

SUMMARY
Transcription factor EB (TFEB) activates lysosomal biogenesis genes in response to environmental cues. Given implications of impaired TFEB signaling and lysosomal dysfunction in metabolic, neurological, and infectious diseases, we aim to systematically identify TFEB-directed circuits by examining transcriptional responses to TFEB subcellular localization and stimulation. We reveal that steady-state nuclear TFEB is sufficient to activate transcription of lysosomal, autophagy, and innate immunity genes, whereas other targets require higher thresholds of stimulation. Furthermore, we identify shared and distinct transcriptional signatures between mTOR inhibition and bacterial autophagy. Using a genome-wide CRISPR library, we find TFEB targets that protect cells from or sensitize cells to lysosomal cell death. BHLHE40 and BHLHE41, genes responsive to high, sustained levels of nuclear TFEB, act in opposition to TFEB upon lysosomal cell death induction. Further investigation identifies genes counter-regulated by TFEB and BHLHE40/41, adding this negative feedback to the current understanding of TFEB regulatory mechanisms.
Graphical Abstract
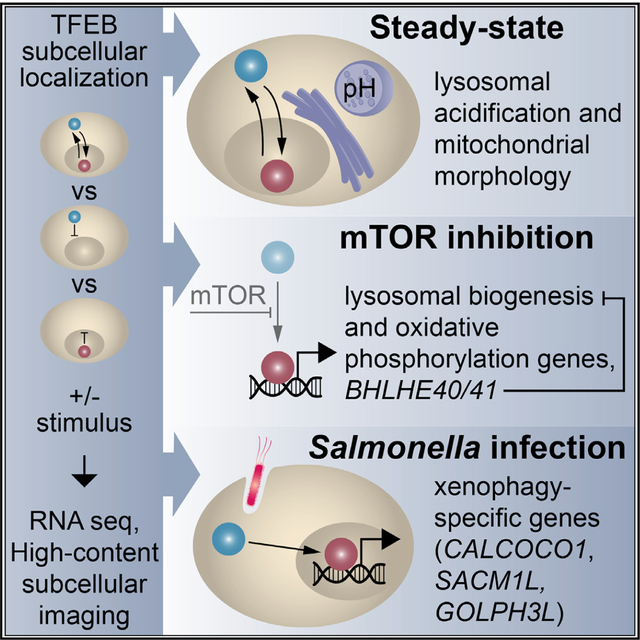
In Brief
Employing RNA sequencing, genome-wide CRISPR screening, and high-content subcellular imaging, Carey et al. systematically unravel localization- and stimulation-specific transcriptional responses to TFEB, including target gene activation at steady state. The authors further uncover a negative feedback loop by BHLHE40 and BHLHE41 that counteracts a TFEB transcriptional signature induced by lysosomal stress.
INTRODUCTION
Lysosomes facilitate recycling of unwanted cytosolic components, including damaged organelles, oxidized lipid aggregates, or pathogens targeted by autophagy (Kuo et al., 2018; Najibi et al., 2016; Visvikis et al., 2014). Digested lysosomal products released by autophagolysosomes satisfy nutritional and energy needs of the cell (Rabinowitz and White, 2010; Singh and Cuervo, 2011). Transcription factor EB (TFEB), a member of the MiT/TFE family of transcription factors expressed widely across cell types, is a master regulator of lysosomal biogenesis, autophagy, and lipid metabolism (Martina et al., 2014; Rusmini et al., 2019; Sardiello et al., 2009; Settembre et al., 2011, 2013; Tan et al., 2019). We previously demonstrated a role for TFEB in maintaining intestinal epithelial-cell-specific functions. Compared with wild type (WT), mice lacking TFEB in the intestinal epithelium were more susceptible to epithelial injury and had reduced expression of antimicrobial peptides required for host defense (Murano et al., 2017). In vivo studies also elucidated functions for TFEB in spatiotemporal control of myelination during central nervous system development and following injury (Goodman et al., 2018; Meireles et al., 2018). Collectively, TFEB activates transcription in response to many physiological signals to maintain cellular homeostasis.
TFEB activity is controlled by its subcellular localization, which is regulated by post-translational modifications, including phosphorylation (Martina et al., 2012; Martina and Puertollano, 2018; Napolitano et al., 2018; Puertollano et al., 2018; Roczniak-Ferguson et al., 2012; Settembre et al., 2011, 2012). In nutrient-rich conditions, mTOR phosphorylates TFEB at S142 and S211, promoting the interaction between TFEB and 14-3-3 proteins that shield its nuclear localization signal (NLS) (Martina et al., 2012; Napolitano et al., 2018; Puertollano et al., 2018; Roczniak-Ferguson et al., 2012; Settembre et al., 2012; Xu et al., 2019). Upon cellular stress signals, such as nutrient deprivation, inhibition of the amino acid sensing mTOR complex 1 (mTORC1) results in accumulation of TFEB dephosphorylated at S142 and S211, which dissociates from 14-3-3 proteins, translocates into the nucleus, and activates its transcriptional program (Martina et al., 2012; Roczniak-Ferguson et al., 2012; Xu et al., 2019). The TFEB dephosphorylated state is also achieved through inhibition of mTOR activity by small molecules such as Torin (Martina and Puertollano, 2018; Napolitano et al., 2018). Studies have further described regulation of TFEB activity in response to glucose deprivation and bacterial pathogens through AMP-activated protein kinase (AMPK) (Eichner et al., 2019; El-Houjeiri et al., 2019; Visvikis et al., 2014).
Additional regulation of TFEB activity occurs through nuclear export. Reports have identified an evolutionarily conserved nuclear export signal and demonstrated that TFEB continuously shuttles between the cytosol and the nucleus at steady state (Li et al., 2018; Napolitano et al., 2018; Silvestrini et al., 2018). Treatment with Torin blocked this shuttling event, indicating that movement of TFEB both in and out of the nucleus may be modulated by nutrient availability in an mTOR-dependent manner (Li et al., 2018; Napolitano et al., 2018). How different stimuli regulate TFEB-dependent gene signatures and what mechanisms govern the magnitude and duration of the transcriptional response remain unknown.
The cellular processes governed by TFEB are complex and require coordinated protein expression; thus, a systematic understanding of how TFEB and its targets are regulated at steady state and in response to stimuli is necessary. In this study, we employed RNA sequencing and a genome-wide CRISPR screen to study TFEB-dependent target genes in response to genetic manipulations and exogenous stimuli. We discovered that a subset of TFEB target genes is activated at steady state, whereas others are stimulation dependent. Further investigation into how TFEB targets affect cellular survival in response to lysosomal stress revealed that BHLHE40 and BHLHE41 counteracted the TFEB response. Here, we demonstrate that these two genes are upregulated in response to stimulus-dependent TFEB activation as part of a negative feedback loop that counter-regulates select TFEB targets involved in lysosomal function.
RESULTS
Engineered Cell Lines Demonstrate Transcriptional Response to TFEB Localization
To study transcriptional responses to TFEB maintained in the nucleus or cytosol, we generated a clonal TFEB-knockout (KO) HeLa cell line using the CRISPR-Cas9 system. TFEB deletion was confirmed by confocal microscopy and immunoblot (Figures 1A and 1B). TFEB was re-expressed in the KO cell line by reconstitution with one of the following constructs: WT TFEB (TFEB-WT); cytosol-restricted TFEB (TFEB-cyto), generated by replacing basic residues in the NLS with alanines (Roczniak-Ferguson et al., 2012); nuclear-restricted TFEB (TFEB-nuc), generated by removing the first 30 amino acids of the N- terminus, which reduces lysosomal targeting and increases nuclear localization in the absence of a stimuli (Roczniak-Ferguson et al., 2012); or a vector control (TFEB-KO). Confocal microscopy established that TFEB-WT and TFEB-cyto constructs were expressed at comparable levels to endogenous TFEB in WT HeLa cells and confirmed expected subcellular localization of TFEB-cyto and TFEB-nuc constructs at steady state (Figure 1A). Upon treatment with Torin, TFEB translocated into the nucleus in TFEB-WT cells, as in WT HeLa cells, whereas TFEB-cyto and TFEB-nuc cells remained in the cytosol and nucleus, respectively (Figure 1A).
Figure 1. Engineered Cell Lines Demonstrate Transcriptional Response to TFEB Localization.
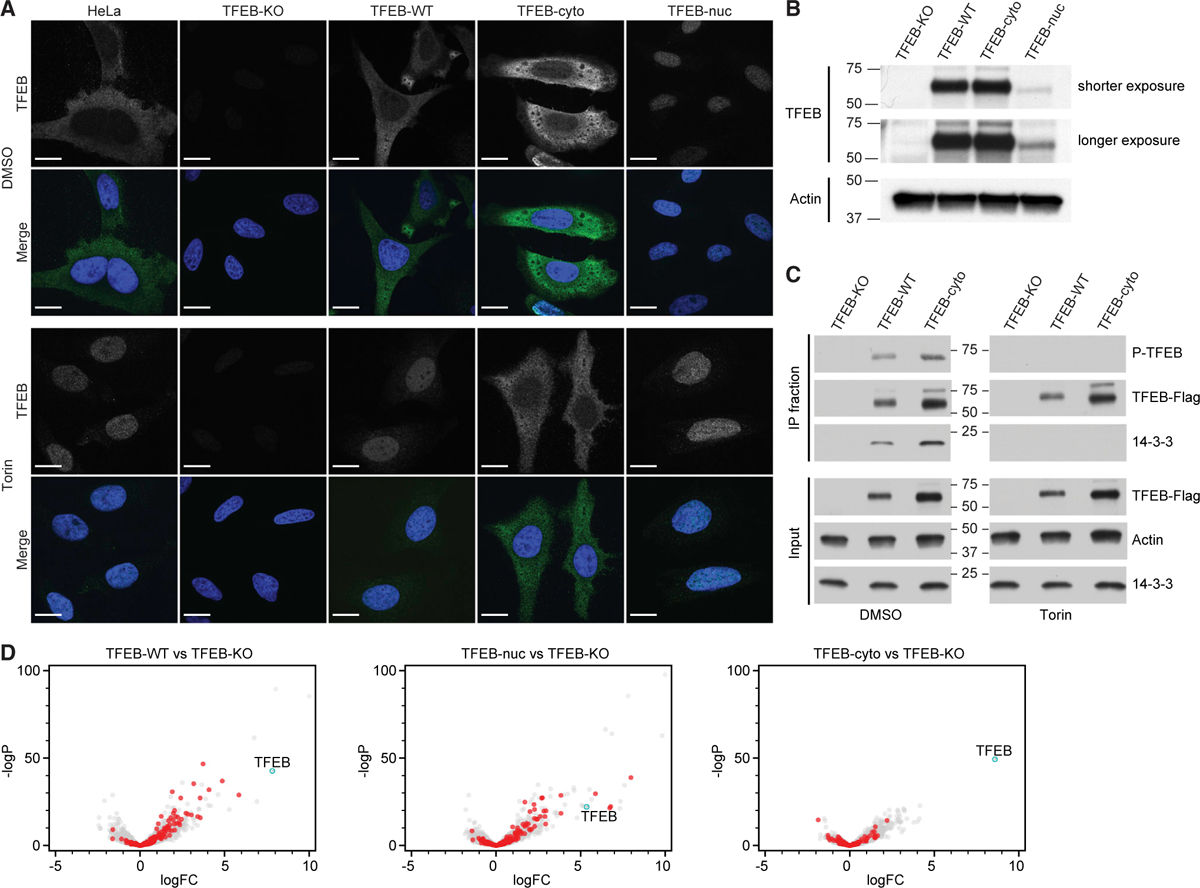
(A) Subcellular localization of TFEB in HeLa and TFEB-knockout HeLa cells reconstituted with empty vector (TFEB-KO), WT TFEB (TFEB-WT), cytosol-restricted TFEB (TFEB-cyto), or nuclear-restricted TFEB (TFEB-nuc) following treatment with DMSO or Torin. Representative confocal microscopy images show TFEB detected with anti-TFEB antibody (green in merged channels; white in single channels) and DNA with Hoechst (blue). Scale bars represent 5 mm.
(B) Representative anti-TFEB immunoblot demonstrates TFEB protein is not detected in TFEB-KO cells. TFEB-WT and TFEB-cyto are expressed at similar levels, whereas detectable TFEB-nuc expression is lower. Actin acts as a loading control.
(C) TFEB-cyto maintains functional interactions in the cytosol. Immunoprecipitation of recombinant Strep/FLAG-tagged TFEB from HeLa cells expressing TFEB-KO, TFEB-WT, or TFEB-cyto following DMSO or Torin treatment. Both TFEB-WT and TFEB-cyto are phosphorylated (P-TFEB) and interact with 14-3-3 proteins before Torin treatment. Upon Torin treatment, TFEB-WT and TFEB-cyto are not detected with the phosphoantibody and do not interact with 14-3-3 proteins. Actin acts as a loading control.
