

Abstract
As the heart matures during embryogenesis from its foetal stages, several structural and functional modifications take place to form the adult heart. This process of maturation is in large part due to an increased volume and work load of the heart to maintain proper circulation throughout the growing body. In recent years, it has been observed that these changes are reversed to some extent as a result of cardiac disease. The process by which this occurs has been characterized as cardiac foetal reprogramming and is defined as the suppression of adult and re‐activation of a foetal genes profile in the diseased myocardium. The reasons as to why this process occurs in the diseased myocardium are unknown; however, it has been suggested to be an adaptive process to counteract deleterious events taking place during cardiac remodelling. Although still in its infancy, several studies have demonstrated that targeting foetal reprogramming in heart failure can lead to substantial improvement in cardiac functionality. This is highlighted by a recent study which found that by modulating the expression of 5‐oxoprolinase (OPLAH, a novel cardiac foetal gene), cardiac function can be significantly improved in mice exposed to cardiac injury. Additionally, the utilization of angiotensin receptor neprilysin inhibitors (ARNI) has demonstrated clear benefits, providing important clinical proof that drugs that increase natriuretic peptide levels (part of the foetal gene programme) indeed improve heart failure outcomes. In this review, we will highlight the most important aspects of cardiac foetal reprogramming and will discuss whether this process is a cause or consequence of heart failure. Based on this, we will also explain how a deeper understanding of this process may result in the development of novel therapeutic strategies in heart failure.
Keywords: foetal gene programme, heart failure

Content List ‐ Read more articles from the symposium: “Cardiovascular program and cardiovascular retreat”.
Introduction
The mammalian heart is the first organ to develop during embryogenesis. As the foetal heart develops, several structural and functional modifications take place to form the four‐chambered adult heart. This process of maturation is in large part a result of increased volume and work load of the heart to maintain proper circulation throughout the growing body [1, 2, 3, 4]. Over the past decade, multiple advances in myocardial cell homeostasis and stem cell biology have enhanced our understanding of cardiac cellular differentiation and maturation. These findings coupled to our knowledge of heart failure (HF) have led to the discovery that cardiac injury in the adult heart leads to a switch in gene expression which to some extent resembles the expression pattern observed in the developing heart, a process known as ‘cardiac foetal reprogramming’. The exact reasons and mechanisms as to why the adult heart reverts back to a foetal‐like expression pattern remain unknown. However, it has been suggested that this process is an adaptive response to cope with adverse remodelling in the heart. Strikingly, it is still uncertain whether the re‐expression of foetal genes is an adaptive response that protects the heart during HF, or a maladaptive response that compounds the insult to an already weakened heart. In the present review, we summarize the current knowledge of the cardiac foetal gene programme, by looking at the expression profiles during cardiac development and disease, with a particular focus on cardiac metabolism, contractile machinery, electrophysiology and neurohormonal expression. Finally, we will explore how a better understanding of this process may lead to novel pathophysiological pathways and treatment targets to improve HF patient outcome.
Foetal reprogramming in cardiac metabolism
Each contraction of the heart requires relatively large amounts of ATP [5]. With very low energy stores and a high ATP turnover, the metabolic activity of the heart is the highest of all organs in the body [6]. To meet energetic needs, the mature myocardium of the adult heart primarily utilizes fatty acids. Under certain conditions, the heart can also use pyruvate, lactate, acetate, amino acids, ketone bodies and phospho‐creatine [7]. Each of these substrates can be metabolized to generate acetyl coenzyme A (acetyl‐CoA), which in turn is essential for the production of substrates used in the oxidative phosphorylation pathway.
From Glycolysis (foetal) to fatty acid oxidation (adult)
Foetal development occurs in a relative hypoxic environment [8]; therefore, ATP is ultimately generated through anaerobic glycolysis during foetal development. This hypoxic state results in high levels of HIF‐1α protein, which induces the transcription of major glycolysis key factors like glucose transporter (GLUT)‐1 and GLUT‐4 [9, 10, 11], hexokinase (HK)‐1 [9], lactate dehydrogenase (LDH)‐A [9, 10] and pyruvate dehydrogenase kinase (PDK)‐1 and PDK‐2 [12, 13]. In the foetal heart, GLUT‐1 is the major transporter of extracellular glucose, which is intracellularly converted to glucose‐6‐phosphate by HK‐1 [14, 15]. Furthermore, high expression levels of LDH‐A greatly contributes to the conversion of glycolysis‐derived pyruvate to lactate, consequently regenerating nicotinamide adenine dinucleotide (NAD+) from its reduced form (NADH), which is needed to sustain glycolysis [16]. Additionally, the foetal heart can also utilize lactate as main energy source [17, 18]. Combined, any additional lactate produced by LDH‐A can be efficiently oxidized in order to produce pyruvate and restore the levels of NADH required for continued ATP production through glycolysis.
After birth, cardiac metabolism does not switch to different substrates until 7 days postpartum, in lambs [18, 19]. In rabbits, it has been observed that circulating lactate levels fall 5‐7 mM to 0.5 mM in the first 2 hours after birth [20]. As such, lactate oxidation contributes notably less to ATP production. Moreover, glycolytic rates decrease from 44% to only 10% by day 7 after birth, in rabbits [21]. In concert with reduced glycolytic rates, fatty acid oxidation rates gradually increase towards levels observed in the hearts of adult animals [22]. Metabolic contribution by the various ATP‐generating pathways stabilizes around 3 weeks after birth with fatty acid oxidation as the main metabolic pathway, contributing to 89% of total ATP production [23, 24].
The transition from glycolysis to fatty acid oxidation is brought about by the shift from a relatively hypoxic environment of the foetus to physiological normoxia attained shortly after birth (Fig. 1) [25]. Subsequently, normalized oxygen tension allows for prolyl hydroxylase‐mediated degradation of HIF‐1α, leading to abrogated expression of the aforementioned HIF‐1α target genes. Amongst these target genes is HAND1, a transcription factor that inhibits lipid oxidation, leading to repression of mitochondrial energy generation [26]. Additionally, HIF‐1α hampers lipid oxidation through inhibition of the peroxisome proliferator‐activated receptor alpha (PPARα)/retinoid X receptor (RXR) heterodimer [27]. The transitions into a more mature heart coincides with a dramatic increase in the expression levels of PPAR‐α and PPAR‐β/δ, which are the key regulators of fatty acid metabolism [28, 29, 30].
Fig. 1.
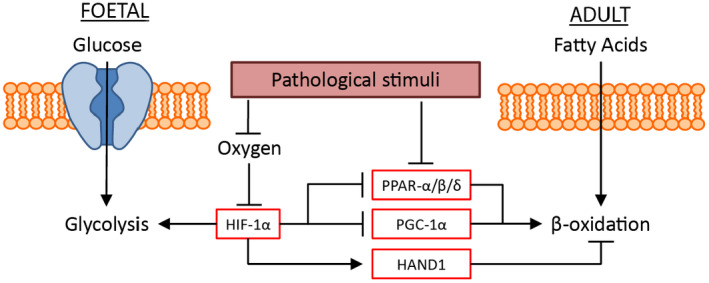
Schematic representation of the metabolic cardiac foetal reprogramming. During cardiogenesis, the cardiac tissue is primarily reliant on glycolysis for its energy requirements. This reliance on glycolysis is regulated by the relative hypoxic environment and therefore the expression of HIF‐1α, which induces the expression of glycolysis‐related genes. HIF‐1α also suppresses fatty acid oxidation (β‐oxidation) by inhibiting the expression of PPAR‐α/β/δ and PGC‐1α, and inducing the expression of HAND1, which actively suppresses β‐oxidation. Following birth, and the influx of oxygen, HIF‐1α is suppressed, leading to an increase in β‐oxidation, and a reduced utilization of glycolysis for energy production. Upon the induction of cardiac injury, there is a re‐expression of HIF‐1α leading to the inhibition of β‐oxidation and an increased reliance on glycolysis.
From fatty acid oxidation (adult) to glycolysis (disease)
In the event of various pathophysiological conditions, genes that have been active during foetal development are re‐expressed (Fig. 1) [31]. Protein levels of glycolysis genes (i.e. GLUTs, PDKs and HK‐1) are lower in healthy mature hearts than in foetal hearts. Conversely, the expression of these genes is increased in failing mature hearts to levels resembling those of the foetal heart [32]. With the re‐emergence of glycolysis as the main ATP‐generating metabolic pathway, fatty acid oxidation rates are greatly reduced (the Randle cycle) [33].
During HF, cardiac metabolism reverts to a foetal pattern in which glycolysis primarily contributes to ATP production as opposed to fatty acid oxidation [34, 35]. It has been shown that glycolysis increases as fatty acid oxidation decreases during pathological hypertrophy [36, 37]. Activity levels of mediating protein change or protein expression is altered in concert with the changes of each metabolic pathway [38, 39, 40, 41]. During HF, PPAR‐α and PGC‐1α levels decrease, consequently reducing fatty acid oxidation [42]. It is hypothesized that this reduction in PPAR‐α and PGC‐1α levels is due to rising levels of HIF‐1α. Most forms of HF result in cardiac hypoxia, for example hypertrophic cardiomyocytes increase in size and consequently oxygen tension per cell decreases. Moreover, HIF‐1α was found to be increased in pressure‐overload hypertrophy [42]. As previously stated, HIF‐1α is a master switch between glycolysis and fatty acid oxidation. Once HIF‐1α levels increase in the adult heart, expression of 6‐phosphofructo‐2‐kinase (PFK2) increases, resulting in increased levels of fructose‐2,6‐biphosphate, thereby activating PFK1 and ultimately glycolysis [43].
