

Peripheral immune responses of Alzheimer’s disease patients are uniquely different from those of healthy and disease controls.
Abstract
Peripheral blood mononuclear cells (PBMCs) may provide insight into the pathogenesis of Alzheimer’s disease (AD) or Parkinson’s disease (PD). We investigated PBMC samples from 132 well-characterized research participants using seven canonical immune stimulants, mass cytometric identification of 35 PBMC subsets, and single-cell quantification of 15 intracellular signaling markers, followed by machine learning model development to increase predictive power. From these, three main intracellular signaling pathways were identified specifically in PBMC subsets from people with AD versus controls: reduced activation of PLCγ2 across many cell types and stimulations and selectively variable activation of STAT1 and STAT5, depending on stimulant and cell type. Our findings functionally buttress the now multiply-validated observation that a rare coding variant in PLCG2 is associated with a decreased risk of AD. Together, these data suggest enhanced PLCγ2 activity as a potential new therapeutic target for AD with a readily accessible pharmacodynamic biomarker.
INTRODUCTION
Many laboratories have tested the hypothesis that peripheral blood mononuclear cells (PBMCs) can provide a window into the pathogenesis of neurodegenerative diseases, either because a subset of them, e.g., specific T cells or monocytes, traffic into the brain and thus may directly participate in disease mechanisms or because an inherited or acquired trait shared between PBMCs and brain cells might serve as a biomarker of neuroinflammation (1, 2). PubMed lists more than 1200 citations for human leukocytes and Alzheimer’s disease (AD) and more than 550 citations for human leukocytes and Parkinson’s disease (PD) (3–5). There has been some success using quantitative traits in leukocytes to validate genetic risk loci for AD or PD (6, 7); however, the outcome of these studies has not yet been broadly reproduced across research laboratories. This could be because of diagnostic misclassification, lack of a validation cohort, or challenges to cell-specific resolution. Hence, none has transitioned to serve as a blood-based clinical biomarker of AD or PD (8).
Although it is difficult to generalize across such a large number of studies, a few themes emerge. Most studies assembled PBMCs from relatively small groups of individuals with variable clinical characterization. Most used unstimulated PBMCs, which means that they likely were in variable states of activation due to site-specific processes of collection and enrichment. A few studies used ex vivo PBMC stimulants to investigate immune responses, but the repertoire of stimulants has been limited thus far to nonspecific stimuli such as propylene glycol monomethyl ether acetate (PMA)/ionomycin or noncanonical stimuli such as β-amyloid (Aβ) or α-synuclein, which activate through incompletely understood mechanisms (9). Relatively few studies have used flow cytometry to isolate specific subsets of PBMCs, and even those that used conventional flow cytometry identified only a limited number of cell types (10). Of those that measured secreted molecules, most measured only one or a small number; measuring a larger number of secreted factors has thus far been irreproducible (11, 12). Last, only a few previous studies (8) have included multiple neurodegenerative diseases to control for features of being chronically ill.
To address these limitations, we designed a study with a more comprehensive (i) cohort, (ii) range of stimuli, (iii) PBMC subtype identification, and (iv) response molecule repertoire. We assembled samples from 132 individuals who had undergone extensive research-quality clinical assessment with diagnosis performed by a consensus panel of experts. Our discovery cohort comprised sporadic (lacking monogenetic cause) AD dementia, sporadic PD, matched healthy controls (HCs), and younger HCs. Our validation cohort comprised AD dementia and matched HCs. PMBCs were stimulated by one of seven canonical activators chosen to provide a range of activation: pro- and anti-inflammatory cytokines that act through specific receptors [interferon-α (IFN-α), interleukin-6 (IL-6), IL-7, IL-10, and IL-21]; a powerful stimulant of innate immune response that activates via Toll-like receptor 4 (TLR4) [lipopolysaccharide (LPS)]; and non–receptor-mediated activation (PMA and ionomycin), along with an unstimulated control. Using time-of-flight mass cytometry (CyTOF), a proteomics technology that assesses the abundance of cell subsets, protein expression, and activation of signaling pathways with single-cell resolution (13–18), we assayed 15 different intracellular signaling responses across 35 PBMC cell types (determined by a combination of 12 cell surface markers). Thus, each individual’s PBMC sample yielded a total of 4200 (8 stimulant conditions × 35 subtypes × 15 responses) intracellular signaling responses. We then used machine learning algorithms and statistical analyses to identify patterns of each diagnosis group from these extensive single-cell data and examine the generalizability and predictive power of the findings.
RESULTS
Single-cell profiling of PBMC response in patients with AD, patients with PD, and HCs
The goal of our study was to determine which components of the peripheral immune response correlated with diagnoses of AD or PD. As outlined in Fig. 1, we performed deep single-cell profiling of PBMCs from our discovery cohort (AD, PD, HC-I, and HC-II; Fig. 1A) in response to a panel of ex vivo stimulants (Fig. 1B) that activate known cell signaling mechanisms. The results from our discovery work (Fig. 1C) could then be used to further test the developed model, without any retraining, on a separate validation cohort comprising patients with AD (AD-V) and matched HCs (HC-V) (Fig. 1A).
Fig. 1. Experimental and analytical workflow from obtaining PBMCs to identifying potential immune cell markers.
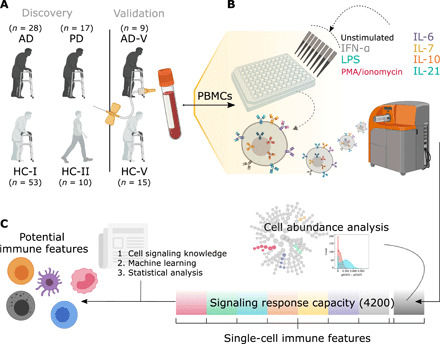
(A) In the discovery cohorts, whole blood was collected from 28 individuals with AD and 17 individuals with PD; AD was compared with the samples from 53 older HCs (HC-I), while PD samples were compared with a subset of those with age- and sex-matched HCs (HC-Isub). A different set of 10 younger HCs (HC-II) was included for examining age effects. In addition, an independent cohort of nine individuals with AD (AD-V) and 15 HCs (HC-V) was used for validation of the developed machine learning models without retraining. (B) PBMCs were either unstimulated or stimulated with IFN-α, IL-6, IL-7, IL-10, IL-21, LPS, or PMA/ionomycin. PBMCs were then bound with 21 metal-conjugated antibodies to surface markers and 15 metal-conjugated antibodies to intracellular signaling molecules before analysis by CyTOF. (C) Cell abundance was evaluated on PBMCs from an unstimulated condition. The stimulations and antibody probes generated a total of 4200 intracellular signaling responses (35 PMBC subtypes under eight stimulating conditions and assayed for 15 intracellular responses), which were used to identify the potential immune features with the aid of cell signaling knowledge, machine learning methods, and statistical analysis.
We first assessed the relative abundance of PBMC subsets before stimulation (fig. S1). Using our panel of 15 cell surface markers [CD3, CD4, CD7, CD8, CD11b and CD11c, CD14, CD16, CD19, CD20, CD24, CD25, CD27, CD38, CD45RA, CD56, CD123, and CD127; immunoglobulin A (IgA) and IgD; and human leukocyte antigen–DR isotype (HLA-DR)], we subtyped them into monocytes, CD8+ T cells, CD4+ T cells, CD3+ (CD4− CD8−) T cells, B cells, natural killer (NK) cells, and dendritic cells (DCs) (fig. S1A). Their relative abundance was characterized by a significance analysis of microarrays (19) with equal events sampled (minimal cluster size of 1% and false discovery rate of 1%). CITRUS (cluster identification, characterization, and regression) was used to compare PBMC subtype abundance for AD/HC-I and for PD/HC-Isub. Unsupervised hierarchical clustering of age- and sex-matched groups identified no significant differences among these well-established PBMC subtypes in paired comparison of AD or PD with appropriate HCs. Although less well matched for age and sex, we found significant differences between AD and PD samples (fig. S1).
