

Summary
Mechanical signals transmitted through the cytoplasmic actin cytoskeleton must be relayed to the nucleus to control gene expression. LIM domains are protein-protein interaction modules found in cytoskeletal proteins and transcriptional regulators. Here we identify three LIM protein families (zyxin, paxillin, and FHL) whose members preferentially localize to the actin cytoskeleton in mechanically-stimulated cells through their tandem LIM domains. A minimal actin-myosin reconstitution system reveals that representatives of all three families directly bind F-actin only in the presence of mechanical force. Point mutations at a site conserved in each LIM domain of these proteins disrupt tensed F-actin binding in vitro and cytoskeletal localization in cells, demonstrating a common, avidity-based mechanism. Finally, we find that binding to tensed F-actin in the cytoplasm excludes the cancer-associated transcriptional co-activator FHL2 from the nucleus in stiff micro-environments. This establishes direct force-activated F-actin binding as a mechanosensing mechanism by which cytoskeletal tension can govern nuclear localization.
eTOC Blurb:
Sun et al. show mechanoresponsive tandem LIM domain proteins directly bind F-actin only in the presence of force through a conserved mechanism, governing their tension-dependent cytoskeletal localization in cells. Binding tensed cytoplasmic F-actin prevents the transcriptional co-activator FHL2 from entering the nucleus, facilitating mechanosensing through the cytoskeleton.
Graphical Abstract
Introduction
The sensing of mechanical stimuli by cells plays a pivotal role in maintaining tissue homeostasis (Jaalouk and Lammerding, 2009), and defects in mechanical signal transduction (“mechanotransduction”) are correspondingly associated with tumorigenesis (Paszek et al., 2005), cancer metastasis (Wolf et al., 2007), and developmental disorders (Krieg et al., 2008). Cells probe the mechanics of their microenvironments through integrin-mediated focal adhesions (FAs) linked to actomyosin stress fibers (SFs) (Case and Waterman, 2015; Kanchanawong et al., 2010). Forces transmitted through these cytoskeletal networks are transduced into gene expression programs by the downstream nuclear localization of transcriptional co-activators (Dupont et al., 2011; Fernández-Sánchez et al., 2015). The upstream mechanically-regulated binding interactions with cytoskeletal partners governing the nuclear localization of these factors remain unknown.
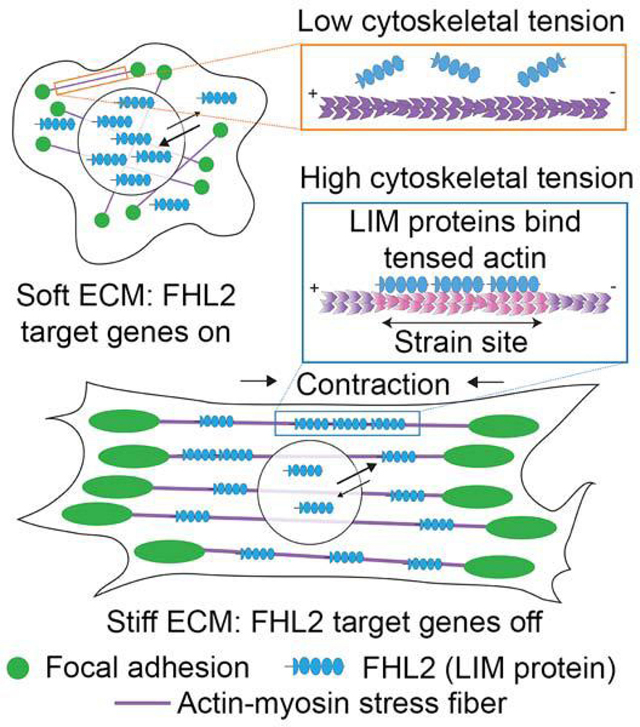
The transcriptional co-activator FHL2 (Four-and-a-Half LIM domains 2, also known as DRAL and SLIM-3) is dysregulated in a wide variety of cancers (Cao et al., 2015; Martin et al., 2007; Ng et al., 2011), cardiomyopathies (Arimura et al., 2007; Friedrich et al., 2014), and developmental disorders (Li et al., 2012; Qian et al., 2009). FHL2 mediates mechanotransduction via enhanced nuclear localization in soft environments (Nakazawa et al., 2016). It is composed solely of LIM domains, protein-protein interaction modules (Schmeichel and Beckerle, 1994) found in diverse actin cytoskeleton-associated proteins (Smith et al., 2014) and homeobox transcription factors (Kadrmas and Beckerle, 2004). Many LIM-domain containing proteins exhibit diminished FA / SF recruitment when actomyosin contractility is suppressed (Kuo et al., 2011; Schiller et al., 2011), suggesting that LIM domains could serve as a mechanical response module. LIM-domain dependent cytoskeletal “mechanoaccumulation” is well-established for the SF repair factor zyxin, defined as recruitment to SFs in response to uniaxial cyclic stretch (Hoffman et al., 2012), as well as rapid localization to micron-scale regions along SFs known as “strain sites” formed spontaneously in contractile cells (Smith et al., 2010). Mechanoaccumulation has also been reported for selected members of the paxillin family of SF / FA proteins (Smith et al., 2013; Watanabe-Nakayama et al., 2013), but it is unknown how widely distributed this activity is in the LIM protein superfamily. Although direct actin-binding has been speculated to play a role in mechanoaccumulation (Schiller and Fässler, 2013; Smith et al., 2014), zyxin exhibits negligible association with F-actin in co-sedimentation assays (Crawford et al., 1992), and a force-dependent molecular mark in SFs recognized by LIM domains remains to be identified.
Here we show that mechanoresponder LIM domains directly bind tensed F-actin through a conserved mechanism. We find that tensed F-actin binding serves as the dominant mechanism to retain FHL2 in the cytoplasm in stiff environments. The degree of FHL2 nuclear localization is quantitatively related to the availability of cytoskeletal binding sites and its capacity to engage them, supporting a simple model where tensed F-actin acts as a cytoplasmic sink for retaining FHL2 in stiff environments to prevent its nuclear shuttling. Our finding that force-activated F-actin binding is widely distributed in the LIM protein superfamily suggests that it could be a general mechanism for coordinating mechanotransduction through the cytoskeleton.
Results
LIM domains from three protein families are sufficient for mechanoaccumulation, which can be negatively regulated by sequence context
To identify mechanoresponsive LIM proteins, we screened a curated list of 28 eGFP-tagged LIM proteins (Figure 1A) identified in FA / SF proteomics studies (Kuo et al., 2011; Schiller et al., 2011) for enhanced localization to the actin cytoskeleton of mouse embryonic fibroblasts (MEFs) mechanically stimulated by uniaxial cyclic stretch (Methods; Figure 1B,C; Figure S1). Mechanical stimulation reoriented SFs perpendicular to the stretch axis (Figures 1B and S1A), as was previously reported (Hoffman et al., 2012; Yoshigi et al., 2005). We found members of the FHL (FHL2 and FHL3), paxillin (HIC5), zyxin (zyxin, FBLIM1, and TRIP6), and ALP (PDLIM1 and PDLIM2) families exhibited significant increases in actin enrichment in stretched cells versus unstretched controls (Figure 1B,C; Figure S1). To investigate which of these LIM protein families could feasibly mechanoaccumulate through a common molecular mechanism, we used live-cell confocal imaging to monitor representative proteins associating with spontaneously generated SF strain sites marked by zyxin-fusionRed (Shemiakina et al., 2012) in a human osteosarcoma cell line (U2OS, Figure S2) plated on glass (Methods). We found that paxillin family member HIC5, zyxin family member FBLIM1, and FHL family members FHL2 and FHL3 are recruited to strain sites with kinetics indistinguishable from zyxin within our 10 s frame rate of imaging, as has previously been reported for paxillin (Figure S2B,C) (Smith et al., 2013; Watanabe-Nakayama et al., 2013), suggesting they likely recognize a common molecular mark. In contrast, an ALP family mechanoresponder, PDLIM1, accumulates at a substantially later time point, comparable to α-actinin (Figure S2B,C), with which the PDZ domain of ALP/Enigma family LIM proteins are known to directly interact (Pomiès et al., 1999; Xia et al., 1997). As this suggests ALP family members mechanoaccumulate by an idiosyncratic mechanism, we focused on the FHL, paxillin, and zyxin families for the remainder of our study.
Figure 1. Three families of LIM-domain proteins mechanoaccumulate on the actin cytoskeleton.

(A) Schematics of LIM-domain proteins examined in this study, drawn to scale by primary sequence. (B) Epifluorescence micrographs of eGFP-labeled FHL-, paxillin-, and zyxin-family proteins in unstretched (−) and stretched (+) MEFs stained with phalloidin to label F-actin. Double-headed arrow indicates the uniaxial stretch direction. Scale bar, 20 μm. (C) Whole-cell actin enrichment of FHL-, paxillin-, and zyxin-family proteins in unstretched (−) and stretched (+) MEFs (20 ≤ n ≤ 63). Bars represent means. Dunnett’s T3 multiple comparison test after Welch’s ANOVA: NS, p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001. See also Figures S1–S2.
Hypothesizing that non-SF actin networks, which are not anchored to the substrate through FAs and are thus not subject to direct mechanical stimulation (Mullins et al., 1998), could mask weak mechanoaccumulation of some LIM proteins, we re-assayed all zyxin, paxillin, and FHL family members for stretch-induced recruitment specifically to substrate-anchored SFs in MEFs (Methods). With the exception of the zyxin-family protein LIMD1, all the examined proteins significantly mechanoaccumulate on SFs (Figures 2A–C, S3A), including endogenous FHL2 (Figure S3B). We next investigated whether their LIM domains are sufficient for mechanoaccumulation, as has previously been reported for paxillin (Smith et al., 2013; Watanabe-Nakayama et al., 2013) and zyxin (Hoffman et al., 2012; Smith et al., 2010). We generated and examined eGFP-tagged LIM-domain-only (LDO) versions of the paxillin- and zyxin-family proteins (FHL proteins only contain LIM domains, Figure 1A), all of which mechanoaccumulate on SFs, including LIMD1 LDO (Figures S3A, 2D). Furthermore, with the exception of the zyxin LDO, all LDOs exhibit enhanced SF enrichment compared to their full-length counterparts (Figure 2E). This suggests that mechanoaccumulation is an intrinsic property of mechanoresponder LIM domains, which can be negatively regulated by non-LIM sequence in cis through auto-inhibition and/or sequestration at other cellular compartments (Kadrmas and Beckerle, 2004).
Figure 2. LIM domains are sufficient for SF mechanoaccumulation, which can be negatively regulated by sequence context.

(A) Confocal slices of phalloidin-stained SFs (top) and eGFP-labeled FHL-family proteins (bottom). Images are cropped around individual SFs. Scale bar, 2 μm. (B) SF enrichments of eGFP-labeled FHL-family proteins in unstretched (−) and stretched (+) MEFs. Each data point is obtained from a single SF (28 ≤ n ≤ 226, N = 2 biological replicates). (C,D) SF enrichments of FL (C) and LDO (D) proteins of paxillin and zyxin families in unstretched (−) and stretched (+) MEFs. Each data point is obtained from a single SF (27 ≤ n ≤ 243, N = 2 biological replicates). Bars represent means. Games-Howell’s multiple comparison test after Welch’s ANOVA: NS, p > 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. Outliers (Methods, Table S1) are not displayed. (E) Difference between mean SF enrichments in stretched and unstretched MEFs of FL (circles) versus LDO (squares) versions of each protein. Error bars represent standard error of the difference between the means. See also Figure S3 and Table S1.