Clinical relevance of targeting metabolic foetal reprogramming in Heart Failure
Cardiac damage leading to HF results in a switch in energy substrate, from fatty acids (postnatal) to carbohydrates (foetal). Several studies have focused on reversing this process of foetal reprogramming, by pharmacological activation of PPAR‐α with the agonist fenofibrate. Fenofibrate was found to prevent the HF‐induced reduction in β‐oxidation with improved cardiac function during the progression of the disease, in dogs with pacing‐induced HF [44]. However, it was unable to prevent end stage heart failure, since eventually filling pressures were elevated in both groups. Interestingly, in pigs with pacing‐induced HF, fenofibrate was found to increase the expression of PPAR‐α‐activated genes, prevent LV hypertrophy, and delayed the development of LV dilation and dysfunction [45]. However, in this study, β‐oxidation was not assessed. Conversely, various studies have also focused on inhibiting β‐oxidation in the failing heart, by pharmacologically targeting carnitine palmitoyltransferase type 1 (CPT‐1), the major source for fatty acid uptake in the mitochondria. In the experimental setting, inhibition of CPT‐1 was found to result in an overall improvement of cardiac function [46, 47]. Following these promising results, several trials were preformed to assess the potential beneficial effects of inhibiting CPT‐1 in the clinical setting [48, 49, 50, 51, 52]. The CPT‐1 inhibitor etomoxir was initially tested in a nonplacebo‐controlled pilot study enrolling 10 patients suffering from congestive HF and was found to improve clinical status, central haemodynamics at rest and during exercise, and left ventricular ejection fraction [48]. The CPT‐1 inhibitor perhexiline was also found to improve overall patient outcome. In a double‐blinded study, 56 chronic HF patients were randomized into either perhexiline or placebo group [51]. Perhexiline was found to improve ventricular ejection fraction, symptoms, and resting and peak stress myocardial function. In an additional study, 46 patients with nonobstructive hypertrophic cardiomyopathy, perhexiline was found to ameliorate cardiac energetic impairment, correct diastolic dysfunction, and increase exercise capacity [52]. Overall, these trials did demonstrate an improvement in patient outcome, however larger clinical trials will have to be conducted in HF patients to truly assess the potency of CPT‐1 inhibition. Based on these studies, it is still uncertain whether the switch in energy substrates resulting from cardiac injury is an adaptive or maladaptive response, and further research should be done to resolve this. However, it does demonstrate the potential of therapeutic intervention targeting foetal reprogramming holds.
Foetal reprogramming in cardiac contractile machinery
The re‐emergence of foetal gene expression in the heart is not only limited to a switch in energy substrate. Maturation from a foetal to an adult heart involves a steady shift from compliant (foetal) to stiffer (adult) contractile proteins. As a result of cardiac disease, the adult heart undergoes a reversion to a more compliant foetal contractile machinery. This turnover has been highly studied in the sarcomere, which gives cardiac muscles their striated appearance and is responsible for the contractile function. The most abundant sarcomeric proteins are myofilament proteins (myosin and actin), regulatory proteins (troponins and tropomyosin) and cytoskeletal proteins (myosin binding protein C and titin). Several isoforms exist of each sarcomeric protein and it is the level of expression of these isoforms that determine the function of the cardiac sarcomere. The turnover of the sarcomeric proteins during cardiac development and disease has been extensively studied in rodent models, and to a lesser extent in the human setting (Table 1). The sarcomeric protein composition and distribution in rodent models is somewhat different from that of the human setting, and it is therefore not always possible to extrapolate the rodent findings to the clinical setting.
Table 1.
Expression of sarcomeric proteins in foetal, adult and diseased hearts
| Foetal | Adult | Disease | |
|---|---|---|---|
| Myosin heavy chain a | |||
| α‐MHC | ↓ | ↑ | ↓ |
| β‐MHC | ↑ | ↓ | ↑ |
| Myosin light chain | |||
| MLC‐1 | ↑ | ↓ | ↑ |
| MLC‐2 | ↓ | ↑ | ↓ |
| Actin b | |||
| α‐Skeletal actin | ↑ | ↓ | ↑ |
| α‐Cardiac actin | ↓ | ↑ | ↓ |
| Troponin | |||
| TnTfoetal | ↑ | ↓ | ↑ |
| TnTadult | ↓ | ↑ | ↓ |
| TnIfoetal | ↑ | ↓ | ↑ |
| TnIadult | ↓ | ↑ | ↓ |
| Titin | |||
| N2BA | ↑ | ↓ | ↑ |
| N2B | ↓ | ↑ | ↓ |
The ratio of α/β‐MHC is different in humans.
It is unknown if this switch occurs in the human setting.
Myosin
Myosin heavy chain (MHC) is the so‐called ‘molecular motor’ protein of the sarcomere, which together with actin is responsible for the contraction of the cardiomyocyte, consuming ATP as the energy source to produce tension. Within cardiomyocytes there are two main isoforms of MHC, the slow twitch, β‐MHC, and the fast α‐MHC. α‐MHC has a higher ATPase activity and shortening velocity, compared to β‐MHC, therefore, hearts expressing α‐MHC possess more rapid contractile velocity than hearts expressing β‐MHC. Besides the MHC isoforms, the motor function of myosin is also regulated by the myosin light chain (MLC). Similar to MHC, the human heart expresses two isoforms of MLC, the essential (MLC‐1) and regulatory (MLC‐2). MLC‐1 has been suggested to act as a MHC/actin tether, whilst MLC‐2 slows the rate of tension development of myosin [53, 54].
The turnover in MHC in the rodent heart has been extensively studied. During cardiac development, the rodent heart switches from MHC‐β to MHC‐α, and upon cardiac injury the heart reverts back to the expression of MHC‐β. On the other hand, in human cardiac development, both α‐MHC and β‐MHC are expressed, and as the heart matures β‐MHC becomes the predominant isoform. As a result of cardiac damage, the expression levels of both isoforms is reduced and reverts back to a foetal‐like expression pattern [36, 54, 55, 56, 57, 58].
Similar to the MHC, the human heart expresses both MLC‐1 and MLC‐2 isoforms. During development, MLC‐1 is primarily expressed in the whole heart. After birth MLC‐1 expression declines rapidly and is replaced by MLC‐2 in the ventricles. However, in response to hypertrophy, ischaemia or dilated cardiomyopathies, MLC‐1 is re‐expressed [54]. Recently, it has been observed that this switch to MLC‐1 expression results in a structural change enabling cardiomyocytes to adjust to enhanced work load, by improving power output and cardiac contractility [53, 54].
These findings suggest that the re‐expression of the foetal‐like myosin, both MHC and MLC, isoforms can be considered a molecular adaptation mechanism to compensate for an increased work demand or impaired sarcomeric function.
Actin
In mammals, actins are encoded by a multigene family, of which two main sarcomeric actin isoforms exist in cardiomyocytes: α‐skeletal and α‐cardiac actin. During cardiac development, α‐skeletal actin is primarily expressed in the foetal and neonatal hearts and as the heart matures α‐skeletal actin is slowly replaced by α‐cardiac actin [57, 59, 60, 61, 62]. Exposure to pressure‐overload hypertrophy in rats was found to result in a turnover from α‐cardiac actin to α‐skeletal actin expression [57, 61, 62, 63]. Similarly, in cultured neonatal cardiomyocytes exposed to α1‐adrenergic agonists or growth factors TGFβ1 and bFGF, α‐skeletal actin mRNA was observed to be significantly increased [64]. Several studies have examined if in humans an actin isoform switch takes place during development and disease; however, the results have been contradictory and as such it still remains unclear if in humans this switch takes place [57, 58, 65, 66, 67]. Noteworthy is the observation that α‐skeletal actin, when compared to α‐cardiac actin, can strongly promote the contractility of the myocardium by activating α‐MHC’s ATPase activity to a larger extend [68]. This suggests the turnover from α‐cardiac to α‐skeletal actin to be an adaptive response to maintain cardiac contractility due to the increased presence of α‐MHC in the myocardium.
Troponin
Troponin is part of the myofibrillar contractile complex, involved in controlling muscular contraction by regulating the myofibrillar responsiveness to calcium and adrenergic stimulation. Troponin consists of 3 subunits, troponin C, T and I (TnC, TnT and TnI, respectively). During normal cardiac development in rats, both TnT and TnI switch from their respective foetal to the adult isovorms [67, 69, 70]. Rat hearts when exposed to cardiac injury demonstrate a re‐expression of the foetal isoforms of TnT and TnI [67, 70, 71]. Several studies have shown that the foetal TnT and TnI isoforms have a lower calcium sensitivity, adrenergic sensitivity, ATPase activity and cardiac muscle relaxation [67, 70]. Suggesting that the re‐expression of the foetal TnT and TnI is either a mechanism of pathologic change or a maladaptive process induced by the pathological changes of HF.
