

Abstract
Antibiotic-resistant Clostridioides difficile is an anaerobic Gram-positive bacterium that colonizes the colon and is responsible for more than 29,000 deaths in the United States each year. Hence, C. difficile infection (CDI) poses an urgent threat to public health. Antibody-mediated neutralization of TcdA and TcdB toxins, the major virulence factors of CDI, represents an effective strategy to combat the disease without invoking antibiotic resistance. However, current antitoxin approaches are mostly based on parenteral infusion of monoclonal antibodies that are costly, narrow spectrum, and not optimized against the intestinal disease. Here, we engineered probiotic Saccharomyces boulardii to constitutively secrete a single tetra-specific antibody that potently and broadly neutralized both toxins and demonstrated protection against primary and recurrent CDI in both prophylactic and therapeutic mouse models of disease. This yeast immunotherapy is orally administered, can be used concurrently with antibiotics, and may have potential as a prophylactic against CDI risk and as a therapeutic for patients with CDI.
INTRODUCTION
Clostridioides difficile infection (CDI) is the most prevalent nosocomial infection in industrial countries and causes a range of diseases from mild diarrhea to fulminant colitis and death. With the emergence of hypervirulent and antibiotic-resistant strains, CDI incidence and disease severity have increased considerably worldwide in recent years (1, 2). Over 450,000 cases of CDI, responsible for more than 29,000 deaths, were reported annually in the United States, and related overall medical costs exceeded $4 billion (3, 4). The standard treatment options for CDI are limited to antibiotics that disrupt gut microflora such as vancomycin, metronidazole, and fidaxomicin, leading to high rates of recurrence (5, 6), morbidity, and mortality (7, 8). Immune-based therapies have been shown to be somewhat effective in clinical trials, including intravenous immunoglobulin (IVIG) (9) against severe CDI and human monoclonal antibodies against recurrent CDI (10, 11). Fecal microbiota transplantation (FMT) is effective against refractory and recurrent CDI, but is difficult to standardize and is associated with the risk of transmitting infectious diseases (12–14). Vaccines as a preventative means against primary and recurrent CDI are also undergoing clinical trials (15); however, the long seroconversion time (15–18) is not ideal for immediate protection. Hence, there is an urgent need for effective prophylactics and therapeutics for both primary and recurrent CDI.
The tissue damage, inflammation, and potential morbidity from CDI is mainly caused by two exotoxins, TcdA and TcdB, because C. difficile strains lacking both are avirulent (19–21). Neutralizing antibodies against the toxins protect against toxigenic CDI in animal models (22–24) and are associated with reduced disease severity and incidence of relapse in humans (25–27). Current therapeutic antitoxin approaches are mostly based on parenteral infusion of purified antibodies (28–31). The underlying mechanism of systemic antibodies protecting against human CDI is not entirely clear, but recent studies showed that the disruption of intestinal barrier function by C. difficile toxins allows the leaking of systemic antitoxins into the intestinal lumen, subsequently neutralizing the toxins (32, 33). Antitoxins delivered directly to the lumen of the intestines where C. difficile colonizes and secretes toxins would offer the first-line protection against CDI.
Live vector-based delivery of antitoxin or therapeutic proteins is an attractive potential alternative because this strategy may achieve a targeted release of these proteins in the intestines at low cost. Probiotics such as Lactobacillus acidophilus, Bifidobacterium bifidum, and Saccharomyces boulardii have been suggested as dietary supplements for CDI (34). Unlike probiotic bacteria, S. boulardii can be taken concurrently with antibiotics such as vancomycin and metronidazole that are typically used to treat patients with CDI. S. boulardii, a U.S. Food and Drug Administration (FDA)–designated Generally Regarded as Safe (GRAS) organism, was clinically tested numerous times for safety and efficacy and is commonly available over the counter for use in promoting intestinal health and amelioration of gastrointestinal (GI) illness due to diarrheal diseases, including CDI (35–44). Unfortunately, there are few reports of S. boulardii being used to express therapeutic proteins. However, Saccharomyces cerevisiae, which is genetically related to S. boulardii, has been used successfully to express single-domain variable fragments of heavy-chain antibodies (VHHs) with high yield (45). Yet, in contrast to S. cerevisiae, S. boulardii grows well at 37°C and is more resistant to acidic environmental conditions (46–48), making this strain particularly well suited for better survival and persistence in the human intestinal tract after oral administration. Moreover, recent studies have found that some of the genetic tools of S. cerevisiae can be used in S. boulardii as well (47, 49). In this study, we engineered an S. boulardii strain to constitutively secrete a tetra-specific fusion of VHHs capable of potently neutralizing both TcdA and TcdB. We demonstrated that the antitoxin-expressing S. boulardii exhibited potent efficacies as a prophylactic and a treatment against both primary and recurrent CDI in mice.
RESULTS
Engineering and optimizing the secretion of a tetra-specific antibody in yeast
We designed a tetra-specific VHH fusion designated as ABAB that consists of four distinctive toxin-neutralizing VHHs (50), two against TcdA and two against TcdB (Fig. 1A). ABAB fully neutralized toxins from a panel of 64 C. difficile clinical isolates (tables S1 and S2), indicating that this combination of VHHs has a broad neutralizing capacity. To compare the neutralizing activities of ABAB with Merck’s actoxumab and bezlotoxumab [human immunoglobulin G1 (IgG1) antibodies against TcdA and TcdB, respectively] (10, 25), we generated Fc-ABAB by fusing ABAB with a human IgG1 Fc fragment. Fc-ABAB had superior neutralizing activities against TcdA- and TcdB-mediated cytotoxicity on cultured Vero cells when compared to Merck antibodies (Fig. 1, B and C). Moreover, Fc-ABAB exhibited 1000-fold more potency than a mixture of Merck anti-TcdA and anti-TcdB in neutralizing the two toxins in mice after intraperitoneal challenge with a lethal dose of mixed TcdA and TcdB (Fig. 1D). Thus, ABAB had potent neutralizing antitoxin activities against cytotoxicity of the two toxins. Next, our strategy focused on delivering ABAB to the site of infection in the GI tract using a probiotic, rather than through parenteral injections.
Fig. 1. Design and optimization of ABAB secretion in yeast.

