

Abstract
Sirtuins are NAD+-dependent protein deacylases that remove acyl modifications from acyl-lysine residues, resulting in essential cellular signaling. Recognized for their role in lifespan extension, humans encode seven sirtuin isoforms (Sirt1–7), and loss of sirtuin deacylase activity is implicated in many aging-related diseases. Despite being intriguing therapeutic targets, cellular studies of sirtuins are hampered by the lack of chemical probes to measure sirtuin activity independent of sirtuin protein levels. Here, we use a modular, peptide-based approach to develop activity-based probes (ABPs) that directly measure Sirt1 activity in vitro and in cell lysates. ABPs were synthesized containing four elements: 1) thioacetyl-lysine for mechanism-based affinity towards only active sirtuins, 2) either histone H3 lysine-14 (H3K14) or p53 sequences for Sirt1 specificity, 3) a diazirine for covalent labeling upon UV irradiation, and 4) an alkyne for bioorthogonal conjugation to a fluorophore for gel-based detection of active Sirt1. Compared to the H3K14 ABP, the p53 ABP showed increased sensitivity and selective labeling of active Sirt1. Acyl-lysine peptide competition, pharmacological inhibition, and inhibitory post-translational modification of Sirt1 resulted in the loss of p53 ABP labeling both in vitro and in HEK293T cell lysates, consistent with the ABP measuring decreased Sirt1 activity. Furthermore, the p53 ABP measured subcellular Sirt1 activity in MCF7 breast cancer cells. The development of a Sirt1-selective ABP that detects Sirt1 activity with an order of magnitude increased sensitivity compared to previous approaches demonstrates the utility of a modular, peptide-based approach for selective-targeting of the sirtuin protein family and provides a framework for further development of sirtuin-selective chemical probes.
Keywords: Activity-based probe, Acylation, Click chemistry, Deacylase, Enzyme catalysis, Peptides, Photoaffinity labeling, Sirtuin, S-nitrosation
Graphical abstract
1. Introduction
Sirtuins are NAD+-dependent protein deacylases that remove acyl modifications from acyllysine residues yielding O-acyl-ADP-ribose and nicotinamide [1,2]. This NAD+-dependence positions sirtuins as important metabolic regulators, coupling the cellular energy state with selective lysine deacylation to regulate a myriad of downstream cellular signaling processes. Humans encode seven sirtuin isoforms (Sirt1–7) that exhibit diverse subcellular localization and deacylase targets [3]. Sirtuins are widely regarded as anti-aging proteins, as increased sirtuin activity is associated with lifespan extension, and decreased activity is associated with the development of multiple aging-related diseases [4,5] including cancer [6], cardiovascular disease [7], neurodegeneration [8], and type-II diabetes [9,10].
Among the seven human sirtuins, Sirt1 plays a particularly important role in many cellular processes including cell survival, apoptosis, stress resistance, insulin production, lipid metabolism, and glucose homeostasis [11]. Sirt1 is primarily nuclear; however, cytosolic Sirt1 localization is observed in certain cell/tissue types and under specific cellular stresses [12]. In addition to histone targets, Sirt1 deacetylates non-histone proteins such as p53, thereby repressing p53 activity upon DNA damage or oxidative stress [13,14]. Taken together, Sirt1 is the most unique and versatile member of the sirtuin protein family, and the development of chemical tools that directly measure Sirt1 activity would be invaluable for elucidating Sirt1-specific roles in human health and disease.
Although the role of Sirt1 in regulating cellular energy status and survival makes it an intriguing therapeutic target, cellular studies have been hampered by the lack of chemical probes to measure sirtuin activity. Sirtuin activity is regulated both transcriptionally and translationally, as well as by protein turnover, protein-protein interactions, subcellular localization, and substrate availability [1,3,15]. Sirtuin deacylase activity can also be inhibited by post-translational modification [16,17], including modification by cellular oxidants under inflammation or metabolic stress conditions [10], and can be activated or inhibited pharmacologically by small molecules [18]. While many sirtuin-activating compounds (STACs) and small molecule inhibitors have been reported, one of the biggest hurdles to overcome in developing sirtuin-targeted compounds is selectivity. For example, EX-527 is used as a selective Sirt1 inhibitor, but EX-527 also inhibits Sirt2 and Sirt3 [19]. STACs have been reported to increase Sirt1 activity, but do so with numerous off-target effects against other enzymes, receptors, transport proteins, and ion channels [20].
Since physiological [1] and pharmacological [18] processes can alter sirtuin activity without impacting transcription, translation, or degradation of sirtuin proteins, techniques are needed to measure sirtuin activity independent of sirtuin protein levels. Current approaches to assay cellular sirtuin activity are largely limited to indirect methods such as immunoblotting or mass spectrometry analysis of acylation levels of sirtuin substrates. However, these techniques fail to account for other factors that may alter protein acylation statuses, such as acyltransferase activity, protein turnover, subcellular localization, protein-protein interactions, and overlapping substrate specificity among sirtuins and/or other histone/protein lysine deacylases. Development of chemical probes that bind only to the active form of a sirtuin and therefore directly report on available sirtuin enzymatic activity would provide novel insights into the role of sirtuin deacylase activity in aging-related disease and healthspan extension. To this end, activity-based probes (ABPs) represent an ideal solution, as ABPs covalently react with enzymes in an activity-based manner. Through labeling an ABP with biotin or a fluorophore, ABPs can quantitatively report on sirtuin activity in normal, diseased, or small molecule treated cells or tissues.
While most sirtuin inhibitors rely solely on competitive binding against acylated protein substrates or NAD+, inhibitors that exploit the unique catalytic mechanism of sirtuins have also been reported [21–24]. Thioacetyl-lysine containing peptides are potent (low nM Ki) mechanism-based sirtuin inhibitors [25,26]. Potent inhibition results from rapid nicotinamide cleavage from NAD+ upon thioacetyl-lysine peptide binding, while the rate of dethioacetylation is ~2 orders of magnitude slower than the rate of deacetylation (Figure 1A) [25] resulting in increased residence time of thioacetyl-lysine peptides on sirtuins. Thioacetyl-lysine peptides are therefore ideal for developing sirtuin ABPs because potent thioacetyl-lysine peptide binding requires an active sirtuin, and thioacetyl-lysine peptides are easily modified during solid-phase peptide synthesis.
