

Abstract
By the condensation of thiosemicarbazide with coumarin aldehyde, two novel substituted thiosemicarbazones with chemical formulae C24H25N3O3S (3a) and C26H23N3O3S (3b) have been synthesized. The synthesized compounds were resolved using SC-XRD, and structure elucidation was carried out using 1H NMR, 13C NMR, UV–visible, and FT-IR spectroscopic analyses. Computational calculations at the B3LYP/6-311+G(d,p) level of theory were performed to countercheck the experimental (UV–vis, FT-IR) findings and explore the electronic (FMO, NBO, MEP) properties of 3a–b. The nonlinear optical (NLO) properties of 3a–b were estimated using B3LYP, HF, LC-BLYP, CAM-B3LYP, M062X, and M06 functionals in combination with the 6-311+G(d,p) basis set. The crystallographic data revealed that compounds were crystallized as an orthorhombic crystal lattice with the Pbcn space group and the triclinic crystal lattice with the P̅1 space group. A good concurrence among experimental SC-XRD-generated bond lengths, bond angles, FT-IR, UV–vis, and corresponding DFT results was found, which confirms the purity of both compounds. The NBO analysis confirmed the presence of intramolecular hydrogen bonding and hyperconjugative interactions, which not only were the pivotal cause of stability of the investigated compounds but also led to an overwhelming NLO response. The energy differences calculated for HOMO/LUMO are 3.053 and 3.118 eV in 3a and 3b, respectively. The crystal 3b showed a higher value of first-order polarizability at all levels of theory than 3a. Overall results show that the crystals under investigation are polarized in nature with a good dipole moment. A comparative analysis with urea molecules clearly indicates that the studied compounds are acceptable NLO candidates and they can be used for future technological applications.
Introduction
Coumarin compounds with reactive oxygen containing heterocycles and a benzopyran backbone are vital classes of compounds that occur naturally.1 The special structure of benzopyran allows its derivatives to easily affect the diversity of enzymes and receptors in organisms with weak binding interactions, demonstrating the wide potential of drugs.2−5 The abundant natural substances used by people worldwide include the coumarin group. Coumarin is a privileged framework for multifarious medical properties and is therefore widely employed as a scaffold for designing novel and potent analogues having physicochemical properties and versatile, easy synthetic transformation into numerous activated coumarins.6,7 Coumarin-containing compounds exhibit various useful biological and pharmaceutical activities liable to substituents present in their parent benzopyran moiety.8,9 Thus, their synthesis has become a focus of interest.10 Important biological applications in which coumarin derivatives are found useful are as antioxidants,11 as antifungals,12,13 as anticoagulants,14−16 in antibacterial activity,17−19 as CNS-stimulants,20 in anti-HIV therapy,21,22 in antitumor activity,23 and in photochemotherapy.
Thiosemicarbazones bearing the CH=N linkage belong to a wide group of thiourea derivatives exhibiting various biological activities as a function of the carbonyl motif. Furthermore, it appears that thiosemicarbazones show improved biological activities when they are attached to an aromatic heterocyclic moiety, thus attracting considerable pharmaceutical interest. Since the last five decades, these derivatives have been scanned and reported to display antibacterial, anticancer, antimalarial, and anti-HIV activities by varying the aldehyde or ketone moiety. Therefore, current research is focused on the identification and characterization of newer thiosemicarbazone derivatives of coumarin aldehyde for existing antimicrobial activity.24
Nonlinear optical (NLO) properties of the chemical building blocks have attracted major consideration because of their potential uses in the vast and important arena of information technology and telecommunication, particularly dynamic image processing, optical computing, and optical communication.25−27 Much attention has been paid by the theoretical as well as experimental communities toward the exploration of NLO properties of the newly synthesized compounds because of their potential applications in the area of biophysics, nuclear science, chemical dynamics, medicine, materials, solid physics, and surface interface.28 The thiosemicarbazones are an important class of compounds that are frequently studied for their role in NLO applications.29,30 Reports are available in the literature in which the potential of thiosemicarbazone as an NLO material is explored.31
Assuming the preceding importance of thiosemicarbazones in the NLO field, herein, thiosemicarbazones having a coumarin moiety were synthesized (Scheme 1) and their electronic properties were studied. Substituted compounds (E)-N-cyclohexyl-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)-hydrazinecarbothioamide (3a) and 2,4(E)-N-(2,4-dimethylphenyl)-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)hydrazinecarbothioamide (3b) were synthesized using a typical condensation reaction. A careful literature survey confirmed that the single-crystal X-ray diffraction (SC-XRD) study, spectroscopic investigation, and computational insights of compounds 3a–b are absent. Therefore, employing our synthetic and computational background, we performed a detailed spectroscopic and density functional theory (DFT)-based computational analysis including frontier molecular orbital (FMO), global reactivity parameters, natural bond orbital (NBO) analysis, molecular electrostatic potential (MEP) surfaces, and nonlinear optical (NLO) exploration of the investigated compounds 3a–b.
Scheme 1. Synthetic Scheme for Targeted Thiosemicarbazones (3a–b).

Experimental Section
Synthesis of Coumarin-Based Thiosemicarbazones
To synthesize targeted thiosemicarbazones (3a–b), 2-((2-oxo-2H-chromen-3-yl)methoxy)benzaldehyde (1) (0.1 mmol) was taken in CH3OH (10 mL) with subsequent addition of a solution of appropriate thiosemicarbazide (2a–b) in methanol (0.1 mmol). The stirred mixture was then heated under reflux for 2–3 h in the presence of p-TsOH (few crystals) as a catalyst. The progression of the reaction was monitored; after completion as checked by TLC, the reflux was stopped and the reaction mixture was allowed to cool down, and light-yellow crystalline solids settled down at the bottom of the flask. The product was filtered with subsequent washings and dried under a vacuum. The products were further purified by recrystallization in MeOH as the solvent.
(Z)-N-Cyclohexyl-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)hydrazinecarbothioamide (3a)
Yield 85%, light-yellow, orthorhombic, mp > 300 °C, IR υmax(cm–1): 1256 (C=S), 1539 (C=N), 1714 (C=O), 3298 (N=CH), 3649 (N=CH), 1H NMR (CDCl3,TMS) δ ppm; 1.23–1.41 (m, 4H, cyclohexyl CH2, H24), 1.65–1.76 (m, 4H,cyclohexyl CH2, H-23), 1.79 (d, 1H, NH-cyclohexyl, J = 2.65 Hz, H-22), 2.10–2.12 (bs, 2H, cyclohexyl CH2, H-25), 4.24 (s, 2H, CH2Ar, H-9), 7.02–7.07 (m, 2H, Ar-H, H-14), 7.32–7.42 (m, 4H, H-12,15, Ar-H), 7.56 (t,1H,J = 7.5 Hz, H6,), 7.69 (d,1H, J = 7.5 Hz, H5), 7.87 (d, 1H, H8, Ar-H, J = 7.4 Hz), 7.91 (s, 1H, H-17), 8.39 (bs, 1H, NH-N, H-19), 9.52 (bs, 1H, NH-CS, H-21); 13C NMR δ ppm; 40.67 (C-24, 25), 65.12 (C22, 23), 113.51 (Ar-C), 113.70 (Ar-C), 116.68 (Ar-C), 120.11 (Ar-C), 121.68 (Ar-C), 123.68 (Ar-C), 124.70 (Ar-C), 125.26 (Ar-C), 127.06 (Ar-C), 128.02 (Ar-C), 129.07 (Ar-C), 132.00 (Ar-C), 132.40 (Ar-C), 132.45 (Ar-C), 138.73 (Ar-C), 140.27 (Ar-C), 153.33 (C8′), 157.11 (C18), 157.39 (C-2), 159.93 (C-11), 176.64 (C-20); Anal calcd for C24H25N3O3S (435.16); C, 66.18; H, 5.79; N, 9.65; found; C, 66.09; H, 5.98; N, 9.78.
(Z)-N-(2,4-Dimethylphenyl)-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)hydrazinecarbothioamide (3b)
Yield 80%, light-yellow, triclinic, mp > 300 °C, IR υmax (cm–1): 1222 (C=S), 1543 (C=N), 1709 (C=O), 3125–3294 (N-H), 1H NMR (DMSO-d6,TMS) δ ppm; 2.18 (s, 3H, CH3), 2.29 (s, 3H, CH3), 5.05 (s, 2H, CH2Ar, H-9), 7.03–7.84 (m, 10H, Ar-H), 8.33 (s,1H, H24, Ar-H), 8.33 (s,1H, H4, Ar-H), 8.66 (s, 1H, H-17), 9.91 (bs, 1H, NH-N, H-19), 11.73 (bs, 1H, NH-CS, H-21); 13C NMR δ ppm; 13.76 (C-CH3), 15.10 (C-CH3), 65.63 (C9), 101.94 (Ar-C), 116.65 (Ar-C), 119.21 (Ar-C), 121.82 (Ar-C), 124.34 (Ar-C), 124.51 (Ar-C), 125.20 (Ar-C), 129.05 (Ar-C), 129.37 (Ar-C), 130.06 (Ar-C), 131.17 (Ar-C), 132.47 (Ar-C), 134.52 (Ar-C), 140.14 (Ar-C), 141.45 (Ar-C), 145.91 (Ar-C), 153.50 (Ar-C), 156.93 (C8′), 159.90 (C18), 160.12 (C-2), 169.46 (C-11), 176.98 (C-20); Anal calcd for C26H23N3O3S (457.15); C, 68.25; H, 5.07; N, 9.18; Found; C, 68.36; H, 5.01; N, 9.28.