(A) Schematic of ABAB. VHHs AH3 and AA6 (blue boxes) target neutralizing epitopes in TcdA; VHHs 5D and E3 (red boxes) target neutralizing epitopes in TcdB. Individual VHHs were joined by glycine-serine linkers. (B and C) In vitro neutralizing activities of Fc-ABAB. TcdA [10 ng/ml (B)] or TcdB [10 pg/ml (C)] was mixed with the indicated concentrations of Merck anti-TcdA (B) or Merck anti-TcdB (C) or Fc-ABAB for 30 min before addition to Vero cell monolayers. Percentage of cell rounding was determined by phase-contrast microscopy. (D) In vivo neutralization by Fc-ABAB and Merck antibodies in a prophylactic murine systemic toxin challenge model. Mice were injected intraperitoneally with PBS, Merck anti-TcdA and anti-TcdB mixture (10 mg/kg each), or Fc-ABAB (10 or 100 μg/kg) and followed by intraperitoneal injection of mixed TcdA and TcdB (1 μg/kg each) 4 hours later. Mouse survival was monitored. P = 0.0041, PBS versus Merck antibodies; P = 0.0015, PBS versus Fc-ABAB at 10 μg/kg; P = 0.0015, PBS versus Fc-ABAB at 100 μg/kg; P = 0.1176, Merck antibodies versus Fc-ABAB at 10 μg/kg; P = 0.0026, Merck antibodies versus Fc-ABAB at 100 μg/kg; P = 0.0025, Fc-ABAB at 10 μg/kg versus Fc-ABAB at 100 μg/kg. (E and F) Relative ABAB secretion in yeast culture supernatant with different secretion signals. E/O value is defined as ELISA optical density (OD)/Optical cell density. (G) Codon optimization for enhanced secretion (y: yeast codon optimized). All in vivo experimental data represent one of at least three separate experiments with n = 5 mice per group and are presented as means ± SEM. Statistical analysis was performed by comparing the indicated groups using Wilcoxon signed-rank tests (B and C), one-way ANOVA (E), or a two-tailed Mann-Whitney test (F and G). Mouse survival (D) was analyzed by Kaplan-Meier survival curves. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
S. boulardii is an ideal probiotic for GI delivery of therapeutic proteins to promote intestinal health. Although no report has used S. boulardii to deliver therapeutic antibodies, this eukaryotic yeast has several advantages in this regard, such as an excellent safety record in humans, baseline probiotic efficacy against CDI, and the capability to be used concurrently with antibiotics (35). Because genetic tools are readily available for S. cerevisiae but less so for S. boulardii (51), we first used S. cerevisiae to test the secretion of tetra-specific VHH ABAB under a full α-mating factor secretion signal. Expression of ABAB was confirmed by Western blotting (fig. S1A), and the neutralizing activities of ABAB against both TcdA and TcdB were verified by a cell-based neutralization assay (fig. S1B). Because ABAB simultaneously binds to TcdA and TcdB, we used a sandwich enzyme-linked immunosorbent assay (ELISA) to measure its secretion by the engineered yeast (fig. S1, C and D). Although α-mating factor is commonly used, studies have shown that other secretion signals from inulinase or invertase could be more suitable for secreting certain heterologous proteins (52, 53). Thus, we genetically fused ABAB with a panel of secretion signals to identify the one best suited for ABAB secretion. The two best secretion signals for ABAB in S. cerevisiae were minimal α-mating factor (AT) and invertase (IV) (Fig. 1E). Next, we examined ABAB secretion in S. boulardii (Sb) under AT and IV signals and found that the AT secretion signal provided a higher amount of secretion, and therefore, we selected it for use in all subsequent constructs (Fig. 1F). In addition, we performed yeast codon optimization (yABAB), which enhanced ABAB secretion in S. boulardii nearly fourfold (Fig. 1G).
Construction of a clinically relevant lead strain for ABAB delivery
For clinical application of ABAB-expressing S. boulardii, antibiotic resistance genes on expression plasmids are not ideal because they potentiate the risk of spreading antibiotic resistance after GI administration. Hence, we generated a uracil auxotrophic strain of S. boulardii and a complementary plasmid for ABAB delivery. To implement this strategy, we optimized the transformation conditions for S. boulardii, which is 100-fold more difficult to transform than S. cerevisiae (figs. S2 to S4). Next, we used two homologous recombination events to delete both chromosomal URA3 alleles (figs. S5 to S8). The URA3−/− S. boulardii strain was validated by the formation of colonies on minimal synthetic media plates supplemented with uracil (MSM + uracil), but not on minimal plates without uracil (MSM) or yeast extract–peptone–dextrose (YPD) plates containing antibiotics (Fig. 2A). Concurrently, the plasmid harboring the AT-yABAB expression cassette was modified to carry a URA3 selection cassette instead of antibiotic resistance genes, and we also generated a control, empty plasmid encoding only URA3 (EP). Both plasmids were then transformed into the uracil auxotrophic S. boulardii strain to generate a lead strain for ABAB delivery (Sb-ABAB) and the control strain (Sb-EP). Western blotting of supernatant from the Sb-ABAB culture detected the corresponding ABAB band as expected (Fig. 2B). Sb-ABAB secreted fully functional ABAB, comparable to Fc-ABAB, as verified by a cell-based neutralization assay (Fig. 2C). We selected an optimal clone by in vitro screening individual Sb-ABAB clones that had comparable growth to the wild-type (Sb) and Sb-EP control strains (Fig. 2D) and stably produced ABAB over multiple passages (Fig. 2E).
Fig. 2. Construction of clinically relevant lead strain Sb-ABAB.
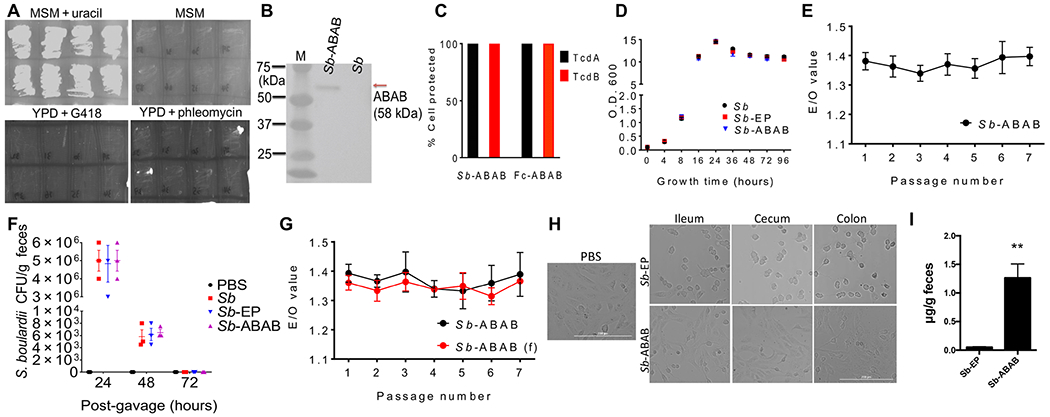
(A) Phenotype of the S. boulardii uracil auxotroph on MSM containing uracil and YPD media containing G418. (B) ABAB antibody in Sb-ABAB culture supernatant detected by Western blotting. (C) Toxin-neutralizing activity of Sb-ABAB culture supernatant compared with purified Fc-ABAB. (D) In vitro growth of Sb, Sb-EP, and Sb-ABAB. (E) ABAB expression in Sb-ABAB culture supernatants over multiple passages. (F) Persistence of Sb, Sb-EP, and Sb-ABAB in antibiotic cocktail–treated mice. (G) ABAB expression in Sb-ABAB culture supernatants over multiple passages in vitro for yeast recovered from fecal (f) samples in (F) versus nonpassaged strains from frozen master stocks. (H) Neutralizing activity of intestinal lavages from mice treated with Sb-EP and Sb-ABAB. (I) ABAB expression in feces from mice treated with Sb-EP and Sb-ABAB. **P < 0.01. Experimental data from in vivo studies represent one of at least three separate experiments with n = 3 mice per group and are presented as means ± SEM.
To examine the in vivo properties of Sb-ABAB and ascertain the highest tolerable dose, we orally gavaged 1010 colony-forming units (CFU) of yeast into antibiotic-treated mice (fig. S9). Mice that were gavaged with Sb, Sb-EP, or Sb-ABAB gained similar amounts of weight as placebo [phosphate-buffered saline (PBS)]–treated mice (fig. S10), and no appearance of adverse effects, such as weight loss, lethargy, hunched posture, ruffled coat, anorexia, or diarrhea, was observed throughout the study. In addition, these strains shared comparable persistence patterns in these mice (Fig. 2F), and Sb-ABAB recovered from mouse feces continued to stably secrete ABAB at amounts similar to the nonpassaged frozen master stock strain (Fig. 2G). Ileal, cecum, and colon lavages collected from Sb-ABAB–treated mice, but not Sb-EP–treated mice, were able to block TcdB-induced cytotoxicity in cultured cells (Fig. 2H). Last, ABAB was detected in the feces of mice dosed with Sb-ABAB, but not Sb-EP (Fig. 2I). These data demonstrated that oral Sb-ABAB led to stable production of ABAB in mouse intestines. Because a dose of 1010 CFU of yeast in 200 μl of PBS is viscous and difficult to gavage, we chose 109 CFU of yeast per dose per day for subsequent preclinical evaluations.
