

Abstract
The fragile X-related (FXR) family proteins FMRP, FXR1, and FXR2 are RNA binding proteins that play a critical role in RNA metabolism, neuronal plasticity, and muscle development. These proteins share significant homology in their protein domains, which are functionally and structurally similar to each other. FXR family members are known to play an essential role in causing fragile X mental retardation syndrome (FXS), the most common genetic form of autism spectrum disorder. Recent advances in our understanding of this family of proteins have occurred in tandem with discoveries of great importance to neurological disorders and cancer biology via the identification of their novel RNA and protein targets. Herein, we review the FXR family of proteins as they pertain to FXS, other mental illnesses, and cancer. We emphasize recent findings and analyses that suggest contrasting functions of this protein family in FXS and tumorigenesis based on their expression patterns in human tissues. Finally, we discuss current gaps in our knowledge regarding the FXR protein family and their role in FXS and cancer and suggest future studies to facilitate bench to bedside translation of the findings.
Keywords: FMRP, FXR1, FXR2, protein domains, RNA binding, fragile X mental retardation syndrome, cancer, post-transcriptional gene regulation, post-translational protein modification
Introduction:
The Fragile X-related (FXR) gene family includes three members that encode the highly homologous RNA binding proteins (RBPs) Fragile X mental retardation 1 (FMR1), FMR1 autosomal homolog 1 (FXR1), and FMR1 autosomal homolog 2 (FXR2), and are located on chromosomes Xq27.3, 3q26.33, and 17p13.1, respectively. The proteins share significant structural similarities, with approximately 60% amino acid sequence identity (Siomi, Zhang et al. 1996). The homology shared among the three protein products is especially pronounced over the first 13 exons, which exhibit a 73–90% amino acid sequence identity (Kirkpatrick, McIlwain et al. 2001). However, the C-termini of these proteins varies substantially. Isoform-specific inclusion and exclusion of specific exons proximal to the C-termini contribute dramatically to this variance. The FXR proteins consist of a nonclassical nuclear localization signal localized at its N-terminal region (Eberhart, Malter et al. 1996, Bardoni, Sittler et al. 1997), a nuclear export signal (Eberhart, Malter et al. 1996), two Tudor domains (Bardoni, Sittler et al. 1997, Ramos, Hollingworth et al. 2006, Adams-Cioaba, Guo et al. 2010), three protein K homology (KH) domains (Zhang, O’Connor et al. 1995), and an RGG box (Zhang, O’Connor et al. 1995, Darnell, Jensen et al. 2001, Schaeffer, Bardoni et al. 2001) (Figure 1). The RGG motif is capable of binding to DNA and RNA structures, including G-quadruplex RNAs (Brown, Small et al. 1998, Darnell, Jensen et al. 2001, Darnell, Fraser et al. 2009). In addition to their RNA-binding capacity, they interact with each other and associate with the ribosomal 40S and 60S subunit (Eberhart, Malter et al. 1996, Khandjian, Corbin et al. 1996, Siomi, Zhang et al. 1996, Tamanini, Meijer et al. 1996). The FXR family of proteins also undergo several post-translational modifications like methylation (Say, Tay et al. 2010), phosphorylation (Stetler, Winograd et al. 2006), sumoylation (Khayachi, Gwizdek et al. 2018), and ubiquitinylation (Qie, Majumder et al. 2017).
Figure 1: Comparison of human FXR family proteins.
