

Abstract
MicroRNA 22 (miR-22) was found in diverse cardiovascular diseases to have a role in regulating multiple cellular processes. However, the regulatory role of miR-22 in aortic dissection (AD) was still unclear. The miR-22 expression in human aorta was explored. A series of mimic, inhibitor, or small interfering RNA (siRNA) plasmids were delivered into vascular smooth muscle cells (VSMCs) to explore the effects of miR-22 and p38 mitogen-activated protein kinase α (p38MAPKα) in controlling VSMC apoptosis in vitro. In addition, a mouse AD model was established, and histopathologic analyses were performed to evaluate the regulatory effects of miR-22. Reduced miR-22 and increased apoptosis of VSMCs was seen in human AD aorta. Downregulation of miR-22 increased the apoptosis of VSMCs in vitro. Bioinformatics analyses revealed that p38MAPKα was a target of miR-22. Inhibiting p38MAPKα expression could reverse the apoptosis of VSMCs induced by miR-22 downregulation. Knockdown of miR-22 in the AD mouse model significantly promoted the development of AD. Our data underscore the importance of vascular remodeling and VSMC function in AD. miR-22 may represent a new therapeutic approach for AD by regulating the apoptosis of VSMCs through the MAPK signaling pathway.
Keywords: aortic dissection, miR-22, microRNA 22, vascular remodeling, apoptosis, vascular smooth muscle cell
Graphical Abstract
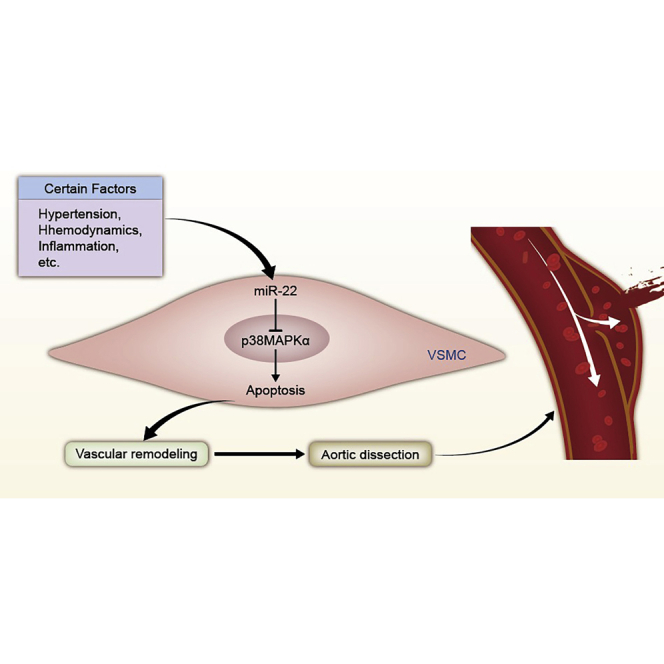
This study indicates miR-22 may represent a new therapeutic approach for aortic dissection by regulating the apoptosis of VSMCs through the MAPK signaling pathway. The study provided a novel insight into the mechanism of AD and the innovative therapeutic strategies in the form of new targets for small molecule therapies.
Introduction
Aortic dissection (AD) is one of the most complex cardiovascular diseases, with high morbidity and mortality.1 Although the prognosis of AD has been improved by the development of new instruments and techniques,2,3 the incidence of postoperative adverse events inducing reintervention needs to be solved by finding new intervention targets of its regulatory mechanisms. The basic pathological change of vascular remodeling in AD is medial degeneration, including cystic medial necrosis.4 The dysfunction of human aortic smooth muscle cells (HASMCs) is deemed to promote the medial degeneration of vascular remodeling.5 It is important to identify novel treatment targets of AD by deeply understanding the regulatory mechanism of vascular remodeling.
MicroRNAs (miRNAs), which are enriched in vascular tissues, participate in many cardiovascular pathologies, such as coronary artery disease (CAD) and heart failure (HF).6,7 It could modulate the expression of DOCK2, transforming growth factor-β genes related to cell proliferation, migration, and apoptosis.8, 9, 10 Although the expression of miRNAs in aortic tissue is known,11 the functional impact of acritical miRNA in regulating AD is generally obscured. Our previous study found that miR-22 was significantly downregulated in AD using miRNA microarrays.12 Whether miR-22 and its potential targets participating in vascular remodeling of AD remains unknown.
This study investigated the miR-22 expression and distribution pattern in AD tissue and explored its role in vascular remodeling by establishing a mouse model of AD in vivo. The regulatory role of miR-22 in the function of HASMCs was investigated in vitro. The molecular mechanisms and pathways regulated by miR-22 in HASMCs were also examined. The study might provide a new regulating strategy in recognizing the pathophysiology of AD and offer a potential therapeutic target for preventing AD.
Results
miR-22 in Human AD Samples
To detect the miR-22 level in AD tissue, expression of miRNAs was evaluated in 10 AD aortas (AD group) and 10 human normal aortas (NA group) by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). As shown in Figure 1A, the expression of miR-22 in the AD group was about 40% of that in the NA group (p < 0.05). Fluorescence in situ hybridization (FISH) was used to investigate the distributing of miR-22 in human aorta tissue, and the result showed that miR-22 was highly expressed in the HASMCs, which located in the media part of the human aorta. Significant downregulation of miR-22 was found in the AD group when compared with the NA group (p < 0.05; Figure 1B and C).
Figure 1.

miR-22 is Downregulated in the Media of Human Aorta Samples with Aortic Dissection
(A) miR-22 was significantly downregulated in AD samples (n = 10 per group, p < 0.05). (B) miR-22 probe (red signal), DAPI (blue signal), and SMA (green signal) staining on the same sections of human aorta in situ hybridization (n = 10 per group). (C) miR-22 was distributed in the media of aorta where VSMCs were located in situ hybridization experiment (p < 0.05). Data were represented as mean ± SD.
