stained sections were dehydrated and cleared in xylene.
Immunohistochemistry
The HistoMouse-Plus Kit (ThermoFisher) was used to detect SETD1B, IL-1β, and IL-6 (ab70378, ab2105, ab6672; Abcam) expression. In brief, sections were first incubated with primary antibodies overnight at 4°C and then with biotinylated secondary antibodies, streptavidin-peroxidase conjugate, and the AEC chromogen following the manufacturer’s instruction. Slides were counterstained with hematoxylin. Negative controls were processed without primary antibodies.
Cell Culture
We used periodontal ligament (PDL) stem cells in our cell culture system to study the effect of inflammation on osteogenesis in periodontal diseases. PDL cells were isolated from human extracted molars and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% antibiotics (ThermoFisher). Periodontitis conditions were mimicked by adding 10 µg/mL Pg-LPS to the culture medium for 2, 4, 6, 8, and 10 h, representing early and late host responses. We chose 6 h culture for chromatin immunoprecipitation (ChIP) and 10 h for gene expression analyses. Exceptions are noted in the figure legends.
Gene Expression Knockdown or Inhibition
SETD1B and p65 small interfering RNAs (siRNA) were purchased from Dharmacon. PDL cells were treated with siRNA using Lipofectamine RNAimax Reagent (ThermoFisher) according to the manufacturer’s instructions. In brief, cells were seeded to be 80% to 90% confluent at transfection. SiRNAs were used at a final concentration of 10 nM. Cells were harvested after 48 h of transfection. For inhibition of NF-κB signaling, the NF-κB activation inhibitor VI, BOT-64 (Santa Cruz), was added to the cell culture medium at a concentration of 1 µM.
ChIP–Polymerase Chain Reaction Analysis
PDL cells grown on 150-mm plates were crosslinked with 1.1% formaldehyde at room temperature. The reaction was stopped with 125 mM glycine. Nuclei were prepared from 1 × 106 cells and ChIP was performed as described previously (Francis et al. 2019). The antibodies used for ChIP analysis were against histone H3K4me3, H3K9me3, H3K27me3, SETD1B, and p65 (ab8580, ab70378, ab8898, ab6147, Abcam; sc-71x, Santa Cruz). ChIP experiments were performed as triplicates.
ChIP assays were performed with the control, LPS, and LPS + siRNA groups. All DNA samples were diluted to a concentration of 2 ng/µL. Real-time quantitative polymerase chain reaction (qPCR) was performed on an ABI 7500 FAST machine with 4 ng ChIP DNA, and the total input was used as an internal reference for normalization. Enrichment for beads alone was used as a negative control. Data presented for each primer pair were obtained after subtracting the values obtained from the corresponding negative controls. The primer sequences are listed in Appendix Table 1.
RNA Extraction and Quantitative Reverse Transcription PCR
Total RNAs were isolated from mouse periodontal tissues or human PDL cells using the RNeasy Plus Mini Kit according to the manufacturer’s instructions (Qiagen). Then, 2 µg total RNA was applied toward complementary DNA (cDNA) generation, and quantitative reverse transcription PCR (RT-qPCR) was performed using sequence specific primers (Appendix Table 1) and SYBR green Master Mix as described previously (Francis et al. 2019). β-Actin was used as an internal control. The comparative CT method (ΔΔCT) was used to determine relative quantity. Values were graphed as the mean ± SD.
Western Blot
Total protein was extracted from the collected cells with RIPA buffer. Equal amounts of protein samples were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer onto PVDF membranes. The membranes were incubated with anti–IL-1b, IL-6, SETD1B, and p65 (ab 32536) antibodies at 4°C overnight and then with the secondary anti-rabbit IgG antibody (ab6721). The enhanced chemiluminescence reagent (PerkinElmer) was used to detect the immunoreactive bands.
Subcutaneous Implantation
In total, 20 µL BOT-64 aqueous solution (1 µM) was dropped on a collagen sponge sheet (3 × 3 × 2 mm2) and incubated at 4°C overnight to coat the surface of the scaffold, and 20 µL PBS was used as a control. The coated collagen sponges were incubated with a suspension of human PDL progenitors at a density of 106 cells/50 µL and followed by incubation for 3 h at 37°C. Two groups (n = 5/group) of collagen sponges (PBS plus PDL cells and BOT-64 plus PDL cells) were implanted into the back of nude mice below the subcutis as described previously (Pan et al. 2013). The experiments were terminated 4 wk after implantation.