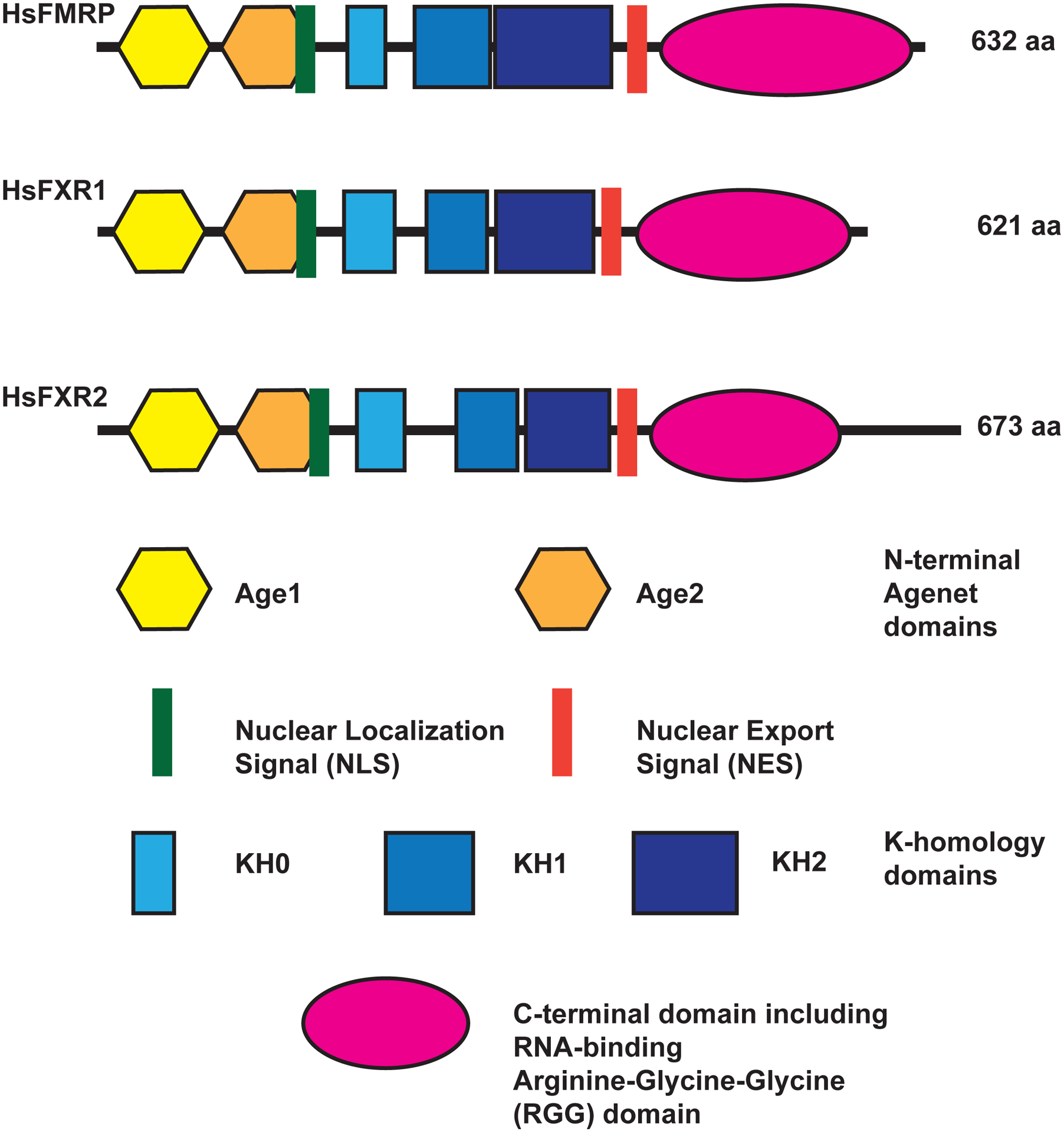
Two N-terminal agenet domains that are often known as protein binding domains. NLS: Nuclear localization signal. Three K-homology domains that are KH0, KH1, and KH2. NES: Nuclear export signal. The C-terminal arginine-glycine-glycine (RGG) RNA-binding domain, known to bind to G-quadruplex RNA structures.
The FXR proteins are found to be highly abundant in the brain where they are mostly localized to the cytoplasm and proximal dendrites of neurons (Bakker, de Diego Otero et al. 2000, Tamanini, Kirkpatrick et al. 2000). Recent studies show that while FMR1 transcripts are primarily present in the neurons and astrocytes, FXR1 and FXR2 transcripts are found in oligodendrocytes, microglia, and endothelial cells in the cortex of mice (Thomsen, Pallesen et al. 2013, Zhang, Chen et al. 2014). Work from the Murai laboratory showed that FXR1 controls local protein synthesis at synapses in the central nervous system as well as in the dendrites and the three homologs exhibit overlying cytoplasmic and dendritic expression patterns in developing and adult mammalian neurons (Cook, Sanchez-Carbente Mdel et al. 2011).