Prophylactic efficacy of Sb-ABAB against CDI in mice and hamsters
Patients who undergo antibiotic treatment, either in preparation for a medical procedure or to treat an ongoing infection, are at high risk of contracting CDI; however, no suitable prophylactics are currently available. Therefore, we tested prevention of CDI by Sb-ABAB in a mouse model featuring antibiotic treatment before challenge with C. difficile spores (fig. S11). Mice were given yeast daily for 7 days starting 3 days before spore challenge. Mice orally dosed with Sb-ABAB were significantly protected from death compared to mice that received either PBS or Sb-EP (P < 0.05) (Fig. 3A). At intestinal mucosa, control mice treated with either PBS or Sb-EP developed typical CDI-associated inflammation and tissue damage, including pseudomembranous colitis, edema, crypt injury, and severe neutrophil infiltration into colonic tissue (Fig. 3B) as well as ceca (fig. S12A), whereas tissues of Sb-ABAB–treated mice had normal or relatively minor observable inflammatory pathology (Fig. 3B and fig. S12A). The average histological scores of colonic tissues (Fig. 3C) or ceca (fig. S12B) from Sb-ABAB–treated mice were significantly lower than those of control mice (P < 0.05). Consistent with the histological results, tissues from mice that received Sb-ABAB also had significantly less myeloperoxidase (MPO) activity, which is indicative of neutrophil infiltration (P < 0.05) (Fig. 3D and fig. S12C) and lower mRNA expression of proinflammatory cytokines (P < 0.05) (Fig. 3, E to G, and fig. S12, D to F), demonstrating a substantial reduction in inflammation in their cecum and colonic tissues compared with those from control mice. Moreover, more C. difficile were recovered from control fecal samples (Fig. 3H), and higher toxin cytotoxicity titers were observed in the feces from control groups compared with Sb-ABAB–treated mice (Fig. 3I), although similar numbers of S. boulardii were recovered from groups of mice that received either Sb-ABAB or Sb-EP (Fig. 3J). These results demonstrate that Sb-ABAB is effective in preventing CDI in antibiotic-treated mice.
Fig. 3. Prophylactic efficacy of Sb-ABAB against CDI in mice.
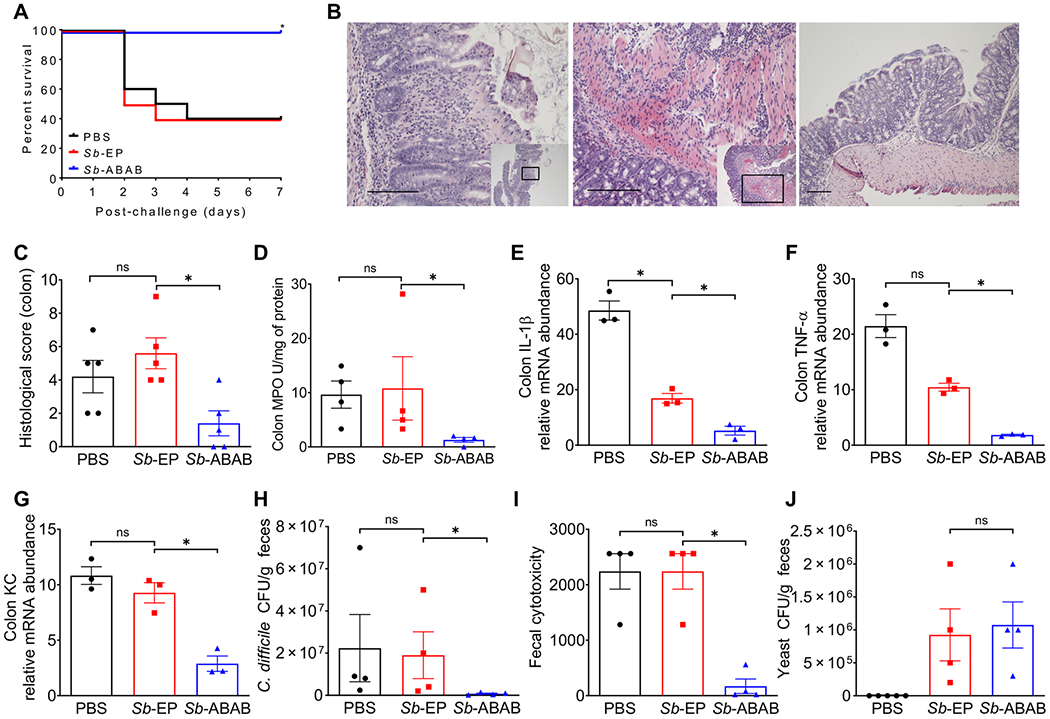
Antibiotic cocktail–treated mice were given Sb-ABAB 3 days before C. difficile spore challenge. (A) Kaplan-Meier survival curves of mice treated with Sb-ABAB compared with controls treated with Sb-EP or PBS. (B) Hematoxylin and eosin (H&E)–stained colonic tissue sections. The insets show where in the tissue the magnified section is located. Left to right: Crypt loss in a representative section from the PBS-treated group (200×), neutrophil infiltration into serosa in a representative section from the Sb-EP–treated group (200×), and a representative tissue sectioning from the Sb-ABAB–treated group (100×). Scale bars, 100 μm. (C) Histological scores for H&E-stained colonic tissue sections. (D) Myeloperoxidase (MPO) assay as a measure of neutrophil infiltration. (E to G) mRNA expression of proinflammatory cytokines interleukin-1β (IL-1β), tumor necrosis factor–α (TNF-α), and cytokine-induced neutrophil-attracting chemokine (KC) in colonic tissues. (H) Fecal C. difficile count from challenged mice. (I) Fecal toxin titers from challenged mice. (J) Fecal S. boulardii CFU counts from prophylactically treated mice. All experimental data in this figure represent one of at least three separate experiments with 10 mice per group (A) or 4 mice per group (C to J) and are presented as means ± SEM. Statistical analysis was performed by comparing the indicated groups using a log-rank (Mantel-Cox) test (A) or a two-tailed Mann-Whitney test (C to J). *P < 0.05; ns, not significant.
Before the establishment of a mouse CDI model (54), hamsters were widely used to evaluate candidate immunotherapies (33, 55). We therefore tested whether oral Sb-ABAB protected hamsters from C. difficile spore challenge. Compared with control animals that were gavaged with Sb-EP, hamsters that were treated with Sb-ABAB only showed a marginal increase of survival (fig. S13). All hamsters died within 7 days of C. difficile spore challenge. Because hamsters are extremely sensitive to CDI and the animals develop toxemia and die rapidly (56), an orally administrated neutralizing antibody delivered by S. boulardii may not be effective in neutralizing the toxins in the bloodstreams of hamsters.
Sb-ABAB shows therapeutic efficacy against primary and recurrent CDI
The antibiotics comprising standard CDI treatment are not optimal because they can allow for CDI recurrence upon cessation. Therefore, we examined the efficacy of Sb-ABAB for treating ongoing CDI and for prevention of recurrence. In a primary acute CDI model (fig. S14), mice rapidly developed CDI symptoms 8 to 12 hours after C. difficile spore challenge and started dying within 48 hours (23, 54). Mice gavaged with Sb-ABAB after spore challenge exhibited a significantly lower mortality rate compared with mice treated with either PBS or Sb-EP (P < 0.05) (Fig. 4A). Moreover, Sb-ABAB–treated mice recovered faster, gaining significantly more weight a day earlier (P < 0.0001 on day 3 and P = 0.021 on day 4 compared to Sb-EP; Fig. 4B) and experiencing fewer diarrheal incidents compared to groups of mice treated with Sb-EP (Fig. 4C). Mice treated with Sb-ABAB had less tissue damage (fig. S15A) and inflammation (fig. S15B) in cecum tissues. Although differences in C. difficile fecal counts were not significant between groups (fig. S15C), significantly lower toxin titers were detected in the feces of Sb-ABAB–treated mice compared with control Sb-EP or PBS treatments (P < 0.05) (fig. S15D). We further examined the efficacy of Sb-ABAB in a recurrent CDI model that mimics a standard clinical course of vancomycin (fig. S16). Consistent with a previous report (57), without interventions, mice developed recurrent CDI after withdrawal of vancomycin (Fig. 4, D to F). Mice that received either PBS or the control Sb-EP strain also developed typical recurrent CDI symptoms such as diarrhea, weight loss, and death, with most weight lost 4 to 5 days after vancomycin withdrawal (Fig. 4, D to F). However, mice that received Sb-ABAB were significantly protected against recurrence-induced death (P < 0.05) (Fig. 4D), suffered no appreciable weight loss (P < 0.0001 on day 4 and P = 0.001 on day 5 compared to Sb-EP; Fig. 4E), and had fewer incidents of diarrhea (Fig. 4F). These results indicate that Sb-ABAB ameliorates ongoing CDI and reduces incidences of CDI recurrence.