Periodontal Implantation
Collagen sponges with or without BOT-64 were prepared as above and freeze-dried. A total of 10 mice (n = 5) were divided into 2 groups (periodontitis/BOT-64-collagen) as well as a defect control (periodontitis/collagen). To mimic a clinical situation and to readily access periodontitis-related bone defects, mice were subjected to gingival flap surgery. For flap surgery, incisions were placed from the mesial surface of the first molar to the distal surface of the third molar on the left maxilla. Full-thickness flaps were elevated, and BOT-64 coated or control scaffolds were applied to the surface of the exposed alveolar bone and sutured in place. Experiments were terminated 6 wk after implantation.
Micro–Computed Tomography
Micro–computed tomography (µCT) images from left maxillae after a 6-wk implantation were captured and reconstructed in 3 dimensions using a Scanco 40 µCT apparatus (Scanco Medical). Bone volume and mineral density were analyzed using a TeraRecon software package as described previously (Lu et al. 2016).
Statistics
Each experiment was repeated at least 3 times, and comparison of the results in each experimental group was performed by an unpaired Student’s t test with Welch’s correction for unequal variances. All calculations were performed using GraphPad Prism 6 (GraphPad Software).
Results
Increased Inflammatory Cell Infiltration and Inflammatory Cytokine Dysregulation in a Mouse Periodontitis Model
Histochemical analysis demonstrated increased infiltration of inflammatory cells and disorganization of periodontal ligament in the periodontitis model (Fig. 1A, B). The distance between alveolar bone ridge and cemento-enamel junction was increased in periodontitis animals when compared to controls (Fig. 1A, B). In the periodontitis mice, the expression of IL-1β (6-fold, P < 0.001), IL-6 (7-fold, P < 0.001), and matrix metalloproteinase MMP2 (4.4-fold, P < 0.01) was significantly increased (Fig. 1C). IL-1β and IL-6 were immunohistochemically detected in periodontal ligament fibroblasts from the periodontitis mouse model (Fig. 1D–G).
Figure 1.
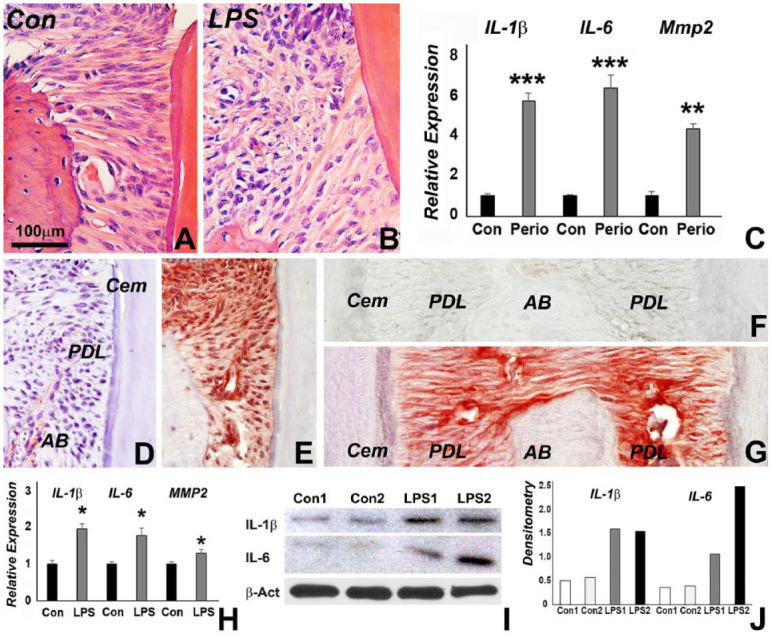
Phenotype and inflammatory gene expression in our periodontitis models. (A, B) Inflammatory infiltrate in periodontitis mouse (B) compared to control (A) as revealed by hematoxylin-eosin staining. (C) Relative IL-1β, IL-6, MMP2 messenger RNA (mRNA) expression in periodontitis mouse model as revealed by quantitative reverse transcription polymerase chain reaction (RT-qPCR). Expression levels were calculated relative to β-actin gene expression levels using the 2–ΔΔCt method, and values were graphed as the mean expression level ± standard deviation. (D–G) Immunohistochemistry revealing red staining for IL-6 (E vs. D) and IL-1β (G vs. F) in our periodontitis mouse model (E, G) compared to the control (D, F). (H) Relative IL-1β, IL-6, and MMP2 mRNA expression in lipopolysaccharide (LPS)–challenged periodontal ligament (PDL) cells versus nontreated control cells as analyzed by RT-qPCR. Expression levels were calculated as in H. (I) Protein levels for IL-1β and IL-6 in 2 representative samples, LPS1 and LPS2, were determined by Western blot using β-actin as a control. (J) Densitometry corresponding to I. *, **, and *** represent a significant difference equivalent to the following P values: P < 0.05, P < 0.01, and P < 0.001.