Downregulation of miR-22 Promoted the Apoptosis of HASMCs via p38MAPKα
By using adenovirus to mediate the expression of miR-22 in HASMCs, we found the interfering effect worked after 48 h of treatment (Figure 2A). Annexin-V antigen-presenting cell (APC) and 7-amino-actinomycin D (7-AAD) staining of HASMCs was used to evaluate the apoptosis and cell death following miR-22 interfering. Apoptosis of HASMCs was significantly induced in the miR-22 downregulated group when compared with negative control, while apoptosis was suppressed in the miR-22 upregulated group (Figure 2B). Mitogen-activated protein kinase (MAPK)-related factors, including p38, extracellular regulated protein kinase (ERK), and c-Jun N-terminal kinases (JNK), were detected using protein antibody array after miR-22 interfered in HASMCs. The results demonstrated that expression of p38 was obviously changed by miR-22 treatment; however, the other MAPK family members, such as ERK and JNK, were not significantly changed (Figure 2C).
Figure 2.

Inhibition of miR-22 Significantly Promoted the Apoptosis of HASMC In Vitro
(A) The significant interference of miR-22 with adenovirus (n = 5 per group). (B) Knockdown of miR-22 could significantly promote HASMC apoptosis, while overexpression inhibited the apoptosis (n = 5 per group). (C) p38MAPK was significantly increased by miR-22 upregulation; however, inhibition of miR-22 had the reverse effect. The expression of other MAPK family members was not affected by miR-22. Data were represented as mean ± SD. All the experiments were performed thrice. ∗p < 0.05.
To identify miR-22’s target gene in humans, the TargetScan algorithm (http://www.targetscan.org) was used to search the overlapping conserved gene set for putative targets with a potential role in MAPK signal pathway. We selected p38MAPKα for further study based on this search strategy. Of the protein obtained from the HASMCs treated by the miR-22 interfering adenovirus, western blotting results showed that p38MAPKα was significantly upregulated when miR-22 was downregulated, whereas miR-22 overexpression by transduction with Ad-miR-22 inhibits p38MAPKα protein expression (Figure 3A). What’s more, the phosphorylation level of p38MAPKα showed the same characteristic as the expression of p38MAPKα (Figure 3A). To confirm p38MAPKα as the direct target of miR-22, we generated a firefly luciferase reporter plasmid fused downstream to a segment of the p38MAPKα 3′ UTR containing either the wild-type putative miR-22 binding sequence or the mutation sequence (Figure 3B). Luciferase reporter assays revealed that the 3′ UTR of p38MAPKα was functional, as evidenced by suppression of the firefly luciferase signal by overexpression of miR-22 but not by overexpression of a control sequence (Figure 3C). In order to further explain the mechanism of miR-22’s regulatory role in HASMC apoptosis, we simultaneously interfered p38MAPKα and miR-22 to detect the apoptosis of HASMCs. The small interfering RNA (siRNA) of p38MAPKα can specifically inhibit its expression (Figure S1). The results indicated that p38MAPKα overexpression can obviously increase HASMC apoptosis. In addition, the promotive effect on HASMC apoptosis can be reversed by inhibiting p38MAPKα expression when miR-22 was downregulated (Figure 3D).
Figure 3.

miR-22 Inhibits p38MAPKα Expression In Vitro
(A) The significant inhibition effect of miR-22 on p38MAPKα expression and the phosphorylation of p38MAPKα (n = 5 per group). (B) Schematic representation of the binding between miR-22-3p and p38MAPKα, with mutated sites labeled with gray shading. (C) The effect of miR-22-3p on p38MAPKα luciferase activity by dual-luciferase assay. (D) The inhibition of p38MAPKα significantly reversed the HASMCs apoptosis, which was caused by miR-22 downregulation (n = 5 per group). Data were represented as mean ± SD. All the experiments were performed thrice. ∗p < 0.05.
p38MAPKα Expression Was Positively Correlated to the Apoptotic Rate of HASMCs in AD Specimens
To investigate the effect of p38MAPKα on apoptosis of HASMCs in AD, the relationship between p38MAPKα expression and the apoptotic rate of HASMCs in AD specimens was analyzed. Terminal deoxytransferase-mediated dUTP nick end labeling (TUNEL) (+) cells were numerous in the media of AD tissues, but not in the normal aortic tissues (p < 0.01, Figure 4A). The western blotting results on human samples showed that p38MAPKα was highly expressed in AD (Figure 4B). What’s more, the expression of p38MAPKα was positively correlated with the rate of TUNEL (+) cells in AD tissues with 0.98 correlation coefficient (r) (p < 0.01; Figure 4C).
Figure 4.

p38MAPKα Expression Was Positively Correlated to the Apoptotic Rate of Aortic VSMCs in Aortic Dissection Specimens
(A) Photographs and quantification of TUNEL in human aorta samples (TUNEL, green signal; DAPI, blue signal; SMA, red signal). Significant difference in the percentage of apoptotic cells was observed between NA and AD (n = 10 per group). (B) Western blotting showed significantly higher p38MAPKα levels (2.5-fold) in AD compared with NA (n = 10 per group). (C) Dot plots represent log10 percentage of apoptotic cells per mm2 against log10 p38MAPKα protein expression level. The lines represent approximated curves. The correlation coefficient (r = 0.98) and the p value (p < 0.05) indicated the statistical significance of the positive correlation between the x and y variables. Data were represented as mean ± SD. The TUNEL experiment was performed twice, and the western blotting experiment was performed thrice. ∗p < 0.05.
Inhibition of miR-22 Promoted the Apoptosis of VSMCs during AD In Vivo
To investigate whether miR-22 inhibited the development of AD in mice, we systemically administered agomir and antagomir to interfere miR-22 expression. Apparent AD was significantly increased in antagomiR-22-treated mice when compared with negative controls, with also a decreased trend in agomiR-22-treated mice (Figure 5A; Table 1). qRT-PCR analysis detected increased miR-22 levels upon AD when treated with agomiR-22 and sharply reduced miR-22 levels after antagomiR-22 treatment (Figure 5C). Histological analysis of the dissected aorta from antagomiR-22-treated mice demonstrated the massive elastin and collagen degradation in the media (Figures 5B and 5C).
Figure 5.

