

Summary
Mitochondrial respiration (oxidative phosphorylation, OXPHOS) is an emerging target in currently refractory cancers such as pancreatic ductal adenocarcinoma (PDAC). However, the variability of energetic metabolic adaptations between PDAC patients has not been assessed in functional investigations. In this work, we demonstrate that OXPHOS rates are highly heterogeneous between patient tumors, and that high OXPHOS tumors are enriched in mitochondrial respiratory complex I at protein and mRNA levels. Therefore, we treated PDAC cells with phenformin (complex I inhibitor) in combination with standard chemotherapy (gemcitabine), showing that this treatment is synergistic specifically in high OXPHOS cells. Furthermore, phenformin cooperates with gemcitabine in high OXPHOS tumors in two orthotopic mouse models (xenografts and syngeneic allografts). In conclusion, this work proposes a strategy to identify PDAC patients likely to respond to the targeting of mitochondrial energetic metabolism in combination with chemotherapy, and that phenformin should be clinically tested in appropriate PDAC patient subpopulations.
Keywords: pancreatic cancer, cancer metabolism, metabolic heterogeneity, energetic metabolism, mitochondria, OXPHOS, mitochondrial Complex I, phenformin, therapeutic strategy, personalized medicine
Graphical Abstract
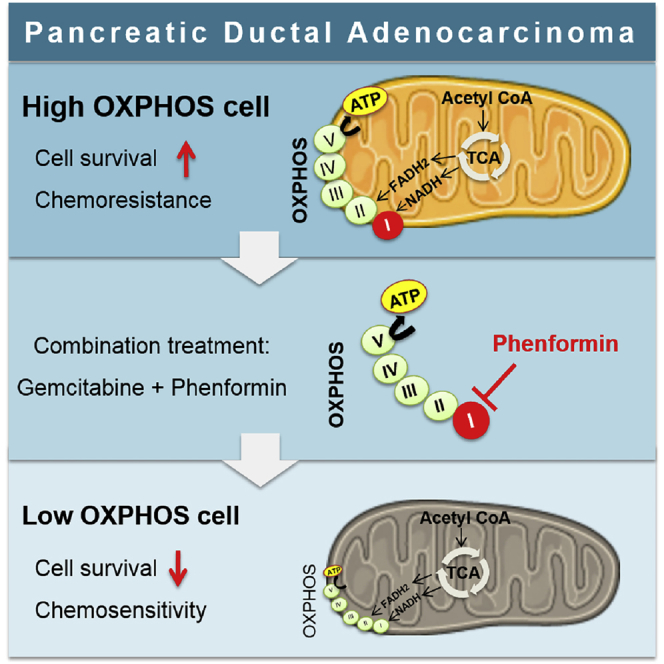
Highlights
Pancreatic ductal adenocarcinoma (PDAC) displays OXPHOS heterogeneity
High OXPHOS PDAC tumors are enriched in mitochondrial respiratory complex I
Complex I inhibitor phenformin synergizes with chemotherapy in high OXPHOS cells
Phenformin cooperates with gemcitabine antitumoral activity in high OXPHOS tumors
Masoud et al. reveal that pancreatic cancer patients can be stratified according to their mitochondrial oxidative phosphorylation (OXPHOS) activity and to the expression of mitochondrial respiratory complex I. Targeting mitochondrial respiration with the complex I inhibitor phenformin cooperates with gemcitabine to eradicate high OXPHOS pancreatic cancer cells.
Introduction
Pancreatic cancer is one of the deadliest human cancers, with a 5-year relative survival rate of 8%.1 Pancreatic cancer is the fourth leading cause of death by cancer in the Western world and expected to become the second by 2030.2 Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer. Because it lacks early symptoms, PDAC is often diagnosed at an advanced stage, when patients are no longer eligible for surgical resection, which is possible in <15% of the cases and increases the 5-year survival rate to only 20%. Chemotherapy and radiation only allow a marginal increase in survival.3 Therefore, new strategies are urgently needed to develop effective treatment options. Recent progress has been made in our understanding of the physiopathology of PDAC, such as the characterization of its large genetic heterogeneity.4 Targeted therapies have not yet had a real impact on this disease; in particular, targeting oncogenic Kras (mutated in most of PDAC) proved to be disappointing.5,6
Recent papers have demonstrated an extensive reprogramming of metabolism in PDAC, as is the case in all cancers, supporting tumor progression and therapeutic resistance.7, 8, 9 Active metabolism is essential for the generation of the energy and biochemical building blocks that are required for tumoral growth. It is now well known that the two pathways generating energy (ATP), glycolysis and mitochondrial respiration through oxidative phosphorylation (OXPHOS), coexist in cancer cells including PDAC.9, 10, 11 The pro-survival role of mitochondria in pancreatic cancer stem cells or dormant cells has been reported,12,13 and OXPHOS is an emerging target in cancer therapy.14, 15, 16, 17 The biguanides metformin and phenformin were reported to inhibit mitochondrial respiratory complex I.18, 19, 20, 21 In PDAC preclinical models, metformin and phenformin are both able to inhibit tumor growth,22,23 and metformin treatment amplifies the gemcitabine-induced delay in tumor growth through a less reactive pancreatic microenvironment.24 Treatment of diabetic patients with metformin was described to lower the risk to develop PDAC,25 motivating clinical trials combining metformin with chemotherapy. However, supplementation with metformin so far has not been found to improve the outcome of PDAC patients treated with gemcitabine and erlotinib.26,27 Drugging OXPHOS still suffers from a lack of knowledge of the tumor context in which OXPHOS is a vulnerability. Although metabolite profiling stratification of PDAC was already reported,28 functional investigation of energetic metabolism in a large cohort of PDAC patients is required. We report here that PDAC tumors are highly heterogeneous in terms of energetic metabolism, and that high OXPHOS is a biomarker for sensitivity to treatment with phenformin (more efficient than metformin) in combination with chemotherapy.
Results
Energetic Metabolism Heterogeneity in PDAC
We used a Seahorse device to measure the cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in PDAC cells, to determine mitochondrial respiration (OXPHOS) and glycolysis, respectively. We started this functional analysis with six classical PDAC cell lines, which showed different levels of basal respiration, ATP production by mitochondria, and respiratory spare capacity, the Panc-1 cell line being the most efficient for mitochondrial respiration (Figures 1A and S1A). Furthermore, different levels of basal glycolysis and glycolytic reserve were observed between cell lines (Figures 1B and S1B). Thus, the cell lines can be classified into three different groups with respect to their basal energetic features (Figure 1C): Panc-1 is mostly respiratory (OXPHOS group); MIA PaCa-2 and BxPC-3 are mainly glycolytic (glycolytic group); and Capan-1, Capan-2, and to a lesser extent SOJ-6, have a relative lower energetic metabolism (less metabolic group). An alternative representation of the cell energetic preference is the ratio OXPHOS:glycolysis (Figure S1C). ATP production was shown to rely more on OXPHOS than glycolysis in all of the cells (Figure S1D). Importantly, the metabolic status of each cell line was found to be independent of its proliferation rate, as shown in Figure S2. Moreover, Seahorse analysis showed that all of the cell lines are plastic (i.e., able to shift to the other energetic pathway, if necessary) at various degrees, Capan-1 and Panc-1 being the most plastic to OXPHOS or glycolysis, respectively (Figures S1E and S1F). Finally, using the Mito Fuel Flex Seahorse analysis, we addressed the dependency and flexibility of mitochondrial respiration toward the three main metabolic pathways feeding the tricarboxylic acid (TCA) cycle: glucose, glutamine, and fatty acids. Respiration was shown to depend on the glucose and glutamine pathways in all of the cell lines except Panc-1 (Figure S1G). Moreover, dependency for the fatty acid pathway was observed in all of the cell lines, including Panc-1, which is the least dependent. Regarding flexibility, the Panc-1 cell line was the only one to be highly flexible toward the three TCA cycle fuels. These data demonstrate that mitochondria are fully able to produce energy in all of the tested PDAC cell lines (at high level in Panc-1), and that all of the cell lines can increase mitochondrial respiration or shift to glycolysis, depending on their needs.
Figure 1.

Stratification of PDAC Tumors According to Their Energetic Metabolism
(A) Oxygen consumption rate (OCR) was measured in the 6 classical PDAC cell lines under basal conditions and following the injection of oligomycin, carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazon (FCCP), and rotenone + antimycin A.
(B) Extracellular acidification rate (ECAR) was measured under basal conditions and following the addition of glucose, oligomycin, and 2-deoxyglucose.
(C) Basal OCR versus basal ECAR plot (both normalized to 10,000 seeded cells) shows the clustering of the 6 PDAC classical cell lines into 3 metabolic groups: Panc-1 is mostly respiratory (OXPHOS); MIA PaCa-2 and BxPC-3 are mostly glycolytic (glycolytic); and Capan-1, Capan-2, and to a lesser extent SOJ-6, have a relative lower energetic metabolism compared to the other cell lines (less metabolic group). The dotted lines indicate the thresholds that we selected between high and low OCR and ECAR.