(D) Cells were processed for RNA sequencing. The TFEB transcript level (cyan) is significantly increased in TFEB-WT, TFEB-nuc, and TFEB-cyto cells relative to TFEB-KO cells. Shown in red is a subset of known TFEB target genes (Table S1: Sardiello et al., 2009), many of which have an increased log fold change (logFC) in TFEB-WT and TFEB-nuc relative to TFEB-KO cells. Most genes in TFEB-cyto cells are not significantly upregulated relative to TFEB-KO cells. See also Figure S1.
Next, we examined TFEB-cyto phosphorylation and interactions with 14-3-3 proteins to verify that mutagenesis of the NLS did not affect other functional domains. Immunoprecipitations of TFEB-WT and TFEB-cyto constructs at steady state confirmed TFEB phosphorylation and interactions with 14-3-3 proteins, whereas neither TFEB nor 14-3-3 protein interactions were detected in the TFEB-KO immunoprecipitated fraction (Figure 1C). Furthermore, the TFEB-cyto construct was no longer detected in the phosphorylated state or interacting with 14-3-3 proteins upon Torin treatment. These data demonstrate that the TFEB-cyto construct maintains functional protein-protein interactions.
To investigate cellular processes that require TFEB nuclear translocation and transcriptional activation, we evaluated LC3 processing as a measure of autophagy initiation. By immunoblot, the ratio of membrane-bound LC3-II to cytosolic LC3-I, which corresponds to autophagosome formation, was similar in TFEB-KO and reconstituted cell lines at steady state, whereas TFEB-WT and TFEB-nuc cells responded to Torin or a combination of Torin and autolysosomal inhibitors E64d/Pepstatin A more robustly than TFEB-KO or TFEB-cyto cells, indicating the reconstituted cell lines behave as expected in a cellular process (Figures S1A and S1B; Settembre et al., 2011).
Previous reports using microarray and chromatin immunoprecipitation sequencing (ChIP-seq) analyses demonstrated that overexpressing epitope-tagged TFEB, in addition to endogenous TFEB, in WT HeLa cells activated target gene transcription (Palmieri et al., 2011; Sardiello et al., 2009; Settembre et al., 2011). Although these identified TFEB-responsive genes, it remains unclear which are controlled by TFEB in the absence of cellular stimulation or stress. We used RNA sequencing to evaluate transcriptional effects of TFEB expression and localization in reconstituted KO cells. Increases in the log fold change (logFC) of the TFEB transcript detected in TFEB-WT, TFEB-nuc, and TFEB-cyto cells relative to TFEB-KO cells served as an internal RNA sequencing control for each cell line (Figure 1D). No steady-state transcriptional changes were detected in other MiT/TFE family members, suggesting there was no compensation in the reconstituted cells. Differential gene expression data demonstrated a robust TFEB-dependent induction of genes, including known targets (Table S1; Sardiello et al., 2009) in TFEB-WT and TFEB-nuc cells and a negligible transcriptional response in TFEB-cyto cells (Figure 1D). Relative to TFEB-WT cells, no significant transcriptional responses were observed in TFEB-nuc cells at steady state, whereas TFEB-cyto cells failed to activate gene transcription (Figure S1C).
TFEB at Steady State Induces a Transcriptional Response that Is Amplified by Sustained Nuclear Localization
To directly compare transcriptional responses between reconstituted cell lines, we calculated the relative expression for all differentially expressed genes at steady state (Figure 2; STAR Methods). The overall transcriptional response of TFEB-nuc cells closely resembled that of TFEB-WT cells, whereas the response of TFEB-cyto cells clustered more closely with TFEB-KO cells (Figure 2A). The absence of a global transcriptional upregulation in TFEB-cyto cells is consistent with the requirement of nuclear localization for TFEB activity. Our data also revealed a subset of genes upregulated in TFEB-cyto relative to TFEB-KO cells, suggesting activation through an indirect mechanism (Figure 2). Importantly, the transcriptional response observed in TFEB-WT relative to TFEB-KO cells indicates the steady-state level of nuclear-localized TFEB is sufficient to induce transcriptional upregulation of TFEB-dependent genes (Figures 1D and 2).
Figure 2. TFEB at Steady State Induces a Transcriptional Response that Is Amplified by Sustained Nuclear Localization.
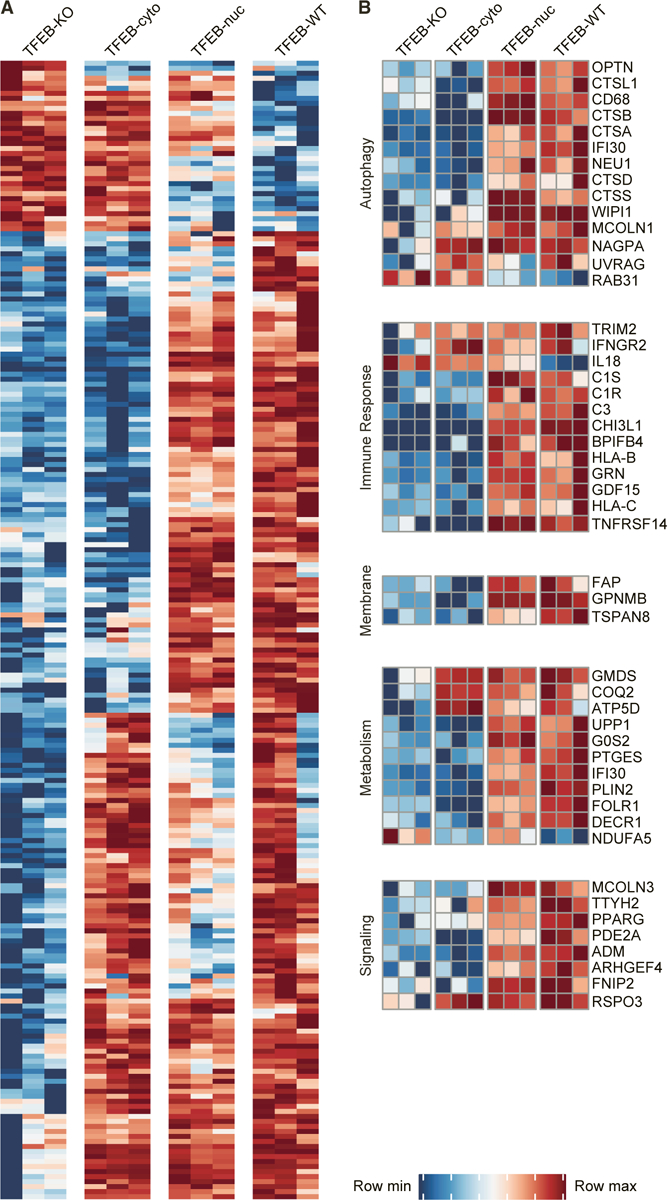
(A) Relative gene expression (CPM-transformed measurements) for genes differentially expressed between TFEB-KO and TFEB-WT cells with fold change (FC) > 1.5 or FC < −1.5 at steady state. Data from three biological replicates for each cell line are shown. For each gene, rows were scaled such that their minimum expression value was 0 and their maximum expression value was 1, and only genes with a relative expression value > 0.7 or a relative expression value < 0.3 in at least 8 of the 12 RNA sequencing samples are shown. Full list in Table S2.
(B) Select genes upregulated in TFEB-WT and TFEB-nuc cells at steady state. See also Figure S2 and Tables S1, S2, and S3.
Among the differentially expressed genes, we detected upregulation of known targets in TFEB-WT and TFEB-nuc cells (Figure 2B; Tables S1 and S2), including genes functioning in lysosomal and autophagy pathways, such as lysosomal enzymes CTSA, CTSB, CTSD, CTSS, and NEU1; WIPI1, a regulator of autophagosome formation; and OPTN, an autophagy adaptor protein (Sardiello et al., 2009). Genes required for cellular metabolism and homeostasis were also selectively upregulated in TFEB-WT and TFEB-nuc cells, such as G0S2, a key regulator of lipid metabolism; IFI30, an interferon g-inducible thiol reductase involved in antigen presentation; and FOLR1, a folate receptor localized to endosomes (Hastings and Cresswell, 2011; Singh et al., 2008; Ward et al., 2016; Wibowo et al., 2013; Zhang et al., 2017). In addition, we identified upregulation of innate immune response genes, including C1S and C3 complement components; CD68, a lysosomal/endosomal-associated transmembrane and lectin binding protein; and GRN, which has reported roles in signaling and inflammatory response (Nguyen et al., 2018; Sorbara et al., 2018; Tanaka et al., 2013; Zhou et al., 2015). GPNMB, a membrane glycoprotein with recently described anti-inflammatory and neuroprotective functions, was among the top differentially expressed genes in TFEB-WT and TFEB-nuc cells at steady state (Budge et al., 2018; Neal et al., 2018; van der Lienden et al., 2018). Using quantitative PCR (qPCR), we validated expression patterns observed by RNA sequencing for selected genes (CTSD, SQSTM1, MCOLN1, IL33, FAP, GPNMB, IFI30, FOLR1, and G0S2). The highest transcript levels for most genes were observed in TFEB-nuc cells, and higher levels of transcription were detected in TFEB-WT cells than in TFEB-cyto or TFEB-KO cells for all genes (Figure S2). Collectively, these data support the role of TFEB in transcriptional regulation of metabolic processes and highlight its importance in sustaining innate immune responses at steady state.
Gene Ontology (GO) analyses of TFEB upregulated genes (logFC > ln2 and q < 0.05) supported our observations that genes classified as functioning in immune system processes and regulation of inflammatory responses were enriched in TFEB-nuc cells (Bonferroni adjusted p < 0.05; Table S3). Similar genes were enriched in TFEB-WT cells but were not statistically significant (Table S3). No GO enrichment was observed in TFEB-cyto cells (Table S3). These data support our findings that nuclear levels in TFEB-WT cells at steady state are sufficient to activate transcription and sustained nuclear localization in TFEB-nuc cells increases this response.
TFEB Expression and Localization Phenotypically Alter Lysosomal and Mitochondrial Compartments
High-content subcellular imaging has been used to characterize responses to genetic or chemical perturbations. Here, we used Cell Painting as an unbiased, image-based profiling approach to detect phenotypic changes of organelles in response to TFEB localization (Bray et al., 2016). In addition to the fluorescent markers used previously in Cell Painting studies to detect DNA, RNA, endoplasmic reticulum (ER), mitochondria, actin, and Golgi and plasma membrane, we included LysoTracker to image effects on lysosomal compartments as a positive control. From captured images, microscopic features, including intensity, radial distribution, granularity, texture, size, and shape of the subcellular structures, were measured and analyzed with CellProfiler to perform illumination correction, quality control, and measurement extraction (Bray et al., 2016).
Similar to transcriptional clustering, the top two principal components of the Cell Painting data illustrated TFEB-cyto and TFEB-nuc cells morphologically resembled TFEB-KO and TFEB-WT cells, respectively, at steady state (Figure 3A). Clustering by genotype remained consistent following treatment with Torin, indicating that reconstitution of TFEB in KO cells had a greater effect on subcellular morphology than treatment with the exogenous stimulus (Figure 3A). Interestingly, TFEB- cyto cells displayed the largest difference between DMSO and Torin treatments, which may be a result of increased dephosphorylated TFEB in the cytosol in response to Torin relative to DMSO treatment (Figures 1C and 3A). Furthermore, using Morpheus software analysis to depict the microscopic features that distinguished TFEB-KO and TFEB-cyto cells from TFEB-WT and TFEB-nuc cells, we observed that 27 of the top 40 differential features by t test analysis were detected in lysosomal or mitochondrial imaging channels (Figure S3). Studies previously detected changes to lysosomal and mitochondrial morphology and positioning (Mansueto et al., 2017; Sardiello et al., 2009; Willett et al., 2017), as well as lysosomal content (Abu-Remaileh et al., 2017), following TFEB stimulation.
Figure 3. TFEB Expression and Localization Phenotypically Alter Lysosomal and Mitochondrial Compartments.