Inhibition of miR-22 In Vivo Strongly Aggravated Pathological Vascular Remodeling during Aortic Dissection
(A) Representative images showing macroscopic features of isolated mouse aorta after the establishment of aortic dissection model with antagomiR-22 or agomiR-22 treatment; rectangular frame indicates location of TAD. (B) Representative images of H&E, EVG, and Masson staining (n = 15 per group). (C–F) Quantification results of (C) miR-22 level, (D) media thickness, (E) media elastic fiber, and (F) media collagen. The interfering of miR-22 in AD model of mice was effective. Downregulation of miR-22 significantly promoted the degradation of elastic fiber (p < 0.05) and media collagen (p < 0.05) and reduced the media thickness (p < 0.05). Data were represented as mean ± SD. All the experiments were performed twice. ∗p < 0.05.
Table 1.
Group Composition, Age, Body Mass, and Occurrence of Aortic Dissection in FVB Mice
| n | Age (Weeks) | Body Mass (g) | Aortic Dissection |
||
|---|---|---|---|---|---|
| % Dissected | p Value | ||||
| Blank | 15 | 7.1 ± 0.0 | 18.8 ± 0.5 | 0 | – |
| Model | 15 | 7.1 ± 0.1 | 18.6 ± 0.5 | 66.7 | <0.01a |
| antagomiR NC | 15 | 7.1 ± 0.1 | 18.7 ± 0.7 | 60 | – |
| antagomiR-22 | 15 | 7.0 ± 0.4 | 18.3 ± 0.8 | 93.3 | 0.03b |
| agomiR NC | 15 | 7.1 ± 0.1 | 18.5 ± 0.5 | 73.3 | – |
| agomiR-22 | 15 | 7.1 ± 0.0 | 18.7 ± 0.6 | 20 | <0.01c |
The mice were untreated (Blank), treated with beta-aminopropionitrile and angiotensin II (Model), treated with beta-aminopropionitrile and angiotensin II, and additionally treated with miR-22 upregulation (agomiR-22), miR-22 downregulation (antagomir-22), and negative control of them. NC, normal control.
Model versus blank.
AntagomiR-22 versus antagomir NC.
AgomiR-22 versus agomiR NC.
To detect whether the VSMC apoptosis was promoted in mouse aorta by miR-22 downregulating, in situ TUNEL staining was analyzed (Figure 6A). There are significantly increased numbers of TUNEL (+) VSMCs in antagomiR-22-treated mice when compared with the negative control (p < 0.05). We also found that the TUNEL (+) VSMCs were significantly decreased in agomiR-22 treated mice (p < 0.05; Figure 6B). Based on the results of in vitro experiments, western blot was used to investigate the expression pattern of p38MAPKα and phosphorylation of p38MAPKα in mouse aorta. As shown in Figure S2, p38MAPKα and phosphorylation of p38MAPKα were obviously overexpressed in the aorta of antagomiR-22-treated mice (p < 0.05). Meanwhile, in agomiR-22 treated mice, p38MAPKα expression was significantly inhibited (p < 0.05). However, the phosphorylation of p38MAPKα was not significantly changed in the agomiR-22-treated group.
Figure 6.

miR-22 Inhibited the Apoptosis of VSMCs in Mouse Aorta
(A) Representative images of TUNEL staining of the mice with the treatment of antagomiR-22 or agomiR-22 (TUNEL, green signal; DAPI, blue signal; SMA, red signal). (B) Quantification results of TUNEL-positive cells showed that knockdown of miR-22 significantly promoted VSMC apoptosis while overexpression inhibited the apoptosis (n = 15 per group). Data were represented as mean ± SD. The TUNEL experiment was performed twice and the western blotting experiment was performed thrice. ∗p < 0.05.
Discussion
The regulatory mechanism of vascular remodeling has become a research focus in AD, which is an important cause of sudden death.2,13 Pathological vascular remodeling and cell or molecular dysfunction are involved in its development.14,15 To block these adverse pathological processes, it is essential to elucidate the underlying mechanisms and identify effective therapeutic targets. In this study, knockdown of miR-22 increased the apoptosis of VSMCs and triggered AD in a mouse model via p38MAPKα production. p38MAPKα was negatively regulated by miR-22 and acted as an activator of apoptosis in VSMCs. To our knowledge, this was the first study to use FISH to examine miR-22 in AD tissue, and it revealed a novel mechanism participating in vascular remodeling of AD.
Dysregulated miRNA expression was observed in many vascular diseases.16 miR-22 contributed to cardiac aging, mediated inflammation response, and was capable of regulating cell proliferation, apoptosis, motility, and invasion by attenuating the expression of SIRT1, caspase, etc.,17,18 suggesting that it played a role in vascular disease. Based on our previous results,12 the primary distribution of miR-22 in aorta media with VSMCs was validated by performing in situ hybridization. This study suggested that miR-22 inhibited the apoptosis of VSMCs, thus leading to media remodeling with weakened mechanical properties of the aorta.
Abnormal vascular remodeling was central to neointimal formation, media degradation, and cellular dysfunction, all of which contributed to vascular diseases such as atherosclerosis and dilatation.19,20 Our previous study demonstrated the regulatory role of FHL1 and SM22 in vascular remodeling of AD.21 Under physiological conditions, VSMCs were highly specialized cells to maintain vascular tone and ensure contractility of the vessel.22 VSMCs had key functions in vascular remodeling in response to injury.23 Vascular remodeling was often characterized by the imbalance between proliferation and apoptosis of VSMCs.24 Abnormal proliferation, apoptosis, and phenotype of VSMCs were involved in the development of AD.25,26 However, the status of VSMCs in AD remained controversial. Our study showed that the apoptosis of VSMCs was commonly found in human aorta with AD as well as in mice aorta with knockdown of miR-22, suggesting that increased VSMC apoptosis was involved in the pathogenesis of vascular remodeling of AD and might be regulated by miR-22. Moreover, in vivo findings indicated that downregulation of miR-22 could significantly promote the degradation of elastin and collagen in mouse aorta. Some proteins, such as matrix metalloproteinases, played a key role in this process. As the main source of extracellular matrix (ECM) proteins in the aorta, the relationship of VSMCs and ECM proteins was critical for the structural and functional integrity of the aortic wall. Furthermore, downregulation of miR-22 significantly promoted apoptosis of VSMCs in vitro. Therefore, protection against pathological vascular remodeling via inhibition of apoptosis of VSMCs by miR-22 could be a potential therapeutic target for the treatment of AD. Whether miR-22 inhibited the degradation of elastin and collagen by regulating the biological functions of VSMCs needed further study.