We detected stimulant (IFN-α; IL-6, IL-7, IL-10, and IL-21; LPS; or PMA/ionomycin) response with additional probes directed at 15 intracellular signaling molecules [pERK-1/2, IκBα, nuclear factor κB (NF-κB), p38, pAKT, pCREB, pLCK, pPLCγ2, pS6, pSTAT1, pSTAT3, pSTAT5, Lamp2, EEA1, and Rab5] (Fig. 2). Signals obtained from AD, PD, and HC-I PBMCs in unstimulated state or in response to IFN-α, IL-6, and IL-7 tended to be more strongly correlated to each other as indicated by proximity and the communities formed by these features in the correlation network (Fig. 2A). Similarly, responses to IL-10, IL-21, LPS, and PMA/ionomycin also tended to group together. Visualizing the pairwise correlation matrix and distributions further confirmed this behavior (fig. S2). To obtain a more meaningful description of each community, we performed dimension reduction (UMAP, uniform manifold approximation and projection) followed by unsupervised clustering (k-means) to segment the network into 24 annotated communities (Fig. 2B). Annotation was based on the PBMC subtype, stimulant, and/or intracellular signal that appeared most frequently in each community. Immune features with the same intracellular signals were likely to belong to the same community. In some communities, features could be further subdivided by cell type and stimulant. Detailed community annotations are presented in table S1.
Fig. 2. Responses from the same intracellular signaling proteins are highly correlated to each other.
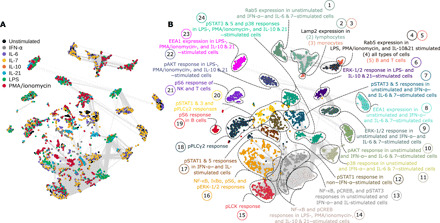
(A) Correlation network (Spearman’s coefficient) of immune features obtained from CyTOF data of HC-I, AD, and PD colored by the type of stimulant. The edges of the network represent features with Spearman’s coefficient higher than 0.8. (B) An unsupervised algorithm clustered the network into 24 communities, where their annotations are based on commonly shared feature attributes (PBMC subtype, stimulation, or signaling property) within the community. ERK-1/2, extracellular signal–regulated kinase–1/2.
iEN model developed from immune responses predicted AD diagnosis
Given these highly correlated immune features, we constructed a multivariate model using immunological Elastic Net (iEN), a recently developed regression algorithm designed for immune signals (20), and examined its predictive power on unseen data as an alternative to multiple univariate testing (Fig. 3). In the discovery cohort, the cross-validated results indicated satisfactory classification of AD/HC-I [P = 1.03 × 10−3, area under receiver operating characteristic (ROC) curve (AUC) = 0.72 ± 0.06; Fig. 3, A and B] but poor performance for PD/HC-Isub (P >> 0.05). The model was then tested on separate validation cohorts: HC-V and AD-V. The model achieved accurate AD-V/HC-V classification (P = 5.42 × 10−3, AUC = 0.84 ± 0.08; Fig. 3, A and B). This suggested generalizability of the model; however, it should be noted that because of the low number of samples in the validation cohort, the higher AUC in the validation cohorts could be by chance, and this AUC was not significantly different from the discovery cohort’s AUC (P = 0.27; Fig. 3B). The power of the resulting iEN predicted values were 0.92 and 0.86, at 0.05 significance level, for the discovery and validation cohort, respectively.
Fig. 3. The iEN model can satisfactorily classify AD/HC-I in both discovery and validation cohorts with the most important model components associated with signals from pPLCγ2 and pSTATs.
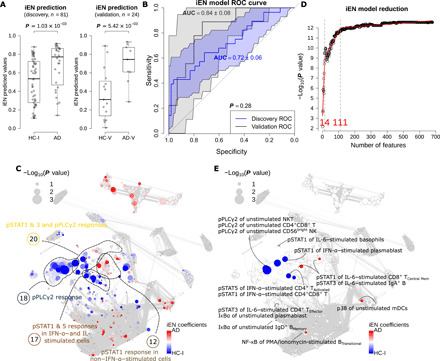
(A) Box plots showing the predicted values from iEN model with Wilcoxon rank sum test P value for discovery and validation cohorts. (B) ROC curves from the iEN model predictions of discovery and validation cohorts with their respective AUC and P values from unpaired t test indicating no significant difference between the discovery and validation’s AUCs. (C) Correlation network colored by iEN model components with red and blue colors highlighting the components that are indicative of AD and HC-I, respectively. The size of the nodes represents the Spearman’s coefficient of the immune feature to the respective ground truths. (D) A model reduction analysis looking at the effect of the number of included features on iEN performance. (E) The correlation network colored and annotated only for the top 14 features that were associated with components selected from model reduction.
Note that one of the inputs to the iEN model was a list of immune features that represent literature-based canonical signaling pathways to prioritize during model optimization. In addition, we tried using signals that were experimentally present in this study and found a ~10% discrepancy compared to canonical features (fig. S3), with no significant effect on the model performance. Other conventional algorithms were also attempted but obtained less accuracy.
Analyses of the model components revealed broader cellular differences between AD and HC-I
Coefficients assigned to the immune features by the iEN model could be used as a proxy to examine biological plausibility. Figure 3C shows these coefficients displayed on a correlation network, with red color indicating immune features that increased with the likelihood of carrying the AD diagnosis and blue indicating features that decreased. Piecewise regression analysis of the model revealed that only about 14 of the features with the highest coefficient magnitudes were necessary, and 111 components were needed to get a comparable performance to using the entire set of immune features (4200 components; Fig. 3D). These top 14 and 111 features were depicted on the correlation networks (Fig. 3E and fig. S4A). We next explored the four communities that contained the top 14 features, since features within the same community were highly correlated, and exhibited a high univariate P value of the immune feature to diagnosis: communities 18, 12, 17, and 20 (Fig. 3C).
Community 18 comprised features with pPLCγ2 intracellular signal. Visualizing the differences in pPLCγ2 signal between the HC-I and the AD participants by PBMC subtypes and stimulant highlighted strong differences from diverse cell types (with the exception of most CD4+ T cells and DCs) under unstimulated and various stimulated conditions (Fig. 4A and fig. S4C), where all of the responses tended to be lower in the AD group (fig. S4B), particularly in NKT cells (Fig. 4, D and E). Among these are the most informative components of the iEN model, including “pPLCγ2 response in unstimulated NKT, CD4+CD8+ T, and CD56bright NK cells” (Fig. 4, D and F, and fig. S4C). These findings provide functional data that buttress the previous association that a variant in the gene that encodes phospholipase C–γ2 (PLCγ2) lowers the risk of AD (21). To determine whether any single pPLCγ2 signal alone could serve as a standalone diagnostic marker without the need of iEN or other machine learning models, we performed cross-validation tests on each of them with evaluation by F1-score (Fig. 4I and see Supplementary Methods). We found that no single feature was sufficiently robust to yield good accuracy in both discovery and validation cohorts and that the iEN method was necessary to help address variations among cohorts (Fig. 4, J and K).
Fig. 4. Heatmaps and box plots of the intracellular response in the PBMC of the selected communities highlight immune features for AD/HC-I classification and examination of the predictive power by F1-score of pPLCγ2 as a standalone biomarker.
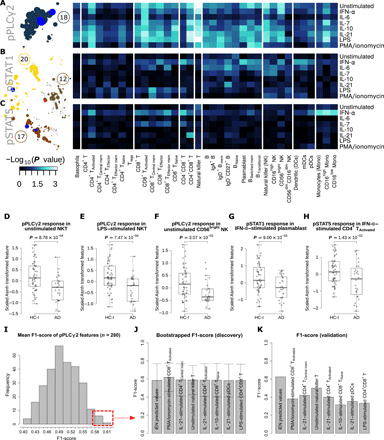
(A to C) Heatmap of the pPLCγ2, pSTAT1, and pSTAT5 responses by PBMC subtypes and stimulations. The color of the heatmap scaled with the Wilcoxon rank sum test P value of the difference in response of the immune feature between HC-I and patients with AD. The network communities annotated with these responses (communities 12, 17, 18, and 20) were depicted on the left-hand side of the heatmap. The size of the nodes in the community corresponds to the Spearman’s coefficient of the immune feature. The features within the communities that were selected by reduced iEN model (14 components) retained their red/blue colors corresponding to the direction of the component. (D to H) Box plots showing the significant difference of the selected immune features from the heatmap. These are mostly features associated with the most informing components of the iEN model. (I) The distribution of the mean F1-score in the test set from 1000 iterations of leave-group out test for each of the 280 pPLCγ2 features from different cell types and stimulating conditions in the discovery set. (J) The mean F1-score and its SD for the top seven performing pPLCγ2 features in the discovery cohort compared to the iEN predicted values. (K) The F1-score for each of the top seven features and iEN predicted values in the validation set. Tregs, regulatory T cells.