LIM domains are functionally swappable mechanoresponder modules
To test whether cytoskeletal mechanoaccumulation by LIM domains is a modular activity, we examined the capacity of FHL2/3 to functionally substitute for zyxin’s LIM domains. Zyxin’s primary sequence is functionally separable into an N-terminal region featuring α-actinin and VASP binding motifs necessary for SF repair, and a C-terminal region composed of three tandem LIM domains which mediates mechanoaccumulation (Hoffman et al., 2012; Smith et al., 2010) (Figure 3A). We generated zyxin-FHL2 and zyxin-FHL3 chimeras by replacing the C-terminal region with the coding sequences of FHL2/3 (Figure 3A), then assayed them for functional rescue in zyxin null MEFs previously reported to feature actin repair defects (Hoffman et al., 2006; Smith et al., 2010) (Figure 3).
Figure 3. The LIM domains of FHL confer actin repair activity when fused to the zyxin N-terminus.
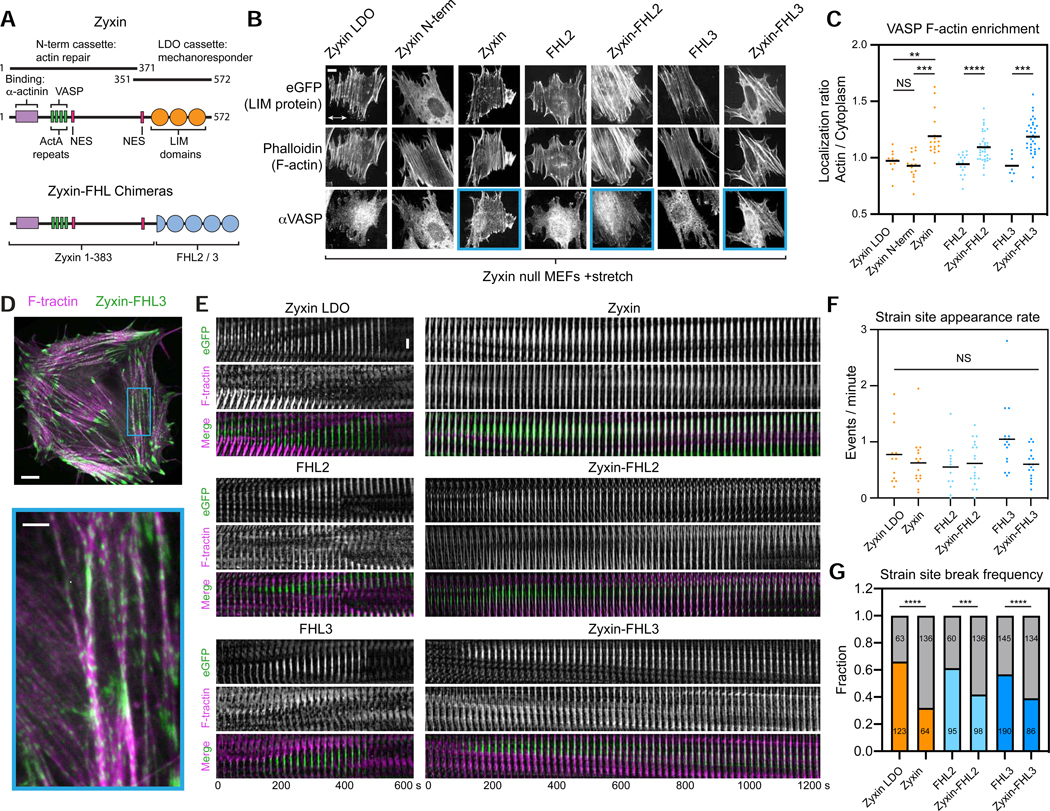
(A) Schematic of zyxin and zyxin-FHL chimeras. Numbers indicate residue ranges. (B) Maximum intensity projections (MIPs) of confocal z-stacks of stretched zyxin null MEFs expressing eGFP-labeled constructs. Top, eGFP. Middle, F-actin (phalloidin). Bottom, endogenous VASP (immunostaining). Blue boxes highlight constructs supporting VASP recruitment to SFs. Double-headed arrow indicates the uniaxial stretch direction. Scale bar, 10 μm. (C) Whole-cell actin enrichment of endogenous VASP in stretched zyxin null MEFs expressing the constructs shown in B. Bars represent means. 7 ≤ n ≤ 39. Dunnett’s T3 multiple comparison test after Welch’s ANOVA: NS, p > 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. (D) Top, spinning-disk confocal snapshot of a zyxin-null MEF co-expressing F-tractin-mApple (magenta) and zyxin-FHL3-eGFP (green). Scale bar, 10 μm. Bottom, zoomed view of boxed region. Arrow heads highlight SF strain sites. Scale bar, 3 μm. (E) Time-lapse montages of SF strain sites labelled by eGFP-labeled constructs. Scale bar, 3 μm. (F) SF strain site appearance rate. Bars represent means. Welch’s ANOVA test: NS, p > 0.05. (G) SF strain site break frequency. Gray bars: number of repairs; colored bars: number of breaks. The number of strain sites in each category is indicated. Fisher’s exact test: *** p < 0.001; **** p < 0.0001.
As anticipated, over-expressed zyxin LDO, FHL2, and FHL3 in zyxin null cells failed to recruit VASP (Figure 3B, mean of localization ratio ≈ 1) in cell-stretching experiments due to the lack of VASP-interacting motifs, and the zyxin N-terminus alone failed to mechanoaccumulate on SFs due to the absence of LIM domains (Figure 3B). Both zyxin-FHL chimeras, however, rescued VASP recruitment to a degree comparable with full-length zyxin (Figure 3C). We next investigated the F-actin repair capacity of these constructs at SF strain sites (Figure 3D,E), whose rate of spontaneous appearance was unaffected by the over-expression of any construct (Figure 3F). Cells expressing zyxin LDO, FHL2, and FHL3 all featured short-lived strain sites (Figure 3E, left, Video S1) which tended to progress to catastrophic SF breaks (Figure 3G). The zyxin-FHL constructs, however, localized to long-lived strain sites that displayed actin recovery (Figure 3E, right, Video S1). Consistently, strain site break frequency was significantly reduced only in zyxin null cells expressing zyxin and zyxin-FHL chimeras (Figure 3G). These findings collectively demonstrate that LIM domains are functionally swappable modules which confer mechanosensitive actin localization.
A conserved phenylalanine in each LIM domain is necessary for mechanoaccumulation
To identify primary sequence features that could confer mechanoaccumulation activity, we performed multiple sequence alignments on the isolated LIM domain sequences from mechanoresponsive and non-mechanoresponsive proteins. This analysis revealed a single invariant phenylalanine residue in mechanoresponder LIM domains (with the exception of LIM3 in zyxin family members) which is variable in non-mechanoresponders, suggesting it could be necessary but not sufficient for mechanoaccumulation (Figures 4A, S4A). We superimposed the structures of mechanoresponder FHL2 LIM2 and non-mechanoresponder LIMS1 LIM4 to examine their chemical environments at this position. F131 (green) in FHL2 LIM2 is simultaneously surface-exposed, where it would be available to mediate protein-protein interactions, and engaged in Van der Waals interactions in the small hydrophobic core of the domain, a hybrid pose also adopted by H238 (red) at the same location in LIMS1 LIM4 (Figure 4B).
Figure 4. Conserved phenylalanines in mechanoresponder LIM domains additively contribute to mechano-accumulation.

(A) Multiple sequence alignment logos highlight the conserved phenylalanine (position 0) in mechanoresponder LIM domains (top), which is variable in non-mechanoresponder LIM domains (bottom). (B) Superimposed ribbon diagrams (left) and space-filling representations (right) of FHL2 LIM2 and LIMS1 LIM4. The conserved phenylalanine in FHL2 LIM2 (F131) and the histidine at the same location in LIMS1 LIM4 (H238) are highlighted in green and red, respectively. (C) Epifluorescence micrographs of MEFs expressing the indicated eGFP-labeled constructs, stained with phalloidin to label F-actin. Scale bar, 20 μm. (D) Top: Primary sequence positions of conserved phenylalanines. The divergent third LIM domain of zyxin is highlighted in bright blue. Bottom: Whole-cell actin enrichment of constructs shown in C. Left, FHL2 (84 ≤ n ≤ 151); middle, HIC5 (60 ≤ n ≤ 87); right, zyxin (90 ≤ n ≤ 101). N = 2 biological replicates. Games-Howell’s multiple comparison test after Welch’s ANOVA: **** p < 0.0001. (E) Whole-cell actin enrichments of FHL2 constructs with zero (blue, n = 151), one (red, 112 ≤ n ≤ 138), two (green, 93 ≤ n ≤ 138), three (purple, 109 ≤ n ≤ 121), and four (orange, n = 112) phenylalanine mutations. N = 2 biological replicates. Welch’s ANOVA test: **** p < 0.0001. (F) Actin enrichments of mutant constructs from E, pooled by number of lesions. Welch’s ANOVA test: **** p < 0.0001. Linear trend test (Altman, 1990): **** p < 0.0001. See also Figure S4.
To investigate the functional role of these conserved phenylalanines in mechanoresponsive proteins, we generated alanine and histidine (mimicking non-mechanoresponder LIMS1 LIM4) point mutants in every LIM domain. For brevity, we indicate LIM domains featuring mutations in parenthesis, e.g., FHL2 with four phenylalanine to alanine substitutions (FHL2 F80A F141A F200A F263A) is named FHL2 F(1–4)A. We found that F(1–4)A and F(1–4)H substitutions in both FHL2 and HIC5 abolish their actin enrichment in MEF cells plated on glass (Figure 4C,D: mean localization ratios ≈ 1), a stiff condition which activates mechanoaccumulation. Zyxin family members have a unique LIM3 featuring two insertions and lacking the conserved phenylalanine (Figure S4A; cysteine in sequence logo position 0 of Figure 4A, top), yet zyxin nevertheless features a phenylalanine adjacent to the conserved position (Figure S4A) which we hypothesized could fulfill an analogous role. Consistent with this hypothesis, zyxin LDO F(1–3)A had more severe impairment of actin enrichment than zyxin LDO F(1,2)A versus wild-type zyxin LDO (Figure S4B,C), and full-length zyxin F(1–3)A and F(1–3)H mutants showed reduced actin enrichment versus wild-type (Figure 4D). This suggests that the tandem LIM domains of zyxin operate by a fundamentally similar mechanism to other mechanoresponder LIM domains. Nevertheless, all full-length and LDO zyxin mutants retain a greater degree of actin enrichment (Figure 4D; Figure S4C: mean localization ratios > 1) than the equivalent mutants of other LIM proteins, suggesting that zyxin LIM3 may be specialized to confer stronger mechanoaccumulation activity through additional mechanisms. Together, these data show that the conserved phenylalanines in mechanoresponder LIM domains are necessary for mechanoaccumulation.
Tandem LIM domains contribute additively to mechanoaccumulation
All mechanoresponsive LIM proteins we identified contain at least three tandem LIM domains, suggesting that mechanoaccumulation could require multiple LIM domains in series. To test this hypothesis, we systematically mutated the conserved phenylalanine to alanine in each LIM domain of FHL2 individually and in all possible combinations, and examined their localizations in MEFs plated on glass (Figure 4E,F).
Mutants with the same number of lesions exhibit distinct degrees of actin enrichment (Figure 4E), suggesting that not all LIM domains possess identical mechanoresponder activity. LIM2 is the greatest contributor to SF localization, as FHL2 F(2)A had the lowest actin enrichment among single mutants and FHL2 F(1,3,4)A had the highest actin enrichment among triple mutants (Figure 4E). LIM4, on the other hand, makes the weakest contribution, indicated by FHL2 F(4)A having the highest actin enrichment among the single mutants, as well as FHL2 F(3,4)A and FHL2 F(1,4)A among the double mutants (Figure 4E). Nevertheless, pooling data from mutants with the same number of lesions reveals a linear trend, where actin enrichment decreases as the number of mutations increases (Figure 4F). This suggests that all LIM domains in a tandem array contribute additively but not equally to mechanoaccumulation.