Titin
Titin is a large protein that has been characterized as molecular spring, with its elastic properties defining the passive mechanical properties of cardiomyocytes. In humans, titin is encoded by a single gene (TTN) containing 363 exons that are differentially spliced, creating the stiffer N2B (short molecular spring) and the more compliant N2BA (long molecular spring) isoforms [67, 72, 73]. Both isoforms are co‐expressed in the sarcomere, and the degree of expression of each isoform adjusts the passive stiffness [67, 72, 73]. When looking at neonatal pig hearts, it was observed that these had a higher abundance of the complaint N2BA titin isoform, whilst adult pig hearts demonstrated a shift towards the stiffer N2B isoform [67, 72]. This observation suggests that as the heart matures there is an increase in passive myocardial stiffness, which could play a role in adjusting for diastolic function during development. Upon cardiac damage, a shift from the N2B to N2BA has been observed, in both the experimental and clinical setting [67, 72, 73, 74]. This shift causes a reduction in titin‐derived myofibrillar stiffness, which can lead to a decrease in cardiac output.
Clinical relevance of targeting contractile foetal reprogramming in heart failure
Several studies have aimed at understanding the therapeutic potential of targeting the foetal reprogramming of the cardiac contractile machinery. These studies have primarily focused on improving the contractility of the cardiomyocytes in the failing heart by targeting myosin. Similar to the foetal human heart, the diseased adult myocardium predominantly expresses more β‐MHC, an MHC isoform characterized for lower ATPase activity and reduced shortening velocity when compared to α‐MHC. As a consequence, the diseased myocardium has a reduced contractile capacity. Omecamtiv mecarbil (OM) is a selective, small‐molecule cardiac myosin activator that binds to the catalytic domain of myosin, thereby increasing cardiac contractility without affecting cardiomyocyte calcium concentrations or myocardial oxygen consumption [75]. OM has been shown in improve cardiac muscle function in animal models for HF [76, 77, 78, 79]. Additionally, OM was found to have properties protecting the heart against ischaemia and reperfusion injury [80]. Whilst OM does not directly reverse cardiac foetal reprogramming of the myosin, it enables the present β‐MHC to have an improved contractile capacity, similar to that of α‐MHC [77, 81]. Following these positive results in the experimental setting, several clinical trials have been performed with the administration of OM to HF patients. In the ATOMIC‐AHF study evaluated the pharmacokinetics, pharmacodynamics, tolerability, safety and efficacy of intravenous OM administration in patients with acute HF [82]. In the 606 patients included, OM administration did not meet the primary end‐point of dyspnoea improvement; however, it was well tolerated, increased systolic ejection time, and it may have improved dyspnoea in the high‐dose group [82]. In the COSMIC‐HF study, a randomized, double‐blinded, placebo‐controlled study, the pharmacokinetics of OM administration were assessed in HF patients, and changes in cardiac function and ventricular diameters [83]. A total of 299 patients were enrolled, 150 received OM whilst 149 were placed in the placebo group. OM dosing achieved plasma concentrations associated with improved cardiac function and decreased ventricular diameter [83]. Finally, the ongoing GALACTIC‐HF trial, a randomized, double‐blinded, placebo‐controlled, event‐driven cardiovascular outcome trial, is assessing the potential of OM treatment for its ability to improve symptoms, prevent clinical HF events and delay cardiovascular death in patients with chronic HF [84]. More than 8,000 chronic HF patients have been enrolled and randomized to either oral placebo or OM treatment. The GALACTIC‐HF trial is the first trial examining whether selectively increasing cardiac contractility in patients with HF with reduced ejection fraction will result in improved clinical outcomes [84].
Foetal reprogramming in cardiac electrophysiology
Besides the switches in metabolism and contractile machinery, the reversion to a more foetal‐like state in response to cardiac injury has also been observed in the mechanisms regulating the electrophysiology of cardiomyocytes. Cardiac electrophysiology is in large part governed by the expression of ion channels, gap junctions and the calcium homeostasis.
Ion channels
Ion channels are essential for the generation and propagation of the current that enables the heart to perform its function. Extensive research has been done to understand the electrophysiological changes that occur during cardiac maturation. It has been observed that the excitability, action potential properties, contractility and relaxation of the foetal and adult heart differ significantly from each other [85]. As a result, the foetal heart expresses different genes involved in the generation and propagation of the action potential then the adult heart. In mice during cardiac development from foetal to adult, there is an up‐regulation of genes involved in the Ik1 (KCNH2), Ito (KCND2 and KCND3), Ikr (KCNH2), Iks (KCNQ1), ICa,L (CACNA1C), INa (SCN5A), If (HCN1 and HCN4) currents, and a down‐regulation of genes involved in the ICa,T (CACNA1H) and NCX (NCX1) currents [85, 86, 87, 88, 89]. The reduced expression of potassium channels in the foetal heart coincides with the observation that these hearts have a less negative resting and longer action potentials compared to adult hearts [85]. The observed lower expression of ICa,L and higher ICa,T and NCX is consistent with the greater importance of alternate calcium‐entry pathways in the foetal versus adult hearts [85]. The increase in sodium channel expression in adult mouse hearts may be necessary for rapid activation of the much larger heart [85]. Similarly, studies comparing human cardiomyocytes derived from human induced pluripotent stem cells (hiPSC) or from human embryonic stem cells (hESC), to adult tissue have shown similar findings [90, 91]. Interestingly, in cardiac tissue of patients diagnosed with end stage HF, the major sodium (INa), potassium (Ito, IK1, IKr and IKs) and calcium (Ica,L) ion channels are significantly repressed, whilst Ica,T, NCX and If (HCN4) are up‐regulated [89, 92, 93]. Therefore, the heart responds to an increased load by decreasing the potassium currents, thereby prolonging the action potential and increasing the calcium within the cardiomyocytes, leading to increased contractility (Fig. 2). These findings suggest that as a result of cardiac injury, the heart undergoes ion channel remodelling, resulting in an expression profile similar to that of the foetal heart. Initially, these changes are adaptive, however in the long run they can become maladaptive by increasing the changes of arrhythmias [94, 95].
Fig. 2.
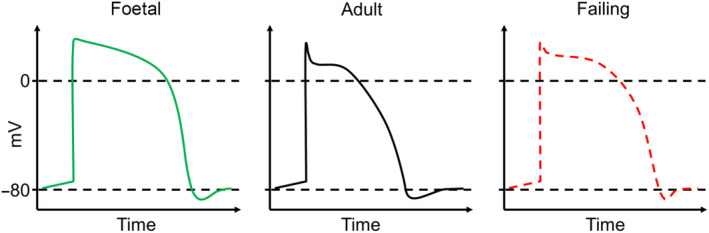
Schematic representations of the foetal, adult and diseased action potential. (LEFT) In the foetal stages of cardiac development, the heart has a prolonged action potential primarily due to a reduced expression of potassium channels. (MIDDLE) A schematic representation of an adult heart action potential. Compared to the foetal heart, the adult heart has an increased expression of potassium channels, sodium channels, and a reduction in calcium channels. (RIGHT) Following cardiac injury, the myocardium has a reduced expression of potassium channels and sodium channels coupled to an increase in the expression of calcium‐sensitive channels. This switch leads to an increase in action potential, reminiscent of the foetal action potential.
Gap junctions
Gap junctions are clusters of intercellular channels, assembled forming connexins. These connexins function as pathways enabling electrical current propagation, which controls the rhythm of the heart. As is the case in most tissues and organs, multiple connexins are expressed in the heart, specifically connexin43, connexin 40 and connexin45 [96, 97]. The presence of each connexin type varies in relative to quantities depending on the functional specialization of each subset of cardiomyocytes. The most predominant gap junction protein in the adult heart is connexin43, expressed highly in all cardiomyocytes subsets of the heart [96, 97]. In the sinoatrial node, the site of impulse generation, and the atrioventricular node, the site where impulse is slowed before being routed to the ventricles, cardiomyocyte gap junctions are formed by connexin43 and connexin45, associated with slow conductance [97]. Cardiomyocytes of the His–Purkinje conduction systems are mainly characterized by the expression of connexin40, a connexin associated with high conductance, which facilitates rapid distribution of the impulse throughout the working ventricular myocardium [97].
It has been well established, in rodents and the human setting, that as the heart matures the expression of connexin43, connexin 40 and connexin45 are progressively increased [96, 97, 98, 99]. This increase in the density of gap junctions in the developing heart is directly linked to an increase in conduction velocity. It has been shown that upon cardiac injury, in both rodent HF models and in the human clinical setting, connexin43 expression is not only drastically reduced (±50%), but the remaining connexin43 gap junctions are also highly disorganized [97, 100, 101]. This decrease in connexin43 expression is also associated with an increase in connexin40 expression [97, 100, 101]. Whether this increase in connexin40 expression is a result of reduced connexin43 levels, or whether it is an adaptive response leading to increased impulse propagation throughout the myocardium is unknown [97, 100, 101]. It has been suggested that the reduction in cell‐to‐cell coupling in HF results in an increase in the QT‐interval, action potential prolongation and increased risk of arrythmias [97, 100, 101].