Communities 12, 17, and 20 were primarily a mix of pSTAT1 and pSTAT5 responses, which were highly correlated with each other. Examining the strength of these signal differences between HC-I and AD by cell type and stimulant using univariate analysis revealed that for both pSTAT1 and pSTAT5 intracellular responses, the strongest signals were mostly from IFN-α–stimulated cells (Fig. 4, B and C). However, unlike pPLCγ2, the trends of both intracellular responses were not monotonic across cell types and stimulations; the responses tended to be higher in HC-I only in IFN-α–stimulated cells or certain types of CD4+ T cells, while other conditions mostly led to an opposite response (fig. S4B). All of the pSTAT features among the top 11 components of the iEN model with a high univariate P value were IFN-α–stimulated cells, including “pSTAT1 response of IFN-α–stimulated plasmablast and CD4+CD8+ T cells” and “pSTAT5 response of IFN-α–stimulated CD4+ TActivated” (Fig. 4, G and H, and fig. S4, D and E). These reflected differential Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling after IFN-α stimulation between the two diagnostic groups. In addition, other pSTAT signals that were expected such as pSTAT1 and pSTAT5 from IL-7–stimulated CD4+ and CD8+ cells also were observed. Conversely, those that were not expected, such as pSTAT signaling from LPS and PMA/ionomycin-stimulated cells, were not observed (fig. S5) (22, 23). These alignments to the existing signaling knowledge further supported the strength of our experimental approach.
AD/HC-I responses were different between sexes and were not driven by aging
The change in univariate P values depicted on the correlation network when separated by sex strongly suggested that most of the differences in pPLCγ2 responses stemmed from male participants, whereas pSTATs were mostly contributed by female participants in both the discovery and validation cohorts (fig. S6, A and B). The pPLCγ2 response was very similar even if only patients with AD were separated by sex and compared to the entire HC-I (fig. S6, C and D), suggesting sex-specific immune responses in patients with AD. Sex-specific immune responses are not uncommon and are being studied actively (23). In addition, for participants with known APOE genotype, separating patients with AD into those with or without an APOE ε4 allele and then comparing both with HC-I with no APOE ε4 implied relevance of pPLCγ2 in both APOE subgroups of AD (fig. S7, A and B). In contrast, the significance of pSTAT signals was reduced in both APOE subgroups. This was potentially due to fewer female participants in the AD group. The results were similar if the entire HC-I cohort was used regardless of APOE ε4 status. As we earlier associated pPLCγ2 with male AD participants, narrowing the APOE analyses only to males revealed that pPLCγ2 signals mostly persisted whether the male HC-I or male AD did or did not have the APOE ε4 allele (fig. S7, C and D), suggesting that pPLCγ2 response is independent of the APOE status. Others have shown repeatedly that macrophage and microglial innate immune responses have apolipoprotein E (apoE) isoform–dependent components in experimental settings (24–26); however, we are aware of only one report from a small number of participants that observed a modest apoE isoform–specific effect on human PBMC response to PMA/ionomycin stimulation (27). The number of observations in each of these subanalyses is limited and consequently more susceptible to potential unknown confounding factors.
Another possibility is that the pPLCγ2 and pSTAT differences observed between AD and HC-I might be due to changes of aging that are accentuated in the AD group. However, correlation networks between HC-II/HC-I and HC-II/AD (fig. S8, A and B) implied that, for both pairs, only a small number of signal differences were contributed by pSTATs, with no significant contribution from pPLCγ2 region. In addition, the specific pSTAT regions were different from those highlighted by HC-I/AD comparison. These results further increase confidence that differential PBMC responses in pPLCγ2 and some pSTATs are specific to AD.
Disease cross-prediction using the AD/HC-I model indicated some similarity between AD and PD
The prediction accuracy of the developed iEN models from AD/HC-I could be used as a proxy to determine the similarities between the diagnostic groups (Fig. 5). As expected, the developed models could perfectly classify the AD diagnostic group on which it was trained (P = 2.59 × 10−13, AUC = 1.00 ± 0.00; Fig. 5, A and B). Disease cross-prediction to PD, i.e., using an AD/HC-I–trained model to classify PD/HC-Isub participants, resulted in a satisfactory performance (P = 1.75 × 10−6, AUC = 0.88 ± 0.06) although not as high as in the original domain. The correlation network of PD/HC-Isub diagnostic groups illustrated potential regions containing features associated with the developed iEN components from AD/HC-I (strong colors) that were also significantly different between PD and HC-Isub (sizable nodes) such as those in community 20 (Fig. 5C). Accurate PD/HC-Isub prediction may indicate comorbidity, shared risk factors, or shared mechanisms between the two neurodegenerative diseases. However, other communities whose features were important in the AD/HC-I iEN model—particularly communities 12, 17 (pSTATs region), and 18 (pPLCγ2 region)—did not overlap with signal differences in PD/HC-Isub. Hence, the model also can separate AD and PD patients (P = 3.99 × 10−3, AUC = 0.75 ± 0.06), although the accuracy decreased when combined with AD-V predictions (P = 1.70 × 10−2, AUC = 0.70 ± 0.07), suggesting that it did not merely classify between HC and those with an unspecified neurodegenerative disease.
Fig. 5. Disease cross-prediction reveals similarities between AD and PD.
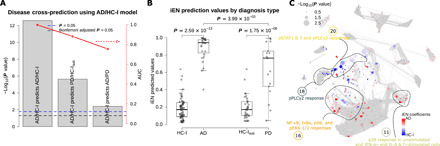
(A) Performance of the disease cross-prediction using iEN components developed from AD/HC-I diagnosis to classify PD/HC-Isub and AD/PD. (B) The iEN predicted values for each diagnostic group. (C) The correlation network with node size corresponding to the Wilcoxon rank sum test P value of each feature for PD/HC-Isub diagnosis, with the color of each node representing the magnitude and direction of the associated iEN components developed from AD/HC-I. The network highlighted possible regions, such as in the labeled clusters, where AD and PD signals can overlap.
DISCUSSION
Our work joins a range of previous publications that have tested whether PBMCs might provide insight into the pathogenesis of neurodegenerative disease, either because some subset of PBMCs traffic into the brain and directly participate in disease mechanisms (1, 2) or because of inherited or acquired traits shared between some PBMCs and some brain-resident immunocompetent cells. As outlined in the Introduction, several groups have attempted to identify subsets of PBMCs or other peripheral immune markers in unstimulated samples that are characteristic of AD, and none of these have been broadly replicated. More recently, colleagues have highlighted an enriched subpopulation of CD8+ T cells in unstimulated samples in roughly equal number of participants diagnosed with AD dementia or mild cognitive impairment compared to matched controls (28). Although we were unable to validate this finding in our unstimulated samples from participants with AD dementia compared to matched controls, we hasten to add that, in addition to the difference in diagnostic groups studied, our focus was on the response of peripheral immune cells to well-characterized stimuli, rather than unstimulated samples, which appear to have greater variability. Our robust study design is distinguished by a large sample set from well-matched participants with research-quality annotation, multiple canonical immune stimulants, flow cytometric identification of 35 PBMC subsets, single-cell quantification of 15 intracellular signaling markers, inclusion of a neurodegenerative disease control in addition to matched HC, and validation of major findings with a completely independent cohort. Our data analysis is distinguished by the application of machine learning, instead of univariate testing, to demonstrate the accuracy and generalizability of the findings from this complex set of diagnostic phenotypes and single-cell quantitative molecular data. Our results showed that 3 of the 15 intracellular signaling pathways were differentially activated in PBMC subsets from people with AD compared to age- and sex-matched HC: muted activation of PLCγ2 across many cell types and stimulations and more selective activation of STAT1 and STAT5 depending on stimulant and cell type. As far as we are aware, none of the previous studies that focused on PBMCs in AD have highlighted altered responses by PLCγ2, STAT1, or STAT5.