Mechanoaccumulation mediates FA localization of LIM proteins
LIM domains are highly enriched in the FA proteome (Zaidel-Bar et al., 2007), and the LIM domains of FHL2 (Samson et al., 2004), FHL3 (Samson et al., 2004), zyxin (Uemura et al., 2011), paxillin (Brown et al., 1996), and HIC5 (Thomas et al., 1999) are necessary for their FA localization. To dissect the role of LIM-domain-mediated cytoskeletal mechanoaccumulation in FA localization, we expressed wild-type and mechanoaccumulation-defective mutants of full-length and LDO versions of eGFP-labeled FHL2, HIC5, and zyxin in MEFs plated on glass, then immunostained for endogenous paxillin to mark FAs (Figure S4D,E).
We found that the phenylalanine mutations greatly diminish FA recruitment, with the FA enrichment of FHL2 F(1–4)A and HIC5 LDO F(1–4)A almost completely abolished (Figure S4E: mean localization ratio ≈ 1). In contrast to HIC5 LDO F(1–4)A, full-length HIC5 F(1–4)A retains a small degree of FA enrichment (Figure S4E: mean localization ratio > 1) despite its overall actin enrichment being abolished, likely conferred by its non-LIM region binding other FA proteins. Unlike their wild-type counterparts, full-length and LDO zyxin F(1–3)A exhibit a positional offset from paxillin. They are depleted from the distal end of FAs, but localize to the thick SFs regions at their proximal ends despite their overall greatly reduced actin enrichment (Fig. 4C,D), thereby maintaining partial FA enrichment (Figure S4E: mean localization ratio > 1). We speculate this is mediated by the divergent zyxin LIM3 (Figures 4D, S4C,E) through two potential, non-exclusive mechanisms: 1) LIM3 interacts with other proteins localized to the region of SF / FA overlap through a mechanism independent of the conserved phenylalanine; 2) The proximal SF segment bears greater load than the distal region of the FA, which full-length/LDO zyxin F(1–3)A bind through their residual mechanoaccumulation activity (Figure 4C,D). HIC5 LDO F(1–4)A also weakly localizes to this region (Figure S4D), consistent with this region of SF / FA overlap featuring particularly high strain. Future studies will be required to dissect the detailed interplay of these mechanisms. Nevertheless, our studies suggest that cytoskeletal mechanoaccumulation plays a prominent role in LIM protein FA localization.
Force along actin filaments is necessary and sufficient for LIM protein binding
We next sought to define the mechanism of LIM domain mechanoaccumulation. Hypothesizing that force across actin filaments is required to activate binding, we adapted the gliding filament assay (Kron and Spudich, 1986) to monitor protein binding to filaments under load. Myosin Va (plus-end directed) and myosin VI (minus-end directed) motor proteins are immobilized on a glass coverslip, where they applied stress along actin filaments in the presence of ATP (Figure 5A). We purified C-terminally Halo-tagged full-length FHL3, HIC5, and zyxin (Figure S5A,B), and labeled them with Janelia Fluor 646 (Grimm et al., 2015). We then examined their F-actin binding in the absence and presence of force (controlled by supplying ATP) by total internal reflection fluorescence (TIRF) microscopy.
Figure 5. Strain in F-actin is necessary and sufficient for direct binding by LIM proteins.

(A) Schematic of in vitro force reconstitution TIRF assay. (B) TIRF snapshots of 10% rhodamine labelled actin filaments (magenta) and FHL3-Halo (green) in the absence (left) and presence (right) of force generation. Arrows (bottom) highlight FHL3 patches. Scale bar, 5 μm. (C) Cumulative projections of detected patches of indicated Halo-tagged wild-type constructs in the absence (top) and presence (middle) of 0.5 mM ATP, as well as mutant constructs in the presence of 0.5 mM ATP (bottom), color-coded by time. Scale bar, 10 μm. (D) Cumulative relative frequency of wild-type construct binding lifetimes. Half-lives are extracted by linear interpolation; errors represent standard deviations obtained from bootstrapping (Methods). (E) Number of patches detected in equal imaging periods across trials for wildtype and mutant constructs. Bars represent means. Dunnett’s T3 multiple comparison test after Welch’s ANOVA: * p < 0.05; *** p < 0.001. One FHL3 wild-type outlier is not shown. See also Figures S5 and S6.
We found that FHL3, HIC5, and zyxin all bind to F-actin in small “patches” only in the presence of ATP (Figure 5B, Video S2). The vast majority of filaments bound by LIM proteins remain static and appear straightened (Videos S2 and S4), indicating that oppositely-directed myosin Va and VI are engaging in a tug-of-war that develops tension in the filaments. This suggests that force is necessary and sufficient to activate this direct binding, further evidenced by cumulative projections of patches detected and tracked over time (Methods), which show many more patches in the presence of force generation (Figures 5C, S6A). FHL3, HIC5, and zyxin patches exhibit indistinguishable lifetime distributions (Figure 5D), and their nearly identical patch half-lives (48.3 ± 1.7 s for FHL3, 42.7 ± 2.9 s for HIC5, and 44.3 ± 1.5 s for zyxin) are consistent with them forming along regions in strained F-actin featuring a common molecular mark.
To test whether the conserved phenylalanine is required for direct binding to strained actin, we purified Halo-tagged FHL3 F(1–4)A, HIC5 F(1–4)A, and zyxin F(1–3)A (Figure S5A,B). All mutants eluted from a size-exclusion column with similar retention volumes to their wild-type counterparts, and also displayed overlapping circular dichroism spectra, suggesting multi-site phenylalanine to alanine mutations do not grossly disrupt tandem LIM domain structure (Figure S5A,C). As illustrated in cumulative projections (Figure 5C), all the mutants were almost completely defective in patch formation (Figure 5E). Zyxin F(1–3)A nevertheless retained a greater residual degree of strained actin binding in vitro than the other two mutants (Figure 5C,E), mirroring its incomplete loss of actin enrichment in cells versus FHL2 F(1–4)A and HIC5 F(1–4)A (Figure 4D). These findings, coupled with the concordant properties of phenylalanine mutants in cells, suggest that direct force activated F-actin binding is the conserved mechanism of LIM domain mechanoaccumulation.
LIM proteins recognize a pre-break state of F-actin which spreads along filaments
To gain further insight into strained F-actin recognition by LIM domains, we examined the detailed dynamics of individual patches. Patches can initiate both in the middle (Figure 6A, top) and in the terminal segments of actin filaments (Figure 6A, bottom), suggesting there is no intrinsic requirement for an exposed filament end to license patch formation. Patches appear along filaments as they straighten, consistent with the presence of mechanical tension, then disassociate abruptly upon load release and filament relaxation (Video S3), or upon filament breakage (Figure 6A, Video S4). To quantify the relationship between unbinding events and filament breakage, we calculated the log ratio of the average actin intensity in the region covered by patches before unbinding vs. immediately after (Methods). A positive log ratio indicates LIM protein dissociation from a filament when it breaks at the patch site, as illustrated by line scans of a representative FHL3 patch (Figure 6B,C). Indeed, the vast majority of the patches formed by all three LIM proteins unbind upon filament breakage (Figure 6D), suggesting this is the major process determining patch lifetime. The residual fraction with negative log ratios presumably results from LIM protein unbinding due to filament relaxation (Video S3).
Figure 6. LIM proteins bind along filament regions featuring a pre-break state of F-actin.

(A) Montages of indicated constructs forming patches in the middle (top) and at the terminal segments (bottom) of actin filaments in the presence of force generation. Scale bars, 2 μm. (B) Kymograph (left, FHL3 channel) and montage (right) of an FHL3 patch. Snapshots correspond to the time points indicated along the kymograph. Scale bar, 2 μm. (C) Intensity line scan along dotted lines shown in B before (top) and after (bottom) FHL3 disappearance. (D) Log ratio histograms of actin intensity at detected patches of the indicated constructs before versus immediately after LIM-protein disappearance. Dashed line denotes equal intensity. See also Figures S5 and S6.
It is also clear that patches are not single molecules, as they are frequently anisotropic (Figure 6B). Kymograph analysis of a representative FHL3 patch shows steady elongation at a rate of 0.3 μm / minute, followed by an absence of growth for several seconds before dissociation in a single frame upon filament breakage (Figure 6B, Video S5). Occasionally, after ruptures at a patch site in the middle of a filament, we observed low-level retention of LIM protein at one end of the two new filament segments, which could then continue to grow as load developed (Figure S6B). We observed no apparent correlation between lifetime and maximum patch area or intensity (Figure S6C), indicating that in the absence of other cytosolic binding partners, LIM proteins do not reinforce strained actin filaments. Patch lifetime is therefore presumably determined by a filament’s mechanical endurance rather than the number of LIM proteins present. Our observations are consistent with patches forming along local regions in filaments that feature a distinct, force-induced F-actin conformation that spreads along filaments and is recognized and bound by mechanoresponder LIM domains prior to filament rupture. Our data do not directly rule out cooperative binding through contacts between LIM proteins that facilitate patch formation. However, the most parsimonious explanation for the lack of a clear positive correlation between patch lifetime and size (Figure S6C top) or intensity (Figure S6C bottom) is that binding is not cooperative, unless stabilizing contacts between LIM proteins are perfectly energetically balanced by mechanical destabilization of the underlying actin filament.
Tensed actin binding retains FHL2 in the cytoplasm in stiff environments, restricting its nuclear shuttling
The inability of wild-type FHL overexpression to rescue the SF repair deficiency of cells lacking zyxin (Figure 3C) suggests that strained actin binding plays a different role in this family. As increasing ECM stiffness upregulates SF-mediated contractility (Gupta et al., 2015) we hypothesized that a concomitant increase in the cytoplasmic pool of tensed actin binding sites could act as a “sink” to retain FHL2 in the cytoplasm and suppress its nuclear translocation (Nakazawa et al., 2016). To test this hypothesis, we expressed eGFP-labeled wild-type FHL2 and FHL2 F(1–4)A in MEFs, and plated the cells on glass, 50-kPa, and 12-kPa hydrogels (Figure 7A). We found that nuclear enrichment of wild-type FHL2 increased as substrate stiffness decreased (Figure 7A,B), consistent with the reported behavior of endogenous FHL2 in Human Foreskin Fibroblasts (HFFs) (Nakazawa et al., 2016). In contrast, FHL2 F(1–4)A was predominantly nuclear regardless of substrate stiffness (Figure 7A,B). Moreover, wild-type FHL2’s actin enrichment showed an inverse trend, diminishing with decreasing stiffness, while FHL2 F(1–4)A always displayed low actin enrichment (Figure 7B).
Figure 7. Tensed F-actin retains FHL2 in the cytoplasm.

(A) Confocal slices of MEFs expressing indicated FHL2-eGFP constructs plated on substrates of varying stiffness and stained with phalloidin (magenta) to label F-actin and DAPI (green) to label nuclei. Scale bars, 10 μm. (B) Quantification of stiffness-dependent nuclear (top) and actin (bottom) enrichment of wild-type and FHL2 F(1–4)A. N = 2 biological replicates; bars represent means. Games-Howell’s multiple comparison test after Welch’s ANOVA: NS, p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. (C) Spinning-disk confocal snapshots of FHL2-eGFP localization in a MEF before (left) and after (middle) cytochalasin D (cyto D) treatment, and after washout (right). Scale bars, 20 μm. (D) Average nuclear (top) and actin (bottom) enrichments (n = 19 cells; N = 13 biological replicates) over time, aligned at the timepoint of cyto D addition (10 min). Error bar represents SEM. Dash lines indicate cyto D addition and washout. (E) Replotting of average nuclear enrichment versus average actin enrichment as a time color-coded scatterplot. (F) Scatter plot of average nuclear enrichment versus average actin enrichment of FHL2 with zero (blue), one (red), two (green), three (purple), and four (orange) phenylalanine mutations in the indicated LIM domains. Data were extracted from experiments displayed in Figure 4E. Trend line and parameters from a linear fit are displayed. (G) Model of rigidity-dependent nuclear localization of FHL2. Magenta, SFs. Green, FAs. Blue, FHL2. See also Figure S7.