(D) Basal OCR versus basal ECAR plot (both normalized to 10,000 seeded cells) shows the clustering of the 21 primary PDAC cancer cells derived from PDX into 4 different groups with respect to their basal energetic features. A fourth group with high rates of both OCR and ECAR was found. High OXPHOS patients are depicted with the red color, and low OXPHOS with blue color. The dotted lines indicate the thresholds that we selected between high and low OCR and ECAR. This plot is representative of at least 3 independent experiments for each patient. For clarity, a short version of the anonymized name of patients is used.
Representative profiles of at least 3 independent experiments are shown in (A) and (B).
See also Figures S1 and S2 and Table S1.
More important, we also analyzed OXPHOS and glycolysis in PDAC cells recently derived from patients enrolled in the Patient-Derived Xenografts (PDX) PaCaOmics program (described in the STAR Methods section). We tested 21 PDAC primary cells of this PDAC cohort using Seahorse. These cells are highly heterogeneous in terms of morphology and size; nevertheless, we were able to compare them by seeding an accurate number of cells to reach the maximum of 70%–80% of confluence in the bottom of the Seahorse plate wells (Table S1), then normalizing the values to 10,000 cells as done for the classical cell lines. This work led to the classification of these cells into four different groups with respect to their basal energetic features (Figure 1D): OXPHOS, glycolytic, less metabolic, and energetic (high OXPHOS and glycolysis). As observed for the PDAC classical cell lines, the metabolic status of the PDAC primary cells is unrelated to their proliferation rate (Figure S2). These data illustrate the high energetic heterogeneity between PDAC tumors, most of them showing a hybrid OXPHOS/glycolysis phenotype. Importantly, this work distinguishes PDAC tumors with a high OXPHOS rate.
Enrichment of Mitochondrial Respiratory Chain Complex I in High OXPHOS PDAC
We then investigated the cellular and molecular mechanisms underlying the high respiratory rate in the high OXPHOS PDAC cells. Mitochondrial mass was monitored by MitoTracker staining followed by flow cytometry analysis in the classical PDAC cell lines (Figure 2A), showing no significant difference between them except a moderately weaker staining in Capan-2 cells. Accordingly, MitoTracker staining followed by fluorescence microscopy analysis did not show any obvious differences of mitochondrial network between cell lines (Figure S3A). Similarly, no difference in mitochondrial membrane potential was detected, and the ratio of mitochondrial membrane potential:mitochondrial mass was independent of the metabolic status (Figures 2B and 2C). Transmission electron microscopy (TEM) analysis was highly informative, showing the distribution of mitochondria in all of the cytoplasm, except for Panc-1, in which mitochondria are close to the nucleus (Figure 2D). Also, many figures of elongated mitochondria were observed in Panc-1 and SOJ-6 cells, contrary to the other cell lines showing round mitochondria (Figure 2E). Cristae appeared normal in all of the cell lines except MIA PaCa-2, which showed unstructured cristae in many swollen mitochondria. These observations suggest good mitochondrial dynamics in Panc-1 and SOJ-6 cells, which could be the basis of their active mitochondrial respiration.
Figure 2.

Structure of Mitochondria and Abundance of Respiratory Complex I Reflect Respiration Activity
(A and B) Flow cytometry analysis of the classical PDAC cells was carried out using (A) MitoTracker DeepRed for mitochondrial mass, and (B) MITO-ID for analysis of the mitochondrial membrane potential. Data are presented as the mean of triplicates ± SEMs. Statistically significant differences between Panc-1 cell line with the 5 other cell lines: ∗p < 0.05, ns = not significant. Data are representative of 3 independent experiments.
(C) The ratio of mitochondrial membrane potential:mitochondrial mass was calculated from (A) and (B).
(D and E) Representative transmission electron microscopy images are shown at different magnifications. The scale is indicated in the lower right part of each image. High magnification is shown in the inset in (E).
(F) Top: Representative immunoblot showing the abundance of 5 mitochondrial proteins (ATP5A, UQCR2, SDHB, COXII, and NDUFB8) belonging to respiratory chain complexes (V, III, II, IV, and I, respectively). Time exposure of the blot was 5 s for ATP5A while the rest was 1 min 30 s. Ponceau red staining was used as the control of equal protein loading (not shown). Bottom: Representative immunoblot showing the abundance of the protein NDUFB8 belonging to mitochondrial complex I. The time exposure was 1 min. Ponceau red staining was used as the control of equal protein loading (not shown). In both blots, an irrelevant lane was spliced out (vertical black line).
(G) Heatmap of differential metabolites between the 6 cell lines. Twelve metabolites were selected according to the results from the OPLS-DA differentiating OXPHOS Panc-1 cells from glycolytic MIA PaCa-2 and BxPC-3 cells (N = 21; 1 predictive + 1 orthogonal OPLS-DA model with R2Y = 0.93; QY = 0.87 ; CV_ANOVA p value = 2.10–6). Data have been Pareto scaled. Each column is the autoscaled mean abundance (isolated NMR signal integration) of each metabolite ranging from dark blue (−1) to dark red (1). Unsat fatty acids, unsaturated fatty acids; GPC, glycerophosphocholine; PC, phosphocholine. Proline is the dominant metabolite in Panc-1, SOJ-6, BxPC-3, and MIA PaCa-2; taurine is the dominant metabolite in Capan-1 and Capan-2.
See also Figures S3 and S4.
We then monitored the level of one protein of each of the five mitochondrial respiratory complexes by western blotting (WB) using an OXPHOS antibodies cocktail (Figures 2F and S3B). Interestingly, we found that complex I is more abundant in Panc-1 and SOJ-6 cells, which have the highest OXPHOS in comparison with all of the other cell lines. This was confirmed using the antibody alone specific to NDUFB8 protein belonging to mitochondrial complex I (Figures 2F and S3B). Moreover, a metabolomics analysis was done by high-resolution magic angle spinning nuclear magnetic resonance (HRMAS-NMR), which is an approach amenable to implementation in the clinic (Figures 2G and S4A). The Panc-1 showed the highest level of metabolites, including those feeding the TCA cycle, such as fatty acids and amino acids. Intriguingly, Panc-1 cells also showed the highest level of the carnitine palmitoyltransferase 1A (CPT1A) protein involved in fatty acid metabolism in mitochondria (Figure S4B). These data, considered in parallel with Mito Fuel Flex Seahorse analysis (Figure S1G), suggest that high respiratory capacities of mitochondria in Panc-1 cells are supported by an active TCA cycle functioning independently of fuel origin. These data illustrate the metabolic specificities of Panc-1 cells, which seem to have a rich metabolic content, making their mitochondrial respiration highly independent and very flexible.
Afterward, we analyzed the RNA sequencing (RNA-seq) data obtained from the 21 primary PDAC cells of the PDX PaCaOmics cohort that we tested by Seahorse (see above). We performed a canonical partial least squares (PLS) analysis between the metabolic variables (OCR and ECAR from the Seahorse analysis) and gene expression. The sample projection on the two-dimensional (2D) space, which comprises the first two components, enables us to distinguish high and low OXPHOS groups in red and blue lettering, respectively (Figure 3A). Moreover, the gene set enrichment analysis (GSEA) on the gene loading values of the second component showed enrichment in mitochondrial electron transport at complex I and complex I assembly genes (normalized enrichment score [NES] = 2.03 and 2.01, respectively, adjusted p value = 3.7E−2), indicating a positive correlation between complex I and the OCR level (Figure 3B).
Figure 3.

Correlation between the High OXPHOS Status and the Abundance of Mitochondrial Complex I, Both at mRNA and Protein Level in the Primary PDAC Cancer Cells
High OXPHOS patients are depicted in red, and low OXPHOS are in blue.
(A) Canonical correlation analysis: graphical representation of PDX using the averaged components 1 and 2 of transcriptomic and metabolic datasets that were retrieved from canonical PLS.
(B) Gene set enrichment analysis: the two plots show the enrichment score for the related mitochondrial pathway on the second canonical PLS component in (A).
(C) Representative immunoblot showing the abundance of the 5 mitochondrial proteins (ATP5A, UQCR2, SDHB, COXII, and NDUFB8) belonging to mitochondrial complexes (V, III, II, IV, and I, respectively). Time exposure of the blot was 1 min. Ponceau red staining was used as the control of equal protein loading. Mitochondrial complexes protein levels were quantified from western blotting (WB) by ImageJ and normalized to Ponceau red staining. p value from Mann-Whitney test.
(D) Representative immunoblot showing the abundance of the mitochondrial protein NDUFB8 belonging to mitochondrial complex I. Time exposure of the blot was 1 min. Complex I protein was quantified by ImageJ and normalized to Ponceau red staining used as control of equal protein loading. p value from Mann-Whitney test.