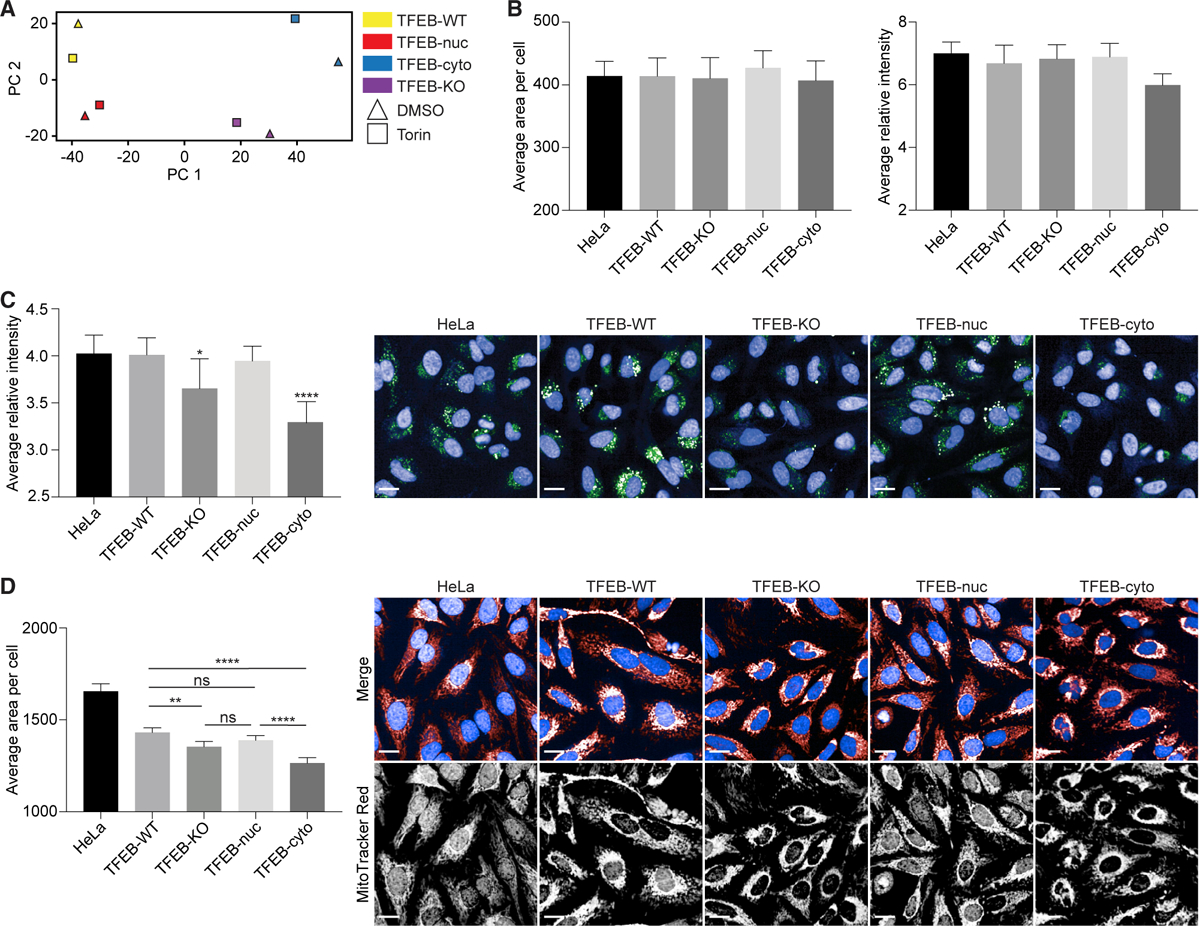
(A) Principal-component analysis (PCA) of Cell Painting subcellular features from TFEB cell lines illustrates that phenotypically TFEB-cyto and TFEB-nuc cells cluster with TFEB-KO and TFEB-WT cells, respectively, following DMSO or Torin treatment.
(B) Quantification of average lysosomal area per cell and relative fluorescence intensity by LysoView staining showed no significant differences among TFEB-KO, reconstituted cell lines, and WT HeLa cells relative to TFEB-WT.
(C) Quantification of average relative intensity of DQ-BSA. Significance values shown are relative to TFEB-WT. In representative confocal images, DNA is detected with Hoechst (blue) and acidified lysosomal compartments by DQ-BSA (green).
(D) Quantification of average mitochondrial area per cell as detected by MitoTracker Red. Significance values are for comparisons as shown. In representative confocal images, DNA is detected with Hoechst (blue in merged channels) and mitochondria with MitoTracker Red (red in merged channels; white in single channels).
For all quantifications, data from at least two independent experiments and at least 500 cells per experiment were analyzed using ordinary one-way ANOVA and Tukey’s multiple comparison test with single pooled variance. Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001. ns, not significant. Scale bars represent 5 mm. See also Figure S3.
To support the Cell Painting data, we assessed lysosomal size and acidification by imaging TFEB-KO and reconstituted cell lines at steady state with LysoView and DQ-BSA markers. Analyses identified no significant difference in lysosomal size (Figure 3B) but did detect decreased lysosomal acidification in TFEB-KO and TFEB-cyto cells compared with TFEB-WT, TFEB-nuc, and WT HeLa cells (Figure 3C). In addition, using MitoTracker Red, we confirmed differences in mitochondrial area per cell between TFEB cell lines at steady state. Compared with TFEB-WT cells, mitochondrial area was reduced in TFEB-KO and TFEB-cyto cells (Figure 3D). Furthermore, representative confocal images of cells stained with MitoTracker Red illustrate that mitochondria in TFEB-KO and TFEB-cyto cells are concentrated in the perinuclear region rather than distributed throughout the cell (Deus et al., 2020). Our data indicate that steady-state levels of nuclear TFEB activate transcripts capable of inducing detectable phenotypic changes to lysosomal and mitochondrial compartments compared with cells lacking nuclear TFEB.
TFEB Target Genes Are Differentially Sensitive to Nuclear TFEB
Our approach using TFEB-KO cells reconstituted with TFEB-WT or TFEB-nuc enabled an evaluation of the global TFEB-dependent transcriptional response at steady state and after mTOR inhibition. We hypothesized that treatment of TFEB-WT cells with Torin would induce transcription of genes not detected at steady state, because increased nuclear translocation in response to exogenous stimuli would increase accessibility or binding of TFEB to the promoter region of its target genes. In contrast, we predicted there would be no significant transcriptional response in TFEB-nuc cells following Torin treatment, because we showed TFEB is restricted to the nucleus (Figure 1A).
We compared RNA sequencing data from TFEB-WT or TFEB-nuc cells relative to TFEB-KO cells at steady state and following Torin treatment. At steady state, TFEB-WT and TFEB-nuc cells activated transcription, including expected target genes (Figure 1D; Tables S1 and S4), whereas Torin treatment predominantly increased the magnitude of expression in TFEB-WT cells but had no significant transcriptional effect on TFEB-nuc cells (Figures 4A and 4B; Table S4). A direct comparison of differential gene expression between TFEB-WT and TFEB-nuc cells confirmed TFEB-WT cells upregulated these genes in response to Torin, whereas TFEB-nuc cells did not (Figure S4). Transcriptional responses in TFEB-WT and TFEB-nuc cells were comparable at steady state (Figures S1C and S4B–S4D). A subset of genes was only upregulated in Torin-treated TFEB-WT cells and was not transcribed at detectable levels in untreated TFEB-WT cells or in TFEB-nuc cells (Figures 4C and 4D; Table S4). Genes responsive to high, sustained levels of nuclear TFEB induced by Torin treatment included CTSF, NPC2, BLOC1S3, and BLOC1S2, which function in lysosomal degradation, transport, and biogenesis; NDUFS4, NDUFA13, NDUFA8, NDUFA1, NDUFB10, and NDUFAF2, subunits of mitochondrial NADH dehydrogenase; PPARG and PPARGC1A, a nuclear re ceptor and co-factor regulating lipid metabolism; and BHLHE40 and BHLHE41, two transcriptional repressors (Figures 4B and 4D; Table S4).
Figure 4. TFEB Target Genes Are Differentially Sensitive to Nuclear TFEB.
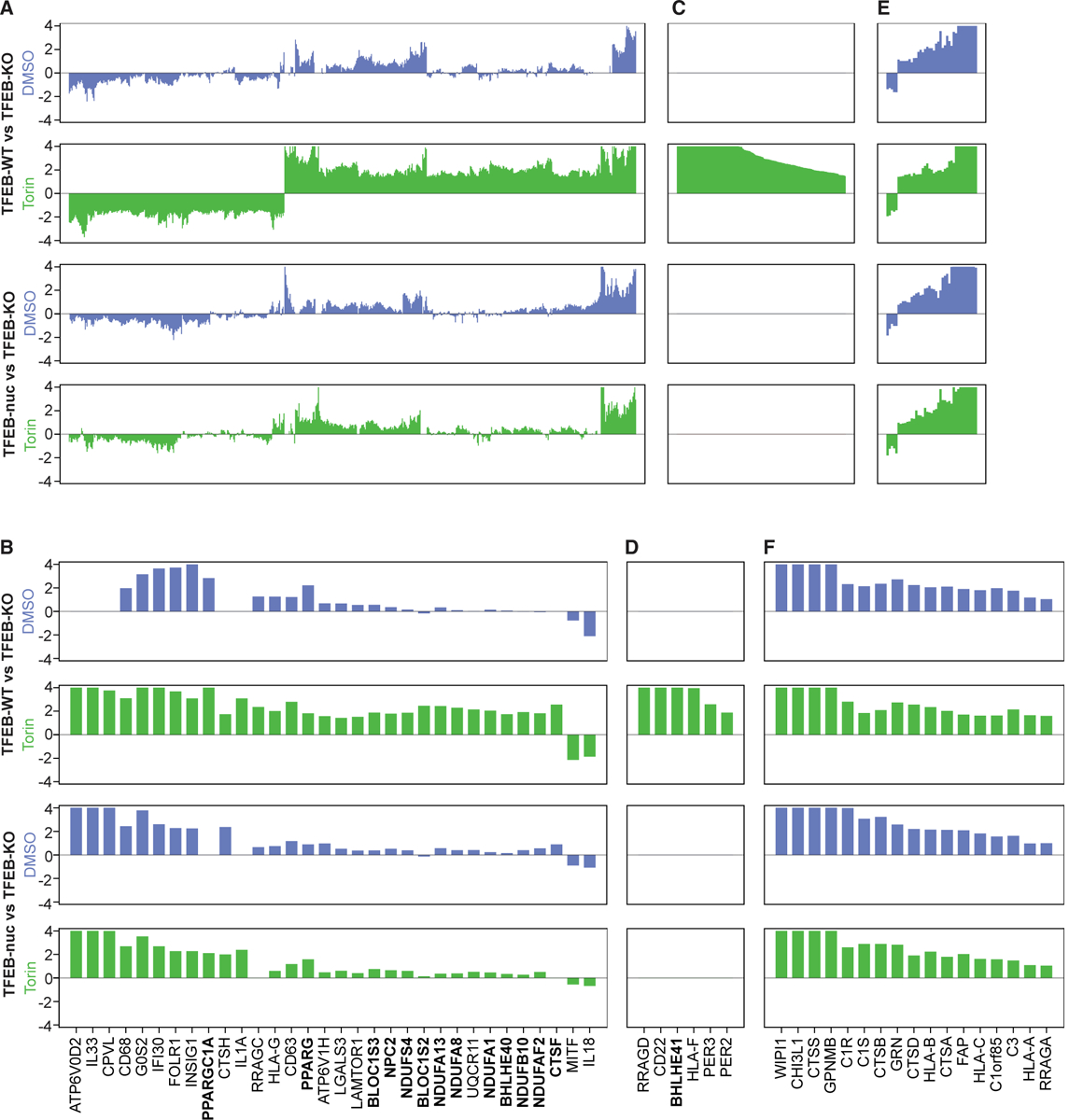
TFEB-KO, TFEB-WT, and TFEB-nuc cells were treated with Torin or DMSO and then processed for RNA sequencing analysis. Panels show differential gene expression from steady state (DMSO, blue) and Torin-treated (green) TFEB-WT versus TFEB-KO cells and TFEB-nuc versus TFEB-KO cells. Each bar corresponds to a gene, and the y axis represents logFC of differential gene expression (truncated logFC ± ln4). Genes represented in the bar plots are all genes (A, C, and E) or select genes (B, D, and F) with significant differential expression (logFC > ln4 or logFC < −ln4 and q < 0.01) in TFEB-WT relative to TFEB-KO cells following Torin treatment. For each differential expression comparison, (A) and (B) represent genes significantly upregulated with Torin treatment, (C) and (D) represents genes significantly upregulated with Torin treatment yet not transcribed at detectable levels without Torin stimulation, and (E) and (F) represent TFEB-dependent genes for which transcription did not significantly change in response to Torin stimulation. Genes in bold indicate those highlighted in Results. See also Figure S4 and Table S4.
A subset of TFEB-dependent genes did not respond to Torin stimulation (Figures 4E and 4F; Table S4), including lysosomal proteases (CTSA, CTSB, CTSD, and CTSS), lysosomal membrane proteins (C1orf85), stress response/tissue repair proteins (FAP and GRN), and complement components (C1S, C1R, and C3). Upregulation of these genes in TFEB-WT and TFEB-nuc cells irrespective of Torin stimulation suggests that low levels of nuclear TFEB are sufficient to maximally induce their transcription.