Fig. 4. Therapeutic efficacy of Sb-ABAB against primary and recurrent CDI in mice.

(A to C) Primary CDI model: After C. difficile spore challenge, mice were orally dosed daily with yeast for 4 days. (D to F) Recurrent CDI model; mice were concurrently given vancomycin and yeast 1 day after C. difficile spore challenge. (A and D) Kaplan-Meier survival curves of mice treated with Sb-ABAB compared with controls treated with Sb-EP or PBS. (B and E) Relative weight change and (C and F) percentage of mice that experienced diarrhea after spore challenge. The experiment was repeated twice with n = 10 mice per group. Data are presented as means ± SEM. Statistical analysis was performed by comparing the indicated groups using Fisher’s exact test (A and D) or two-way ANOVA (B, C, E, and F). *P < 0.05, ***P < 0.001, and ****P < 0.0001.
DISCUSSION
Antibiotic resistance is a global public health crisis (58, 59). The recent rapid increase in the incidence of CDI and the associated disease severity stemming from the emergence of hypervirulent and antibiotic-resistant strains is alarming (60). The use of antibiotics is a major risk factor for CDI, and hence, the current standard antibiotic treatments for CDI are not ideal and induce high rates of recurrent CDI approaching up to 35% (61–63). Ideal CDI therapeutics should inhibit CDI pathogenesis or bacterial proliferation without disturbing gut microbiota. Antibody-based strategies targeting the major C. difficile virulence factors have been developed and represent a valid alternative to antibiotics, because these antitoxin treatments should not induce dysbiosis or antibiotic resistance (64, 65). Merck is leading the effort of developing antitoxin therapies, and its bezlotoxumab (anti-TcdB) has been approved by the FDA for use concurrently with standard antibiotics for preventing recurrence (66); its actoxumab (anti-TcdA) alone displayed no effect on lowering CDI recurrence and, in combination with bezlotoxumab, did not improve on the results of bezlotoxumab alone (10). Although the reason for this result is unclear, the observed, relatively low efficacy of this therapeutic antibody may be linked to its narrow specificity or low neutralizing activity. To overcome these potential deficiencies, several companies have used a cocktail of antibodies to target multiple distinctive epitopes, showing improved efficacies over Merck’s antibodies (67–70). Unfortunately, this approach would appreciably increase the complexity of development and costs. We have designed a tetra-specific, toxin-neutralizing molecule ABAB that is a single 58-kDa molecule that targets four distinctive toxin epitopes, is 1000-fold more potent than bezlotoxumab and actoxumab combined, and broadly neutralizes toxins from a variety of clinical C. difficile isolates.
It is desirable to deliver antitoxins to intestines where C. difficile colonizes and produces the toxins. Our probiotic yeast-based immunotherapy allows the delivery of highly potent antitoxin ABAB to the site of CDI in contrast to parenterally infused purified antibodies. Moreover, oral delivery is more convenient and reduces cost because parenteral intravenous administration requires trained personnel and healthcare facilities. There are other less costly alternatives to antibody injections, such as FMT or other bacteria-based biotherapeutics, but they cannot be concurrently coadministered with antibiotics. In contrast, S. boulardii can be taken concurrently with vancomycin or metronidazole. In our therapeutic efficacy studies, mice recovered faster and experienced much less recurrence when Sb-ABAB was administered concurrently with vancomycin, making S. boulardii an ideal delivery vehicle in the context of the current standard antibiotic therapy for CDI.
The use of S. boulardii as a delivery vehicle offers several potential advantages for CDI therapeutics: clinical safety, probiotic effects, cost-effective to manufacture, and ease of administration. Although S. boulardii is generally used as diet supplement for healthy populations, because of the increasing incidence and severity of CDI, S. boulardii has been prescribed to reduce the rate of recurrent CDI in patients. S. boulardii was shown to help patients with CDI (35, 36), and we observed that the control strain Sb-EP did elicit a decrease in some proinflammatory cytokines. However, such effects were not sufficient to protect the mice from disease symptoms. A possible reason for the insufficient protection from disease symptoms seen in the Sb-EP group is that the hypervirulent C. difficile strain used here produces severe disease symptoms or that the parental S. boulardii strain that we used is not the same as those used in the clinical trials. Despite the advantages of using engineered S. boulardii as therapeutic vehicles, in particular individuals, treatment with S. boulardii should be avoided or well managed because previous reports showed that S. boulardii might cause fungal infections in certain patient populations (71, 72).
Currently, S. boulardii has not been used for delivering therapeutic proteins. Only now the molecular genetic tools, previously developed for use in S. cerevisiae, are being used for S. boulardii (47, 49, 73). Because the FDA no longer supports inclusion of antibiotic genes, and also to avoid fungal microscale evolution under antibiotic pressure (74), we generated our own auxotrophic S. boulardii strain through homologous recombination. The ultraviolet random mutagenesis approach (47, 49) and CRISPR-Cas9–based (51, 75, 76) approach had been previously used in S. boulardii to generate auxotrophic strains. However, both approaches typically generate point mutations or short deletions that could potentially allow phenotypic reversion. We adapted existing homologous recombination strategies from S. cerevisiae and, considering its sporulation-deficient phenotype (77), developed an auxotrophic S. boulardii strain. We then optimized the ABAB expression and secretion to generate a lead strain. Sb-ABAB stably expressed fully functional ABAB both in vitro and in vivo because functional ABAB was detected in mouse intestines and feces. In addition, Sb-ABAB colonies recovered from mouse feces continued to express ABAB at quantities similar to those before inoculation in the mouse. In vivo persistence and in vitro growth patterns were similar among wild-type S. boulardii, Sb-EP, and Sb-ABAB, suggesting that the engineering of S. boulardii did not alter the wild-type persistence phenotype and that the expression of ABAB did not cause considerable metabolic burden in the strains that were chosen. These strains were safe when given to the mice that received antibiotics, because the mice remained free of clinical signs of illness and maintained steady weight gain. In this study, we created an S. boulardii strain capable of consistently producing therapeutic proteins and optimized expression of our antitoxin ABAB for a clinically relevant strain to deliver therapeutics in situ to either prevent or treat ongoing CDI.
CDI causes immense suffering, and currently, there is no approved preventative approach. Hospitalized patients undergoing antibiotic treatment are at high risk of CDI. The toxoid vaccine previously under phase 3 clinical trial required multiple immunizations and about 2 months for a robust immune response (15) and was thus not ideal for protecting high-risk patients from an imminent threat of CDI. Antibody-based therapy such as Zinplava (Merck’s bezlotoxumab) is expensive and impractical for use as prevention against primary CDI. In our prophylactic efficacy study, mice administered Sb-ABAB after antibiotic treatment but before C. difficile challenge were protected against CDI-induced deaths, had substantially less CDI-related histopathology, and reduced MPO units and down-regulation of mRNA expression of proinflammatory cytokines in their cecal and colonic tissues. In addition, less C. difficile were recovered from their fecal samples, suggesting that Sb-ABAB reduces inflammation and promotes faster recovery of normal microbiota, which, in turn, increases host resistance to C. difficile colonization (78–81). Because S. boulardii is more economical to manufacture and easier to administer than therapeutic antibodies, the yeast immunotherapy would be more likely used for protecting high-risk patients from CDI.