(E) Metabolic analysis by HRMAS-NMR for 6 primary PDAC cancer cells derived from PDX, 3 high OXPHOS (27, 74, and 84) and 3 low OXPHOS (22, 32, and 85). OPLSDA score plot showing the discrimination between high OXPHOS and low OXPHOS.
(F) Kaplan-Meier survival curve using transcriptomic analysis on patient-derived xenografts, divided into high and low NDUFB8 gene expression groups (n = 40 and n = 35, respectively; the cutpoint was 13.22). p value was calculated by the log-rank test.
See also Figures S5 and S6 and Table S2.
We also monitored the level of one protein of each of the five mitochondrial respiratory complexes by WB (Figure 3C), demonstrating that complex I specifically is significantly more abundant in high OXPHOS tumors than in low OXPHOS tumors. This was confirmed using anti-NDUFB8 antibody alone (Figure 3D). Furthermore, transcriptomic data mining showed a highly significant enrichment in mitochondrial pathways in the 3 highest OXPHOS patients (27, 74, and 84) compared to 3 of the lowest OXPHOS patients (22, 32, and 85) (Table S2). In particular, 24 genes over 44 that encode proteins belonging to complex I29 were found enriched in the high OXPHOS cells, including NDUFB8 (Figure S5A; Table S2). Importantly, these 24 genes encode complex I protein subunits located in the 4 functional modules of the complex (Figure S5A).
Metabolomic analysis by HRMAS-NMR was done for these 3 high OXPHOS patients compared to the 3 low OXPHOS patients, confirming that they segregate into 2 different groups in the orthogonal PLS-discriminant analysis (OPLSDA) score plot (Figure 3E). Differential analysis highlighted the abundance of specific metabolites in each OXPHOS category: high OXPHOS cells were enriched in the metabolites choline (precursor of phosphocholine), phosphocholine (major lipid in membranes), glutamate (feeding the TCA cycle, and one of the 3 amino acids constituting glutathione), and glutathione (antioxidant), whereas low OXPHOS cells showed an accumulation of glucose (Figures S6A and S6B).
Finally, by examining the patient clinical data, we observed no link between the cell energetic preference and the histopathological differentiation, the basal and classical tumors classification,30 or Kras and p53 mutations (most of the tumors are mutated for both genes; Table S1). In contrast, we observed that the overall survival of high OXPHOS patients (functional analysis) was lower compared to low OXPHOS, even if the difference was not reaching statistical significance, probably because of the small number of patients (Figure S5B). To further address this point, we examined the expression of the gene encoding NDUFB8 belonging to complex I that we quantified by western blotting (Figures 3C and 3D). For this purpose, we used RNA-seq data from the 75 PDX of the PaCaOmics cohort. RNA-seq data from xenografts instead of cells present the obvious advantage that tumor cells are in an environment closer to the context found in patients than in 2D cultures. Importantly, RNA-seq analysis can discriminate human tumor genes from murine stromal genes. Figure 3F shows with strong evidence (p = 0.028) that patients with high NDUFB8 expression have a lower survival rate than patients with low NDUFB8 expression, independently whether or not the patients were resected (Figure S5C).
These data show a correlation between mitochondrial respiratory genes expression and high OXPHOS status in PDAC, strongly suggesting that the transcriptomic analysis is a potential tool to identify high OXPHOS patients in the clinic. Moreover, these data point to mitochondrial respiratory complex I as a vulnerability of high OXPHOS PDAC.
Synergy of Phenformin with Gemcitabine in High OXPHOS PDAC In Vitro
We observed different sensitivities of the six classical PDAC cell lines to gemcitabine in vitro, with the Panc-1 cell line being the least sensitive (Figure 4A). We wondered whether treating cells with a drug inhibiting mitochondrial respiratory complex I could counter the resistance to gemcitabine in the high OXPHOS Panc-1 cell line. We first treated cells with phenformin, metformin, and rotenone, which are known to target complex I. Dose-response curves show that these three drugs are able to decrease pancreatic cancer cell viability, and that phenformin is more efficient than metformin (Figures 4B, S7A, and S7B). More important, we showed that combining phenformin at 0.5 mM (greater than the half-maximal inhibitory concentration [IC50] of the Panc-1 line, which is the least sensitive; Figure 4B) with increasing doses of gemcitabine specifically sensitizes Panc-1 cells (Figures 4C and S7C). Combination indexes lower than 0.5 for low gemcitabine concentrations show a strong synergy between phenformin and gemcitabine specifically in the high OXPHOS Panc-1 cells (Figure S7D).
Figure 4.

Targeting Mitochondrial Respiratory Complex I with Phenformin Synergizes with Gemcitabine Antitumoral Activity in High OXPHOS Cells
(A and B) Dose-response curves for the 6 classical PDAC cell lines treated with different concentrations of gemcitabine (A) or phenformin (B) for 72 h. Live cells are indicated as a percentage of the control (vehicle treated). Data are means of triplicates ± SEMs. Data are representative of 3 independent experiments. The 0.5 mM concentration of phenformin is highlighted by a vertical dotted line in (B). The IC50 of phenformin was calculated and is shown on the right of the dose-response curve.
(C) Representative dose-response curves for the 6 PDAC cell lines treated with low concentrations of gemcitabine alone (blue curves) and gemcitabine in combination with 0.5 mM phenformin (red curves) for 72 h. Cell viability is indicated as a percentage of the control (vehicle treated). Data are means of triplicates ± SEMs.
(D) Representative dose-response curves for 6 primary PDAC cancer cells derived from PDX, 3 high OXPHOS (27, 74, and 84) and 3 low OXPHOS (22, 32, and 85). Cells were treated for 72 h with low concentrations of gemcitabine alone (blue curves) and in combination with 0.5 mM phenformin (red curves). Cell viability is indicated as the percentage of the control (vehicle treated). Data are means of triplicates ± SEMs.
See also Figures S7 and S8.
We then addressed the possibility that combining gemcitabine with phenformin could also be synergistic in primary high OXPHOS PDAC cells. For this purpose, we performed chemograms with the same 6 patients than for the transcriptomic data mining and metabolomic analyses (shown above): 3 high OXPHOS patients (27, 74, and 84) compared to 3 low OXPHOS patients (22, 32, and 85). We treated the cells with phenformin, showing its ability to decrease cell viability for the 6 tested patients irrespective of their OXPHOS status (Figure S8A). More important, our data shown in Figures 4D and S8B demonstrate that combining phenformin at 0.5 mM with increasing doses of gemcitabine specifically sensitizes high OXPHOS cells to gemcitabine, with combination indexes lower than 0.5, indicating a strong synergy (Figure S8C). These data show that targeting complex I with phenformin in the high OXPHOS PDAC cells potentiates their sensitivity to gemcitabine.
The importance of the OXPHOS status in the gemcitabine response was also assessed by OXPHOS shift assays (Figure 5). Pharmacologic manipulation of high OXPHOS cell Panc-1 toward a low OXPHOS phenotype by inhibiting mitochondrial protein synthesis with tigecycline15 was shown to increase cell sensitivity to gemcitabine (Figure 5A). Conversely, culturing low OXPHOS cells (MIA PaCa-2 and BxPC-3) in a medium containing galactose instead of glucose as the sole sugar source, which shifts the energetic metabolism from glycolysis to mitochondrial OXPHOS,15 induced resistance to low concentrations of gemcitabine (Figure 5B). Thus, manipulating the mitochondrial energetic status toward low OXPHOS or high OXPHOS confers sensitivity or resistance to gemcitabine, respectively, confirming the potential of inhibiting OXPHOS in combination with gemcitabine to sensitize PDAC cells to chemotherapy.
Figure 5.

OXPHOS Shift Experiments Demonstrating That Gemcitabine Sensitivity Is a Feature of OXPHOS Low Cells
(A) Panc-1 cells shift from high OXPHOS to low OXPHOS by treatment with tigecycline (Tig) enhances gemcitabine (Gem) cytotoxicity efficacy. Cell viability was monitored after 72 h of treatment with or without Gem (Left: 1 nM; right: 4 nM) and Tig (50 μM). Live cells are indicated as a percentage of the control (vehicle treated). Data are means of triplicates ± SEMs (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗, p < 0.001; symbol over the bar, versus non-treated control; ns, not significant). Data are representative of 3 independent experiments.
(B) MIA PaCa-2 and BxPC-3 cells shift from low OXPHOS to high OXPHOS by culture in galactose instead of glucose induces Gem resistance. Cell viability was monitored after 72 h of treatment with Gem (4 nM). Live cells are indicated as a percentage of the control (vehicle treated). Data are means of triplicates ± SEMs (∗∗∗p < 0.001 versus non-treated). Data are representative of 3 independent experiments.
Effect of Phenformin/Gemcitabine Combination in High OXPHOS PDAC In Vivo
We further assessed the synergistic impact of phenformin with gemcitabine in high OXPHOS PDAC in vivo in two different orthotopic mouse models: xenografts (Figures 6A–6C) and syngeneic allografts (Figures 6D and 6E).