Calcium homeostasis
Intracellular calcium homeostasis (release and uptake) plays an essential role in regulating excitation–contraction coupling and in modulating systolic and diastolic function in the heart. Calcium ions are initially imported into the cell through the plasma membrane by means of the ICa,L current, which is generated as a result of the depolarization of the plasma membrane. Calcium entry from the plasma membrane activate the ryanodine receptors (RyR), which results in an efflux of calcium ions from the sarcoplasmic reticulum (SR), in a process known as calcium‐induced calcium release. The released cytoplasmic calcium interacts with calcium‐sensitive proteins (i.e. TnC) controlling the force and rate of contraction. The cytoplasmic calcium is then pumped back into the SR, by SR calcium‐ATPases (SERCA) activity, and out through the plasma membrane, by NCX activity. The expression of the ICa,L current, TnC and NCX in cardiac development and disease has been covered in the previous section; here, we will focus on the expression of RyR and SERCA during cardiac development and disease.
Calcium homeostasis has been extensively studies in both cardiac development and cardiac disease, especially in terms of the expression of RyR and SERCA. There are three major isoforms of the RyR, of which RyR2 is the major SR calcium‐release channel involved in excitation–contraction coupling in the heart. SERCA functions by transferring calcium from the cytosol to the SR at the expense of ATP during muscle contraction, in the cardiomyocytes the major SERCA isoform is SERCA2, encoded by ATP2A2. In studies exploring cardiac development in rodents and in hESC/hIPCS cardiac differentiation, it has been observed that the expression of both RyR2 and SERCA2 is up‐regulated [85, 102, 103, 104]. The greater expression of the calcium handling proteins (ATP2A2, RyR2 and CACNA1C) during cardiac development may be essential for the stronger mechanical function required to provide the blood supply to much larger adult bodies [85, 102, 103, 104].
The expression of RyR2 in HF has been controversial, several studies have shown a reduction in the expression levels, back to foetal levels, in rodent and human HF [105, 106], however numerous studies have also shown no change in the expression of RyR2 in HF [105, 106]. Therefore, the exact expression and involvement of RyR2 during HF remains uncertain. Interestingly, studies have demonstrated that during HF, RyR2 are hyperphosphorylated resulting in a leaky RyR2 channel and reduced SR calcium content [105, 107]. On the other hand, SERCA2 expression, which is increased during cardiac developing, has been shown to be substantially reduced in HF, in both rodent and human models [85, 108, 109]. Furthermore, a decrease in phospholamban (PLN), a regulator of SERCA2 activity, phosphorylation in HF has been described, which further depressing the function of SERCA2 [107]. Combined, the reduction of SERCA2 expression/activity and SR leakage by hyperphosphorylated RyR2 channels lead to reduced SR calcium content, resulting in reduced SR calcium release, myofilament activation and contractility [107].
Clinical relevance of targeting electrical physiology foetal reprogramming in Heart Failure
To help extrapolate the observations demonstrating the process of foetal reprogramming on the electrical physiological level in HF, a number of studies have explored modulating ion channels and calcium handling in the failing heart. Such studies have demonstrated that by overexpressing SERCA2, thus reverting cardiac foetal reprogramming, in transgenic rodent models for HF, these animals have improved cardiac function and are less prone to develop HF following myocardial injury [110, 111, 112]. Similarly, overexpression of SERCA2 by means of adenoviral gene transfer, in HF models, has also demonstrated beneficial effects [113, 114]. Following these positive results observed from modulating the cardiac foetal reprogramming of SERCA2 in the experimental setting, clinical trials with SERCA2 gene therapy have been performed. The initial findings were encouraging, demonstrating improved cardiac function, decreased HF symptoms and reduced mortality in patients with advanced HF [115, 116]. However, a recent study utilizing the same SERCA2 gene therapy showed no improvement on ventricular remodelling in patients with advanced systolic HF [117]. In contrast to boosting gene expression, the application of antisense oligonucleotide therapy is becoming more feasible (technically and financially) to intervene in pathways at different levels [118, 119]. Consequently, SERCA2 function has been successfully enhanced by antisense oligonucleotide therapy in mice targeting PLN, a regulator of SERCA2 activity. Besides utilizing gene therapy as a therapeutic strategy, pharmacological agents that restore SERCA2 function have also been explored. One such agent is istaroxime, which functions by stimulating SERCA2 activity and indirectly inhibiting NCX function by increasing intracellular sodium levels [120]. Treatment with istaroxime in animal models of HF have shown improved cardiac function with no adverse effects [121, 122]. Following these animal studies, a clinical trial evaluating the effects of acute istaroxime administration in HF patients has demonstrated an improvement in cardiac function in these patients [123, 124, 125]. Although such a pharmacological intervention does not directly target cardiac foetal reprogramming, like direct gene therapy does, it does ensure that the remaining SERCA2 is more active, therefore compensating for the lack of SERCA2 expression. Taken together, these studies have further demonstrated the potential of targeting foetal reprogramming in cardiac electrical physiology in HF.
Cardiac neurohormonal foetal reprogramming
Cardiac foetal reprogramming is not only limited to the metabolism, contractile machinery and electrophysiology, but also occurs in the expression of cardiac neurohormones. Specifically, foetal reprogramming has been observed in the expression of atrial and brain natriuretic peptides.
The atrial natriuretic peptide (ANP) was the first natriuretic peptide identified in 1981 [126]. Since then extensive research has been done on ANP, whose primary function has been identified to reduce plasma volume, and therefore blood pressure, by increased renal excretion of salt and water, vasodilation, increased vascular permeability [127]. In the adult murine and human heart, the atrium is the major source of ANP expression; however, during cardiac development, ANP expression is primarily localized to the ventricles [128]. As the heart matures, the expression of ANP in the ventricles is significantly reduced in the ventricles [128]. The primary stimuli for ANP expression is stretch, thus as a result of cardiac hypertrophy, remodelling and HF, ANP is significantly expressed in the ventricles returning to foetal expression levels [128, 129].
Following the discovery of ANP, a second natriuretic peptide was identified in the brain, brain natriuretic peptide (BNP) [128]. Although initially isolated and characterized in the brain, BNP was later identified as being predominantly expressed in the heart ventricles [128]. Furthermore, BNP has a similar mode of action as ANP, that is to lower blood volume, reduce cardiac output and systemic blood pressure. Additionally, BNP mimics the expression profile of ANP during cardiac development, with BNP levels being significantly reduced in the adult compared to the foetal myocardium [128, 129]. Upon myocardial stretch, BNP, like ANP, is re‐expressed by the ventricles [128, 129].
In recent years, it has been well established that the re‐expression of both ANP and BNP has a cardioprotective effect in the failing myocardium [130]. In the human setting, the actions of BNP are of particular interest. BNP not only helps unload the failing heart, by reducing preload, facilitate renal excretion of salt and water, and to inhibit the renin–angiotensin system, but it is also involved in the inhibition of the sympathetic drive to the heart, enhancement of the parasympathetic cardiac reflex, and inhibition of pathological cardiac hypertrophy [130]. Thus, the re‐expression of ANP and BNP in the failing heart is an adaptive response that helps to protect the failing heart.
Clinical relevance of targeting neurohormonal foetal reprogramming in Heart Failure
The notion that neurohormonal foetal reprogramming occurs in the failing heart has been widely adapted in the form of biomarkers, where ANP and BNP have been characterized to associate with HF disease severity and progression [131]. However, it has recently also become evident that neurohormonal foetal reprogramming can also serve as an ideal handle for intervention, specifically BNP. As previously mentioned, the re‐expression of BNP results in several cardioprotective effects. Based on these, two main therapeutic strategies have been employed to further increase the levels of natriuretic peptides in HF patients. The first approach has been to target neprilysin, the enzyme responsible for the degradation of natriuretic peptides. A prodrug that strongly inhibits the activity of neprilysin, sacubitril, has been shown to not only have beneficial effects in models for HF, but also in the clinical setting [132, 133, 134, 135]. The combination of valsartan/sacubitril has now become a class I recommendation in the treatment of heart failure with a reduced ejection fraction [136]. The second approach has been to administer engineered recombinant natriuretic peptides, which mimic the effects of the endogenous natriuretic peptides. These recombinant natriuretic peptides have also demonstrated beneficial effects in both HF animal models and in the clinical setting [137, 138, 139].
Exploring unknown elements of the foetal programme to uncover new therapeutic avenues
The heart exposed to stress undergoes physiological changes bringing it back to a more foetal‐like state, in other words cardiac foetal reprogramming. Cardiac damage leading to HF results in a switch in energy substrate, from fatty acids (postnatal) to carbohydrates (foetal). Similarly, the contractile machinery of the heart reverts back to a more compliant state, as observed in the foetal heart. Finally, cardiac electrophysiology, governed by ion channels, gap junctions and calcium homeostasis, also switches to a state similar to that observed in the foetal heart. Initially, the process of cardiac foetal reprogramming seems to be an adaptive response to cope with adverse remodelling in the heart, and as such a consequence of cardiac injury. However, as time progresses, these changes are detrimental to the myocardium and add further insult leading to disease progression. Therefore, targeting cardiac foetal reprogramming could be ideal for therapeutic interventions.