To test if the cytoplasmic tensed actin sink model could quantitatively explain mechanosensitive FHL2 nuclear-cytoplasmic partitioning, we performed live-cell imaging of Hoechst-treated MEFs co-expressing FHL2-eGFP and LifeAct-mApple while reversibly disrupting the actin cytoskeleton with low doses of cytochalasin D. Cytochalasin D treatment triggered rapid FHL2 dissociation from the actin cytoskeleton and nuclear shuttling, which was reversed upon washout (Figure 7C–E, Video S6). Nuclear and actin enrichments tracked on a frame-by-frame basis show opposite trends (Figure 7D). A time-coded scatter plot of average nuclear enrichment versus average actin enrichment reveals two trajectories with opposing directionality that essentially overlap (Figure 7E), suggesting that FHL2 nuclear localization is dictated by the number of available cytoplasmic tensed actin binding sites.
To specifically reduce contractility in SFs, we repeated the live-cell imaging while reversibly inhibiting ROCK, thereby reducing myosin light chain phosphorylation, with 5 μM Y-27632. Compromised myosin II activity led to a loss of tension, partial disassembly of SFs, and a consequent boost of the monomeric actin pool, evidenced by enhanced lamellipodial activity during Y-27632 treatment (Figure S7A, Video S6), consistent with prior studies (Uehata et al., 1997). FHL2 preferentially localized to the lamella vs. the lamellipodia (Figure S7A), suggesting that branched F-actin is not sufficient to recruit FHL2. SFs that were preserved (Figure S7A, “SF 1”), as well as SFs that transiently formed during myosin inhibition (Figure S7A, “SF 2”) were completely depleted of FHL2. Robust nuclear enrichment and depletion of FHL2 were observed upon Y-27632 addition and washout, respectively (Figure S7A, Video S6). The opposing trends in the nuclear and actin enrichments suggest that FHL2 localization is directly dictated by myosin II-mediated contractility in SFs (Figure S7B, C). We next examined our allelic series of phenylalanine mutants (Figure 4E,F), and found a negative linear relationship between nuclear enrichment and actin enrichment (Figure 7F). This finding suggests that FHL2’s nuclear localization is also dictated by its affinity for tensed actin.
Phosphorylation of FHL2 Y93 by FAK has previously been reported to be required for FHL2 nuclear translocation in soft environments in HFFs (Nakazawa et al., 2016); a counterintuitive finding, as tensile force and stiff ECM collectively upregulate FAK activity (Bauer et al., 2019; Bell and Terentjev, 2017; Zhou et al., 2015). To dissect the interplay of tensed actin binding and FAK phosphorylation, we generated phospho-resistant FHL2 mutants and pharmacologically inhibited FAK, which did not impact mechanosensitive FHL2 localization in MEFs (Figure S7D–F), demonstrating that strained actin binding is the dominant upstream process mediating FHL2 mechanotransduction in these cells.
Discussion
We find that mechanoresponsive LIM proteins directly bind tensed F-actin through a conserved mechanism. This interaction excludes FHL2 from the nucleus in stiff environments (Figure 7G), providing, to our knowledge, the first direct link between upstream forces transmitted through the cytoskeleton and downstream nuclear localization of a transcriptional co-activator. Our strategy for generating point mutants specifically deficient in tensed F-actin binding will facilitate the dissection of mechanoregulated gene expression controlled by FHL2, including in disease contexts, as well as by other transcriptional co-activators in the FHL family (Ding et al., 2009; Fimia et al., 2000). Dynamic nuclear localization of members of the zyxin and paxillin families has also been reported (Kadrmas and Beckerle, 2004; Wang and Gilmore, 2003), the physiological role of which is not clear. We speculate that their nuclear localization is coordinately regulated by their LIM domains’ intrinsic activity to bind tensed F-actin, as well as by tissue and cell-type specific mechanisms which operate through their non-LIM sequence elements. This coordinate regulation may be a general mechanism for the evolution of specialized signaling networks incorporating LIM domains as actin force-sensing modules.
We propose mechanoresponsive LIM proteins constitute a novel class of actin-binding proteins which only associate with F-actin in the presence of mechanical load. Recognition of filament regions featuring a specific molecular mark is the most parsimonious explanation for the formation of LIM protein patches, suggesting that mechanical load can evoke a specialized conformation of F-actin which spreads along local domains within a filament. We speculate that the conserved phenylalanine residues likely play an essential role at the binding interface between mechanoresponder LIM domains and strained actin. However, it is also clear that the presence of a phenylalanine residue at this conserved position is insufficient to confer force-activated actin binding activity, suggesting that other structural elements of LIM domains are also required to form this interface (Figures 4A, S4A). Structural studies of LIM proteins bound to F-actin in the presence of mechanical load will be necessary to establish a detailed mechanism for their strained actin recognition, which could potentially be realized by adapting the force reconstitution platform we describe here for cryo-electron microscopy.
Our observation that CRP1, which directly binds F-actin in the absence of load through its LIM domains (Tran et al., 2005; Jang and Greenwood, 2009), is mechanodepleted suggests there must be multiple modes by which LIM domains can engage F-actin. One possibility is that CRP1 vs. mechanoresponder LIM domains employ distinct surfaces to engage the filament. Another, non-exclusive possibility is that the spacing imposed by the length of the linkers between tandem LIM domains is important for measuring the distance between actin protomers, which could be modulated by mechanical load as has previously been speculated (Schiller and Fässler, 2013). Contemporary with our studies, complementary work from the Gardel and Kovar groups have shown that increasing the length of the linkers between LIM domains in zyxin LDO abrogates its recruitment to SF strain sites (Winkelman et al., 2020), consistent with this speculated role as a ruler. Furthermore, they found that CRP family members are the oldest LIM-domain proteins found in plants and animals, and proposed that divergence from an ancestral CRP-like LIM domain produced mechanoresponder LIM domains that specifically recognize tensed F-actin, which may have co-evolved with myosin II and α-actinin as cellular reporters for forces generated by the actomyosin machinery (Winkelman et al., 2020).
Beyond force-activated actin binding, additional mechanisms may reinforce LIM protein mechanoaccumulation in cells. Due to the absence of cross-linkers (Gardel et al., 2004), crowding agents (Hosek and Tang, 2004), or a high concentration of multivalent counterions ([Mg2+] > 20 mM) (Huber et al., 2012), the vast majority of the actin filaments in our reconstitution assays should remain single as opposed to bundled. There is a qualitative disparity between robust LIM-protein mechanoaccumulation in cells (Figures 1, 2, 3, S2, S3) and the limited number of patches formed in vitro (Figure 5B, C). In addition, zyxin patch dynamics (44.3 ± 1.5 s half-life) in vitro arise from slow growth (patch elongation) and fast dissociation (nearly instantaneous unbinding upon filament failure or breakage) processes. This contrasts with the previously reported fast accumulation (28.6 ± 3.7 s half-life) and slow dissociation (112 ± 12 s half-life) kinetics of zyxin at SF strain sites (Smith et al., 2010). While the FHL2 F(3)A mutation results in a lower average actin enrichment than F(4)A (Figure 4E), the F(2,3)A and F(2,4)A double mutants exhibit comparable average actin enrichments (Figure 4E). A plausible explanation is that separated LIM domains that maintain the tandem-LIM spatial registration are more effective for mechanoaccumulation on SFs, which contain F-actin bundles, than adjacent LIM domains. This suggests that mechanoaccumulation could be reinforced by LIM proteins bridging adjacent actin filaments within bundles, in addition to their recognition of strain within single filaments.
Regardless, our studies establish direct strained actin binding as the obligatory first step in the LIM protein mechanoresponse. As there are dozens to hundreds of actin-binding proteins in the cell (Pollard, 2016), it is likely that other factors also recognize force-evoked actin states, providing an attractive mechanism for coordinating diverse mechanotransduction pathways through direct mechanical regulation of F-actin structure.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gregory M. Alushin (galushin@rockefeller.edu).
Materials Availability
Plasmids generated in this study are available from the lead contact upon reasonable request.
Data and Code Availability
All custom code used in data analysis is available at: https://github.com/alushinlab/LIM-domain. All data supporting the findings of this study are available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Immortalized mouse embryonic fibroblast (MEF) cell line was generated from tissue (torso) explant from newborn mice in the Beckerle lab (Hoffman et al., 2006). The sex of the cell line is unknown because it has not been karyotyped. MEFs were cultured in DMEM (Gibco, 11995–065) supplemented with 4.5 g/L D-glucose and L-glutamine, 110 mg/L sodium pyruvate, 10% fetal bovine serum (FBS), and 1% antibiotic-antimycotic (ThermoFisher, 15240062), and grown on polystyrene tissue culture dishes (ThermoFisher, FB0875712, diameter 10 cm) at 37 °C in 5% CO2.
U2OS cells (ATCC, HTB-96, Female) were cultured in McCoy’s 5A medium supplemented with 10% FBS and 1% Penicillin/Streptomycin, and grown on polystyrene tissue culture dishes (diameter 10 cm) at 37 °C in 5% CO2.
FreeStyle 293-F cells (ThermoFisher, R79007) were cultured in FreeStyle 293 expression medium (ThermoFisher, 12338026) on an orbital shaker at 37 °C in 8% CO2.
METHODS DETAILS
Chemical reagents were purchased from Sigma-Aldrich unless otherwise specified.
Molecular biology
Plasmids and cloning
Plasmids encoding C-terminal eGFP-labeled full-length human LIM proteins were purchased from GeneCopoeia in the m98 vector, where expression is driven by the CMV promoter. Plasmids encoding eGFP-labeled LDOs, zyxin N-term, and all phenylalanine mutants, were generated via Gibson assembly (NEB) in the same vector (Gibson et al., 2009). The coding sequences for the zyxin-FHL2 and zyxin-FHL3 chimeras were generated by gene synthesis (Blue Heron), then subcloned into the m98 vector by Gibson assembly.
For heterologous expression and purification, full-length human FHL3 (AA 1–280), HIC5 (AA 1–461), and zyxin (AA 1–572), coding sequences were subcloned into a modified pCAG mammalian expression vector with a C-terminal Halo-tag, PreScission protease cleavage site, and GFP-tag. Human myosin Va (AA 1–1091, Dharmacon BC172485) and myosin VI constructs (AA 1–1021, a generous gift from James Sellers, NHLBI) were subcloned into a modified pCAG mammalian expression vector with a C-terminal GFP-tag and Flag-tag. Human full-length calmodulin (AA 1–149, a generous gift from Roderick Mackinnon, The Rockefeller University) was subcloned into a modified pCAG mammalian expression vector with no tag.
All plasmids were propagated in NEB 5-alpha competent E. coli cells (NEB, C2987U), and purified by QIAprep spin miniprep kit (Qiagen, 27106) or PureYield plasmid maxiprep system (Promega, A2392) before transfection.
Cell maintenance and mechanobiology
Transient transfection
MEFs were harvested at 80% confluency for transient transfection. MEFs used in the cytochalasin D and Y-27632 treatment (Figures 7C, S7A) were transfected with lipofectamine. Briefly, 2 μg of FHL2-eGFP plasmid and 2 μg of LifeAct-mApple plasmid were diluted in 250 μL of Opti-MEM I Reduced Serum Medium without serum. 10 μL of lipofectamine 2000 were diluted in 250 μL of Opti-MEM I Reduced Serum Medium without serum. The diluted plasmids and diluted lipofectamine were mixed gently, incubated at room temperature for 20 minutes, and then mixed with the cells. MEFs used in other studies were transfected with 7–10 μg of LIM-protein plasmid using SE Cell Line 4D-Nucleofector X Kits (Lonza, V4XC-1024, program CM-137). Each plate of MEFs (2.0 ⋅ 106 cells) were used for two transfection reactions. U2OS cells (Figure S2) were harvested at 70% confluency for transfection. 1–2 μg of LIM-protein plasmids was used to transfect U2OS cells.