Figure 6.

Targeting Mitochondrial Respiratory Complex I with Phenformin Enhances Gemcitabine Antitumoral Activity in High OXPHOS Tumors in 2 Preclinical Mouse Models
(A–C) Orthotopic xenografts.
(A) Experimental schematic representation. One or 2 million MIA PaCa-2 and Panc-1 cells, respectively, were surgically implanted into the pancreas of 6-week-old female Swiss nude mice. The presence of a pancreatic tumor was confirmed by exploratory laparotomy at day 16 (MIA PaCa-2) or day 33 (Panc-1). After surgery recovery, treatments were started and lasted 4 weeks.
(B) Representative gross photographs of pancreatic tumors for each treatment category.
(C) Weight of orthotopic xenografts. Vehicle (control, n = 6); gemcitabine alone (Gem, n = 5 for MIA-PaCa-2 and n = 9 for Panc-1); combo gemcitabine + phenformin (Gem + Phen, n = 8 for MIA-PaCa-2 and n = 11 for Panc-1); and phenformin alone (Phen, n = 5). The mean in each group is shown as a horizontal line. p values were calculated from the Mann-Whitney test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(D and E) Orthotopic syngeneic allografts.
(D) Experimental scheme. One million KPC luc2 cells were intraperitoneally injected into 6-week-old female C57BL/6 mice. The presence of a pancreatic tumor was confirmed by bioluminescence (day 10), and the treatments were started at day 11 for 6 days.
(E) Weight of orthotopic allografts. Vehicle (control, n = 6); gemcitabine alone (Gem, n = 5); combo gemcitabine + phenformin (Gem + Phen, n = 6); and phenformin alone (Phen, n = 6). The mean in each group is shown as a horizontal line. p values were calculated from the Mann-Whitney test. ∗p < 0.05 and ∗∗p < 0.01.
See also Figure S9.
For xenografts, Panc-1 and MIA PaCa-2 cells were implanted into the pancreas of immunodeficient mice, and treatments were administrated using either a combination of phenformin and gemcitabine, or each drug alone. As expected, phenformin markedly increased the antitumoral effect of gemcitabine in the high OXPHOS Panc-1 xenografts, whereas no impact of phenformin on gemcitabine antitumoral activity was observed in the low OXPHOS MIA PaCa-2 xenografts (Figures 6B and 6C).
For the syngeneic allografts, we used KPC luc2 cells, which showed high OXPHOS status and moderate plasticity by Seahorse analysis (Figure S9A). In this immunocompetent context as well, the combination of the two drugs was more potent than gemcitabine alone to induce tumor regression (Figures 6E and S9B). These preclinical data demonstrate that targeting complex I with phenformin in high OXPHOS PDAC enhances the anticancer effect of gemcitabine, independently of the host immune system.
Discussion
This study shows clearly that PDAC tumors can be functionally stratified according to their energetic metabolism, pointing to tumors with high OXPHOS activity. Concerning classical PDAC cell lines, our data are consistent with other reports, showing high OCR in Panc-1 and high glycolytic activity in BxPC-3 and MIA PaCa-2 cells.28,31 Furthermore, our study shows that PDAC cells are highly plastic for energy production, as they can shift to OXPHOS or glycolysis when necessary, and that they can use any fuel to feed the TCA cycle. The most frequently mutated genes in PDAC are Kras and TP53, both known to play a role in metabolism.32, 33, 34, 35 We show that the OXPHOS status in PDAC cells is independent of the main genetic alterations. Therefore, we propose that mitochondrial metabolism could be targeted in therapy independently of the “Kras addiction.”
Moreover, our study provides insight into the cellular and molecular mechanisms underlying active mitochondrial respiration in PDAC cells. We show that high OXPHOS status is correlated with the abundance of metabolites fueling the TCA cycle, and elongated morphology of mitochondria, consistent with the importance of mitochondrial quality with a healthy metabolic machinery.36
Of foremost importance, we demonstrate a correlation between high OXPHOS status and the abundance of mitochondrial complex I, both at the protein level (WB) and mRNA level (transcriptomic analysis showing the gene enrichment of 24 complex I subunits). Of great relevance is that we found lower overall survival of high OXPHOS patients compared to low OXPHOS patients, both functionally and according to NDUFB8 expression, the latter reaching statistical significance due to the high number of PDX (75). Importantly, this observation is independent whether or not the patients were resected (in our PaCaOmics cohort as in others, resected patients have a better prognosis than non-resected patients). This suggests an association between high OXPHOS status and poor prognosis of PDAC patients.
Furthermore, we demonstrate a synergy between standard chemotherapy (gemcitabine) and phenformin (targeting mitochondrial complex I) in high OXPHOS PDAC cells, regardless of whether they are long-standing established cell lines or recently established primary cells from PDX. Targeting mitochondria with phenformin induces an energetic shift toward low OXPHOS status,15 which markedly enhances gemcitabine’s antitumoral effect.
The cooperation between phenformin and gemcitabine in high OXPHOS PDAC cells was also demonstrated in vivo. Here, we show that phenformin noticeably enhances the antitumoral effect of gemcitabine in high OXPHOS PDAC tumors, in two different preclinical assays. One mode of action of the combination therapy could be that highly active mitochondrial respiration better sustains resistance to stress induced by the chemotherapeutic agent (DNA synthesis inhibition by gemcitabine), this pro-survival process being reduced by OXPHOS inhibition.
In this study, we used phenformin instead of metformin because of its higher efficiency on PDAC cells in vitro. This could result from the fact that metformin relies on members of the organic cation transporter (OCT) family for entering into cells, contrary to phenformin, which is readily transported into them. This capacity suggests that lower plasmatic concentrations of phenformin than metformin are clinically needed to successfully induce effect on the tumors (and our in vivo data are in favor of phenformin influence in tumors). Although phenformin was withdrawn from the US market because of its predisposition to induce lactic acidosis in diabetic patients, this side effect may be manageable with proper clinical vigilance. The cooperative action of phenformin on gemcitabine antitumoral activity observed specifically in high OXPHOS tumors, which we demonstrated in vitro and in vivo, suggests that phenformin should be clinically tested as an anticancer agent for the treatment of PDAC with an appropriate patient selection (high OXPHOS).
In conclusion, our study reveals a strong correlation between functional (Seahorse experiments) and molecular (RNA and protein) OXPHOS levels. This strong association opens the possibility of stratifying patients in the clinic according to mitochondrial respiration gene expression, and to identify the patients most likely to respond to mitochondrial respiration targeting in combination with standard chemotherapy.
Limitations of Study
This study involves both the demonstration of OXPHOS functional and molecular stratification of PDAC patients and the proposal of drug combination for some of them endowed with high OXPHOS activity. The first limitation is that the method to stratify the patients in the clinic still needs to be developed further. The second limitation is that the mechanism of the cooperative action of phenformin on gemcitabine antitumoral activity, specifically in high OXPHOS tumors, must be investigated further. It is reported that the inhibition of mitochondrial activity induces an energy crisis and apoptosis in cancer cells, including PDAC. We therefore suggest that phenformin-driven inhibition of mitochondrial activity in the high OXPHOS PDAC cells induces a shift toward a low OXPHOS status, with the consequence of an energetic stress, which enhances the antitumoral activity of gemcitabine. Nevertheless, we cannot exclude other hypotheses, such as the possibility that phenformin could modify the metabolism of gemcitabine (entry into the cells, conversion into active metabolites).