In the previous sections, we have demonstrate that targeting various aspects of the cardiac foetal gene programme holds great promise in improving patients’ outcome. Therefore, it is our opinion that attaining a better understanding of this process may eventually lead to better therapeutic options for HF patients. To obtain a better understanding of cardiac foetal reprogramming, studies in the field of cardiovascular research should focus on further elucidating this process. There are several ways one could go about characterizing the cardiac foetal gene programme, especially with the current improvements in the ‘omics’ techniques (i.e. genomics, proteomics and metabolomics). These techniques could lead to a better understanding of how diseased myocardium mimics the developing heart. Furthermore, pathophysiological pathways implicated in cardiac foetal reprogramming could be uncovered, leading to novel therapeutic targets. Recently, we focused on characterizing the murine cardiac foetal gene programme, by means of RNA sequencing, this has led to the identification of several new cardiac foetal genes, including genes associated with oxidative stress, extra cellular matrix composition, metabolism and signal transduction (Table 2) [140]. Of particular interest was OPLAH, a gene that encodes for 5‐oxoprolinase, an antioxidant enzyme involved in the γ‐glutamyl cycle, where it is responsible for the conversion of 5‐oxoproline, a degradation product of glutathione, into glutamate [141, 142]. OPLAH was found to be expressed during cardiac development and repressed in cardiac disease, in both the experimental and clinical settings [140, 143]. By overexpressing OPLAH, and reversing cardiac foetal reprogramming of the gene, mice were found to have improved cardiac function following myocardial infarction [140]. The improvement in cardiac function was found to result from a reduction in oxidative stress, due to a decrease in 5‐oxoproline, a strong mediator of oxidative stress [140, 144, 145]. These findings suggest OPLAH may be an interesting candidate for therapeutic intervention. Identifying drugs and or small molecules capable of enhancing OPLAH expression and/or activity may therefore lead to novel therapeutic strategies for HF patients. Interestingly, 5‐oxoproline was also found to be a potential novel biomarker for HFyy 140 . Combined, this study demonstrates the potential of characterizing cardiac foetal reprogramming to help uncover novel pathophysiological pathways which may lead to new therapeutic strategies and improved patient outcome (Fig. 3).
Table 2.
Several known and novel members of the cardiac foetal gene program recently identified
| Gene | Annotation | Developmental | Diseased |
|---|---|---|---|
| Know members of the cardiac foetal gene program | |||
| RYR2 | Ryanodine receptor 2, cardiac | ↑ | ↓ |
| CACNA2D1 | Calcium channel, voltage‐dependent, alpha2/delta subunit 1 | ↑ | ↓ |
| ATP2A2 | ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | ↑ | ↓ |
| Novel members of the cardiac foetal gene program | |||
| OPLAH | 5‐Oxoprolinase (ATP‐hydrolysing) | ↑ | ↓ |
| ANXA11 | Annexin A11 | ↑ | ↓ |
| HADH | Hydroxyacyl‐Coenzyme A dehydrogenase | ↑ | ↓ |
| CD300LG | CD300 antigen like family member G | ↑ | ↓ |
| MCCC1 | Methylcrotonyl‐Coenzyme A carboxylase 1 (alpha) | ↑ | ↓ |
| LGALS4 | Lectin, galactose binding, soluble 4 | ↑ | ↓ |
| CYYR1 | Cysteine and tyrosine‐rich protein 1 | ↑ | ↓ |
| SLCO2B1 | Solute carrier organic anion transporter family, member 2b1 | ↑ | ↓ |
| RALGAPA2 | Ral GTPase activating protein, alpha subunit 2 (catalytic) | ↑ | ↓ |
| DNAJB1 | DnaJ (Hsp40) homolog, subfamily B, member 1 | ↓ | ↑ |
| THEM6 | Thioesterase superfamily member 6 | ↑ | ↓ |
| ETL4 | Enhancer trap locus 4 | ↑ | ↓ |
| ABHD14B | Abhydrolase domain containing 14b | ↑ | ↓ |
| VPS13C | Vacuolar protein sorting 13C (yeast) | ↑ | ↓ |
| OSTC | Oligosaccharyltransferase complex subunit | ↓ | ↑ |
| FREM1 | Fras1 related extracellular matrix protein 1 | ↓ | ↑ |
| DENND4C | DENN/MADD domain containing 4C | ↑ | ↓ |
| SNX6 | Sorting nexin 6 | ↓ | ↑ |
| HSP90AA1 | Heat shock protein 90, alpha (cytosolic), class A member 1 | ↓ | ↑ |
| PSMC3IP | Proteasome (prosome, macropain) 26S subunit, ATPase 3, interacting protein | ↓ | ↑ |
| DNAJA1 | DnaJ (Hsp40) homolog, subfamily A, member 1 | ↓ | ↑ |
| ITM2A | Integral membrane protein 2A | ↓ | ↑ |
| VPS13D | Vacuolar protein sorting 13 D (yeast) | ↑ | ↓ |
| PPRC1 | Peroxisome proliferative activated receptor, gamma, coactivator‐related 1 | ↓ | ↑ |
| SLC25A22 | Solute carrier family 25 (mitochondrial carrier, glutamate), member 22 | ↑ | ↓ |
| LSM1 | LSM1 homolog, U6 small nuclear RNA associated (S. cerevisiae) | ↓ | ↑ |
| BANP | BTG3 associated nuclear protein | ↓ | ↑ |
| KIF26B | Kinesin family member 26B | ↓ | ↑ |
| GRB10 | Growth factor receptor bound protein 10 | ↓ | ↑ |
Fig. 3.
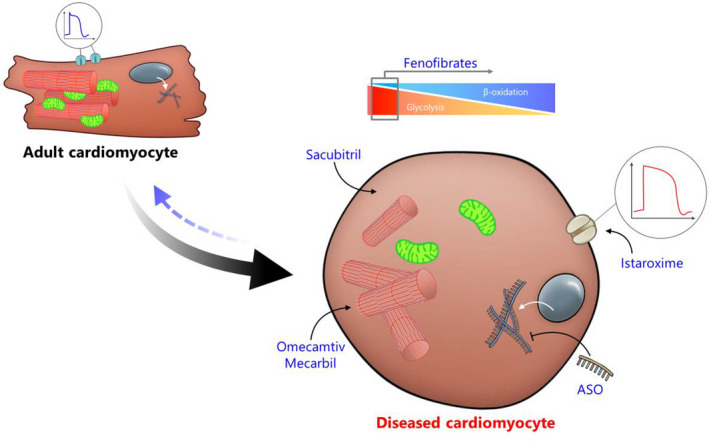
Therapeutic strategies to revert foetal reprogramming in disease. Regained foetal characteristics in diseased adult cardiomyocytes are potential therapeutic targets based on specific dysfunctional aspects. Therapeutic strategies marked in blue may improve cardiomyocyte function and may, therefore, revert pathological foetal reprogramming (blue arrow). Fenofibrate, a PPAR‐α agonist, can prevent HF‐induced switch from β‐oxidation to glycolysis. Omecamtiv Mecarbil, a small‐molecule cardiac myosin activator, improves cardiac contractility, thereby reversing foetal reprogramming. A reduction of SERCA2 expression is a hallmark of cardiac electrical physiological foetal reprogramming, and istaroxime has been found to reverse this by improving SERCA2 activity. Re‐expressing natriuretic peptides, a prime example of cardiac foetal reprogramming, has a positive effect on cardiac outcome, Sacubitril has been found to strongly improve the presence of natriuretic peptides by inhibiting the activity of neprilysin. Antisense oligonucleotides (ASO) have demonstrated their potential to intervene in cardiac foetal reprogramming by improving the activity of SERCA2, however this approach could be extrapolated to target various other aspects of foetal reprogramming.
Author Contribution
Atze Van der Pol: Visualization (equal); Writing‐original draft (lead); Writing‐review & editing (lead). Martijn Hoes: Visualization (supporting); Writing‐original draft (supporting); Writing‐review & editing (supporting).
van der Pol A, Hoes MF, de Boer RA, van der Meer P (University Medical Center Groningen, University of Groningen, Groningen, the Netherlands; University of Oldenburg, Oldenburg, Germany). Cardiac foetal reprogramming: a tool to exploit novel treatment targets for the failing heart (Review Symposium). J Intern Med 2020;288:491–506. 10.1111/joim.13094
Content List ‐ Read more articles from the symposium: “Cardiovascular program and cardiovascular retreat”.