Computational Procedure
In this current investigation, the Gaussian 09 program32 was utilized to perform all computations. From the chemical structures determined through SC-XRD, primary geometries of 3a and 3b had been obtained. Compounds 3a and 3b were optimized using the B3LYP/6-311+G(d,p) functional. No imaginary frequency as calculated from the vibrational analysis at the B3LYP/6-311+G(d,p) functional existed, which points out the completion of geometry optimization. FMO and MEP surfaces were estimated at the B3LYP/6-311+G(d,p) functional, while UV–vis spectra were estimated using the time-dependent density functional theory (TD-DFT) and at the aforesaid functional. The NBO study was computed using the NBO program33 at the B3LYP/6-311+G(d,p) functional. NLO properties were estimated at M06, M062X, CAM-B3LYP, LC-BLYP, HF, and B3LYP functionals in combination with the 6-311+G(d,p) basis set. Moreover, GaussView 5.034 was used to make input files and visualize output vibrational results. Avogadro35 was used to make HOMO–LUMO orbital diagrams and exclude corresponding energy values. Chemcraft36 programs were used for the interpretation of NBO output results.
Chemistry of Compounds (3a–b)
Thiosemicarbazones having a coumarin moiety were synthesized to determine the potential of 2-((2-oxo-2H-chromen-3-yl)methoxy)benzaldehyde-hybrid thiosemicarbazones as biologically active scaffolds. Chromen-aldehyde (1) was prepared by following the Baylis–Hillman reaction of salicylaldehyde with methyl acrylate.37,38 Substituted thiosemicarbazones (3a–b) were synthesized using a typical condensation reaction of the NH2– group of thiosemicarbazide and C=O of the aldehyde functional group. Equimolar amounts of corresponding thiosemicarbazide (2a–b) and aldehyde (1) were reacted. The reaction was performed in CH3OH using a catalytic amount of p-TsOH (1–2 crystals). The reaction conditions were optimized by carrying out the reaction in various solvents with variable polarities (methanol, CH2Cl2, dioxane) and different catalysts (AcOH, H2SO4). The optimum conditions were attained by refluxing the reactants in methanol as the solvent and catalyzing under p-TsOH acid. Desired thiosemicarbazones (3a–b) were acquired in excellent yield (80–85%).
Spectroscopic data, i.e., IR, 1H NMR, and 13C NMR, provided the support for the structure confirmation for the targeted coumarin-hybrid thiosemicarbazones. In FT-IR, C=N stretching emerged in the range of 1539–1543 cm–1, while the C=S band was found between 1212 and 1256 cm–1. NH stretching was from 3125 to 3294 cm–1, and C=O stretching was detected between 1709 and 1714 cm–1. In 1H NMR, CH2 emerged as a singlet in the range from δ 4.24 to 5.05 ppm, while the NH–N signal appeared at δ 8.35 ppm in 3a and at δ 9.91 ppm in compound 3b. On the other hand, a broad singlet was observed for NH–C=S at δ 9.52 and 11.73 ppm for 3a and 3b, respectively. The spectral data of the remaining protons were also confirmed by the above-mentioned analysis and found to be in good accordance with the anticipated structure. CHN analysis results also confirm the structures (3a–b), and the observed values were in good conformity with the resulting values.
Crystallographic Discussion
To prove the structure of coumarin-pendant thiosemicarbazones 3a and 3b unambiguously, the X-ray-quality single crystals (Table 1) were grown by slow evaporation of their solution in choloroform and dioxan solvent, respectively, at room temperature. Compounds 3a and 3b were crystallized as the orthorhombic crystal lattice with the Pbcn space group and the triclinic crystal lattice with the P̅1 space group, respectively. Interestingly, in a unit cell of compound 3a, there are two different/independent molecules (named conformer A and conformer B) having slightly dissimilar bond lengths, bond angles, and dihedral angles. The molecular structures of coumarin-pendant thiosemicarbazones 3a and 3b having crystallographic numbering are shown in Figure 1.
Table 1. X-ray Crystallographic Data of Compounds 3a and 3b.
| crystal data | 3a | 3b |
|---|---|---|
| CCDC | 2026874 | 2026873 |
| chemical formula | C24H25N3O3S | C26H23N3O3S |
| Mr | 435.53 | 457.53 |
| crystal system, space group | orthorhombic, Pbcn | triclinic, P̅1 |
| temperature (K) | 296 | 296 |
| a, b, c (Å) | 17.8975 (13), 11.9162 (8), 41.526 (3) | 8.2958 (13), 11.681 (3), 12.753 (3) |
| α, β, γ (°) | 88.841 (6), 89.295 (6), 71.562 (6) | |
| V (Å3) | 8856.3 (10) | 1172.1 (4) |
| Z | 16 | 2 |
| radiation type | Mo Kα | Mo Kα |
| μ (mm–1) | 0.18 | 0.17 |
| crystal size (mm) | 0.44 × 0.38 × 0.32 | 0.38 × 0.32 × 0.24 |
| data collection | ||
| diffractometer | Bruker Kappa APEXII CCD | Bruker Kappa APEXII CCD |
| absorption correction | multiscan (SADABS; Bruker, 2005) | multiscan (SADABS; Bruker, 2005) |
| Tmin, Tmax | 0.895, 0.965 | 0.915, 0.970 |
| no. of measured, independent, and observed [I > 2σ(I)] reflections | 48611, 8699, 4680 | 15784, 4588, 3254 |
| Rint | 0.084 | 0.056 |
| (sin θ/λ)max (Å–1) | 0.617 | 0.617 |
| refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.064, 0.175, 1.02 | 0.051, 0.152, 1.02 |
| no. of reflections | 8699 | 4588 |
| no. of parameters | 545 | 300 |
| no. of restraints | 24 | |
| H-atom treatment | H-atom parameters constrained | H-atom parameters constrained |
| Δ⟩max, Δ⟩min (e Å–3) | 0.58, −0.33 | 0.33, −0.21 |
Figure 1.

View of the atom-labeled structures of compounds 3a and 3b.
The central core of coumarin-pendant thiosemicarbazones 3a and 3b comprising the N-iminothiourea moiety is nearly planar in the crystal structures of both compounds (Figure 1). The planarity of the central N-iminothiourea core can be ascribed to some double-bond nature of C–N of the NH–C=S moiety due to substantial delocalization of the lone pair of N at the C=S moiety, which is clearly depicted by shortening bond lengths of N(2)/(5)–C(7)/(31)–N(1)/(4) in 3a [N2–C7 1.349(4) Å (conformer A), N5–C31 1.350(4) Å (conformer B), N1–C7 1.349(4) Å (conformer A), N4–C31 1.325(4) Å (conformer B)] and N2–C18–N3 in 3b [N2–C18 1.350(3) Å and N3–C18 1.338(3) Å]. The slightly longer bond lengths of N(2)/(5)–C(7)/(31) in 3a and N2–C18 in 3b show less delocalization at thiocarbonyl (C=S), presumably due to the attached nearby sp2-hybridized N atom. This character of the central N-iminothiourea core offers two different possibilities of its conformational arrangements. Nevertheless, it preferably adopts only one conformation, i.e., cis, trans conformation in both 3a and 3b. The formation of this preferred conformation is attributed to comparatively stronger intramolecular hydrogen bonding in 3a [N(1)–H(1)···N(3) 2.226 Å (conformer A) and N(4)–H(4E)···N(6) 2.211 Å (conformer B)] and in 3b [N(3)–H(3A)···N(1) 2.234]. Notably, these findings are in complete agreement with our previously reported similar compounds.39−42
Owing to this preferred conformation around the central N-iminothiourea core, a thioamide R22 (8) [···H–N–C=S]2 synthon was observed in the packing of both the coumarin-pendant thiosemicarbazones 3a and 3b. This thioamide synthon is observed to be the major supramolecular force that stabilizes the solid-state molecular packing in 3a [N(2)–H(2)···S(1) 2.501 Å and N(5)–H(5)···S(2) 2.507 Å] and 3b [N(2)–H(2A)···S(1) 2.522 Å] (Figure 2). The other notable supramolecular interactions involved in the packing of 3a are N–H···O [N(1)–H(1)···O(5) 2.509 Å] and C–H···O [C(10)–H(10)···O(5) 2.719 Å, C(6A)–H(6A)···O(5) 2.714 Å, and C(30)–H(30A)···O(3) 2.475 Å] interactions. Similarly, the N–H···O [N(3)–H(3A)···O(2) 2.388 Å] and C-H···O [C(15)–H(15)···O(2) 2.635 Å, C(2)–H(2)···O(2) 2.657 Å] interactions are also found to be responsible for the 3D packing of molecules in 3b along with thioamide synthon (Figure 3).
Figure 2.