Reduced pPLCγ2 activation (community 18) was a strong feature of AD in all major subclasses of cells except DCs and most CD4+ T cells and under all stimulation conditions except IL-6 (Fig. 4A). Although pPLCγ2 differential response was observed across most stimulants, it appeared strongest with LPS. pSTAT1 (community 20) and pSTAT5 (community 17) differential responses were more selective and largely restricted to IFN-α stimulation. pSTAT5 differential response was focused strongly in monocytes (Fig. 4C), while pSTAT1 differential response was weaker and more broadly observed in CD8+ T cells, B cells, and NK cells but not in monocytes (Fig. 4B). These differential immune responses have potential pathogenetic relevance to AD. Aβ peptides directly and indirectly activate TLR4, the pattern recognition receptor for LPS (29, 30). STAT1 tissue concentration is increased in diseased regions of AD brain (31) and can regulate expression of β secretase 1, one of the endoproteases that sequentially catalyzes the hydrolysis of amyloid precursor protein to generate Aβ peptides (32). STAT5 activation regulates microglial activation and is required for monocyte-mediated synaptic degradation (33). A recent pathway analysis that combined multiple GWAS (genome-wide association study) data with clinical annotation nominated JAK-STAT signaling abnormalities as prominent contributors to AD etiology or pathogenesis (34). Last, both T cells and monocytes can traffic from peripheral blood into the brain, at least raising the possibility that the differential responses observed in these subsets of PBMCs might access the brain, rather than simply being peripheral biomarkers of brain immune responses.
A rare variant in the gene that encodes pPLCγ2 (PCLG2; rs72824905, P522R) recently has been linked to a decreased risk of AD (21), a finding that was recently validated in three other diverse cohorts (35–37). One of these validation studies also demonstrated that rs72824905 is associated with decreased risk of other forms of dementia but showed no risk modification for PD despite adequate sample size (36). None of these four genetic association studies evaluated the influence of sex or APOE genotype on PLCG2 risk modification for AD. Three groups have published results from human and mouse brain showing that expression of PLCG2 is limited to microglia among the major cell types in the brain and that the protective P522R polymorphism modestly increases enzyme activity (35, 36, 38). These genetic risk findings implicate PLCG2-dependent pathways as being important in AD pathogenesis. Our functional results from the peripheral immune system are consistent with these earlier findings; however, this rare variant in PLCG2 cannot be the mechanism underlying the common functional changes we observed in AD participants. We speculate that other yet-to-be-clarified epigenetic or environmental mechanisms are affecting activation of PLCγ2 in AD. Together, these data raise the possibility that increasing PLCγ2 activity may be a new therapeutic target for AD and perhaps other forms of dementia. Moreover, our data suggest that a PBMC biomarker may be developed to aid in screening for therapeutics that enhances PLCγ2 activity. Although PLCγ2 activation alone does not appear to be a diagnostic biomarker in PBMCs, it might have a potential application as a pharmacodynamic biomarker. However, it should be kept in mind that other PLCG2 variants have been asssociated with immune dysregulation (39), warranting caution in attempting to modulate its activity.
The role of microglia and the immune response in AD remains unclear but is an area of very active investigation highlighted by single-cell approaches (40). On the basis of epidemiologic observations and results from transgenic mouse models, initial hypotheses focused on increased immune response in AD brain; however, recent observational and experimental data support a more nuanced view of both pro- and anti-inflammatory facets to AD progression (41). Our work fits well within this contemporary construct by revealing reduced and increased peripheral immune responses as characteristic of AD.
The iEN algorithm was unable to develop a predictive model for PD despite a similarly sized sample set as AD. Although disappointing, these results enhance our confidence in the predictive model for AD by controlling for nonspecific features of age-related neurodegenerative disease, like reduced physical activity. Our disease cross-prediction analysis showed that the AD/HC-I model much more weakly, but significantly, predicted PD/HC-Isub. There are several possible explanations for this outcome including partially shared genetic or environmental risk factors or partially shared neurodegenerative mechanisms for AD and PD. The latter is supported by the now well-established observation that the neuropathologic hallmark of PD (Lewy body disease) is present in about one-third to one-half of people diagnosed clinically with AD despite using the most rigorous research criteria. Alternatively, this disease cross-prediction may indicate partially shared alterations in the peripheral immune response between people with AD or PD. In this respect, disease cross-prediction of PD was driven largely by community 20 and not by community 18 (pPLCγ2 region; Fig. 5C), consonant with the finding that the genetic variant in PLCG2 that lowers risk for AD does not modulate risk of PD (36).
Our extensive and unbiased investigation and robust analysis of the peripheral immune response strongly implicates reduced activation of PLCγ2 as a molecular characteristic of AD but not PD or advancing age. Our experimental data from patient samples functionally buttress the now multiply-validated observations that a rare coding variant in PLCG2 is associated with decreased risk of AD (36). Last, our analyses suggest that altered PLCγ2 activity in PBMCs is more commonly a feature of men with AD and that it is not strongly influenced by APOE genotype or aging. Together, these data point to enhancing PLCγ2 activity as a potentially new therapeutic target for AD that has a readily accessible pharmacodynamic biomarker.
MATERIALS AND METHODS
Study design
The aim of this study was to determine whether differences in peripheral immune responses between healthy participants and participants with neurodegenerative diseases are detectable by CyTOF analysis of PBMCs. Participants were research volunteers at Stanford University in the Alzheimer’s Disease Research Center or the Pacific Udall Center. All participants provided written informed consent to participate in the study, which followed protocols approved by the Stanford Institutional Review Board. Clinical diagnosis was made by consensus criteria (42–44) (see the Supplementary Materials for details).
Blood was collected from volunteers after obtaining informed consent. We assembled a discovery cohort (n = 108) and a completely separate validation cohort (n = 24; summary statistics is shown in Table 1, and individual information is shown in tables S2 and S3). The discovery cohort consisted of four groups: AD, PD, and two different HC groups. The first HCs (HC-I) were older and were matched for AD with a subgroup of these people (HC-Isub; ages between 63 and 80 for male and 67 and 73 for female) matched for PD. The second HCs (HC-II) were younger. A separate validation cohort also was assembled: AD-V and HC-V. No validation cohort was assembled for PD because there was no generalizable model identified from the discovery cohort. A separate technical quality control of PBMCs was prepared from a single healthy individual (70-year-old man) not included in the discovery or validation cohorts and frozen into multiple aliquots and included in every CyTOF run.
Table 1. Summary of participants in the discovery (n = 108) and validation cohorts (n = 24) diagnosed with AD, PD, or HCs.
| Cohort | Group | n | Age | |
| Discovery | HC-I | M | 29 | 75 ± 8 |
| F | 24 | 68 ± 6 | ||
| AD | M | 14 | 74 ± 10 | |
| F | 14 | 67 ± 9 | ||
| HC-Isub | M | 22 | 73 ± 5 | |
| F | 12 | 71 ± 2 | ||
| PD | M | 11 | 70 ± 5 | |
| F | 6 | 71 ± 3 | ||
| HC-II | M | 4 | 40 ± 9 | |
| F | 6 | 40 ± 10 | ||
| Validation | HC-V | M | 6 | 71 ± 10 |
| F | 9 | 70 ± 6 | ||
| AD-V | M | 4 | 74 ± 12 | |
| F | 5 | 71 ± 10 |
Metal-tagged monoclonal antibodies
A panel of 37 metal-tagged monoclonal antibodies was used to probe PBMCs (table S4). All preconjugated antibodies were purchased from Fluidigm. All other antibodies were purchased in carrier protein–free phosphate-buffered saline (PBS) and conjugated in-house with the respective metal isotope using the MaxPar antibody conjugation kit (Fluidigm). Metal-labeled antibodies were diluted to 0.5 mg/ml in a Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH) for storage at 4°C.