Numerous mechanisms were proposed to explain the development of AD, including apoptosis of VSMCs, oxidative stress, and inflammation.27,28 Our previous study indicated that oxidative stress was closely related to AD’s pathological process, especially via the MAPK signal pathway.29,30 The p38MAPKα signal pathway was proved to impact the HASMCs apoptosis.31 For each of these pathogenic mechanisms, the MAPK signaling pathway was identified as the downstream target. Extracellular signal-regulated kinase 1/2, c-Jun N-terminal protein kinase, and p38MAPKα were involved in the MAPK pathway, which regulated various physiological and pathological processes.32 The regulatory effect of p38MAPK on the apoptosis occurred through upregulation of signal transducer and activator of CEBP homologous protein, focal adhesion kinase, transcription 1, cytochrome c, nuclear factor-κB and p53 pathways.33 Apoptosis was caused by oxidative stress and nitric oxide through activation of the p38MAPK pathway.31 Moreover, inflammation was also involved in the apoptosis mediated by p38MAPK.34 p38MAPKα was also predicted to be a target gene of miR-22. In our study, p38MAPKα expression was obviously induced by knockdown of miR-22 both in vitro and in vivo. Inhibition of p38MAPKα dampened the pro-apoptosis response induced by downregulation of miR-22 in VSMCs. Taken together, these findings revealed the pro-apoptotic and vascular remodeling modulatory properties of miR-22.
Given the advancements in understanding the genetics of AD, gene-tailored management became a research hotspot. Gene therapy for human disease was feasible and considered as a viable alternative treatment for some cardiovascular diseases.35,36 This study revealed the therapeutic potential of miRNAs in early intervention and AD mouse model and the possible application in AD patients.
This study had some limitations. First, other potential targets of miR-22 were not identified in VSMCs. Moreover, the upstream and downstream signaling pathways of p38MAPKα that may contribute to miR-22-mediated cellular effects were not identified. Second, the action mediated by p38MAPKα, which could further illustrate the mechanism of apoptosis of VSMCs regulated by miR-22, was not explored. Third, the mechanisms underlying the role of miR-22 in matrix degradation by regulating the biological functions of VSMCs was not examined. Future studies should address these limitations.
Conclusions
Vascular remodeling and apoptosis of VSMCs induced by miR-22 knockdown played a key role in AD. MAPK signaling pathway might participate in this process. This study provided a novel insight into the mechanism of AD and the innovative therapeutic strategies in the form of new targets for small molecule therapies.
Materials and Methods
Ethics Statement
This study was performed following the principles outlined in the Declaration of Helsinki and approved by the Changhai Hospital Medical Ethics Committee. Written informed consent was obtained from all patients and direct relatives of organ donors. The animal experiments strictly adhered to the principle of minimizing the pain, suffering, and discomfort of experimental animals.
Patient Recruitment and Tissue Collection
The AD tissues were acquired from 10 thoracic aortic dissection (TAD) patients who underwent the open surgery of prosthetic aorta replacement at Changhai Hospital during October 2012 and January 2016. Patients with smoking history, Ehlers-Danlos syndrome, Marfan syndrome, aortic aneurysm and other connective tissue disorders were excluded according to the inclusion and exclusion criteria. Controlling thoracic aortic tissues were obtained from 10 organ donors who died from nonvascular diseases. All the specimens were collected within 30 min after aorta excision. All the samples were rinsed many times with precooled saline solution to remove the blood and mural thrombus attaching to the vascular wall. All the tissue samples were cut into three parts. One part was diced into approximately 2 mm3 pieces, which were then placed into sterile tubes. These samples were frozen in liquid nitrogen, and then the frozen specimens were transported to the laboratory and stored at −80°C until RNA extraction. The second part was fixed in 4% paraformaldehyde and embedded in paraffin. The third part was frozen embedded for in situ hybridization. The demographic and clinical characteristic of the included patients and controls were presented in Table 2.
Table 2.
The Demographic and Clinical Characteristics of the Included Patients and Controls
| Patients (n = 10) | Controls (n = 10) | |
|---|---|---|
| Age, years | 47.30 ± 6.91 | 46.90 ± 8.16 |
| Sex, male:female | 10:0 | 10:0 |
| Hypertension, n (%) | 7 (70.00) | 2 (20.00) |
| Hyperlipidemia, n (%) | 1 (10.00) | 0 (0.00) |
| Diabetes mellitus, n (%) | 0 (0.00) | 0 (0.00) |
| Smoking history, n (%) | 0 (0.00) | 0 (0.00) |
| Stanford Classification, n (%) | ||
| Type A | 10 (100.00) | – |
| Type B | 0 (0.00) | – |
RNA Isolation and qRT-PCR
Total RNA was extracted from the frozen samples or cultured cells in accordance with the manufacturer’s instruction for the miRNease Mini Kit (QIAGEN, Hilden, Germany) and checker for a RIN number to inspect RNA integration by an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Complementary DNA was synthesized with 500 ng of total RNA using the PrimeScript RT reagent Kit (TaKaRa, Tokyo, Japan). The primers used are listed in Table 3. qRT-PCR analyses were performed using LightCycler 480 SYBR Green I Master (Roche, Welwyn Garden, Swiss). Each SYBR Green reaction (total volume, 20 μL) contained 2 μL of cDNA as template and each primer at 0.25 μL. Controls without template DNA were negative. The reactions were incubated at 95°C for 30 s followed by 40 cycles at 95°C for 5 s, at 55°C for 10 s, and at 72°C for 15 s. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the loading control. To verify that only one qRT-PCR product was detected by the SYBR Green dye, the samples were subjected to the heat dissociation protocol after the final cycle of qRT-PCR to examine the presence of only peak. Moreover, mature miRNAs were quantified with specific primers using miScript II RT Kit (QIAGEN) for reversing transcription and miScript SYBR Green PCR Kit (QIAGEN) for the sake of the subsequent qRT-PCR, as instructed by the manufacturer. U6b small nuclear RNA was used as internal reference gene for those experiments.