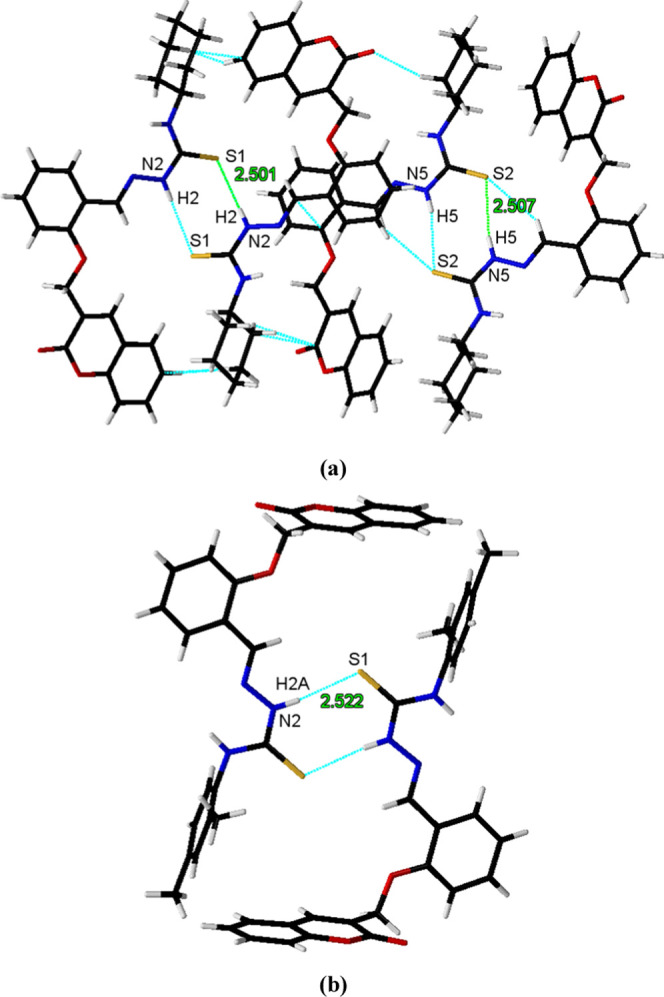
Supramolecular thioamide R22 (8) [···H–N–C=S]2 synthon in the packing of coumarin-pendant thiosemicarbazones: (a) 3a and (b) 3b.
Figure 3.

Solid-state molecular packing of coumarin-pendant thiosemicarbazones: (a) 3a along the c-axis and (b) 3b along the c-axis.
Molecular Geometric Parameters
For comparative evaluation of experimental results with DFT, the optimized structural parameters (bond angles, bond lengths) are estimated by the B3LYP level, and the results listed in Tables S1 and S2 (Supporting Information) are compared with structural parameters obtained from SC-XRD-generated geometries. The results obtained demonstrated that XRD-determined geometrical parameters and DFT-determined bond angles and lengths have good agreement. The infinitesimal difference between SC-XRD and DFT calculated parameters is constantly anticipated because SC-XRD generated results obtained from solid state, while DFT results are coupled to the gaseous state. Identical calculated SC-XRD and DFT bond lengths values were found: 1.459 Å in 3a for C15–C16 and 1.435 Å for O4–17 in 3b. The maximum deviation in bond length was observed to be 0.365 Å for N36–C45 and 0.028 Å for N6–25 in both 3a and 3b, respectively. In the same way, identical bond angles were found for O33–C52–C47, C46–C47–C52, and C25–C26–C27 in 3a and for O3–C16–C15, C8–C9–C10, and C30–C29–C33 in 3b. The greater deviation in bond angles was 11.4 and 2.6° in 3a for N36–C45–N37 and C25–N7–C26, respectively. Nevertheless, deviations in bond lengths were seen in the ranges of 0.365 ± 0.288 and 0.028 ± 0.022 Å in 3a and 3b, while for bond angles, these were calculated to be in the ranges of 11.4 ± 4.1 and 2.6 ± 1.5° in 3a and 3b, respectively.
In summary, certain DFT values deviate from XRD values, possibly because of intramolecular interactions in the solid state. Despite these small differences, both structural parameters are in good agreement and are able to provide a starting point for calculating other properties.
Natural Bond Orbital (NBO) Analysis
NBO analysis is an efficient technique that effectively describes the delocalization of electron density and intramolecular interactions. It offers suitable foundations to study intramolecular hydrogen bonding and charge transfer among the filled and vacant orbitals by the second-order Fock matrix.43,44 With the help of eq 1, the stabilization energy of the analyzed compounds (3a and 3b) is calculated, and the results are tabulated in Tables S3 and S4 (Supporting Information).
| 1 |
Herein, E(2) is the stabilization energy, F(i,j) is the diagonal, έj and έi are the off-diagonal NBO Fock matrix elements, and qi is the occupancy of the contributor orbital. In this NBO study, electron acceptor–donor interactions are exposed by the E(2) value. Usually, four major transitions—σ → σ*, π → π*, LP → σ*, and LP → π*—are observed for any chemical compound. The transitions (π → π*) were seen to have an extra projection, whereas LP → σ* and LP → π* transitions displayed reasonable E(2) values. Besides, the lowest E(2) values originate from σ → σ* transitions.
The peak values for π → π* electronic transitions are π(C43–C44) → π*(O4–C56) and π(C44–C45) → π*(C40–C47) with stabilization energies of 25.15 and 23.27 kcal/mol in 3a and 3b, respectively (see Tables S3 and S4 (Supporting Information)). However, some other transitions with significant values are also observed such as π(C37–C39) → π*(C33–C35), π(C30–C31) → π*(N9–C28), and π(C46–C55) → π*(C51–C53) in 3a with 21.85, 18.52, and 16.47 kcal/mol and π(C34–C36) → π*(N5–C37), π(C27–C28) → π*(C30–C32), and π(C34–C36) → π*(C30–C32) in 3b with 18.02, 14.96, and 12.22 kcal/mol stabilization energy, respectively. The π → π* transitions that offer minimum energy to the system are (C37–C39) → π*(C37–C39) and π(C27–C28) → π*(C27–C28) exhibiting 0.6 and 0.62 kcal/mol stabilization energy in both 3a and 3b, respectively (Tables S3 and S4 (Supporting Information)).
Some of the other significant transitions that are responsible for the resonance are as follows: LP(2)O3 → π*(O4–C56) and LP(2) O4 → ∂* with stabilization energies 41.09 and 35.24 kcal/mol in 3a and LP(2)O3 → σ*(O2–C23) and LP(2)O3 → σ*(C22–C23) with 37.01 and 15.67 kcal/mol in 3b. Some intramolecular hydrogen-bonding interactions are also observed, among which LP(1)S1 → ∂*(N7–H8) having 2.5 kcal/mol energy in 3a deserves special mention (Figure 4 and Table S3). Similarly in 3b, LP(2)S1 → ∂*(C10–H11), LP(1)O2 → ∂*(C40–H41), and LP(1)O2 → ∂*(C40–H42) transitions with 1.55, 1.12, and 1.20 kcal/mol stabilization energy values confirmed the stabilization of the system 3b (Figure 4 and Table S4) due to successful charge transfer. It is confirmed by the NBO investigation that noteworthy intramolecular interactions and extended hyperconjugation exist in the studied compounds, which provide them more stability and also give vital explanation of charge-transfer properties of these compounds, which are significant for charge transfer and potential NLO features.
Figure 4.

Frontier molecular orbitals describing charge density distribution in studied compounds 3a and 3b.
Vibrational Analysis
The theoretical and experimental communities use vibrational spectroscopy as a method to investigate the molecular vibrations.45 Similarly, DFT analysis for vibrational absorption spectroscopy is utilized to examine the existence of the vibrational modes for compounds 3a and 3b using the DFT/B3LYP/6-311+G(d,p) functional, and the results are listed in Tables S5 and S6 (Supporting Information), while their experimentally determined absorption frequencies and spectra are shown in Figures S1 and S2. In general, due to a well-known systematic error of anharmonicity and basis set deficiencies, the DFT hybrid B3LYP functional overrates the vibrational wavenumbers compared to the experimental results. Thus, the vibrational wavenumbers were scaled down by multiplying them with the scaling factor of 0.9617.46
C–H Vibrations
In the literature, 3100–3000 cm–1 is the characteristic area in which aromatic rings’ C–H vibrations typically arise.47 In our studied compounds, the stimulated C–H antisymmetric and symmetrical stretching modes in benzene rings are studied in the range of 3124–3096 cm–1 in 3a and at 3098–3084 cm–1 in 3b, which correlated with the experimental absorption frequencies at 3298 and 3295 cm–1, respectively, in both compounds. Similarly, stimulated C–H stretching vibrations other than aromatic rings are calculated at 3114 in 3a, which is in good agreement with their experimentally determined absorption frequencies noted at 3114 cm–1. Moreover, some of the symmetrical modes in C–H in 3b are also analyzed in the range of 2970–2923 cm–1.
C=C–C=C Vibrations
Both in aliphatic and aromatic (benzene and its derivatives) hydrocarbons, bands arising due to stretching C=C and C–C vibrations are highly important. Such vibrational bands are noted in the range of 1650–1400 cm–1 48,49 typically with aromatic derivatives. The stimulated absorption band is located at 1748–1567 cm–1 for 3a and at 1594–1304 cm–1 for 3b, which are in satisfactory agreement with experimentally determined absorption bands found at 1714 and 1455 cm–1, respectively.
C=O Vibrations
The absorption frequencies for the C=O band in 3a are located at 1748 and 1725 cm–1, which agreed with the experimental absorption band found at 1714 cm–1. In the same manner, the stimulated aforesaid absorption band for 3b is seen at 1739 cm–1, which was in good agreement at 1709 cm–1 with the experimental determination. Some other C–O symmetrical absorption bands are present in the range of 1395–1249 cm–1 in 3a and at 1217–995 cm–1 in 3b (see Tables S5 and S6).
C–N Vibrations
In our studied systems, the C–N stimulated band absorption frequencies are located at 1614–1573 and 1602–1560 cm–1 in 3a and 3b, respectively. Nevertheless, some C–N symmetrical vibrational frequencies are also observed at 1527–1478 cm–1 in 3a, which agreed with experimental absorption frequencies found at 1596, 1539, and 1506 cm–1. Similarly in 3b, the aforesaid C–N absorption frequencies are located at 1496–1477 cm–1, and their corresponding experimental absorption band is determined at 1595 cm–1.