Figure 1.

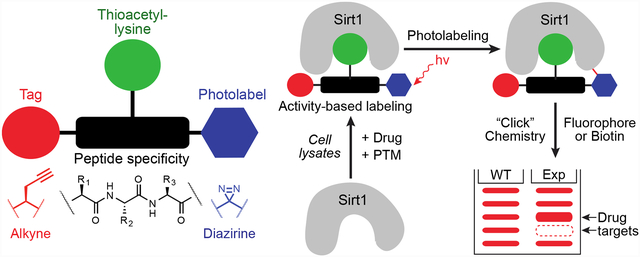
Sirt1 activity-based probe development, design, and workflow. (A) Mechanism-based inhibition of sirtuin deacylases by thioacetyl-lysine. Rates shown are from the yeast sirtuin, Hst2 [25]. (B) Sirtuin activity-based probe components: sirtuin-specific peptide sequence, thioacetyl-lysine, diazirine photolabel, and bioorthogonal alkyne tag. (C) H3K14 (top) and p53 (bottom) ABP peptide chemical structures targeting Sirt1. (D) Sirtuin ABP workflow. ABPs bind active sirtuin either in vitro or in cell lysates via thioacetyl-lysine mechanism-based inhibition, UV-irradiation of diazirine yields covalently-labeled sirtuin, sirtuin interacting proteins and non-specific targets, click chemistry (CuAAC) appends a fluorophore or biotin (e.g. 5-TAMRA-azide or biotin azide) to the ABP alkyne, and in-gel fluorescence or biotin immunoblot provides a direct readout of sirtuin activity along with the potential to label sirtuin interacting proteins.
Inspired by work from the Cravatt laboratory [27,28], we first proposed an overall strategy to develop sirtuin-targeted ABPs over a decade ago [21], and encouragingly, initial efforts to develop sirtuin ABPs using this strategy have been met with some success [29,30]. Cen and colleagues developed sirtuin ABPs with activities against Sirt2, Sirt5, and Sirt6 [29]. More recently, the Olsen laboratory reported a series of thioamide and hydrazide acyl-lysine ABPs that exhibit labeling of each sirtuin isoform [30]. Despite these initial efforts, there remains a need for sirtuin ABPs with increased selectivity and sensitivity to meaningfully deconvolute isoform-specific activities in the cell. To this end, we developed a Sirt1-selective ABP through a modular, peptide-based approach that directly measures Sirt1 activity with enhanced sensitivity compared to the previous studies. We show activity-based labeling of Sirt1 in response to acyl-lysine peptide substrate competition, pharmacological inhibition, and post-translational modification of Sirt1 in vitro and in cell lysates. Furthermore, cellular fractionation coupled with activity-based labeling measured subcellular Sirt1 activity in MCF7 breast cancer cells. Taken together, our study further demonstrates the utility of a modular, peptide-based approach for developing selective activity-based probes of the sirtuin protein family.
2. Materials and Methods
2.1. Peptide synthesis
A 9-mer thioacetyl-lysine H3K14 peptide [H3K14 ABP; (diazirine)-(propargylglycine)-TGGK(thioacetyl)APRY-NH2], 8-mer thioacetyl-lysine p53 peptide [p53 ABP; (diazirine)-RHKK(thioacetyl)LMF-(propargylglycine)-NH2], and 7-mer trifluoroacetyl-lysine p53 peptide (p53tfa; H2N-RHKK(trifluoroacetyl)LMF-NH2) were synthesized on a 0.5 mmol scale using standard tert-butyl/Fmoc solid-phase peptide synthesis techniques [31]. Fmoc amino acids were conjugated to 100–200 mesh Rink amide resin (Chem-Impex International, 12662) and protecting groups were as follows: Boc for lysine, trityl for histidine, tert-butyl for tyrosine, and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl for arginine. Dde was used as a protecting group for the thioacetyl-lysine residue in H3K14 ABP which was derivatized after amino acid coupling. The resin was swollen in DCM overnight at 4 °C, then Dde-lysine was deprotected with 2% v/v hydrazine in DMF for 10 min. Deprotection was repeated twice. The resin was then washed 3 × 5 mL DCM and 3 × 5 mL NMP. Deprotection of Dde-lysine was confirmed via positive Kaiser test [32]. Thioacetylation of H3K14 ABP was achieved by incubating resin 3× with 5 equiv ethyl dithioacetate and 10% v/v DIPEA in NMP for 1 h. The resin was then washed 3 × 5 mL DCM and 3 × 5 mL NMP. The p53tfa peptide was synthesized using commercially available Fmoc-Lys(trifluoroacetyl)-OH. H3K14 ABP and p53 ABP were N-terminally appended with a diazirine by deprotecting the N-terminal Fmoc amino acid with 20% v/v piperidine in NMP (3 × 5 mL), followed by washing 3 × 5 mL NMP, and the diazirine group coupled (protecting from light) with 5 equiv of 3-methyl-diazirine-3-propanoic acid and HBTU (0.095 g, 250 μmol) with 5% v/v DIPEA in NMP (5 mL). Complete couplings were confirmed by negative Kaiser tests [32]. Peptides were then cleaved from the resin and globally deprotected with 95% v/v TFA, 2.5% v/v H2O, and 2.5% v/v triisopropylsilane. Peptides were precipitated with ice-cold diethyl ether, pelleted by centrifugation, and washed 2× with ice-cold diethyl ether. The precipitates were dried, resuspended in water, and lyophilized. Crude peptides were purified by semipreparative HPLC on a μBondapak C18 column (Waters, 3.9×300 mm) using an Agilent 1100 series HPLC, using a gradient of 0–80% v/v acetonitrile in water with 0.1% v/v TFA. Fractions were collected and lyophilized (protected from light for H3K14 ABP and p53 ABP) to yield final peptides as dry white powders. Purified peptide masses were confirmed by direct injection ESI mass spectrometry (Finnigan LTQ, Thermo Scientific). Expected and observed masses (Table S1) and HPLC traces (Figure S1) of purified peptides are shown in the Supplementary Information. Peptide concentrations were determined from the mass of the peptide as a TFA salt of basic residues, assuming 1 equivalent of TFA per cationic group.