Table 3.
The Sequence of Primers, RNA Oligo, AgomiRs, and AntagomiRs in This Study
| Sequence (5′ to 3′) | |
|---|---|
| Primers | |
| hsa-miR-22-3p | forward: CACCGACAGTTCTTCAACTGGCAGCTT |
| reverse: AAAAAAGCTGCCAGTTGAAGAACTGTC | |
| U6b | forward: CTCGCTTCGGCAGCACA |
| reverse: AACGCTTCACGAAYYYGCGT | |
| MAPK14 | forward: GGGACCTCCTTATAGATGATGAGTGG |
| reverse: GGACTCCATCTCTTCTTGGTCA | |
| GAPDH | forward: CAGGCATAATGGTTAAAGTTGGTA |
| reverse: CATGTAATCAAGGTCAATGAATGG | |
| RNA Oligo | |
| miR-22 mimics | AAGCUGCCAGUUGAAGAACUGU |
| ACAGUUCUUCAACUGGCAGCUU | |
| miR-22 mimics NC | GCAACCUGUUAGACUAUGGAGA |
| UCUCCAUAGUCUAACAGGUUGC | |
| MAPK14 | GUCCAUCAUUCAUGCGAAATT |
| UUUCGCAUGAAUGAUGGACTT | |
| Negative control | UUCUCCGAACGUGUCACGUTT |
| ACGUGACACGUUCGGAGAATT | |
| GAPDH positive control | UGACCUCAACUACAUGGUUTT |
| AACCAUGUAGUUGAGGUCATT | |
| AntagomiRs and AgomiRs | |
| mmu-miR-22-3p agomiR | AAGCUGCCAGUUGAAGAA CUGUAGUUCUUCAACUGG CAGCUUUU |
| agomiR NC | UUCUCCGAACGUGUCAC GUTTACGUGACACGUUC GGAGAATT |
| mmu-miR-22-3p antagomiR | ACAGUUCUUCAACUGGCAGCUU |
| antagomiR NC | CAGUACUUUUGUGUAGUACAA |
FISH
LNA-modified probes for miR-22 (5′- and 3′-DIG-labeled), miRNA in situ hybridization (ISH) buffer, and Proteinase K were purchased from Exiqon (Vedbaek, Denmark). This experiment was performed on frozen sections following the manufacturer’s protocol (Exiqon). Briefly, tissue slides were warmed first and then washed with diethyl pyrocarbonate (DEPC)-treated 1× PBS 3 times for 5 min each, followed by acetylating in 100 mM triethanolamine buffer plus 0.25% acetic anhydride for 10 min, then permeabilized in PBST (1× PBS plus 0.1% Triton X-100 in DEPC-treated water) for 30 min, and finally washed 3 times for 5 min each at RT in 1× PBS. Hybridization was conducted at 56°C overnight in the same hybridization buffer containing 100 nM of miR-22 (5′-ACAGTTCTTCAACTGGCAGCTT-3′) DIG-labeled LNA probes, after 1.5 h prehybridization was finished (10 mM Tris-HCl [pH 8.0], 600 mM NaCl, 50% formamide, 200 μg/mL tRNA, 1× Denhardt’s solution, 10% dextran sulfate, 1 mM EDTA, and 0.25% SDS) at room temperature. The tissue slides were sequentially washed by hybridization buffer at 56°C for 15 min. Next, hybridization buffer and 2× SSC at the ratio 1:1 were used to rinse the tissue slides in sequence at 56°C 3 times for 5 min each time. Then, MABT buffer (100 mM maleic acid, 150 mM NaCl, 0.1% Tween 20 [pH 7.5]) was washed at room temperature twice for 10 min each time. In the end, the slides were then incubated in blocking solution (MABT plus 10% horse serum) for 2 h at room temperature and then were incubated with Anti-Digoxigenin-POD (1:200, Roche) overnight at 4°C. After the slides were washed in MABT 7 times for 20 min each time and in 1× PBS twice for 10 min each time at indoor temperature, signals were then developed using Alexa Fluor 633 Dye-conjugated secondary antibody (Roche).
TUNEL Assay
To detect the apoptosis, the TUNEL technique was employed to stain the paraffin-embedded sections by using an in situ apoptosis detection kit (Roche, Mannheim, Germany) as instructed by the manufacturer. The localized red fluorescence of the apoptotic cells was tested by a Pannoramic Confocal Scanner (3DHISTECH, Budapest, Hungary) in scanner software 1.20 Sp1 and Pannoramic Viewer 1.15.4, which were developed for Windows system. In addition, the media area in human aorta sections and the numbers of TUNEL-positive cells were also measured in 5 random microscopic fields.
Immunofluorescence (IF)
To conduct double-staining immunofluorescence, paraffin sections (5 μm) were washed in PBS and then blocked by nonimmune serum for 1 h at room temperature, followed by incubation with primary antibodies (α-smooth muscle actin [α-SMA], 1:1,000, Abcam) overnight at 4°C. Alexa Fluor 488, 568, and 647 conjugated secondary antibodies (Signalway Antibody, MD, USA, 1:200 dilution) were used to visualize signals. Semiquantitative analyses were conducted on positive signals in samples by using Image-ProPlus software version 6.0 (Media Cybernetics).
Cell Culture
Prime HASMCs were purchased from Sciencell and cultured in smooth muscle cell media (Sciencell, Carlsbad, CA, USA) at 37°C in a humidified atmosphere with 5% CO2. HEK293 T cells were cultured in DMEM (GIBCO, New York, NY, USA) supplemented with fetal bovine serum (FBS) (10%). Cells used in the experiments were 3–8 passages.