Cell stretching
For the initial screen (Figures 1, S1), MEFs transfected with LIM-protein plasmids were plated in 4-well Strex chambers (STB-CH-4W, 2 cm2 ⋅ 4 chambers) coated with 50 μg/mL fibronectin (Millipore, FC010), and allowed to recover overnight. A Strex STB-140–10 cell stretching system was used to apply cyclical uniaxial stretch (program 14, 10% extension, 0.5 Hz) in an incubator (37 °C, 5% CO2) for 2 hours. For individual SF analysis (Figures 2, S3), MEFs transfected with mechanoresponder LIM-protein plasmids were allowed to recover on polystyrene tissue culture dishes overnight, then re-plated on Flexcell UniFlex culture plates coated with ProNectin (Flexcell UF-4001P) supplemented with 10 μg/mL fibronectin, allowed to recover for 2 hours, then subjected to cyclical uniaxial stretch on a Flexcell FX6000-T apparatus (Program FX-5000, 10% extension, 0.5 Hz) in an incubator (37 °C, 5% CO2) for 1 hour.
Cell fixation and staining for fluorescent imaging
Cells were fixed in Cytoskeleton Buffer (100 mM MES pH 6.1, 1.38 M KCl, 30 mM MgCl2, 20 mM EGTA) with the indicated supplements. All the permeabilizing and staining steps were performed in DPBS (Gibco, 14190–144). Both unstretched and stretched MEFs were immediately fixed using 4% formaldehyde (ThermoFisher, 28908) for 20 minutes, permeabilized in 0.1% Triton X-100 for 10 minutes, incubated in 0.1 M glycine for 10 minutes, and washed in DPBS. Cells were than blocked in 2% Bovine serum albumin (BSA, Gemini Bio-Products, 700–101P) for 1 hour, stained with 165 nM Alexa Fluor 568 phalloidin (ThermoFisher, A12380) for 20 minutes and 300 nM DAPI (ThermoFisher, D1306) for 15 minutes, and washed with DPBS. The membrane of the stretching chamber was excised and mounted onto a No. 1.5 glass coverslip using DAKO mounting medium (Agilent, S3023) for imaging.
The endogenous FHL2 in MEFs was stained with anti-FHL2 monoclonal antibody (1:100, rabbit host, Abcam, ab202584) at 4 °C overnight, and Alexa Fluor 488 goat anti-rabbit secondary antibody (1:200, ThermoFisher, A32731) at room temperature for 1 hour, followed by staining with Alexa Fluor 568 phalloidin and DAPI, and mounting in DAKO mounting medium as described above.
MEFs used in the focal adhesion analysis were stained with anti-paxillin monoclonal antibody (1:200, mouse host, BD, 610052) at 4 °C overnight, and Alexa Fluor 568 goat anti-mouse secondary antibody (1:200, ThermoFisher, A11031) at room temperature for 1 hour, followed by staining with Alexa Fluor 647 phalloidin (ThermoFisher, A22287) and DAPI as described above.
For experiments that involve plating cells on glass or hydrogel substrates (Matrigen), the substrates were coated with 10 μg/mL fibronectin. Cells were plated on those substrates after transfection and were allowed to recover overnight. Cells were fixed and stained using the approach described above, and were then transferred to DPBS for imaging.
Pharmacological treatments
For live-cell imaging experiments visualizing nuclear localization dynamics of FHL2 in the presence of actin disruption (Figure 7C–E), MEFs co-expressing FHL2-eGFP and Lifeact-mApple were incubated with 5 μg/mL Hoechst (Invitrogen, H1399) for 1 hour, then treated with 250 nM cytochalasin D (Millipore, 504776) or 5 μM Y-27632 (Sigma-Aldrich, 688000).
FAK activity of MEFs (Figure S7E, F) was inhibited by incubating the cells with 1 μM PF573228 (Sigma-Aldrich, PZ0117) for 1 hours. Cells were then fixed and stained for confocal imaging.
Cellular imaging
Fixed cell imaging
MEFs used in the initial screen (Figures 1, S1) and combinatorial mutation studies (Figure 4E,F) were imaged via epifluorescence on a Nikon Ti-E microscope using a CFI Apo 100X TIRF oil immersion objective (NA 1.49). Epifluorescence illumination was provided by a PEKA LED illuminator (Lumencor, 300 mW). Images were captured at a depth of 12 bit using a Zyla 4.2 sCMOS camera (Andor). Image acquisition was performed using the NIS-Elements software (Nikon). A z-stack that spaned 1.4–1.8 μm with a step size of 0.2 μm was acquired for each cell to ensure that the epifluorescence image with the maximum amount of actin cytoskeleton in focus (visible as maximum sharpness in the actin channel) was captured for the subsequent image analysis. Excitation power and exposure time were maintained constant to ensure fair comparisons between unstretched and stretched cells.
MEFs used in the individual SF analysis (Figures 2, S3) and zyxin-FHL chimera studies (Figure 3), were imaged on Spinning Disk System 1: a Nikon TE2000E2 microscope equipped with a spinningdisk confocal scan head (Yokogawa) using an Apo 60X TIRF oil immersion objective (NA 1.49). Illumination was provided by solid-state 488-nm (100 mW) and 561-nm (550 mW) lasers. Images were captured at a depth of 16 bit using an Myo CMOS camera (Photometrics). Image acquisition was performed using the MetaMorph software (MDS Analytical Technologies).
MEFs used in the substrate stiffness (Figure 7A,B), FAK-phospho-resistant mutant (Y93F) (Figure S7D), and FAK inhibition (Figure S7E,F) studies were imaged on a Leica TCS SP8 confocal microscope using a Plan Apo 40X water immersion objective (NA 1.10) with motorized correction collar. Illumination was provided by a UV diode laser (405 nm, 50 mW) and a fully-tunable white-light laser (470–670 nm, 1.5 mW) with an acousto-optical modulator. Images were captured at a depth of 16 bit using HyD and PMT detectors. A z-stack was acquired for each cell with the system-optimized step size. Each image was sampled at the Nyquist frequency. Image acquisition was performed using the LAS-AF software (Leica).
Live cell imaging
MEFs used in zyxin-FHL chimera studies (Figure 3) as well as U2OS cells used in the LIM-protein binding kinetics studies (Figure S2) were imaged on Spinning Disk System 1 as described above. Time-lapse confocal images were captured at an interval of 10 s with the cells incubated in DMEM (10% FBS, 25 mM HEPES pH 7.2, 30 U/mL oxyrase) for MEFs or McCoy’s 5a (10% FBS, 25 mM HEPES pH 7.2, 30 U/mL oxyrase) for U2OS cells without phenol red at 37 °C.
MEFs used for visualizing nuclear localization dynamics of FHL2 in the presence of actin disruption (Figure 7C, Video S6) were imaged on an Andor spinning disk confocal on a Nikon TiE microscope using a Plan Apo 60X oil immersion objective (NA 1.40). Illumination was provided by 405-nm, 488-nm, and 568-nm lasers, switched by an acousto-optic tunable filter-based laser combiner (Andor), and delivered to the spinning-disk confocal scan head (CSU-10, Yokogawa). Images were captured at a depth of 8 bit every minute using an iXon885 EMCCD camera (Andor). Image acquisition was performed using the Andor IQ imaging software (Andor). Cells were incubated in DMEM/F12 (15 mM HEPES) without phenol red at 37 °C. 250 nM cytochalasin D was added at 10 minutes and washed out 60 minutes after the time-lapse imaging started. Cells were imaged for another 2 hours following washout.
MEFs used for visualizing nuclear localization dynamics of FHL2 in the presence of myosin inhibition (Figure S7A, Video S6) were imaged on a Leica SP8 laser scanning confocal microscope using an HC Plan Apo 63X oil immersion objective (NA 1.40). Illumination was provided by a white light laser coupled to an acousto-optical beam splitter. Cells were incubated in DMEM/F12 (15 mM HEPES) without phenol red at 37 °C. The environment was controlled by an OkoLab water jacket stage-top incubator. Images were acquired at a depth of 8 bit every minute using the Leica LASX software (Leica). 5 μM Y-27632 was added at 10 minutes and washed out 100 minutes after the time-lapse imaging started. Cells were imaged for another 2 hours following washout.
In vitro protein studies
Protein expression and purification
The cells were transfected when cell density reached 1.8×106 cells/mL. For 400 mL of cell culture, 400 μg of plasmid was premixed with 1.2 mL of 1 mg/mL PEI MAX (Polysciences) in 15 mL of FreeStyle 293 expression medium and incubated at room temperature for 20 minutes before transfection. Myosin Va and myosin VI were co-transfected with calmodulin at a mass ratio of 1:6. Cells were grown on an orbital shaker at 37 °C in 8% CO2 then harvested 60 hours after transfection.
All subsequent steps of protein purification were conducted at 4 °C or on ice unless otherwise specified. To purify Halo-tagged wild-type and mutant FHL2, HIC5, and zyxin, cells were resuspended in Lysis Buffer A (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.2% NP40, 3 mM DTT, 2 mM ATP, 1 mM PMSF, 1 μg/mL aprotinin, leupeptin, and pepstatin), and were incubated on a rocker for 40 minutes. Cell lysate was clarified by centrifugation (Sorvall) at 20,000 g for 30 minutes. The supernatant was mixed with NHS-activated Sepharose 4 Fast Flow resin (GE Healthcare) coupled to a GFP nanobody expressed and purified by published protocols (Kirchhofer et al., 2010) and incubated on a rocker for 1.5 hours. The resin was then washed three times with Lysis Buffer A, and incubated with 0.5 mg/mL PreScission protease that was expressed and purified by published protocols (Cordingley et al., 1989) on a rocker overnight to cleave the C-terminal GFP tag. Protein was then eluted with Elution Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.2% NP40, and 3 mM DTT). The eluted protein was sequentially purified by anion exchange chromatography utilizing a Mono Q column (GE Healthcare) followed by size exclusion chromatography on a Superdex 200 increase column (GE Healthcare) in Gel Filtration Buffer (10 mM Tris-HCl pH 8.0, 100 mM NaCl, and 3 mM DTT). Peak fractions were concentrated (Amicon Ultra-4, MWCO 10 kD) to 1–2 mg/mL, snap-frozen in liquid nitrogen, and stored at −80 °C until use.
To purify myosin Va and myosin VI, cells were resuspended in Lysis Buffer B (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, 0.2% CHAPS, 3 mM DTT, 2 mM ATP, 1 mM PMSF, 1 μg/mL aprotinin, leupeptin, and pepstatin), and were incubated on a rocker for 40 minutes. Cell lysate was clarified by centrifugation (Sorvall) at 20,000 g for 30 minutes. The supernatant was mixed with anti-Flag M2 affinity beads (Sigma-Aldrich) and incubated on a rocker for 1.5 hours. The protein-bound beads were washed three times with Myosin Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM MgCl2, and 2 mM ATP). Protein was eluted with Myosin Buffer supplemented with 100 μg/mL Flag peptide (Sigma-Aldrich). The eluent was buffer-exchanged to Storage Buffer (10 mM Tris-HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, and 3 mM DTT) using a concentrator (Amicon Ultra-4, MWCO 50 kD). Myosin Va and myosin VI were snap-frozen in liquid nitrogen and stored at −80 °C until use.
Actin was purified from chicken skeletal muscle as previously described (Pardee and Spudich, 1982), with all steps performed at 4 °C. Briefly, 1 g of chicken skeletal muscle acetone powder was resuspended in 20 mL of G-Ca buffer (2 mM Tris-Cl pH 8.0, 0.5 mM DTT, 0.2 M ATP, 0.01% NaN3, 0.1 mM CaCl2) and mixed by inversion for 30 minutes. The suspension was centrifuged in a Ti70 rotor at 42,500 RPM for 30 minutes. 50 mM KCl and 2 mM MgCl2 was added to the supernatant, which contains actin monomers, to initiate actin polymerization. The actin was allowed to polymerize for 1 hour. 0.8 M KCl was then added to the solution and incubated for 30 minutes to facilitate the dissociation of contaminating factors from actin filaments. The solution was then centrifuged in a Ti70 rotor at 42,500 RPM for 3 hours. The pellet was resuspended in 2 mL of G-Ca buffer and incubated overnight. The mixture was transferred to a Dounce chamber and homogenized for 10–15 passes, sheared through 26G and 30G needles consecutively, then dialyzed in Spectra/Por 1 dialysis tubing (MWCO 6–8 kDa) in 1 L of G-Ca buffer overnight. The actin solution was then sheared through a 30G needle again before dialysis in 1L of fresh G-Ca buffer for another day. It was then centrifuged in a Ti90 rotor at 70,000 RPM for 3 hours. The upper 2/3 of the supernatant was purified by size-exclusion chromatography using a HiLoad 16/600 Superdex 200 column (GE Healthcare). The purified actin was maintained in G-Ca buffer at 4 °C before use.