Upregulated Inflammatory Gene and Protein Expression in LPS-Treated Cells
To investigate the effect of LPS on inflammatory gene expression and protein presentation, PDL cells were treated with LPS. Real-time RT-qPCR analysis demonstrated that in response to LPS challenge, the expression of IL-1β (1.9-fold, P < 0.05), IL-6 (1.8-fold, P < 0.05), and MMP2 (1.4-fold, P < 0.05) was significantly increased (Fig. 1H). Western Blot analysis demonstrated increased protein levels for IL-1β and IL-6 when treated with LPS (Fig. 1I, J).
Altered H3K4 Trimethylation and SETD1B Recruitment to Promoter Regions of Inflammatory Genes in LPS-Treated Cells
To systematically assess epigenetic changes in response to inflammation in cultured PDL cells, the occupancy of H3K4me3 and SETD1B on individual inflammatory gene promoter regions in our LPS-treated PDL cells was examined. Chromatin immunoprecipitation analysis with H3K4me3 and SETD1B-specific antibodies demonstrated that promoter regions of the inflammatory cytokines IL-6 and IL-1β, as well as of the matrix metalloproteinase MMP2, were predominantly occupied by the active H3K4me3 histone modification in LPS-treated PDL cells (Fig. 2A). However, SETD1B was only enriched on the promoters of IL-1β and IL-6 genes (Fig. 2B) and not of MMP2.
Figure 2.
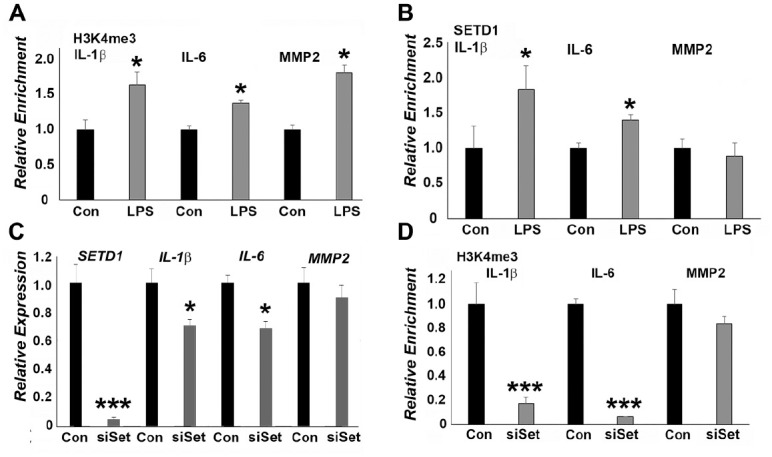
SETD1B mediates upregulation of inflammatory genes in lipopolysaccharide (LPS)–treated periodontal ligament (PDL) cells. (A, B) Chromatin immunoprecipitation (ChIP)–quantitative polymerase chain reaction (qPCR) analysis of SETD1 and H3K4me3 enrichment on the IL-1β, IL-6, and MMP2 promoters in LPS-treated PDL cells compared to control cells. LPS significantly increased both SETD1 and H3K4me3 occupancy on the IL-1β and IL-6 promoter regions. (C) Relative messenger RNA (mRNA) expression of IL-1β, IL-6, and MMP2 in PDL cells treated either with LPS or with LPS plus SETD1 small interfering RNA (siRNA). SETD1B siRNA treatment significantly reduced LPS-induced upregulation of IL-1β and IL-6 expression when compared to LPS-exposed cells. (D) ChIP-qPCR analysis of H3K4me3 enrichment on the IL-1β, IL-6, and MMP2 promoters in PDL cells treated either with LPS or with LPS plus SETD1B siRNA. There was a highly significant reduction of H3K4me3 occupancy on the IL-1β and IL-6 promoters when comparing a combination of SETD1 siRNA and LPS treatment versus LPS treatment alone. Changes in the MMP2 group in B, C, and D were not significant. *P < 0.05. ***P < 0.001.
