

Abstract
Strokes are feared complications of sickle cell disease (SCD) and yield significant neurologic and neurocognitive deficits. However, even without detectable strokes, SCD patients have significant neurocognitive deficits in domains of learning and memory, processing speed and executive function. In these cases, mechanisms unrelated to major cerebrovascular abnormalities likely underlie these deficits. While oxidative stress and stress-related signaling pathways play a role in SCD pathophysiology, their role in cerebral injury remains unknown. We have shown that Townes and BERKs SCD mice, while not having strokes, recapitulate neurocognitive deficits reported in humans. We hypothesized that cognitive deficits in SCD mice are associated with cerebral oxidative stress. We showed that SCD mice have increased levels of reactive oxygen species, protein carbonylation, and lipid peroxidation in hippocampus and cortex, thus suggesting increased cerebral oxidative stress. Further, cerebral oxidative stress was associated with caspase-3 activity alterations and vascular endothelial abnormalities, white matter changes, and disruption of the blood brain barrier, similar to those reported after ischemic/oxidative injury. Additionally, after repeated hypoxia/reoxygenation exposure, homozygous Townes had enhanced microglia activation. Our findings indicate that oxidative stress and stress-induced tissue damage is increased in susceptible brain regions, which may, in turn, contribute to neurocognitive deficits in SCD mice.
Keywords: cognitive, anemia, hypoxia, memory, learning, sickle cell
Introduction
Sickle cell disease (SCD) is an inherited blood disorder that affects millions of individuals of predominantly African heritage worldwide [1, 2]. In the United States, it affects over 100,000 Americans and has an incidence of one in 365 African-American births. The disease encompasses a group of complex hemoglobinopathies and results from a mutation in the β subunit hemoglobin gene leading to the formation of an abnormal hemoglobin (HbS). Upon deoxygenation, HbS polymerizes and leads to “sickling” of erythrocytes resulting in hemolysis, chronic anemia, vascular endothelial injury, vaso-occlusion, and chronic inflammation [2, 3]. As childhood survival has improved, SCD has become a chronic degenerative disease with lifetime complications including recurrent acute pain crises, chronic pain, and multisystem organ damage [2, 3]. In turn, because of these complications, SCD patients can have severe disabilities, decreased productivity, and significant reduction (decades) in life expectancy [4].
Some of the most serious SCD complications are those involving the central nervous system [5–8]. Throughout life and as early as in childhood, SCD patients can present clinical and silent cerebral infarcts, headaches (acute and chronic), seizures, and transient ischemic attacks [5–8]. In these patients, silent and clinical strokes can yield severe neurologic and neurocognitive deficits and significant disability [5, 9, 10]. However, a growing body of evidence indicates that even without detectable strokes on imaging studies, SCD patients can present cognitive deficits in domains of learning and memory, processing speed and executive function [6, 11–13]. Together, these reports suggest that mechanisms unrelated to major cerebrovascular abnormalities or strokes underlie some of these neurocognitive deficits.
Several studies in SCD patients [14–18] and animal models [19–22] support the notions that SCD is associated with oxidative stress and that oxidative changes contribute to the pathobiology of SCD-related microvascular injury, recurrent vaso-occlusion, ischemia/reperfusion events, pain, and organ dysfunction [14, 15, 18, 22, 23]. In fact, in SCD, several events contribute to increased production of reactive oxygen species and promote vaso-occlusion and ischemia/reperfusion episodes [18, 24–26]. For example, hemoglobin released from erythrocytes during hemolysis is oxidized to free heme, which acts as a damage-associated molecular pattern molecule (DAMP) and triggers endothelial cell activation, inflammation, and oxidative stress responses [24–26]. Further supporting the role of oxidative stress on SCD pathobiology are studies showing that SCD mice treated with dimethyl fumarate, a drug that activates the main defense mechanism against oxidative stress [nuclear factor erythroid-2-related factor-2 (Nrf2)], have less vaso-occlusion and are protected from hepatic damage [27]. Thus, mounting evidence suggests that oxidative stress contributes to the pathobiology of SCD-induced organ damage.
The central nervous system, given its high oxygen demand and protein and lipid content, is highly susceptible to oxidative stress-induced injury [28]. Indeed, the contribution of oxidative stress-induced neuronal damage has been demonstrated in models of brain ischemia and of neurodegenerative disorders including Huntington, Alzheimer, and Parkinson diseases [29, 30]. There is also evidence that cerebral oxidative stress plays a critical role in neuropsychiatric disorders such as anxiety, depression, autism spectrum disorder, and schizophrenia [31, 32]. In brain ischemia models, researchers have shown that excessive production of reactive oxidative species by mitochondria underlies ischemia-induced hippocampal neuronal damage [30]. Therefore, given the evidence demonstrating that SCD is associated with oxidative stress [19–22, 33] and repeated episodes of ischemia/reperfusion [25, 34], it is reasonable to hypothesize that SCD is associated with cerebral oxidative stress.
Two strains of humanized SCD mice, Townes [35, 36] and BERK [37], recapitulate severe phenotypes observed in humans with SCD and allow for relevant studies of its pathobiology. Homozygotes from both strains express the mutated human sickle hemoglobin and have severe anemia, ongoing hemolysis, severe multi-organ dysfunction including liver necrosis [38], renal and cardiac dysfunction[39, 40], as well as altered nociception [41–43] and high spontaneous mortality. We have recently shown that homozygous (sickling) mice display profound learning and memory deficits, depression- and anxiety-like behavior, decreased exercise capacity, abnormal gait and balance, and impaired gait adaptability, all in the absence of histopathologic evidence of strokes [44–46]. Thus, these models are suitable for studies of mechanisms underlying neurocognitive deficits unrelated to strokes. In this investigation, we tested the hypothesis that SCD is associated with oxidative stress in the brain, which in turn could contribute to the occurrence of cerebral damage and lead to neurocognitive deficits.
Materials and Methods
SCD Mouse Models
The National Institutes of Health Clinical Center and Children’s Research Institute Institutional Animal Care and Use Committees approved all animal studies. We examined male and female, age 20-25-week-old mice from two SCD mouse models, the Townes [35, 36] and BERK [37] strains. Mouse breeding schemes and genotyping were carried out as described [45]. These mice carry knockout mutations of mouse hemoglobin genes and knock-in mutations (Townes) or a transgene containing human hemoglobin genes. Townes controls, heterozygotes and homozygotes express no mouse hemoglobin, as those genes were knocked out and replaced with human α, ß, and/or ßS gene (heterozygotes and homozygotes). Homozygous BERK mice express human α and ßS genes and hemizygotes, in addition to expressing a human ßS gene, express a mouse ß gene, thus having an attenuated phenotype. For the BERK strain, we used C57BL/6J animals as controls as they share >50% genetic identity with hemizygous and homozygous BERK mice [37].
We also evaluated microglia activation pattern during normoxia and after hypoxia exposure. We utilized a chronic intermittent hypoxia/reoxygenation exposure, which is regarded as a model of obstructive sleep apnea [47]. For seven consecutive days, animals were placed in a chamber for three consecutive hours where they were exposed to an 8% or 21% fraction of inspired oxygen. For the remainder of each day animals were returned to their cages and exposed to room air. Seventy-two hours after the last hypoxia or normoxia exposure, animals were euthanized and brains collected as describe below.
Transmission electron microscopy
Hippocampi and prefrontal cortices sections (1 mm3) were fixed at 4°C (2.5% glutaraldehyde and 1% paraformaldehyde in 0.1M cacodylate buffer) for 48 h and washed three times with cacodylate buffer. Hippocampus and prefrontal cortex sections were then fixed with 1% osmium tetroxide for two hours, washed with 0.1M cacodylate buffer. Tissues were quickly rinsed with water and placed in 1% uranyl acetate for one hour. Samples were then dehydrated in ethanol and propylene oxide and embedded in EMBed 812 resin (Electron Microscopy Sciences, Hatfield, PA, USA). Tissue sections (approximately 80 nm) were made using the Leica ultracut-UCT ultramicrotome (Leica, Deerfield, IL, USA), placed onto copper grids, and stained with saturated uranyl acetate in 50% methanol and with lead citrate. Grids were examined with a JEM-1200EXII electron microscope (JEOL Ltd, Tokyo, Japan) at 80kV and images recorded with the XR611M, mid mounted, 10.5M pixel, charge-coupled device camera (Advanced Microscopy Techniques Corp, Danvers, MA, USA). Pathologists (MMQ, IH-S), unaware of animals genotypes examined the images.
Markers of oxidative stress
In order to examine the production of reactive oxygen species, we measured reactive dihydroethidium in hippocampi slices using fluorescent microscopy, as an indication of superoxide production in hippocampus. Briefly, brains were collected and immediately frozen in liquid nitrogen. Frozen brains were embedded in optimal cutting temperature compound (OCT), to aid sectioning, and kept at −80°C. At the time of analysis, brains were cryo-sectioned at −20°C, dried at room temperature (30 min) and incubated with dihydroethidium for 10 min at 37°C. Sections then were rinsed once with ddH2O, air dried in the dark, and cover-slipped with 4′,6-diamidino-2-phenylindole (DAPI) antifade mounting solution (Vector Laboratories, Burlingame, CA). Samples were then incubated with dihydroethidium (Sigma-Aldrich, St. Louis, MO) prepared as follows. One mg of dihydroethidium was diluted in 200 μl of DMSO and then diluted to 1 ml with phosphate buffered saline (PBS, 3 mM). This stock solution was prepared freshly and further diluted with PBS 1: 80000 for incubation of the tissue (final concentration 37.5 nM). Slides were examined under a fluorescent microscope with the same exposure settings for all studied samples by an investigator blinded to sample genotype. After images were obtained, they were digitally combined to recreate a full hippocampus. Five investigators (blinded to the genotype of each image) rated the degree of fluorescence from one (minimal or no fluorescence) to four (maximum fluorescence).
We then examined lipid and protein oxidative modification markers in hippocampi and cerebral cortex homogenates of SCD mice. In hippocampi and cerebral cortices homogenates, using the thiobarbituric acid reactive substance (TBARS) fluorometric assay (BioAssay Systems, Hayward, CA)[48], we measured malondialdehyde formation, an indicator of free radical oxidation of polyunsaturated fatty acid. Samples (20 ± 2 mg tissue) were homogenized in 350μ1 of ice-cold PBS using rapid agitation with zirconium beads (2.3 mm) and insoluble materials were precipitated with centrifugation (10,000g, 5 min, 4°C). We then measured protein carbonylation, an indicator of oxidative modification of proteins by reactive species, in hippocampi and cerebral cortices homogenates using a fluorometric assay kit (Cayman Chemical, cat #701530) per manufacturer’s instructions. Protein concentration was measured by NanoDrop One (Thermo Scientific) on the final pellet as to account for protein loss during sample preparation.
We then examined antioxidative protein and enzyme levels in hippocampus and brain cortex of SCD mice. Heme oxygenase-1 levels were measured using an ELISA kit (BioVision, #E4524-100) per manufacturer’s instructions. During sample optimization phase, standard curve for the hippocampus assay was adjusted two additional standard points were added to the lower end of the curve. We also measured catalase activity using a fluorometric assay (BioAssay Systems, Hayward, CA).
Apoptosis and Caspase 3 activity.
For terminal nucleotidyl transferase-mediated nick end labeling (TUNEL) assay, the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit from Chemicon (Billerica, MA) was used according to manufacturer’s instructions. The sections were counterstained with Methyl Green and mounted using Permount.