C–S Vibrations
The absorption frequencies for the C–S group are present at 1490–1478 cm–1 and 1477, 1464, and 1189 cm–1 in 3a and 3b, respectively, which is in excellent agreement with the experimental absorption band at 1540 cm–1 in 3a and at 1223 cm–1 in 3b.
N–H Vibrations
In compound 3a, the stimulated symmetrical N–H vibrations are located in the span of 3460–3418 cm–1, which excellently correlated with the experimental band at 3649 cm–1. For 3b, these N–H frequencies are found at 3426–3407 cm–1. Moreover, some other frequencies like rocking are also observed at the 1527–1478 cm–1 range in 3a and at 1496–1237 cm–1 in 3b. Their experimentally determined absorption bands are present at 1596, 1540, and 1506 in 3a and at 1595, 1544, and 1455 cm–1 in 3b (see Tables S5 and S6).
In a nutshell, a good agreement is noted between DFT-computed and experimentally recorded vibrational frequencies.
UV–Vis Spectral Analysis
UV–visible spectroscopy provides a useful understanding of the charge-transfer prospects of compounds under investigation.50−52 The absorption spectra of 3a and 3b are calculated, and experimentally determined graphs are shown in Figures S3 and S4 (Supporting Information). Figures S3 and S4 reveal that 3a has the maximum wavelength of 335 nm with the absorbance value nearly equal to 1. On the other hand, the λmax calculated for 3b is found to be almost similar to that of 3a and has the same absorbance value.
Frontier Molecular Orbital (FMO) Analysis
The electronic transitions produced owing to the dipole moment that occurred between the excited and ground states of molecules are responsible for the optical properties. Generally, a transition occurs between the LUMO and HOMO.53−55 Moreover, the energy difference (ΔE) between these orbitals is a vital factor to describe the reactivity and stability of the molecules.56 For newly synthesized molecules 3a and 3b, the LUMO, HOMO, LUMO+1, HOMO-1, LUMO+2, and HOMO-2 energies and their band gaps are calculated and displayed in Table 2.
Table 2. EHOMO, ELUMO, and Energy Gap (ELUMO–EHOMO) of Investigated Compounds 3a–ba.
|
3a |
3b |
||||
|---|---|---|---|---|---|
| MO(s) | E | ΔE | MO(s) | E | ΔE |
| LUMO | –2.318 | 3.053 | LUMO | –2.579 | 3.118 |
| HOMO | –5.371 | HOMO | –5.697 | ||
| LUMO+1 | –2.046 | 3.364 | LUMO+1 | –1.942 | 3.762 |
| HOMO-1 | –5.410 | HOMO-1 | –5.704 | ||
| LUMO+2 | –1.729 | 3.83 | LUMO+2 | –1.106 | 5.285 |
| HOMO-2 | –5.559 | HOMO-2 | –6.391 | ||
E(HOMO), energy of HOMO; E(LUMO), energy of LUMO; ΔE, E(LUMO) – E(HOMO); MO, molecular orbital; energy is in eV.
It is analyzed that the band gaps calculated for different orbitals—HOMO → LUMO, HOMO-1 → LUMO+1, and HOMO-2 → LUMO+2—in 3b are greater than those in 3a. The band gap calculated for HOMO–LUMO is 3.118 eV in 3b, which narrows to 3.053 eV in 3a. The ΔE for HOMO-1 → LUMO+1 is 3.762 eV in 3b, which lessens to 3.364 eV in 3a, and the analyzed energy for HOMO-2 → LUMO+2 in 3b is 5.285 eV, which then further decreases to 3.83 eV in 3a. This decrease in band gap in 3a might be due to the existence of extended conjugation and noncovalent attraction as compared to 3b. The orbital showing the electron charge density distribution is expressed in Figure 4.
It can be seen from Figure 4 that the electron density for HOMO in 3a is concentrated at central N–N- or S-linked atoms, while LUMO is populated mainly on terminal cyclohexane units. In 3b, the same situation is noticed where the HOMO charge density is concentrated on central N–N- or S-linked atoms, while the LUMO charge density is found on the side 2H-chromen-2-one unit. In both studied structures, intramolecular charge transfer from the central unit (HOMO) to the terminal unit (LUMO) is evident, hence providing suitable reasons to utilize these molecules in charge-transfer-related phenomenon like NLO properties.
Global Reactivity Parameters
The band energy (ΔE) of FMOs is an excellent tool for the investigation of global reactivity parameters describing the donating, accepting, hardness, and softness of an investigated specie.57 Compounds with a huge energy difference are chemically hard in nature, which makes them kinetically more stable and least reactive.58−60 On the other hand, with less energy difference, molecules are soft in nature and show more polarizability, which makes them able to show excellent NLO response. Global reactivity parameters61−64 include ionization energy (IA), electron affinity (EA), electronegativity (X), global hardness (η), chemical potential (μ), global electrophilicity (ω), and global softness (σ), which can be calculated with the help of the energy difference of FMOs and eqs 2–8 and are listed in Table 3.
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
Table 3. Global Reactivity Descriptors of Investigated Compoundsa.
| compounds | I | A | X | η | μ | ω | σ |
|---|---|---|---|---|---|---|---|
| 3a | 5.371 | 2.318 | 3.844 | 1.526 | –3.844 | 4.841 | 0.327 |
| 3b | 5.697 | 2.579 | 4.138 | 1.559 | –4.138 | 5.491 | 0.320 |
Ionization potential (I), electron affinity (A), electronegativity (X), global hardness (η), chemical potential (μ), global electrophilicity (ω), and global softness (σ). Units are in eV.
The most significant chemical property is electronegativity, which explains the capability of any compound to attract electrons. The stability of molecules is indicated by the negative values of the chemical potential (μ). These attractive investigations can play an important role in the area of experimental research and especially in biological activity of compounds. Thus, for scientists, it becomes alluring to perform a large-scale study of global reactivity parameters. From Table 3, it is seen that the value of the ionization potential in 3b (5.697 eV) is greater, having higher values of electronegativity (4.138 eV) and electron affinity (2.579 eV), with a more negative value of the chemical potential (−4.138 eV) than 3a, which indicates that 3b is more stable. Furthermore, there is a direct relationship between the hardness and the band gap, so the molecule with a greater value of band gap is the least reactive chemically. Thus, the values of band gap and hardness calculated for 3b are higher than those for 3a with a lower value of softness (see Table 3), indicating that 3a is less stable and more reactive. The increasing order of chemical reactivity is 3b < 3a.
Nonlinear Optical (NLO) Analysis
Nonlinear optical organic materials are being increasingly taken into account in promising optoelectronic technologies due to their easy synthesis, less cost, facile manufacturing, and structural modifications. Quantum chemical evaluation of polarizability <α> and hyperpolarizability (β) is a field of extensive analysis since they are linked to the new optical nonlinear material architectures.65−70 Thus, DFT calculations were performed to explore the possible NLO properties of investigated molecules 3a and 3b. The dipole moment (D), polarizability <α>, and frequency-dependent second hyperpolarizability (γ) at different levels of theory include the HF method, metahybrid GGA methods M06, M062X, range-corrected methods LC-BLYP, CAM-B3LYP, and exchange correlation functional B3LYP in conjunction with the 6-311+G(d,p) basis set. Furthermore, the second harmonic generation (SHG) form γ(−2ω,ω,ω,0) and the EOKO (electro-optic Kerr effect) form γ(−ω,ω,0,0) are also computed at the commonly exercised wavelength 0.02389 nm, and the results are tabulated in Tables 4–7.
Table 4. Dipole Polarizabilities with Major Contributing Tensors (a.u.) for Investigated Compounds 3a and 3b.
| compounds | methods | αxx | αyy | αzz | αtotal |
|---|---|---|---|---|---|
| 3a | B3LYP | 782.52 | 791.21 | 385.61 | 653.12 |
| CAM-B3LYP | 748.19 | 761.07 | 377.50 | 628.92 | |
| LC-BLYP | 714.98 | 731.58 | 368.70 | 605.09 | |
| HF | 690.83 | 708.49 | 365.00 | 588.11 | |
| M06 | 779.83 | 790.16 | 386.70 | 652.23 | |
| M062X | 755.33 | 766.45 | 381.91 | 634.56 | |
| 3b | B3LYP | 489.42 | 450.24 | 289.30 | 409.65 |
| CAM-B3LYP | 465.44 | 429.48 | 285.05 | 393.32 | |
| LC-BLYP | 441.85 | 409.03 | 278.80 | 376.56 | |
| HF | 427.51 | 395.28 | 276.11 | 366.30 | |
| M06 | 487.43 | 446.67 | 289.54 | 407.88 | |
| M062X | 464.10 | 427.45 | 282.65 | 391.40 |
Table 7. Frequency-Dependent Second Hyperpolarizability (a.u.) of Investigated Compounds 3a and 3b.