Finally, it is important to mention that the therapeutic efficacy of the proposed combination treatment (phenformin and gemcitabine) has not yet been tested clinically.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Total OXPHOS Human WB Antibody Cocktail | Abcam | Cat# ab110411; RRID: AB_2756818 |
| anti-human NDUFB8 | Abcam | Cat# ab110242; RRID: AB_10859122 |
| anti-CPT1A | Abcam | Cat# ab128568; RRID: AB_11141632 |
| Peroxidase-conjugated secondary antibody (Mouse) | Santa Cruz Biotechnology | Cat# sc-2005; RRID: AB_631736 |
| Biological Samples | ||
| PaCaOmics patient’s primary cells | 37 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Serum-free ductal media (SFDM) | 37 | N/A |
| DMEM | Life Technologies | Cat# 11995 |
| RPMI | Life Technologies | Cat# 21870076 |
| Fetal Bovine Serum | Lonza | Cat# 12103C |
| DMEM | Thermo Fisher Scientific | Cat# 11966025 |
| DMEM for Seahorse | Sigma-Aldrich | Cat# D5030 |
| Glucose | Sigma Aldrich | Cat# G8644 |
| Galactose | Sigma Aldrich | Cat# G0750 |
| Sodium pyruvate | Thermo Fisher Scientific | Cat# 11360070 |
| L-glutamine | Thermo Fisher Scientific | Cat# 25030081 |
| Hygromycine B | Thermo Fisher Scientific | Cat# H7772 |
| Accutase | GIBCO | Cat# A6964 |
| MitoTracker Deep Red | Molecular Probes | Cat# M22426 |
| Paraformaldehyde | Sigma Aldrich | Cat# P6148 |
| ProLong™ Gold antifade reagent with DAPI | Thermo Fisher Scientific | Cat# P36931 |
| Glutaraldehyde | Sigma Aldrich | Cat# G5882 |
| Agarose | Sigma Aldrich | Cat# A2576 |
| Osmium tetroxide | Electron Microscopy Science | Cat# 19100 |
| Phenformin | Sigma-Aldrich | Cat# P7045 |
| Metformin | Sigma-Aldrich | Cat# D150959 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Gemcitabine (Gemzar) | Eli Lilly & Co. | N/A |
| Tigecycline | Sigma-Aldrich | Cat# PZ0021 |
| Luciferin | Promega | Cat# E6552 |
| Nitrocellulose membranes | Pall Gelman Laboratory | Cat# 60208 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Ponceau red | Sigma-Aldrich | Cat# BI-PB0437-25G |
| Triton X-100 | EUROMEDEX | Cat# 2000-B |
| Tween 20 | EUROMEDEX | Cat# 2001-B |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Cat# 103015-100 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent Technologies | Cat# 103020-100 |
| Seahorse XF Mito Fuel Flex Test | Agilent Technologies | Cat# 103260-100 |
| MITO-ID Membrane potential detection | ENZO | Cat# 51018 |
| Bio-Rad Protein Assay | Bio-Rad laboratories | Cat# 5000001 |
| Deposited Data | ||
| Original and analyzed data | This paper | https://doi.org/10.17632/57vwny5g7j.1 |
| PaCaOmics RNA-seq data | 38 | Accession number E-MTAB-5039 |
| NMR metabolites in-house and online databases | 39 | N/A |
| Experimental Models: Cell Lines | ||
| BxPC-3 | ATCC | Cat# CRL-1687 |
| Capan-1 | ATCC | Cat# HTB-79 |
| Capan-2 | ATCC | Cat# HTB-80 |
| MIA PaCa-2 | ATCC | Cat# CRM-CRL-1420 |
| Panc-1 | ATCC | Cat# CRL-1469 |
| SOJ-6 | Dr. Eric Mas | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Female Swiss nude (4 week old) | Charles River | Cat# Crl:NU(Ico)-Foxn1nu |
| Mouse: Female C57BL/6J (4 week old) | Charles River | Cat# C57BL/6 J |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij | N/A |
| FlowJo version 10.0.7 | https://www.flowjo.com/solutions/flowjo | N/A |
| GraphPad Prism software | https://www.graphpad.com | N/A |
| NMRprocflow online software | https://link.springer.com/article/10.1007/s11306-017-1178-y | N/A |
| SIMCA-P + v.14 | Umetrics | N/A |
| lixOmics package | 40 | N/A |
| Limma package | Bioconductor | N/A |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alice Carrier (alice.carrier@inserm.fr).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Raw data were deposited on Mendeley at https://doi.org/10.17632/57vwny5g7j.1. All data used for this study are available from the lead contact alice.carrier@inserm.fr upon request.
Experimental Model and Subject Details
Pancreatic Ductal Adenocarcinoma (PDAC) Cell Lines
The classical PDAC cell lines (BxPC-3, Capan-1, Capan-2, MIA PaCa-2, and Panc-1) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The SOJ-6 cell line was kindly provided by Dr. Eric Mas (INSERM U1068, Marseille, France). The classical PDAC cell lines BxPC-3, Capan-1, Capan-2 and SOJ-6 were maintained in RPMI media (GIBCO, Life Technologies) supplemented with 10% fetal bovine serum (FBS; Lonza); MIA PaCa-2 and Panc-1 were maintained in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO, Life Technologies) supplemented with 10% FBS. The authentication was performed by the ATCC for all cell lines except SOJ-6.
The murine PDAC cell line KPC luc2 (LSL-KrasG12D/+; Trp53R172H/+; Elas-CreER, transfected with a luciferase-encoding vector) was a gift from Dr. Nathalie Auphan-Anezin (CIML, Marseille, France). The KPC cell line was originally obtained by S.F. Konieczny (West Lafayette, USA) from a pancreatic tumor in a KPC mouse on C57BL/6 background.41 KPC luc2 cells were maintained in RPMI medium supplemented with 10% FBS and 600 μg/ml Hygromycine B (ThermoFisher Scientific) for selection of cells containing the luciferase-encoding vector.
All cell lines were cultured at 37°C with 5% CO2 in a humidified atmosphere. Cells were harvested using Accutase (GIBCO) and reseeded once or twice per week. Cell lines were regularly tested for Mycoplasma contamination and found to be negative.
Xenografts and allografts mouse models
The orthotopic xenografts were generated using the human PDAC classical cell lines Panc-1 and MIA PaCa-2. Cells were maintained as described above and harvested at exponential growth for implantation into the pancreas of recipient mice. Six-week-old athymic female Swiss Nude mice, SOPF (Specific and Opportunistic Pathogen Free) health status, strain Crl:Nu(lco)-Foxn1nu (Charles River France), were used. Grafting experiments were implemented by surgery under isoflurane anesthesia to perform intrapancreatic parenchymal injection of one and two millions of MIA PaCa-2 and Panc-1 cells, respectively.
The presence of a pancreatic tumor was confirmed by exploratory laparotomy on day 16 and day 33 post-grafting in MIA PaCa-2 and Panc-1 xenografts, respectively. After surgery recovery, randomization prior to treatment was carried out and mice were assigned to four groups (at least n = 5 per condition) and injected intraperitoneally with PBS (vehicle control), Gemcitabine (120 mg/kg twice a week), Phenformin (50 mg/kg daily), and combination Gemcitabine and Phenformin at the same dose. Four weeks after the start of treatment, mice were sacrificed by cervical dislocation, necropsy was performed, and pancreatic tumors were weighed.
Orthotopic syngeneic allografts were generated by intraperitoneal injection of one million of the murine cells KPC luc2 into 6-week-old female C57BL/6 mice (immunocompetent strain, SOPF health status, Charles River, France). Tumoral growth was followed by bioluminescence upon injection of 3 mg luciferin-EF (Promega) using a Photon Imager device (Biospace Lab). On day 11 post-grafting, mice were randomized to four treatments cohorts (at least n = 5 per condition) as done for xenografts. Mice were sacrificed by cervical dislocation when the non-treated mice (vehicle-injected as controls) reached the ethical limit point on day 17, necropsy was performed and pancreatic tumors were weighed.
All mice were kept under specific pathogen-free conditions and according to the current European regulation; the experimental protocol was approved by the Institutional Animal Care and Use Committee (#16711).
PaCaOmics patient’s cohort
This cohort has been well described.30,37, 38, 42 Briefly, 75 patients of this cohort with a confirmed PDAC diagnosis were included in this study. Tumor samples were obtained from pancreatectomy, Endoscopic ultrasound-guided fine needle aspiration (EUS-FNA) biopsy, carcinomatosis or liver metastasis during exploratory laparotomy. All samples were xenografted in immunocompromised mice to generate Patient-Derived Xenografts (PDX) samples as described previously.37,38 Briefly, PDAC tumors were fragmented into pieces, mixed with Matrigel, and subcutaneously implanted in 5- to 6-week-old immunocompromised male mice (Swiss Nude; strain Crl: NU(lco)-Foxn1nu; Charles River, Wilmington, MA). Fourty-four primary cells were obtained from these PDX as described.37 Briefly, the xenograft pieces were minced, treated with collagenase and trypsin/EDTA, suspended in DMEM supplemented with Penicillin/Streptomycin and FBS, then in Serum Free Ductal Media (SFDM). These PaCaOmics primary cells were maintained in SFDM at 37°C with 5% CO2 in a humidified atmosphere. Cells were harvested and reseeded similarly to the classical PDAC cell lines and maintained for a maximum of 10 passages. Next Generation Sequencing of RNA (RNA-Seq) from the 75 PDX and the 44 primary cells is published elsewhere (accession number E-MTAB-5039).38
In the present study, 21 of the primary cells were used for the Seahorse analysis, RNA-Seq data of these 21 primary cells were used for transcriptomics analysis, and RNA-Seq data of the 75 PDX were used for the survival curve shown in Figure 3F.
Method Details
Real-time metabolic analysis
Measurements were performed using the Seahorse Bioscience XFe24 Extracellular Flux Analyzer (Agilent). This device allows the measurement of the cellular oxygen consumption rate (OCR in pmoles/min) and of the extracellular acidification rate (ECAR in mpH/min), for mitochondrial respiration (OXPHOS) and glycolysis, respectively. Sixteen hours before the assay, cells at exponential growth were seeded into Seahorse 24-well plates and cultured at 37°C with 5% CO2. The number of seeded cells was optimized to ensure 70%–80% confluence the day of analysis (Table S1).