Caspase-3 activity in young (8-12 weeks) and older (20-25 weeks) mice was examined by measuring caspase-3-induced hydrolysis of the substrate acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC) into 7-amino-4-methylcoumarin (AMC) fluorometrically (Sigma-Aldrich cat# CASP3F). In order to document non-specific Ac-DEVD-AMC metabolism, measurements included duplicates with the caspase-3 antagonist Ac-DEVD-CHO; only the fluorometric difference between samples with and without Ac-DEVD-CHO were considered specific caspase-3 activity. Fluorescence measurements were acquired every 15 min in a plate reader (PerkinElmer EnSpire 2300) for a total of 90 min; for standardization, caspase-3 activity was calculated after 60 min incubation
Anatomic pathology, immunohistochemistry, and immunofluorescence
Mice were anesthetized and perfused (trans-cardiac) with ice-cold PBS followed by 4% of paraformaldehyde. Paraformaldehyde-fixed tissue was embedded in paraffin for H&E, cluster of differentiation 68 (CD68, an activated microglia marker [49]), and glial fibrillary acidic protein (GFAP, an astroglia marker) staining or processed for free-floating sections for heat shock protein 27 (Hsp27) or CD68 analyses. Five-micron thick paraffin sections stained with hematoxylin and eosin were assessed for morphometric analysis, using two to three sections from each animal.
GPAP and CD68 immunohistochemistry.
Paraffin sections of mouse brain were deparaffinized, rehydrated, blocked in 1% H2O2 for 10 min and in 15% normal rabbit (for CD68) or goat (for GFAP) serums in PBS for 30 min; then incubated in a primary antibody overnight, on a shaker at 4°C (Rabbit anti-mouse GFAP antibody, Sigma-Aldrich, St. Louis, MO, 1:1000 dilution or rat anti-mouse CD68 antibody, Bio-Rad Laboratories, Hercules, CA, 1:2000 dilution). ImmPRESS Immunohistochemistry Kit (Vector Laboratories, Burlingame, CA) was used according to the manufacture’s protocol. After antigenic sites were visualized with DAB chromogen, sections were counterstained with hematoxylin.
Heat shock protein 27 (Hsp27) immunohistochemistry.
After 24h incubation at 4°C in 4% paraformaldehyde, tissue was moved to 10% glycerin for 24 hours and then 20% glycerin for 24 hours at 4°C. Cryoprotected tissue was sectioned at 30 microns on a freezing sliding microtome. Immunohistochemical staining of free-floating sections was performed with goat anti-mouse Hsp27 antibody at 1:500 dilution (Hsp 27 Antibody (M-20) from Santa Cruz Biotechnology, Dallas, TX) and ImmPRESS Immunohistochemistry Kit (Vector Laboratories, Burlingame, CA) according to the manufacture’s protocol. After antigenic sites were visualized with DAB chromogen, sections were counterstained with hematoxylin. Hippocampus on both sides of the coronal sections was semi-quantitatively evaluated for Hsp27 expression, using a 0–3 grading of the stained intensity (grading scale used was: 0 for no staining, 1 for low staining, 2 for moderate staining, and 3 for intense staining). An investigator blind to mouse genotype collected images for analysis. For all immunohistochemistry stainings, negative control sections were similarly processed, except for omission of the respective primary antibody.
CD68 immunofluorescence staining, a general marker of activated phagocytic microglia/macrophages, the main inflammatory cells in the central nervous system, was performed after exposure to normoxia and hypoxia/reoxygenation exposures. Seventy-two hours after the seventh day of normoxia or hypoxia exposure, mice were euthanized and their brains fixed in 4% paraformaldehyde for 24h at 4°C, followed by incubation with 30% sucrose supplemented with 0.1% sodium azide (all from Sigma-Aldrich, St. Louis, MO) overnight at 4°C and then frozen in OTC. Fixed brains were sectioned in coronal plane at 30-μm thickness and mounted on gelatin-covered slides. Samples were blocked with 15% of normal rabbit serum (Invitrogen division of Thermo Fisher Scientific, Waltham, MA) and incubated in 1:2000 solution of rat anti-mouse CD68 antibody (Bio-Rad Laboratories, Hercules, CA). Negative control slides prepared similarly had primary antibody omitted. After labeling the slides with Texas Red-labeled secondary antibody, slides were cover slipped using a DAPI-containing mounting solution (both from Vector Laboratories, Burlingame, CA). The staining was visualized using Olympus FV1000 confocal microscope with 20X magnification. We sampled three areas of the cortex adjacent to the midline on each hemisphere. A 2D projection from five slices with the depth of 1 μm was obtained for further analysis. The resulting images were analyzed using Image J, where we calculated areas of Texas Red positive pixels that were located near DAPI-stained nuclei, the areas of nuclei that had adjacent green staining, as well as a number of all nuclei in the section. During analysis, we considered a total number of Texas Red+ cells in the section, the area of Texas Red+ pixels per nucleus area as an indicator of the expression levels per cell, and the total area of Texas Red+ pixels in the section.
Bromodeoxyuridine / 5-bromo-2′-deoxyuridine (BrDU)
BrDU, a thymidine analog used to identify proliferating cells, was administered via intraperitoneal (ip) injection at a dose of 50 mg/kg 2 hours before euthanasia. Mice were euthanized with isoflurane inhalation, cardiac-perfused with 10 unit/ml heparin in cold PBS, followed by 4% paraformaldehyde. Brains were dissected out and further fixed in 4% PFA for 24 hours at 4°C. After that, tissue was cryo-protected first with 10% glycerol (24 hours, 4°C) and 20% glycerol (24 hours, 4°C, kept in this condition afterward). Free-floating sections were cut at 30 μM on a freeze-sliding microtome and stored in PBS with 0.01% of sodium azide. Nuclear DNA was denatured into single-stranded form by treating floating sections with 2M HC1 for 2 h at room temperature on a rocker. After washing floating sections in PBS, sections were blocked in 15% of normal rabbit serum (NRS) for 30 min and incubated overnight in 1:400 dilution of rat anti-BrDU (dissolved in 10% NRS and 0.1 % of Triton-X in PBS (PBS-T) antibody on a rocker at 4°C. Sections then were incubated for 45 min in a biotinylated rabbit anti-rat antibody (1:1000) HRP (1:1000). Between each incubations, sections were washed twice in PBS-T. Staining was visualized with a 3,3’-diaminobenzidine (DAB) chromogen. Five controls and five homozygous Townes mice were used in this study. We cut five sections per brain at different bregma distances and hippocampus areas were imaged on each hemisphere. Images were acquired at 20X magnification and BrDU positive cells were counted in each area of hippocampus, as well as in subventricular zone underlying hippocampus.
Statistical Analysis
For the analysis of the reactive dihydroethidium studies, we compared the ratings between genotypes and examined the concordance among ratings. To compare the ratings (from one to four) between the two genotypes (control vs. homozygotes) from the five raters, a repeated-measures ordinal logistic regression (via the SAS procedure PROC GENMOD) was used. To assess concordance of ratings between the five raters, a multi-rater with ordered categorical scale kappa measure was calculated (via the MAGREE SAS macro). This kappa measure (denoted by κma) is based on a weighted cumulative probit generalized linear mixed model [50]. Both of these analyses were performed with SAS Version 9.4 software (SAS Institute, Inc., Cary, North Carolina).
Protein carbonyl and heme oxygenase-1 were modeled separately as a response (dependent) variable. Genotype (control, homozygote), region (hippocampus, cerebral cortex), and genotype-by-region interactions were used as explanatory (independent) variables. Because both hippocampus and cortex regions were from the same mouse, a repeated-measures mixed model was used to analyze those variables. For each model, the “best” variance-covariance matrix structure was determined based on the lowest Bayesian Information Criterion value. Model assumptions were checked based on model fit statistics, e.g. studentized residuals, normality of the errors.
All other data were analyzed using one or two way analysis of variance (ANOVA) with SigmaPlot 14.0 (Chicago, IL). For all ANOVAs, residuals were examined for normality and homogeneity of variances. The Holm-Sidak multiple comparison test was used for post hoc pairwise comparisons when appropriate. P-values < 0.05 were considered to reflect evidence of an effect or a difference.
Results
Sickle cell mice have abnormal ultrastructural morphology and vascular endothelial cells and blood brain barrier abnormalities
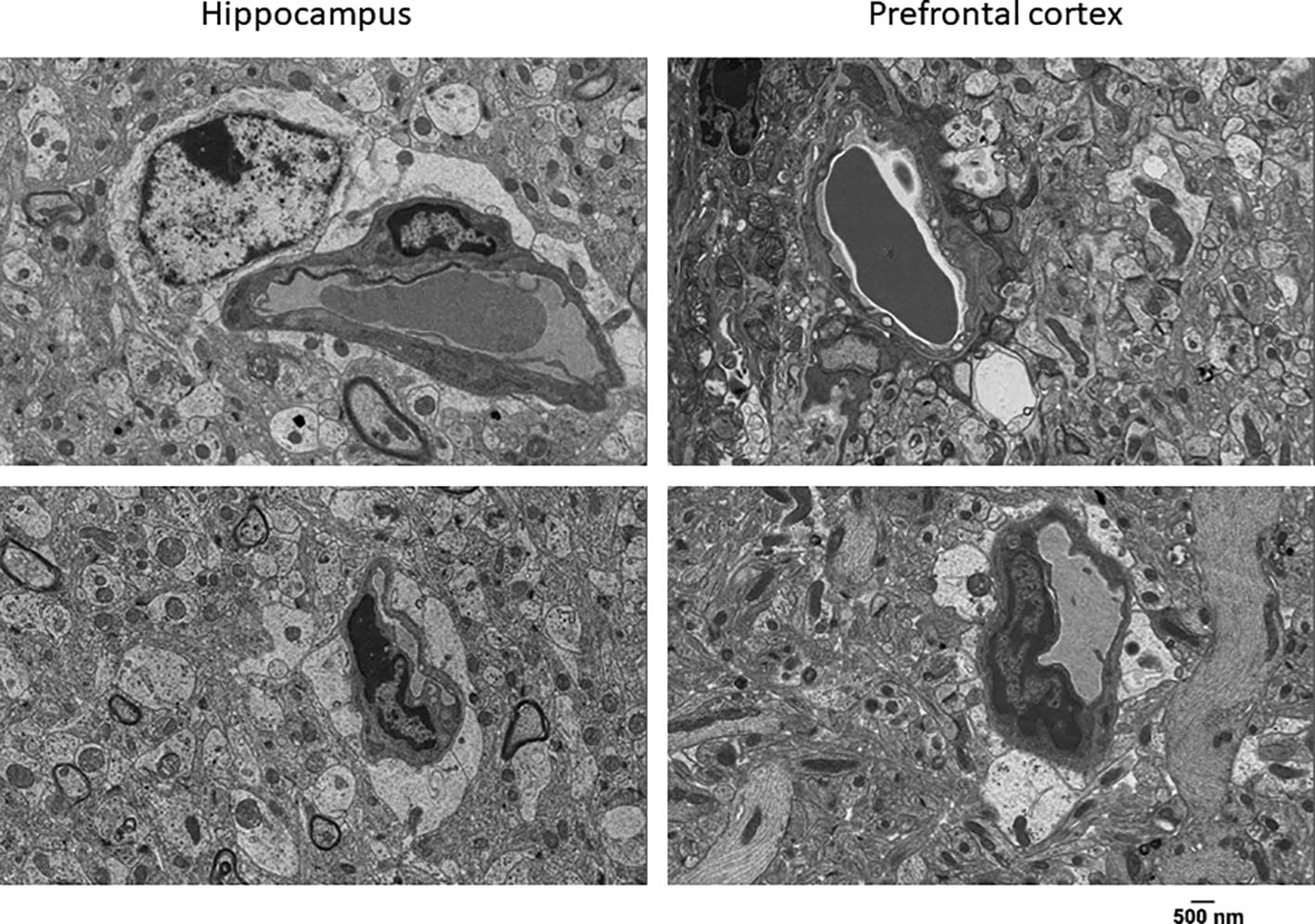
We recently showed that Townes SCD mice while having no strokes, have behavior and learning and memory deficits as well as gait, balance and gait adaptability abnormalities, which are associated with neuropathologic changes in hippocampus and cerebellum [45, 46]. Here we examined biological correlates of those reported deficits. We first performed an ultrastructural analysis of hippocampus and prefrontal cerebral cortex in Townes mice using transmission electron microscopy. We found that homozygotes had cytoplasmic vacuolization in vascular endothelial cells and extensive pseudopodia formation projecting into the vascular lumen (Figure 1), which were not detected in control mice (Supplemental Figure 1). Additionally, homozygous Townes, compared to controls, had significantly distended astrocyte foot processes as well as attenuation/loss of tight/adherens junctions (Figure 1). Such findings in homozygotes are strongly suggestive of damage to blood brain barrier integrity and are similar to those described following hypoxic/ischemic injury [51, 52].