| parameters | frequency ω | B3LYP |
CAM-B3LYP |
LC-BLYP |
|||
|---|---|---|---|---|---|---|---|
| compound | 3a | 3b | 3a | 3b | 3a | 3b | |
| γ(−ω,ω,0,0) | 0.000 | 2.435 × 105 | 2.764 × 105 | 1.534 × 105 | 1.835 × 105 | 1.106 × 105 | 1.333 × 105 |
| 0.02389 nm | 2.609 × 105 | 2.973 × 105 | 1.604 × 105 | 1.925 × 105 | 1.143 × 105 | 1.380 × 105 | |
| γ(−2ω,ω,ω,0) | 0.000 | 2.435 × 105 | 2.764 × 105 | 1.534 × 105 | 1.835 × 105 | 1.106 × 105 | 1.333 × 105 |
| 0.02389 nm | 3.030 × 105 | 3.439 × 105 | 1.760 × 105 | 2.110 × 105 | 1.230 × 105 | 1.484 × 105 | |
| HF |
M06 |
M062X |
|||||
|---|---|---|---|---|---|---|---|
| 3a | 3b | 3a | 3b | 3a | 3b | ||
| γ(−ω,ω,0,0) | 0.000 | 8.129 × 104 | 1.061 × 105 | 2.157 × 105 | 2.498 × 105 | 1.615 × 105 | 1.861 × 105 |
| 0.02389 nm | 8.358 × 104 | 1.090 × 105 | 2.340 × 105 | 2.585 × 105 | 1.714 × 105 | 1.933 × 105 | |
| γ(−2ω,ω,ω,0) | 0.000 | 8.129 × 104 | 1.061 × 105 | 2.157 × 105 | 2.498 × 105 | 1.615 × 105 | 1.861 × 105 |
| 0.02389 nm | 8.852 × 104 | 1.155 × 105 | 2.662 × 105 | 2.955 × 105 | 1.885 × 105 | 2.126 × 105 | |
The following equation (eq 9) is used to calculate average polarizability:
| 9 |
The dynamic frequency-dependent second hyperpolarizability γ is calculated using the following equation (eq 10):
| 10 |
It can be seen from Table 4 that the polarizability tensor calculated in the y-axis is dominant among all tensors in 3a and the x-axis in 3b at all levels of theory and contributed well to the total linear polarization values.
The maximum <α> value for both compounds 3a and 3b was investigated, which were found to be 653.12 and 409.65 (a.u.), respectively, at B3LYP; then, this value reduces to 652.23 and 407.88 (a.u.), as analyzed at M06, respectively. This average polarizability value further narrows down to 634.56 (a.u.) at M062X in 3a and to 393.32 (a.u.) at the CAM-B3LYP level in 3b. Further decline in the value of <α> from 605.09 to 588.11 (a.u.) in 3a and from 376.56 to 366.30 (a.u.) in 3b was seen, as studied at LC-BLYP and HF, respectively. The lowest polarizability values in both compounds among all levels of theory were found using the HF method. Overall, the highest value of <α> was studied in 3a at all levels of theory.
Table 5 reveals that for both crystals 3a and 3b the highest dipole moment values were analyzed using the HF method as 2.3953 D and 3.3752 D, respectively. The lowest value for 3a was 2.1180 D obtained using the B3LYP method, while in 3b, 2.7907 D was calculated at the M06 level. Overall, the maximum value of the dipole moment was investigated in 3b (3.3752 D) at all levels of theory and also shows a higher value than that of the reference molecule (urea) (1.3732 D).71 Nevertheless, it is generally seen that molecules with a greater dipole moment show higher hyperpolarizability properties. Thus, the maximum γ value was investigated in 3b, and this value becomes narrow in 3a (see Table 5).
Table 5. Computed Dipole Moments D of Investigated Compounds 3a and 3b.
| compounds | methods | μx | μy | μz | μtotal |
|---|---|---|---|---|---|
| 3a | B3LYP | 0.2444 | 1.0410 | 1.8281 | 2.1180 |
| CAM-B3LYP | 0.2384 | 1.0545 | 1.8689 | 2.1591 | |
| LC-BLYP | 0.2328 | 1.0691 | 1.9109 | 2.2020 | |
| HF | 0.2848 | 1.1217 | 2.0971 | 2.3953 | |
| M06 | 0.2396 | 1.0206 | 1.8503 | 2.1267 | |
| M062X | 0.2332 | 1.0272 | 1.8534 | 2.1318 | |
| 3b | B3LYP | –1.8213 | 0.9229 | –1.9517 | 2.8246 |
| CAM-B3LYP | –1.8933 | 1.0585 | –2.0075 | 2.9555 | |
| LC-BLYP | –1.9710 | –1.9710 | –2.0792 | 3.1056 | |
| HF | –2.1419 | 1.3572 | –2.2276 | 3.3752 | |
| M06 | –1.8360 | 0.8490 | –1.9225 | 2.7907 | |
| M062X | –1.8227 | 1.0309 | –1.9750 | 2.8785 |
The investigation performed to calculate the second hyperpolarizability values of entitled compounds at different levels of theory results shown in Table 6 indicates that between both compounds, the highest value of <γ> was found to be 2.435 × 105 (a.u.) in 3a and 2.764 × 105 (a.u.) in 3b at the B3LYP level of theory. Contrarily, the lowest value was 8.129 × 104 in 3a and 1.060 × 105 (a.u.) in 3b obtained from the HF method. So overall, B3LYP and HF methods exhibit the highest and lowest average second hyperpolarizability values, respectively. Moreover, it was also seen that crystal 3b showed a higher value of the second-order polarizability at all levels of theory than that of 3a, except for the HF method in 3a. Our findings have been compared with the reference urea molecule for comparative NLO analysis. The values of αtotal, dipole moment, and γ of the studied compounds are larger in contrast to the reference molecule.71
Table 6. Computed Second Hyperpolarizability γ and Major Contributing Tensors (a.u.) for Investigated Compounds 3a and 3b.
| compounds | methods | γx | γy | γz | average <γ> | magnitude of γ |
|---|---|---|---|---|---|---|
| 3a | B3LYP | 1.148 × 105 | 1.035 × 105 | 2.519 × 105 | 2.435 × 105 | 1.566 × 105 |
| CAM-B3LYP | 7.112 × 104 | 6.626 × 104 | 1.607 × 104 | 1.534 × 105 | 9.852 × 104 | |
| LC-BLYP | 4.907 × 104 | 4.988 × 104 | 1.164 × 104 | 1.106 × 105 | 7.093 × 104 | |
| HF | 3.301 × 104 | 3.926 × 104 | 9.016 × 103 | 8.129 × 104 | 5.208 × 104 | |
| M06 | 1.004 × 105 | 9.273 × 104 | 2.254 × 104 | 2.157 × 105 | 1.385 × 105 | |
| M062X | 7.492 × 104 | 6.983 × 104 | 1.677 × 104 | 1.615 × 105 | 1.037 × 105 | |
| 3b | B3LYP | 1.366 × 105 | 1.156 × 105 | 2.404 × 104 | 2.764 × 105 | 1.806 × 105 |
| CAM-B3LYP | 8.398 × 104 | 8.013 × 104 | 1.941 × 104 | 1.835 × 105 | 1.176 × 105 | |
| LC-BLYP | 5.894 × 104 | 5.909 × 104 | 1.531 × 104 | 1.333 × 105 | 8.485 × 104 | |
| HF | 4.365 × 104 | 4.785 × 104 | 1.456 × 104 | 1.060 × 105 | 6.638 × 104 | |
| M06 | 1.220 × 105 | 1.054 × 105 | 2.240 × 104 | 2.497 × 105 | 1.627 × 105 | |
| M062X | 8.554 × 104 | 8.220 × 104 | 1.842 × 104 | 1.861 × 105 | 1.200 × 105 |
The second harmonic generation (SHG) form γ(−2ω,ω,ω,0) and the EOKO (electro-optic Kerr effect) form γ(−ω,ω,0,0) computed at zero and 0.02389 nm wavelengths are shown in Table 7.
Moreover, it was also seen that crystal 3b showed a higher value of second-order hyperpolarizability at all levels of theory than that of 3a. Overall results show that the crystals under investigation are polarized in nature with a good dipole moment. The comparative analysis with urea molecules clearly indicates that the studied compounds are acceptable NLO candidates, and they can be used for future technological applications.
Molecular Electrostatic Potential (MEP)
MEP is associated with electron density (ED), and through it we can illustrate the noncovalent interactions and also comprehend reactivity, such as electrophilic and nucleophilic attack sites.72,73 MEP is actually the elucidation of the three-dimensional electron density in the form of a graph, and with the help of this three-dimensional map, the chemical and physical characteristics of a chemical structure can also be revealed. There are different types of standard colors on the MEP map: blue, green, yellow, orange, and red, which represent the electrostatic potential magnitude.74 The electrostatic potential magnitude in increasing order is red < orange < yellow < green < blue.75 On the MEP plot, the blue part represents the highest positive potential and is a more appropriate place for the attack of a nucleophile, while the red color indicates the highest negative potential and is a suitable site for electrophile attack. MEP can be calculated with the help of eq 11.
| 11 |
where V(r) is the electrostatic potential, ρ(r′) is the electron density, and ZA is the nuclear charge that is placed at RA.76,77 The MEP maps of studied compounds (3a and 3b) are displayed in Figure 5, which explain that the red color in maps is due to the O and S atoms in 3a and 3b. Thus, it is an electron-rich area and a suitable site for electrophilic attack. On the other hand, the blue color, which describes the electron-deficient part on the plot, appears due to some of the H and C atoms and is a potential place for nucleophile attack. The green areas show the mean potential, i.e., the area between two extremes. From red, yellow, and blue colors, it is clear that different reaction sites are present in all molecules.
Figure 5.