These findings define their expression patterns and functionally link the proteins to the diseases associated with the Central Nervous System (CNS). Transcriptional silencing of the FMR1 gene, due to an unstable hypermethylated CGG repeat expansion in its 5’-untranslated region (UTR), is often linked to human fragile X syndrome and autism (Verkerk, Pieretti et al. 1991). Recent findings also demonstrate that both FXR1 and FMR1 are directly or indirectly linked to schizophrenia, bipolar disorders, and mood regulation, suggesting a broader function of these proteins in adult-onset mental diseases (Del’Guidice, Latapy et al. 2015, Takata, Matsumoto et al. 2017). Genome-wide association studies (GWAS) in 36,989 schizophrenia cases and 113,075 controls have found single nucleotide polymorphisms (SNPs) in the FXR1 locus only, that may possibly contribute to the disease by affecting alternative splicing(Schizophrenia Working Group of the Psychiatric Genomics 2014, Takata, Matsumoto et al. 2017). Significant reduction in FMRP was reported in the lateral cerebella of subjects with schizophrenia, bipolar disorder, and major depression (Fatemi, Kneeland et al. 2010) while no change was observed in the FMR1 levels (Fatemi and Folsom 2015). Moreover, a study of the FMRP regulon from superior frontal cortex (Brodmann Area 9 (BA9)) of subjects with schizophrenia and bipolar disorder found mutations in FMRP targets (Folsom, Thuras et al. 2015). A noteworthy study demonstrated that a GSK3β (glycogen synthase kinase 3 beta) mediated phosphorylation of FXR1 promotes its downregulation and contributes to the regulation of mood and emotional processing (Del’Guidice, Latapy et al. 2015). Notably, patients with significant psychosis disorders show an interaction between SNPs GSK3β rs12630592 and FXR1 rs496250 genotypes (Bureau, Beaulieu et al. 2017). Thus, both FMRP and FXR1, along with GSK3β, are considered therapeutic targets for mental disorders and neurogenerative diseases (Kaytor and Orr 2002, Guo, Murthy et al. 2012, Del’Guidice, Latapy et al. 2015). Several groups in the last decade have used GWAS to link the FXR family proteins to anxiety, bipolar disorders, schizophrenia, and mood regulation pointing toward a greater involvement in mental illnesses (Bontekoe, McIlwain et al. 2002, Hamilton, Esseltine et al. 2014, Renoux, Carducci et al. 2014, Borreca, Gironi et al. 2016, Khlghatyan, Evstratova et al. 2018).
In recent years, FXR1, more so than the two other proteins, has surfaced as an oncogenic protein, or at least a facilitator, that promotes carcinomas in various tissues. Our laboratory and others have observed that FXR1 is capable of promoting tumorigenesis and blocking senescence in lung and head and neck squamous cell carcinoma, respectively (Qian, Hassanein et al. 2015, Majumder, House et al. 2016). Both reports demonstrate that the FXR1 transcript is highly abundant in tumor tissues and correlates with poor prognosis and survival. Several more recent publications support our finding that FXR1 overexpression is a strong predictor of poor prognosis in multiple cancers (Fan, Yue et al. 2017, Jo, Kim et al. 2017, Nordio, Marques et al. 2018, Cao, Gao et al. 2019, Cao, Zheng et al. 2019, Phelps, Pierce et al. 2019). Collectively, these studies demonstrated that high FXR1 expression at the RNA and protein levels promotes oncogenesis, in contrast to the trend observed in FXS, where FXR1 and FMR1 are functionally silenced or suppressed. In this review, we will focus on the structural features of the FXR family proteins, their differential expression in tissues, and the diverse functions of these proteins in several neurological disorders and cancer.