Figure 1. SCD mice have vascular endothelial cells and blood brain barrier abnormalities.
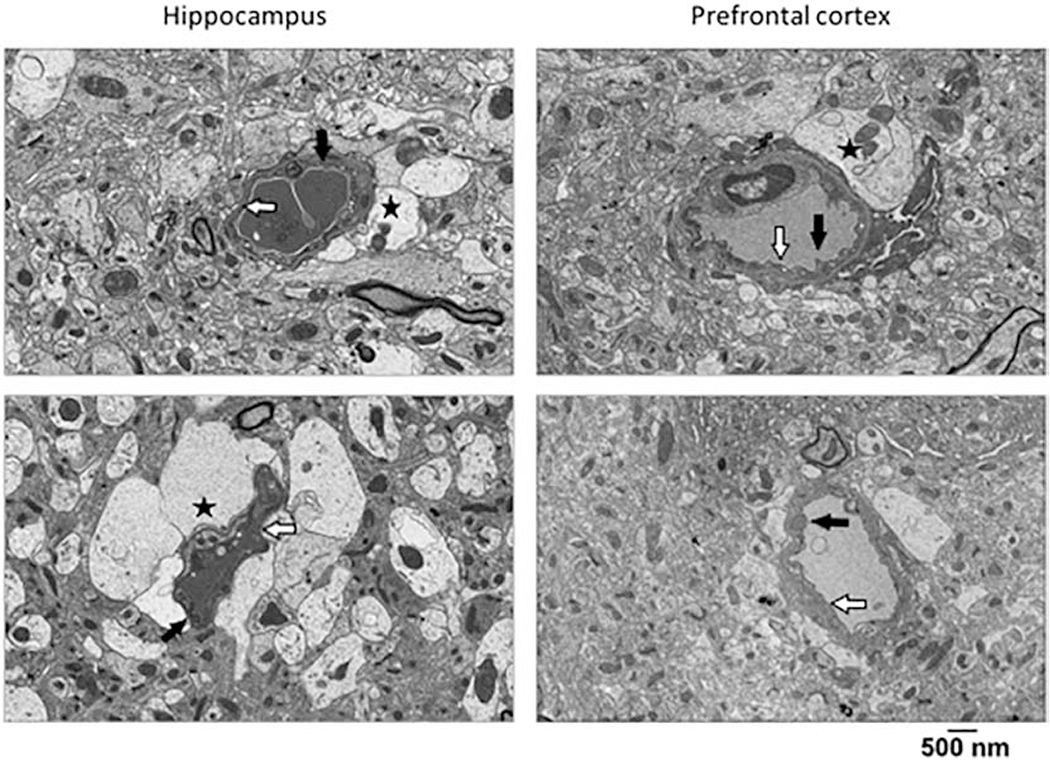
Representative ultrastructural images of hippocampus (left panels) and prefrontal cortex (right panels) from homozygous Townes mice demonstrating cytoplasmic vacuolization in vascular endothelial cells and extensive pseudopodia formation into the vascular lumen black arrow). Additionally, homozygous Townes had significantly distended astrocytes foot processes (black star) as well as attenuation/loss of tight junctions/adherens junctions (white arrow). These alterations in integrity of blood brain barrier are suggestive of hypoxic/ischemic injury. Two control and two homozygous mice were examined.
Townes SCD mice have upregulated superoxide content and oxidative modification of lipids and proteins in the brain
Given these findings suggestive of hypoxic/ischemic injury in the blood brain barrier, we then investigated the state of oxidative stress in hippocampus and cerebral cortex in Townes mice. We first examined superoxide levels by quantifying oxidized dihydroethidium in hippocampus slices from Townes mice. In this method, dihydroethidium is oxidized by superoxide to a red fluorescent material thus serving as a redox-responsive probe and a surrogate measure of reactive oxygen species production (Figure 2). The images of hippocampi immunofluorescence were rated for intensity/quantity of fluorescence (1 to 4) by five independent raters who were unaware of samples’ genotype. There was high rating concordance among the five raters (κma=0.72, SE=0.081, 95% CI: 0.57, 0.88). We found that oxidized dihydroethidium staining (intense bright red color) in hippocampi from homozygous Townes, was greater than in those from Townes controls (p=0.043, Figure 2). The intense bright red color staining was morphologically granular and reminiscent of the pyknotic neuronal morphology on H&E stained sections.
Figure 2. Townes sickle cell mice have increased superoxide levels in hippocampus.
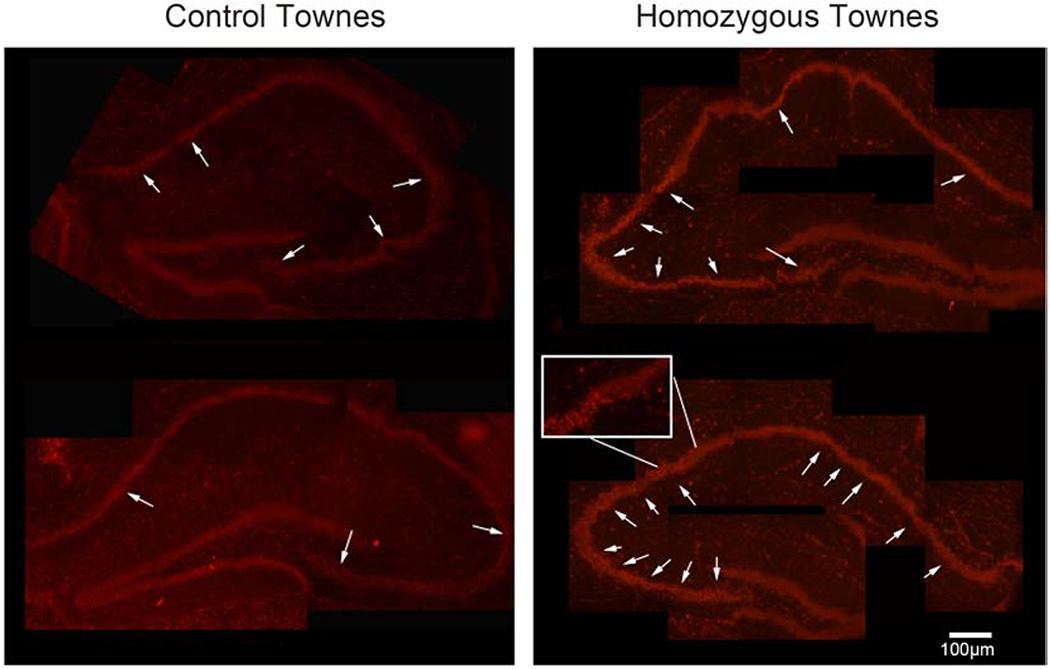
Representative image of fluorescence indicating the presence of oxidized dihydroethidium (arrows) in hippocampi slices, a surrogate measure of reactive oxygen species production. Dihydroethidium is oxidized by superoxide to a red fluorescent material thus serving as a redox-responsive probe. Five independent raters who were unaware of samples’ genotype rated samples of hippocampi immunofluorescence (between one and four). There was high concordance in rating among the five raters (κma=0.72, SE=0.081, 95% CI: 0.57, 0.88). We found that in hippocampi from homozygous Townes, oxidized dihydroethidium staining (red color) was more intense compared to those from Townes controls (p=0.043). The intense staining was morphologically granular and reminiscent of the pyknotic neuronal morphology on H&E stained sections. Five mice were examined, three controls and two homozygotes.
We then examined lipid and protein oxidative modification in hippocampi and cerebral cortices of SCD mice (Figure 3). In hippocampus, homozygous Townes had higher malondialdehyde formation (a measurement indicative of lipid peroxidation) compared to controls (p<0.001) and heterozygous mice (p=0.024), Figure 3A. Moreover, malondialdehyde formation was higher in heterozygous Townes compared to controls (p=0.017, Figure 3A). In cerebral cortex, homozygous Townes had higher malondialdehyde formation compared to controls (p=0.004) and heterozygotes (p=0.004), Figure 3B.
Figure 3. Townes sickle cell mice have increased oxidative modifications in lipid and protein in hippocampus and cerebral cortex.