For SDS-PAGE analysis, 3 μg of each protein was loaded on a NuPAGE 12% Bis-Tris gel (Invitrogen, NP0343BOX) and stained with Coomassie Brillant Blue R-250 (Figure S5B). The gel was run at 140 V for 90 minutes. All protein concentrations were estimated by Bradford colorimetric assay (ThermoFisher, 1856209), calibrated with BSA.
Circular dichroism spectroscopy
Halo-tagged wild-type and mutant FHL3, HIC5, and zyxin were buffer-exchanged into 100 mM NaF (Alfa Aesar, A13019) dissolved in phosphate buffer (pH 7.2) using a concentrator (Amicon Ultra-4, MWCO 10 kDa), and diluted to 0.01–0.1 mg/mL using the same solution. Spectra were collected in an AVIV circular dichroism spectrometer (model 62DS) using a cuvette with an optical path length of 1 mm. Each sample was scanned twice from 180 nm to 260 nm with a step size of 0.5 nm. The average of the two measurements is plotted in Figure S5C.
Force reconstitution assay and TIRF microscopy
No. 1.5 24 X 60 mm glass coverslips (Corning, CLS2980246) were sonicated (Branson 3800) in 100% acetone for 30 minutes, 100% ethanol for 10 minutes, and 2% Hellmanex for 2 hours. They were then rinsed with MilliQ water and incubated in a solution that contains 1 mg/mL mPEG-silane (Laysan bio, MPEG-SIL-5000), 10 mM HCl, and 96% ethanol on a shaker overnight at room temperature. Individual coverslips were sequentially rinsed in ethanol and water, air-dried, and stored under nitrogen in a sealed container at 4 °C before use.
10% rhodamine-labeled F-actin was prepared by co-polymerizing 0.9 μM unlabeled actin monomers and 0.1 μM rhodamine-labeled actin monomers (Cytoskeleton, AR05) in the presence of G-Mg (2 mM Tris-HCl pH 8.0, 0.5 mM DTT, 0.2 mM ATP, 0.01% NaN3, 0.1 mM MgCl2) and KMEI (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole pH 7.0) at room temperature for 1 hour. 20 μg of rhodamine-labeled actin monomer was resuspended in 18 μL of G-Ca buffer and 2 μL of milli-Q water, incubated at 4 °C for at least 1 hour, and clarified by ultracentrifugation at 100,000 rpm in a TLA100 rotor for 20 minutes before use. F-actin was freshly polymerized for each experiment.
Myosin Va-GFP and myosin VI-GFP were premixed and diluted to 0.02 μM and 0.04 μM, respectively, with Motility Buffer (MB: 20 mM MOPS pH 7.4, 5 mM MgCl2, 0.1 mM EGTA, 50 mM KCl, 1 mM DTT). Halo-tagged wild-type and mutant FHL3, HIC5, and zyxin were fluorescently labelled by incubation with Janelia Fluor 646 HaloTag ligand (JF646, Promega, GA1121) at a 1:2 molar ratio on ice for 2 hours. Proteins were freshly labeled for each experiment. Excess dye was removed using Pierce dye removal columns (ThermoFisher, 22858). JF646-labeled proteins were then diluted to 500 nM with Imaging Buffer (MB supplemented with 15 mM glucose, 100 μg/mL glucose oxidase, 20 μg/mL catalase, and 1 μM calmodulin). Proteins were clarified by ultracentrifugation at 60,000 rpm in a TLA100 rotor for 15 minutes at 4 °C immediately before imaging.
Imaging wells were prepared by attaching a CultureWell reusable gasket (GraceBio 103280, diameter 6 mm, well depth 1 mm) to the PEG-coated coverslip. Before imaging, each well was treated with the following sequence: coated with 0.02 μM GFP-myosin Va and 0.08 μM GFP-myosin VI in MB for 2 minutes, blocked with 0.1% polyvinylpyrrolidone (Sigma-Aldrich, PVP10, Mw = 10,000) in MB for 1 minute, coated with 0.8 μM 10% rhodamine-labeled F-actin in MB for 30 s, rinsed with MB, then immersed in 20 μL of imaging buffer without ATP for TIRF microscopy. Sequential solutions were added and removed by pipetting.
The imaging well was mounted on a Nikon Ti-E microscope equipped with an H-TIRF motorized module. Time-lapse dual-color TIRF imaging was then initiated to visualize basal level background, followed by addition of 20 μL of 500 nM JF646-labeled LIM protein in the presence or absence of 1 mM ATP (final concentrations: 0.5 mM ATP, 250 nM JF646-labeled LIM protein) in the “+ Force” and “– Force” conditions, respectively. This ensured that the onset of binding events was captured in the “+ Force” condition. Images were acquired every 2 s through a CFI Apo 60X TIRF oil immersion objective (NA 1.49), a quad filter (Chroma), and an iXon EMCCD camera (Andor) with the Perfect Focus System (Nikon) engaged. Illumination was provided by 561-nm (50 mW) and 640-nm (45 mW) lasers (Agilent) switched by an acousto-optic tunable filter. Image acquisition was performed using the NIS-Elements software (Nikon).
Data analysis
Image analysis was performed with custom Python scripts utilizing functions from the scikit-image package (Walt et al., 2014) unless otherwise specified.
Actin enrichment
To identify mechanoresponder LIM proteins from the screen (Figures 1, S1), actin enrichment of the LIM protein in each cell was computed from a z-stack of epifluorescence images. The maximum intensity projection (MIP) of the eGFP channel was thresholded (li method), binarized, and subsequently applied as a cell mask to the DAPI and phalloidin channels to exclude the interference from the surrounding cells with negligible levels of eGFP-labeled LIM protein expression. To find the slice with the largest fraction of the actin cytoskeleton in focus in the z-stack, the Sobel transform was used on the phalloidin channel to find the edge magnitude (gradient of the raw image). The sharpness of each image in the phalloidin channel is defined as the sum of the edge magnitude normalized by the sum of the pixel intensity of the raw image. eGFP, phalloidin, and DAPI images obtained at the z-height that conveys the maximum sharpness in the phalloidin channel were then used to compute actin enrichment.
The actin and nuclear masks were generated by thresholding and binarizing the phalloidin (mean method) and DAPI (li method) channels, respectively. To partition the contribution of LIM proteins localized in the nucleus, a “clean” actin mask was generated by excluding the nuclear region from the actin mask. A cytosolic mask was generated by excluding the nuclear and actin region from the cell mask. The clean actin mask and cytosolic mask were individually applied to the eGFP image obtained at the same z-height. The actin enrichment was computed as the ratio of the average intensity of eGFP on actin cytoskeleton to that in the cytosol. eGFP images with more than 500 saturated pixels covered by the cell mask were not considered for quantification. The same approach was used to quantify the actin enrichments of combinatorial point mutants (Figure 4D–F).
Cell and actin masks were generated by thresholding and binarizing the MIP of confocal z-stacks of the eGFP (LIM protein, mean method) and phalloidin (actin, li method) channels, respectively. The cytosolic mask was generated by excluding the actin region from the cell mask. The actin enrichment of VASP (Figure 3C) was computed using the same approach described above.
FA enrichment
To find the slice with the largest fraction of paxillin in focus in the z-stack, the Sobel transform was applied on the paxillin channel to find the edge magnitude (gradient of the raw image). The sharpness of each image in the paxillin channel is defined as the sum of the edge magnitude normalized by the sum of the pixel intensity of the raw image. eGFP and paxillin images obtained at the z-height that conveys the maximum sharpness in the paxillin channel were then used to compute FA enrichment. The FA mask was generated by thresholding and binarizing the paxillin channel (triangle method). The FA enrichment was computed as the ratio of the average intensity of eGFP in the FAs to that in the cytosol (Figure S4E).
SF enrichment
To quantify the enrichment of LIM proteins on individual SFs (Figure 2B–D), a polygonal ROI that encompasses the targeted SF and a sub-stack of confocal slices that contain the targeted SF were extracted manually in FIJI (Schindelin et al., 2012). The nuclear region was intentionally avoided in the ROIs. A custom FIJI Macro script was used to generate ROI masks and MIPs of the eGFP (LIM protein) and phalloidin (actin) channels with background subtraction (rolling ball radius, 50 pixels) through batch processing in FIJI. The SF mask was generated by thresholding (yen method) and binarizing the MIP of the phalloidin channel within the ROI mask. The local cytosolic mask was generated by excluding the SF region from the ROI mask. The SF and local cytosolic masks were individually applied to the MIP of the eGFP channel. The SF enrichment was computed as the ratio of the average intensity of eGFP on SF to that in the local cytosol.
Nuclear enrichment
To quantify the actin and nuclear enrichments of eGFP-labeled wild-type and F(1–4)A FHL2 in MEFs plated on substrates of various stiffnesses (Figure 7B), the MIP of the eGFP channel was thresholded (li method), binarized and subsequently applied as a cell mask to the DAPI and phalloidin channels. The confocal slices of the DAPI channel were binarized using a single threshold value, which was obtained by thresholding (isodata method) a 2-dimensional array constructed by concatenating all the slices of the DAPI channel. The area of the nucleus was computed for each slice using the binarized DAPI z-stack. Slices captured at the z-height with the largest nuclear area were used to compute the actin and nuclear enrichments. The actin enrichment was obtained using the same approach described in the initial screen. To quantify the nuclear enrichment, the nuclear mask was dilated, bounded by the cell mask, until the area of the dilated mask is twice that of the original mask. A local cytosolic mask was generated by excluding the actin and nuclear region from the dilated mask. Nuclear enrichment was computed as the ratio of the average intensity of eGFP in the nucleus to that in the local cytosol. eGFP images with more than 500 saturated pixels covered by the cell mask were not considered for quantification.
Live-cell imaging quantification
To quantify the timing of the mechanoaccumulation of LIM proteins relative to zyxin-FusionRed (Figure S2C), flashes were identified in the zyxin-FusionRed channel. ROIs were manually delineated based on each frame in eGFP and zyxin-FusionRed channels. The integral intensities within each ROI throughout the lifetime of the flash were extracted for both channels using FIJI, normalized to the maximum value in the corresponding channel, and plotted against time. The differential between the timing of the peak intensity of eGFP and that of zyxin-FusionRed were extracted from the intensity plots (Figure S2B).
To quantify the actin and nuclear enrichment of FHL2 in the presence of actin disruption (Figure 7C), cell, actin and nuclear masks were generated by thresholding and binarizing the FHL2-eGFP (triangle method), Lifeact-mApple (li method), and Hoechst (isodata method) channels, respectively. Cytosolic masks were generated by excluding the actin and nuclear regions from the cell masks. Actin and nuclear enrichments were computed as described above. The colored scatter plot of nuclear versus actin enrichment was constructed in matplotlib (Hunter, 2007).
To quantify the actin and nuclear enrichment of FHL2 in the presence of ROCK inhibition (Figure S7A), MIPs were constructed from three confocal z-slices. Actin and nuclear enrichments were then computed as described above.