2.2. Recombinant sirtuin expression and purification
Recombinant Sirt1 (pET24), Sirt2 (pET28a), Sirt3 (pET28a), Sirt5 (pET28a), and Sirt6 (pQE80) were purified from BL21(DE3) and Sirt7 (pET28a) from ArcticExpress (DE3) E. coli by nickel-affinity chromatography. BL21(DE3) cells were transformed and grown at 37 °C in 2XYT media (ArcticExpress (DE3) cells were grown at 30 °C) supplemented with 50 mg/L kanamycin (pET24 or pET28a) or ampicillin (pQE80) to an OD of ~0.7 at 600 nm. Protein expression was induced for 20 h with 0.5 mM IPTG at 18 °C (ArcticExpress (DE3) cells were induced at 13 °C). Cells were harvested via centrifugation at 5,000 × g and cell pellets frozen at −80 °C until lysis. Frozen cells were thawed on ice, resuspended in lysis buffer, and lysed via sonication. Lysates were cleared via centrifugation for 30 min at 30,000 × g. Cleared lysate was incubated with Ni-NTA resin (0.75 mL resin/L culture) at 4 °C for a minimum of 1 h. The bound resin was pelleted via centrifugation, resuspended in 10× resin volume of lysis buffer (Table S2), and packed into a column. The packed resin was washed with 10 column volumes of wash buffer (Table S2) and purified enzymes eluted with 5 column volumes of elution buffer (Table S2). Enzymes were further purified and exchanged into storage buffer via size exclusion chromatography using an ENrich SEC 650 10×300 mm (Sirt1) or ENrich SEC 70 10×300 mm column (Sirt2, Sirt3, Sirt5, Sirt6, Sirt7). Concentrations of purified enzymes were determined by the method of Bradford using BSA as a standard [33], aliquoted, and stored at −80 °C.
2.3. ABP labeling of recombinant sirtuins
Labeling reactions of recombinant sirtuins contained equimolar concentrations of enzyme and ABP (0.2 μM) and 500 μM NAD+ in 50 mM HEPES, pH 7.4 unless denoted otherwise. For treatments that decrease Sirt1 activity, recombinant Sirt1 was treated with 0.2–500 μM of the p53tfa peptide or the Sirt1 inhibitor EX-527 [19] or pretreated with 100 μM S-nitrosoglutathione (GSNO) as previously described [17]. After mixing reaction components, samples were immediately irradiated (365 nm) for 30 min in a Fisher Scientific FB-UVXL-1000 UV Crosslinker. Following irradiation, alkyne-labeled samples were conjugated to 5-TAMRA or biotin azide via a copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) reaction containing 50 μM 5-TAMRA azide, 2 mM tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) or tris((1-benzyl-4-triazolyl)methyl)amine (TBTA), 1 mM CuSO4, and 1 mM TCEP in 50 mM HEPES, pH 7.4. Samples were protected from light and incubated with shaking for 1 h at 25 °C. Following CuAAC, reactions were quenched with ice-cold acetone and stored at −80 °C overnight. Labeled samples were pelleted by centrifugation (20,000 × g, 30 min, 4 °C) and the supernatant removed. Residual acetone was removed by a Savant SpeedVac SC100 (10 min). Dried pellets were resuspended in PBS containing 1% w/v SDS and prepared for SDS-PAGE. Following gel electrophoresis, gels containing ABP samples appended with 5-TAMRA azide were washed in destain (50:40:10 water:methanol:glacial acetic acid) for 30 min and equilibrated in water (2 × 10 min) before measuring in-gel rhodamine fluorescence on a GE Amersham 9400 Variable Mode Gel Imager or ChemiDoc MP Imaging System (BioRad). Following in-gel rhodamine fluorescence imaging, gels were washed in SYPRO Ruby destain (83:10:7 water:methanol:glacial acetic acid) for 30 min and incubated overnight with SYPRO Ruby Protein Gel Stain (Invitrogen S12000). Gels were washed for 30 min in SYPRO Ruby destain and equilibrated in water (2 × 10 min) before measuring SYPRO Ruby fluorescence on a GE Amersham 9400 Variable Mode Gel Imager or ChemiDoc MP Imaging System. For gels containing ABP samples appended with biotin azide, gels were transferred to a nitrocellulose membrane, blocked with 5% w/v milk in PBST, washed (3 × 5 min, PBST), and probed for biotin using the Vectastain ABC-HRP kit (PK-4000). Next, blots were washed (3 × 5 min, PBST), developed by ECL, and imaged on a ChemiDoc MP Imaging System.
2.4. Continuous enzyme-coupled sirtuin deacetylase assays
Deacetylase activity of Sirt1, Sirt2, and Sirt3 were monitored under initial rate conditions using a continuous enzyme-coupled assay for nicotinamide-producing enzymes as previously described [34]. The assay was performed at 25 °C in a reaction mixture containing 20 mM potassium phosphate pH 7.5, 1 mM NAD+, 50 μM acetylated H3K14 peptide, 1–100 μM H3K14 ABP or p53 ABP, 2.5 μM MBP-PncA, 3.3 mM alpha-ketoglutarate, 200 μM NADH, 2.5 units of (L)-glutamic dehydrogenase, and 1 μM sirtuin. Reactions were initiated by addition of sirtuin and NAD+. Rates were monitored continuously for 15 min in a clear flat-bottom 96-well plate (Greiner Bio-One) on a BioTek Cytation 5 multi-mode microplate reader (Winooski, VT) and initial rates were determined using our Interactive Continuous Enzyme Kinetics Analysis Tool (ICEKAT) [35].