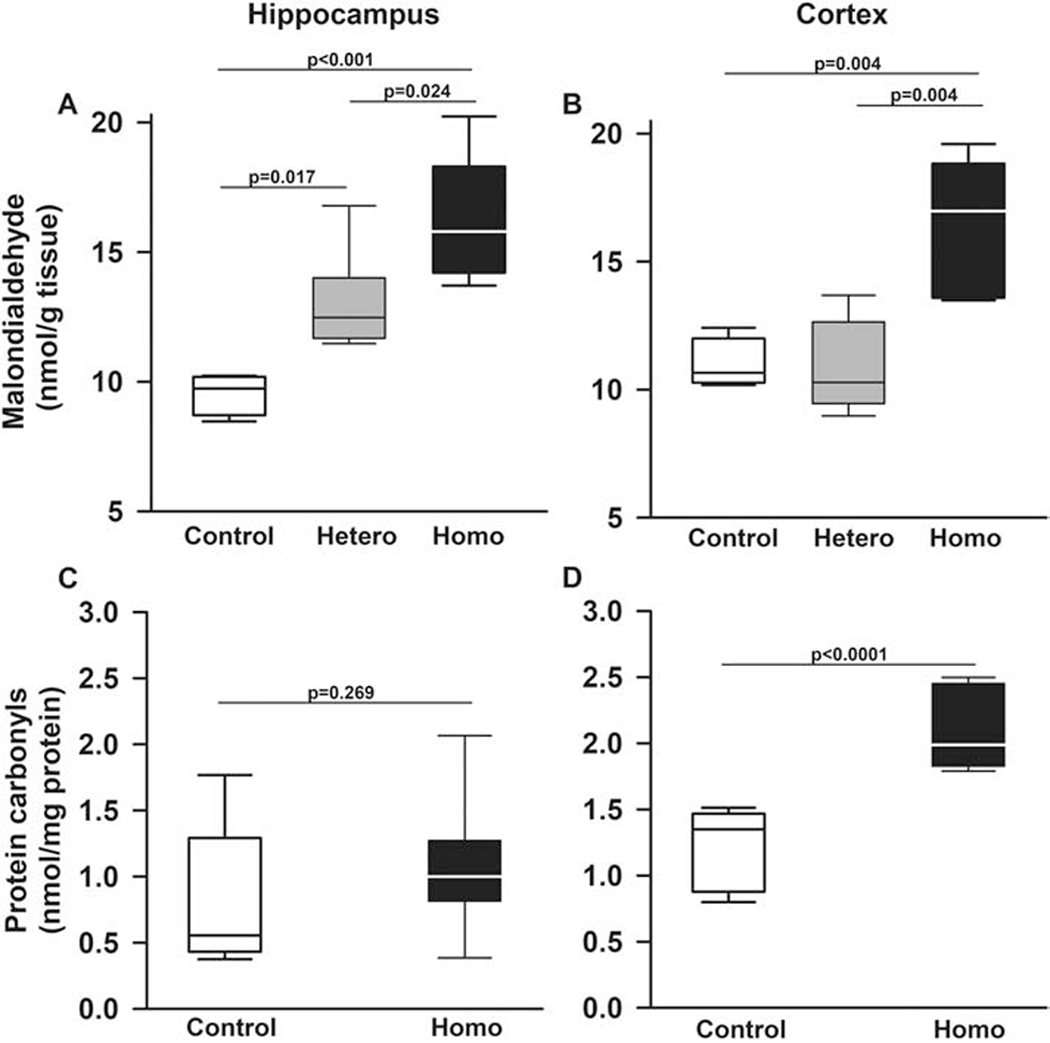
Box plots of respective variables indicate the variable’s median and first and third quartiles and the whiskers the 5th and 95th percentiles. A. In hippocampi of homozygous Townes mice, malondialdehyde formation, a surrogate measurement of lipid peroxidation and oxidative stress, there was higher malondialdehyde formation compared to controls (p<0.001) and heterozygous (p=0.024). Interestingly heterozygous Townes also had higher levels of malondialdehyde formation compared to controls (p=0.017). B. In cerebral cortex, homozygous Townes had higher malondialdehyde formation compared to controls (p=0.004) and heterozygotes (p=0.004). C and D. Protein carbonyl content (a measurement indicative of post translational oxidative modification producing protein-carbonyl adducts) varied by genotype and brain region (p=0.034 for genotype by region interaction, Figures 3C and 3D). Specifically, in cerebral cortex, but not in hippocampus (p=0.269, C), homozygotes had significantly higher protein carbonyls content compared to control mice (p<0.0001, D). N=4-8 mice per genotype.
Regarding protein carbonylation, we found that overall, protein carbonyl content (a measurement indicative of post translational oxidative modification producing protein-carbonyl adducts) varied by genotype and brain region (p=0.034 for genotype by region interaction, Figures 3C and 3D). Specifically, in cerebral cortex, but not in hippocampus (p=0.269, Figure 3C), homozygotes had significantly higher protein carbonyls content compared to control mice (p<0.0001, Figure 3D).
Sickle cell mice expression of antioxidant markers in the brain vary by region and marker
We next measured components of the antioxidative response (heme oxygenase-1 protein and catalase activity) levels in hippocampus and cerebellum of Townes mice. Heme oxygenase-1 levels varied according to genotype and brain region (p=0.031 for genotype by region interaction, Figures 4A and 4B). Specifically, in hippocampus, homozygotes had lower (p=0.015), whereas in cortex, a trend towards higher levels (p=0.061) of heme oxygenase-1 compared to control mice (Figures 4A and 4B). Overall, regardless of genotype, heme oxygenase-1 levels were higher in cortex than in hippocampus (p<0.0001, Figures 4A and 4B). Regarding catalase, in hippocampus and cerebral cortex of Townes mice, mean catalase levels were similar comparing homozygotes, heterozygotes, and controls, Figures 4C and 4D.
Figure 4. Sickle cell mice expression of antioxidant markers in the brain vary by region and marker.
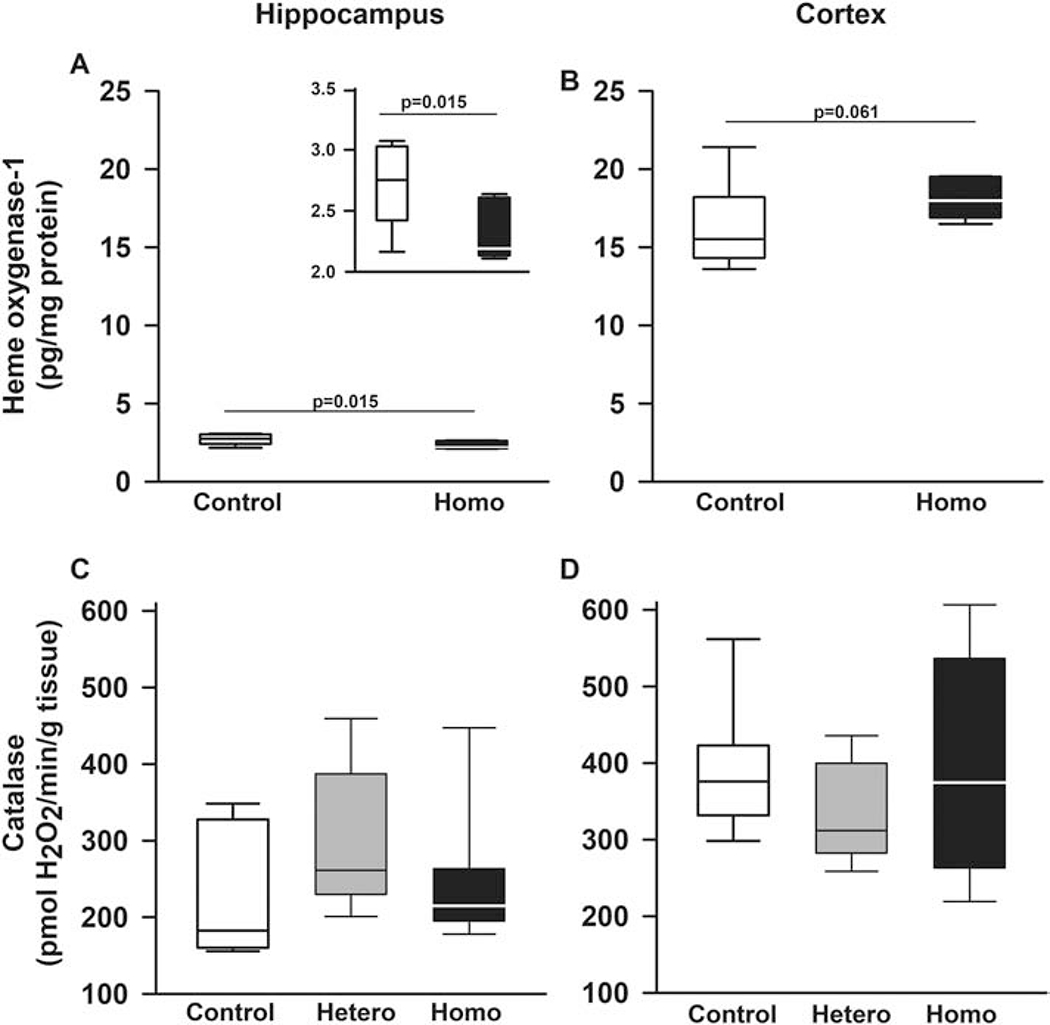
Box plots of respective variables indicate the variable’s median and first and third quartiles and the whiskers the 5th and 95th percentiles. A and B. Heme oxygenase-1 levels varied according to genotype and brain region (p=0.031 for genotype by region interaction). Specifically, in hippocampus, homozygotes had lower (p=0.015, insert in A), whereas in cortex, a trend towards higher levels (p=0.061) of heme oxygenase-1 protein compared to control mice. Additionally, regardless of genotype, heme oxygenase-1 levels were higher in cortex than in hippocampus (p<0.0001, A and B). C. In hippocampi and D. cerebral cortex of Townes mice, mean catalase levels were similar comparing homozygotes, heterozygotes, and controls. N=4-8 mice per genotype.
Given the findings of changes suggestive of cerebral ischemia and increased oxidative stress in hippocampus and cortex, we then tested the hypothesis that the expression of Hsp27 would be elevated. Hsp27 is a heat shock protein, which is induced in the brain after cerebral ischemia and other injury states such as oxidative stress [53, 54]. We found that during basal conditions, Hsp27 expression in the cornu amonis (CA) 1, CA2, and CA3 subfields of hippocampus of homozygous Townes was markedly higher than that observed in control animals (representative images of Hsp27 immunohistochemical assay are shown in Figure 5).
Figure 5. Sickle cell mice have increased expression of HSP 27 in hippocampi.
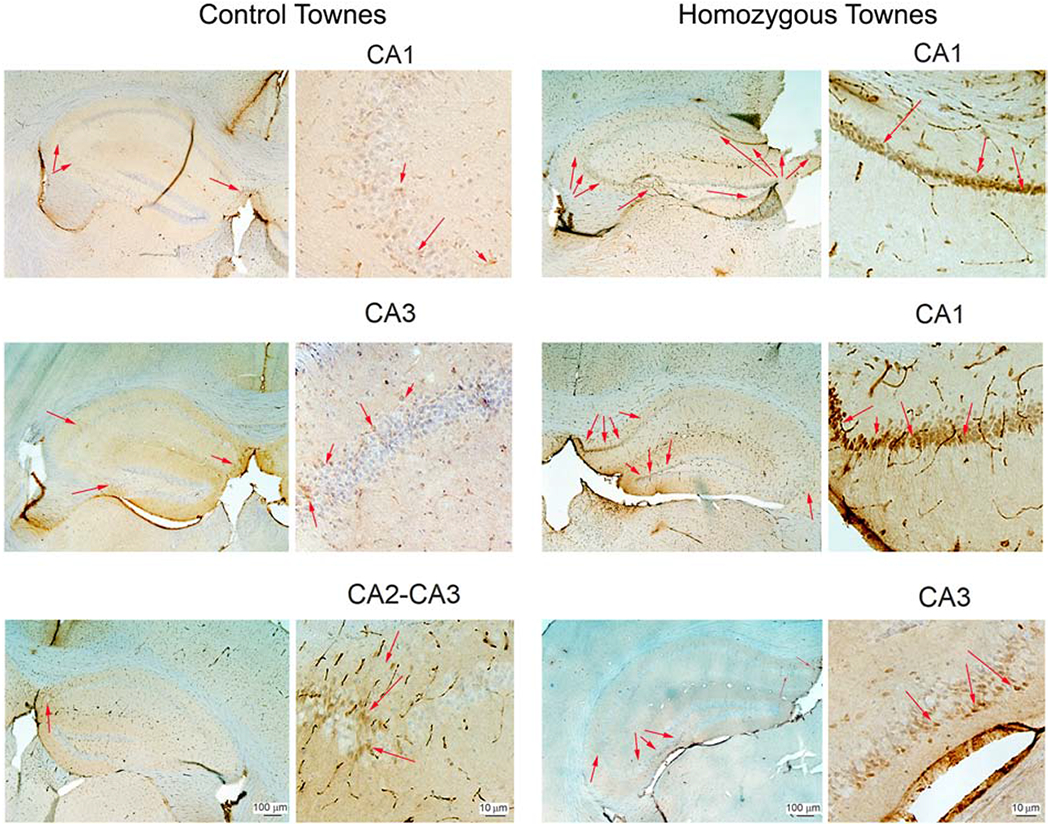
Representative images of Hsp27 immunohistochemical assay in Townes control (left two columns) and homozygotes (right two columns). During basal conditions, Hsp27 expression (arrows) in the cornu amonis (CA) 1, CA2, and CA3 subfields of hippocampus of homozygous Townes was markedly higher than that observed in control animals. N=5 for each genotype.