The FXR family proteins
The FMR1 gene was first identified in 1991 (Verkerk, Pieretti et al. 1991) followed by FMRP autosomal homolog 1 (FXR1) (Zhang, O’Connor et al. 1995), and FMRP autosomal homolog 2 (FXR2) (Zhang, O’Connor et al. 1995). FMR1 plays a vital role in fragile X syndrome (FXS), and FXS patients harbor long hypermethylated CGG repeats in the promoter region of the FMR1 gene (Verkerk, Pieretti et al. 1991). Following this observation, FXR1 was identified as a cytoplasmic RNA binding protein (RBP) (Zhang, O’Connor et al. 1995) expressed independently of the FMR1 gene at the mRNA and protein levels. By the use of a yeast two-hybrid system, Zhang et al. identified another FMRP autosomal homolog, FXR2 (Zhang, O’Connor et al. 1995). They showed that FXR2, FXR1, and FMRP form heteromers with each other in addition to the formation of homodimers by themselves. The authors further demonstrated that the KH1 domain or the region between KH1 and KH2 is responsible for the heteromers, drawing attention to the KH domains’ structural features and their implications for a coordinated protein-protein network.
The FXR family protein domains
All three FXR proteins contain two Tudor domains (also known as agenet domains) proximal to the N-terminus (Bardoni, Sittler et al. 1997, Ramos, Hollingworth et al. 2006, Adams-Cioaba, Guo et al. 2010, D’Annessa, Cicconardi et al. 2019, McClure and Palanisamy 2019) (Figure 1). The first Tudor domain spans exons 1–3, while the second spans exons 3–5. These Tudor domains are relatively dissimilar; however, they share high structural similarity to the Tudor domains of the Survival motor neuron protein (SMN) (Ramos, Hollingworth et al. 2006). The Tudor domains of Fragile X proteins serve as sites for protein-protein interaction by recognition of methylated lysine residues of other proteins (Adams-Cioaba, Guo et al. 2010). Subsequent studies demonstrated that Tudor Domain 2 is primarily responsible for interacting with proteins containing methylated lysine residues (Ramos, Hollingworth et al. 2006).
A non-classical Nuclear Localization Sequence (NLS) domain is present between the C-terminus and the Tudor domain (Figure 1). The specific amino acids comprising the NLS domain lie between residues 114 and 150, spanning exons 5 and 6 (Eberhart, Malter et al. 1996, Bardoni, Sittler et al. 1997, D’Annessa, Cicconardi et al. 2019, McClure and Palanisamy 2019). Despite the presence of this NLS, the protein products of FMRP, FXR1, and FXR2 are predominantly in the cytoplasm, suggesting that alternative sequences present in the proteins prevent nuclear accumulation (Verheij, Bakker et al. 1993, Zhang, O’Connor et al. 1995). The discovery by Adinolfi et al. of the propensity of an area overlapping the NLS to bind with RNA shed some light on this (Adinolfi, Bagni et al. 1999). They showed that the NLS exhibits RNA binding activity and binds to mRNA in the nucleus. Once attached, the NLS is “masked,” leading to export from the nucleus to cytoplasm. The mRNA-bound FMRP transfers the mRNA to the polysomes for translation, then dissociates itself (Adinolfi, Bagni et al. 1999). After dissociation from FMRP, mRNA exposed to NLS is subjected to a further round of intracellular shuttling (Adinolfi, Bagni et al. 1999). This hypothesis gained more attention with the identification of the KH0 domain that is also a single-stranded nucleotide binding motifs (Myrick, Hashimoto et al. 2015, D’Annessa, Cicconardi et al. 2019). Unlike the KH1 and 2 domains in this protein family and from other KH domains identified in other proteins, the sequence of the KH0 domain lacks the canonical G-X-X-G motif (Myrick, Hashimoto et al. 2015, D’Annessa, Cicconardi et al. 2019). Counteracting the NLS, exon 12 of FXR1 and FXR2, and exon 14 of FMR1, encode the Nuclear Export Sequence (NES) (Eberhart, Malter et al. 1996). This sequence shares high similarity with the NES found in the HIV-1 Rev protein and Protein Kinase Inhibitor-α (PKIA) (Fridell, Benson et al. 1996, Bardoni, Sittler et al. 1997). The presence of a NES, coupled with the nuclear export protein, Exportin 1, mediates the export of these proteins from the nucleus to the cytoplasm. However, inhibition of nuclear export with leptomycin B leads to an accumulation of all three proteins in the nucleus (Tamanini, Bontekoe et al. 1999), demonstrating the necessity of the NES domain and cytoplasmic function of FXR proteins.