2.5. ABP IC50 Analysis
Concentration-dependent inhibition of Sirt1–3 by H3K14 and p53 ABP was determined using the assay described above. The data were plotted as percentages of deacetylase activity in the absence of ABP versus ABP concentration and fit to the following equation using GraphPad Prism (La Jolla, CA, USA):
2.6. Cell culture
HEK293T cells were obtained from ATCC (#CRL-3216) and were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. MCF7 cells were obtained from ATCC (#HTB-22) and were cultured in MEM supplemented with 10% FBS, 0.2% human insulin solution (Sigma I9278), 1% sodium pyruvate, 1% non-essential amino acids, and 1% antibiotic-antimycotic. Cells were cultured as subconfluent monolayers before harvesting for cell lysis. For Sirt1 overexpression, 1.92 × 106 HEK293T cells were transfected with 5.5 μg of pcDNA3.1 Sirt1 His/Myc-tagged plasmid using Avalanche®-Omni transfection reagent (EZT-OMNI-1). These cells were supplemented with fresh media at 12 h and cultured for a total of 36 h. Overexpression was confirmed by immunoblot (Figure S2).
2.7. Cell lysis and fractionation
HEK293T and MCF7 cells were washed with and scraped in ice-cold PBS, and collected by centrifugation (a ‘pop-spin’ for 10 sec in a tabletop microfuge) [36]. HEK293T cells were immediately resuspended in 50 mM HEPES, pH 7.4 containing 1% NP-40 and a protease inhibitor cocktail (CST 5871). HEK293T lysates were sonicated for 10 sec and cleared by centrifugation (20,000 × g, 30 min, 4 °C) to obtain a soluble whole-cell lysate. MCF7 cytoplasmic and nuclear fractionation was carried out as previously described [36]. Soluble lysate fractions were prepared fresh before each experiment and quantitated by the method of Bradford using BSA as the standard [33].
2.8. Immunoblotting
Recombinant sirtuins were labeled as described in section 2.3 and resolved by SDS-PAGE, transferred to a nitrocellulose membrane, blocked with 5% w/v milk in PBST, and probed with an anti-His6 antibody (1:1000 dilution, Abgent AM1010a, lot SG100323A) at 4 °C overnight. The membrane was washed (3 × 5 min, PBST), incubated with secondary antibody for 1 h at ambient temperature, and washed again (3 × 5 min, PBST) before being developed by ECL and imaged on a ChemiDoc MP Imaging System. HEK293T whole-cell lysates and MCF7 subcellular fractions (50 μg) were generated as described in section 2.5 and processed by the same immunoblot protocol. In HEK293T lysates, Sirt1 was detected with anti-Sirt1 antibody (1:1000 dilution, Cell Signaling Technology, 1F3-CST8469). For MCF7 cells, anti-H3 antibody (1:5000 dilution, Abcam 1791, lot GR3236360–1) detected subcellular fractions. Secondary antibodies used were goat anti-rabbit (1:5000 dilution, Thermo A10549, lot UH2818652) or goat anti-mouse (1:5000 dilution, Invitrogen 31437, lot UG2814213). All antibodies were diluted with 5% w/v milk in PBST.
2.9. ABP labeling of cell lysates
Labeling reactions of HEK293T and MCF7 soluble lysate fractions contained 50 μg protein, 500 μM NAD+, and 5 μM ABP in 50 mM HEPES, pH 7.4, unless denoted otherwise. HEK293T lysates were treated with 500 μM p53tfa, EX-527, SirReal2, TM (a thiomyristoyl-lysine compound), or 100 μM GSNO as in section 2.3. Samples were incubated for 10 min at ambient temperature and then subjected to UV-irradiation and CuAAC followed by in-gel fluorescence analysis as described in section 2.3.
3. Results and Discussion
3.1. Sirt1 activity-based probe development, design, workflow, and characterization.
Using a modular peptide-based approach, two potential sirtuin activity-based probes (ABPs) were synthesized each consisting of four elements: 1) Sirt1 substrate peptide sequences based on either histone 3 (H3K14) [37] or p53 [13,14,17]; 2) a thioacetyl-lysine residue for mechanism-based sirtuin inhibition [25,26]; 3) a diazirine photolabeling group for efficient crosslinking to Sirt1 when bound [38,39]; and 4) an alkyne reporter tag amenable to copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) labeling [40] with either a fluorophore or biotin for detection and measurement of Sirt1 activity (Figure 1B–D). The development of the sirtuin ABP workflow into an efficient and reproducible methodology required multiple rounds of optimization. Steps of the workflow requiring optimization included, but were not limited to, ABP binding of active sirtuin, photolabeling time, CuAAC ligand identity and reagent ratios, sample preparation for gel-based signal detection, and detergent and buffer lysis conditions for cellular experiments. The aspects found most critical are highlighted below.
To examine the relative ability of the H3K14 and p53 ABPs (Figure 1C) to efficiently and selectively label only active forms of Sirt1, the H3K14 (Figure 2A) and p53 (Figure 2B) ABPs were incubated with Sirt1 in the presence or absence of the necessary sirtuin cosubstrate, NAD+, and irradiated with UV light followed by CuAAC with 5-TAMRA-azide. Detection of activity-based labeling was dependent on the presence of alkyne/ABP (see e.g. Figure 4D), azide (see e.g. Figure S3), sirtuin, and UV crosslinking inclusion and duration (see e.g. Figure S4). As anticipated, a greater extent of Sirt1 labeling was observed in the presence of NAD+ in a concentration-dependent manner, consistent with ABP labeling favoring active catalysis by Sirt1. The p53 ABP exhibited robust labeling and enhanced sensitivity compared to the H3K14 ABP; therefore, the p53 ABP was selected for further in vitro and cellular studies. The use of equimolar amounts (0.2 μM) of Sirt1 and the p53 ABP resulted in a 4-fold increase (ImageJ) in the labeling of enzymatically active Sirt1 as compared to Sirt1 in the absence of NAD+, thus, these concentrations were used for remaining in vitro experiments. To determine the limit of detection at this concentration of the p53 ABP (0.2 μM), the concentration of Sirt1 was further decreased and activity-based labeling was examined. Activity-based labeling of Sirt1 was detectable at concentrations as low as 0.05 μM Sirt1 (Figure 2C). This sensitivity was remarkably improved compared to previous in vitro studies that used 10 μM probe to detect 10 μM sirtuin [29] or ≥12.5 μM probe to detect 1 μM of sirtuin [30] when optimized.