SETD1B Inhibition Decreased H3K4me3 Enrichment and Inflammatory Gene Expression in LPS-Treated PDL Cells
To determine the effect of SETD1B on inflammatory gene expression in PDL cells, SETD1B expression was knocked down using SETD1B siRNA. SETD1B knockdown resulted in decreased gene expression levels for SETD1B (96%), IL-1β (31%, P < 0.05) IL-6 (33%, P < 0.05), and MMP2 (11%) in LPS-treated PDL cells (Fig. 2C). The SETD1B siRNA reduced occupancy of the H3K4me3 mark on the IL-1β (83%, P < 0.001) and IL-6 (94%, P < 0.001) promoter regions but not on the MMP2 promoter (17%) (Fig. 2D). Our study further established a correlation between histone modification levels at the promoter and downstream gene expression.
LPS Treatment Affected SETD1B Expression and Histone Trimethylation on the SETD1B Promoter
RT-qPCR analysis revealed that that both SetD1a and SetD1b expression was significantly increased in periodontitis tissues in vivo and that SETD1A and SETD1B expression was significantly upregulated 6, 8, and 10 h after LPS treatment (Fig. 3A). Chromatin immunoprecipitation studies with H3K4me3, H3K9me3, and H3K27me3-specific antibodies demonstrated that SETD1B was predominantly marked with the active H3K4me3 histone modification and displayed reduced enrichment for H3K9me3 and H3K27me3 repressive marks (Fig. 3B). Western blots demonstrated that the level of SETD1B protein expression was 2-fold increased in the nucleus of LPS-treated PDL cells when compared to untreated controls (Fig. 3C). Confirming the nuclear positioning of SETD1B under inflammatory conditions, immunofluorescence imaging revealed significantly enhanced SETD1B nuclear localization (Fig. 3D, E), indicating that SETD1B is active under inflammatory conditions.
Figure 3.

SETD1B promoter histone methylation enrichment profile and nuclear localization in lipopolysaccharide (LPS)–treated periodontal ligament (PDL) cells. (A) Relative expression of SETD1A and 1B in periodontal tissues as well as in PDL cells 2, 4, 6, 8, and 10 h after LPS challenge (LPS2, LPS4, LPS6, LPS8, and LPS10). (B) Chromatin immunoprecipitation (ChIP)–quantitative polymerase chain reaction (qPCR) analysis of the SETD1B gene promoter in PDL cells. Representative graphs show relative enrichment compared to input DNA for H3K4me3, H3K9me3, and H3K27me3. ChIP-qPCRs were performed at least 3 times with similar results, and 1 representative experiment is presented (± standard deviation). *P < 0.05. **P < 0.01. ***P < 0.001. (C) Western blot analysis of 2 representative samples and corresponding densitometry reveal the level of SETD1B protein expression in the nucleus of PDL cells. (D, E) Immunofluorescent staining of SETD1B nuclear localization as a result of LPS exposure. LPS treatment resulted in higher fluorescence levels in LPS-treated PDL nuclei (E) when compared to the nontreated control group (D).
p65 Was Recruited to Promoter Regions and Regulated the Expression of Inflammatory Genes in LPS-Treated Cells
To verify whether NF-κB regulates inflammatory gene expression in our model system, p65 occupancy on inflammatory gene promoter regions in LPS-treated PDL cells was examined. ChIP-qPCR analysis demonstrated that the promoter regions of the inflammatory genes IL-1β and IL-6 elicited increased p65 occupancy (Fig. 4A). To determine the effect of p65 on inflammatory gene expression, p65 expression was knocked down using p65 siRNA. The p65 siRNA decreased p65 expression by 97%, P < 0.001 (Fig. 4B) in LPS-treated cells. Knocking down p65 expression with p65 siRNA was followed by decreased expression of IL-1β (69%, P < 0.001) and IL-6 (80%, P < 0.001, Fig. 4B). The P65 inhibitor BOT-64 also decreased gene expression of IL-1β (23%, P < 0.05) and IL-6 (27%, P < 0.05) in LPS-treated PDL cells (Fig. 4B).
Figure 4.