PBMC processing and stimulations
PBMCs were isolated from freshly drawn whole blood using density gradient centrifugation (Ficoll-Paque PLUS; GE Healthcare) in Sepmate tubes (45). The isolated and washed whole-blood PBMCs were resuspended in 10% dimethyl sulfoxide and 90% fetal bovine serum and cryopreserved in liquid nitrogen. Frozen aliquots of PBMCs (batch of ~10 samples and an aliquot of technical control) were washed twice in RPMI at 37°C. The samples were brought up in 1 ml of RPMI, and viability was checked. One hundred microliters of each sample was aliquoted and rested for 1 hour at 37°C. PBMCs were then incubated in RPMI (unstimulated) or one of the seven specific stimulants, either a cytokine (IFN-α, IL-6, IL-7, IL-10, and IL-21), LPS, or PMA/ionomycin for 15 min at 37°C as shown in table S5 and described previously (46). Following incubation under these eight conditions, PBMCs were fixed for 15 min with 4% paraformaldehyde (PFA) at room temperature, washed, permeabilized, and barcoded exactly according to published methods to facilitate processing and minimize batch effects (47).
All eight stimulation conditions from a barcoded sample were pooled and incubated with titrated metal-labeled antibodies directed at cell surface markers designed to identify 35 immune cell subsets (48, 49). Cell types were identified by surface antibody signal for the following lineage markers: CD3, CD4, CD7, CD8, CD11b, CD11c, CD14, CD16, CD19, CD20, CD24, CD25, CD27, CD38, CD45RA, CD56, CD123, CD127, IgA, IgD, and HLA-Dr. Pooled sample was then permeabilized with methanol and stored at −80°C. Frozen samples were washed and incubated with metal-labeled antibodies directed at 15 intracellular signaling markers, followed by Ir-intercalator staining and resuspended in 0.1 EQ Four Element Calibration Beads (50). Intracellular signaling markers were pERK-1/2, IκBα, NF-κB, p38, pAKT, pCREB, pLCK, pPLCγ2, pS6, pSTAT1, pSTAT3, pSTAT5, endosomal proteins, Lamp2, EEA1, and Rab5 (51–53). Single-cell data were acquired by CyTOF (model: Helios, software version 6.5.358, Fluidigm, South San Francisco, CA) at 300 to 400 events per second. Initiation and tuning were performed according to the manufacturer’s recommendation. Post-acquisition normalization and debarcoding were done with CyTOF software version 6.7.1014. From these, about 1.2 million individual cells were collected, unassigned events were then removed, and subsampled at a maximum of 100,000 cells for subsequent analyses. Counts in each gating stage are shown in table S6. Gating was performed using FlowJo-10 (fig. S9) and Cytobank. More details on data processing and gating can be found in the Supplementary Materials.
Statistical analysis
Application and evaluation of iEN for multivariate modeling of mass cytometry data
The iEN model was used as a multivariate model to examine generalizability and predictive power. The iEN added on to the commonly used EN algorithm the capability to incorporate knowledge of intracellular signal transduction on the generation of the mass cytometry data. We have recently reported in detail that this model demonstrates increased predictive power relative to multiple traditional methods (20). Briefly, the iEN algorithm optimized the coefficient (β) for each associated feature by minimizing the cost function
Here, X is a matrix of size m × n, where m is the number of samples, n is the number of all immune features (intracellular signals from cells under different stimulating conditions), Y is a vector of ground truths (diagnostic groups) with length n, β is a vector of the model’s coefficients with length n, and λ and α together control the magnitude of the model regularization. Last, Φ is a diagonal matrix of size n × n containing a prioritization value of 1 for elements associated with canonical immune feature or e−φ for other lower priority (noncanonical) features. The list of canonical signals that were prioritized is tabulated in table S7.
To determine objectively the value of the hyperparameters and the generalizability of the iEN model from the discovery cohort, 250 iterations of a two-layered cross-validation scheme were used. In each iteration, the outer layer randomly held out one-third of the samples for performance evaluation on unseen data, and the inner layer used the rest of the samples for parameter optimization (grid search of λ, α, and φ). At the completion of the 250 iterations, the mean values of the predictions were used for model evaluation by Wilcoxon rank sum test P value and AUC. The ROC curve’s 95% CI and AUC’s SD was calculated by bootstrapped replicates as described in previous studies (54, 55). The statistical tests related to the ROC comparison (unpaired t test) was also calculated (54). The power of the model’s predicted value was calculated using an established formula (54, 56). The weights and φ determined from all iterations were averaged to obtain a final model. The final model was used for feature component analyses, prediction of validation cohort, and disease cross-prediction.
Supplementary Material
Acknowledgments
We would like to acknowledge E. J. Fox for putting together the prior matrix for iEN and for help in harmonizing of raw CyTOF data and K. Montine for editorial assistance. The CyTOF files in this work were generated at the Human Immune Monitoring Center (HIMC) at Stanford. Funding: This work was supported by grants from the NIH (NS062684, GM138353, and AG047366) and the Scully Initiative Fund. Author contributions: T.J.M. and R.F. conceived and supervised the execution of the study. N.A. supervised all statistical analyses and machine learning aspects. N.A., B.G., and A.C. developed the iEN analytical approach. K.L.P. was responsible for clinical diagnosis, patient recruitment, and clinical data collection. R.F. fabricated reagents, processed samples, and performed mass cytometry experiments, gatings, and extracted statistical values. T.P. preprocessed data and executed statistical analyses and machine learning algorithms. D.M. and A.M.W. assisted in data preprocessing. R.F. interpreted the mass cytometry data. N.S. assisted with unsupervised data clustering. T.J.M., R.F., C.R.G., and T.P. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: T.J.M., T.P., R.F., N.A., and B.G. are inventors on a provisional patent related to this work filed by Leland Stanford Junior University (no. 63/001,195, filed on 27 March 2020). Data for reproduction of the results are available at http://flowrepository.org/experiments/3020 and http://flowrepository.org/experiments/3023
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabd5575/DC1
REFERENCES AND NOTES
- 1.Butovsky O., Koronyo-Hamaoui M., Kunis G., Ophir E., Landa G., Cohen H., Schwartz M., Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. U.S.A. 103, 11784–11789 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiala M., Liu Q. N., Sayre J., Pop V., Brahmandam V., Graves M. C., Vinters H. V., Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 32, 360–371 (2002). [DOI] [PubMed] [Google Scholar]
- 3.T. J. Lewis, C. L. Trempe, The End of Alzheimer’s: The Brain and Beyond (Academic Press, 2017). [Google Scholar]
- 4.R. Perneczky, Biomarkers for Alzheimer’s Disease Drug Development (Humana Press, 2018). [Google Scholar]