References
- 1. Jonker SS, Giraud MK, Giraud GD et al Cardiomyocyte enlargement, proliferation and maturation during chronic fetal anaemia in sheep. Exp Physiol 2010; 95: 131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J Appl Physiol 2007; 102: 1130–42. [DOI] [PubMed] [Google Scholar]
- 3. Smolich JJ, Walker AM, Campbell GR, Adamson TM. Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. Am J Physiol Circ Physiol 1989; 257: H1–9. [DOI] [PubMed] [Google Scholar]
- 4. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol 2013; 14: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kolwicz SC, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 2013; 113: 603–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z, Ying Z, Bosy‐Westphal A et al Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 2010; 92: 1369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kodde IF, van der Stok J, Smolenski RT, de Jong JW. Metabolic and genetic regulation of cardiac energy substrate preference. Comp Biochem Physiol Part A Mol Integr Physiol 2007; 146: 26–39. [DOI] [PubMed] [Google Scholar]
- 8. Fischer B, Bavister BD. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil 1993; 99: 673–9. [DOI] [PubMed] [Google Scholar]
- 9. Iyer NV, Kotch LE, Agani F et al Cellular and developmental control of O2 homeostasis by hypoxia‐inducible factor 1 alpha. Genes Dev 1998; 12: 149–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ryan HE, Lo J, Johnson RS. HIF‐1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17: 3005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wood SM, Wiesener MS, Yeates KM et al Selection and analysis of a mutant cell line defective in the hypoxia‐inducible factor‐1 alpha‐subunit (HIF‐1alpha). Characterization of hif‐1alpha‐dependent and ‐independent hypoxia‐inducible gene expression. J Biol Chem 1998; 273: 8360–8. [DOI] [PubMed] [Google Scholar]
- 12. Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF‐1‐mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–85. [DOI] [PubMed] [Google Scholar]
- 13. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF‐1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–97. [DOI] [PubMed] [Google Scholar]
- 14. Santalucía T, Camps M, Castelló A et al Developmental regulation of GLUT‐1 (erythroid/Hep G2) and GLUT‐4 (muscle/fat) glucose transporter expression in rat heart, skeletal muscle, and brown adipose tissue. Endocrinology 1992; 130: 837–46. [DOI] [PubMed] [Google Scholar]
- 15. Postic C, Leturque A, Printz RL et al Development and regulation of glucose transporter and hexokinase expression in rat. Am J Physiol 1994; 266: E548–59. [DOI] [PubMed] [Google Scholar]
- 16. Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol 1970; 218: 153–9. [DOI] [PubMed] [Google Scholar]
- 17. Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol 1974; 36: 413–59. [DOI] [PubMed] [Google Scholar]
- 18. Werner JC, Sicard RE. Lactate metabolism of isolated, perfused fetal, and newborn pig hearts. Pediatr Res 1987; 22: 552–6. [DOI] [PubMed] [Google Scholar]
- 19. Bartelds B, Gratama JW, Knoester H et al Perinatal changes in myocardial supply and flux of fatty acids, carbohydrates, and ketone bodies in lambs. Am J Physiol 1998; 274: H1962–9. [DOI] [PubMed] [Google Scholar]
- 20. Medina JM. The role of lactate as an energy substrate for the brain during the early neonatal period. Biol Neonate 1985; 48: 237–44. [DOI] [PubMed] [Google Scholar]
- 21. Lopaschuk GD, Spafford MA, Marsh DR. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol 1991; 261: H1698–705. [DOI] [PubMed] [Google Scholar]
- 22. Itoi T, Lopaschuk GD. The contribution of glycolysis, glucose oxidation, lactate oxidation, and fatty acid oxidation to ATP production in isolated biventricular working hearts from 2‐week‐old rabbits. Pediatr Res 1993; 34: 735–41. [DOI] [PubMed] [Google Scholar]
- 23. Fukushima A, Alrob OA, Zhang L et al Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol 2016; 311: H347–63. [DOI] [PubMed] [Google Scholar]
- 24. Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 2010; 56: 130–40. [DOI] [PubMed] [Google Scholar]
- 25. Rabi Y, Yee W, Chen SY, Singhal N. Oxygen saturation trends immediately after birth. J Pediatr 2006; 148: 590–4. [DOI] [PubMed] [Google Scholar]
- 26. Breckenridge RA, Piotrowska I, Ng KE et al Hypoxic regulation of Hand1 controls the fetal‐neonatal switch in cardiac metabolism. PLoS Biol 2013; 11: 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Belanger AJ, Luo Z, Vincent KA et al Hypoxia‐inducible factor 1 mediates hypoxia‐induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator‐activated receptor alpha/retinoid X receptor. Biochem Biophys Res Commun 2007; 364: 567–72. [DOI] [PubMed] [Google Scholar]
- 28. Panadero M, Herrera E, Bocos C. Peroxisome proliferator‐activated receptor‐alpha expression in rat liver during postnatal development. Biochimie 2000; 82: 723–6. [DOI] [PubMed] [Google Scholar]
- 29. Wang Y‐X, Lee C‐H, Tiep S et al Peroxisome‐proliferator‐activated receptor delta activates fat metabolism to prevent obesity. Cell 2003; 113: 159–70. [DOI] [PubMed] [Google Scholar]
- 30. Gilde AJ, van der Lee KAJM, Willemsen PHM et al Peroxisome proliferator‐activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res 2003; 92: 518–24. [DOI] [PubMed] [Google Scholar]
- 31. Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program. Ann N Y Acad Sci 2010; 1188: 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: role of thyroid hormone status and lipid supply. Biochem J 2000; 352(Pt 3): 731–8. [PMC free article] [PubMed] [Google Scholar]
- 33. Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 1998; 14: 263–83. [DOI] [PubMed] [Google Scholar]
- 34. Clerk A, Cullingford TE, Fuller SJ et al Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol 2007; 212: 311–22. [DOI] [PubMed] [Google Scholar]
- 35. van Bilsen M, Smeets PJH, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn‐out syndrome? Cardiovasc Res 2004; 61: 218–26. [DOI] [PubMed] [Google Scholar]
- 36. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation 2001; 104: 2923–31. [DOI] [PubMed] [Google Scholar]
- 37. Depre C, Shipley GL, Chen W et al Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med 1998; 4: 1269–75. [DOI] [PubMed] [Google Scholar]
- 38. el Alaoui‐Talibi Z, Landormy S, Loireau A, Moravec J. Fatty acid oxidation and mechanical performance of volume‐overloaded rat hearts. Am J Physiol 1992; 262: H1068–74. [DOI] [PubMed] [Google Scholar]
- 39. El Alaoui‐Talibi Z, Guendouz A, Moravec M, Moravec J. Control of oxidative metabolism in volume‐overloaded rat hearts: effect of propionyl‐L‐carnitine. Am J Physiol 1997; 272: H1615–24. [DOI] [PubMed] [Google Scholar]
- 40. Wambolt RB, Henning SL, English DR, Dyachkova Y, Lopaschuk GD, Allard MF. Glucose utilization and glycogen turnover are accelerated in hypertrophied rat hearts during severe low‐flow ischemia. J Mol Cell Cardiol 1999; 31: 493–502. [DOI] [PubMed] [Google Scholar]
- 41. Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 1994; 267: H742–50. [DOI] [PubMed] [Google Scholar]
- 42. Young ME, Patil S, Ying J et al Uncoupling protein 3 transcription is regulated by peroxisome proliferator‐activated receptor (alpha) in the adult rodent heart. FASEB J 2001; 15: 833–45. [DOI] [PubMed] [Google Scholar]
- 43. Minchenko O, Opentanova I, Caro J. Hypoxic regulation of the 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase gene family (PFKFB‐1‐4) expression in vivo. FEBS Lett 2003; 554: 264–70. [DOI] [PubMed] [Google Scholar]
- 44. Labinskyy V, Bellomo M, Chandler MP et al Chronic activation of peroxisome proliferator‐activated receptor‐α with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing‐induced heart failure. J Pharmacol Exp Ther 2007; 321: 165–71. [DOI] [PubMed] [Google Scholar]
- 45. Brigadeau F, Gelé P, Wibaux M et al The PPARα activator fenofibrate slows down the progression of the left ventricular dysfunction in porcine tachycardia‐induced cardiomyopathy. J Cardiovasc Pharmacol 2007; 49: 408–15. [DOI] [PubMed] [Google Scholar]
- 46. Wolff AA, Rotmensch HH, Stanley WC, Ferrari R. Metabolic approaches to the treatment of ischemic heart disease: the clinicians’ perspective. Heart Fail Rev 2002; 7: 187–203. [DOI] [PubMed] [Google Scholar]
- 47. Lionetti V, Linke A, Chandler MP et al Carnitine palmitoyl transferase‐I inhibition prevents ventricular remodeling and delays decompensation in pacing‐induced heart failure. Cardiovasc Res 2005; 66: 454–61. [DOI] [PubMed] [Google Scholar]
- 48. Schmidt‐Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci 2000; 99: 27–35. [PubMed] [Google Scholar]
- 49. Bristow M. Etomoxir: A new approach to treatment of chronic heart failure. Lancet 2000; 356: 1621–2. [DOI] [PubMed] [Google Scholar]
- 50. Holubarsch CJF, Rohrbach M, Karrasch M et al A double‐blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) 2007; 113: 205–12. [DOI] [PubMed] [Google Scholar]
- 51. Lee L, Campbell R, Scheuermann‐Freestone M et al Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short‐term use of a novel treatment. Circulation 2005; 112: 3280–8. [DOI] [PubMed] [Google Scholar]