Detecting and tracking LIM protein patches
Images of the LIM-protein channel were sequentially Sobel-transformed, thresholded (triangle method), and binarized. The holes of the resulting rings (regions with large edge magnitude) were filled, producing “patch masks” that potentially represent LIM protein patches. Each candidate patch was labeled and tracked based on the overlap of pixels between patch masks in neighboring frames. Due to the low signal-to-noise ratio of the LIM-protein channel, dim patches in the raw frames tended to be lost during tracking. Therefore, overlap was searched among the previous and future 20 mask frames (ft-20 to ft+20). Based on the overlap, depth first traversal was then implemented to assign masks to patches and extract the entire trajectory of each patch.
Actin masks were generated by thresholding (mean method) and binarizing the actin channel, then were applied to the corresponding patch masks. Patches featuring no pixels which overlap with the actin mask for at least one time point likely represent non-specific binding of protein aggregates to the coverslip and therefore were excluded from further analysis. Patches that persisted to the last frame were also removed, as their lifetimes could not be measured. Furthermore, any patch that persisted for less than 5 frames was considered indistinguishable from noise and was not considered in the analysis (lifetime cutoff, 10 s).
To account for the intrinsic non-uniform illumination of TIRF, the average intensities of each patch throughout its trajectory were normalized by the corresponding average intensities of its local background. Dim patches tended to fluctuate in intensity during the time-lapse, compromising the accuracy of tracking. Therefore, an intensity cutoff was also imposed on the lifetime analysis. Trajectories with a maximum normalized patch intensity of less than 1.2 were considered too dim to be tracked accurately and were excluded from further analysis (normalized intensity cutoff, 1.2).
Quantifying lifetime of LIM-protein patches and half-life bootstrapping
The lifetime of each patch was then computed from the number of frames in which it was detected. The reported average half-lives (Figure 5D) were extracted by linear interpolation on the observed cumulative probability distributions (CPDs) of lifetime. Bootstrapping was performed to estimate the standard deviations of the half-lives. Briefly, the CPD of the lifetime population was estimated by the CPD of the observed lifetimes. Samples were drawn with replacement from the observed CPD. The size of each sample equals the size of the observed data set. Resampling was performed 50,000 times, resulting in 50,000 average half-lives extracted by linear interpolation. The standard deviation of the 50,000 half-lives was reported for each LIM protein (Figure 5D).
The maximum area and maximum normalized intensity of each trajectory were extracted by the regionprops function in scikit-image package. The scatter plots of maximum area and maximum normalized intensity versus lifetime were constructed using matplotlib (Figure S6C).
Quantifying actin-intensity change upon LIM-protein patch unbinding
Patch masks were applied to the actin channel to extract the average actin intensity for each patch. The mean of all the average actin intensities throughout the entire trajectory (f[t0] to f[tn]) of the patch was defined as the actin intensity before patch unbinding (Ibefore unbinding). To calculate the actin intensity after patch unbinding, the last patch mask was applied to the actin channel for the future 20 frames after patch unbinding, resulting in 20 average actin intensities (f[tn+1] to f[tn+20]) covered by the last patch mask. The mean of those 20 extracted average actin intensities was defined as the actin intensity after the patch unbinding (Iafter unbinding). The ratio of the actin intensity before to after patch unbinding was then computed. A ratio greater than 1 indicates a decrease in actin intensity upon patch unbinding. The histogram of the logarithm of the ratio (to ensure unbiased distribution) was plotted for each LIM protein (Figure 6D).
Sequence alignment and molecular graphics
Multiple sequence alignment was performed using Clustal Omega (Sievers and Higgins, 2018) provided by the European Bioinformatics Institute (EMBL-EBI) web server (Madeira et al., 2019) (https://www.ebi.ac.uk/Tools/msa/). The aligned sequences (Figure S4A) were colored in Jalview (Waterhouse et al., 2009) (http://www.jalview.org/). Alignment logos (Figure 4A) were created using the web-based application WebLogo (Crooks et al., 2004) (https://weblogo.berkeley.edu/).
The superimposed ribbon diagrams and space-filling surface representations of FHL2 LIM2 and LIMS1 LIM4 (Figure 4B) were created using UCSF ChimeraX (Goddard et al., 2018) (http://www.rbvi.ucsf.edu/chimerax/).
QUANTIFICATION AND STATISTICAL ANALYSIS
To identify mechanoresponder LIM proteins from the screen, Dunnett’s T3 multiple comparison test after Welch’s ANOVA was performed by comparing the actin enrichments of each LIM protein in unstretched versus stretched cells simultaneously for all the LIM proteins covered in the screen (Figures 1, S1). NS (not significant), p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001.
In the individual SF analysis, Games-Howell’s multiple comparison test after Welch’s ANOVA was performed by comparing the actin enrichments of each LIM protein in unstretched versus stretched cells simultaneously for all the LIM proteins belonging to the same family. Given the large number of SF ROIs extracted (nmin = 28, nmax = 244), outliers were identified by ROUT method (Motulsky and Brown, 2006) using GraphPad Prism. 0 ≤ n ≤ 14 outliers were identified in each data set: for the numbers of data points and outliers in each data set, see Table S1. Statistical tests were performed on the cleaned data sets with outliers removed. NS (not significant), p > 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Unless otherwise noted, all statistical analysis and plotting was performed in GraphPad Prism. The figure legends contain details of other standard statistical analyses which were employed as indicated throughout the study.
Supplementary Material
Video S1. Spinning-disk confocal live-cell imaging of SF strain sites in MEFs co-expressing F-tractin-mApple and the indicated eGFP-labeled constructs. Related to Figure 3. Scale bar, 3 μm. Time is minutes : seconds.
Video S2. TIRF imaging of in vitro force reconstitution assay in the absence (left) and presence (right) of ATP. Related to Figure 5. Scale bar, 10 μm. Time is minutes : seconds.
Video S3. FHL3 dissociates from an actin filament upon filament failure, and rebinds to the filament as load develops. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S4. FHL3 patches bind to the middle and to the terminal segments of actin filaments, and unbind upon filament breakage. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S5. An FHL3 patch elongates along a tensed actin filament, and unbinds upon filament breakage. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S6. Confocal live-cell imaging of a MEF co-expressing LifeAct-mApple and FHL2-eGFP pre-treated with Hoechst to label the nucleus. Related to Figure 7. In the first clip, 250 nM cytochalasin D was added at 10 minutes and washed out 60 minutes after the time-lapse imaging started. In the second clip, 5 μM Y-27632 was added at 10 minutes and washed out 100 minutes after the time-lapse imaging started. Scale bar, 20 μm. Time is minutes : seconds.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-FHL2 | Abcam | Cat# ab202584 |
| Mouse monoclonal anti-paxillin | BD Biosciences | Cat# 610052 |
| Alexa Fluor 488 goat anti-rabbit secondary antibody | ThermoFisher | Cat# A32731 |
| Alexa Fluor 568 goat anti-mouse secondary antibody | ThermoFisher | Cat# A11031 |
| Bacterial Strains | ||
| NEB 5-alpha competent E. coli | NEB | C2987U |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fibronectin | EMD Millipore | Cat# FC010 |
| Alexa Fluor 568 phalloidin | ThermoFisher | Cat# A12380 |
| Alexa Fluor 647 phalloidin | ThermoFisher | Cat# A22287 |
| DAPI | ThermoFisher | Cat# D1306 |
| Hoechst | Invitrogen | Cat# H1399 |
| Y-27632 | Sigma-Aldrich | Cat# 688000 |
| Cytochalasin D | EMD Millipore | Cat# 504776 |
| PF573228 | Sigma-Aldrich | Cat# PZ0117 |
| mPEG-silane | Laysan Bio | Cat# MPEG-SIL-5000 |
| Rhodamine-labeled G-actin (Rabbit skeletal muscle) | Cytoskeleton | Cat# AR05 |
| Janelia Fluor 646 HaloTag ligand | Promega | Cat# GA1121 |
| Polyvinylpyrrolidone | Sigma-Aldrich | Cat# PVP10 |
| Critical Commercial Assays | ||
| QIAprep spin miniprep kit | Qiagen | 27106 |
| PureYield plasmid maxiprep system | Promega | A2392 |
| SE Cell Line 4D-Nucleofector X Kit L | Lonza | V4XC-1024 |
| Experimental Models: Cell Lines | ||
| MEF | Mary Beckerle Lab Huntsman Cancer Institute University of Utah | N/A |
| U2OS | ATCC | HTB-96 |
| FreeStyle 293-F | ThermoFisher | R79007 |
| Oligonucleotides | ||
| DNA primers, see Table S2 | This study | N/A |
| Recombinant DNA | ||
| mApple-F-tractin | Michael Davidson Lab Florida State University | N/A |
| Zyxin-fusionRed | Michael Davidson Lab Florida State University | N/A |
| Myosin Va (AA 1–1091) | Dharmacon | BC172485 |
| Myosin VI (AA 1–1021) | James Sellers Lab NHLBI, NIH | N/A |
| LifeAct-mApple | Mary Beckerle Lab Huntsman Cancer Institute University of Utah | N/A |
| Other plasmid DNAs, see Table S2 | This study | N/A |
| Software and Algorithms | ||
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji |
| ChimeraX | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| JalView | Waterhouse et al., 2009 | https://www.jalview.org/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Custom Python scripts computing actin and nuclear enrichments | This paper | https://github.com/alushinlab/LIM-domain |
| Custom Python scripts for patch detection and analysis | This paper | https://github.com/alushinlab/LIM-domain |
| Other | ||
| 4-well stretch chamber | Strexcell | STB-CH-4W |
| Flexcell UniFlex culture plates | Flexcell | UF-4001P |
| 50-kPa softwell 12G plate | Matrigen | 50-kPa |
| 12-kPa softwell 12G plate | Matrigen | 12-kPa |
| CultureWell reusable gasket | GraceBio | 103280 |
| NuPAGE 12% Bis-Tris gel | Invitrogen | NP0343BOX |
Also see: Table S2. Additional oligonucleotides and recombinant DNAs used in this paper. Related to the Key Resources Table.
Highlights:
Tension recruits many tandem LIM domain proteins to the actin cytoskeleton.
A conserved phenylalanine in each LIM domain is required for this localization.
Mechanoresponsive LIM proteins directly bind F-actin only in the presence of force.
This binding governs the ECM-rigidity dependent nuclear localization of FHL2.