TFEB Transcriptional Signature Is Modulated by Different Exogenous Stimuli
Our data show that intracellular Salmonella defense requires autophagy and lysosomal pathways, induction of which activates a TFEB transcriptional response (Kuo et al., 2018; Ravenhill et al., 2019; Verlhac et al., 2015; Wang et al., 2018; Figure S5A). To address whether different stimuli activate unique TFEB transcriptional circuits, we examined responses in TFEB-KO and TFEB-WT cells infected with Salmonella enterica serovar Typhimurium. Similar to Torin-treated cells (Figures 4 and 5A), infected TFEB-WT cells induced a TFEB-dependent transcriptional response that included many known targets (Figures 5A, S5B, and S5C; Table S1; Sardiello et al., 2009). By comparing stimulus-dependent differential expression patterns, we identified genes that (1) displayed a greater magnitude of transcriptional response following Torin treatment than following Salmonella infection (e.g., CTSF, BLOC1S2, BLOC1S3, and LGALS3); (2) shared a similar magnitude of transcriptional response following either Torin treatment or Salmonella infection (e.g., CTSS, WIPI1, C1R, C1S, and C3); or (3) were differentially expressed in either Torin-treated or Salmonella-infected cells (e.g., BHLHE40, BHLHE41, and PER2) (Figures 5B, 5C, S5B, and S5C).
Figure 5. TFEB Transcriptional Signature Is Modulated by Different Exogenous Stimuli.
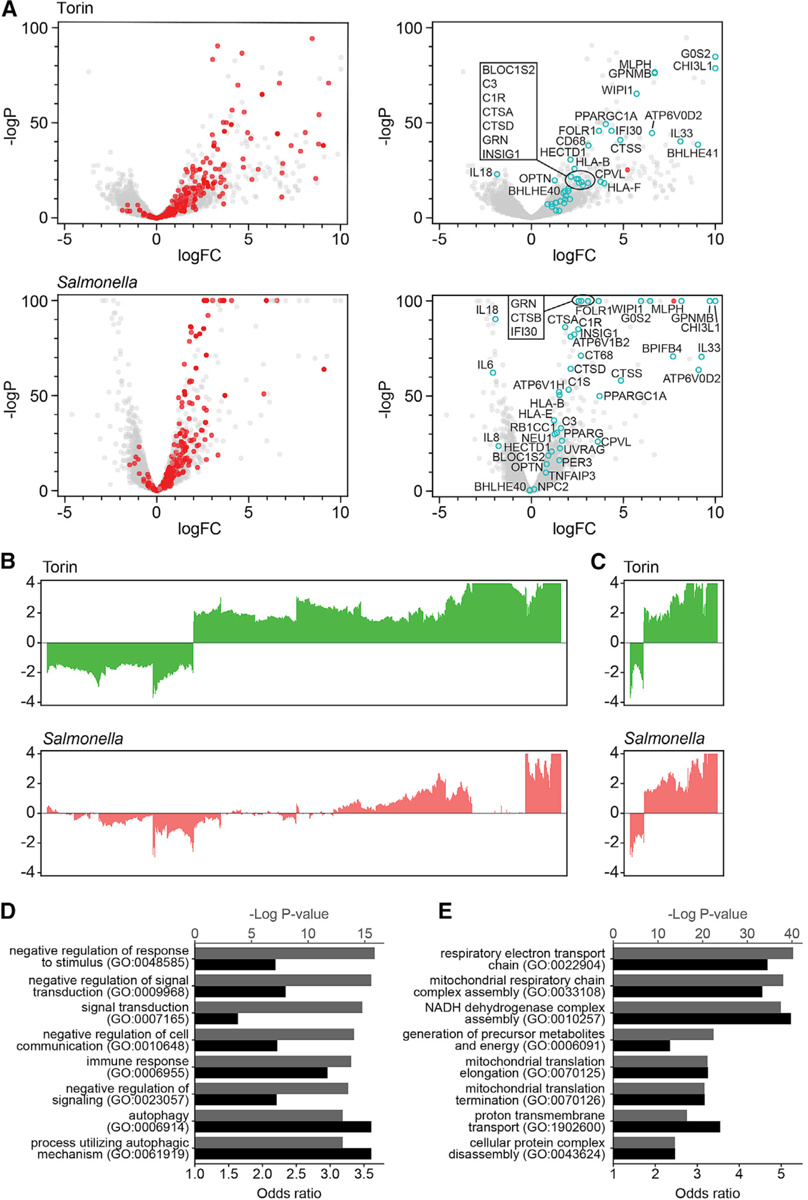
(A) Volcano plots illustrate differential gene expression from TFEB-KO and TFEB-WT cell lines treated with Torin (upper) or infected with S. enterica (lower). Genes shown in red in left panels are previously reported TFEB target genes (Table S1: Sardiello et al., 2009), many of which are significantly upregulated in response to both Torin and intracellular bacteria based on logP and logFC. Select genes functioning in autophagy, lysosomal, and immune responses are highlighted in cyan in the right panels. Compared with TFEB-KO cells, TFEB transcript level is significantly increased in response to stimuli, as denoted in red in the right panels.
(B) Comparison of TFEB-dependent transcriptional profiles in response to Torin and S. enterica infection. Genes shown are differentially expressed in TFEB-WT versus TFEB-KO cells in response to Torin treatment (logFC > ln4 or logFC < −ln4 and q < 0.01).
(C) Bar plots of genes differentially expressed in response to both Torin treatment and Salmonella infection (logFC > ln4 or logFC < −ln4 and q < 0.01).
(D) Gene Ontology enrichment analysis of genes upregulated in response to both Torin and Salmonella infection (logFC > ln2 and q < 0.05). Black bars represent −log p values, and gray bars represent odds ratios.
(E) Gene Ontology enrichment analysis of genes upregulated in response to Torin (logFC > ln2 and q < 0.05), but not in response to Salmonella (logFC < ln1.5 and q > 0.9). Black bars represent −log p values, and gray bars represent odds ratios. See also Figures 4 and S5 and Tables S1 and S5.
TFEB-dependent genes upregulated at steady state were identified among those maximally expressed following both Torin treatment and Salmonella infection, confirming that these genes require low levels of nuclear TFEB for transcription (Figures 4, 5B, 5C, S5B, and S5C). Based on GO analyses, these genes were enriched in immune response and autophagy pathways (Figures 5D and S5D; Table S5). Enrichment of autophagy genes following Torin treatment and bacterial infection was expected, because both mTOR inhibition and intracellular pathogens are known to induce a TFEB-dependent autophagy response (Li et al., 2018; Murano et al., 2017; Najibi et al., 2016; Napolitano et al., 2018; Roczniak-Ferguson et al., 2012). We also detected Salmonella-specific upregulated genes, including genes related to autophagy and lysosomes (CALCOCO1, TBK1, DRAM1, GNS, HPS3, HPS4, and HPS4), innate immune response (BFIFB4, IL1B, IL24, and IL6R), and phosphoinositide lipid signaling and vesicular trafficking (MTM1, SACM1L, ANKFY1, GOLPH3L, WDFY1, WDFY3, and COPB2) (Table S5). These data provide the first evidence linking xenophagy with upregulation of TFEB target genes. When investigating TFEB- and Torin-dependent genes that did not respond to Salmonella infection by GO analyses, we identified enrichment in genes functioning in mitochondrial processes (Figures 5E, S5B, and S5D; Table S5), including glutaredoxins and subunits of the NADH:ubiquinone oxidoreductase, cytochrome c oxidase, and ubiquinol-cytochrome c reductase complexes. These findings suggest that transcriptional regulation of a subset of targets depends on the pathway by which TFEB is activated (Figures 5 and S5; Table S5). Notably, BHLHE40 and BHLHE41 were upregulated only in response to Torin treatment, indicating a response to the strength or nature of the stimulus (Figures 5A and S5B).
TFEB Protects and BHLHE40 and BHLHE41 Sensitize Cells to Lysosomal Cell Death
We next sought to determine how TFEB target genes directly influence lysosomal functions. Traditionally thought to function predominantly in cellular degradative processes, lysosomes are viewed as intracellular hubs integrating signals required for both catabolic and anabolic pathways (Lawrence and Zoncu, 2019; Perera and Zoncu, 2016). To identify genes involved in lysosomal functions or regulation, we used L-leucyl-L-leucine methyl ester (LLME) in a genome-wide CRISPR-Cas9 screen. LLME induces lysosomal membrane permeabilization and subsequent cell death by the release of lysosomal enzymes (Repnik et al., 2017; Thiele and Lipsky, 1990). We reasoned that CRISPR-mediated disruption of genes such as cathepsin C (CTSC), a lysosomal protease critical for LLME-induced cell death, would protect cells from LLME treatment, whereas disruption of lysosomal biogenesis or cellular homeostasis genes would sensitize cells (Brojatsch et al., 2015; Jacobson et al., 2013). Because of their high levels of lysosomal activity and sensitivity to LLME treatment (Jin et al., 2018), BV2 microglial cells stably expressing Cas9 were transduced with a pooled CRISPR library, followed by treatment with LLME or a mock control. Sequencing data for both positively (protective) and negatively (sensitized) enriched guide RNAs were deconvolved, and STARS score rankings and false discovery rates (FDRs) were determined (Table S6; Doench et al., 2016). Validating our results, CTSC was the top-ranked positively enriched gene (Figure 6A; Table S6). TFEB was negatively enriched, suggesting that its role in lysosomal biogenesis is required to protect cells from LLME-induced cell death (Figure 6A; Table S6).
Figure 6. TFEB Protects and BHLHE40 and BHLHE41 Sensitize Cells to Lysosomal Cell Death.
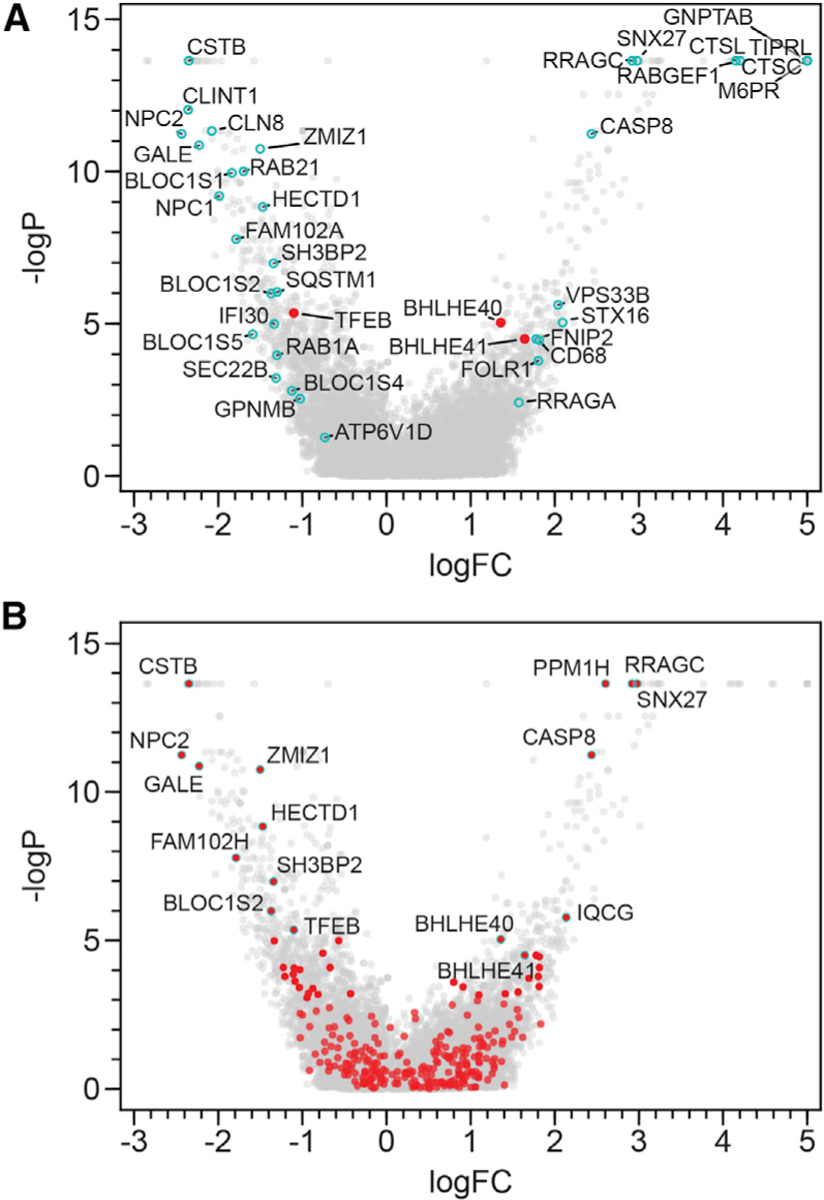
Visualization of STARS analysis from a genome-wide CRISPR screen in BV2 cells identifying genes that sensitize cells to (negative logFC) or protect cells from (positive logFC) LLME treatment.
(A) TFEB, BHLHE40, and BHLHE41 (red) and select genes of interest (cyan).
(B) Data from the genome-wide CRISPR screen are shown, with genes in red representing all those found by RNA sequencing to be significantly upregulated in TFEB-WT versus TFEB-KO cells following Torin treatment (logFC > ln4 and q < 0.01). Genes of particular interest are outlined with cyan. See also Table S6.