In SCD mice, oxidative stress is associated with changes in caspase-3 activity but not increased apoptosis in hippocampus and cerebral cortex
Given the evidence of oxidative stress in hippocampus and cerebral cortex in Townes mice, we then examined caspase-3 activity and degree of apoptosis in those brain regions in young (8-12 weeks) and older (20-25 weeks) mice. In hippocampus, caspase-3 activity in Townes mice varied according to age and genotype (p<0.001 for genotype by age interaction, Figure 6A and 6B). In hippocampus of young mice, caspase-3 activity in homozygous (p<0.001) and heterozygous (p<0.001) Townes was lower compared to control mice (Figure 6A). Conversely, among old mice, homozygous (p=0.046) and heterozygous (p=0.041) Townes had higher caspase-3 activity levels compared to controls (Figure 6B). Additionally, as animals aged, in controls, there were decreases (p=0.003), whereas in heterozygotes (p<0.001) and homozygotes (p=0.002) there were increases in caspase-3 activity levels in hippocampus and these patterns of caspase-3 activity changes over time, were different (p<0.001 for genotype by age interaction, Figure 6A and 6B).
Figure 6. In Townes SCD mice, increased oxidative stress is associated with changes in caspase activity.
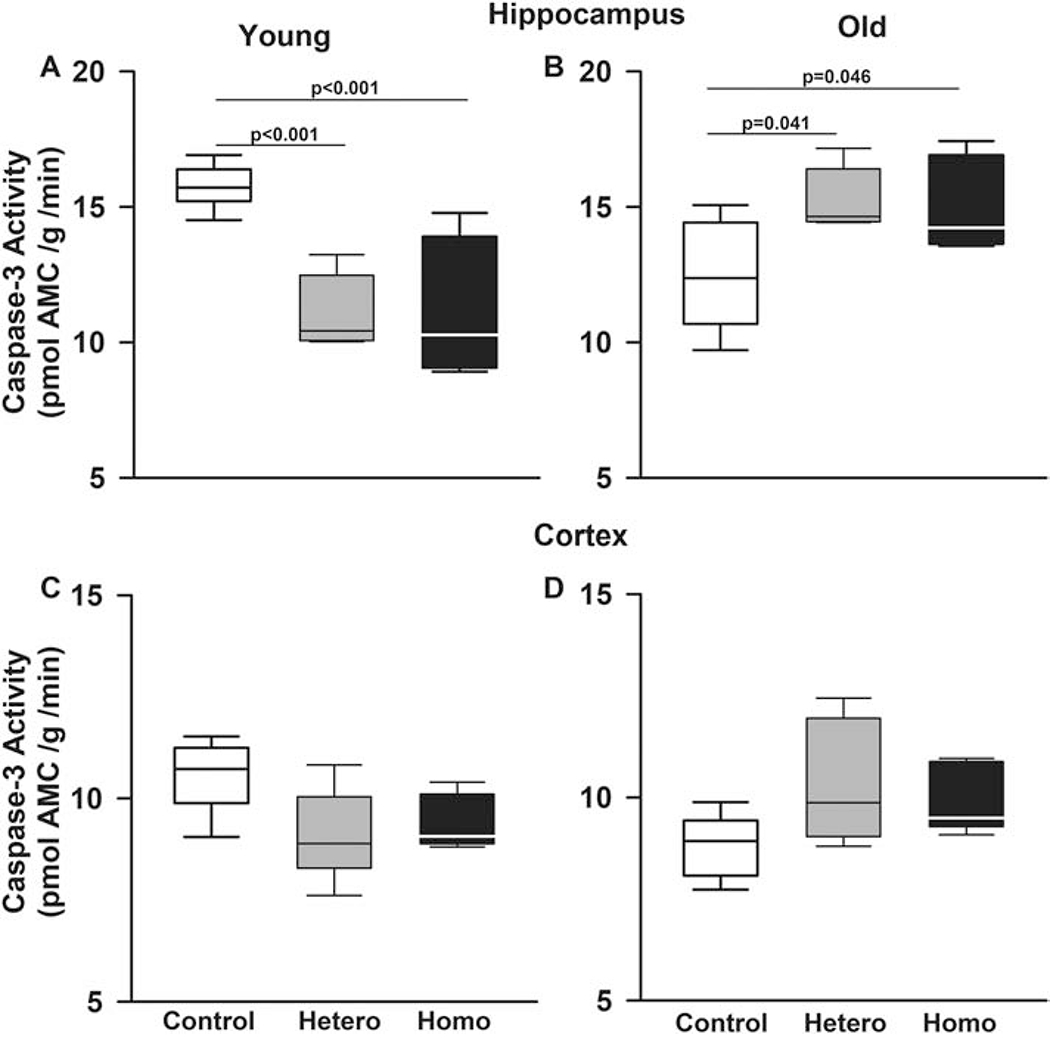
Box plots of respective variables indicate the variable’s median and first and third quartiles and the whiskers the 5th and 95th percentiles. Given the evidence of increased oxidative stress in hippocampi and cerebral cortex in Townes mice, we tested the hypothesis that there would be alteration in caspase-3 activity. In hippocampi, caspase-3 activity in Townes mice varied according to mouse age and genotype (p<0.001 for genotype by age interaction). A. In hippocampi, among young mice, homozygous (p<0.001) and heterozygous (p<0.001) Townes had significantly lower caspase-3 activity compared to controls. B. Conversely, among old mice, homozygous (p=0.046) and heterozygous (p=0.041) Townes had significantly higher caspase-3 activity compared to controls. As animals aged (A and B), in controls, there were decreases (p=0.003), whereas in heterozygotes (p<0.001) and homozygotes (p=0.002) there were increases in caspase-3 activity levels and these patterns of changes were significantly different (p<0.001 for genotype by age interaction). In cerebral cortex (C and D), we also observed that there were genotype by age interaction (p=0.005). Specifically, as animals aged, in controls, there were decreases (p=0.008), whereas in heterozygotes (p=0.049) increases, and in homozygotes (p=0.409) no changes in caspase-3 activity levels in cerebral cortex and these patterns of change comparing young and old animals were significantly different (p=0.005). N=4-6 mice per genotype and age group
In cerebral cortex, there was also a genotype by age interaction (p=0.005, Figure 6C and 6D). Specifically, as animals aged, caspase-3 activity levels in cerebral cortex decreased in controls (p=0.008), increased in heterozygotes (p=0.049), and remained unchanged in homozygotes (p=0.409) and these patterns of caspase-3 activity changes over time were different (p=0.005, Figures 6C and 6D).
Interestingly, these alterations in caspase-3 activity were not associated with changes in apoptotic cell death as control and homozygous Townes mice had similar number of TUNEL-positive cells in hippocampus (Supplemental Figure 2).
Sickle cell disease mice have enhanced microglia activation after hypoxia/reoxygenation exposure
We also examined microglia in Townes mice while exposed to normoxia (room air) and after repeated intermittent hypoxia (8% fraction of inspired oxygen)/reoxygenation exposures. We found that during basal conditions (exposure to normoxia/room air) homozygous Townes had similar percentage of CD68 (a marker of activated microglia and of cells of monocyte lineage) positive cells (p=0.801) and similar CD68+ staining/nucleus area ratio (p=0.693) compared to controls, Figure 7A and 7B. The effect of repeated hypoxia/reoxygenation exposures on the percentage of CD68-positive cells varied according to genotype as there was a genotype by exposure interaction (p=0.026, Figure 7C–7E). Specifically, hypoxia-exposed homozygotes, had a higher percentage of CD68 positive cells compared to normoxia-exposed animals (p=0.002, Figure 7E). In contrast, hypoxia- and normoxia-exposed controls had similar percentage of CD68-positive cells (p=0.823, Figure 7E).
Figure 7. Hypoxia exposure is associated with microglia activation is SCD mice.
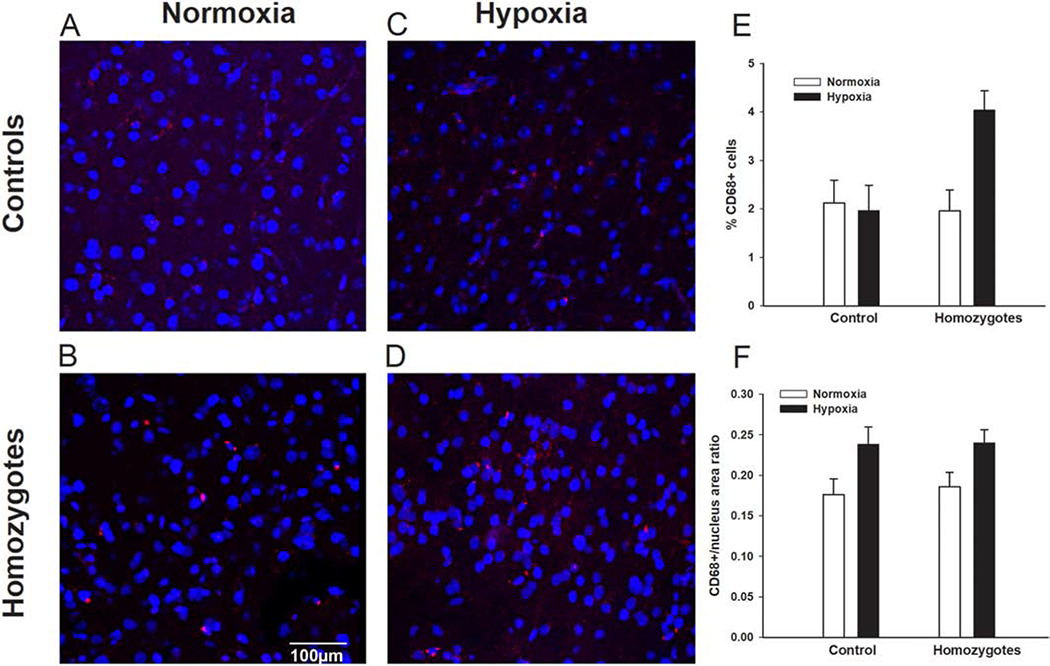
Representative images of CD68 immunofluorescence staining of cortex samples form controls and homozygous Townes after exposures to normoxia (room air) and chronic intermittent hypoxia (8% fraction of inspired oxygen)/reoxygenation for seven days. We found that during basal conditions (exposure to normoxia/room air, A and B) homozygous Townes had similar percentage of CD 68 positive cells (intense red dots, p=0.801) and similar CD68+ staining/nucleus area ratio (p=0.693) compared to controls, C, D, E, F. The effect of chronic intermittent hypoxia/reoxygenation exposures on the percentage of CD68-positive cells varied according to genotype as there was a genotype by exposure interaction (p=0.026, E). Specifically, hypoxia-exposed homozygotes, had a higher percentage of CD68 positive cells compared to normoxia-exposed animals (p=0.002). In contrast, hypoxia- and normoxia-exposed controls had similar percentage of CD68-positive cells (p=0.823). F. Overall, there was a significant effect of repeated hypoxia/reoxygenation exposures in that both controls and homozygotes had greater CD68+ staining/nucleus area ratio compared to normoxia-exposed animals (p=0.006, for effect of exposure). N=4-7 per genotype (control, homozygote) and exposure (normoxia, hypoxia).