SETD1B is involved in p65-mediated upregulation of inflammatory gene expression in lipopolysaccharide (LPS)–treated periodontal ligament (PDL) cells. (A) Chromatin immunoprecipitation (ChIP)–quantitative polymerase chain reaction (qPCR) analysis of p65 enrichment on the IL-1β and IL-6 promoters in LPS-treated PDL cells compared to control. LPS increased p65 occupancy on IL-1β and IL-6 promoter regions. (B) Relative messenger RNA (mRNA) expression of p65, IL-1β, and IL-6 in PDL cells treated with LPS only, with LPS plus p65 siRNA, or with BOT-64. p65 siRNA decreased the expression of p65, IL-1β, and IL-6 in LPS-treated cells. BOT-64 decreased IL-1β and IL-6 expression in LPS-treated cells. (C) p65 binding on the IL-1β and IL-6 promoters after SETD1B depletion as revealed by ChIP. Knockdown of SETD1B suppressed the occupancy of P65 on these promoters. (D–G) Immunofluorescence for p65 and SETD1B expression in the nucleus of LPS-treated (D, F), LPS plus SETD1 siRNA-treated (E), or LPS plus p65 siRNA-treated PDL cells using antibodies against p65 (D, E) and SETD1B (F, G). Knockdown of p65 decreased SETD1B nuclear localization while knockdown of SETD1B reduced p65 nuclear localization (E, G). (H) Western blot analysis of p65 and SETD1B expression after knockdown of p65 or SETD1B and corresponding densitometry. (I) Effect of p65 or SETD1B knockdown on nuclear factor κB (NF-κB) activity in PDL cells in control and in LPS-treated cells. LPS treatment significantly increased NF-κB activity. Knockdown of either p65 or SETD1B reduced NF-κB reporter activity. *P < 0.05. **P < 0.01. ***P < 0.001.
SETD1B Was Involved in the Regulation NF-κB Activity in LPS-Treated PDL Cells
Earlier experiments in this study demonstrated that SETD1B and p65 were enriched on the same regions of the IL-1β and IL-6 promoters (Figs. 2, 4) and regulated their expression. To probe whether SETD1B and p65 cooperated to regulate interleukin gene expression, we examined the binding of p65 on IL-1β and IL-6 promoters after SETD1B depletion via ChIP assay. Knockdown of SETD1B suppressed the occupancy of p65 on these promoters (Fig. 4C), and immunofluorescence using antibodies against p65 and SETD1B revealed that p65 and SETD1B were colocalized in the nucleus in LPS-treated PDL cells. Knockdown of p65 decreased SETD1B nuclear localization while knockdown of SETD1B reduced p65 nuclear localization (Fig. 4D–G). Western blot analysis confirmed that knockdown of p65 reduced the level of SETD1B protein expression (17%) while knockdown of SETD1B decreased the level of p65 protein expression (45%) in the nucleus of PDL cells (Fig. 4H), reducing the regulatory control of NF-κB on downstream gene expression. NanoLuc Luciferase-NF-κB reporter assays were used to verify the effect of p65 and SETD1 on NF-κB activity (Fig. 4I). These studies demonstrated not only that LPS treatment significantly increased NF-κB activity but also that knockdown of either p65 or SETD1B reduced NF-κB reporter activity in the LPS-treated cells (Fig. 4I).
NF-κB Inhibitor Reduced SETD1B Expression and Enhanced Osteogenesis In Vivo
The suppression of NF-κB via the BOT-64 inhibitor revealed reduced SETD1 expression as well as enlarged alizarin red–positive areas in subcutaneous collagen implants compared to untreated controls (Fig. 5A–C), indicative of increased bone remodeling and new bone formation (Fig. 5D–F). Three-dimensional reconstruction of µCT images demonstrated significantly decreased distances between the cervical alveolar bone ridges and the cementum-enamel junction (CE junction) of maxillary molars in the BOT-64 treatment group versus the control (Fig. 5G, H). Alveolar bone volume to tissue density ratio was 1.9-fold increased in the BOT-64 treatment group when compared to the periodontitis control group (Fig. 5I), indicating that BOT-64 facilitated periodontal tissue regeneration.
Figure 5.