To pinpoint additional TFEB-dependent genes functioning in lysosomal biology, we compared the list of genes upregulated in a TFEB- and Torin-dependent manner with the output from the genome-wide CRISPR screen. Several genes were negatively enriched, including ones with higher STARS rankings than TFEB: CSTB, NPC2, GALE, ZMIZ1, HECTD1, FAM102A, SH3BP2, and BLOC1S2 (Figure 6B; Table S6). Additional components of the biogenesis of lysosomal organelle complex 1—BLOC1S1, BLOC1S5, and Snapin, which are thought to initiate lysosomal biogenesis (Lee et al., 2012; Luzio et al., 2014)—were among the top 5% of negatively enriched genes by STARS ranking (Table S6). These data indicate that to survive the stress of lysosomal damage inflicted by LLME treatment, cells respond by translocating TFEB into the nucleus and initiating lysosomal biogenesis pathways. TFEB target genes (BHLHE40, BHLHE41, RRAGC, CASP8, SNX27, PPM1H, and IQCG) were also among the top 10% of positively enriched genes, indicating that deletion of these genes protects cells from LLME-induced cell death (Figure 6B; Table S6). Although caspase-8 depletion may prevent caspase-dependent cell death upon LLME treatment, the protective roles that other TFEB targets play require further investigation.
BHLHE40 and BHLHE41 Repress Expression of Select TFEB Target Genes through a Negative Feedback Loop
Considering that BHLHE40 and BHLHE41 (1) are TFEB target genes (Palmieri et al., 2011), (2) bind to the same consensus E box motif as TFEB (Chung et al., 2015; Kanda et al., 2016; Nakashima et al., 2008; Nishiyama et al., 2012), (3) are only upregulated in response to high levels of nuclear TFEB (Figures 4B and 4D; Table S4), and (4) act in opposition to TFEB in response to lysosomal damage (Figure 6; Table S6), we hypothesized that BHLHE40 and BHLHE41 function as counter-regulators of TFEB target genes through competitive DNA binding in a negative feedback loop. To test this, we used the CRISPR-Cas9 system to generate a BHLHE40/41 double-knockout (dKO) HeLa cell line, which we reconstituted with empty vector (BHLHE40/41-dKO) or with WT BHLHE40 and BHLHE41 (BHLHE40/41-WT). Deletion and reconstitution of the BHLHE40/41 genes were confirmed by DNA sequencing and immunoblot (Figure S6A). Increases in logFC of BHLHE40 and BHLHE41 transcripts in BHLHE40/41-WT relative to BHLHE40/41-dKO cells were observed and served as internal controls for the RNA sequencing data (Figure S6B). Volcano plots representing BHLHE40/41-dependent gene regulation revealed that BHLHE40/41 both induced and repressed gene transcription (Figure 7A)—in contrast to TFEB, which predominantly induced gene transcription (Figure 1D). In addition, the analysis demonstrated that TFEB- and Torin-dependent upregulated genes were both up- and downregulated in response to BHLHE40/41 reconstitution (Figure 7A).
Figure 7. BHLHE40 and BHLHE41 Repress Expression of Select TFEB Target Genes through a Negative Feedback Loop.
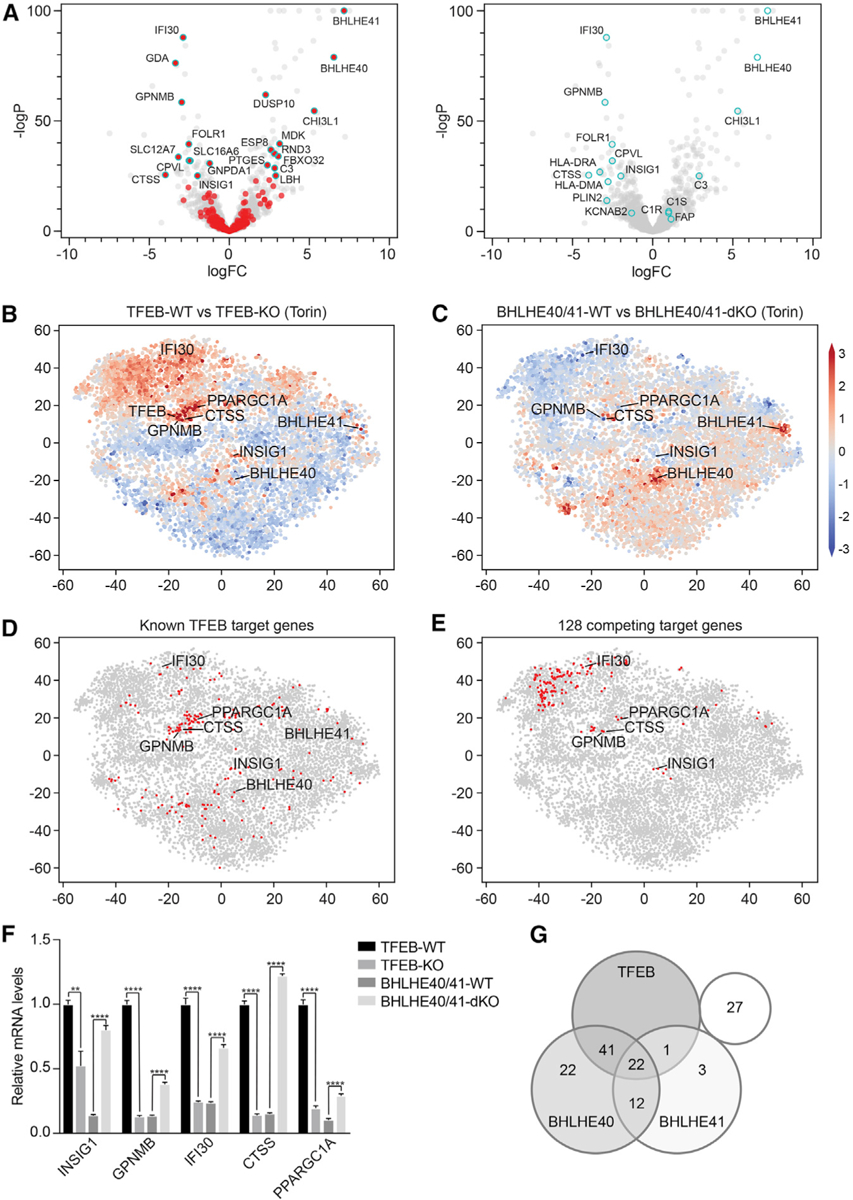
(A) Volcano plots depicting differential gene expression in BHLHE40/41-WT versus BHLHE40/41-KO cells reconstituted cells at steady state. Highlighted in red (left panel) are genes significantly upregulated in TFEB-WT versus TFEB-KO cells following Torin treatment (logFC > ln4 and q < 0.01). Highlighted in cyan (right panel) are select TFEB target genes.
(B–E) tSNE plots representing all RNA sequencing datasets in our study. Comparison of RNA sequencing datasets from (B) TFEB-WT and TFEB-KO cells or (C) BHLHE40/41-WT and BHLHE40/41-dKO cells treated with Torin, in which each gene (dot) is colored by the logFC. (D) Known TFEB target genes or (E) strongest putative TFEB and BHLHE40/41 competing target genes are highlighted in red.
(F) Gene expression of select TFEB and BHLHE40/41 target genes in TFEB-KO, TFEB-WT, BHLHE40/41-dKO, and BHLHE40/41-WT cells as quantified by qRT-PCR. By two-tailed t test analyses, all genes were significantly upregulated in cells reconstituted with TFEB and downregulated in those reconstituted with BHLHE40/41. Data are representative from three independent experiments (mean ± SEM [standard error of the mean]). **p < 0.006, ****p < 0.0001.
(G) Venn diagram represents the number of the 128 strongest putative competing target genes (logFC > ln2 and < −ln2 in TFEB- and BHLHE40/41-WT versus TFEB- and BHLHE40/41-KO cells, respectively, and q < 0.05 in both) bound by TFEB, BHLHE40, and/or BHLHE41 based on the published ChIP-seq datasets GEO: GSM2354032, GSE106000, GSM2797493, and GSM2461743. See also Figure S6 and Tables S7 and S8.
Next, to identify genes upregulated by TFEB and downregulated by BHLHE40/41 in an unbiased manner, we dissected different branches of TFEB-dependent response pathways that are preferentially activated upon different cellular conditions and external stimuli using an unsupervised gene clustering method (t-stochastic neighborhood embedding [tSNE]) on all RNA sequencing datasets in our study. Validating this approach, we successfully recovered known target genes that clustered around TFEB (Figure 7B; Table S1; Sardiello et al., 2009). Moreover, we observed a large cluster of genes that were upregulated in TFEB-WT cells and trended toward downregulation in BHLHE40/41-WT cells following Torin treatment, including genes that we identified as sensitive to low levels of nuclear TFEB (CTSS, PPARGC1A, IFI30, and GPNMB) (Figures 4, 7B–7D, S5B, and S5C). Limiting our analysis to the strongest putative competing targets from all genes (logFC > ln2 and logFC < −ln2 in TFEB- and BHLHE40/41-WT versus TFEB- and BHLHE40/41-KO cells, respectively, and q < 0.05 in both) (Figure 7E; Table S7), we searched for their overlap with known motifs in published ChIP-seq datasets with HOMER (Heinz et al., 2010) and found predominantly basic-helix-loop-helix (bHLH) transcription factor binding site enrichment in their promoter regions. Controlling the false discovery only among bHLH transcription factor binding sites highlighted overenrichment of TFE3/TFEB binding motifs (Palmieri et al., 2011; Sardiello et al., 2009) and the overlapping BHLHE40 and BHLHE41 binding motifs (Table S8). Published TFEB, BHLHE40, and BHLHE41 ChIP-seq datasets provided additional evidence that these transcription factors are able to bind promoter regions of competing target genes in cellular contexts (GEO: GSM2354032, Doronzo et al., 2019; GEO: GSE106000, ENCODE Project Consortium, 2012; GEO: GSM2797493 and GSM2461743, Kreslavsky et al., 2017; Table S8).
To confirm the RNA sequencing data and analyses, several of the genes we identified as most significantly upregulated by TFEB and downregulated by BHLHE40/41 were examined by qPCR: transcript levels of CTSS, IFI30, INSIG1, GPNMB, and PPARGC1A were elevated in TFEB-expressing cells and reduced in BHLHE40/41-expressing cells (Figure 7F). These data validate previously reported TFEB, BHLHE40, and BHLHE41 target genes from ChIP-seq analyses (ENCODE Project Consortium, 2012; Doronzo et al., 2019; Kreslavsky et al., 2017) and identify putative common targets that may be cell type or stimulation specific (Figure 7G; Table S8). Altogether, our data provide evidence for a BHLHE40/41-dependent negative feedback loop not previously described that counter-regulates select TFEB target genes.
DISCUSSION
As a master regulator of essential cellular processes, TFEB affects a range of lysosomal storage, metabolic, neurodegenerative, and cardiac diseases. Here, we used RNA sequencing and genome-wide CRISPR approaches to discover TFEB localization- and stimulus-specific responses. Clustering target genes based on responsiveness to nuclear-localized TFEB and evaluating their effects in response to lysosomal membrane permeabilization revealed a BHLHE40/41-dependent counter-regulatory mechanism capable of downregulating a subset of TFEB targets.
By comparing transcriptional signatures at steady state or after stimulation, we observed that most TFEB-dependent genes were upregulated in response to mTOR inhibition, including known and previously unknown targets, such as mitochondrial respiratory chain complex subunits. These observations indicated that most TFEB targets are induced in response to increased cellular demand and demonstrated that the level of nuclear TFEB titrates the magnitude of transcription, providing an effective mechanism by which cells tightly control responses to cellular stimuli (Kribelbauer et al., 2019). In contrast, we observed that select components of the complement, lysosomal, and autophagy pathways were transcriptionally upregulated at steady state in TFEB-WT cells. The magnitude of expression of these genes did not change in response to nuclear-localized TFEB or Torin treatment, suggesting that their enhancer motif is highly accessible and requires low levels of nuclear TFEB for activation. Furthermore, deficiencies in the lysosomal protease and classical complement component genes are associated with lysosomal storage, neurodegenerative, and autoimmune diseases (Butler et al., 2019; Lintner et al., 2016; Macedo and Isaac, 2016; Marques et al., 2020; Nakajima et al., 2019; Prada et al., 2014; Tang et al., 2006). Thus, cells likely require a constant level of transcription of these innate immune response genes to maintain homeostasis or rapidly respond to exogenous stimuli. Our data illustrate that TFEB target genes are regulated on multiple levels to differentiate their responsiveness to the intensity and duration of stimulation.
Little is known about how different stimuli or cellular contexts affect TFEB-dependent transcriptional signatures. We investigated TFEB response to mTOR inhibition and bacterial infection. Although both stimuli induced upregulation of genes in TFEB-WT cells, GO analyses identified common and stimulation-specific transcriptional signatures. This unbiased analysis identified shared upregulation of genes classified as functioning in autophagic processes. Both Torin treatment and Salmonella infection induced TFEB-dependent transcriptional regulation of signal transduction and immune response genes, such as tumor necrosis factor alpha (TNF-α)-induced proteins (TNFAIP3 and TNFAIP6), complement components (C1S, C1R, and C3), and interleukin-1 (IL-1) cytokine family members (IL1a, IL1R, and IL33). We also discovered autophagy, lysosomal, and membrane trafficking genes (CALCOCO1, TBK1, DRAM1, GNS, HPS3, HPS4, HPS4, MTM1, SACM1L, ANKFY1, GOLPH3L, WDFY1, WDFY3, and COPB2) that were specifically upre gulated in response to Salmonella infection, which provides new insights into host innate response to restrict bacteria and maintain cellular homeostasis through xenophagy (El-Houjeiri et al., 2019; Murano et al., 2017; Visvikis et al., 2014).