Adenovirus-Mediated Expression of miR-22
To construct miR-22 expression vector, the human miR-22 gene was synthesized and oligonucleotide sequences were annealed and cloned into the pAdeno-U6-Mir30-CMV-EGFP vector (Shanghai OBiO Biotech). DNA sequencing and named pAdeno-U6-miR-22-CMV-EGFP were employed to confirm the adenovirus-based miR-22-expressing vectors. Tough-decoy (TuD) RNA was a new strategy that can efficiently suppress a specific endogenous miR-22. The miR-22 TuD cassette was designed as suggested and synthesized according to the GenScript cassette protocol (Piscataway, NJ, USA). The sense sequence of hsa-miR-22 control small hairpin RNA (shRNA) was 5′-GGCCTAAGGTTAAGTCGC-3′. The synthesized sequence was inserted into the pDKD-CMV-eGFP-U6-shRNA vector (Shanghai OBiO Biotech), namely pDKD-CMV-eGFP-U6-shRNA (miR-22). Adenoviruses were generated by transfecting plasmids and packaging vector carriers Pshuttle-CMV with Lipo 2000 into 293 cells at 80% confluence. The virus-containing medium was gathered 7–15 days after transfection. For adenovirus infections, target cells were cultured into 6-well plates for infection by moderate virus-contained DMEM medium with polybrene (Sigma-Aldrich, 4 μg/mL), incubated at 37°C for 2 h, and replaced by DMEM medium with FBS. After incubation for 36–48 h, expression of EGFP above 90% was considered successful, and extraction of RNA and protein was performed.
Apoptosis Assay by Flow Cytometer
The cells were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and resuspended in 1× binding buffer (KeyGEN BioTECH, Nanjing, China) at a concentration of 1 × 106 cells/mL. The Annexin V apoptosis detection kit (KeyGen Biotech) was used to stain the cells with Annexin V-APC and 7-AAD, which was then detected by flow cytometry. HASMCs without any treatment were used as control. The experiments were repeated at least 3 times.
Transfection for p38MAPKα Silencing
A siRNA specific for p38MAPKα and control non-silencing siRNAs were bought from GenePharma (Shanghai, China) (sense: 5′-GGUCUCUGGAGGAAUUCAATTUUGAAUUCCUCCAGAGACCTTsiRNAs were transfected into HASMC cells at 40 μm using Lipofectamine 2000. After 48 h, the siRNA of p38MAPKα suppressed the expression of p38MAPKα mRNA by 74% in comparison to the control siRNA, according to qRT-PCR analysis.
Luciferase Reporter Assay
The miRNA mimics were synthesized from GenePharma. The sequences of miR-22 mimics were listed in Table 2. HEK293T cells were cultured in 24-well plates and co-transfected with 200 ng psiCHECK-2 vector containing 3′ UTR of Bcl-2 or 3′ UTR of Bid and 40 nM miRNA mimics per well. Transfections were performed using Lipofectamine 2000. The luciferase analysis was performed 24 h later using Dual-Luciferase Reporter Assay (Promega, Manheim, Germany) according to the manufacturer’s protocol. The relative firefly luciferase activity was obtained after normalizing to renilla luciferase activity.
Western Blotting
HASMCs were washed 3 times in PBS and 100 μL RIPA buffer (Cell Signaling Technology, MA, USA) per well (6-well plate) applied to the cells. Then, a cell lifter (Corning Costar) was used to mix and lyse the cells, which were transferred to tubes (Eppendorf) in sequence. For tissues, 100 mg of tissue was homogenized in 1 mL RIPA buffer. The bicinchoninic acid (BCA) method was applied to determine protein concentration. All the proteins were standardized to 1.0 mg/mL and then 1× SDS sample buffer was added to the tubes at the 25% concentration. The cells were sonicated for 10–15 s and then heated for 5 min at 95°C, after which the cells were cooled on ice for 2 min and finally centrifuged at 3,000 × g for 1 min. A total of 20 mL was loaded on a 6%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis plate and then was transferred onto a polyvinylidene difluoride membrane, as a result of which the amount of each specimen for one western blot gel totaled 20 μg. In the following step, protein concentration was blocked by using 5% bovine serum albumin (BSA) in Tris-buffered saline (100 mM NaCl, 10 mM Tris, pH 7.6) with 0.1% Tween 20 (TBST). The primary antibodies were diluted in 5% BSA, and one antibody was used to incubate membranes overnight at 4°C.
After being washed 3 times in TBST, membranes were incubated with HRP-linked secondary antibodies (Signalway Antibody) for 2 h at indoor temperature. Relative band intensities were assessed in Image-ProPlus software version 6.0. The primary antibodies were p38MAPKα (1:1,000; Abcam, Cambridge, MA, USA) and β-Tubulin (1:500; Abcam, Cambridge, MA, USA).
Animals
All animal procedures were approved by and performed in line with the Experimental Animal Ethics Committee of Navy Medical University and the Institute of Laboratory Animal Science of China (A5655-01). All procedures were conducted conforming to the Directive 2010/63/EU of the European Parliament. 3-week-old male mice on a FVB background were bought from the Animal Centre of the Navy Medical University (Shanghai, China).
AD Murine Model
The mice were housed in a room with temperature controlled at 23°C ± 2°C and with 50% ± 5% relative humidity in a 12-h light-dark cycle (lights were turned on at 7:00 a.m.), and were allowed free access to food and water. All the animals were fed with water containing beta-aminopropionitrile (1 g/kg/day) and normal food to 7 weeks old. At 6 weeks old, the male FVB mice were divided into blank group (n = 15), model group (n = 15), agomiR-22 group (n = 15), agomiR-NC group (n = 15), antagomiR-22 group (n = 15), and antagomiR-NC group (n = 15). A 200 μL solution of agomiR-22 (15 optical density [OD]), agomiR-NC (15 OD), antagomiR-22 (15 OD), and antagomiR-NC (15 OD) was injected, respectively, through tail vein in 4 groups (except the model group) via a 30-gauge needle. The sequences of the antagomiRs and agomiRs are shown in Table 3. At 7 weeks old, all manipulated mice (n = 100) were implanted with a 100 μL osmotic pump (model Alzet 1003D; Durect, Cupertino, CA, USA) filled with a solution of angiotensin II (1 μg/kg/min) (Sigma Aldrich, St. Louis, MO, USA) for 24 h in the back subcutaneously by local anesthesia. At the endpoint of the experiments, the mice were anaesthetized by phenobarbital (40 mg/kg, respectively, intraperitoneal) in sequence, and the sacrifice was resolved after tissue collection.