- 5.Rezai-Zadeh K., Gate D., Szekely C. A., Town T., Can peripheral leukocytes be used as Alzheimer’s disease biomarkers? Expert Rev. Neurother. 9, 1623–1633 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj T., Rothamel K., Mostafavi S., Ye C., Lee M. N., Replogle J. M., Feng T., Lee M., Asinovski N., Frohlich I., Imboywa S., Von Korff A., Okada Y., Patsopoulos N. A., Davis S., McCabe C., Paik H.-I., Srivastava G. P., Raychaudhuri S., Hafler D. A., Koller D., Regev A., Hacohen N., Mathis D., Benoist C., Stranger B. E., De Jager P. L., Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan K. J., White C. C., Patel K., Xu J., Olah M., Replogle J. M., Frangieh M., Cimpean M., Winn P., McHenry A., Kaskow B. J., Chan G., Cuerdon N., Bennett D. A., Boyd J. D., Imitola J., Elyaman W., De Jager P. L., Bradshaw E. M., A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 9, eaai7635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delvaux E., Mastroeni D., Nolz J., Chow N., Sabbagh M., Caselli R. J., Reiman E. M., Marshall F. J., Coleman P. D., Multivariate analyses of peripheral blood leukocyte transcripts distinguish Alzheimer’s, Parkinson’s, control, and those at risk for developing Alzheimer’s. Neurobiol. Aging 58, 225–237 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Schmidbauer G., Hancock W. W., Wasowska B., Badger A. M., Kupiec-Weglinski J. W., Abrogation by rapamycin of accelerated rejection in sensitized rats by inhibition of alloantibody responses and selective suppression of intragraft mononuclear and endothelial cell activation, cytokine production, and cell adhesion. Transplantation 57, 933–941 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Davies R., Vogelsang P., Jonsson R., Appel S., An optimized multiplex flow cytometry protocol for the analysis of intracellular signaling in peripheral blood mononuclear cells. J. Immunol. Methods 436, 58–63 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ray S., Britschgi M., Herbert C., Takeda-Uchimura Y., Boxer A., Blennow K., Friedman L. F., Galasko D. R., Jutel M., Karydas A., Kaye J. A., Leszek J., Miller B. L., Minthon L., Quinn J. F., Rabinovici G. D., Robinson W. H., Sabbagh M. N., So Y. T., Sparks D. L., Tabaton M., Tinklenberg J., Yesavage J. A., Tibshirani R., Wyss-Coray T., Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat. Med. 13, 1359–1362 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Soares H. D., Chen Y., Sabbagh M., Rohrer A., Schrijvers E., Breteler M., Identifying early markers of Alzheimer’s disease using quantitative multiplex proteomic immunoassay panels. Ann. N. Y. Acad. Sci. 1180, 56–67 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Cosma A., Nolan G., Gaudilliere B., Mass cytometry: The time to settle down. Cytometry A 91, 12–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baca Q., Cosma A., Nolan G., Gaudilliere B., The road ahead: Implementing mass cytometry in clinical studies, one cell at a time. Cytometry B Clin. Cytom. 92, 10–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kvistborg P., Gouttefangeas C., Aghaeepour N., Cazaly A., Chattopadhyay P. K., Chan C., Eckl J., Finak G., Hadrup S. R., Maecker H. T., Maurer D., Mosmann T., Qiu P., Scheuermann R. H., Welters M. J. P., Ferrari G., Brinkman R. R., Britten C. M., Thinking outside the gate: Single-cell assessments in multiple dimensions. Immunity 42, 591–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendall S. C., Nolan G. P., Roederer M., Chattopadhyay P. K., A deep profiler’s guide to cytometry. Trends Immunol. 33, 323–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aghaeepour N., Chattopadhyay P., Chikina M., Dhaene T., Van Gassen S., Kursa M., Lambrecht B. N., Malek M., McLachlan G. J., Qian Y., Qiu P., Saeys Y., Stanton R., Tong D., Vens C., Walkowiak S., Wang K., Finak G., Gottardo R., Mosmann T., Nolan G. P., Scheuermann R. H., Brinkman R. R., A benchmark for evaluation of algorithms for identification of cellular correlates of clinical outcomes. Cytometry A 89, 16–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanley N., Stelzer I., Fallahzadeh R., Ganio E., Tsai A., Becker M., Phongpreecha T., Nassar H., Ghaemi S., Culos A., Han X., Rumer K., Peterson L., Chang A., Maric I., Gaudilliere D., Tsai E., Ando K., Leipold M., Bermoser G., Cross D., Pollard A., Maecker H., Shaw G., Stevenson D., Angst M., Gaudilliere B., Aghaeepour N., VoPo leverages cellular heterogeneity for predictive modeling of single-cell data. Nat. Commun. 11, 3738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tusher V. G., Tibshirani R., Chu G., Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culos A. E., Tsai A. S., Stanley N., Becker M., Ghaemi M. S., Mcilwain D. R., Fallahzadeh R., Tanada A., Nassar H., Ganio E., Peterson L., Han X., Stelzer I., Ando K., Gaudilliere D., Phongpreecha T., Marić I., Chang A. L., Shaw G. M., Stevenson D. K., Bendall S., Davis K. L., Fantl W., Nolan G. P., Hastie T., Tibshirani R., Angst M. S., Gaudilliere B., Aghaeepour N., Integration of mechanistic immunological knowledge into a machine learning pipeline improves predictions. Nat. Mach. Intell. 2, 619–628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sims R., van der Lee S. J., Naj A. C., Bellenguez C., Badarinarayan N., Jakobsdottir J., Kunkle B. W., Boland A., Raybould R., Bis J. C., Martin E. R., Grenier-Boley B., Heilmann-Heimbach S., Chouraki V., Kuzma A. B., Sleegers K., Vronskaya M., Ruiz A., Graham R. R., Olaso R., Hoffmann P., Grove M. L., Vardarajan B. N., Hiltunen M., Nöthen M. M., White C. C., Hamilton-Nelson K. L., Epelbaum J., Maier W., Choi S.-H., Beecham G. W., Dulary C., Herms S., Smith A. V., Funk C. C., Derbois C., Forstner A. J., Ahmad S., Li H., Bacq D., Harold D., Satizabal C. L., Valladares O., Squassina A., Thomas R., Brody J. A., Qu L., Sánchez-Juan P., Morgan T., Wolters F. J., Zhao Y., Garcia F. S., Denning N., Fornage M., Malamon J., Naranjo M. C. D., Majounie E., Mosley T. H., Dombroski B., Wallon D., Lupton M. K., Dupuis J., Whitehead P., Fratiglioni L., Medway C., Jian X., Mukherjee S., Keller L., Brown K., Lin H., Cantwell L. B., Panza F., McGuinness B., Moreno-Grau S., Burgess J. D., Solfrizzi V., Proitsi P., Adams H. H., Allen M., Seripa D., Pastor P., Cupples L. A., Price N. D., Hannequin D., Frank-García A., Levy D., Chakrabarty P., Caffarra P., Giegling I., Beiser A. S., Giedraitis V., Hampel H., Garcia M. E., Wang X., Lannfelt L., Mecocci P., Eiriksdottir G., Crane P. K., Pasquier F., Boccardi V., Henández I., Barber R. C., Scherer M., Tarraga L., Adams P. M., Leber M., Chen Y., Albert M. S., Riedel-Heller S., Emilsson V., Beekly D., Braae A., Schmidt R., Blacker D., Masullo C., Schmidt H., Doody R. S., Spalletta G., Longstreth W. T. Jr., Fairchild T. J., Bossù P., Lopez O. L., Frosch M. P., Sacchinelli E., Ghetti B., Yang Q., Huebinger R. M., Jessen F., Li S., Kamboh M. I., Morris J., Sotolongo-Grau O., Katz M. J., Corcoran C., Dunstan M., Braddel A., Thomas C., Meggy A., Marshall R., Gerrish A., Chapman J., Aguilar M., Taylor S., Hill M., Fairén M. D., Hodges A., Vellas B., Soininen H., Kloszewska I., Daniilidou M., Uphill J., Patel Y., Hughes J. T., Lord J., Turton J., Hartmann A. M., Cecchetti R., Fenoglio C., Serpente M., Arcaro M., Caltagirone C., Orfei M. D., Ciaramella A., Pichler S., Mayhaus M., Gu W., Lleó A., Fortea J., Blesa R., Barber I. S., Brookes K., Cupidi C., Maletta R. G., Carrell D., Sorbi S., Moebus S., Urbano M., Pilotto A., Kornhuber J., Bosco P., Todd S., Craig D., Johnston J., Gill M., Lawlor B., Lynch A., Fox N. C., Hardy J., Consortium A. R. U. K., Albin R. L., Apostolova L. G., Arnold S. E., Asthana S., Atwood C. S., Baldwin C. T., Barnes L. L., Barral S., Beach T. G., Becker J. T., Bigio E. H., Bird T. D., Boeve B. F., Bowen J. D., Boxer A., Burke J. R., Burns J. M., Buxbaum J. D., Cairns N. J., Cao C., Carlson C. S., Carlsson C. M., Carney R. M., Carrasquillo M. M., Carroll S. L., Diaz C. C., Chui H. C., Clark D. G., Cribbs D. H., Crocco E. A., DeCarli C., Dick M., Duara R., Evans D. A., Faber K. M., Fallon K. B., Fardo D. W., Farlow M. R., Ferris S., Foroud T. M., Galasko D. R., Gearing M., Geschwind D. H., Gilbert J. R., Graff-Radford N. R., Green R. C., Growdon J. H., Hamilton R. L., Harrell L. E., Honig L. S., Huentelman M. J., Hulette C. M., Hyman B. T., Jarvik G. P., Abner E., Jin L.