Figure 2.

Comparison of H3K14 and p53 ABP potency and selectivity. (A/B) Concentration-dependent labeling of recombinantly purified full-length Sirt1 by the (A) H3K14 and (B) p53 ABPs (0.1–1 μM) in the absence or presence of NAD+ (500 μM). SYPRO staining is shown as a loading control. (C) p53 ABP (0.2 μM) labeling of recombinantly purified full-length Sirt1 (0.01–0.2 μM) in the absence or presence of NAD+ (500 μM). SYPRO staining is shown as a loading control. (D) p53 ABP labeling (0.2 μM) of recombinantly purified sirtuins shows preferential Sirt1 labeling with weak labeling of Sirt2 and Sirt3 in the presence of NAD+ (500 μM). The anti-His6 immunoblot shows the relative loading of His6-tagged sirtuins. (E) Concentration-dependent inhibition of Sirt1 (red), Sirt2 (blue), and Sirt3 (black) by 1–100 μM H3K14 ABP (left) and p53 ABP (right) was assessed via a continuous enzyme-coupled assay [34]. Percentage of deacetylase activity in the absence of ABP was plotted versus log10 of the ABP concentration (n = 3 ± S.E.M.)
Figure 4.

ABPs measure cellular Sirt1 activity. (A) p53 ABP labeling of Sirt1 is decreased upon treatment with p53tfa (500 μM), EX-527 (500 μM), GSNO (100 μM), SirReal2 (500 μM), or the thiomyristoyl-lysine compound TM (500 μM) in HEK293T cells overexpressing Sirt1 in the presence of NAD+ (500 μM). (B) The p53 ABP (1–5 μM) shows concentration-dependent labeling of endogenous Sirt1 in HEK293T cell lysate (10 μg) in the presence of NAD+ (500 μM). (C) The p53 ABP (5 μM) exhibits differential labeling of endogenous Sirt1 in MCF7 cellular fractions (WC, whole-cell lysate; C, cytosolic fraction; and N, nuclear fraction) in the presence of NAD+ (500 μM). (D) Comparison of p53 and H3K14 ABP (5 μM) labeling in HEK293T lysates overexpressing Sirt1 (50 μg) reveal similar but distinct banding patterns in the presence of NAD+ (500 μM).
Since human sirtuins harbor overlapping substrate specificity [1,3], the selectivity of the p53 ABP for Sirt1 compared to the other human sirtuin isoforms was examined. When comparing p53 ABP labeling of recombinant sirtuins, selective and strong labeling of Sirt1 was observed (Figure 2D). Among the rest of the human sirtuins tested, only the other sirtuins that exhibit strong deacetylase activity, Sirt2 and Sirt3, showed weak labeling compared to Sirt1 (Figure 2D). The selectivity of the p53 ABP for Sirt1 is likely a result of the p53 peptide sequence, indicating that molecules that target the unique substrate specificity of sirtuins can be effective, sirtuin-selective chemical probes. Examination of p53 ABP selectivity toward other sirtuin isoforms required further optimization of the sirtuin ABP workflow. Following the CuAAC reaction, methanol precipitation resulted in sample loss for samples containing sirtuin isoforms that were not labeled by the p53 ABP leading to unequal loading controls. To solve this issue, acetone precipitation coupled with SpeedVac evaporation resulted in undetectable sample loss from input to output, allowing for maximal signal detection (data not shown). To confirm ABP selectivity for Sirt1, we examined concentration-dependent inhibition of Sirt1–3 deacetylase activity by H3K14 ABP and p53 ABP via a continuous enzyme-coupled assay [34] (Figure 2E). Consistent with the increased sensitivity of the p53 ABP compared to the H3K14 ABP, the p53 ABP (IC50 = 0.11 ± 0.01 μM) exhibited 7.5-fold increased inhibition of Sirt1 deacetylase activity relative to the H3K14 ABP (IC50 = 0.83 ± 0.09 μM). In agreement with the observed increased ABP labeling of recombinant Sirt1 compared to Sirt2 and Sirt3 (Figure 2D), both ABPs showed >4-fold selectivity for Sirt1 over Sirt2 (p53 ABP: IC50 = 0.64 ± 0.12 μM; H3K14 ABP: IC50 = 3.3 ± 0.6 μM;) and >7-fold selectivity for Sirt1 over Sirt3 (p53 ABP: IC50 = 4.6 ± 0.5 μM; H3K14 ABP: IC50 = 6.2 ± 0.8 μM) (Figure 2E).
3.2. Peptide competition, small molecule inhibition, and post-translational modification of Sirt1 decreases p53 ABP labeling
Since the p53 ABP exhibited robust, selective labeling of Sirt1, the ability of the p53 ABP to measure Sirt1 activity upon peptide competition, pharmacological inhibition, and inhibitory post-translational modification was examined. First, to ensure that the observed Sirt1 labeling was due to specific targeting of the p53 ABP to the Sirt1 active site where acetyl-lysine substrates bind, and not non-specific diazirine crosslinking, a competition experiment was performed using a trifluoroacetyl-lysine containing p53 peptide (p53tfa). Trifluoroacetylated peptides bind sirtuins with tighter affinity compared to the corresponding acetylated peptides and with similar affinity to the corresponding thioacetylated peptides [25,41]. The p53tfa peptide was titrated against recombinant Sirt1 in the presence of the p53 ABP, and the relative Sirt1 labeling measured. A concentration-dependent decrease in Sirt1 labeling was observed in response to increasing concentrations of p53tfa peptide, consistent with competition of the p53tfa and p53 ABP peptides for the Sirt1 active site (Figure 3A).
Figure 3.