Acknowledgements
We gratefully acknowledge Pinar Gurel (RU) for initial assistance with setting up the TIRF force reconstitution assays. We also thank Laura Hoffman and Chris Jensen (HCI) for the gift of the zyxin−/− MEF cell line, Yasuharu Takagi and James Sellers (NHLBI) for the gift of myosin motor proteins and Luke Lavis (HHMI Janelia) for the gift of JF-646 dye for pilot TIRF studies, Laura Yen (NYSBC) for technical assistance with cloning, Matthew Reynolds (RU) for insightful discussion on statistical analysis, and Jonathan Winkelman, Ronen Zaidel-Bar, and Margaret Gardel for discussion of unpublished data. We acknowledge use of the Bio-imaging Resource Center (RU) for confocal imaging. X.S. is supported by a National Institutes of Health Cancer Cell Biology training grant (CA009673-40) and a Pels Family Foundation Fellowship. R.G. is supported by a H. Li. Memorial Fellowship. L.A., R.C.C, C.M.W., and G.M.A. were supported by the National Heart Lung and Blood Institute’s Division of Intramural Research. This work was additionally supported by grants from the Irma T. Hirschl / Monique Weill-Caulier Trust and the Pew Charitable Trusts to G.M.A, and National Institutes of Health grants to M.B. (RO1GM050877) and G.M.A. (DP5OD017885).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman D. (1990). Practical Statistics for Medical Research (Chapman & Hall; ). [Google Scholar]
- Arimura T, Hayashi T, Matsumoto Y, Shibata H, Hiroi S, Nakamura T, Inagaki N, Hinohara K, Takahashi M, Manatsu S-I, et al. (2007). Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy. Biochemical and Biophysical Research Communications 357, 162–167. [DOI] [PubMed] [Google Scholar]
- Bauer MS, Baumann F, Daday C, Redondo P, Durner E, Jobst MA, Milles LF, Mercadante D, Pippig DA, Gaub HE, et al. (2019). Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. PNAS 116, 6766–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S, and Terentjev EM (2017). Focal Adhesion Kinase: The Reversible Molecular Mechanosensor. Biophysical Journal 112, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Perrotta JA, and Turner CE (1996). Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol 135, 1109–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao CY, Mok SW-F, Cheng VW-S, and Tsui SK-W (2015). The FHL2 regulation in the transcriptional circuitry of human cancers. Gene 572, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LB, and Waterman CM (2015). Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 17, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordingley MG, Register RB, Callahan PL, Garsky VM, and Colonno RJ (1989). Cleavage of small peptides in vitro by human rhinovirus 14 3C protease expressed in Escherichia coli. Journal of Virology 63, 5037–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford AW, Michelsen JW, and Beckerle MC (1992). An interaction between zyxin and alpha-actinin. The Journal of Cell Biology 116, 1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: A Sequence Logo Generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Wang Z, Yan J, Yang X, Liu A, Qiu W, Zhu J, Han J, Zhang H, Lin J, et al. (2009). Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-β–like signaling pathway. J Clin Invest 119, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J-M, Tay FP-L, Swa HL-F, Gunaratne J, Leung T, Burke B, and Manser E. (2016). Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci. Signal. 9, rs4–rs4. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Fernández-Sánchez ME, Barbier S, Whitehead J, Béalle G, Michel A, Latorre-Ossa H, Rey C, Fouassier L, Claperon A, Brullé L, et al. (2015). Mechanical induction of the tumorigenic β-catenin pathway by tumour growth pressure. Nature 523, 92–95. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Cesare DD, and Sassone-Corsi P. (2000). A Family of LIM-Only Transcriptional Coactivators: Tissue-Specific Expression and Selective Activation of CREB and CREM. Molecular and Cellular Biology 20, 8613–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich FW, Reischmann S, Schwalm A, Unger A, Ramanujam D, Münch J, Müller OJ, Hengstenberg C, Galve E, Charron P, et al. (2014). FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res Cardiol 109, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel ML, Shin JH, MacKintosh FC, Mahadevan L, Matsudaira P, and Weitz DA (2004). Elastic Behavior of Cross-Linked and Bundled Actin Networks. Science 304, 1301–1305. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature Methods 12, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Sarangi BR, Deschamps J, Nematbakhsh Y, Callan-Jones A, Margadant F, Mège R-M, Lim CT, Voituriez R, and Ladoux B. (2015). Adaptive rheology and ordering of cell cytoskeleton govern matrix rigidity sensing. Nature Communications 6, 7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Kloeker S, Wang C-LA, Yoshigi M, and Beckerle MC (2006). Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J. Cell Biol. 172, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, and Beckerle MC (2012). Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol. Biol. Cell 23, 1846–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosek M, and Tang JX (2004). Polymer-induced bundling of F actin and the depletion force. Phys. Rev. E 69, 051907. [DOI] [PubMed] [Google Scholar]
- Huber F, Strehle D, and Käs J. (2012). Counterion-induced formation of regular actin bundle networks. Soft Matter 8, 931–936. [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Computing in Science Engineering 9, 90–95. [Google Scholar]
- Jaalouk DE, and Lammerding J. (2009). Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HS, and Greenwood JA (2009). Glycine-rich region regulates cysteine-rich protein 1 binding to actin cytoskeleton. Biochem. Biophys. Res. Commun. 380, 484–488. [DOI] [PubMed] [Google Scholar]
- Kadrmas JL, and Beckerle MC (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol 5, 920–931. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, and Waterman CM (2010). Nanoscale architecture of integrin-based cell adhesions. Nature 468, nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, et al. (2010). Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Krieg M, Arboleda-Estudillo Y, Puech P-H, Käfer J, Graner F, Müller DJ, and Heisenberg C-P (2008). Tensile forces govern germ-layer organization in zebrafish. Nature Cell Biology 10, 429–436. [DOI] [PubMed] [Google Scholar]
- Kron SJ, and Spudich JA (1986). Fluorescent actin filaments move on myosin fixed to a glass surface. PNAS 83, 6272–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J-C, Han X, Hsiao C-T, Yates Iii JR, and Waterman CM (2011). Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nature Cell Biology 13, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Ponten F, and Remedios CG dos (2012). The interactome of LIM domain proteins: The contributions of LIM domain proteins to heart failure and heart development. PROTEOMICS 12, 203–225. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BT, Kleiber K, Wixler V, Raab M, Zimmer B, Kaufmann M, and Strebhardt K. (2007). FHL2 Regulates Cell Cycle-Dependent and Doxorubicin-Induced p21Cip1/Waf1 Expression in Breast Cancer Cells. Cell Cycle 6, 1779–1788. [DOI] [PubMed] [Google Scholar]
- McGough A, Pope B, Chiu W, and Weeds A. (1997). Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J. Cell Biol. 138, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HJ, and Brown RE (2006). Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, and Pollard TD (1998). The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. PNAS 95, 6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa N, Sathe AR, Shivashankar GV, and Sheetz MP (2016). Matrix mechanics controls FHL2 movement to the nucleus to activate p21 expression. PNAS 113, E6813–E6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C-F, Zhou WJ-W, Ng PK-S, Li M-S, Ng Y-K, Lai PB-S, and Tsui SK-W (2011). Characterization of human FHL2 transcript variants and gene expression regulation in hepatocellular carcinoma. Gene 481, 41–47. [DOI] [PubMed] [Google Scholar]
- Pardee JD, and Spudich JA (1982). Purification of muscle actin. Meth. Enzymol. 85 Pt B, 164–181. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Pollard TD (2016). Actin and Actin-Binding Proteins. Cold Spring Harb Perspect Biol 8, a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomiès P, Macalma T, and Beckerle MC (1999). Purification and Characterization of an α-Actininbinding PDZ-LIM Protein That Is Up-regulated during Muscle Differentiation. J. Biol. Chem. 274, 29242–29250. [DOI] [PubMed] [Google Scholar]
- Qian Z, Mao L, Fernald AA, Yu H, Luo R, Jiang Y, Anastasi J, Valk PJ, Delwel R, and Le Beau MM (2009). Enhanced expression of FHL2 leads to abnormal myelopoiesis in vivo. Leukemia 23, 1650–1657. [DOI] [PubMed] [Google Scholar]
- Samson T, Smyth N, Janetzky S, Wendler O, Müller JM, Schüle R, Mark H. von der Mark, von der K, and Wixler V. (2004). The LIM-only Proteins FHL2 and FHL3 Interact with α- and β-Subunits of the Muscle α7β1 Integrin Receptor. J. Biol. Chem. 279, 28641–28652. [DOI] [PubMed] [Google Scholar]
- Schiller HB, and Fässler R. (2013). Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 14, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller HB, Friedel CC, Boulegue C, and Fässler R. (2011). Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Reports 12, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeichel KL, and Beckerle MC (1994). The LIM domain is a modular protein-binding interface. Cell 79, 211–219. [DOI] [PubMed] [Google Scholar]
- Shemiakina II, Ermakova GV, Cranfill PJ, Baird MA, Evans RA, Souslova EA, Staroverov DB, Gorokhovatsky AY, Putintseva EV, Gorodnicheva TV, et al. (2012). A monomeric red fluorescent protein with low cytotoxicity. Nat Commun 3, 1204. [DOI] [PubMed] [Google Scholar]
- Sievers F, and Higgins DG (2018). Clustal Omega for making accurate alignments of many protein sequences. Protein Science 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Gardel ML, Luettjohann L, Waterman CM, and Beckerle MC (2010). A Zyxin-Mediated Mechanism for Actin Stress Fiber Maintenance and Repair. Developmental Cell 19, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Blankman E, Deakin NO, Hoffman LM, Jensen CC, Turner CE, and Beckerle MC (2013). LIM Domains Target Actin Regulators Paxillin and Zyxin to Sites of Stress Fiber Strain. PLOS ONE 8, e69378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Hoffman LM, and Beckerle MC (2014). LIM proteins in actin cytoskeleton mechanoresponse. Trends Cell Biol. 24, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C, Roland J, Boujemaa-Paterski R, Kang H, McCullough BR, Reymann A-C, Guérin C, Martiel J-L, De la Cruz EM, and Blanchoin L. (2011). Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr. Biol. 21, 862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Hagel M, and Turner CE (1999). Characterization of a focal adhesion protein, Hic-5, that shares extensive homology with paxillin. Journal of Cell Science 112, 181–190. [DOI] [PubMed] [Google Scholar]
- Tran TC, Singleton C, Fraley TS, and Greenwood JA (2005). Cysteine-rich protein 1 (CRP1) regulates actin filament bundling. BMC Cell Biol. 6, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, et al. (1997). Calcium sensitization of smooth muscle mediated by a Rhoassociated protein kinase in hypertension. Nature 389, 990–994. [DOI] [PubMed] [Google Scholar]
- Uemura A, Nguyen T-N, Steele AN, and Yamada S. (2011). The LIM domain of zyxin is sufficient for force-induced accumulation of zyxin during cell migration. Biophys. J. 101, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walt S. van der, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, and Yu T. (2014). scikit-image: image processing in Python. PeerJ 2, e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Gilmore TD (2003). Zyxin and paxillin proteins: focal adhesion plaque LIM domain proteins go nuclear. Biochim. Biophys. Acta 1593, 115–120. [DOI] [PubMed] [Google Scholar]
- Watanabe-Nakayama T, Saito M, Machida S, Kishimoto K, Afrin R, and Ikai A. (2013). Requirement of LIM domains for the transient accumulation of paxillin at damaged stress fibres. Biol Open 2, 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, and Barton GJ (2009). Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelman JD, Anderson CA, Suarez C, Kovar DR, and Gardel ML (2020). Evolutionarily diverse LIM domain-containing proteins bind stressed actin filaments through a conserved mechanism. BioRxiv 2020.03.06.980649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, and Friedl P. (2007). Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nature Cell Biology 9, 893–904. [DOI] [PubMed] [Google Scholar]
- Xia H, Winokur ST, Kuo W-L, Altherr MR, and Bredt DS (1997). Actinin-associated LIM Protein: Identification of a Domain Interaction between PDZ and Spectrin-like Repeat Motifs. The Journal of Cell Biology 139, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshigi M, Hoffman LM, Jensen CC, Yost HJ, and Beckerle MC (2005). Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J Cell Biol 171, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, and Geiger B. (2007). Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Aponte-Santamaría C, Sturm S, Bullerjahn JT, Bronowska A, and Gräter F. (2015). Mechanism of Focal Adhesion Kinase Mechanosensing. PLOS Computational Biology 11, e1004593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Spinning-disk confocal live-cell imaging of SF strain sites in MEFs co-expressing F-tractin-mApple and the indicated eGFP-labeled constructs. Related to Figure 3. Scale bar, 3 μm. Time is minutes : seconds.
Video S2. TIRF imaging of in vitro force reconstitution assay in the absence (left) and presence (right) of ATP. Related to Figure 5. Scale bar, 10 μm. Time is minutes : seconds.
Video S3. FHL3 dissociates from an actin filament upon filament failure, and rebinds to the filament as load develops. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S4. FHL3 patches bind to the middle and to the terminal segments of actin filaments, and unbind upon filament breakage. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S5. An FHL3 patch elongates along a tensed actin filament, and unbinds upon filament breakage. Related to Figure 6. Scale bar, 2 μm. Time is minutes : seconds.
Video S6. Confocal live-cell imaging of a MEF co-expressing LifeAct-mApple and FHL2-eGFP pre-treated with Hoechst to label the nucleus. Related to Figure 7. In the first clip, 250 nM cytochalasin D was added at 10 minutes and washed out 60 minutes after the time-lapse imaging started. In the second clip, 5 μM Y-27632 was added at 10 minutes and washed out 100 minutes after the time-lapse imaging started. Scale bar, 20 μm. Time is minutes : seconds.
Data Availability Statement
All custom code used in data analysis is available at: https://github.com/alushinlab/LIM-domain. All data supporting the findings of this study are available from the lead contact upon reasonable request.