In this study, we found that oral Sb-ABAB failed to protect hamsters from severe CDI and death. This is somewhat expected because hamster CDI is extremely acute and severe, and all our animals died within days of spore inoculation. Many hamsters died without the appearance of any typical clinical symptoms such as weight loss and diarrhea. In our previous study, we found that C. difficile toxins rapidly entered into blood circulation in hamsters after spore inoculation (56), which is likely the major cause of animal death of severe systemic CDI (82). Therefore, delivering neutralizing antibody to hamster intestines would be less effective to protect from death induced by toxins leaking into the blood circulation. Although the hamster model had been the only available C. difficile challenge model for decades before the successful development of mouse CDI models and has been used extensively, we believe that the mouse primary (54) and recurrent (57) CDI models resemble more closely the clinical causes of human disease and are better models to evaluate oral Sb-ABAB therapy against the disease.
There are limitations to the current study. The physiology of mouse intestines is appreciably different from that of humans. Therefore, the doses, dosing schedules, administration and subsequent S. boulardii colonization, and antitoxin secretion may be different for humans. In this study, we used 109 CFU of yeast per gavage and found that S. boulardii was not detectable 3 days after gavage, consistent with data showing that the yeast is transiently colonized in humans for 3 to 5 days (43, 83). The Sb-ABAB colonies recovered from mouse feces maintained their ability to produce ABAB, suggesting that the ABAB expression plasmids were stable in the yeast cells at least during the period of the gastrointestinal passage. Fecal yeast CFU counts were reduced, suggesting that most of the live yeast did not survive the stomach after gavage. However, we envision that future yeast products for human use would be enteric-coated capsules to protect the live yeast from stomach acidity and would only release the yeast in the lower GI tract. In this case, we expect that the efficacy of a therapeutic dose of Sb-ABAB would be higher if the yeast were administered as oral capsules. Another limitation is that it is difficult to compare therapeutic efficacy of our approach with parenterally delivered antitoxin antibodies. Although systemically delivered neutralizing antibody bezlotoxumab has been proven efficacious in clinical trials, the underlying mechanism of action is not entirely clear. In this study, Sb-ABAB groups were substantially protected in both prophylactic and therapeutic models as compared with groups of mice gavaged with S. boulardii carrying an empty plasmid, suggesting that the amount of ABAB secreted by Sb-ABAB was sufficient to neutralize intestinal TcdA and TcdB and block toxin-induced pathogenesis. Therapeutic doses, dosing schedules, and efficacy in humans, however, need to be determined in clinical trials.
In summary, we described an oral yeast-based immunotherapeutic approach using probiotic S. boulardii engineered to secrete a tetra-specific antitoxin that potently and broadly neutralized the two major virulence factors of C. difficile at the site of infection in preclinical models. We demonstrated the utility of this approach in preventing and treating CDI in these clinically relevant animal models. Standard antibiotics have several advantages such as broad effectiveness against different clinical isolates, reasonable cost to patients, and easy oral administration. Sb-ABAB offers the same advantages, but unlike standard antibiotic therapy, it does not disrupt the intestinal microbiota and subsequently reduces the recurrence rate and is thus an attractive potential therapeutic agent in the fight against the challenge of CDI.
MATERIALS AND METHODS
Study design
The overall objective of this study was to produce a clinically relevant yeast immunotherapy, Sb-ABAB, and determine its efficacy in treating and preventing CDI. Mice and hamsters were administered antibiotics to disrupt the microbiota and then challenged with C. difficile. Animals were gavaged with the yeast immunotherapy or controls for several days either before or after C. difficile challenge. Mouse weights, diarrhea score, and survival were monitored. Mouse fecal samples were analyzed to assess C. difficile colonization and toxin titers, as well as S. boulardii colonization and persistence. Histology, MPO, and cytokine expression in mouse samples were analyzed to assess severity of infection and inflammation. End points were determined by the recovery of mice to their prechallenge weight. Hamsters were monitored for survival. Animals were randomly assigned to treatment groups. Sampling and replicates differed between experiments and are stated in the figure legends. Blinding was carried out for histological samples. No data were excluded.
In vitro neutralizing assays
ABAB-neutralizing activity was determined by a cell-based neutralization assay as previously described (23). To assess the ability of ABAB from Sb-ABAB culture supernatants to neutralize toxins from a broad range of clinical isolates, we used two collections of C. difficile strains. Most of the first collection (table S1, 14 strains) was provided by T. Lawley, except for UK1 that was provided by D. Gerding; this represents an assortment of genetically and geographically diverse clinical isolates from Europe (84, 85). The second collection (table S2, 50 strains), from the Centers for Disease Control and Prevention, was obtained through Biodefense and Emerging Infections (BEI) Resources, National Institute of Allergy and Infectious Diseases, National Institutes of Health; isolates were selected to represent the diversity of strain types and geographical locations circulating in the United States during 2010–2011 (https://www.cdc.gov/hai/eip/clostridium-difficile.html). The C. difficile strains were cultured under anaerobic conditions for 2 days, and supernatants were collected and diluted 100 times in cell culture medium before being applied to Vero cell monolayers in a 96-well plate. The supernatants caused cell rounding within 4 hours or after overnight incubation. In some wells, 10 μl of Sb-ABAB supernatant (final dilution 10×) was added to the monolayers before applying C. difficile culture supernatants, and cell rounding was monitored after overnight culture. Neutralization was defined as protection of 100% of the cells from C. difficile toxin–induced cell rounding. To determine the neutralization activity of ABAB secreted in animal intestines, the intestinal lavages from ilea, ceca, and colons were diluted with PBS (10×) and filtered before mixing with TcdB (final concentration was 10 pg/ml) and applying to Vero cell monolayers in a 96-well plate. After 24 hours of incubation, cell rounding was observed by phase-contrast microscopy.
Mouse systemic toxin challenge
CD-1 mice (five per group) were administered PBS, Merck antibodies, or Fc-ABAB by intraperitoneal injection 4 hours before intraperitoneal challenge with a mixture of TcdA and TcdB (1 μg/kg of each toxin). Both purified recombinant toxins were from the VPI 10463 strain (86). Mice were monitored hourly for signs of illness, including hunched posture, ruffled coat, and rapid breathing. Animals that became moribund were euthanized.
Mouse primary and recurrent CDI models
C57BL/6 mice (10 per group) were challenged with 105 spores of UK1 [027/B1/NAP1 strain (23, 50)], which were generated in the laboratory. Both primary and recurrent models have been described previously (23, 54, 57, 87), and the timeline of each is outlined in figs. S11, S14, and S15. The yeast dosage was 109 CFU per mouse per day. For prevention of primary infection, mice were treated daily with yeast starting 3 days before spore challenge for a total of 7 days. For treatment of primary infection, mice were treated with yeast at 6, 24, 48, and 72 hours after spore challenge. For prevention of recurrent infection, mice were treated daily with yeast 1 day after spore challenge for a total of 13 days. Four mice were sacrificed on day 3 or 4 after challenge from each group of mice in the primary CDI prevention study. Before sacrifice, mouse fecal samples were collected. After sacrifice, intestinal lavages from ilea, ceca, and colons were collected. Cecum and tissue samples were collected and appropriately prepared for histology, MPO, and proinflammatory cytokine mRNA studies as described in Supplementary Materials and Methods.