With repeated hypoxia/reoxygenation exposures, both controls and homozygotes had greater CD68+ staining/nucleus area ratio compared to normoxia-exposed animals (p=0.006, for effect of exposure, Figure 7A–7F).
A second strain of severe SCD mice, the BERK strain, also has neuropathologic changes and oxidative stress in hippocampus and cerebral cortex
We also examined the brains of BERK mice, another model of severe SCD, and found that akin to Townes, BERKs, did not have strokes but had significant neuropathologic changes displaying large bands of dark/pyknotic neurons in hippocampus, a finding highly suggestive of hypoxic injury (Supplemental Figure 3).
We then examined the presence of lipid peroxidation and catalase activity in BERK mice. Homozygotes and hemizygotes BERKs and controls had similar malondialdehyde formation in hippocampus (p=0.078 for effect of genotype, Figure 8A). In contrast, in cerebral cortex, (Figure 8B), homozygous BERKs had higher malondialdehyde formation compared to controls (p<0.001) and hemizygotes (p=0.015). Interestingly, akin to heterozygous Townes, hemizygous BERKs also had elevated levels of malondialdehyde formation compared to controls (p<0.001, Figure 8B).
Figure 8. The BERK strain of SCD mice also has increased oxidative stress in hippocampi and cerebral cortex.
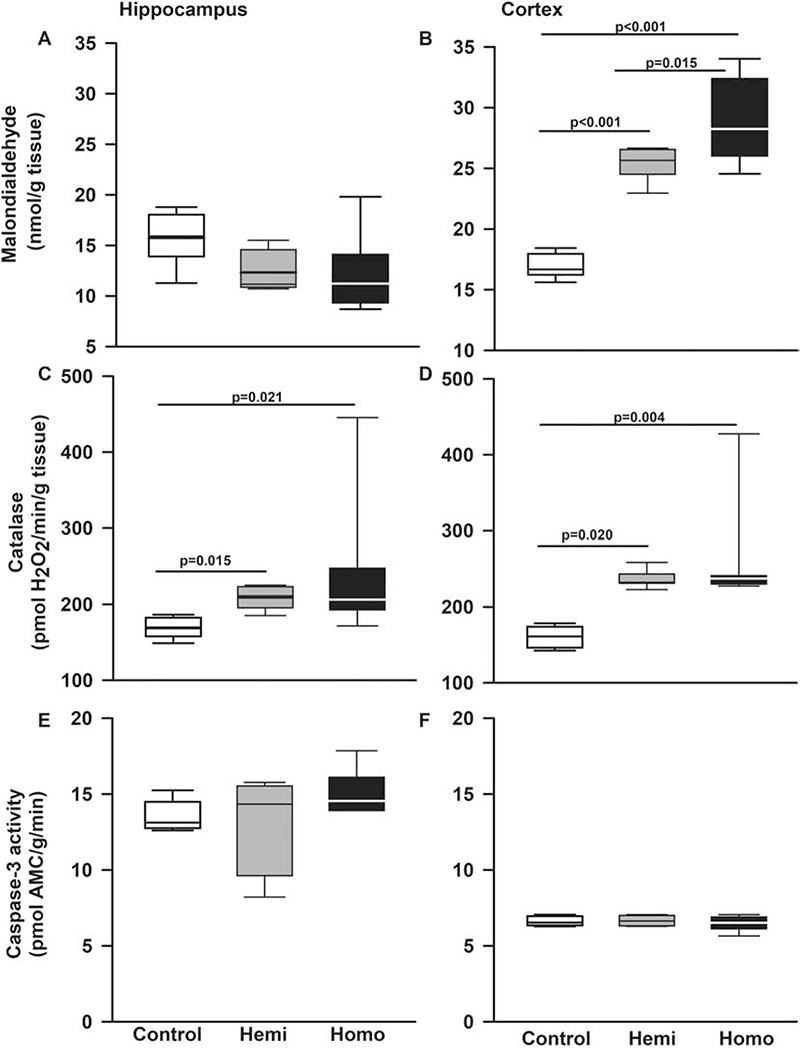
Box plots of respective variables indicate the variable’s median and first and third quartiles and the whiskers the 5th and 95th percentiles. We also examined the state of oxidative stress in a second strain of SCD mice, the BERK strain. A. In hippocampi, malondialdehyde formation, a surrogate measurement of lipid peroxidation and oxidative stress, was similar in BERKs homozygotes, hemizygotes, and controls (p=0.078 for effect of genotype). B. In cerebral cortex, homozygous BERKs had significantly higher malondialdehyde formation compared to controls (p<0.001) and hemizygotes (p=0.015). Interestingly, hemizygous Berks also had elevated levels of malondialdehyde formation compared to controls. C. In hippocampi, homozygous (p=0.021) and hemizygous (p=0.015) BERKs had significantly higher catalase levels compared to control mice. D. In cerebral cortex, compared to control mice, homozygous (p=0.004) and hemizygous (p=0.020) BERKs had significantly higher catalase levels, Figure 6D. Contrary to Townes mice, in BERKs we found no alterations in caspase-3 activity levels in hippocampus (p=0.301 for genotype effect) or cerebral cortex (p=0.391 for genotype effect), (E and F). N=5-8 mice per genotype
Regarding catalase, in hippocampus, compared to control mice, homozygous (p=0.021) and hemizygous (p=0.015) BERKs had higher catalase activity levels, Figure 8C. Similar findings were observed in cerebral cortex, in that homozygous (p=0.004) and hemizygous (p=0.020) BERKs had higher catalase activity levels compared to control mice, Figure 8D. Contrary to Townes mice, BERKs exhibited no alterations in caspase-3 activity levels in hippocampus (p=0.301 for effect of genotype) or cerebral cortex (p=0.391 for effect of genotype), Figure 8E and 8F.
Sickle cell disease mice lack evidence of increased astrogliosis, microgliosis, impaired neurogenesis, and neuronal cell death in hippocampus
Immunohistochemistry analysis indicated that homozygous Townes and controls had similar expression of GFAP (Supplemental Figure 4), CD68, BrDU, and Fluoro C Jade (data not shown).
Discussion
In two clinically relevant mouse models of severe SCD, Townes and BERKs, we found vascular endothelial abnormalities, white matter changes, and disruption of the blood brain barrier, in a pattern suggestive of ischemic/oxidative stress injury in hippocampus and cortex. Further, these changes in blood brain barrier integrity were associated with oxidative stress as shown by increased superoxide production and oxidative changes in lipids (increased lipid peroxidation) and proteins (increased protein carbonylation) in hippocampus and cerebral cortex. These oxidative changes were associated with variable protein (increase in heme oxygenase-1) and enzyme (catalase) antioxidant response. Additionally, SCD mice displayed increased cerebral susceptibility to hypoxia/reoxygenation as evidenced by increased activation of microglia in cerebral cortex after exposure to chronic intermittent hypoxia. It is noteworthy that at the ages studied here (20-25) weeks, these animals do not have evidence of strokes on optic microscopy or ultrastructural studies. Therefore, despite the absence of cerebral strokes, our findings suggest that in SCD mice, there is evidence of ischemic injury and oxidative stress in susceptible areas of the brain. One could postulate that these biochemical and ultrastructural abnormalities possibly underlie the neurocognitive deficits observed in this model [45, 46].
Our findings of increased oxidation of dihydroethidium in pyramidal neurons in hippocampus slices coupled with increased lipid peroxidation and protein carbonylation suggest that reactive oxygen species production in homozygous SCD mice is upregulated and that lipids and proteins undergo oxidative changes in hippocampus and cortex. Noticeably, this increase in reactive oxygen species and protein and lipid oxidative changes was not uniformly associated with increases in components of the protein (heme oxygenase-1) and enzymatic (catalase) antioxidant system. In fact, heme oxygenase-1 was down regulated in hippocampus but trended higher in cortex of Townes mice compared to controls. We also observed increases in catalase in brains from BERKs but not in Townes mice. While measuring all components of the antioxidant system in the brain was beyond the scope of this work, we saw evidence that there might be an overall insufficient anti-oxidant response in Townes and BERKs. Taken together these findings suggest that free radical species production in SCD mouse brains exceeds the defensive antioxidant response, thus yielding oxidative stress in susceptible brain regions.
What might be the relevance the findings of oxidative stress in brain from SCD mice? Several investigations in humans [14–18] and animals [19–22, 27] indicate that SCD is associated with oxidative stress in blood and organ systems. Additionally, while still a matter of debate, several studies support the hypothesis that oxidative stress contributes to organ damage and morbidity in SCD. In fact, some have shown that in SCD, oxidative stress contributes to the development of acute chest syndrome, pulmonary hypertension, and vaso-occlusion in SCD patients [14, 15, 23] and pain [22] and liver necrosis in SCD mice [27, 38]. Our findings of oxidative stress and associated changes in the blood brain barrier in Townes mice are in keeping with the findings of increased oxidative stress in NY1DD mice, a SCD model that displays no anemia and a only shows a mild SCD phenotype. In that study, using magnetic resonance imaging and magnetic resonance spectroscopy, the NY1DD mouse had increased cerebral blood flow, depleted glutathione levels, and cerebral oxidative stress [55]. These findings of oxidative stress in SCD models with varying degree of severity and independent of the presence of anemia suggests that oxidative stress occurs in SCD independently of its severity.
We found that in brain of homozygous Townes mice there was increased expression of Hsp27 at baseline. Hsp27, a member of the heat shock protein family, functions as a protein chaperone, an antioxidant, and inhibits apoptosis and actin cytoskeletal remodeling. Cerebral ischemia in mice, yields increases in Hsp 27 expression [53] and its upregulation has a protective role in ischemia-induced brain damage [54, 56]. Researchers have shown that mice overexpressing Hsp27 are protected from cerebral ischemia and that this protective role of Hsp27 is due to its anti-apoptotic effect resulting from inhibition of ASK1-dependent MKK4/JNK activation, upstream of mitochondria-dependent apoptosis pathways [54]. In SCD, in vitro, Hsp27 recruitment to deoxygenated SCD erythrocytes, is much greater than to control erythrocytes [57]. In vivo, during normoxic conditions, Hsp27 binds to erythrocytes from SCD mice and controls similarly [57]. In contrast, after hypoxia exposure, there is marked increase in Hsp27 membrane binding to erythrocytes from SCD mice (SAD model) but not from control animals [57]. In other studies, researchers showed that during basal conditions, Hsp27 expression in the liver was similar in controls and in SAD mice [58], whereas after exposure to hypoxia and reoxygenation, hepatic expression of Hsp27 was significantly greater in SCD mice compared to control animals [57, 58]. Together, these data suggest that hypoxia exposure and ischemia in SCD mice trigger increases in Hsp27 expression [57, 58]. Here we found that at baseline, SCD mice have oxidative stress, which is associated with increased expression of Hsp27 in the brain. One could postulate that increased expression of Hsp27 in the Townes mice brain reflects a response to cerebral ischemia possibly due to chronic anemia, as these animals do not have strokes. Nevertheless, to our knowledge, this is the first report of increased Hsp27 expression in the brain of SCD mice during basal conditions. Future studies will determine whether Hsp27 is a biomarker of disease severity, response to chronic anemia and or recurrent sickling, and whether its upregulation might play a protective role in the setting of chronic anemia, recurrent episodes of ischemia and reperfusion, and increased oxidative stress in SCD brain.