XF Cell Mito Stress Test (OXPHOS experiment)
OCR was measured using the Seahorse XF Cell Mito Stress Test Kit. Culture medium was replaced with OXPHOS assay medium (DMEM without phenol red [Sigma-Aldrich reference D5030], 143 mM NaCl, 2 mM glutamine, 1 mM sodium pyruvate and 10 mM glucose, pH 7.4) and the plate was pre-incubated for 1h at 37°C in a non-CO2 incubator. OCR was measured under basal conditions, and then after sequential injections of different reagents: 1 μM oligomycin (respiratory Complex V inhibitor that allows to calculate ATP production by mitochondrion), carbonyl cyanide-p-trifluoromethox- yphenyl-hydrazon (FCCP; an uncoupling agent allowing determination of the maximal respiration and the spare capacity; the concentration was optimized for each cell line, Table S1), and finally 0.5 μM rotenone + 0.5 μM antimycin A (Complex I and III inhibitors, respectively) to stop mitochondrial respiration enabling the calculation of the background (i.e., non-mitochondrial respiration driven by processes outside the mitochondria). Levels of OCR were normalized to 10,000 seeded cells, which we found as the most accurate way to normalize when comparing different cell types.
XF Glycolysis Stress Test (Glycolysis experiment)
ECAR was measured using the Seahorse XF Glycolysis Stress Test Kit. Culture medium was replaced with glycolysis assay medium (DMEM without phenol red, 143 mM NaCl, 2 mM glutamine, pH 7.4) and the plate was pre-incubated for 1h at 37°C in a non-CO2 incubator. ECAR was first measured from non-glycolytic acidification (background), and then after sequential injections of different reagents. For basal glycolysis calculation, 10 mM glucose was injected, and glycolytic capacity was then calculated following the injection of 1 μM oligomycin. Glycolytic reserve was calculated as the difference between glycolytic capacity and basal glycolysis. At the end of the experiment, glycolysis was stopped by adding 2-Deoxyglucose (100 mM). Levels of ECAR were normalized to 10,000 seeded cells.
The contribution of OXPHOS and glycolysis to ATP production was calculated using the OCR and proton production rate (PPR) as previously described.43 Glycolysis was also measured using the recently developed XF Glycolytic Rate Assay (no glucose starvation required for background determination) with the same outcomes, suggesting that glucose starvation has no impact on glycolysis in PDAC cell lines.
XF Mito Fuel Flex Test
The Seahorse XF Mito Fuel Flex Test Kit was used to determine dependency and flexibility of cells to oxidize three critical mitochondrial fuels: glucose, glutamine, and fatty acids. Culture medium was replaced by OXPHOS assay medium and the plate was pre-incubated for 1h at 37°C in a non-CO2 incubator. Inhibitors of mitochondrial pyruvate carrier (UK5099 2 μM), glutaminase (BPTES 3 μM), and carnitine palmitoyl-transferase 1A (Etomoxir 4 μM) were used. The rate of oxidation of each fuel was determined by measuring OCR in the presence or absence of fuel pathway inhibitors according to the manufacturers’ instructions.
Flow cytometry
Mitochondrial mass measurement
Cells were seeded in 12-well plates (175,000 cells per well). The day after, MitoTracker Deep Red (Molecular Probes) was added in culture medium to a final concentration of 200 nM for 10 min. After incubation, cells were washed with warm PBS, detached with Accutase, and resuspended in HBSS (GIBCO, Life Technologies) for flow cytometry. Ten thousand events per sample were acquired in a MACSQuant-VYB (Miltenyi Biotec) and data analysis was performed using FlowJo software.
Mitochondrial membrane potential
Measurement was performed using the MITO-ID Membrane potential detection kit (ENZ-51018) according to the manufacturer’s protocol. Briefly, cells were collected, washed, and preincubated in 500 μL of the Assay Solution containing 5 μL of MITO-ID MP Detection Reagent for 15 min. Then, 10,000 events per sample were acquired in a MACSQuant-VYB and orange fluorescence data analysis was performed using the FlowJo software.
Fluorescence microscopy
Mitochondrial network
Cells were seeded on coverslips in 12-well plates (200,000 cells per well). The day after, the mitochondrial network was analyzed by incubation of cells in the presence of MitoTracker DeepRed FM (200 nM, Molecular Probes) at 37°C for 30 min. Then, cells were washed twice with PBS and fixed with warm 4% paraformaldehyde. Finally, samples were mounted using the ProLong™ Gold antifade reagent with DAPI. Confocal images were acquired using an inverted microscope equipped with LSM 880 with Airyscan detector controlled by Zeiss Zen Black, 63x lens.
Transmission Electron Microscopy (TEM)
The cells at exponential growth were fixed in 37°C-warm 4% paraformaldehyde and 2.5% Glutaraldehyde in PBS overnight. The next day, the plates were washed three times in PBS at RT. The cells were then scraped in PBS, transferred in a 2 mL microtube and pelleted by centrifugation for 5 min at 5000 rpm. The supernatant was removed, the cells were resuspended in 2% 37°C-warm low-melting-point agarose and transferred into microvettes (Sarstedt) and pelleted by centrifugation for 5 min at 5000 rpm. The microvettes were then placed on ice for 10 minutes. The microvettes tips were then removed with a razor blade and the agarose-embedded cell pellet was processed through a classical protocol for TEM. Briefly, the pellets were post-fixed in aqueous 1% OsO4 for one hour, and left overnight in aqueous 1% uranyl acetate. The next day, the pellets were dehydrated in graded series of ethanol baths (10 minutes each) and infiltrated with epon resin in ethanol (1:3, 2:2, 3:1, 2 hours each and pure resin overnight). The next day the pellets were embedded in fresh pure epon resin and cured for 48h at 60°C. Seventy nm-ultrathin sections were performed on a Leica UCT Ultramicrotome (Leica, Austria) and deposited on formvar-coated slot grids. The grids were contrasted using lead citrate and observed in an FEI Tecnai G2 at 200 KeV. Acquisition was performed on a Veleta camera (Olympus, Japan).
Immunoblotting
For each cell line, at least two different culture passages were analyzed by western blotting (WB), and at least 3 immunoblots were made for each tested protein. Cells at exponential growth were resuspended in lysis buffer (HEPES 50 mM, NaCl 150 mM, Triton X-100 1%, EDTA 1mM, EGTA 1 mM, glycerol 10%, NaF 25 mM, ZnCl2 10 μM) with a cocktail of protease and phosphatase inhibitors added freshly. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad laboratories, France). Proteins (50 μg) were resolved by SDS-PAGE, and transferred to nitrocellulose membranes. All membranes were systematically stained with Ponceau red to confirm equal protein loading and transfer, since usual normalization with so-called house-keeping proteins cannot be made as these proteins are variable in between PDAC cells (data not shown). Then membranes were blocked 1h at room temperature with TBS 5% milk, and incubated overnight in TBS 5% milk 0.1% Tween containing appropriate primary antibodies: CPT1A (1:500; Abcam #ab128569), Total OXPHOS Human WB Antibody Cocktail (1:100; Abcam #ab110411), NDUFB8 (1:2000; Abcam #ab110242). After three washes in TBS 0.1% Tween, membranes were incubated 1h at RT with a HRP-conjugated secondary antibody at 1:5000 before being revealed with ECL. Acquisition was performed with a Fusion FX7 imager (Vilber-Lourmat, France).
1H High Resolution Magic Angle Spinning-Nuclear Magnetic Resonance (HRMAS NMR) spectroscopy
For each sample, ten microliters of D2O were added to the cell pellet (5x106 cells), mixed and placed into a 30 μL disposable insert. The insert was then placed into a 4 mm ZrO2 HRMAS rotor. All NMR experiments were carried out on a Bruker Advance III spectrometer operating at 400 MHz for the 1H frequency equipped with a 1H/13 C/ 13P HRMAS probe. Spectra were recorded at 277K with a spin rate of 4 kHz. A Carr-Purcell-Meiboom-Gill (CPMG) NMR spin echo sequence [90° — (τ — 180° — τ)n] with an effective spin echo time of 37.5 ms, preceded by a water presaturation pulse during a relaxation time of 2 s to reduce the signal intensities of lipids and macromolecules. For each spectrum, 380 free induction decays (FID) of 26624 complex data points were collected using a spectral width of 8000 Hz. Each FID was then multiplied by an exponential weighting function corresponding to a line broadening of 0.3 Hz and zero-filled prior to Fourier transformation. Subsequently, each spectrum was phased and referenced to the alanine signal (δ = 1.46 ppm). Assignments of the NMR signals were performed using 1H−1H TOCSY spectrum,44 1H−13C HSQC spectrum,45 in-house and online databases.39 1H HRMAS NMR spectra were exported to NMRprocflow online software (10.1007/s11306-017-1178-y) to be baseline and signal shift corrected, and divided into 0.005 ppm-width buckets. To remove the effect of water suppression, the region between 4.70 and 5.27 ppm was discarded. The dataset was then normalized to the number of cells. Finally, the matrix was exported to the SIMCA-P + v.14 software (Umetrics, Umea, Sweden) for multivariate statistical analysis. First, Principal Component Analysis (PCA) was performed in order to check the homogeneity of the dataset. Orthogonalized Projections on Latent Structure Discriminant Analysis (OPLSDA) was then applied to a subset of the data matrix composed of Panc-1, BxPC-3 and MIA PaCa-2 samples in order to target metabolic differences between OXPHOS and glycolytic samples. The resulting score and loading plots were used to visualize the discriminant features. A leave-one out internal cross-validation was performed in order to calculate Q2 and R2Y values representing, respectively, the predictive capability and the sensitivity of the model, as well as the CV-ANOVA p value and to ensure the robustness of the statistical model. The integration of each discriminant signal, which represents the relative concentration of the corresponding metabolites, was then exported to Metaboanalyst online software (https://doi.org/10.1093/nar/gky310) and a heatmap was calculated based on autoscaled features.