- 52. Abozguia K, Elliott P, McKenna W et al Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010; 122: 1562–9. [DOI] [PubMed] [Google Scholar]
- 53. Ritter O, Peter H, Hannelore L et al Expression of atrial myosin light chains but not α ‐myosin heavy chains is correlated in vivo with increased ventricular function in patients with hypertrophic obstructive cardiomyopathy. J Mol Med 1999; 77: 677–85. [DOI] [PubMed] [Google Scholar]
- 54. Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med 1999; 77: 544–55. [DOI] [PubMed] [Google Scholar]
- 55. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res 2000; 86: 386–90. [DOI] [PubMed] [Google Scholar]
- 56. Lompre AM, Schwartz K, d’Albis A, Lacombe G, Van Thiem N, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature 1979; 282: 105–7. [DOI] [PubMed] [Google Scholar]
- 57. Schwartz K, Carrier L, Chassagne C, Wisnewsky C, Boheler KR. Regulation of myosin heavy chain and actin isogenes during cardiac growth and hypertrophy. Symp Soc Exp Biol 1992; 46: 265–72. [PubMed] [Google Scholar]
- 58. Schwartz K, Boheler KR, de la Bastie D, Lompre AM, Mercadier JJ. Switches in cardiac muscle gene expression as a result of pressure and volume overload. Am J Physiol 1992; 262: R364–9. [DOI] [PubMed] [Google Scholar]
- 59. Mayer Y, Czosnek H, Zeelon PE, Yaffe D, Nudel U. Expression of the genes coding for the skeletal muscle and cardiac actions in the heart. Nucleic Acids Res 1984; 12: 1087–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tondeleir D, Vandamme D, Vandekerckhove J, Ampe C, Lambrechts A. Actin isoform expression patterns during mammalian development and in pathology: Insights from mouse models. Cell Motil Cytoskeleton 2009; 66: 798–815. [DOI] [PubMed] [Google Scholar]
- 61. Suurmeijer AJ, Clément S, Francesconi A et al α‐Actin isoform distribution in normal and failing human heart: a morphological, morphometric, and biochemical study. J Pathol 2003; 199: 387–97. [DOI] [PubMed] [Google Scholar]
- 62. Franco D, Lamers WH, Moorman AFM. Patterns of expression in the developing myocardium: towards a morphologically intergrated transcriptional model. Cardiovasc Res 1998; 38: 25. [DOI] [PubMed] [Google Scholar]
- 63. Schwartz K, de la Bastie D, Bouveret P, Oliviéro P, Alonso S, Buckingham M. Alpha‐skeletal muscle actin mRNA’s accumulate in hypertrophied adult rat hearts. Circ Res 1986; 59: 551–5. [DOI] [PubMed] [Google Scholar]
- 64. Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest 1990; 85: 507–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boheler KR, Carrier L, de la Bastie D et al Skeletal actin mRNA increases in the human heart during ontogenic development and is the major isoform of control and failing adult hearts. J Clin Invest 1991; 88: 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuwahara K, Nishikimi T, Nakao K. Transcriptional regulation of the fetal cardiac gene program. J Pharmacol Sci 2012; 119: 198–203. [DOI] [PubMed] [Google Scholar]
- 67. Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim Biophys Acta ‐ Mol Basis Dis 2015; 1852: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hewett TE, Grupp IL, Grupp G, Robbins J. Alpha‐skeletal actin is associated with increased contractility in the mouse heart. Circ Res 1994; 74: 740–6. [DOI] [PubMed] [Google Scholar]
- 69. Ausoni S, De Nardi C, Moretti P, Gorza L, Schiaffino S. Developmental expression of rat cardiac Troponin‐I messenger‐RNA. Development 1991; 112: 1041–51. [DOI] [PubMed] [Google Scholar]
- 70. Schiaffino S, Gorza L, Ausoni S. Troponin isoform switching in the developing heart and its functional consequences. Trends Cardiovasc Med 1993; 3: 12–7. [DOI] [PubMed] [Google Scholar]
- 71. Kim S‐H, Kim H‐S, Lee M‐M. Re‐expression of fetal troponin isoforms in the postinfarction failing heart of the rat. Circ J 2002; 66: 959–64. [DOI] [PubMed] [Google Scholar]
- 72. Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of Titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 2004; 94: 505–13. [DOI] [PubMed] [Google Scholar]
- 73. Neagoe C, Kulke M, Del Monte F et al Titin isoform switch in ischemic human heart disease. Circulation 2002; 106: 1333–41. [DOI] [PubMed] [Google Scholar]
- 74. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev 2007; 12: 331–43. [DOI] [PubMed] [Google Scholar]
- 75. Planelles‐Herrero VJ, Hartman JJ, Robert‐Paganin J, Malik FI, Houdusse A. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat Commun 2017; 8: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu LC, Dorhout B, van der Meer P, Teerlink JR, Voors AA. Omecamtiv mecarbil: a new cardiac myosin activator for the treatment of heart failure. Expert Opin Investig Drugs 2016; 25: 117–27. [DOI] [PubMed] [Google Scholar]
- 77. Winkelmann DA, Forgacs E, Miller MT, Stock AM. Structural basis for drug‐induced allosteric changes to human β‐cardiac myosin motor activity. Nat Commun 2015; 6: 7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bakkehaug JP, Kildal AB, Engstad ET et al Myosin activator Omecamtiv Mecarbil increases myocardial oxygen consumption and impairs cardiac efficiency mediated by resting myosin ATPase activity clinical perspective. Circ Hear Fail 2015; 8: 766–75. [DOI] [PubMed] [Google Scholar]
- 79. Shen Y‐T, Malik FI, Zhao X et al Improvement of cardiac function by a cardiac myosin activator in conscious dogs with systolic heart failure. Circ Hear Fail 2010; 3: 522–7. [DOI] [PubMed] [Google Scholar]
- 80. Stroethoff M, Behmenburg F, Meierkord S et al Cardioprotective properties of omecamtiv mecarbil against ischemia and reperfusion injury. J Clin Med 2019; 8: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu Y, White HD, Belknap B, Winkelmann DA, Forgacs E. Omecamtiv mecarbil modulates the kinetic and motile properties of porcine β‐cardiac myosin. Biochemistry 2015; 54: 1963–75. [DOI] [PubMed] [Google Scholar]
- 82. Teerlink JR, Felker GM, McMurray JJV et al Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: the ATOMIC‐AHF study. J Am Coll Cardiol 2016; 67: 1444–55. [DOI] [PubMed] [Google Scholar]
- 83. Teerlink JR, Felker GM, McMurray JJV et al Chronic oral study of myosin activation to increase contractility in heart failure (COSMIC‐HF): a phase 2, pharmacokinetic, randomised, placebo‐controlled trial. Lancet 2016; 388: 2895–903. [DOI] [PubMed] [Google Scholar]
- 84. Teerlink JR, Diaz R, Felker GM et al Omecamtiv mecarbil in chronic heart failure with reduced ejection fraction. JACC Hear Fail 2020; 8: 329–40. [DOI] [PubMed] [Google Scholar]
- 85. Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large‐scale analysis of ion channel gene expression in the mouse heart during perinatal development. Genomics 2007; 28: 273–83. [DOI] [PubMed] [Google Scholar]
- 86. Schweizer PA, Thomas D, Zehelein J et al Transcription profiling of HCN‐channel isotypes throughout mouse cardiac development. Basic Res Cardiol 2009; 104: 621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Domínguez JN, de la Rosa A, Navarro F, Franco D, Aránega AE. Tissue distribution and subcellular localization of the cardiac sodium channel during mouse heart development. Cardiovasc Res 2008; 78: 45–52. [DOI] [PubMed] [Google Scholar]
- 88. Despa S, Bers DM. Na+ transport in the normal and failing heart ‐ remember the balance. J Mol Cell Cardiol 2013; 61: 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nattel S, Frelin Y, Gaborit N, Louault C, Demolombe S. Ion‐channel mRNA‐expression profiling: Insights into cardiac remodeling and arrhythmic substrates. J Mol Cell Cardiol 2010; 48: 96–105. [DOI] [PubMed] [Google Scholar]
- 90. Robertson C, Tran D, George S. Concise review: maturation phases of human pluripotent stem cell‐derived cardiomyocytes. Stem Cells 2013; 31: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. van den Berg CW, Okawa S, de Sousa Chuva et al Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015; 142: 3231–8. [DOI] [PubMed] [Google Scholar]
- 92. Borlak J, Thum T. Hallmarks of ion channel gene expression in end‐stage heart failure. FASEB J 2003; 17: 1592–608. [DOI] [PubMed] [Google Scholar]
- 93. Nattel S, Maguy A, Le Bouter S, Yeh Y‐H. Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 2007; 87: 425–56. [DOI] [PubMed] [Google Scholar]
- 94. Tomaselli GF, Beuckelmann DJ, Calkins HG et al Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation 1994; 90: 2534–9. [DOI] [PubMed] [Google Scholar]
- 95. Wickenden AD, Kaprielian R, Kassiri Z et al The role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc Res 1998; 37: 312–23. [DOI] [PubMed] [Google Scholar]
- 96. Hertzberg EL, Spray DC, Leinwand LA. Expression of Connexin43 in the developing rat heart. Circ Res 1991; 68: 782–7. [DOI] [PubMed] [Google Scholar]
- 97. Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res 2004; 62: 368–77. [DOI] [PubMed] [Google Scholar]
- 98. Van Kempen MJA, Fromaget C, Gros D, Moorman AFM, Lamers WH. Spatial distribution of Connexin43, the major cardiac gap junction protein, in the developing and adult rat. Heart 1991; 68: 1638–51. [DOI] [PubMed] [Google Scholar]