Peptide competition, small molecule inhibition, and inhibitory post-translational modification of Sirt1 decrease p53 ABP labeling consistent with activity-based labeling. (A) p53 ABP labeling of Sirt1 decreases with the competition of a trifluoroacetylated p53 peptide (p53tfa; 0.2–500 μM) in the presence of NAD+ (500 μM). (B) Treatment with the Sirt1 inhibitor EX-527 (0.2–500 μM) attenuates p53 ABP labeling in the presence of NAD+ (500 μM). (C) Inhibition of Sirt1 by S-nitrosation upon GSNO treatment ablates p53 ABP labeling in the presence and absence of NAD+ (500 μM).
To determine if the p53 ABP can report on the relative activity of pharmacologically or physiologically inhibited Sirt1, recombinant Sirt1 was treated with known pharmacological or physiological inhibitors of Sirt1. First, recombinant Sirt1 was treated with increasing concentrations of the widely used indole EX-527, which inhibits Sirt1 with nanomolar potency [19], and relative Sirt1 activity was measured by p53 ABP labeling. A concentration-dependent decrease in Sirt1 labeling by the p53 ABP was observed upon EX-527 treatment, indicating the p53 ABP can measure attenuated Sirt1 activity upon pharmacological inhibition (Figure 3B). It is important to note that in both the p53tfa (Figure 3A) and EX-527 (Figure 3B) competition assays, the concentration needed to compete with p53 ABP labeling exceeds the Kd values for these interactions as p53tfa and EX-527 are non-covalent and reversible whereas p53 ABP labeling is covalent and irreversible.
We and others have previously shown that Sirt1 can be transnitrosated by the nitric oxide-derived cysteine oxidant S-nitrosoglutathione (GSNO) [17,42–44], resulting in post-translational modification of the Sirt1 Zn2+-tetrathiolate by S-nitrosation and complete loss of deacetylase activity [17]. Therefore, recombinant Sirt1 was treated with GSNO as previously described [17], and relative Sirt1 activity measured by p53 ABP labeling. A dramatic loss of Sirt1 labeling by p53 ABP upon treatment with GSNO in the presence and absence of NAD+ was observed, indicating the p53 ABP can measure the loss of Sirt1 activity by an inhibitory post-translational modification (Figure 3C). The loss of p53 ABP labeling is consistent with the inability of S-nitrosated Sirt1 to bind acetylated substrates, as we have previously shown [17]. Taken together, these data indicate that the p53 ABP can measure relative changes in Sirt1 activity modulated by pharmacological or physiological processes and not by changes in Sirt1 protein levels.
3.3. p53 ABP measures cellular Sirt1 activity
Encouraged by our in vitro experiments, we next determined if the p53 ABP could measure relative Sirt1 activity in a cellular context. The more complex cellular environment required further optimization of the sirtuin ABP workflow. In particular, buffer selection for the CuAAC reaction proved to be critical. In agreement with a previous report [45], we found that the use of Tris-based buffers resulted in decreased efficiency of the CuAAC reaction, as tris(hydroxymethyl)aminomethane can act as an inhibitory ligand in the Cu(I) complex [46]. Furthermore, use of hydrophilic Cu(I)-stabilizing ligand THPTA instead of hydrophobic TBTA improved click efficiency, and increasing ligand concentration (2:1 THPTA:CuSO4 ratio) while decreasing azide concentration (10:1 azide:alkyne/ABP ratio) decreased background and nonspecific labeling in cell lysates. Additionally, certain buffers and detergents can have deleterious effects on activity-based protein profiling in cell lysates [45]. Indeed, a significant increase in Sirt1 labeling was observed when cells were lysed in buffers containing only non-ionic detergents (Triton-X100 or NP-40), compared to lysis buffers containing ionic detergents (RIPA). This observation is consistent with the ability of ionic detergents to denature proteins [47] and attenuate sirtuin enzymatic activity as a result. To this end, non-ionic detergent-containing HEPES buffers were superior to phosphate buffers for maintaining Sirt1 activity and maximizing p53 ABP labeling (Figure S5). After optimization, as little as 1 μM p53 ABP could label endogenous Sirt1 using 10 μg of HEK293T lysate (Figure 4B). However, the maximal signal was observed using 5 μM of the p53 ABP, 50 μg of cell lysate, and 30 minutes UV crosslinking (Figure S4). Therefore, further cell lysate experiments were performed using 5 μM of the p53 ABP and 50 μg of cell lysate.
The experiments performed with recombinant Sirt1 in section 3.2 were repeated in HEK293T whole cell lysates overexpressing Sirt1. HEK293T lysates were treated with p53tfa, EX-527, or GSNO at concentrations of maximal in vitro response, and labeling of Sirt1 by the p53 ABP was measured. Consistent with our in vitro experiments (Figure 3), Sirt1 labeling by the p53 ABP in HEK293T lysates was decreased upon peptide competition, pharmacological inhibition, or inhibitory post-translational modification. A ≥2-fold reduction (ImageJ) in activity-based labeling was observed upon competition with the p53tfa peptide, and inhibition of Sirt1 activity by EX-527 or GSNO when compared to the untreated control (Figure 4A). To further examine the utility of the p53 ABP in a cellular context, we treated HEK293T lysates with the Sirt2-selective inhibitors SirReal2 [48] and TM (a thiomyristoyl-lysine compound) [22] and observed no reduction in activity-based labeling of Sirt1 (Figure 4A). SirReal2 treatment resulted in a slight increase in activity-based labeling of Sirt1, likely due to abrogation of the p53 ABP-Sirt2 interaction that allowed increased “free” p53 ABP to target Sirt1. Furthermore, the p53 ABP labeled endogenous Sirt1 in a concentration-dependent manner in HEK293T lysates (Figure 4B). Next, to examine the efficacy of the p53 ABP in an alternative cell line, Sirt1 activity in MCF7 cells was measured. Additionally, since Sirt1 functions as both a nuclear and cytoplasmic deacetylase, Sirt1 activity was examined as a function of subcellular localization. MCF7 cells were fractionated into whole-cell, cytosolic, and nuclear lysate fractions and treated with the p53 ABP (Figure 4C). Labeling of endogenous Sirt1 was observed in each fraction, but enhanced Sirt1 labeling was observed in the nucleus, consistent with Sirt1 functioning primarily as a nuclear deacetylase (Figure 4C) [24]. To determine if the p53 and H3K14 ABPs may label (possibly different) Sirt1-interacting proteins, HEK293T lysates overexpressing Sirt1 were incubated with both the p53 and H3K14 ABPs, and their banding patterns were compared (Figure 4D). Interestingly, similar but distinct banding patterns were observed, indicating possible ABP-dependent detection of Sirt1-interacting proteins. In cellular studies, the dynamic range of Sirt1 activity-based labeling was decreased relative to in vitro experiments, likely due to interaction of the p53 ABP with the broader proteome of a cellular lysate. Consistent with this hypothesis, inhibition of Sirt1 in a cellular context led to increased background labeling of the broader proteome by the p53 ABP.