Canonical correlation analysis
The canonical correlation analysis between transcriptome of primary PDAC cells derived from PDX and OCR+ECAR data (from Seahorse analysis) was performed by PLS approach (Partial least square) using the 2 first component (lixOmics package).40 To focus on genes more specifically correlated with OCR, we chose to focus on the component that shows opposite loading values of OCR and ECAR. For biological interpretation of the component of interest, we performed a gene set enrichment analysis on its loading values. The p values were adjusted for false discovery rate (FDR).
Differential gene expression analysis
The differential expression analysis between high and low OXPHOS groups was performed with Limma package (Bioconductor). GSEA analysis was performed to assess the enriched pathways using the logFold change statistic with the package fGSEA (Bioconductor).
Chemograms
Cells were seeded in 96-well plates (5,000 cells per well). Twenty-four hours later, the medium was supplemented with increasing concentrations of selected drugs in triplicates: Phenformin, Metformin, Rotenone (all provided by Sigma-Aldrich, Saint-Quentin Fallavier, France), and Gemcitabine. Rotenone that has to be dissolved in chloroform was prepared as 200x stock solution. Cell viability was determined 72h later by Crystal violet viability assay which is independent from cell metabolism. Briefly, cells were fixed in Glutaraldehyde 1%, washed twice with PBS, stained with crystal violet 0.1% for 10 min, then washed three times with PBS. Crystals were solubilized in SDS 1%, and absorbance was measured at 600 nm with Epoch-Biotek spectrophotometer.
For the combination treatment, we used Phenformin at 0.5 mM (IC50) with increasing doses of Gemcitabine. Combination index (CI) values were calculated for all tested drug concentrations according to the Chou and Talalay method46,47 using the following equation: CI = (D)1 / (DX)1 + (D)2 / (DX)2 where (D)1 and (D)2 represent the dose of agent 1 and 2 used in combination to induce X% growth inhibition, and (DX)1 and (DX)2 represent the dose of agent 1 and 2 required to reach X% growth inhibition when used alone. The CI theorem then provides quantitative definition for additive effects (0.8 ≤ CI ≤ 1.2), synergism (CI < 0.8) and antagonism (CI > 1.2) in drug combinations. Calculations were done with Graphpad Prism software (GraphPad Software Inc., La Jolla, CA).
OXPHOS shift assays
Panc-1 cells were forced to shift from high OXPHOS to low OXPHOS status by disruption of mitochondrial metabolism by treatment with Tigecycline (Sigma-Aldrich; inhibiting mitochondrial protein synthesis), as reported.15 Panc-1 cells were seeded in 96-well plates (5,000 cells per well). Twenty-four hours later, the medium was supplemented with Gemcitabine (1 or 4 nM) and Tig (50 μM). Cell viability was determined 72h later by Crystal violet viability assay.
MIA PaCa-2 and BxPC-3 cells were forced to shift from low OXPHOS to high OXPHOS status by culture in Galactose instead of Glucose, as reported.15 MIA PaCa-2 and BxPC-3 cells were cultivated for 3 weeks (equivalent to 6 consecutive passages) in DMEM (ThermoFisher Ref 11966025) supplemented with either Glucose (25 mM) or galactose (10 mM) as sole source of carbon. In galactose, proliferation of the cells is first decreased, then gradually after 4-5 passages cells recover their regular (i.e., in media containing glucose) proliferation rate.
After this adaptation, the cells were seeded in 96-well plates (5,000 cells per well), Gemcitabine (4 nM) was added in the medium 24 hours later, and cell viability was determined 72h later by Crystal violet viability assay.
In vivo experiments
Orthotopic xenografts were made using the classical human PDAC cell lines Panc-1 and MIA PaCa-2 (ATCC, Manassas, VA, USA). Cells were cultured and maintained with DMEM supplemented with 10% FBS at 37°C with 5% CO2 in a humidified atmosphere. After early passages, cells at exponential growth were harvested with Accutase (GIBCO) and two million Panc-1 or one million MIA PaCa-2 cell suspensions (in a 50 μl volume of DMEM without FBS), were implanted into the pancreas of recipient mice. Six-week-old (at the time of transplant) athymic female Swiss Nude mice, SOPF (Specific and Opportunistic Pathogen Free) health status, strain Crl:Nu(lco)-Foxn1nu (Charles River, France) were used. Grafting experiments were implemented by surgery under isoflurane anesthesia to perform intrapancreatic parenchymal injection of PDAC cells. The presence of a pancreatic tumor was confirmed by exploratory laparotomy on day 16 (MIA PaCa-2) or day 33 (Panc-1). After surgery recovery (one week), randomization prior to treatment was carried out and mice were assigned to four groups (at least n = 5 per condition) and injected intraperitoneally with PBS (vehicle control), Gemcitabine (120 mg/kg twice a week), Phenformin (50 mg/kg daily), and combination Gemcitabine and Phenformin at the same dose. Drugs were freshly prepared in PBS prior injection. Four weeks after the start of treatment, mice were sacrificed by cervical dislocation, necropsy was performed, and pancreatic tumors were weighed.
Orthotopic syngeneic allografts were obtained by intraperitoneal injection of one million KPC luc2 cell suspension (in a 100 μl volume of PBS) in 6-week-old female C57BL/6 mice (immunocompetent strain, SOPF health status, Charles River, France). The KPC luc2 cells are endowed with a high capacity to home into the pancreas and develop a pancreatic tumor. Tumoral growth was followed by bioluminescence (starting on day 3 post-inoculation and continuing on days 10, 14 and 17) upon injection of 3 mg luciferin-EF (Promega) using a Photon Imager device (Biospace Lab). On day 11 post-grafting, mice were randomized to four treatments cohorts (at least n = 5 per condition) as done for xenografts. Mice were sacrificed by cervical dislocation when the non-treated mice (vehicle-injected as controls) reached the ethical limit point on day 17, necropsy was performed and pancreatic tumors were weighed.
All mice were kept under specific pathogen-free conditions and according to the current European regulation; the experimental protocol was approved by the Institutional Animal Care and Use Committee (#16711).
Quantification and Statistical Analysis
Results are expressed as the mean ± SEM of triplicates, and at least three independent experiments were done for each analysis. Statistical analysis of data was performed by one-way analysis of variance (ANOVA) except for the immunoblot data (two-tailed unpaired Student’s t test) and in vivo data (Mann-Whitney U test). The log-rank statistic test was applied to the Kaplan-Meier survival curves. p values < 0.05 were considered statistically significant.
Acknowledgments
We thank Eric Mas (CRCM) for the SOJ-6 cells, Nathalie Auphan-Anezin (CIML) for the KPC luc2 cells, the cell culture platform (PCC, TPR2, Marseille, France) for technical assistance, Karim Sari and Régis Vitestelle for assistance with the use of the PSEA animal housing facility, Jérémy Nigri and Victoire Gouirand for advice on the orthotopic xenografts experiments, Jean-Emmanuel Sarry for helpful discussion, and Valérie Depraetere-Ferrier for editing the manuscript. We are grateful to members of the PiCSL-FBI core facility (IBDM, AMU-Marseille) belonging to the France-BioImaging national research infrastructure for the electron microscopy experiments. This work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Institut National Du Cancer (INCa), Fondation ARC pour la Recherche sur le Cancer (PJA 20151203544), La Ligue Nationale contre le Cancer (LNCC), DGOS (labelization SIRIC), Fondation Amidex, and Fondation de France. This work also benefited from grants from Canceropôle Provence-Alpes-Côte d’Azur (PACA) and Fédération GEFLUC for the acquisition of the Seahorse XFe24 device. R.M. was supported by the Fondation ARC pour la Recherche sur le Cancer, S.L. and J.G. were supported by the Fondation pour la Recherche Médicale, G.R.-C. was supported by CONACYT (Mexico, grant 339091/471717), S.D. was funded by IMODI, N.A.H. was funded by Association Azm et Saadé, T.G. was supported by the French Agence Nationale de la Recherche, and B.D. was funded by Canceropôle PACA.