- 99. Fromaget C, el Aoumari A, Dupont E, Briand JP, Gros D. Changes in the expression of connexin 43, a cardiac gap junctional protein, during mouse heart development. J Mol Cell Cardiol 1990; 22: 1245–58. [DOI] [PubMed] [Google Scholar]
- 100. Wang Y, Hill JA, Akar FG, Tomaselli GF. Electrophysiological remodeling in heart failure. Electr Dis Hear Vol 1 Basic Found Prim Electr Dis 2013; 48: 369–86. [Google Scholar]
- 101. Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res 2008; 80: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Itzhaki I, Schiller J, Beyar R, Satin J, Gepstein L. Calcium handling in embryonic stem cell‐derived cardiac myocytes: of mice and men. Ann N Y Acad Sci 2006; 1080: 207–15. [DOI] [PubMed] [Google Scholar]
- 103. Tyser RCV, Miranda AMA, Chen CM, Davidson SM, Srinivas S, Riley PR. Calcium handling precedes cardiac differentiation to initiate the first heartbeat. Elife 2016; 5: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu J, Fu JD, Siu CW, Li RA. Functional sarcoplasmic reticulum for calcium handling of human embryonic stem cell‐derived cardiomyocytes: insights for driven maturation. Stem Cells 2007; 25: 3038–44. [DOI] [PubMed] [Google Scholar]
- 105. Ather S, Respress JL, Li N, Wehrens XHT. Alterations in ryanodine receptors and related proteins in heart failure. Biochim Biophys Acta ‐ Mol Basis Dis 2013; 1832: 2425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Scoote M, Williams AJ. The cardiac ryanodine receptor (calcium release channel): emerging role in heart failure and arrhythmia pathogenesis. CardiovascRes 2002; 56: 359–72. [DOI] [PubMed] [Google Scholar]
- 107. Dobrev D, Wehrens XHT. Role of RyR2 phosphorylation in heart failure and arrhythmiasresponse to Dobrev and Wehrens. Circ Res 2014; 114: 1311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Balke CW, Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res 1998; 37: 290–9. [DOI] [PubMed] [Google Scholar]
- 109. Lou Q, Janardhan A, Efimov IR. Remodeling of calcium handling in human heart failure. Adv Exp Med Biol 2012; 740: 1145–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. He H, Giordano FJ, Hilal‐Dandan R et al Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest 1997; 100: 380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen Y, Escoubet B, Prunier F et al Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation 2004; 109: 1898–903. [DOI] [PubMed] [Google Scholar]
- 112. Müller OJ, Lange M, Rattunde H et al Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res 2003; 59: 380–9. [DOI] [PubMed] [Google Scholar]
- 113. del Monte F, Williams E, Lebeche D et al Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)‐ATPase in a rat model of heart failure. Circulation 2001; 104: 1424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sakata S, Lebeche D, Sakata N et al Restoration of mechanical and energetic function in failing aortic‐banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol 2007; 42: 852–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jessup M, Greenberg B, Mancini D et al Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+‐ATPase in patients with advanced heart failure. Circulation 2011; 124: 304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zsebo K, Yaroshinsky A, Rudy JJ et al Long‐term effects of AAV1/SERCA2a gene transfer in patients with severe heart failurenovelty and significance. Circ Res 2014; 114: 101–8. [DOI] [PubMed] [Google Scholar]
- 117. Hulot J‐S, Salem J‐E, Redheuil A et al Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT‐HF randomized phase 2 trial. Eur J Heart Fail 2017; 19: 1534–41. [DOI] [PubMed] [Google Scholar]
- 118. Grund A, Szaroszyk M, Döppner JK et al A gene therapeutic approach to inhibit calcium and integrin binding protein 1 ameliorates maladaptive remodelling in pressure overload. Cardiovasc Res 2019; 115: 71–82. [DOI] [PubMed] [Google Scholar]
- 119. Laina A, Gatsiou A, Georgiopoulos G, Stamatelopoulos K, Stellos K. RNA therapeutics in cardiovascular precision medicine. Front Physiol 2018; 9: 953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Roe AT, Frisk M, Louch WE. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des 2015; 21: 431–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lo Giudice P, Mattera GG, Gagnol J‐P, Borsini F. Chronic istaroxime improves cardiac function and heart rate variability in cardiomyopathic hamsters. Cardiovasc Drugs Ther 2011; 25: 133–8. [DOI] [PubMed] [Google Scholar]
- 122. Mattera GG, Lo Giudice P, Loi FMP et al Istaroxime: A New Luso‐inotropic agent for heart failure. Am J Cardiol 2007; 99: S33–40. [DOI] [PubMed] [Google Scholar]
- 123. Gheorghiade M, Blair JEA, Filippatos GS et al Hemodynamic, echocardiographic, and neurohormonal effects of istaroxime, a novel intravenous inotropic and lusitropic agent. J Am Coll Cardiol 2008; 51: 2276–85. [DOI] [PubMed] [Google Scholar]
- 124. Shah SJ, Blair JEA, Filippatos GS et al Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON‐HF) trial. Am Heart J 2009; 157: 1035–41. [DOI] [PubMed] [Google Scholar]
- 125. Carubelli V, Zhang Y, Metra M et al Treatment with 24 hour istaroxime infusion in patients hospitalised for acute heart failure: a randomised, placebo‐controlled trial. Eur J Heart Fail 2020. 10.1002/ejhf.1743 [DOI] [PubMed] [Google Scholar]
- 126. de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci 1981; 28: 89–94. [DOI] [PubMed] [Google Scholar]
- 127. Curry F‐RE. Atrial natriuretic peptide: an essential physiological regulator of transvascular fluid, protein transport, and plasma volume. J Clin Invest 2005; 115: 1458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Cameron VA, Ellmers LJ. Minireview: Natriuretic peptides during development of the fetal heart and circulation. Endocrinology 2003; 144: 2191–4. [DOI] [PubMed] [Google Scholar]
- 129. Sergeeva IA, Christoffels VM. Regulation of expression of atrial and brain natriuretic peptide, biomarkers for heart development and disease. Biochim Biophys Acta ‐ Mol Basis Dis 2013; 1832: 2403–13. [DOI] [PubMed] [Google Scholar]
- 130. Woods RL. Cardioprotective functions of atrial natriuretic peptide and B‐type natriuretic peptide: a brief review. Clin Exp Pharmacol Physiol 2004; 31: 791–4. [DOI] [PubMed] [Google Scholar]
- 131. Davidovski FS, Goetze JP. ProANP and proBNP in plasma as biomarkers of heart failure. Biomark Med 2019; 13: 1129–35. [DOI] [PubMed] [Google Scholar]
- 132. Almufleh A, Marbach J, Chih S et al Ejection fraction improvement and reverse remodeling achieved with Sacubitril/Valsartan in heart failure with reduced ejection fraction patients. Am J Cardiovasc Dis 2017; 7: 108–13. [PMC free article] [PubMed] [Google Scholar]
- 133. Suematsu Y, Jing W, Nunes A et al LCZ696 (Sacubitril/Valsartan), an angiotensin‐receptor neprilysin inhibitor, attenuates cardiac hypertrophy, fibrosis, and vasculopathy in a rat model of chronic kidney disease. J Card Fail 2018; 24: 266–75. [DOI] [PubMed] [Google Scholar]
- 134. McMurray JJV, Packer M, Desai AS et al Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371: 993–1004. [DOI] [PubMed] [Google Scholar]
- 135. Velazquez EJ, Morrow DA, DeVore AD et al Angiotensin‐neprilysin inhibition in acute decompensated heart failure. N Engl J Med 2019; 380: 539–48. [DOI] [PubMed] [Google Scholar]
- 136. Ponikowski P, Voors AA, Anker SD et al 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 137. Chen B‐Y, Chen J‐K, Zhu M‐Z et al A novel chimeric peptide with natriuretic and vasorelaxing actions. Zoccali C, ed. PLoS One 2011; 6: e20477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. From AHL. Do engineered natriuretic peptides have greater therapeutic potential than do native peptides? Cardiovasc Res 2010; 88: 391–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wylie JV, Tsao L. Nesiritide for the treatment of decompensated heart failure. Expert Rev Cardiovasc Ther 2004; 2: 803–13. [DOI] [PubMed] [Google Scholar]
- 140. van der Pol A, Gil A, Silljé HHW et al Accumulation of 5‐oxoproline in myocardial dysfunction and the protective effects of OPLAH. Sci Transl Med 2017; 9: eaam8574. [DOI] [PubMed] [Google Scholar]
- 141. Meister A, Anderson ME. Glutathione. Annu Rev Biochem 1983; 52: 711–60. [DOI] [PubMed] [Google Scholar]
- 142. Liu Y, Hyde AS, Simpson MA, Barycki JJ. Emerging regulatory paradigms in glutathione metabolism. Adv Cancer Res 2014; 122: 69–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yang K‐C, Yamada KA, Patel AY et al Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation 2014; 129: 1009–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Pederzolli CD, Sgaravatti AM, Braum CA et al 5‐Oxoproline reduces non‐enzymatic antioxidant defenses in vitro in rat brain. Metab Brain Dis 2007; 22: 51–65. [DOI] [PubMed] [Google Scholar]
- 145. Pederzolli CD, Mescka CP, Zandoná BR et al Acute administration of 5‐oxoproline induces oxidative damage to lipids and proteins and impairs antioxidant defenses in cerebral cortex and cerebellum of young rats. Metab Brain Dis 2010; 25: 145–54. [DOI] [PubMed] [Google Scholar]