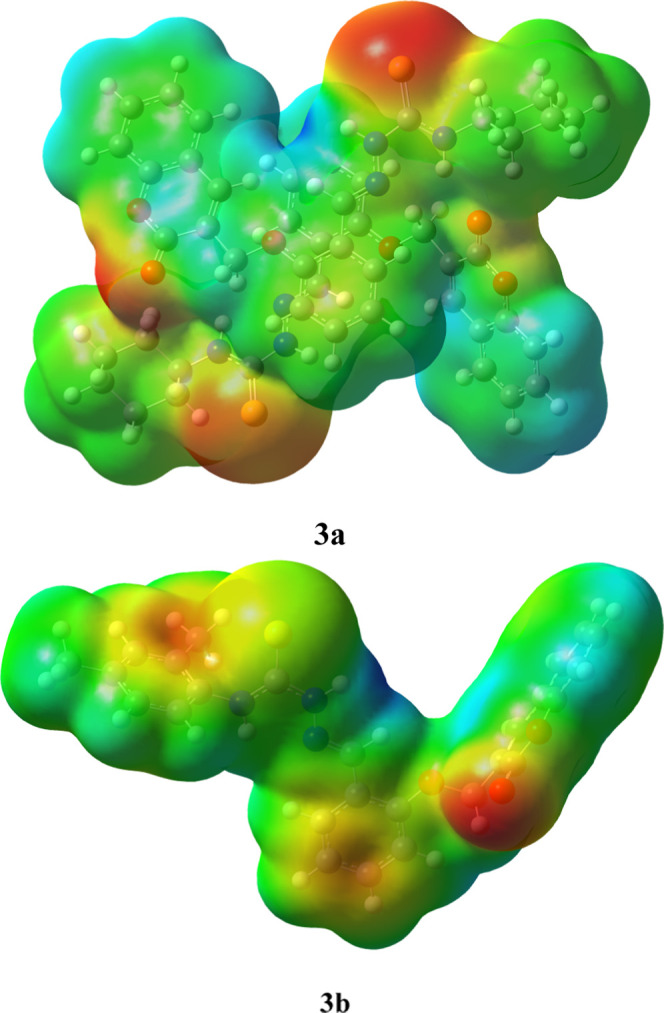
MEPs and color scheme of the studied compounds 3a and 3b.
Conclusions
By the condensation of thiosemicarbazide with coumarin aldehyde, two novel thiosemicarbazone substituted (E)-N-cyclohexyl-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)hydrazinecarbothioamide (3a) and 2,4(E)-N-(2,4-dimethylphenyl)-2-(2-((2-oxo-2H-chromen-3-yl)methoxy)benzylidene)hydrazinecarbothioamide (3b) were synthesized in excellent 80% yield, exhibiting good spectral as well as single-crystal X-ray investigations. The SC-XRD analysis reveals that the compounds were crystallized as an orthorhombic crystal lattice with the Pbcn space group and the triclinic crystal lattice with the P̅1 space group. Roles of several noncovalent interactions were discussed via SC-XRD, which stabilizes both compound’s solid-state structures. Aside from trial SC-XRD, the molecular geometries of the investigated molecules (3a and 3b) have also been concentrated by the computational calculations. The calculated results were compared with the experimental structural parameters, vibrational frequencies, and UV–vis findings. As analyzed by the NBO investigation, the most significant transitions have been found in 3a with a larger stabilization energy of 25.15 kcal/mol and in 3b with 23.27 kcal/mol. The energy differences calculated for HOMO/LUMO are 3.053 and 3.118 eV in 3a and 3b, respectively. Thus, the values of band gap and hardness calculated for 3b are higher than those for 3a with a lower value of softness, indicating that 3a is less stable and more reactive than 3b. The UV–visible analysis findings at 345 and 340 nm for experimentally determined values in 3a and 3b, respectively, indicated that compounds have a bathochromic shift. The highest <α> values for both compounds 3a and 3b were found to be 653.12 and 409.65 (a.u.), respectively, at the B3LYP level. The lowest polarizability values in both compounds among all levels of theory were found from the HF method. The highest values of <γ> were 2.435 × 105 (a.u.) in 3a and 2.764 × 105 (a.u.) in 3b at the B3LYP level of theory. Contrarily, the lowest value of <γ> was 8.129 × 104 in 3a and 1.060 × 105 (a.u.) in 3b obtained from the HF method. Moreover, it was also found that crystal 3b showed a higher value of second-order polarizability at all levels of theory than that of 3a. The comparative analysis with urea molecules clearly indicates that the studied compounds are acceptable NLO candidates and they can be used for future technological applications.
Acknowledgments
Z.S. is thankful to Higher Education Commission (HEC), Islamabad, Pakistan through Project No. 6975/NRPU/R&D for the financial support. A.A.C.B. (grants # 2011/07895-8, 2015/01491-3, and 2014/25770-6) is thankful to Fundação de Amparo à Pesquisa do Estado de São Paulo for financial support. A.A.C.B. (grant 309715/2017-2) also thanks the Brazilian National Research Council (CNPq) for financial support and fellowships. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brazil (CAPES)—Finance Code 001.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04653.
Comparison of selected bond lengths (Å) and angles (°) for 3a and 3b calculated using B3LYP/6-311+G(d,p) and the X-ray diffraction method; natural bond orbital (NBO) analysis data; experimental and DFT-calculated vibrational frequencies; and experimental calculated UV–vis, FT-IR, and NMR spectra of investigated compounds 3a and 3b (PDF)
Crystallographic data of 3a (CIF)
Crystallographic data of 3b (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Borges F.; Roleira F.; Milhazes N.; Santana L.; Uriarte E. Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]
- Nicolaou K.; Pfefferkorn J.; Mitchell H.; Roecker A.; Barluenga S.; Cao G.-Q.; Affleck R.; Lillig J. Natural product-like combinatorial libraries based on privileged structures. 2. Construction of a 10 000-membered benzopyran library by directed split-and-pool chemistry using NanoKans and optical encoding. J. Am. Chem. Soc. 2000, 122, 9954–9967. 10.1021/ja002034c. [DOI] [Google Scholar]
- Nicolaou K.; Pfefferkorn J.; Barluenga S.; Mitchell H.; Roecker A.; Cao G.-Q. Natural product-like combinatorial libraries based on privileged structures. 3. The “libraries from libraries” principle for diversity enhancement of benzopyran libraries. J. Am. Chem. Soc. 2000, 122, 9968–9976. 10.1021/ja0020355. [DOI] [Google Scholar]
- Cragg G. M.; Newman D. J. Natural products: a continuing source of novel drug leads. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 3670–3695. 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jashari A.; Imeri F.; Ballazhi L.; Shabani A.; Mikhova B.; Dräger G.; Popovski E.; Huwiler A. Synthesis and cellular characterization of novel isoxazolo-and thiazolohydrazinylidene-chroman-2, 4-diones on cancer and non-cancer cell growth and death. Bioorg. Med. Chem. 2014, 22, 2655–2661. 10.1016/j.bmc.2014.03.026. [DOI] [PubMed] [Google Scholar]
- Peng X.-M.; LV Damu G.; Zhou H. Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des. 2013, 19, 3884–3930. 10.2174/1381612811319210013. [DOI] [PubMed] [Google Scholar]
- Yang J.; Liu G.-Y.; Dai F.; Cao X.-Y.; Kang Y.-f.; Hu L.-M.; Tang J.-J.; Li X.-Z.; Li Y.; Jin X.-L.; et al. Synthesis and biological evaluation of hydroxylated 3-phenylcoumarins as antioxidants and antiproliferative agents. Bioorg. Med. Chem. Lett. 2011, 21, 6420–6425. 10.1016/j.bmcl.2011.08.090. [DOI] [PubMed] [Google Scholar]
- Musa M. A.; Badisa V. L.; Latinwo L. M.; Cooperwood J.; Sinclair A.; Abdullah A. Cytotoxic activity of new acetoxycoumarin derivatives in cancer cell lines. Anticancer Res. 2011, 31, 2017–2022. [PMC free article] [PubMed] [Google Scholar]
- Borah P.; Naidu P. S.; Bhuyan P. J. Synthesis of some tetrazole fused pyrido [2, 3-c] coumarin derivatives from a one-pot three-component reaction via intramolecular 1, 3-dipolar cycloaddition reaction of azide to nitriles. Tetrahedron Lett. 2012, 53, 5034–5037. 10.1016/j.tetlet.2012.07.060. [DOI] [Google Scholar]
- El Ansary S. Synthesis and biological activity of some new coumarins, Egypt. J. Pharm. Sci. 1992, 33, 639–650. [Google Scholar]
- Mazzone G.; Malaj N.; Galano A.; Russo N.; Toscano M. Antioxidant properties of several coumarin–chalcone hybrids from theoretical insights. RSC Adv. 2015, 5, 565–575. 10.1039/C4RA11733F. [DOI] [Google Scholar]
- Montagner C.; de Souza S. M.; Groposo C.; Delle Monache F.; Smânia E. F.; Smânia A. Jr. Antifungal activity of coumarins. Z. Naturforsch. C 2008, 63, 21–28. 10.1515/znc-2008-1-205. [DOI] [PubMed] [Google Scholar]
- De Araújo R. S.; Guerra F. Q.; Lima D. O.; De Simone C. A.; Tavares J. F.; Scotti L.; Scotti M. T.; De Aquino T. M.; De Moura R. O.; Mendonça F. J.; et al. Synthesis, structure-activity relationships (SAR) and in silico studies of coumarin derivatives with antifungal activity. Int. J. Mol. Sci. 2013, 14, 1293–1309. 10.3390/ijms14011293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J.-C.; Kim J.-C.; Park O.-S. Simple and cost effective syntheses of 4-hydroxycoumarin. Synth. Commun. 1999, 29, 3587–3595. 10.1080/00397919908085993. [DOI] [Google Scholar]
- Barker W. M.; Hermodson M. A.; Link K. 4-Hydroxycoumarins. Synthesis of the metabolites and some other derivatives of warfarin. J. Med. Chem. 1971, 14, 167–169. 10.1021/jm00284a022. [DOI] [PubMed] [Google Scholar]