4. Conclusion
We developed a Sirt1-selective ABP with unprecedented sensitivity that measures both in vitro and cellular Sirt1 activity. The p53 ABP detects attenuated Sirt1 activity upon pharmacological or physiological inhibition in vitro and in HEK293T cell lysates, and subcellular Sirt1 activity in MCF7 breast cancer cells. This study provides a novel chemical probe for the direct measurement of relative Sirt1 activity, irrespective of Sirt1 protein levels or acylation status of Sirt1 substrates. Future studies will interrogate disease- and cell-type specific roles of Sirt1 activity. The modular nature of these thioacyl-lysine based ABPs can be extended to all sirtuin isoforms, and properties optimized for cell penetrance, subcellular localization, and cytotoxicity using a similar peptide-based approach. The development of such probes will greatly advance the sirtuin field by providing a more direct means to measure sirtuin deacylase activity in cells and elucidate yet undiscovered roles for the sirtuin deacylase family in human biology.
Supplementary Material
Highlights.
Development of a Sirt1-selective activity-based probe (ABP)
Activity-based labeling measured Sirt1 activity in vitro and in HEK293T lysates
Sirt1 ABP detected decreased Sirt1 activity upon treatment with inhibitory stimuli
Sirt1 ABP measured subcellular Sirt1 activity in MCF7 breast cancer cells
Acknowledgements
We thank Kelsey Kalous for assistance in recombinant protein production and valuable discussion. This work was supported by the National Institutes of Health [grant number R35 GM128840]; the American Heart Association [grant number 15SDG25830057], the Michael Keelan, Jr., MD, Research Foundation, and the Advancing a Healthier Wisconsin endowment.
Abbreviations
- ABP
activity-based probe
- BSA
bovine serum albumin
- CuAAC
copper(I)-catalyzed alkyne-azide cycloaddition
- DCM
dichloromethane
- DIPEA
N,N-Diisopropylethylamine
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DMF
N,N-Dimethylformamide
- ECL
enhanced chemiluminescence
- ESI
electrospray ionization
- GSNO
S-nitrosoglutathione
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- NAD+
nicotinamide adenine dinucleotide
- NMP
N-Methyl-2-pyrrolidone
- NTA
nitrilotriacetic acid
- PBS
phosphate-buffered saline
- RIPA
radioimmunoprecipitation assay buffer
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- STAC
sirtuin-activating compound
- TAMRA
tetramethyl rhodamine
- TCEP
tris(2-carboxyethyl)phosphine
- TFA
trifluoroacetic acid
- TBTA
tris((1-benzyl-4-triazolyl)methyl)amine
- THPTA
tris(3-hydroxypropyltriazolylmethyl)amine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Feldman JL, Dittenhafer-Reed KE, Denu JM, Sirtuin catalysis and regulation, J. Biol. Chem 287 (2012) 42419–42427. doi: 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jackson MD, Denu JM, Structural identification of 2“- and 3-”O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta -NAD+-dependent histone/protein deacetylases, J. Biol. Chem 277 (2002) 18535–18544. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- [3].Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I, Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins, Mol. Biol. Cell 16 (2005) 4623–4635. doi: 10.1091/mbc.e05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sebastián C, Satterstrom FK, Haigis MC, Mostoslavsky R, From sirtuin biology to human diseases: an update, J. Biol. Chem 287 (2012) 42444–42452. doi: 10.1074/jbc.R112.402768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bonkowski MS, Sinclair DA, Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds, Nat. Rev. Mol. Cell Biol 17 (2016) 679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chalkiadaki A, Guarente L, The multifaceted functions of sirtuins in cancer, Nat. Rev. Cancer 15 (2015) 608–624. doi: 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- [7].Winnik S, Auwerx J, Sinclair DA, Matter CM, Protective effects of sirtuins in cardiovascular diseases: from bench to bedside, Eur. Heart J 36 (2015) 3404–3412. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Donmez G, Outeiro TF, SIRT1 and SIRT2: emerging targets in neurodegeneration, EMBO Mol Med. 5 (2013) 344–352. doi: 10.1002/emmm.201302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kitada M, Koya D, SIRT1 in Type 2 Diabetes: Mechanisms and Therapeutic Potential, Diabetes Metab J. 37 (2013) 315–325. doi: 10.4093/dmj.2013.37.5.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kitada M, Ogura Y, Monno I, Koya D, Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function, Front Endocrinol (Lausanne). 10 (2019) 187. doi: 10.3389/fendo.2019.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guarente L, Sirtuins in aging and disease, Cold Spring Harb. Symp. Quant. Biol 72 (2007) 483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- [12].Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y, Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1, J. Biol. Chem 282 (2007) 6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- [13].Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. , Negative control of p53 by Sir2alpha promotes cell survival under stress, Cell. 107 (2001) 137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- [14].Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. , hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase, Cell. 107 (2001) 149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- [15].Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. , Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase, Science. 305 (2004) 390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- [16].Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, et al. , Phosphorylation regulates SIRT1 function, PLoS ONE. 3 (2008) e4020. doi: 10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kalous KS, Wynia-Smith SL, Olp MD, Smith BC, Mechanism of Sirt1 NAD+-dependent Protein Deacetylase Inhibition by Cysteine S-Nitrosation, J. Biol. Chem 291 (2016) 25398–25410. doi: 10.1074/jbc.M116.754655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dai H, Sinclair DA, Ellis JL, Steegborn C, Sirtuin activators and inhibitors: Promises, achievements, and challenges, Pharmacol. Ther 188 (2018) 140–154. doi: 10.1016/j.pharmthera.