Author Contributions
Conceptualization and Methodology, R.M., S.L., and A.C.; Investigation, R.M., S.L., G.R.-C., J.G., L.S., N.A.H., T.G., B.D., F.T., L.C., and L.B.; Formal Analysis, R.M., S.L., G.R.-C., J.G., S.D., L.S., N.A.H., A.E.K., E.P., and A.C.; Resources, O.G., N.D., and J.I.; Writing – Original Draft, R.M., G.R.-C., and A.C.; Writing – Review & Editing, R.M., G.R.-C., and A.C.; Visualization, R.M., G.R.-C., and A.C.; Supervision, A.C.; Project Administration and Funding Acquisition, A.C.
Declaration of Interests
The authors declare no competing interests.
Published: November 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100143.
Contributor Information
Rawand Masoud, Email: masoud.rawand@gmail.com.
Alice Carrier, Email: alice.carrier@inserm.fr.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Saluja A.K., Dudeja V., Banerjee S. Evolution of novel therapeutic options for pancreatic cancer. Curr. Opin. Gastroenterol. 2016;32:401–407. doi: 10.1097/MOG.0000000000000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J., Quinn M.C., Australian Pancreatic Cancer Genome Initiative Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 5.Eser S., Schnieke A., Schneider G., Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drosten M., Barbacid M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell. 2020;37:543–550. doi: 10.1016/j.ccell.2020.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Biancur D.E., Kimmelman A.C. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim. Biophys. Acta Rev. Cancer. 2018;1870:67–75. doi: 10.1016/j.bbcan.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbrook C.J., Lyssiotis C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell. 2017;31:5–19. doi: 10.1016/j.ccell.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Vaziri-Gohar A., Zarei M., Brody J.R., Winter J.M. Metabolic Dependencies in Pancreatic Cancer. Front. Oncol. 2018;8:617. doi: 10.3389/fonc.2018.00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia D., Park J.H., Jung K.H., Levine H., Kaipparettu B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells. 2018;7:21. doi: 10.3390/cells7030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J., DeBerardinis R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019;30:434–446. doi: 10.1016/j.cmet.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sancho P., Burgos-Ramos E., Tavera A., Bou Kheir T., Jagust P., Schoenhals M., Barneda D., Sellers K., Campos-Olivas R., Graña O. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashton T.M., McKenna W.G., Kunz-Schughart L.A., Higgins G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018;24:2482–2490. doi: 10.1158/1078-0432.CCR-17-3070. [DOI] [PubMed] [Google Scholar]
- 15.Farge T., Saland E., de Toni F., Aroua N., Hosseini M., Perry R., Bosc C., Sugita M., Stuani L., Fraisse M. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017;7:716–735. doi: 10.1158/2159-8290.CD-16-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gentric G., Kieffer Y., Mieulet V., Goundiam O., Bonneau C., Nemati F., Hurbain I., Raposo G., Popova T., Stern M.H. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019;29:156–173.e10. doi: 10.1016/j.cmet.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentric G., Mieulet V., Mechta-Grigoriou F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017;26:462–485. doi: 10.1089/ars.2016.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bridges H.R., Jones A.J., Pollak M.N., Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Owen M.R., Doran E., Halestrap A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 20.Wheaton W.W., Weinberg S.E., Hamanaka R.B., Soberanes S., Sullivan L.B., Anso E., Glasauer A., Dufour E., Mutlu G.M., Budigner G.S., Chandel N.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollak M. Overcoming Drug Development Bottlenecks With Repurposing: Repurposing Biguanides to Target Energy Metabolism for Cancer Treatment. Nat. Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 22.Rajeshkumar N.V., Yabuuchi S., Pai S.G., De Oliveira E., Kamphorst J.J., Rabinowitz J.D., Tejero H., Al-Shahrour F., Hidalgo M., Maitra A., Dang C.V. Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017;23:5639–5647. doi: 10.1158/1078-0432.CCR-17-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kisfalvi K., Moro A., Sinnett-Smith J., Eibl G., Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas. 2013;42:781–785. doi: 10.1097/MPA.0b013e31827aec40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian W., Li J., Chen K., Jiang Z., Cheng L., Zhou C., Yan B., Cao J., Ma Q., Duan W. Metformin suppresses tumor angiogenesis and enhances the chemosensitivity of gemcitabine in a genetically engineered mouse model of pancreatic cancer. Life Sci. 2018;208:253–261. doi: 10.1016/j.lfs.2018.07.046. [DOI] [PubMed] [Google Scholar]
- 25.Evans J.M., Donnelly L.A., Emslie-Smith A.M., Alessi D.R., Morris A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kordes S., Pollak M.N., Zwinderman A.H., Mathôt R.A., Weterman M.J., Beeker A., Punt C.J., Richel D.J., Wilmink J.W. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- 27.Bhaw-Luximon A., Jhurry D. Metformin in pancreatic cancer treatment: from clinical trials through basic research to biomarker quantification. J. Cancer Res. Clin. Oncol. 2016;142:2159–2171. doi: 10.1007/s00432-016-2178-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daemen A., Peterson D., Sahu N., McCord R., Du X., Liu B., Kowanetz K., Hong R., Moffat J., Gao M. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA. 2015;112:E4410–E4417. doi: 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giachin G., Bouverot R., Acajjaoui S., Pantalone S., Soler-López M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016;3:43. doi: 10.3389/fmolb.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bian B., Bigonnet M., Gayet O., Loncle C., Maignan A., Gilabert M., Moutardier V., Garcia S., Turrini O., Delpero J.R. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: implications for individualized medicine efforts. EMBO Mol. Med. 2017;9:482–497. doi: 10.15252/emmm.201606975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovalenko I., Glasauer A., Schöckel L., Sauter D.R., Ehrmann A., Sohler F., Hägebarth A., Novak I., Christian S. Identification of KCa3.1 Channel as a Novel Regulator of Oxidative Phosphorylation in a Subset of Pancreatic Carcinoma Cell Lines. PLOS ONE. 2016;11:e0160658. doi: 10.1371/journal.pone.0160658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryant K.L., Mancias J.D., Kimmelman A.C., Der C.J. KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27:2065–2071. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blum R., Kloog Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014;5:e1065. doi: 10.1038/cddis.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vousden K.H., Ryan K.M. p53 and metabolism. Nat. Rev. Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 36.Bulthuis E.P., Adjobo-Hermans M.J.W., Willems P.H.G.M., Koopman W.J.H. Mitochondrial Morphofunction in Mammalian Cells. Antioxid. Redox Signal. 2019;30:2066–2109. doi: 10.1089/ars.2018.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gayet O., Loncle C., Duconseil P., Gilabert M., Lopez M.B., Moutardier V., Turrini O., Calvo E., Ewald J., Giovannini M. A subgroup of pancreatic adenocarcinoma is sensitive to the 5-aza-dC DNA methyltransferase inhibitor. Oncotarget. 2015;6:746–754. doi: 10.18632/oncotarget.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicolle R., Blum Y., Duconseil P., Vanbrugghe C., Brandone N., Poizat F., Roques J., Bigonnet M., Gayet O., Rubis M., BACAP Consortium Establishment of a pancreatic adenocarcinoma molecular gradient (PAMG) that predicts the clinical outcome of pancreatic cancer. EBioMedicine. 2020;57:102858. doi: 10.1016/j.ebiom.2020.102858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wishart D.S., Jewison T., Guo A.C., Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lê Cao K.A., Martin P.G., Robert-Granié C., Besse P. Sparse canonical methods for biological data integration: application to a cross-platform study. BMC Bioinformatics. 2009;10:34. doi: 10.1186/1471-2105-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi G., DiRenzo D., Qu C., Barney D., Miley D., Konieczny S.F. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene. 2013;32:1950–1958. doi: 10.1038/onc.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolle R., Blum Y., Marisa L., Loncle C., Gayet O., Moutardier V., Turrini O., Giovannini M., Bian B., Bigonnet M. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017;21:2458–2470. doi: 10.1016/j.celrep.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu H., Ying M., Hu X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget. 2016;7:40621–40629. doi: 10.18632/oncotarget.9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavanagh J., Rance M. Sensitivity improvement in isotropic mixing (Tocsy) experiments. J. Magn. Reson. 1990;88:72–85. [Google Scholar]
- 45.Schleucher J., Schwendinger M., Sattler M., Schmidt P., Schedletzky O., Glaser S.J., Sørensen O.W., Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- 46.Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 47.Chou T.C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were deposited on Mendeley at https://doi.org/10.17632/57vwny5g7j.1. All data used for this study are available from the lead contact alice.carrier@inserm.fr upon request.