-W., Jun G., Karydas A., Kaye J. A., Kim R., Kowall N. W., Kramer J. H., LaFerla F. M., Lah J. J., Leverenz J. B., Levey A. I., Li G., Lieberman A. P., Lunetta K. L., Lyketsos C. G., Marson D. C., Martiniuk F., Mash D. C., Masliah E., McCormick W. C., McCurry S. M., McDavid A. N., McKee A. C., Mesulam M., Miller B. L., Miller C. A., Miller J. W., Morris J. C., Murrell J. R., Myers A. J., O’Bryant S., Olichney J. M., Pankratz V. S., Parisi J. E., Paulson H. L., Perry W., Peskind E., Pierce A., Poon W. W., Potter H., Quinn J. F., Raj A., Raskind M., Reisberg B., Reitz C., Ringman J. M., Roberson E. D., Rogaeva E., Rosen H. J., Rosenberg R. N., Sager M. A., Saykin A. J., Schneider J. A., Schneider L. S., Seeley W. W., Smith A. G., Sonnen J. A., Spina S., Stern R. A., Swerdlow R. H., Tanzi R. E., Thornton-Wells T. A., Trojanowski J. Q., Troncoso J. C., Van Deerlin V. M., Van Eldik L. J., Vinters H. V., Vonsattel J. P., Weintraub S., Welsh-Bohmer K. A., Wilhelmsen K. C., Williamson J., Wingo T. S., Woltjer R. L., Wright C. B., Yu C.-E., Yu L., Garzia F., Golamaully F., Septier G., Engelborghs S., Vandenberghe R., De Deyn P. P., Fernadez C. M., Benito Y. A., Thonberg H., Forsell C., Lilius L., Kinhult-Stählbom A., Kilander L., Brundin R., Concari L., Helisalmi S., Koivisto A. M., Haapasalo A., Dermecourt V., Fievet N., Hanon O., Dufouil C., Brice A., Ritchie K., Dubois B., Himali J. J., Keene C. D., Tschanz J., Fitzpatrick A. L., Kukull W. A., Norton M., Aspelund T., Larson E. B., Munger R., Rotter J. I., Lipton R. B., Bullido M. J., Hofman A., Montine T. J., Coto E., Boerwinkle E., Petersen R. C., Alvarez V., Rivadeneira F., Reiman E. M., Gallo M., O’Donnell C. J., Reisch J. S., Bruni A. C., Royall D. R., Dichgans M., Sano M., Galimberti D., George-Hyslop P. S., Scarpini E., Tsuang D. W., Mancuso M., Bonuccelli U., Winslow A. R., Daniele A., Wu C.-K.; GERAD/PERADES, CHARGE, ADGC, EADI, Peters O., Nacmias B., Riemenschneider M., Heun R., Brayne C., Rubinsztein D. C., Bras J., Guerreiro R., Al-Chalabi A., Shaw C. E., Collinge J., Mann D., Tsolaki M., Clarimón J., Sussams R., Lovestone S., O’Donovan M. C., Owen M. J., Behrens T. W., Mead S., Goate A. M., Uitterlinden A. G., Holmes C., Cruchaga C., Ingelsson M., Bennett D. A., Powell J., Golde T. E., Graff C., De Jager P. L., Morgan K., Ertekin-Taner N., Combarros O., Psaty B. M., Passmore P., Younkin S. G., Berr C., Gudnason V., Rujescu D., Dickson D. W., Dartigues J.-F., DeStefano A. L., Ortega-Cubero S., Hakonarson H., Campion D., Boada M., Kauwe J. K., Farrer L. A., Van Broeckhoven C., Ikram M. A., Jones L., Haines J. L., Tzourio C., Launer L. J., Escott-Price V., Mayeux R., Deleuze J.-F., Amin N., Holmans P. A., Pericak-Vance M. A., Amouyel P., van Duijn C. M., Ramirez A., Wang L.-S., Lambert J.-C., Seshadri S., Williams J., Schellenberg G. D., Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Saout C., Luckey M. A., Villarino A. V., Smith M., Hasley R. B., Myers T. G., Imamichi H., Park J.-H., O’Shea J. J., Lane H. C., Catalfamo M., IL-7-dependent STAT1 activation limits homeostatic CD4+ T cell expansion. JCI Insight 2, e96228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fragiadakis G. K., Bjornson-Hooper Z. B., Madhireddy D., Sachs K., Spitzer M. H., Bendall S. C., Nolan G. P., Variation of immune cell responses in humans reveals sex-specific coordinated signaling across cell types. bioRxiv, 567784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch J. R., Tang W., Wang H., Vitek M. P., Bennett E. R., Sullivan P. M., Warner D. S., Laskowitz D. T., APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 278, 48529–48533 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Vitek M. P., Brown C. M., Colton C. A., APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahley R. W., Rall S. C. Jr., Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Olgiati P., Politis A., Malitas P., Albani D., Dusi S., Polito L., De Mauro S., Zisaki A., Piperi C., Stamouli E., Mailis A., Batelli S., Forloni G., De Ronchi D., Kalofoutis A., Liappas I., Serretti A., APOE epsilon-4 allele and cytokine production in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 25, 338–344 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Gate D., Saligrama N., Leventhal O., Yang A. C., Unger M. S., Middeldorp J., Chen K., Lehallier B., Channappa D., De Los Santos M. B., McBride A., Pluvinage J., Elahi F., Tam G. K.-Y., Kim Y., Greicius M., Wagner A. D., Aigner L., Galasko D. R., Davis M. M., Wyss-Coray T., Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart C. R., Stuart L. M., Wilkinson K., van Gils J. M., Deng J., Halle A., Rayner K. J., Boyer L., Zhong R., Frazier W. A., Lacy-Hulbert A., El Khoury J., Golenbock D. T., Moore K. J., CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujita K., Motoki K., Tagawa K., Chen X., Hama H., Nakajima K., Homma H., Tamura T., Watanabe H., Katsuno M., Matsumi C., Kajikawa M., Saito T., Saido T., Sobue G., Miyawaki A., Okazawa H., HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 6, 31895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitamura Y., Shimohama S., Ota T., Matsuoka Y., Nomura Y., Taniguchi T., Alteration of transcription factors NF-κB and STAT1 in Alzheimer’s disease brains. Neurosci. Lett. 237, 17–20 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Lee W. J., Ham S. A., Lee G. H., Choi M., Yoo H., Paek K. S., Lim D., Hong K., Hwang J. S., Seo H. G., Activation of peroxisome proliferator-activated receptor delta suppresses BACE 1 expression by up-regulating SOCS 1 in a JAK2/ STAT1-dependent manner. J. Neurochem. 151, 370–385 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Di Liberto G., Pantelyushin S., Kreutzfeldt M., Page N., Musardo S., Coras R., Steinbach K., Vincenti I., Klimek B., Lingner T., Salinas G., Lin-Marq N., Staszewski O., Costa Jordão M. J., Wagner I., Egervari K., Mack M., Bellone C., Blümcke I., Prinz M., Pinschewer D. D., Merkler D., Neurons under T cell attack coordinate phagocyte-mediated synaptic stripping. Cell 175, 458–471.e19 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Nevado-Holgado A. J., Ribe E., Thei L., Furlong L., Mayer M.-A., Quan J., Richardson J. C., Cavanagh J., Consortium N., Lovestone S., Genetic and real-world clinical data, combined with empirical validation, nominate JAK-STAT signaling as a target for Alzheimer’s disease therapeutic development. Cell 8, 425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conway O. J., Carrasquillo M. M., Wang X., Bredenberg J. M., Reddy J. S., Strickland S. L., Younkin C. S., Burgess J. D., Allen M., Lincoln S. J., Nguyen T., Malphrus K. G., Soto A. I., Walton R. L., Boeve B. F., Petersen R. C., Lucas J. A., Ferman T. J., Cheshire W. P., van Gerpen J. A., Uitti R. J., Wszolek Z. K., Ross O. A., Dickson D. W., Graff-Radford N. R., Ertekin-Taner N., ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol. Neurodegener. 