The predominant difference observed between stimulation-dependent transcriptional signatures was the activation of mitochondrial genes functioning in oxidative respiration following Torin treatment. The overwhelming number of genes encoded subunits for mitochondrial complexes I, III, and IV of the electron transport chain. These data suggested that Torin-induced TFEB translocation stimulates transcriptional activation of genes required for mitochondrial oxidative phosphorylation to maintain cellular ATP levels, in addition to stimulating mitochondrial biogenesis through peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) induction (Mansueto et al., 2017). TFEB-dependent activation of oxidative phosphorylation genes further establishes a role for TFEB in mitochondrial energy production and metabolic homeostasis (Mansueto et al., 2017). Deficiencies in mitochondrial complex genes have been linked to immune-mediated T cell activation and differentiation (Baixauli et al., 2015; Brady et al., 2018; Mansueto et al., 2017; Nabar and Kehrl, 2017), as well as classical mitochondrial diseases, cancer, and neurodegenerative diseases (Fernández-Mosquera et al., 2017; Lynch et al., 2019). Intracellular bacterial infection is also expected to alter cellular metabolism (Cornejo et al., 2017; Eisenreich et al., 2019). Our data may highlight a cellular context-dependent difference, whereby a nutritional stress response results in intense and rapid TFEB-dependent transcriptional activation. Alternatively, a low steady-state level of transcripts may be sufficient to maintain cellular homeostasis until later time points during an infection as the intracellular bacterial burden increases. Therefore, it is possible that the selective upregulation of mitochondrial oxidative phosphorylation genes could depend on the stimulus, its intensity, or a combination of these factors.
Finally, we discovered evidence suggesting that BHLHE40 and BHLHE41 counteract TFEB transcriptional activation. First, these two genes are significantly upregulated upon mTOR inhibition, suggesting that neither gene is required at steady state. We also noted that neither BHLHE40 nor BHLHE41 was differentially expressed following Salmonella infection, which may be a consequence of the pathway by which TFEB is activated or of the intensity of stimulation. Second, TFEB protects and BHLHE40/41 sensitizes cells to LLME-induced cell death. Third, BHLHE40/41-dKO cells showed transcriptional effects opposite to those of TFEB-KO cells. Specifically, transcript levels of select genes were higher in TFEB-WT cells lacking BHLHE40/41 and lower in BHLHE40/41-WT cells lacking TFEB. These data suggest a negative feedback mechanism, whereby BHLHE40 and BHLHE41 transcription is upregulated in response to stimulus-dependent TFEB activation and subsequently represses transcription of select TFEB target genes that influence lysosomal function.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ramnik J. Xavier (xavier@molbio.mgh.harvard.edu).
Materials availability
Materials generated in this study will be provided upon request.
Data and code availability
The accession number for the RNA sequencing data reported in this paper is database of Genotypes and Phenotypes (dbGaP) HeLa Cell Genome Sequencing Studies: phs002099 and listed in Table S2.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HeLa cells were cultured in Iscove’s Modified Dulbecco’s Medium supplemented with GlutaMAX, 10% Fetal Bovine Serum and 15 μg/ml gentamicin. TFEB-KO HeLa cells were generated by targeting exons 1, 4 and 5 of the coding region with TFEB-sgRNA 20-nucleotide guide sequences in the lentiCRISPR v2 backbone (Ran et al., 2013) and transducing low-passage HeLa cells. Two days post-transduction, cells were placed under selection with 2 μg/ml of puromycin, and 4-days post-transduction, single cell clones were generated by limiting dilution in 96-well plates. Similarly, double knockouts of BHLHE40 and BHLHE41 were generated by simultaneously targeting exon 1 of BHLHE40 and exon 3 of BHLHE41 with sgRNA guides. Clones were screened for successful knockout via western blot and Sanger sequencing.
Vector construction
To complement the knockout cells, lentivirus was made based on N-terminal Flag-StrepII-tagged CSGW-T2A-blasticidin (TFEB and BHLHE41; GFP-LC3-CSGW backbone was a generous gift from Dr. Christian Münz, University of Zürich) or N-terminal CSGW-T2A-puromycin (BHLHE40) backbones. TFEB cDNA was sub-cloned out of EGFP-N1-TFEB (Roczniak-Ferguson et al., 2012). The TFEB nuclear localized mutant (TFEB-nuc) was made by deleting the first 30 amino acids of the protein sequence containing the lysosomal targeting sequence (Roczniak-Ferguson et al., 2012), and the TFEB cytoplasmic mutant (TFEB-cyto) was generated by mutating basic residues found within the predicted nuclear localization signal (R245-R248) to alanine residues, as previously described (Roczniak-Ferguson et al., 2012). BHLHE40 and BHLHE41 were subcloned from OriGene’s RC210294L1 and RC206882L1 plasmids into the CSGW backbones. In addition to reconstituting the BHLHE40/41 double knockout cell line with BHLHE40 and BHLHE41, the double knockout and reconstituted lines were transduced with TFEB-WT and corresponding empty vector to induce higher levels of TFEB induced transcription. After reconstitution with empty vector, wild-type or mutant cDNA, cells were cultured under selection (2 μg/ml puromycin and/or 5 μg/ml blasticidin). When cells were plated for experiments, they were plated in the absence of antibiotics (bacterial or mammalian).
Bacterial strains
Salmonella enterica serovar Typhimurium strain SL1344 expressing the Photorhabdus luminescens lux operon (Xen26) (Conway et al., 2013) and S. Typhimurium SL1344 DsRed2 (Rioux et al., 2007) were grown on Luria-Bertani (LB) agar or in LB media containing 30 μg/ml kanamycin.
METHOD DETAILS
Immunofluorescence microscopy
Cells were seeded onto 18mm glass coverslips in 12-well plates. The following day, cells were treated with DMSO or 2 μM Torin-1 for 3 h prior to processing for microscopy. Briefly, cells were washed with phosphate buffered saline (PBS) then fixed for 15min at RT with PBS containing 4% (v/v) paraformaldehyde. After fixation, cells were washed with PBS, blocked and permeabilized for 1 h with PBS containing 5% (v/v) goat serum and 0.1% (v/v) Triton X-100 then incubated overnight at 4°C in PBS containing 5% (v/v) goat serum and mouse anti-FLAG antibody (1/1000). The following day, cells were washed with PBS, incubated for 1 h at RT with goat anti-mouse secondary antibody then washed and mounted on glass slides. Confocal images were captured with an Andor Zyla 4.2 plus digital camera at 40× using a Nikon Ti2-E inverted microscope with W1 spinning disk confocal and Nikon NIS-Elements. For LysoView 488, DQ-BSA or MitoTracker Red, cells were stained as per manufacturer’s instructions, and live confocal images were captured using a Perkin Elmer Opera Phenix system equipped with a high NA 20x air objective and Perkin Elmer Harmony High-Content Imaging and Analysis Software.
Immunoprecipitation
TFEB knockout HeLa cells reconstituted with empty vector (TFEB-KO), TFEB-WT or TFEB-cyto were seeded into 10cm dishes. The following day, cells were treated with DMSO or 2 μM Torin-1 for 2 h prior to scraping cells and lysing on ice for 30min. Lysate was centrifuged for 15min at 4°C. From the supernatant, an input sample was collected, treated with 6× sample buffer, boiled, and stored at −20°C. To immunoprecipitate TFEB, the remainder of the supernatant was transferred to a tube containing pre-washed Strep-Tactin Sepharose resin and incubated on a rotator for 2 h at 4°C. Resin was washed three times with lysis buffer before boiling in 1x sample buffer. The input and immunoprecipitated samples were separated by SDS-Page, transferred to PVDF and detected by immunoblot with antibodies recognizing the Phospho-(ser) 14-3-3 binding motif (1/1000), anti-FLAG M2 epitope (1/1000) and Pan 14-3-3 (1/100). β-actin served as a loading control (1/1000).
LC3 turnover
TFEB knockout HeLa cells reconstituted with empty vector (TFEB-KO), TFEB-WT, TFEB-cyto, or TFEB-nuc were seeded in 24-well plates at 6 ×104 cells/well. The following day, cells were treated with 2 μM Torin-1 ± 10 μg/mL E64d/Pepstatin A for 6 h at 37°C. Cells were washed and lysed with RIPA buffer containing protease inhibitor cocktail on ice for 30min. Prior to SDS-PAGE, protein concentrations were determined by BCA assay. Equal amounts of protein samples were loaded and separated by 4%–20% bis-tris SDS-PAGE gel, transferred onto PVDF membranes and detected by immunoblot with rabbit anti-LC3B (1/1000). β-actin to serve as a loading control.
RNA sequencing sample preparation
TFEB and BHLHE40/41 cell lines were seeded in 12-well plates at 1.5×105 cells/well or 24-well plates at 7.5×104 cells/well, respectively. The following day, cells were treated with DMSO (6 h), 2 μTFEB knockout HeLa cells reconstituted with empty M Torin-1 (6 h) or infected with S. Typhimurium SL1344 DsRed2 (MOI 200:1; 6 h). After treatment, TFEB and BHLHE40/41 cells were lysed in 400 μL or 150 μL TCL-buffer (QIAGEN) containing 1% (v/v) beta-mercaptoethanol, respectively, and stored at −80°C until sequenced.
Full-length cDNA libraries were prepared with lysate from approximately 200 cells per sample with a modified version of the SmartSeq2 protocol previously described (Picelli et al., 2013). Post SmartSeq2, double stranded cDNA was purified with Agencourt AMPure XP beads and tagged using the Nextera XT DNA Library Prep Kit and the Nextera XT index kit. Post reaction purification was performed with Agencourt AMPure XP beads. The samples were pooled, and size selection was performed by gel extraction with Zymoclean gel DNA recovery column after a 2% E-Gel EX Agarose Gel. Samples were prepared and loaded onto a NextSeq 500 (Illumina) per the manufacturer’s instructions.
Quantitative PCR
Total RNA was extracted using RNeasy Plus Mini Kit after which cDNA was generated by reverse transcription using the iSCRIPT cDNA synthesis kit according to manufacturer’s instructions. Real-time PCR was performed with gene specific primers using the iQ SYBR Green Supermix kit as per manufacturer’s instructions. Relative mRNA abundance was calculated with the ΔΔCt method where samples were normalized to GAPDH or B2M.
Cell Painting
TFEB-KO, TFEB-WT, TFEB-nuc and TFEB-cyto HeLa cells were plated at a density of 1,500 cells/well in a 384-well plate (Perkin Elmer; 384 CellCarrier Ultra Microplate) with 6 replicates per plate 48 h prior to staining. Cell Painting procedure followed the previously published protocol (Bray et al., 2016). Briefly, nine different cell components and organelles were stained with fluorescent dyes: nucleus, endoplasmic reticulum (concanavalin A/AlexaFluor488 conjugate), nucleoli and cytoplasmic RNA (SYTO14 green fluorescent nucleic acid stain), Golgi apparatus and plasma membrane (wheat germ agglutinin/AlexaFluor594 conjugate), F-actin (phalloidin/AlexaFluor594 conjugate) and mitochondria (MitoTracker Deep Red) or lysosomes (LysoTracker Deep Red). WGA and Mito-Tracker/LysoTracker were added to living cells, with the remaining stains carried out after cell fixation with PBS containing 3.2% (v/v) formaldehyde. Images from five fluorescent channels were captured at 20x magnification on an Opera Phenix High Content Screening System (Perkin Elmer): DAPI (387/447 nm), GFP (472/520 nm), Cy3 (531/593 nm), Texas Red (562/624 nm), Cy5 (628/692 nm). Nine sites per well were acquired, with laser based autofocus using the DAPI channel at the first site of each well.
Bacterial replication assay
Cells were plated in 96-well plates at a density of 1.5×104 cells/well in antibiotic-free media 18 h prior to infection (8 replicates per condition). An overnight culture of bioluminescent S. Typhimurium SL1344 Xen26 was subcultured for 4 h and then diluted 1:200 in antibiotic-free media for infection of the cells. After a 30min infection, plates were washed 4 times with IMDM media containing 10% (v/v) FBS and 50 μg/ml gentamycin. At 2 h post-infection, culture media was replaced with IMDM media containing 10% FBS and 20 μg/ml gentamycin. Luciferase counts per second were read every hour from 2–10 h post-infection using a PerkinElmer TopCount NXT. Fold replication was calculated at each time point per well.