- Greaves M. Pharmacogenetics in the management of coumarin anticoagulant therapy: the way forward or an expensive diversion?. PLoS Med. 2005, 2, e342 10.1371/journal.pmed.0020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum N. F.; Aleem A.; Nayeem N.; Asdaq S. Synthesis and antibacterial activity of substituted 2-phenyl-4-chromones. Pharma Chem. 2011, 3, 56–62. [Google Scholar]
- de Souza S. M.; Delle Monache F.; Smânia A. Antibacterial activity of coumarins. Z. Naturforsch. C 2005, 60, 693–700. 10.1515/znc-2005-9-1006. [DOI] [PubMed] [Google Scholar]
- Behrami A. Antibacterial Activity of Coumarine Derivatives Synthesized from 4-Chloro-chromen-2-one. The Comparison with Standard Drug. Orient. J. Chem. 2014, 30, 1747–1752. 10.13005/ojc/300433. [DOI] [Google Scholar]
- McKee T. C.; Fuller R. W.; Covington C. D.; Cardellina J. H.; Gulakowski R. J.; Krepps B. L.; McMahon J. B.; Boyd M. R. New pyranocoumarins isolated from Calophyllum lanigerum and Calophyllum teysmannii. J. Nat. Prod. 1996, 59, 754–758. 10.1021/np9603784. [DOI] [PubMed] [Google Scholar]
- Wattenberg L. W.; Lam L. K.; Fladmoe A. V. Inhibition of chemical carcinogen-induced neoplasia by coumarins and α-angelicalactone. Cancer Res. 1979, 39, 1651–1654. [PubMed] [Google Scholar]
- Kashman Y.; Gustafson K. R.; Fuller R.; Cardellina J. 2nd; McMahon J.; Currens M.; Buckheit R. Jr; Hughes S.; Cragg G.; Boyd M. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum. J. Med. Chem. 1992, 35, 2735–2743. 10.1021/jm00093a004. [DOI] [PubMed] [Google Scholar]
- Manfredini S.; Simoni D.; Ferroni R.; Bazzanini R.; Vertuani S.; Hatse S.; Balzarini J.; De Clercq E. Retinoic acid conjugates as potential antitumor agents: synthesis and biological activity of conjugates with Ara-A, Ara-C, 3 (2 H)-furanone, and aniline mustard moieties. J. Med. Chem. 1997, 40, 3851–3857. 10.1021/jm9602322. [DOI] [PubMed] [Google Scholar]
- Sayed M.; Kamal El-Dean A. M.; Ahmed M.; Hassanien R. Synthesis of some heterocyclic compounds derived from indole as antimicrobial agents. Synth. Commun. 2018, 48, 413–421. 10.1080/00397911.2017.1403627. [DOI] [Google Scholar]
- Peng Z.; Yu L. Second-order nonlinear optical polyimide with high-temperature stability. Macromolecules 1994, 27, 2638–2640. 10.1021/ma00087a039. [DOI] [Google Scholar]
- Tsutsumi N.; Morishima M.; Sakai W. Nonlinear optical (NLO) polymers. 3. NLO polyimide with dipole moments aligned transverse to the imide linkage. Macromolecules 1998, 31, 7764–7769. 10.1021/ma9803436. [DOI] [Google Scholar]
- Breitung E. M.; Shu C.-F.; McMahon R. J. Thiazole and thiophene analogues of donor– acceptor stilbenes: molecular hyperpolarizabilities and structure– property relationships. J. Am. Chem. Soc. 2000, 122, 1154–1160. 10.1021/ja9930364. [DOI] [Google Scholar]
- Eaton D. F.Nonlinear Optical Materials: The Great and Near Great; ACS Publications, 1991. [Google Scholar]
- Hanumantharao R.; Kalainathan S.; Bhagavannarayana G. Growth, spectral, optical, thermal, crystallization perfection and nonlinear optical studies of novel nonlinear optical crystal—Urea thiosemicarbazone monohydrate. Spectrochim. Acta, Part A 2012, 91, 345–351. 10.1016/j.saa.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Jawaria R.; Hussain M.; Khalid M.; Khan M. U.; Tahir M. N.; Naseer M. M.; Braga A. A. C.; Shafiq Z. Synthesis, crystal structure analysis, spectral characterization and nonlinear optical exploration of potent thiosemicarbazones based compounds: A DFT refine experimental study. Inorg. Chim. Acta 2019, 486, 162–171. 10.1016/j.ica.2018.10.035. [DOI] [Google Scholar]
- Santhakumari R.; Ramamurthi K.; Vasuki G.; Yamin B. M.; Bhagavannarayana G. Synthesis and spectral characterization of acetophenone thiosemicarbazone—A nonlinear optical material. Spectrochim. Acta, Part A 2010, 76, 369–375. 10.1016/j.saa.2010.03.030. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. J.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Weinhold F.; Glendening E. D.. NBO 5.0 Program Manual: Natural Bond Orbital Analysis Programs. In Theoretical Chemistry Institute and Department of Chemistry; University of Wisconsin: Madison, WI, 2001; p 53706. [Google Scholar]
- Dennington R.; Keith T.; Millam J.. GaussView, Version 5; Semichem, Inc.: Shawnee Mission, KS, 2009.
- Hanwell M. D.; Curtis D. E.; Lonie D. C.; Vandermeersch T.; Zurek E.; Hutchison G. R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrienko G. A.Chemcraft. Graphical Software for Visualization of Quantum Chemistry Computations, 2010.
- Kaye P. T.; Musa M. A.; Nocanda X. W.; Robinson R. S. Does the DABCO-catalysed reaction of 2-hydroxybenzaldehydes with methyl acrylate follow a Baylis–Hillman pathway?. Org. Biomol. Chem. 2003, 1, 1133–1138. 10.1039/b300360d. [DOI] [PubMed] [Google Scholar]
- Hameed A.; Shafiq Z.; Yaqub M.; Hussain M.; Ahmad H. B.; Tahir M. N.; Naseer M. M. Robustness of a thioamide {··· H–N–C [double bond, length as m-dash] S} 2 synthon: synthesis and the effect of substituents on the formation of layered to cage-like supramolecular networks in coumarin–thiosemicarbazone hybrids. New J. Chem. 2015, 39, 6052–6061. 10.1039/C5NJ00734H. [DOI] [Google Scholar]
- Hameed A.; Shafiq Z.; Yaqub M.; Hussain M.; Hussain M. A.; Afzal M.; Tahir M. N.; Naseer M. M. Me 3 N-promoted synthesis of 2, 3, 4, 4 a-tetrahydroxanthen-1-one: preparation of thiosemicarbazone derivatives, their solid state self-assembly and antimicrobial properties. New J. Chem. 2015, 39, 9351–9357. 10.1039/C5NJ01879J. [DOI] [Google Scholar]
- Hameed A.; Shafiq Z.; Yaqub M.; Hussain M.; Ahmad H.; Tahir M.; Naseer M. Robustness of a thioamide synthon: synthesis and the effect of substituents on the formation of layered to cage-like supramolecular networks in coumarin–thiosemicarbazone hybrids. New J. Chem. 2015, 39, 6052–6061. 10.1039/C5NJ00734H. [DOI] [Google Scholar]
- Jawaria R.; Hussain M.; Shafiq Z.; Ahmad H. B.; Tahir M. N.; Shad H. A.; Naseer M. M. Robustness of thioamide dimer synthon, carbon bonding and thioamide–thioamide stacking in ferrocene-based thiosemicarbazones. CrystEngComm 2015, 17, 2553–2561. 10.1039/C4CE02566K. [DOI] [Google Scholar]
- Hussain M.; Jawaria R.; Shafiq Z.; Abbas G.; Naseer M. M. Ferrocene-based thiosemicarbazones: Solvent effect on thiol-thione tautomerism and conformational polymorphism. J. Organomet. Chem. 2017, 846, 121–128. 10.1016/j.jorganchem.2017.05.005. [DOI] [Google Scholar]
- Haroon M.; Khalid M.; Akhtar T.; Tahir M. N.; Khan M. U.; Muhammad S.; Al-Sehemi A. G.; Hameed S. Synthesis, crystal structure, spectroscopic, electronic and nonlinear optical properties of potent thiazole based derivatives: Joint experimental and computational insight. J. Mol. Struct. 2020, 1202, 127354 10.1016/j.molstruc.2019.127354. [DOI] [Google Scholar]
- Khan B.; Khalid M.; Shah M. R.; Tahir M. N.; Khan M. U.; Ali A.; Muhammad S. Efficient Synthesis by Mono-Carboxy Methylation of 4, 4′-Biphenol, X-ray Diffraction, Spectroscopic Characterization and Computational Study of the Crystal Packing of Ethyl 2-((4′-hydroxy-[1, 1′-biphenyl]-4-yl) oxy) acetate. ChemistrySelect 2019, 4, 9274–9284. 10.1002/slct.201901422. [DOI] [Google Scholar]
- Ali A.; Khalid M.; Rehman M. A.; Anwar F.; Zain-Ul-Aabidin H.; Akhtar M. N.; Khan M. U.; Braga A. A. C.; Assiri M. A.; Imran M. An Experimental and Computational Exploration on the Electronic, Spectroscopic, and Reactivity Properties of Novel Halo-Functionalized Hydrazones. ACS Omega 2020, 5, 18907–18918. 10.1021/acsomega.0c02128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M. P.; Uvdal P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis set 6-311+ G (d, p). J. Phys. Chem. A 2005, 109, 2937–2941. 10.1021/jp045733a. [DOI] [PubMed] [Google Scholar]