Hamster CDI model
Golden Syrian hamsters (five per group) were treated with clindamycin (30 mg/kg) 1 day before challenge with 104 spores of UK1 on day 0. Hamsters were orally gavaged with 1010 CFU of yeast daily from day −3 to day 7, as outlined in fig. S13A. Hamsters were monitored for survival.
Statistical analysis
Data were analyzed by Fisher’s exact test, Wilcoxon signed-rank tests, and one-way or two-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test using the Prism statistical software program. Mouse survival was analyzed by Kaplan-Meier survival curve. P < 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We thank J. E. Heath and S. Shyu for assistance with histopathological analysis.
Funding: This study was supported by the NIH (U19AI142725 and U19AI109776 to H.F. and R01AI132207 and R43AI129044 to Z.Y.).
Footnotes
stm.sciencemag.org/cgi/content/full/12/567/eaax4905/DC1
Materials and Methods
Fig. S1. ABAB secretion in S. cerevisiae.
Fig. S2. Transformation competency of S. cerevisiae and S. boulardii.
Fig. S3. Diagram of optimization procedure for S. boulardii transformation.
Fig. S4. Transformation of yeasts under different conditions.
Fig. S5. Diagram of targeted deletion of URA3 loci by homologous recombination in chromosome V of S. boulardii.
Fig. S6. Phenotypic characteristics of S. boulardii URA3Δ::aphA1/Δ::ble.
Fig. S7. PCR validation of S. boulardii URA3Δ::aphA1/Δ::ble.
Fig. S8. PCR validation of pPL5071 spin-off and curing of 2-μm plasmid from S. boulardii URA3−/−.
Fig. S9. Diagram of S. boulardii tolerance assessment in antibiotic-treated mice.
Fig. S10. Relative weight change after oral administration of S. boulardii to antibiotic-treated mice.
Fig. S11. Diagram of oral administration of Sb-ABAB for CDI prevention in mice.
Fig. S12. Cecal histology and inflammation in mice in the CDI prevention study.
Fig. S13. Efficacy of oral Sb-ABAB against CDI in hamsters.
Fig. S14. Diagram of oral administration of Sb-ABAB for treating primary CDI.
Fig. S15. Intestinal histopathology, inflammation, and fecal bacterial counting after treatment with Sb-ABAB against primary CDI.
Fig. S16. Diagram of oral administration of Sb-ABAB for preventing recurrent CDI.
Table S1. Tetra-specific ABAB is a broadly neutralizing antibody.
Table S2. A summary of ABAB-neutralizing activity against toxins produced by C. difficile clinical isolates from the Emerging Infections Program Clostridium difficile Surveillance Project.
Data file S1. Raw data.
Competing interests: The technology reported in this study has been under patent application (US2018/0319872A1: Yeast-based immunotherapy against Clostridium difficile infection), and the intellectual property rights have been licensed to FZata Inc. H.F. has been a consultant and chief scientific officer of the company. Z.Y. and H.F. hold equity of the company. H.F., J.G., K.C., and Y. Zhu are listed inventors.
Data and materials availability: All data associated with this study are available in the main text or the Supplementary Materials. Materials are available from H.F. under a material transfer agreement with University of Maryland, Baltimore.
REFERENCES AND NOTES
- 1.Kelly CP, LaMont JT, Clostridium difficile—More difficult than ever. N. Engl. J. Med 359, 1932–1940 (2008). [DOI] [PubMed] [Google Scholar]
- 2.McDonald LC, Owings M, Jernigan DB, Clostridium difficile infection in patients discharged from US short-stay hospitals, 1996-2003. Emerg. Infect. Dis 12, 409–415 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC, Burden of Clostridium difficile infection in the United States. N. Engl. J. Med 372, 825–834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lessa FC, Winston LG, McDonald LC; Emerging Infections Program C difficile Surveillance Team, Burden of Clostridium difficile infection in the United States. N. Engl. J. Med 372, 2369–2370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly CP, Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin. Microbiol. Infect 18 (suppl. 6), 21–27 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Johnson S, Recurrent Clostridium difficile infection: Causality and therapeutic approaches. Int. J. Antimicrob. Agents 33 (suppl. 1), S33–S36 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Leffler DA, Lamont JT, Clostridium difficile infection. N. Engl. J. Med 372, 1539–1548 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Leffler DA, Lamont JT, Clostridium difficile infection. N. Engl. J. Med 373, 287–288 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Abougergi MS, Kwon JH, Intravenous immunoglobulin for the treatment of Clostridium difficile infection: A review. Dig. Dis. Sci 56, 19–26 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Wilcox MH, Gerding DN, Poxton IR, Kelly C, Nathan R, Birch T, Cornely OA, Rahav G, Bouza E, Lee C, Jenkin G, Jensen W, Kim Y-S, Yoshida J, Gabryelski L, Pedley A, Eves K, Tipping R, Guris D, Kartsonis N, Dorr M-B, Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med 376, 305–317 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Kociolek LK, Gerding DN, Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol 13, 150–160 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Rao K, Young VB, Fecal microbiota transplantation for the management of Clostridium difficile infection. Infect. Dis. Clin. North Am 29, 109–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vyas D, Aekka A, Vyas A, Fecal transplant policy and legislation. World J. Gastroenterol 21, 6–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeFilipp Z, Bloom PP, Soto MT, Mansour MK, Sater MRA, Huntley MH, Turbett S, Chung RT, Chen Y-B, Hohmann EL, Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N. Engl. J. Med 381, 2043–2050 (2019). [DOI] [PubMed] [Google Scholar]
- 15.de Bruyn G, Saleh J, Workman D, Pollak R, Elinoff V, Fraser NJ, Lefebvre G, Martens M, Mills RE, Nathan R, Trevino M, van Cleeff M, Foglia G, Ozol-Godfrey A, Patel DM, Pietrobon PJ, Gesser R, Defining the optimal formulation and schedule of a candidate toxoid vaccine against Clostridium difficile infection: A randomized phase 2 clinical trial. Vaccine 34, 2170–2178 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Kotloff KL, Wasserman SS, Losonsky GA, Thomas WJ, Nichols R, Edelman R, Bridwell M, Monath TP, Safety and immunogenicity of increasing doses of a Clostridium difficile toxoid vaccine administered to healthy adults. Infect. Immun 69, 988–995 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sougioultzis S, Kyne L, Drudy D, Keates S, Maroo S, Pothoulakis C, Giannasca PJ, Lee CK, Warny M, Monath TP, Kelly CP, Clostridium difficile toxoid vaccine in recurrent C. difficile-associated diarrhea. Gastroenterology 128, 764–770 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Greenberg RN, Marbury TC, Foglia G, Warny M, Phase I dose finding studies of an adjuvanted Clostridium difficile toxoid vaccine. Vaccine 30, 2245–2249 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Seal D, Borriello SP, Barclay F, Welch A, Piper M, Bonnycastle M, Treatment of relapsing Clostridium difficile diarrhoea by administration of a non-toxigenic strain. Eur. J. Clin. Microbiol 6, 51–53 (1987). [DOI] [PubMed] [Google Scholar]
- 20.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP, The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Lyras D, O’Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI, Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernie DS, Thomson RO, Batty I, Walker PD, Active and passive immunization to protect against antibiotic associated caecitis in hamsters. Dev. Biol. Stand 53, 325–332 (1983). [PubMed] [Google Scholar]