Genome-wide CRISPR knockout screen
The microglia-like cell line BV2 (kindly provided by Dr. Yuanan Lu, University of Hawai’i at Mānoa) stably expressing Cas9 (Addgene, 52962; Sanjana et al., 2014) was transduced with the Brie mouse CRISPR knockout library (Addgene, 73632; Doench et al., 2016) as previously described (Orchard et al., 2016). For the L-Leucyl-L-Leucine methyl ester (LLME) challenge, 500 cells per guide were plated in duplicate for each treatment condition and after 16 h were treated with 2.5mM LLME or DMSO for mock condition. After 24 h treatment, cells were washed with growth media and new media was added. Surviving cells were allowed to propagate over the next 7 days. Cells were harvested, re-plated then re-challenged with 2.5mM LLME. Cells were washed with growth media after 24 h treatment and new media was added. Surviving cells were allowed to propagate over the next 48 h. Cells were harvested and DNA was isolated using the DNeasy Blood & Tissue Kit according to manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses not described in detail below were performed and visualized using GraphPad Prism8. Descriptions of statistical tests used, number of replicates, mean and SEM can be found in figure legends.
RNA sequencing analysis
After sequencing, reads were aligned to the human reference genome hg19 using Tophat2 (Kim et al., 2013). Htseq-count (Anders et al., 2015) summarized read counts for each gene, and R/Bioconductor packages edgeR (Robinson et al., 2010) was used for differential expression (DE) analysis. Differential gene expression determined using edgeR was restricted to genes with average counts per million (CPM) values greater than 1 in at least one of the two conditions compared. p values were obtained from edgeR. FDR and q-value for the remaining genes were calculated using Benjamini-Hochberg procedure. Differential gene expression thresholds can be found in the figure legends.
Relative gene expression
The relative expression was calculated from the log2 CPM-transformed measurements. Specifically, for each gene, log2 CPMs were scaled such that their minimum expression value was 0 and maximum value was 1. Only genes with relative expression values > 0.7 or < 0.3 in 8 of the 12 RNA sequencing samples were included in the analysis. The hierarchical clustering was done in pheatmap using the complete linkage with Euclidean distance (default settings).
Cell Painting
Workflow for image processing and cellular feature extraction has been previously described (Bray et al., 2016). In summary, CellProfiler (Carpenter et al., 2006) software version 2.1.0 was used to correct the image channels for uneven illumination, and identify, segment, and measure the cells. An image quality workflow (Bray et al., 2012) was applied to exclude saturated and/or out of focus wells. Cellular morphological, intensity, textural, and adjacency statistics were then measured for the cell, nuclei, and cytoplasmic sub-compartments. Details for the complete list of features and their meaning can be found here: https://github.com/carpenterlab/2016_bray_natprot/wiki/What-do-Cell-Painting-features-mean%3F
Cellular features extracted were normalized as follows: for each feature, the median and median absolute deviation were calculated across all untreated cells within a plate; feature values for all the cells in the plate were then normalized by subtracting the median and dividing by the median absolute deviation (MAD) times 1.4826 (Chung et al., 2008). Features having MAD = 0 in any plate were excluded. Morpheus was used to visualize and analyze the data (https://software.broadinstitute.org/morpheus).
Principal components analysis was completed by first removing cell painting features with single or missing values. After averaging wells for each cell line and stimulation, each feature was normalized to zero mean and unit variance. The top 2 principal components were obtained using sklearn.
CRISPR screen analysis
Illumina sequencing and data deconvolution was performed at the Broad Institute as previously described (Orchard et al., 2016). For analysis, read counts were log-normalized for each guide using the following formula: log-normalized reads per million for guide = log2((# of reads for guide / total reads in condition × 1e6) +1). Log-normalized reads were averaged for each sample, and untreated average was subtracted from LLME treated average to achieve the log2 fold changes for each sgRNA, which were then averaged to gene-level log2 fold changes (https://github.com/mhegde/volcano_plots) as previously described (Table S6) (Orvedahl et al., 2019). The STARS program (https://portals.broadinstitute.org/gpp/public/software/stars) was used to obtain gene-level p values (Table S6) (Doench et al., 2016).
Gene identifier conversion
Mygene was used to convert gene identifiers between identifier systems (Xin et al., 2016). One-to-many maps/conversions are resolved as follows: we regarded two gene identifiers in any identifier system(s) as the same gene if their converted ensembl IDs have any overlap. For enrichment analyses, all converted identifiers are included (in background or test sets).
Gene ontology enrichment
Gene names were converted into uniprot IDs, then goatools (Klopfenstein et al., 2018) was used to compute gene ontology enrichment and to obtain Bonferroni adjusted p values. Enrichments with Bonferroni adjusted p values < 0.05 are shown. Background genes are limited to those expressed (average CPM > = 1) in either TFEB knockout or reconstituted cells in Torin-1 treatment. As described in figure legends, GO enrichment is defined as logFC > ln2 and q-value < 0.05.
TSNE visualization
From all genes across all TFEB knockout and reconstituted (including overexpression) conditions and all BHLHE40/41 conditions, across all stimulation (DMSO, Torin-1 or Salmonella) and all replicates, we removed low expressing genes (CPM < 1 in over 80% of all samples). Log CPM of each gene was normalized to zero mean and unit variance across samples. We performed tSNE dimension reduction on the top 20 principal components of every gene using sklearn.
Known motif enrichment
HOMER analysis (Heinz et al., 2010) was used to compute known motif enrichment and to obtain enrichment p values. Background genes were limited to those expressed (average CPM > = 1) in at least one of the four conditions (TFEB knockout or reconstituted, or BHLHE40/41 knockout or reconstituted cells) in Torin-1 treatment. For bHLH-specific q-values, only motifs with “bHLH” annotations were selected to re-run for a separate FDR control.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG | Cell Signaling Technology | Cat #2368S; RRID: AB_2217020 |
| Phospho-(ser) 14-3-3 binding motif | Cell Signaling Technology | Cat #9601S; RRID: AB_330306 |
| Goat anti-mouse IgG | Thermo Fisher | Cat #A11029; RRID: AB_138404 |
| Pan 14-3-3 | Santa Cruz | Cat #SC-133233; RRID: AB_2016726 |
| Monoclonal anti-β-actin | Millipore Sigma | Cat #A1978; RRID: AB_476692 |
| Monoclonal anti-FLAG M2 | Millipore Sigma | Cat #F3165; RRID: AB_259529 |
| Rabbit anti-LC3B | Millipore Sigma | Cat #L7543; RRID: AB_796155 |
| Bacterial and Virus Strains | ||
| S. Typhimurium SL1344 Xen26 | Conway et al., 2013 | N/A |
| S. Typhimurium SL1344 DsRed2 | Rioux et al., 2007 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Torin-1 | Millipore Sigma | Cat #475991 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat #15714 |
| LysoView 488 | Biotium | Cat #70067-T |
| DQ-BSA | Thermo Fisher | Cat #D12051 |
| MitoTracker Red | Thermo Fisher | Cat #M22415 |
| MitoTracker Deep Red | Thermo Fisher | Cat #M22426 |
| Concanavalin A, Alexa Fluor 488 Conjugate | Thermo Fisher | Cat #C11252 |
| SYTO14 green fluorescent nucleic acid stain | Thermo Fisher | Cat #S7576 |
| Wheat Germ Agglutinin, Alexa Fluor 647 Conjugate | Thermo Fisher | Cat # W32466 |
| Alexa Fluor 594 Phalloidin | Thermo Fisher | Cat #A12381 |
| LysoTracker Deep Red | Thermo Fisher | Cat #L12492 |
| L-Leucyl-L-Leucyl methyl ester (LLME) | Cayman Chemicals | Cat #16008 |
| Critical Commercial Assays | ||
| Strep-Tactin Sepharose resin | IBA Life Sciences | Cat #2-1201-010 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat #A63881 |
| Nextera XT DNA Library Prep Kit | Illumina | Cat #FC-131–1096 |
| Nextera XT Index Kit | Illumina | Cat #TG-131–2001 |
| Zymoclean gel DNA recovery column | Zymo Research Corp | Cat #D4008 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat #1708891 |
| iQ SYBR Green Supermix Kit | Bio-Rad | Cat # 1708884 |
| RNeasy Plus Mini Kit | QIAGEN | Cat #74136 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat #69506 |
| Deposited Data | ||
| RNA-seq data, see Table S2 | Submitted to dbGaP | phs002099 |
| Experimental Models: Cell Lines | ||
| Human: HeLa | ATCC | Cat #CCL-2 |
| Human: HeLa TFEB-KO | This paper | N/A |
| Human: HeLa TFEB-WT | This paper | N/A |
| Human: HeLa TFEB-cyto | This paper | N/A |
| Human: HeLa TFEB-nuc | This paper | N/A |
| Human: HeLa BHLHE40/BHLHE41 dKO | This paper | N/A |
| Human: HeLa BHLHE40/41 dKO/BHLHE40/41 WT | This paper | N/A |
| Murine: BV-2 | Laboratory of Yuanan Lu | N/A |
| Oligonucleotides | ||
| qPCR primers | This paper | See Table S2 |
| Recombinant DNA | ||
| GFP-LC3-CGSW | Laboratory of Christian Munz | N/A |
| EGFP-N1-TFEB | Roczniak-Ferguson et al., 2012 | Addgene #38119 |
| BHLHE40 Human Tagged ORF clone | OriGene | RC210294L1 |
| BHLHE41 Human Tagged ORF clone | OriGene | RC206882L1 |
| lentiCRISPR v2 | Ran et al., 2013 | Addgene #52961 |
| lentiCas9-Blast | Sanjana et al., 2014 | Addgene #52962 |
| Mouse sgRNA library Brie in lentiCRISPRv2 | Doench et al., 2016 | Addgene #73632 |
| Software and Algorithms | ||
| NIS-Elements | Nikon | N/A |
| Harmony High-Content Imaging and Analysis Software | Perkin Elmer | N/A |
| CellProfiler software version 2.1.0 | Carpenter et al., 2006 | https://cellprofiler.org/ |
| Morpheus | https://software.broadinstitute.org/morpheus | N/A |
| STARS software | Doench et al., 2016 | https://portals.broadinstitute.org/gpp/public/software/stars |
| Mygene 3.1.0 | Xin etal., 2016 | https://pypi.org/project/mygene/ |
| Goatools | Klopfenstein et al., 2018 | https://github.com/tanghaibao/goatools |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GraphPad Prism8 | GraphPad Software, Inc. | N/A |
Highlights.
TFEB transcriptional programs are defined by subcellular localization and stimulation
Nuclear TFEB maintains lysosomal and mitochondrial compartments at steady state
BHLHE40 and BHLHE41 counter-regulate stimulation-specific TFEB target genes
ACKNOWLEDGMENTS
We thank Dr. Christian Münz (University of Zürich) for the gift of GFP-LC3-CSGW and Dr. Yuanan Lu (University of Hawai’i at Mānoa) for the BV2 cell line, Ariel Lefkovith and Bihua Li for RNA sequencing library generation, Kyle Karhohs for imaging analysis of the Cell Painting data, Mukund Varma and Gautam Goel for the initial RNA sequence analysis, and Theresa Reimels for help with manuscript preparation and editing. This work was supported by National Institutes of Health grants R01 DK097485 (to R.J.X.) and U19 AI109725 and U19 AI142784 (to H.W.V. and R.J.X.) and the Center for the Study of Inflammatory Bowel Disease (P30 DK043351 to R.J.X.). S.S. was supported by R35 GM122547 to Anne E. Carpenter. The genome sequence described and used in this research was derived from a HeLa cell line. Henrietta Lacks, and the HeLa cell line that was established from her tumor cells without her knowledge or consent in 1951, have made significant contributions to scientific progress and advances in human health. We are grateful to Lacks, now deceased, and to her surviving family members for their contributions to biomedical research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108371.
DECLARATION OF INTERESTS
P.B., N.R., and B.N. are employees of Novartis. D.R.B. and H.W.V. are employees of Vir Biotechnology. R.J.X. is a consultant to Novartis and a cofounder of Jnana Therapeutics and Celsius Therapeutics. H.W.V. is a founder of Casma Therapeutics and PierianDx. These organizations did not participate in funding this work.