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Napper AD, Hixon J, McDonagh T, Keavey K, Pons J-F, Barker J, et al. , Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1, J. Med. Chem 48 (2005) 8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- [20].Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, et al. , SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1, J. Biol. Chem 285 (2010) 8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Smith BC, Hallows WC, Denu JM, Mechanisms and molecular probes of sirtuins, Chem. Biol 15 (2008) 1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jing H, Hu J, He B, Negrón Abril YL, Stupinski J, Weiser K, et al. , A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity, Cancer Cell. 29 (2016) 297–310. doi: 10.1016/j.ccell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rajabi N, Auth M, Troelsen KR, Pannek M, Bhatt DP, Fontenas M, et al. , Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight, Angew. Chem. Int. Ed. Engl 56 (2017) 14836–14841. doi: 10.1002/anie.201709050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Farooqi AS, Hong JY, Cao J, Lu X, Price IR, Zhao Q, et al. , Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model, J. Med. Chem 62 (2019) 4131–4141. doi: 10.1021/acs.jmedchem.9b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith BC, Denu JM, Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide, Biochemistry. 46 (2007) 14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- [26].Fatkins DG, Monnot AD, Zheng W, Nepsilon-thioacetyl-lysine: a multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation, Bioorg. Med. Chem. Lett 16 (2006) 3651–3656. doi: 10.1016/j.bmcl.2006.04.075. [DOI] [PubMed] [Google Scholar]
- [27].Salisbury CM, Cravatt BF, Activity-based probes for proteomic profiling of histone deacetylase complexes, Proc. Natl. Acad. Sci. U.S.a 104 (2007) 1171–1176. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Salisbury CM, Cravatt BF, Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes, J. Am. Chem. Soc 130 (2008) 2184–2194. doi: 10.1021/ja074138u. [DOI] [PubMed] [Google Scholar]
- [29].Graham E, Rymarchyk S, Wood M, Cen Y, Development of Activity-Based Chemical Probes for Human Sirtuins, ACS Chem. Biol 13 (2018) 782–792. doi: 10.1021/acschembio.7b00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bæk M, Martín-Gago P, Laursen JS, Madsen JLH, Chakladar S, Olsen CA, Photo Cross-Linking Probes Containing ϵ-N-Thioacyllysine and ϵ-N-Acyl-(δ-aza)lysine Residues, Chemistry. 26 (2020) 3862–3869. doi: 10.1002/chem.201905338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].White PD, Chan WC, Fmoc Solid Phase Peptide Synthesis, 2004. doi: 10.1021/ol300592w. [DOI] [Google Scholar]
- [32].Kaiser E, Colescott RL, Bossinger CD, Cook PI, Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides, Anal. Biochem 34 (1970) 595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- [33].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem 72 (1976) 248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- [34].Smith BC, Hallows WC, Denu JM, A continuous microplate assay for sirtuins and nicotinamide-producing enzymes, Anal. Biochem 394 (2009) 101–109. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Olp MD, Kalous KS, Smith BC, ICEKAT: an interactive online tool for calculating initial rates from continuous enzyme kinetic traces, BMC Bioinformatics. 21 (2020) 186–12. doi: 10.1186/s12859-020-3513-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, Riabowol K, REAP: A two minute cell fractionation method, BMC Res Notes. 3 (2010) 294. doi: 10.1186/1756-0500-3-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D, Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin, Molecular Cell. 16 (2004) 93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- [38].Dubinsky L, Krom BP, Meijler MM, Diazirine based photoaffinity labeling, Bioorg. Med. Chem 20 (2012) 554–570. doi: 10.1016/j.bmc.2011.06.066. [DOI] [PubMed] [Google Scholar]
- [39].Yang T, Liu Z, Li XD, Developing diazirine-based chemical probes to identify histone modification ‘readers’ and ‘erasers’, Chem Sci. 6 (2015) 1011–1017. doi: 10.1039/c4sc02328e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Thirumurugan P, Matosiuk D, Jozwiak K, Click chemistry for drug development and diverse chemical-biology applications, Chem. Rev 113 (2013) 4905–4979. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]
- [41].Smith BC, Denu JM, Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases, J. Biol. Chem 282 (2007) 37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- [42].Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, et al. , Redox regulation of sirtuin-1 by S-glutathiolation, Antioxidants & Redox Signaling. 13 (2010) 1023–1032. doi: 10.1089/ars.2010.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JVK, Snowman AM, et al. , GAPDH mediates nitrosylation of nuclear proteins, Nature Cell Biology. 12 (2010) 1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shinozaki S, Chang K, Sakai M, Shimizu N, Yamada M, Tanaka T, et al. , Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase SIRT1 to increase acetylation and activation of p53 and p65, Sci Signal. 7 (2014) ra106–ra106. doi: 10.1126/scisignal.2005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yang Y, Yang X, Verhelst SHL, Comparative analysis of click chemistry mediated activity-based protein profiling in cell lysates, Molecules. 18 (2013) 12599–12608. doi: 10.3390/molecules181012599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hong V, Presolski SI, Ma C, Finn MG, Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation, Angew. Chem. Int. Ed. Engl 48 (2009) 9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Otzen D, Protein-surfactant interactions: a tale of many states, Biochim. Biophys. Acta 1814 (2011) 562–591. doi: 10.1016/j.bbapap.2011.03.003. [DOI] [PubMed] [Google Scholar]
- [48].Rumpf T, Schiedel M, Karaman B, Roessler C, North BJ, Lehotzky A, et al. , Selective Sirt2 inhibition by ligand-induced rearrangement of the active site, Nature Communications. 6 (2015) 6263–13. doi: 10.1038/ncomms7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.