While our understanding of the impact of increased oxidative stress on execution of apoptosis is incomplete, researchers have shown that ischemia/reperfusion and reactive oxygen species can have a direct effect on caspase activation [59–62]. Here we found that, compared to control mice, the pattern of caspase-3 activation in hippocampus and cerebral cortex in Townes mice was altered in an age dependent manner. In homozygotes, at young age, caspase-3 activity was decreased, whereas at older age, it was increased compared to control mice. Interestingly, while capase-3 activity was increased, we found no indication of increased apoptosis in hippocampus from SCD mice. One could postulate that increased caspase-3 activation could be associated with non-apoptotic, rather than apoptotic, functions in SCD brain. In fact, others have shown that activated caspases have a role in axon and dendrite pruning, structural remodeling, synaptic plasticity, as well as long-term potentiation and long-term depression [63, 64]. As we previously showed that SCD mice have special learning and memory deficits [45], it is reasonable to hypothesize that alterations in the pattern of brain caspase activation might contribute to the cognitive deficits observed in SCD mice.
A great deal of literature demonstrates that oxidative stress increases susceptibility to brain injury and plays a significant role in neurodegenerative and neuropsychiatric disorders [28, 32]. While the mechanisms underlying oxidative stress-induced brain damage are incompletely understood, reactive oxygen species are known to trigger a cascade of pathophysiologic events that result in blood brain barrier disruption, inflammation, and brain tissue damage [28, 32, 52, 65]. Further, oxidative stress is known to upregulate pattern recognition receptor signaling such as toll-like receptor 4 (TLR4) [66] and nucleotide-binding domain leucine-rich repeat containing protein 3 (NLRP3) [67]. Additionally, activation of TLR4 and NLRP3 in microglia has been shown to play a critical role in mediating neuroinflammation and brain tissue damage in both neurodegenerative and neuropsychiatric disorders [68–70]. Interestingly, others and we have recently shown that TLR4 and NLRP3 signaling are critical regulators of inflammation and abnormal platelet aggregation in SCD mice [71, 72]. Therefore, the hypothesis that TLR4 and NLRP3 signaling may mediate oxidative stress-dependent abnormal brain morphology and blood brain barrier damage in SCD will be tested in future studies and may provide the critical link between upregulated oxidative stress responses and brain abnormalities and stroke independent neurocognitive deficits in SCD.
In the central nervous system, the presence and trafficking of immune cells during normal states is minimal [73]. With injuries resulting from stroke, global ischemia, chronic intermittent hypoxia, enhanced reactive oxygen species, the microglia are activated and become a pivotal element of neuroinflammation in the brain [47, 74]. Here we found that after repeated chronic intermittent hypoxia and reperfusion exposure (a model of obstructive sleep apnea), but not at baseline (normoxia), SCD mice have enhanced microglia activation as shown by increased percentage of CD68 (a marker of activated microglia and of cells of monocyte lineage) positive cells in the cerebral cortex. While it is possible that this increase in CD68-positive cells reflect the migration of monocytes from the bloodstream into the cortex, rather than proliferation of microglia in situ, this determination was beyond the scope of this investigation. Nevertheless, the finding of enhanced microglia activation after long-term chronic intermittent hypoxia exposure supports the idea that SCD mouse brains are highly susceptible to hypoxia and oxidative stress-induced injury even in the absence of strokes. While one should be circumspect about extrapolating these date clinically, our findings suggest that in SCD, exposure to chronic intermittent hypoxia, such as observed in settings of obstructive sleep apnea, can pose significant risks of increased microglia activation in the brain.
Collectively, the results of this investigation clearly suggest that there is increased reactive oxygen species in the brain, which in turn is associated with downstream oxidative changes in lipid and protein as well as damage to blood bran barrier. However, we did not answer the question of how and where those radicals are formed. One possibility is that reactive oxygen species formed in the blood stream could diffuse into the central nervous system given observed abnormalities in blood brain barrier. Another possibility is that oxidative side reactions of cell-free hemoglobin (HbS) in the brain tissues due to disruption of blood brain barrier takes place. Yet another possibility is that sickle Hb, either free or within red blood cell or red blood cell-derived microparticles, which are known to greatly contribute to oxidative stress in SCD [18, 19], could be one source of increased production of reactive oxygen species that could be transferred to the brain or produced in situ. Nevertheless, the Townes mouse model is certainly a suitable model in which to test these hypotheses.
That different brain regions and different neuron populations have differential susceptibility to oxidative stress is well known [28]. Others have shown that CA1 and CA2 neurons are significantly more susceptible to damage than CA3 neurons in hippocampus during exposure to global cerebral ischemia and oxidative stress [75, 76]. Here, we found increased oxidative stress, disruption of blood brain barrier, and increased neuronal damage in hippocampi from SCD mice. In that context, SCD mice, despite the absence of strokes, display impaired spatial learning and anxiety- and depression-like behaviors compared to controls [45]. Therefore, it is reasonable to hypothesize that behavior deficits in domains reflective of hippocampus injury, such as learning and memory deficits, and mood disorders [77] could possibly be related to oxidative stress and inflammation in vulnerable brain regions such as the hippocampus and cerebral cortex. Collectively, our results point to the hippocampus and cortex as areas of great vulnerability to oxidative stress injury in SCD. Future studies will determine whether the role of antioxidant strategies (pharmacological or nutritional) are effective in ameliorating oxidative stress and cognitive deficits in SCD.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Sickle cell (SCD) patients without brain abnormalities can have cognitive deficits
SCD mice have evidence of neurocognitive deficits as described in SCD patients
Behavior deficits in SCD mice are associated with white matter abnormalities
Behavior deficits are associated with vascular injury and brain oxidative stress
SCD mice are more susceptible to chronic hypoxia/reoxygenation exposure
Acknowledgements
The authors wish to express their gratitude to Paulette Price for technical support throughout the study, to Taylor Baugh, MS, Lillian Hallmark, and Sherouk Hassan for editorial comments, and to Sirsendu Jana, PhD and Christos D. Georgiou, Ph.D for technical advice on the protein carbonylation assay. The authors are also eternally grateful to Nicholas Kenyon (in memoriam) for technical support during normoxia/hypoxia experiments.
Funding
The Intramural Program from the National Institutes of Health Clinical Center, NIH (grant numbers 1ZIACL090052-01, 1ZIACL090053-01, and 1ZIACL090054-01) supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
The authors have no conflict of interest to declare
References
- [1].Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, Vichinsky EP, Sickle cell disease, Nat Rev Dis Primers, 4 (2018) 18010. [DOI] [PubMed] [Google Scholar]
- [2].Piel FB, Steinberg MH, Rees DC, Sickle Cell Disease, New England Journal of Medicine, 376 (2017) 1561–1573. [DOI] [PubMed] [Google Scholar]
- [3].Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J, Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members, JAMA, 312 (2014) 1033–1048. [DOI] [PubMed] [Google Scholar]
- [4].Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, Gleeson M, Halperin M, Lanzkron S, Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease, JAMA Netw Open, 2 (2019) e1915374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stotesbury H, Kawadler JM, Hales PW, Saunders DE, Clark CA, Kirkham FJ, Vascular Instability and Neurological Morbidity in Sickle Cell Disease: An Integrative Framework, Front Neurol, 10 (2019) 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, Truran D, Kasten J, Eggleston B, Kesler K, McMahon L, Orringer EP, Harrington T, Kalinyak K, De Castro LM, Kutlar A, Rutherford CJ, Johnson C, Bessman JD, Jordan LB, Armstrong FD, Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia, JAMA, 303 (2010) 1823–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Farooq S, Testai FD, Neurologic Complications of Sickle Cell Disease, Curr Neurol Neurosci Rep, 19 (2019) 17. [DOI] [PubMed] [Google Scholar]
- [8].DeBaun MR, Kirkham FJ, Central nervous system complications and management in sickle cell disease, Blood, 127 (2016) 829–838. [DOI] [PubMed] [Google Scholar]
- [9].DeBaun MR, Schatz J, Siegel MJ, Koby M, Craft S, Resar L, Chu JY, Launius G, Dadash-Zadeh M, Lee RB, Noetzel M, Cognitive screening examinations for silent cerebral infarcts in sickle cell disease, Neurology, 50 (1998) 1678–1682. [DOI] [PubMed] [Google Scholar]
- [10].Mackin RS, Insel P, Truran D, Vichinsky EP, Neumayr LD, Armstrong FD, Gold JI, Kesler K, Brewer J, Weiner MW, Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition, Neurology, 82 (2014) 835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Steen RG, Miles MA, Helton KJ, Strawn S, Wang W, Xiong X, Mulhern RK, Cognitive impairment in children with hemoglobin SS sickle cell disease: relationship to MR imaging findings and hematocrit, AJNR Am J Neuroradiol, 24 (2003) 382–389. [PMC free article] [PubMed] [Google Scholar]
- [12].Wang W, Enos L, Gallagher D, Thompson R, Guarini L, Vichinsky E, Wright E, Zimmerman R, Armstrong FD, Neuropsychologic performance in school-aged children with sickle cell disease: a report from the Cooperative Study of Sickle Cell Disease, J Pediatr, 139 (2001) 391–397. [DOI] [PubMed] [Google Scholar]
- [13].Newby RF, Epping A, Geiger JA, Miller MS, Scott JP, Visual Motor Integration in Children With Sickle Cell Disease, J Pediatr Hematol Oncol, 40 (2018) 495–498. [DOI] [PubMed] [Google Scholar]
- [14].Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, Vichinsky EP, Shigenaga M, Ames B, Kuypers FA, Klings ES, Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease, Blood, 111 (2008) 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Klings ES, Farber HW, Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease, Respir Res, 2 (2001) 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Renoux C, Joly P, Faes C, Mury P, Eglenen B, Turkay M, Yavas G, Yalcin O, Bertrand Y, Garnier N, Cuzzubbo D, Gauthier A, Romana M, Mockesch B, Cannas G, Antoine-Jonville S, Pialoux V, Connes P, Association between Oxidative Stress, Genetic Factors, and Clinical Severity in Children with Sickle Cell Anemia, J Pediatr, 195 (2018) 228–235. [DOI] [PubMed] [Google Scholar]
- [17].Gizi A, Papassotiriou I, Apostolakou F, Lazaropoulou C, Papastamataki M, Kanavaki I, Kalotychou V, Goussetis E, Kattamis A, Rombos I, Kanavakis E, Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status, Blood Cells Mol Dis, 46 (2011) 220–225. [DOI] [PubMed] [Google Scholar]
- [18].Strader MB, Jana S, Meng F, Heaven MR, Shet AS, Thein SL, Alayash AI, Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease, Sci Rep, 10 (2020) 14218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jana S, Strader MB, Meng F, Hicks W, Kassa T, Tarandovskiy I, De Paoli S, Simak J, Heaven MR, Belcher JD, Vercellotti GM, Alayash AI, Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles, JCI Insight, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Charrin E, Ofori-Acquah SF, Nader E, Skinner S, Connes P, Pialoux V, Joly P, Martin C, Inflammatory and oxidative stress phenotypes in transgenic sickle cell mice, Blood Cells Mol Dis, 62 (2016) 13–21. [DOI] [PubMed] [Google Scholar]