REFERENCES
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E, Ledesma MD, Blázquez A, Martin MA, Falcón-Pérez JM, et al. (2015). Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 22, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Martina JA, and Puertollano R (2018). Emerging roles for TFEB in the immune response and inflammation. Autophagy 14, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MA, Fraser AN, Hasaka TP, and Carpenter AE (2012). Workflow and metrics for image quality control in large-scale high-content screens. J. Biomol. Screen 17, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray MA, Singh S, Han H, Davis CT, Borgeson B, Hartland C, Kost-Alimova M, Gustafsdottir SM, Gibson CC, and Carpenter AE (2016). Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc 11, 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brojatsch J, Lima H Jr., Palliser D, Jacobson LS, Muehlbauer SM, Furtado R, Goldman DL, Lisanti MP, and Chandran K (2015). Distinct cathepsins control necrotic cell death mediated by pyroptosis inducers and lyso-some-destabilizing agents. Cell Cycle 14, 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budge KM, Neal ML, Richardson JR, and Safadi FF (2018). Glycoprotein NMB: an Emerging Role in Neurodegenerative Disease. Mol. Neurobiol 55, 5167–5176. [DOI] [PubMed] [Google Scholar]
- Butler VJ, Cortopassi WA, Argouarch AR, Ivry SL, Craik CS, Jacobson MP, and Kao AW (2019). Progranulin Stimulates the In Vitro Maturation of Pro-Cathepsin D at Acidic pH. J. Mol. Biol 431, 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung N, Zhang XD, Kreamer A, Locco L, Kuan PF, Bartz S, Linsley PS, Ferrer M, and Strulovici B (2008). Median absolute deviation to improve hit selection for genome-scale RNAi screens. J. Biomol. Screen 13, 149–158. [DOI] [PubMed] [Google Scholar]
- Chung SY, Kao CH, Villarroya F, Chang HY, Chang HC, Hsiao SP, Liou GG, and Chen SL (2015). Bhlhe40 Represses PGC-1a Activity on Metabolic Gene Promoters in Myogenic Cells. Mol. Cell. Biol 35, 2518–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. (2013). Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145, 1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo E, Schlaermann P, and Mukherjee S (2017). How to rewire the host cell: A home improvement guide for intracellular bacteria. J. Cell Biol 216, 3931–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deus CM, Yambire KF, Oliveira PJ, and Raimundo N (2020). Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med 26, 71–88. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronzo G, Astanina E, Corà D, Chiabotto G, Comunanza V, Noghero A, Neri F, Puliafito A, Primo L, Spampanato C, et al. (2019). TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 38, e98250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, Ross DS, et al. (2019). Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 29, 285–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenreich W, Rudel T, Heesemann J, and Goebel W (2019). How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Houjeiri L, Possik E, Vijayaraghavan T, Paquette M, Martina JA, Kazan JM, Ma EH, Jones R, Blanchette P, Puertollano R, et al. (2019). The Transcription Factors TFEB and TFE3 Link the FLCN-AMPK Signaling Axis to Innate Immune Response and Pathogen Resistance. Cell Rep. 26, 3613–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Mosquera L, Diogo CV, Yambire KF, Santos GL, Luna Sánchez M, Bénit P, Rustin P, Lopez LC, Milosevic I, and Raimundo N (2017). Acute and chronic mitochondrial respiratory chain deficiency differentially regulate lysosomal biogenesis. Sci. Rep 7, 45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RP, Calvo SE, and Mootha VK (2018). Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J. Biol. Chem 293, 7508–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings KT, and Cresswell P (2011). Disulfide reduction in the endocytic pathway: immunological functions of gamma-interferon-inducible lysosomal thiol reductase. Antioxid. Redox Signal 15, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson LS, Lima H Jr., Goldberg MF, Gocheva V, Tsiperson V, Sutterwala FS, Joyce JA, Gapp BV, Blomen VA, Chandran K, et al. (2013). Cathepsin-mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)-associated adjuvants. J. Biol. Chem 288, 7481–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, Yang YP, Zhao Z, Hu LF, and Liu CF (2018). A Critical Role of Autophagy in Regulating Micro-glia Polarization in Neurodegeneration. Front. Aging Neurosci 10, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda M, Yamanaka H, Kojo S, Usui Y, Honda H, Sotomaru Y, Harada M, Taniguchi M, Suzuki N, Atsumi T, et al. (2016). Transcriptional regulator Bhlhe40 works as a cofactor of T-bet in the regulation of IFN-g production in iNKT cells. Proc. Natl. Acad. Sci. USA 113, E3394–E3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DV, Zhang L, Pedersen BS, Ramírez F, Warwick Vesztrocy A, Naldi A, Mungall CJ, Yunes JM, Botvinnik O, Weigel M, et al. (2018). GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep 8, 10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreslavsky T, Vilagos B, Tagoh H, Poliakova DK, Schwickert TA, Wöhner M, Jaritz M, Weiss S, Taneja R, Rossner MJ, and Busslinger M (2017). Essential role for the transcription factor Bhlhe41 in regulating the development, self-renewal and BCR repertoire of B-1a cells. Nat. Immunol 18, 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kribelbauer JF, Rastogi C, Bussemaker HJ, and Mann RS (2019). Low-Affinity Binding Sites and the Transcription Factor Specificity Paradox in Eukaryotes. Annu. Rev. Cell Dev. Biol 35, 357–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CJ, Hansen M, and Troemel E (2018). Autophagy and innate immunity: Insights from invertebrate model organisms. Autophagy 14, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RE, and Zoncu R (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol 21, 133–142. [DOI] [PubMed] [Google Scholar]
- Lee HH, Nemecek D, Schindler C, Smith WJ, Ghirlando R, Steven AC, Bonifacino JS, and Hurley JH (2012). Assembly and architecture of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem 287, 5882–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Friedrichsen HJ, Andrews S, Picaud S, Volpon L, Ngeow K, Ber-ridge G, Fischer R, Borden KLB, Filippakopoulos P, and Goding CR (2018). A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat. Commun 9, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, Atkinson JP, and Yu CY (2016). Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Hackmann Y, Dieckmann NM, and Griffiths GM (2014). The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol 6, a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MR, Tran MT, Ralto KM, Zsengeller ZK, Raman V, Bhasin SS, Sun N, Chen X, Brown D, Rovira II, et al. (2019). TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 5, e126749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo AC, and Isaac L (2016). Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansueto G, Armani A, Viscomi C, D’Orsi L, De Cegli R, Polishchuk EV, Lamperti C, Di Meo I, Romanello V, Marchet S, et al. (2017). Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 25, 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques ARA, Di Spiezio A, Thießen N, Schmidt L, Grötzinger J, Lüllmann-Rauch R, Damme M, Storck SE, Pietrzik CU, Fogh J, et al. (2020). Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy 16, 811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, and Puertollano R (2018). Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem 293, 12525–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, and Puertollano R (2012). MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Li H, and Puertollano R (2014). Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life Sci 71, 2483–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meireles AM, Shen K, Zoupi L, Iyer H, Bouchard EL, Williams A, and Talbot WS (2018). The Lysosomal Transcription Factor TFEB Represses Myelination Downstream of the Rag-Ragulator Complex. Dev. Cell 47, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano T, Najibi M, Paulus GLC, Adiliaghdam F, Valencia-Guerrero A, Selig M, Wang X, Jeffrey K, Xavier RJ, Lassen KG, and Irazoqui JE (2017). Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci. Rep 7, 13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabar NR, and Kehrl JH (2017). The Transcription Factor EB Links Cellular Stress to the Immune Response. Yale J. Biol. Med 90, 301–315. [PMC free article] [PubMed] [Google Scholar]
- Najibi M, Labed SA, Visvikis O, and Irazoqui JE (2016). An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense. Cell Rep. 15, 1728–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Ueno M, Adachi K, Nanba E, Narita A, Tsukimoto J, Itoh K, and Kawakami A (2019). A new heterozygous compound mutation in the CTSA gene in galactosialidosis. Hum. Genome Var 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Kawamoto T, Honda KK, Ueshima T, Noshiro M, Iwata T, Fujimoto K, Kubo H, Honma S, Yorioka N, et al. (2008). DEC1 modulates the circadian phase of clock gene expression. Mol. Cell. Biol 28, 4080–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott-Schwartz J, and Ballabio A (2018). mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun 9, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal ML, Boyle AM, Budge KM, Safadi FF, and Richardson JR (2018). The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation 15, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AD, Nguyen TA, Zhang J, Devireddy S, Zhou P, Karydas AM, Xu X, Miller BL, Rigo F, Ferguson SM, et al. (2018). Murine knockin model for progranulin-deficient frontotemporal dementia with nonsense-mediated mRNA decay. Proc. Natl. Acad. Sci. USA 115, E2849–E2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y, Goda N, Kanai M, Niwa D, Osanai K, Yamamoto Y, Senoo-Matsuda N, Johnson RS, Miura S, Kabe Y, and Suematsu M (2012). HIF-1a induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol 56, 441–447. [DOI] [PubMed] [Google Scholar]
- Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, and Virgin HW (2016). Discovery of a proteinaceous cellular receptor for a norovirus. Science 353, 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, McAllaster MR, Sansone A, Dunlap BF, Desai C, Wang YT, Balce DR, Luke CJ, Lee S, Orchard RC, et al. (2019). Autophagy genes in myeloid cells counteract IFNg-induced TNF-mediated cell death and fatal TNF-induced shock. Proc. Natl. Acad. Sci. USA 116, 16497–16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, and Ballabio A (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet 20, 3852–3866. [DOI] [PubMed] [Google Scholar]
- Perera RM, and Zoncu R (2016). The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol 32, 223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Prada CE, Gonzaga-Jauregui C, Tannenbaum R, Penney S, Lupski JR, Hopkin RJ, and Sutton VR (2014). Clinical utility of whole-exome sequencing in rare diseases: Galactosialidosis. Eur. J. Med. Genet 57, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Ferguson SM, Brugarolas J, and Ballabio A (2018). The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37, e98804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, and White E (2010). Autophagy and metabolism. Science 330, 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenhill BJ, Boyle KB, Von Muhlinen N, Ellison CJ, Masson GR, Otten EG, Foeglein A, Williams R, and Randow F (2019). The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 74, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U, Borg Distefano M, Speth MT, Ng MYW, Progida C, Hoflack B, Gruenberg J, and Griffiths G (2017). L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J. Cell Sci 130, 3124–3140. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. (2007). Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet 39, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, and Ferguson SM (2012). The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V, Vezzoli G, Tedesco B, Meroni M, Messi E, et al. (2019). Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. (2013). TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol 15, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, Marion SW, St Amand V, Reenan RA, and Lapierre LR (2018). Nuclear Export Inhibition Enhances HLH-30/TFEB Activity, Autophagy, and Lifespan. Cell Rep. 23, 1915–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, and Cuervo AM (2011). Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Jamieson A, and Cresswell P (2008). GILT is a critical host factor for Listeria monocytogenes infection. Nature 455, 1244–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbara MT, Foerster EG, Tsalikis J, Abdel-Nour M, Mangiapane J, Sir-luck-Schroeder I, Tattoli I, Van Dalen R, Isenman DE, Rohde JR, et al. (2018). Complement C3 Drives Autophagy-Dependent Restriction of Cytoinvasive Bacteria. Cell Host Microbe 23, 644–652. [DOI] [PubMed] [Google Scholar]
- Tan HWS, Anjum B, Shen HM, Ghosh S, Yen PM, and Sinha RA (2019). Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 15, 1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Matsuwaki T, Yamanouchi K, and Nishihara M (2013). Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 250, 8–19. [DOI] [PubMed] [Google Scholar]
- Tang CH, Lee JW, Galvez MG, Robillard L, Mole SE, and Chapman HA (2006). Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease. Mol. Cell. Biol 26, 2309–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele DL, and Lipsky PE (1990). Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc. Natl. Acad. Sci. USA 87, 83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lienden MJC, Gaspar P, Boot R, Aerts JMFG, and van Eijk M (2018). Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages. Int. J. Mol. Sci 20, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlhac P, Viret C, and Faure M (2015). Handcuffs for bacteria—NDP52 orchestrates xenophagy of intracellular Salmonella. Microb. Cell 2, 214–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AF, Wollenberg AC, Stuart LM, Stormo GD, and Irazoqui JE (2014). Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 40, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yan J, Niu H, Huang R, and Wu S (2018). Autophagy and Ubiquitination in Salmonella Infection and the Related Inflammatory Responses. Front. Cell. Infect. Microbiol 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C, Martinez-Lopez N, Otten EG, Carroll B, Maetzel D, Singh R, Sarkar S, and Korolchuk VI (2016). Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 1861, 269–284. [DOI] [PubMed] [Google Scholar]
- Wibowo AS, Singh M, Reeder KM, Carter JJ, Kovach AR, Meng W, Ratnam M, Zhang F, and Dann CE 3rd. (2013). Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifo-late recognition. Proc. Natl. Acad. Sci. USA 110, 15180–15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett R, Martina JA, Zewe JP, Wills R, Hammond GRV, and Puertollano R (2017). TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat. Commun 8, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin J, Mark A, Afrasiabi C, Tsueng G, Juchler M, Gopal N, Stupp GS, Putman TE, Ainscough BJ, Griffith OL, et al. (2016). High-performance web services for querying gene and variant annotation. Genome Biol. 17, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ren J, He X, Chen H, Wei T, and Feng W (2019). YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization. Autophagy 15, 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Heckmann BL, Campbell LE, and Liu J (2017). G0S2: A small giant controller of lipolysis and adipose-liver fatty acid flux. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (10 Pt B), 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown WJ, Smolka MB, Sun Y, and Hu F (2015). Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J. Cell Biol 210, 991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing data reported in this paper is database of Genotypes and Phenotypes (dbGaP) HeLa Cell Genome Sequencing Studies: phs002099 and listed in Table S2.