All three FXR family proteins contain a coiled-coil domain encoded within exon 7 (Siomi, Zhang et al. 1996). This domain is responsible for interactions between the Fragile X proteins and the ribosomal machinery, as well as for homo- and hetero-dimerization between members of the Fragile X family (Zhang, O’Connor et al. 1995, Siomi, Zhang et al. 1996, Tamanini, Bontekoe et al. 1999). The coiled-coil domain was also reported to be important for protein-protein interactions; it is necessary for proper binding of FMR1 to cytoplasmic FMRP-interacting protein 1/2 (CYFIP1/2) (Schenck, Bardoni et al. 2001).
Exon 8 encodes the K Homology, KH1 domain in all three Fragile X family members (Siomi, Choi et al. 1994, Zhang, O’Connor et al. 1995, D’Annessa, Cicconardi et al. 2019). The KH1 domain is not unique to the Fragile X family: Several different families of RNA binding proteins possess motifs with similar amino acid sequences (Siomi, Siomi et al. 1993). This motif confers upon proteins the ability to bind to DNA/RNA structures that are typically four unpaired bases in length (Sjekloca, Konarev et al. 2009). Based on their secondary structure and folding, the FXR proteins’ KH1 and C-terminal KH2 domains are considered to be Type 1 (Dube, Huot et al. 2000). Exons 9–11 of FXR1 encode the KH2 domain (Siomi, Choi et al. 1994, Zhang, O’Connor et al. 1995). FXR1 differs in sequence from the FMR1 variant, which contains additional exons that encode a lengthy variable loop within the domain (Darnell, Fraser et al. 2009). Like the KH1 domain, the KH2 also binds to RNA (Sung, Conti et al. 2000). Moreover, the KH2 domain is uniquely capable of binding RNA containing a kissing-complex or loop-loop pseudoknot (Darnell, Fraser et al. 2009). Finally, the KH2 domain of FXR1 is uniquely capable of binding to the Auto-Inhibitory Domain (AID) of PAK1 that in turn, leads to a decrease in PAK1 concentration in cells (Say, Tay et al. 2010).
The final signature RNA binding motif of the FXR family is the RGG box (Figure 1) (Zhang, O’Connor et al. 1995, Darnell, Jensen et al. 2001, Schaeffer, Bardoni et al. 2001). Exon 15 encodes this motif in both FMRP and FXR1. FMRP and FXR1 share high amino acid sequence similarity in the RGG box and can bind G-quadruplex containing DNA/RNA structures (Brown, Small et al. 1998, Darnell, Jensen et al. 2001, Darnell, Fraser et al. 2009). The selectivity and the propensity of the RGG box to bind G-quadruplex-containing DNA/RNA structures have been the subject of much experimental scrutiny resulting in a plethora of evidence. Brown et al. were the first to report that the RGG box of FMRP was the dominant RNA binding motif (Brown, Small et al. 1998). Later the Darnell group published a series of manuscripts firmly establishing FMRP’s ability to bind RNA via the RGG box (Brown, Small et al. 1998, Darnell, Jensen et al. 2001, Darnell, Fraser et al. 2009). A recent review (D’Annessa, Cicconardi et al. 2019) sheds light on the structural features of the FXR proteins, mainly FMRP, and the network of their interactions with RNAs and other proteins that is helpful in reaching the deeper level of understanding which is so critical for generation of FXS and cancer therapeutics.