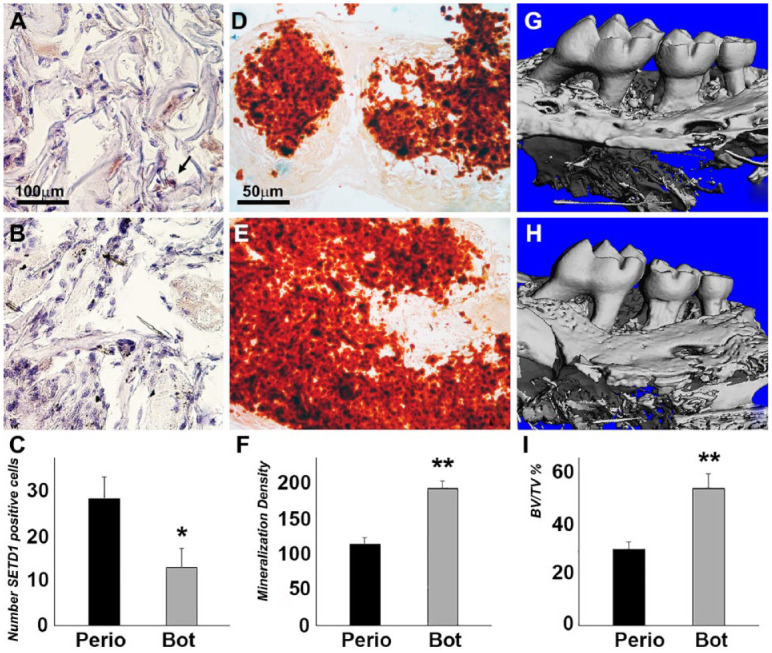
The effect of IKK2/nuclear factor κB (NF-κB) inhibition on SETD1B expression and osteogenesis in vivo. BOT-64 was used to coat collagen sponge implants seeded periodontal ligament (PDL) cells (bottom row) compared to control collagen sponge implants seeded PDL cells (top row). (A–C) Immunohistochemistry revealed brown staining for SETD1B (B) and the numbers of SETD1B-positive cells (C). (D, E) Alizarin red staining showed mineralization in collagen implants (D, E), and the mineral nodule density was analyzed by ImageJ (F). *P < 0.05. **P < 0.01. (G, I) Micro–computed tomography 3-dimensional reconstruction of the left maxillary mouse molars after a 6-wk implantation of PBS-COL and BOT-64-COL scaffolds subjected to periodontitis treatment and alveolar bone volume to tissue volume analysis (I).
Discussion
In the present study, we have examined the effect of periodontal bacterial toxins on the methyltransferase SETD1B as it regulates the chromatin state on inflammatory gene promoters by altering their histone methylation state. In our studies, we have used P. gingivalis LPS to induce periodontitis in mice and to mimic inflammatory conditions in vitro. Specifically, we have focused on the effect of LPS on SETD1B-facilitated H3K4me3 histone methylation on the promoters of the proinflammatory cytokines IL-1β and IL-6 and of the collagen degrading metalloproteinase MMP2 as mediators of the inflammatory sequelae associated with periodontal disease. Our data demonstrate that SETD1B plays a significant role in the LPS-induced upregulation of IL-1β and IL-6. LPS also induced the nuclear localization of either SETD1 or p65 in an interdependent fashion, verifying the relationship between SETD1 and p65 in the mediation of inflammation. Confirming the role of SETD1 in p65-mediated periodontal inflammation, the IKK2 inhibitor BOT-64 reduced SETD1-positive cells in periodontal inflammation and restored periodontal tissue integrity following periodontal inflammation. Together, our study demonstrates that the inflammation-induced and methyltransferase-mediated chromatin opening represents a key mechanism for the tissue destruction associated with periodontal disease.
In our PDL progenitor cell culture model, P. gingivalis LPS increased H3K4me3 histone methylation on IL-1β, IL-6, and MMP2 gene promoters, while SETD1B inhibition decreased H3K4me3 enrichment and inflammatory gene expression in LPS-treated PDL cells. These studies indicate that exposure to LPS from the walls of periodontal pathogens leads to increased H3K4me3 enrichment and resulting open chromatin on inflammatory gene promoters. IL-1β and IL-6 are potent stimulators of extracellular matrix–degrading MMPs in connective tissue cells (Kusano et al. 1998) and lymphoid cell lines (Kossakowska et al. 1999). The involvement of signaling molecules from the IL-1β or IL-6 pathway in the regulation of MMP gene expression as shown here may explain the different regulatory mechanisms for inflammation-related MMP2 upregulation when compared to the SETD1-modulated IL-1β or IL-6 upregulation.