- Socrates G. Infrared and Raman characteristic group frequencies: table and charts. Ltd WJS 2001, 1–347. [Google Scholar]
- Perkampus H. H. LJ Bellamy: The Infrared Spectra of Complex Molecules, Vol. 1, 3. Auflage, Chapman and Hall Ltd., London 1975, 433 Seiten, 32 Abb., 22 Tabellen, Preis:£ 8.—. Ber. Bunsenges. Phys. Chem. 1976, 80, 99–100. 10.1002/bbpc.19760800121. [DOI] [Google Scholar]
- Sathiyanarayanan D.Vibrational Spectroscopy Theory and Application; New Age International Publishers: New Delhi, 2004. [Google Scholar]
- Khan M. U.; Khalid M.; Arshad M. N.; Khan M. N.; Usman M.; Ali A.; Saifullah B. Designing Star-Shaped Subphthalocyanine-Based Acceptor Materials with Promising Photovoltaic Parameters for Non-fullerene Solar Cells. ACS Omega 2020, 23039–23052. 10.1021/acsomega.0c02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. U.; Hussain R.; Yasir Mehboob M.; Khalid M.; Shafiq Z.; Aslam M.; Al-Saadi A. A.; Jamil S.; Janjua M. R. S. A. In Silico Modeling of New “Y-Series”-Based Near-Infrared Sensitive Non-Fullerene Acceptors for Efficient Organic Solar Cells. ACS Omega 2020, 24125–24137. 10.1021/acsomega.0c03796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. U.; Hussain R.; Mehboob M. Y.; Khalid M.; Ehsan M. A.; Rehman A.; Janjua M. R. S. A. First theoretical framework of Z-shaped acceptor materials with fused-chrysene core for high performance organic solar cells. Spectrochim. Acta, Part A 2020, 118938. [DOI] [PubMed] [Google Scholar]
- Khan M. U.; Iqbal J.; Khalid M.; Hussain R.; Braga A. A. C.; Hussain M.; Muhammad S. Designing triazatruxene-based donor materials with promising photovoltaic parameters for organic solar cells. RSC Adv. 2019, 9, 26402–26418. 10.1039/C9RA03856F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M. U.; Mehboob M. Y.; Hussain R.; Afzal Z.; Khalid M.; Adnan M. Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high-efficiency organic solar cells. Int. J. Quantum Chem. 2020, e26377 10.1002/qua.26377. [DOI] [Google Scholar]
- Khan M. U.; Mehboob M. Y.; Hussain R.; Fatima R.; Tahir M. S.; Khalid M.; Braga A. A. C. Molecular designing of high-performance 3D star-shaped electron acceptors containing a truxene core for nonfullerene organic solar cells. J. Phys. Org. Chem. 2020, e4119 10.1002/poc.4119. [DOI] [Google Scholar]
- Ahmed M.; Imran M.; Muddassar M.; Hussain R.; Khan M. U.; Ahmad S.; Mehboob M. Y.; Ashfaq S. Benzenesulfonohydrazides inhibiting urease: Design, synthesis, their in vitro and in silico studies. J. Mol. Struct. 2020, 128740 10.1016/j.molstruc.2020.128740. [DOI] [Google Scholar]
- Khalid M.; Ali M.; Aslam M.; Sumrra S. H.; Khan M. U.; Raza N.; Kumar N.; Imran M. Frontier molecular, Natural bond orbital, UV-Vis spectral stduy, Solvent influence on geometric parameters, Vibrational frequencies and solvation energies of 8-Hydroxyquinoline. Int. J. Pharm. Sci. Res. 2017, 8, 457. [Google Scholar]
- Sinha L.; Prasad O.; Narayan V.; Shukla S. R. Raman, FT-IR spectroscopic analysis and first-order hyperpolarisability of 3-benzoyl-5-chlorouracil by first principles. Mol. Simul. 2011, 37, 153–163. 10.1080/08927022.2010.533273. [DOI] [Google Scholar]
- Lewis D.; Ioannides C.; Parke D. Interaction of a series of nitriles with the alcohol-inducible isoform of P450: Computer analysis of structure—activity relationships. Xenobiotica 1994, 24, 401–408. 10.3109/00498259409043243. [DOI] [PubMed] [Google Scholar]
- Kosar B.; Albayrak C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim. Acta, Part A 2011, 78, 160–167. 10.1016/j.saa.2010.09.016. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Szentpaly Lv.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Parr R. G.; Donnelly R. A.; Levy M.; Palke W. E. Electronegativity: the density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. 10.1063/1.436185. [DOI] [Google Scholar]
- Chattaraj P. K.; Sarkar U.; Roy D. R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. 10.1021/cr040109f. [DOI] [PubMed] [Google Scholar]
- Lesar A.; Milošev I. Density functional study of the corrosion inhibition properties of 1, 2, 4-triazole and its amino derivatives. Chem. Phys. Lett. 2009, 483, 198–203. 10.1016/j.cplett.2009.10.082. [DOI] [Google Scholar]
- Khan M. U.; Khalid M.; Ibrahim M.; Braga A. A. C.; Safdar M.; Al-Saadi A. A.; Janjua M. R. S. A. First Theoretical Framework of Triphenylamine–Dicyanovinylene-Based Nonlinear Optical Dyes: Structural Modification of π-Linkers. J. Phys. Chem. C 2018, 122, 4009–4018. 10.1021/acs.jpcc.7b12293. [DOI] [Google Scholar]
- Janjua M. R. S. A.; Khan M. U.; Bashir B.; Iqbal M. A.; Song Y.; Naqvi S. A. R.; Khan Z. A. Effect of π-conjugation spacer (C C) on the first hyperpolarizabilities of polymeric chain containing polyoxometalate cluster as a side-chain pendant: A DFT study. Comput. Theor. Chem. 2012, 994, 34–40. 10.1016/j.comptc.2012.06.011. [DOI] [Google Scholar]
- Janjua M. R. S. A.; Amin M.; Ali M.; Bashir B.; Khan M. U.; Iqbal M. A.; Guan W.; Yan L.; Su Z. M. A DFT Study on The Two-Dimensional Second-Order Nonlinear Optical (NLO) Response of Terpyridine-Substituted Hexamolybdates: Physical Insight on 2D Inorganic–Organic Hybrid Functional Materials. Eur. J. Inorg. Chem. 2012, 2012, 705–711. 10.1002/ejic.201101092. [DOI] [Google Scholar]
- Khan M. U.; Ibrahim M.; Khalid M.; Qureshi M. S.; Gulzar T.; Zia K. M.; Al-Saadi A. A.; Janjua M. R. S. A. First theoretical probe for efficient enhancement of nonlinear optical properties of quinacridone based compounds through various modifications. Chem. Phys. Lett. 2019, 715, 222–230. 10.1016/j.cplett.2018.11.051. [DOI] [Google Scholar]
- Khan M. U.; Ibrahim M.; Khalid M.; Braga A. A. C.; Ahmed S.; Sultan A. , Prediction of Second-Order Nonlinear Optical Properties of D–p–A Compounds Containing Novel Fluorene Derivatives: A Promising Route to Giant Hyperpolarizabilities. J. Cluster Sci. 2019, 30, 415–430. 10.1007/s10876-018-01489-1. [DOI] [Google Scholar]
- Khan M. U.; Ibrahim M.; Khalid M.; Jamil S.; Al-Saadi A. A.; Janjua M. R. S. A. Quantum Chemical Designing of Indolo [3, 2, 1-jk] carbazole-based Dyes for Highly Efficient Nonlinear Optical Properties. Chem. Phys. Lett. 2019, 719, 59–66. 10.1016/j.cplett.2019.01.043. [DOI] [Google Scholar]
- Prasad P. N.; Williams D. J.. Introduction to Nonlinear Optical Effects in Molecules and Polymers; Wiley: New York, 1991; Vol. 1. [Google Scholar]
- Scrocco E.; Tomasi J. Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar]
- Luque F. J.; López J. M.; Orozco M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000, 343–345. [Google Scholar]
- Murray J. S.; Sen K.. Molecular Electrostatic Potentials: Concepts and Applications; Elsevier, 1996; Vol. 3. [Google Scholar]
- Mahalakshmi G.; Balachandran V. NBO, HOMO, LUMO analysis and vibrational spectra (FTIR and FT Raman) of 1-Amino 4-methylpiperazine using ab initio HF and DFT methods. Spectrochim. Acta, Part A 2015, 135, 321–334. 10.1016/j.saa.2014.06.157. [DOI] [PubMed] [Google Scholar]
- Okulik N.; Jubert A. H. Theoretical analysis of the reactive sites of non-steroidal anti-inflammatory drugs. Internet Electron. J. Mol. Des. 2005, 4, 17–30. [Google Scholar]
- Muthu S.; Prabhakaran A. Vibrational spectroscopic study and NBO analysis on tranexamic acid using DFT method. Spectrochim. Acta, Part A 2014, 129, 184–192. 10.1016/j.saa.2014.03.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.