- 23.Wang H, Sun X, Zhang Y, Li S, Chen K, Shi L, Nie W, Kumar R, Tzipori S, Wang J, Savidge T, Feng H, A chimeric toxin vaccine protects against primary and recurrent Clostridium difficile infection. Infect. Immun 80, 2678–2688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steele J, Mukherjee J, Parry N, Tzipori S, Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis 207, 323–330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WDJ, Leney M, Sloan S, Hay CA, Ambrosino DM, Treatment with monoclonal antibodies against Clostridium difficile toxins. N. Engl. J. Med 362, 197–205 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Katchar K, Taylor CP, Tummala S, Chen X, Sheikh J, Kelly CP, Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin. Gastroenterol. Hepatol 5, 707–713 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, Ambrosino D, Molrine D, Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28, 965–969 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Saylor C, Dadachova E, Casadevall A, Monoclonal antibody-based therapies for microbial diseases. Vaccine 27 (suppl. 6), G38–G46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chow S-K, Casadevall A, Monoclonal antibodies and toxins—A perspective on function and isotype. Toxins 4, 430–454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greig SL, Obiltoxaximab: First global approval. Drugs 76, 823–830 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Kufel WD, Devanathan AS, Marx AH, Weber DJ, Daniels LM, Bezlotoxumab: A novel agent for the prevention of recurrent Clostridium difficile infection. Pharmacotherapy 37, 1298–1308 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Yang Z, Ramsey J, Hamza T, Zhang Y, Li S, Yfantis HG, Lee D, Hernandez LD, Seghezzi W, Furneisen JM, Davis NM, Therien AG, Feng H, Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun 83, 822–831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Chen X, Hernandez LD, Lipari P, Flattery A, Chen S-C, Kramer S, Polishook JD, Racine F, Cape H, Kelly CP, Therien AG, Toxin-mediated paracellular transport of antitoxin antibodies facilitates protection against Clostridium difficile infection. Infect. Immun 83, 405–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau CS, Chamberlain RS, Probiotics are effective at preventing Clostridium difficile-associated diarrhea: A systematic review and meta-analysis. Int. J. Gen. Med 9, 27–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Surawicz CM, McFarland LV, Greenberg RN, Rubin M, Fekety R, Mulligan ME, Garcia RJ, Brandmarker S, Bowen K, Borjal D, Elmer GW, The search for a better treatment for recurrent Clostridium difficile disease: Use of high-dose vancomycin combined with Saccharomyces boulardii. Clin. Infect. Dis 31, 1012–1017 (2000). [DOI] [PubMed] [Google Scholar]
- 36.McFarland LV, Surawicz CM, Greenberg RN, Fekety R, Elmer GW, Moyer KA, Melcher SA, Bowen KE, Cox JL, Noorani Z, A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 271, 1913–1918 (1994). [PubMed] [Google Scholar]
- 37.Pothoulakis C, Kelly CP, Joshi MA, Gao N, O’Keane CJ, Castagliuolo I, Lamont JT, Saccharomyces boulardii inhibits Clostridium difficile toxin A binding and enterotoxicity in rat ileum. Gastroenterology 104, 1108–1115 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Castagliuolo I, LaMont JT, Nikulasson ST, Pothoulakis C, Saccharomyces boulardii protease inhibits Clostridium difficile toxin A effects in the rat ileum. Infect. Immun 64, 5225–5232 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C, Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect. Immun 67, 302–307 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qamar A, Aboudola S, Warny M, Michetti P, Pothoulakis C, LaMont JT, Kelly CP, Saccharomyces boulardii stimulates intestinal immunoglobulin A immune response to Clostridium difficile toxin A in mice. Infect. Immun 69, 2762–2765 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, Kokkotou EG, Mustafa N, Bhaskar KR, Sougioultzis S, O’Brien M, Pothoulakis C, Kelly CP, Saccharomyces boulardii inhibits ERK1/2 mitogen-activated protein kinase activation both in vitro and in vivo and protects against Clostridium difficile toxin A-induced enteritis. J. Biol. Chem 281, 24449–24454 (2006). [DOI] [PubMed] [Google Scholar]
- 42.McFarland LV, Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World J. Gastroenterol 16, 2202–2222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elmer GW, McFarland LV, Surawicz CM, Danko L, Greenberg RN, Behaviour of Saccharomyces boulardii in recurrent Clostridium difficile disease patients. Aliment. Pharmacol. Ther 13, 1663–1668 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Czerucka D, Piche T, Rampal P, Review article: Yeast as probiotics—Saccharomyces boulardii. Aliment. Pharmacol. Ther 26, 767–778 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Gorlani A, de Haard H, Verrips T, Expression of VHHs in Saccharomyces cerevisiae. Methods Mol. Biol 911, 277–286 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Fietto JLR, Araujo RS, Valadao FN, Fietto LG, Brandao RL, Neves MJ, Gomes FCO, Nicoli JR, Castro IM, Molecular and physiological comparisons between Saccharomyces cerevisiae and Saccharomyces boulardii. Can. J. Microbiol 50, 615–621 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Hudson LE, Fasken MB, McDermott CD, McBride SM, Kuiper EG, Guiliano DB, Corbett AH, Lamb TJ, Functional heterologous protein expression by genetically engineered probiotic yeast Saccharomyces boulardii. PLOS ONE 9, e112660 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hudson LE, McDermott CD, Stewart TP, Hudson WH, Rios D, Fasken MB, Corbett AH, Lamb TJ, Characterization of the probiotic yeast Saccharomyces boulardii in the healthy mucosal immune system. PLOS ONE 11, e0153351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamedi H, Misaghi A, Modarressi MH, Salehi TZ, Khorasanizadeh D, Khalaj V, Generation of a uracil auxotroph strain of the probiotic yeast Saccharomyces boulardii as a host for the recombinant protein production. Avicenna J. Med. Biotechnol 5, 29–34 (2013). [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Z, Schmidt D, Liu W, Li S, Shi L, Sheng J, Chen K, Yu H, Tremblay JM, Chen X, Piepenbrink KH, Sundberg EJ, Kelly CP, Bai G, Shoemaker CB, Feng H, A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect. Dis 210, 964–972 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J-J, Kong II, Zhang G-C, Jayakody LN, Kim H, Xia P-F, Kwak S, Sung BH, Sohn J-H, Walukiewicz HE, Rao CV, Jin Y-S, Metabolic engineering of probiotic Saccharomyces boulardii. Appl. Environ. Microbiol 82, 2280–2287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rakestraw JA, Sazinsky SL, Piatesi A, Antipov E, Wittrup KD, Directed evolution of a secretory leader for the improved expression of heterologous proteins and full-length antibodies in Saccharomyces cerevisiae. Biotechnol. Bioeng 103, 1192–1201 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang HA, Nam SW, Kwon KS, Chung BH, Yu MH, High-level secretion of human α1-antitrypsin from Saccharomyces cerevisiae using inulinase signal sequence. J. Biotechnol 48, 15–24 (1996). [DOI] [PubMed] [Google Scholar]
- 54.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP, A mouse model of Clostridium difficile-associated disease. Gastroenterology 135, 1984–1992 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD Jr., Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun 74, 6339–6347 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartlett JG, Chang T, Moon N, Onderdonk AB, Antibiotic-induced lethal enterocolitis in hamsters: Studies with eleven agents and evidence to support the pathogenic role of toxin-producing Clostridia. Am. J. Vet. Res 39, 1525–1530 (1978). [PubMed] [Google Scholar]
- 57.Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H, Mouse relapse model of Clostridium difficile infection. Infect. Immun 79, 2856–2864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frieden T, U.S. Centers for Disease Control and Prevention, Antibiotic resistance threats in the United States (2013); https://www.cdc.gov/drugresistance/biggest_threats.html.