Our study revealed a synergistic effect between SETD1 and p65 in the regulation of the inflammatory response to LPS. In support of a synergism between SETD1 and NF-κB, previous studies have associated methyltransferase-dependent histone methylation with increased mRNA expression of inflammatory genes and correlated H3K4 methylation states with the recruitment of NF-κB on the promoters of these genes (Natoli et al. 2005; Medzhitov and Horng 2009; Bhatt and Ghosh 2014). Earlier studies have also reported that the H3-lysine 4 mono-methyltransferase SET7/9 was required for the activation of NF-κB–dependent gene transcription and colocalized in the same cellular complex with p65, the transactivation subunit of NF-κB (Li et al. 2008). In the present study, we have demonstrated that SETD1B nuclear localization was increased upon LPS challenge, and SETD1B as well as p65 occupied the same promoter regions of IL-1β and IL-6 genes. Knockdown of either the SETD1B or the p65 gene resulted in a significant reduction of NF-κB activity and downregulation of IL-1β and IL-6 gene expression. These results suggest that SETD1B might be a novel coactivator of NF-κB in PDL cells.
Confirming the role of SETD1 in p65-mediated periodontal inflammation, the IKK2 inhibitor BOT-64 reduced the number of SETD1-positive cells in inflamed periodontal tissues, restored periodontal tissue integrity, and enhanced osteogenesis in a periodontal inflammation model in vivo. Based on the inhibitory effect of inflammation on osteogenic potential of mesenchymal stem cells (Lacey et al. 2009, Graves et al. 2019), we expect that NF-κB signaling inhibitors such as BOT-64 display similar effects on SETD1 function in terms of gene regulation. In the present study, we successfully introduced the IKK2/NF-κB activation inhibitor BOT-64 to enhance osteogenic differentiation of PDL cells in the subcutaneously implanted collagen sponges and to facilitate alveolar bone regeneration. We now propose a model in which LPS stimulates NF-κB to translocate to the nucleus, where it recruits the SETD1B enzyme to place the H3K4me3 active mark on inflammatory gene promoter regions, resulting in the upregulation of inflammatory genes. Application of the IKK2/NF-κB activation inhibitor BOT-64 decreases SETD1B expression while enhancing collagen fiber remodeling and osteogenesis in collagen sponge implants and periodontal defects. These results suggest that the small-molecule mediator BOT-64 has high potential as a therapeutic for periodontal disease.
Author Contributions
M. Francis, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; G. Gopinathan, contributed to design, data analysis, and interpretation, drafted and critically revised the manuscript; A. Salapatas, contributed to conception, data acquisition, and analysis, drafted and critically revised the manuscript; S. Nares, contributed to conception and data interpretation, drafted and critically revised the manuscript; M. Gonzalez, contributed to design and data interpretation, drafted and critically revised the manuscript; T.G.H. Diekwisch, contributed to conception, design, and data acquisition, drafted and critically revised the manuscript; X. Luan, contributed to conception or design, data acquisition, analysis, or interpretation of data, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, DS_10.1177_0022034520939029 for SETD1 and NF-κB Regulate Periodontal Inflammation through H3K4 Trimethylation by M. Francis, G. Gopinathan, A. Salapatas, S. Nares, M. Gonzalez, T.G.H. Diekwisch and X. Luan in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Generous funding for these studies was provided by National Institute of Dental and Craniofacial Research (NIDCR) grants DE026198 and DE027930 to T.G.H. Diekwisch, DE019463 to X. Luan, and F30 DE 024352 to M. Francis.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
ORCID iD: T.G.H. Diekwisch  https://orcid.org/0000-0003-3356-9677
https://orcid.org/0000-0003-3356-9677
References
- Bartold PM, Van Dyke TE. 2013. Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts: unlearning learned concepts. Periodontol 2000. 62(1):203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D, Ghosh S. 2014. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front Immunol. 5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF, et al. 2014. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development. 141(5):1022–1035. [DOI] [PubMed] [Google Scholar]
- Busslinger M, Tarakhovsky A. 2014. Epigenetic control of immunity. Cold Spring Harb Perspect Biol. 6(6):a019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangaria SJ, Ito Y, Luan X, Diekwisch TGH. 2011. Differentiation of neural-crest-derived intermediate pluripotent progenitors into committed periodontal populations involves unique molecular signature changes, cohort shifts, and epigenetic modifications. Stem Cells Dev. 20(1):39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R. 2007. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 447(7147):972–978. [DOI] [PubMed] [Google Scholar]