13, 53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Lee S. J., Conway O. J., Jansen I., Carrasquillo M. M., Kleineidam L., van den Akker E., Hernández I., van Eijk K. R., Stringa N., Chen J. A., Zettergren A., Andlauer T. F. M., Diez-Fairen M., Simon-Sanchez J., Lleó A., Zetterberg H., Nygaard M., Blauwendraat C., Savage J. E., Mengel-From J., Moreno-Grau S., Wagner M., Fortea J., Keogh M. J., Blennow K., Skoog I., Friese M. A., Pletnikova O., Zulaica M., Lage C., de Rojas I., Riedel-Heller S., Illán-Gala I., Wei W., Jeune B., Orellana A., Bergh F. T., Wang X., Hulsman M., Beker N., Tesi N., Morris C. M., Indakoetxea B., Collij L. E., Scherer M., Morenas-Rodríguez E., Ironside J. W., van Berckel B. N. M., Alcolea D., Wiendl H., Strickland S. L., Pastor P., Rodríguez Rodríguez E.; DESGESCO (Dementia Genetics Spanish Consortium), EADB (Alzheimer Disease European DNA biobank); EADB (Alzheimer Disease European DNA biobank); IFGC (International FTD-Genomics Consortium); IPDGC (The International Parkinson Disease Genomics Consortium); IPDGC (The International Parkinson Disease Genomics Consortium); RiMod-FTD (Risk and Modifying factors in Fronto-Temporal Dementia); Netherlands Brain Bank (NBB), Boeve B. F., Petersen R. C., Ferman T. J., van Gerpen J. A., Reinders M. J. T., Uitti R. J., Tárraga L., Maier W., Dols-Icardo O., Kawalia A., Dalmasso M. C., Boada M., Zettl U. K., van Schoor N. M., Beekman M., Allen M., Masliah E., de Munain A. L., Pantelyat A., Wszolek Z. K., Ross O. A., Dickson D. W., Graff-Radford N. R., Knopman D., Rademakers R., Lemstra A. W., Pijnenburg Y. A. L., Scheltens P., Gasser T., Chinnery P. F., Hemmer B., Huisman M. A., Troncoso J., Moreno F., Nohr E. A., Sørensen T. I. A., Heutink P., Sánchez-Juan P., Posthuma D.; GIFT (Genetic Investigation in Frontotemporal Dementia and Alzheimer’s Disease) Study Group, Clarimón J., Christensen K., Ertekin-Taner N., Scholz S. W., Ramirez A., Ruiz A., Slagboom E., van der Flier W. M., Holstege H., A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 138, 237–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dalmasso M. C., Brusco L. I., Olivar N., Muchnik C., Hanses C., Milz E., Becker J., Heilmann-Heimbach S., Hoffmann P., Prestia F. A., Galeano P., Avalos M. S. S., Martinez L. E., Carulla M. E., Azurmendi P. J., Liberczuk C., Fezza C., Sampaño M., Fierens M., Jemar G., Solis P., Medel N., Lisso J., Sevillano Z., Bosco P., Bossù P., Spalletta G., Galimberti D., Mancuso M., Nacmias B., Sorbi S., Mecocci P., Pilotto A., Caffarra P., Panza F., Bullido M., Clarimon J., Sánchez-Juan P., Coto E., Sanchez-Garcia F., Graff C., Ingelsson M., Bellenguez C., Castaño E. M., Kairiyama C., Politis D. G., Kochen S., Scaro H., Maier W., Jessen F., Mangone C. A., Lambert J.-C., Morelli L., Ramirez A., Transethnic meta-analysis of rare coding variants in PLCG2, ABI3, and TREM2 supports their general contribution to Alzheimer’s disease. Transl. Psychiatry 9, 55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magno L., Lessard C. B., Martins M., Cruz P., Katan M., Bilsland J., Chakrabaty P., Golde T. E., Whiting P., Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res. Ther. 11, 16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milner J. D., PLAID: A syndrome of complex patterns of disease and unique phenotypes. J. Clin. Immunol. 35, 527–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nott A., Holtman I. R., Coufal N. G., Schlachetzki J. C. M., Yu M., Hu R., Han C. Z., Pena M., Xiao J., Wu Y., Keulen Z., Pasillas M. P., O’Connor C., Nickl C. K., Schafer S. T., Shen Z., Rissman R. A., Brewer J. B., Gosselin D., Gonda D. D., Levy M. L., Rosenfeld M. G., McVicker G., Gage F. H., Ren B., Glass C. K., Brain cell type–specific enhancer-promoter interactome maps and disease risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golde T. E., Harnessing immunoproteostasis to treat neurodegenerative disorders. Neuron 101, 1003–1015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Litvan I., Bhatia K. P., Burn D. J., Goetz C. G., Lang A. E., McKeith I., Quinn N., Sethi K. D., Shults C., Wenning G. K.; Movement Disorders Society Scientific Issues Committee , Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for parkinsonian disorders. Mov. Disord. 18, 467–486 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Cholerton B. A., Zabetian C. P., Quinn J. F., Chung K. A., Peterson A., Espay A. J., Revilla F. J., Devoto J., Watson G., Hu S. C., Edwards K. L., Montine T. J., Leverenz J. B., Pacific Northwest Udall Center of excellence clinical consortium: Study design and baseline cohort characteristics. J. Parkinsons Dis. 3, 205–214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKhann G. M., Knopman D. S., Chertkow H., Hyman B. T., Jack C. R., Kawas C. H., Klunk W. E., Koroshetz W. J., Manly J. J., Mayeux R., Mohs R. C., Morris J. C., Rossor M. N., Scheltens P., Carrillo M. C., Thies B., Weintraub S., Phelps C. H., The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grievink H. W., Luisman T., Kluft C., Moerland M., Malone K. E., Comparison of three isolation techniques for human peripheral blood mononuclear cells: Cell recovery and viability, population composition, and cell functionality. Biopreserv. Biobank. 14, 410–415 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Fernandez R., Maecker H., Cytokine-stimulated phosphoflow of PBMC using CyTOF mass cytometry. Bio Protoc. 5, e1496 (2015). [PMC free article] [PubMed] [Google Scholar]
- 47.Zunder E. R., Finck R., Behbehani G. K., Amir E.-A. D., Krishnaswamy S., Gonzalez V. D., Lorang C. G., Bjornson Z., Spitzer M. H., Bodenmiller B., Fantl W. J., Pe’er D., Nolan G. P., Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 10, 316–333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi J., Fernandez R., Maecker H. T., Butte M. J., Systems approach to uncover signaling networks in primary immunodeficiency diseases. J. Allergy Clin. Immunol. 140, 881–884.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bendall S. C., Simonds E. F., Qiu P., Amir E.-A. D., Krutzik P. O., Finck R., Bruggner R. V., Melamed R., Trejo A., Ornatsky O. I., Balderas R. S., Plevritis S. K., Sachs K., Pe’er D., Tanner S. D., Nolan G. P., Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011).21551058 [Google Scholar]
- 50.Finck R., Simonds E. F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe’er D., Nolan G. P., Bendall S. C., Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Gorman W. E., Huang H., Wei Y.-L., Davis K. L., Leipold M. D., Bendall S. C., Kidd B. A., Dekker C. L., Maecker H. T., Chien Y.-H., Davis M. M., The split virus influenza vaccine rapidly activates immune cells through Fcγ receptors. Vaccine 32, 5989–5997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galligan C. L., Siebert J. C., Siminovitch K. A., Keystone E. C., Bykerk V., Perez O. D., Fish E. N., Multiparameter phospho-flow analysis of lymphocytes in early rheumatoid arthritis: Implications for diagnosis and monitoring drug therapy. PLOS ONE 4, e6703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu W., Fang F., Ding J., Wu C., Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic 19, 253–262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xavier R., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J. C., Müller M., pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.James C., Bithell J., Bootstrap confidence intervals: When, which, what? A practical guide for medical statisticians. Stat. Med. 19, 1141–1164 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Obuchowski N. A., Lieber M. L., Wians F. H. Jr., ROC curves in Clinical Chemistry: Uses, misuses, and possible solutions. Clin. Chem. 50, 1118–1125 (2004). [DOI] [PubMed] [Google Scholar]
- 57.M. D. Leipold, E. W. Newell, H. T. Maecker, Multiparameter Phenotyping of Human PBMCs using Mass Cytometry in Immunosenescence (Humana Press, 2015), pp. 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson W. E., Li C., Rabinovic A., Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 59.A. Scherer, Batch Effects and Noise in Microarray Experiments: Sources and Solutions (John Wiley & Sons, 2009). [Google Scholar]
- 60.Di Mitri D., Azevedo R. I., Henson S. M., Libri V., Riddell N. E., Macaulay R., Kipling D., Soares M. V. D., Battistini L., Akbar A. N., Reversible senescence in human CD4+ CD45RA+ CD27− memory T cells. J. Immunol. 187, 2093–2100 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Muggeo V. M. R., Estimating regression models with unknown break-points. Stat. Med. 22, 3055–3071 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabd5575/DC1