Histopathological Staining and Analysis
After euthanasia was done, the aortas were obtained from the ascending aorta to the iliac artery and then fixed in 10% buffered formalin. Next, the fixed and paraffin-embedded tissues were cut into 5 μm thickness, stained with hematoxylin and eosin (H&E), Elastica van Gieson (EVG), and Masson following standard procedures.
Statistical Analysis
Average data were presented as mean and standard deviation (SD) unless otherwise stated. Two groups in statistical comparison were subject to 2-tailed, unpaired Student’s t test. For the comparison of three or more groups, ANOVA was performed, which was followed by Tukey post hoc tests. All the results were obtained by doing at least three independent experiments, and all statistical analyses were carried out by using Empower (R) (http://www.empowerstats.com, X&Y Solutions, Boston, MA, USA) and R (http://www.R-project.org). In addition, p <0.05 was considered significant.
Author Contributions
Z.J., J.Z., Y.X., and Q.J. designed the study. Y.S., L.Z., G.W., C.W., Z.L., and X.M. collated the data, carried out data analyses, and produced the initial draft of the manuscript. Y.S., J.W., and Z.Z. contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors thank Shanghai Institutes for Biological Sciences (SIBS), and Chinese Academy of Sciences (CAS) laboratory researchers Han Xiaoshuai, Chen Jian, Li Fangfang, for their great help in molecular cell experiments. This study was financed by the National Natural Science Foundation of China (NSFC) (grant nos. 81570425, 81770482, 91739110, 81870366), the Young Yangtze Scholar Program project (grant no. Q2017219), and the Foundation of Shanghai Science and Technology Commission (grant nos. 17441901900 and 17411954100).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.08.018.
Contributor Information
Qing Jing, Email: qjing@sibs.ac.cn.
Jian Zhou, Email: zhoujian1-2@163.com.
Zaiping Jing, Email: jingzaiping_endo@126.com.
Supplemental Information
References
- 1.Erbel R., Alfonso F., Boileau C., Dirsch O., Eber B., Haverich A., Rakowski H., Struyven J., Radegran K., Sechtem U., Task Force on Aortic Dissection, European Society of Cardiology Diagnosis and management of aortic dissection. Eur. Heart J. 2001;22:1642–1681. doi: 10.1053/euhj.2001.2782. [DOI] [PubMed] [Google Scholar]
- 2.Lu Q., Feng J., Zhou J., Zhao Z., Bao J., Feng R., Yuan L., Feng X., Qu L., Pei Y. Endovascular repair of ascending aortic dissection: a novel treatment option for patients judged unfit for direct surgical repair. J. Am. Coll. Cardiol. 2013;61:1917–1924. doi: 10.1016/j.jacc.2012.08.994. [DOI] [PubMed] [Google Scholar]
- 3.Li Z., Lu Q., Feng R., Zhou J., Zhao Z., Bao J., Feng X., Feng J., Pei Y., Song C., Jing Z. Outcomes of Endovascular Repair of Ascending Aortic Dissection in Patients Unsuitable for Direct Surgical Repair. J. Am. Coll. Cardiol. 2016;68:1944–1954. doi: 10.1016/j.jacc.2016.08.031. [DOI] [PubMed] [Google Scholar]
- 4.Isselbacher E.M., Lino Cardenas C.L., Lindsay M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation. 2016;133:2516–2528. doi: 10.1161/CIRCULATIONAHA.116.009762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu D., Shen Y.H., Russell L., Coselli J.S., LeMaire S.A. Molecular mechanisms of thoracic aortic dissection. J. Surg. Res. 2013;184:907–924. doi: 10.1016/j.jss.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R., Ding X., Zhou S., Li M., Sun L., Xu X., Fei G. Microrna-26b attenuates monocrotaline-induced pulmonary vascular remodeling via targeting connective tissue growth factor (CTGF) and cyclin D1 (CCND1) Oncotarget. 2016;7:72746–72757. doi: 10.18632/oncotarget.10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi X.-Y., Huang H., Ordog B., Luo X., Naud P., Sun Y., Wu C.-T., Dawson K., Tadevosyan A., Chen Y. Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial remodeling. Circ. Res. 2015;116:836–845. doi: 10.1161/CIRCRESAHA.116.305326. [DOI] [PubMed] [Google Scholar]
- 8.Guo X., Shi N., Cui X.B., Wang J.N., Fukui Y., Chen S.Y. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ. Res. 2015;116:e71–e80. doi: 10.1161/CIRCRESAHA.116.305863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iaconetti C., Gareri C., Polimeni A., Indolfi C. Non-coding RNAs: the “dark matter” of cardiovascular pathophysiology. Int. J. Mol. Sci. 2013;14:19987–20018. doi: 10.3390/ijms141019987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Rooij E. The art of microRNA research. Circ. Res. 2011;108:219–234. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 11.Liao M., Zou S., Weng J., Hou L., Yang L., Zhao Z., Bao J., Jing Z. A microRNA profile comparison between thoracic aortic dissection and normal thoracic aorta indicates the potential role of microRNAs in contributing to thoracic aortic dissection pathogenesis. J. Vasc. Surg. 2011;53:1341–1349.e3. doi: 10.1016/j.jvs.2010.11.113. [DOI] [PubMed] [Google Scholar]
- 12.Nienaber C.A., Clough R.E. Management of acute aortic dissection. Lancet. 2015;385:800–811. doi: 10.1016/S0140-6736(14)61005-9. [DOI] [PubMed] [Google Scholar]