- Foster SL, Medzhitov R. 2009. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 130(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis M, Pandya M, Gopinathan G, Lyu H, Ma W, Foyle D, Nares S, Luan X. 2019. Histone methylation mechanisms modulate the inflammatory response of periodontal ligament progenitors. Stem Cells Dev. 28(15):1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, et al. 2010. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 32(3):317–328. [DOI] [PubMed] [Google Scholar]
- Gopinathan G, Kolokythas A, Luan X, Diekwisch TG. 2013. Epigenetic marks define the lineage and differentiation potential of two distinct neural crest–derived intermediate odontogenic progenitor populations. Stem Cells Dev. 22(12):1763–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Ding Z, Yang Y. 2019. The impact of diabetes on periodontal diseases. Periodontol 2000. 8(1):214–224. [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y. 2012. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 13(5):343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R, Nakamura Y. 2004. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 6(8):731–740. [DOI] [PubMed] [Google Scholar]
- Keating S, El-Osta A. 2013. Transcriptional regulation by the Set7 lysine methyltransferase. Epigenetics. 8(4):361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossakowska AE, Edwards DR, Prusinkiewicz C, Zhang MC, Guo D, Urbanski SJ, Grogan T, Marquez LA, Janowska-Wieczorek A. 1999. Interleukin-6 regulation of matrix metalloproteinase (MMP-2 and MMP-9) and tissue inhibitor of metalloproteinase (TIMP-1) expression in malignant non-Hodgkin’s lymphomas. Blood. 94(6):2080–2089. [PubMed] [Google Scholar]
- Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, Kamoi K, Suda T. 1998. Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology. 139(3):1338–1345. [DOI] [PubMed] [Google Scholar]
- Lacey DC, Simmons PJ, Graves SE, Hamilton JA. 2009. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: implications for bone repair during inflammation. Osteoarthritis Cartilage. 17(6):735–742. [DOI] [PubMed] [Google Scholar]
- Li C, Li B, Dong Z, Gao L, He X, Liao L, Hu C, Wang Q, Jin Y. 2014. Lipopolysaccharide differentially affects the osteogenic differentiation of periodontal ligament stem cells and bone marrow mesenchymal stem cells through Toll-like receptor 4 mediated nuclear factor κB pathway. Stem Cell Res Ther. 5(3):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Reddy MA, Miao F, Shanmugam N, Yee JK, Hawkins D, Ren B, Natarajan R. 2008. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes: relevance to diabetes and inflammation. J Biol Chem. 283(39):26771–26781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Fukumoto S, Yamada Y, Evans CA, Diekwisch TG, Luan X. 2016. Ameloblastin, an extracellular matrix protein, affects long bone growth and mineralization. J Bone Miner Res. 31(6):1235–1246. [DOI] [PubMed] [Google Scholar]
- Luan X, Zhou X, Naqvi A, Francis M, Foyle D, Nares S, Diekwisch TGH. 2018. MicroRNAs and immunity in periodontal health and disease. Int J Oral Sci. 10(3):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Horng T. 2009. Transcriptional control of the inflammatory response. Nat Rev Immunol. 9(10):692–703. [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A. 2001. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA. 98(23):12902–12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G, Saccani S, Bosisio D, Marazzi I. 2005. Interactions of NF-κB with chromatin: the art of being at the right place at the right time. Nat Immunol. 6(5):439–445. [DOI] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. 2010. Suppression of inflammation by a synthetic histone mimic. Nature. 468(7327):1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, Dangaria S, Gopinathan G, Yan X, Lu X, Kolokythas A, Niu Y, Luan X. 2013. SCF promotes dental pulp progenitor migration, neovascularization, and collagen remodeling—potential applications as a homing factor in dental pulp regeneration. Stem Cell Rev Rep. 9(5):655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LC, Prince LR, Sabroe I. 2007. Translational mini-review series on Toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity: networks regulated by TLRs mediate innate and adaptive immunity. Clin Exp Immunol. 147(2):199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SV, Krebs W, Ulas T, Xue J, Baßler K, Günther P, Hardt AL, Schultze H, Sander J, Klee K, et al. 2016. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. 26(2):151–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, DS_10.1177_0022034520939029 for SETD1 and NF-κB Regulate Periodontal Inflammation through H3K4 Trimethylation by M. Francis, G. Gopinathan, A. Salapatas, S. Nares, M. Gonzalez, T.G.H. Diekwisch and X. Luan in Journal of Dental Research