- [21].Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, Hebbel RP, Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease, Am J Pathol, 158 (2001) 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Valverde Y, Benson B, Gupta M, Gupta K, Spinal glial activation and oxidative stress are alleviated by treatment with curcumin or coenzyme Q in sickle mice, Haematologica, 101 (2016) e44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Antwi-Boasiako C, Dankwah GB, Aryee R, Hayfron-Benjamin C, Donkor ES, Campbell AD, Oxidative Profile of Patients with Sickle Cell Disease, Med Sci (Basel), 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM, Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease, Blood, 123 (2014) 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hebbel RP, Belcher JD, Vercellotti GM, The multifaceted role of ischemia/reperfusion in sickle cell anemia, J Clin Invest, 130 (2020) 1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, Ofori-Acquah SF, Extracellular hemin crisis triggers acute chest syndrome in sickle mice, J Clin Invest, 123 (2013) 4809–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Belcher JD, Chen C, Nguyen J, Zhang P, Abdulla F, Nguyen P, Killeen T, Xu P, O’Sullivan G, Nath KA, Vercellotti GM, Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate, Antioxid Redox Signal, 26 (2017) 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang X, Michaelis EK, Selective neuronal vulnerability to oxidative stress in the brain, Front Aging Neurosci, 2 (2010) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gandhi S, Abramov AY, Mechanism of oxidative stress in neurodegeneration, Oxid Med Cell Longev, 2012 (2012) 428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Friberg H, Wieloch T, Castilho RF, Mitochondrial oxidative stress after global brain ischemia in rats, Neurosci Lett, 334 (2002) 111–114. [DOI] [PubMed] [Google Scholar]
- [31].Maes M, Galecki P, Chang YS, Berk M, A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness, Prog Neuropsychopharmacol Biol Psychiatry, 35 (2011) 676–692. [DOI] [PubMed] [Google Scholar]
- [32].Salim S, Oxidative Stress and the Central Nervous System, J Pharmacol Exp Ther, 360 (2017) 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hebbel RP, Eaton JW, Balasingam M, Steinberg MH, Spontaneous oxygen radical generation by sickle erythrocytes, The Journal of Clinical Investigation, 70 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hebbel RP, Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain, Hematol Oncol Clin North Am, 28 (2014) 181–198. [DOI] [PubMed] [Google Scholar]
- [35].Wu L-C, Sun C-W, Ryan T, Pawlik K, Ren J, Townes T, Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J and Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108: 1183-1188, Blood, 108 (2006) 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, Beard C, Brambrink T, Wu LC, Townes TM, Jaenisch R, Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin, Science, 318 (2007) 1920–1923. [DOI] [PubMed] [Google Scholar]
- [37].Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM, Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease, Science, 278 (1997) 876–878. [DOI] [PubMed] [Google Scholar]
- [38].Almeida LEF, Damsker JM, Albani S, Afsar N, Kamimura S, Pratt D, Kleiner DE, Quezado M, Gordish-Dressman H, Quezado ZMN, The corticosteroid compounds prednisolone and vamorolone do not alter the nociception phenotype and exacerbate liver injury in sickle cell mice, Sci Rep, 8 (2018) 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Khaibullina A, Adjei EA, Afangbedji N, Ivanov A, Kumari N, Almeida LEF, Quezado ZMN, Nekhai S, Jerebtsova M, RON kinase inhibition reduces renal endothelial injury in sickle cell disease mice, Haematologica, 103 (2018) 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bakeer N, James J, Roy S, Wansapura J, Shanmukhappa SK, Lorenz JN, Osinska H, Backer K, Huby AC, Shrestha A, Niss O, Fleck R, Quinn CT, Taylor MD, Purevjav E, Aronow BJ, Towbin JA, Malik P, Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology, Proc Natl Acad Sci U S A, 113 (2016) E5182–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kohli DR, Li Y, Khasabov SG, Gupta P, Kehl LJ, Ericson ME, Nguyen J, Gupta V, Hebbel RP, Simone DA, Gupta K, Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids, Blood, 116 (2010) 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Khaibullina A, Almeida LE, Wang L, Kamimura S, Wong EC, Nouraie M, Maric I, Albani S, Finkel J, Quezado ZM, Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice, Blood Cells Mol Dis, 55 (2015) 363–372. [DOI] [PubMed] [Google Scholar]
- [43].Kenyon N, Wang L, Spornick N, Khaibullina A, Almeida LE, Cheng Y, Wang J, Guptill V, Finkel JC, Quezado ZM, Sickle cell disease in mice is associated with sensitization of sensory nerve fibers, Exp Biol Med (Maywood), 240 (2015) 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang L, Almeida LEF, Kamimura S, van der Meulen JH, Nagaraju K, Quezado M, Wakim P, Quezado ZMN, The role of nitrite in muscle function, susceptibility to contraction injury, and fatigability in sickle cell mice, Nitric Oxide, 80 (2018) 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang L, Almeida LEF, de Souza Batista CM, Khaibullina A, Xu N, Albani S, Guth KA, Seo JS, Quezado M, Quezado ZMN, Cognitive and behavior deficits in sickle cell mice are associated with profound neuropathologic changes in hippocampus and cerebellum, Neurobiol Dis, 85 (2016) 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Almeida LEF, Wang L, Kamimura S, Zerfas PM, Smith ML, Neto OLA, Vale T, Quezado MM, Horkayne-Szakaly I, Wakim P, Quezado ZMN, Locomotor mal-performance and gait adaptability deficits in sickle cell mice are associated with vascular and white matter abnormalities and oxidative stress in cerebellum, Brain Res, 1746 (2020) 146968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kiernan EA, Smith SM, Mitchell GS, Watters JJ, Mechanisms of microglial activation in models of inflammation and hypoxia: Implications for chronic intermittent hypoxia, J Physiol, 594 (2016) 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yagi K, Simple assay for the level of total lipid peroxides in serum or plasma, Methods Mol Biol, 108 (1998) 101–106. [DOI] [PubMed] [Google Scholar]
- [49].Korzhevskii DE, Kirik OV, Brain Microglia and Microglial Markers, Neuroscience and Behavioral Physiology, 46 (2016) 284–290. [Google Scholar]
- [50].Nelson KP, Edwards D, A measure of association for ordered categorical data in population-based studies, Stat Methods Med Res, 27 (2018) 812–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hawkins BT, Davis TP, The blood-brain barrier/neurovascular unit in health and disease, Pharmacol Rev, 57 (2005) 173–185. [DOI] [PubMed] [Google Scholar]
- [52].Salameh TS, Shah GN, Price TO, Hayden MR, Banks WA, Blood-Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate, J Pharmacol Exp Ther, 359 (2016) 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dhodda VK, Sailor KA, Bowen KK, Vemuganti R, Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis, J Neurochem, 89 (2004) 73–89. [DOI] [PubMed] [Google Scholar]
- [54].Stetler RA, Cao G, Gao Y, Zhang F, Wang S, Weng Z, Vosler P, Zhang L, Signore A, Graham SH, Chen J, Hsp27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling, The Journal of neuroscience : the official journal of the Society for Neuroscience, 28 (2008) 13038–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cui MH, Suzuka SM, Branch NA, Ambadipudi K, Thangaswamy S, Acharya SA, Billett HH, Branch CA, Brain neurochemical and hemodynamic findings in the NY1DD mouse model of mild sickle cell disease, NMR Biomed, 30 (2017). [DOI] [PubMed] [Google Scholar]
- [56].Behdarvandy M, Karimian M, Atlasi MA, Azami Tameh A, Heat shock protein 27 as a neuroprotective biomarker and a suitable target for stem cell therapy and pharmacotherapy in ischemic stroke, Cell Biol Int, 44 (2020) 356–367. [DOI] [PubMed] [Google Scholar]
- [57].Biondani A, Turrini F, Carta F, Matte A, Filippini A, Siciliano A, Beuzard Y, De Franceschi L, Heat-shock protein-27, -70 and peroxiredoxin-II show molecular chaperone function in sickle red cells: Evidence from transgenic sickle cell mouse model, Proteomics Clin Appl, 2 (2008) 706–719. [DOI] [PubMed] [Google Scholar]
- [58].Siciliano A, Malpeli G, Platt OS, Lebouef C, Janin A, Scarpa A, Olivieri O, Amato E, Corrocher R, Beuzard Y, De Franceschi L, Abnormal modulation of cell protective systems in response to ischemic/reperfusion injury is important in the development of mouse sickle cell hepatopathy, Haematologica, 96 (2011) 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Circu ML, Aw TY, Reactive oxygen species, cellular redox systems, and apoptosis, Free Radic Biol Med, 48 (2010) 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Borutaite V, Brown GC, Mitochondrial regulation of caspase activation by cytochrome oxidase and tetramethylphenylenediamine via cytosolic cytochrome c redox state, J Biol Chem, 282 (2007) 31124–31130. [DOI] [PubMed] [Google Scholar]
- [61].Borutaite V, Brown GC, Caspases are reversibly inactivated by hydrogen peroxide, FEBS Lett, 500 (2001) 114–118. [DOI] [PubMed] [Google Scholar]
- [62].Kim GW, Sugawara T, Chan PH, Involvement of oxidative stress and caspase-3 in cortical infarction after photothrombotic ischemia in mice, J Cereb Blood Flow Metab, 20 (2000) 1690–1701. [DOI] [PubMed] [Google Scholar]
- [63].Hollville E, Deshmukh M, Physiological functions of non-apoptotic caspase activity in the nervous system, Seminars in cell & developmental biology, 82 (2018) 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mukherjee A, Williams DW, More alive than dead: non-apoptotic roles for caspases in neuronal development, plasticity and disease, Cell death and differentiation, 24 (2017) 1411–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Salameh TS, Mortell WG, Logsdon AF, Butterfield DA, Banks WA, Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate, Fluids Barriers CNS, 16 (2019) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Manček-Keber M, Frank-Bertoncelj M, Hafner-Bratkovič I, Smole A, Zorko M, Pirher N, Hayer S, Kralj-Iglič V, Rozman B, lie N, Horvat S, Jerala R, Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles, Sci Signal, 8 (2015) ra60. [DOI] [PubMed] [Google Scholar]
- [67].Yang Y, Wang H, Kouadir M, Song H, Shi F, Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors, Cell Death Dis, 10 (2019) 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yang J, Wise L, Fukuchi KI, TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease, Front Immunol, 11 (2020) 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Feng X, Zhao Y, Yang T, Song M, Wang C, Yao Y, Fan H, Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors, Front Mol Neurosci, 12 (2019) 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK, Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease, Mov Disord, 35 (2020) 20–33. [DOI] [PubMed] [Google Scholar]
- [71].Xu H, Wandersee NJ, Guo Y, Jones DW, Holzhauer SL, Hanson MS, Machogu E, Brousseau DC, Hogg N, Densmore JC, Kaul S, Hillery CA, Pritchard KA Jr., Sickle cell disease increases high mobility group box 1: a novel mechanism of inflammation, Blood, 124 (2014) 3978–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, Shet AS, Combs CA, Quezado ZMN, Thein SL, The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase, Blood Adv, 2 (2018) 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ransohoff RM, Cardona AE, The myeloid cells of the central nervous system parenchyma, Nature, 468 (2010) 253–262. [DOI] [PubMed] [Google Scholar]
- [74].Fumagalli S, Perego C, Pischiutta F, Zanier ER, De Simoni M-G, The ischemic environment drives microglia and macrophage function, Frontiers in neurology, 6 (2015) 81–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Olsson T, Wieloch T, Smith ML, Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade, Brain Res, 982 (2003) 260–269. [DOI] [PubMed] [Google Scholar]
- [76].Wang X, Pal R, Chen XW, Limpeanchob N, Kumar KN, Michaelis EK, High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region, Brain Res Mol Brain Res, 140 (2005) 120–126. [DOI] [PubMed] [Google Scholar]
- [77].Huang Y, Coupland NJ, Lebel RM, Carter R, Seres P, Wilman AH, Malykhin NV, Structural changes in hippocampal subfields in major depressive disorder: a high-field magnetic resonance imaging study, Biol Psychiatry, 74 (2013) 62–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.