- 13.Tieu B.C., Lee C., Sun H., Lejeune W., Recinos A., 3rd, Ju X., Spratt H., Guo D.-C., Milewicz D., Tilton R.G. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 2009;119:3637–3651. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuler G., Adams V., Goto Y. Role of exercise in the prevention of cardiovascular disease: results, mechanisms, and new perspectives. Eur. Heart J. 2013;34:1790–1799. doi: 10.1093/eurheartj/eht111. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y., Dong C.-Q., Peng G.-Y., Huang H.Y., Yu Y.S., Ji Z.-C., Shen Z.-Y. MicroRNA-134-5p Regulates Media Degeneration through Inhibiting VSMC Phenotypic Switch and Migration in Thoracic Aortic Dissection. Mol. Ther. Nucleic Acids. 2019;16:284–294. doi: 10.1016/j.omtn.2019.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Small E.M., Olson E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W., You R., Yuan X., Yang T., Samuel E.L., Marcano D.C., Sikkema W.K., Tour J.M., Rodriguez A., Kheradmand F., Corry D.B. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat. Immunol. 2015;16:1185–1194. doi: 10.1038/ni.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S., Zhang D., Yi C., Wang Y., Wang H., Wang J. MicroRNA-22 functions as a tumor suppressor by targeting SIRT1 in renal cell carcinoma. Oncol. Rep. 2016;35:559–567. doi: 10.3892/or.2015.4333. [DOI] [PubMed] [Google Scholar]
- 19.Li J., Zhang Y., Zhao J., Kong F., Chen Y. Overexpression of miR-22 reverses paclitaxel-induced chemoresistance through activation of PTEN signaling in p53-mutated colon cancer cells. Mol. Cell. Biochem. 2011;357:31–38. doi: 10.1007/s11010-011-0872-8. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y., Zhao Z., Hou L., Xiao Y., Qin F., Yan J., Zhou J., Jing Z. The regulatory role of smooth muscle 22 on the proliferation of aortic smooth muscle cells participates in the development of aortic dissection. J. Vasc. Surg. 2017;66:875–882. doi: 10.1016/j.jvs.2016.02.063. [DOI] [PubMed] [Google Scholar]
- 21.Gareri C., De Rosa S., Indolfi C. MicroRNAs for Restenosis and Thrombosis After Vascular Injury. Circ. Res. 2016;118:1170–1184. doi: 10.1161/CIRCRESAHA.115.308237. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz S.M. Smooth muscle migration in atherosclerosis and restenosis. J. Clin. Invest. 1997;100(11, Suppl):S87–S89. [PubMed] [Google Scholar]
- 23.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 24.McMurtry M.S., Bonnet S., Wu X., Dyck J.R.B., Haromy A., Hashimoto K., Michelakis E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 25.Liao W.-L., Tan M.-W., Yuan Y., Wang G.-K., Wang C., Tang H., Xu Z.-Y. Brahma-related gene 1 inhibits proliferation and migration of human aortic smooth muscle cells by directly up-regulating Ras-related associated with diabetes in the pathophysiologic processes of aortic dissection. J. Thorac. Cardiovasc. Surg. 2015;150:1292. doi: 10.1016/j.jtcvs.2015.08.010. 301.e2. [DOI] [PubMed] [Google Scholar]
- 26.Wang L., Zhang J., Fu W., Guo D., Jiang J., Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J. Vasc. Surg. 2012;56:1698–1709.e1. doi: 10.1016/j.jvs.2012.05.084. [DOI] [PubMed] [Google Scholar]
- 27.Liao M., Liu Z., Bao J., Zhao Z., Hu J., Feng X., Feng R., Lu Q., Mei Z., Liu Y. A proteomic study of the aortic media in human thoracic aortic dissection: implication for oxidative stress. J. Thorac. Cardiovasc. Surg. 2008;136:65–72. doi: 10.1016/j.jtcvs.2007.11.017. 72.e1–3. [DOI] [PubMed] [Google Scholar]
- 28.Anzai A., Shimoda M., Endo J., Kohno T., Katsumata Y., Matsuhashi T., Yamamoto T., Ito K., Yan X., Shirakawa K. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ. Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L., Liao M.F., Tian L., Zou S.L., Lu Q.S., Bao J.M., Pei Y.F., Jing Z.P. Overexpression of interleukin-1β and interferon-γ in type I thoracic aortic dissections and ascending thoracic aortic aneurysms: possible correlation with matrix metalloproteinase-9 expression and apoptosis of aortic media cells. Eur. J. Cardiothorac. Surg. 2011;40:17–22. doi: 10.1016/j.ejcts.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L., Zhou J., Jing Z., Xiao Y., Sun Y., Wu Y., Sun H. Glucocorticoids Regulate the Vascular Remodeling of Aortic Dissection Via the p38 MAPK-HSP27 Pathway Mediated by Soluble TNF-RII. EBioMedicine. 2018;27:247–257. doi: 10.1016/j.ebiom.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato K., Hamanoue M., Takamatsu K. Inhibitors of p38 mitogen-activated protein kinase enhance proliferation of mouse neural stem cells. J. Neurosci. Res. 2008;86:2179–2189. doi: 10.1002/jnr.21668. [DOI] [PubMed] [Google Scholar]
- 32.Kyriakis J.M., Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 33.Yokota T., Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–376. doi: 10.1016/j.gene.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J., Chen J., Wang X., Wang C., Cao W., Zhao Y., Zhang B., Cui M., Shi Q., Zhang G. Ligustrazine alleviates acute pancreatitis by accelerating acinar cell apoptosis at early phase via the suppression of p38 and Erk MAPK pathways. Biomed. Pharmacother. 2016;82:1–7. doi: 10.1016/j.biopha.2016.04.048. [DOI] [PubMed] [Google Scholar]
- 35.Salmon F., Grosios K., Petry H. Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera®) Expert Rev. Clin. Pharmacol. 2014;7:53–65. doi: 10.1586/17512433.2014.852065. [DOI] [PubMed] [Google Scholar]
- 36.Zsebo K., Yaroshinsky A., Rudy J.J., Wagner K., Greenberg B., Jessup M., Hajjar R.J. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014;114:101–108. doi: 10.1161/CIRCRESAHA.113.302421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.