- 59.Ventola CL, The antibiotic resistance crisis: Part 1: Causes and threats. P. T 40, 277–283(2015). [PMC free article] [PubMed] [Google Scholar]
- 60.Rupnik M, Wilcox MH, Gerding DN, Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Microbiol 7, 526–536 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Wenisch C, Parschalk B, Hasenhundl M, Hirschl AM, Graninger W, Comparison of vancomycin, teicoplanin, metronidazole, and fusidic acid for the treatment of Clostridium difficile-associated diarrhea. Clin. Infect. Dis 22, 813–818 (1996). [DOI] [PubMed] [Google Scholar]
- 62.Zar FA, Bakkanagari SR, Moorthi KMLST, Davis MB, A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin. Infect. Dis 45, 302–307 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Cornely OA, Miller MA, Louie TJ, Crook DW, Gorbach SL, Treatment of first recurrence of Clostridium difficile infection: Fidaxomicin versus vancomycin. Clin. Infect. Dis 55 (Suppl. 2), S154–S161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lyerly DM, Bostwick EF, Binion SB, Wilkins TD, Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect. Immun 59, 2215–2218 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tjellstrom B, Stenhammar L, Eriksson S, Magnusson KE, Oral immunoglobulin A supplement in treatment of Clostridium difficile enteritis. Lancet 341, 701–702 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Markham A, Bezlotoxumab: First global approval. Drugs 76, 1793–1798 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, Donovan GP, Magargal WW, Maddon PJ, Olson WC, Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J. Infect. Dis 206, 706–713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies NL, Compson JE, Mackenzie B, O’Dowd VL, Oxbrow AKF, Heads JT, Turner A, Sarkar K, Dugdale SL, Jairaj M, Christodoulou L, Knight DEO, Cross AS, Herve KJM, Tyson KL, Hailu H, Doyle CB, Ellis M, Kriek M, Cox M, Page MJT, Moore AR, Lightwood DJ, Humphreys DP, A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin. Vaccine Immunol 20, 377–390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu H, Cassan R, Johnstone D, Han X, Joyee AG, McQuoid M, Masi A, Merluza J, Hrehorak B, Reid R, Kennedy K, Tighe B, Rak C, Leonhardt M, Dupas B, Saward L, Berry JD, Nykiforuk CL, Novel Clostridium difficile anti-toxin (TcdA and TcdB) humanized monoclonal antibodies demonstrate in vitro neutralization across a broad spectrum of clinical strains and in vivo potency in a hamster spore challenge model. PLOS ONE 11, e0157970 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anosova NG, Cole LE, Li L, Zhang J, Brown AM, Mundle S, Zhang J, Ray S, Ma F, Garrone P, Bertraminelli N, Kleanthous H, Anderson SF, A combination of three fully human toxin A- and toxin B-specific monoclonal antibodies protects against challenge with highly virulent epidemic strains of Clostridium difficile in the hamster model. Clin. Vaccine Immunol 22, 711–725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leonardi I, Li X, Semon A, Li D, Doron I, Putzel G, Bar A, Prieto D, Rescigno M, McGovern DPB, Pla J, Iliev ID, CX3CR1+ mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cassone M, Serra P, Mondello F, Girolamo A, Scafetti S, Pistella E, Venditti M, Outbreak of Saccharomyces cerevisiae Subtype boulardii fungemia in patients neighboring those treated with a probiotic preparation of the organism. J. Clin. Microbiol 41, 5340–5343 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Douradinha B, Reis VCB, Rogers MB, Torres FAG, Evans JD, Marques ETA Jr., Novel insights in genetic transformation of the probiotic yeast Saccharomyces boulardii. Bioengineered 5, 21–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, Bennett RJ, Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc. Natl. Acad. Sci. U.S.A 115, E8688–E8697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM, Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41, 4336–4343 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bao Z, Xiao H, Liang J, Zhang L, Xiong X, Sun N, Si T, Zhao H, Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol 4, 585–594 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Edwards-Ingram L, Gitsham P, Burton N, Warhurst G, Clarke I, Hoyle D, Oliver SG, Stateva L, Genotypic and physiological characterization of Saccharomyces boulardii, the probiotic strain of Saccharomyces cerevisiae. Appl. Environ. Microbiol 73, 2458–2467 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barc M-C, Charrin-Sarnel C, Rochet V, Bourlioux F, Sandre C, Boureau H, Dore J, Collignon A, Molecular analysis of the digestive microbiota in a gnotobiotic mouse model during antibiotic treatment: Influence of Saccharomyces boulardii. Anaerobe 14, 229–233 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Swidsinski A, Loening-Baucke V, Verstraelen H, Osowska S, Doerffel Y, Biostructure of fecal microbiota in healthy subjects and patients with chronic idiopathic diarrhea. Gastroenterology 135, 568–579 (2008). [DOI] [PubMed] [Google Scholar]
- 80.Kabbani TA, Pallav K, Dowd SE, Villafuerte-Galvez J, Vanga RR, Castillo NE, Hansen J, Dennis M, Leffler DA, Kelly CP, Prospective randomized controlled study on the effects of Saccharomyces boulardii CNCM I-745 and amoxicillin-clavulanate or the combination on the gut microbiota of healthy volunteers. Gut Microbes 8, 17–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dzunkova M, D’Auria G, Xu H, Huang J, Duan Y, Moya A, Kelly CP, Chen X, The monoclonal antitoxin antibodies (actoxumab-bezlotoxumab) treatment facilitates normalization of the gut microbiota of mice with Clostridium difficile infection. Front. Cell. Infect. Microbiol 6, 119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steele J, Chen K, Sun X, Zhang Y, Wang H, Tzipori S, Feng H, Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J. Infect. Dis 205, 384–391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blehaut H, Massot J, Elmer GW, Levy RH, Disposition kinetics of Saccharomyces boulardii in man and rat. Biopharm. Drug Dispos 10, 353–364 (1989). [DOI] [PubMed] [Google Scholar]
- 84.Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, Riley TV, Songer JG, Kuijper EJ, Dingle KE, Wren BW, Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLOS ONE 7, e31559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HMB, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J, Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. U.S.A 107, 7527–7532 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang G, Zhou B, Wang J, He X, Sun X, Nie W, Tzipori S, Feng H, Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol 8, 192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang Z, Shi L, Yu H, Zhang Y, Chen K, Saint Fleur A, Bai G, Feng H, Intravenous adenovirus expressing a multi-specific, single-domain antibody neutralizing TcdA and TcdB protects mice from Clostridium difficile infection. Pathog. Dis 74, ftw078 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He X, Sun X, Wang J, Wang X, Zhang Q, Tzipori S, Feng H, Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect. Immun 77, 2294–2303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vickers CE, Bydder SF, Zhou Y, Nielsen LK, Dual gene expression cassette vectors with antibiotic selection markers for engineering in Saccharomyces cerevisiae. Microb. Cell Fact 12, 96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gottschling DE, High efficiency LiAc transformation (modified from Gietz); https://research.fhcrc.org/gottschling/en/protocols/yeast-protocols/transformation.html.
- 91.Gietz RD, Schiestl RH, High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 31–34 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Konishi T, Harata M, Improvement of the transformation efficiency of Sacchaaromyces cerevisiae by altering carbon sources in pre-culture. Biosci. Biotechnol. Biochem 78, 1090–1093 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Panchal CJ, Whitney GK, Stewart GG, Susceptibility of Saccharomyces spp. and Schwanniomyces spp. to the aminoglycoside antibiotic G418. Appl. Environ. Microbiol 47, 1164–1166 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gatignol A, Baron M, Tiraby G, Phleomycin resistance encoded by the ble gene from transposon Tn 5 as a dominant selectable marker in Saccharomyces cerevisiae. Mol. Gen. Genet 207, 342–348 (1987). [DOI] [PubMed] [Google Scholar]
- 95.MacDonald C, Piper RC, Puromycin- and methotrexate-resistance cassettes and optimized Cre-recombinase expression plasmids for use in yeast. Yeast 32, 423–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boeke JD, LaCroute F, Fink GR, A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-Fluoro-orotic acid resistance. Mol. Gen. Genet 197, 345–346 (1984). [DOI] [PubMed] [Google Scholar]
- 97.Tsalik EL, Gartenberg MR, Curing Saccharomyces cerevisiae of the 2 micron plasmid by targeted DNA damage. Yeast 14, 847–852 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.