Post-translational modification of FXR family proteins
Methylation:
Ong et al. first demonstrated that methylation of FXR1 occurred in vivo on the arginine residues in the RGG box (Ong, Mittler et al. 2004). This finding was later validated by several groups (Dolzhanskaya, Merz et al. 2006, Stetler, Winograd et al. 2006), who demonstrated that Protein Arginine Methyltransferase 1 (PRMT1) is likely responsible for such methylation. Methylation of the RGG box residues determines the protein’s ability to associate with polysomes (Blackwell, Zhang et al. 2010). Further methylation alters the heterodimerization between the FXR family members (Dolzhanskaya, Merz et al. 2006). As FXR1 methylation occurs mainly within its RGG box, many groups have studied its effects on RNA binding and substrate recognition. Stetler et al. concluded that methylation on critical amino acid residues within the RGG box of FXR1 leads to a decrease in RNA binding capability (Stetler, Winograd et al. 2006). This finding remains mostly unchallenged concerning FXR1; however, Blackwell et al. studied the methylation state of FMRP and concluded that methylation does not affect FMRP’s ability to bind RNA but does affect the protein’s ability to associate with the mRNA translational machinery (Blackwell, Zhang et al. 2010). How methylation impacts FMRP- and FXR1-mediated RNA metabolism and other biological processes remains understudied.
Phosphorylation:
Several studies have identified and characterized kinases that are responsible for site-specific phosphorylation of FXR proteins. About two decades ago, Siomi et al. showed that residue S406 (S499 in mouse, S500 in human) of Drosophila melanogaster FMRP is phosphorylated by Casein Kinase II (CK2) (Siomi, Higashijima et al. 2002). This phosphorylation is proximal to the RGG box and appears to control RNA recognition. This region is highly conserved in FXR1 and FXR2, which prompted others to conduct further studies on FXR proteins’ phosphorylation. Later, Bartley et al. established a connection between CK2 and mouse and mammalian FMRP (Bartley, O’Keefe et al. 2014, Bartley, O’Keefe et al. 2016), providing evidence that CK2 phosphorylates residue S499 of FMRP, “unlocking” the ability of FMRP to be further phosphorylated at nearby sites. The CK2 enzyme has been shown in the past to be constitutively active (Ruzzene, Di Maira et al. 2010), which may suggest that FMRP S499 can be regulated by dephosphorylation at that site alone more than phosphorylation. Interestingly, Bartley et al. showed that S499 phosphorylation levels are unaffected by the putative FMRP phosphatase, PP2A (Bartley, O’Keefe et al. 2014). FMRP residues 496–503 are shown to interact with Dicer protein in their dephosphorylated form (Cheever and Ceman 2009), but phosphorylation at the S499 residue of FMRP is found to be required for an RNA-dependent FMRP-AGO2 interaction (Muddashetty, Nalavadi et al. 2011). Bartley et al. also demonstrated that a difference in the phosphorylation of FMRP influences glutamate receptor (mGluR) activation. They further identified that burst (<5 minutes) activation of mGluR leads to phosphorylation of FMRP; however, prolonged mGluR activation leads to activation of Protein Phosphatase 2a (PP2A) that subsequently dephosphorylates FMRP (Bartley, O’Keefe et al. 2014, Bartley, O’Keefe et al. 2016). A recent comprehensive study succeeded in solving to solve this puzzle by showing that although CK2 is constitutively active, it can stimulate secondary phosphorylation by other kinases, many of which are regulated in a signal-dependent manner (St-Denis, Gabriel et al. 2015). The S499 of FMR1 (S438, isoform X1) is present at a highly conserved region between FMR1 and FXR1. Thus, a feedback regulatory pathway operates to control the function of FMRP. Given the high level of homology between FMRP and FXR1 in this region, and the near-perfect homology of conserved serine and threonine residues, we postulate that a similar mechanism of FXR1 dephosphorylation may determine its functionality. Reports indicate that FXR1 is phosphorylated at S449 by a p21-activated serine/threonine kinase 1 (PAK1) (Say, Tay et al. 2010). The impact of this phosphorylation remains poorly understood; however, only the activated form of PAK1, unlike its inactivated precursor protein, can perform this phosphorylation (Say, Tay et al. 2010). Protein Kinase C Iota also phosphorylates FXR1 at an unknown threonine residue (Qian, Hassanein et al. 2015). Other kinases such as ERK2 and GSK3β are reported to phosphorylate FXR1 sequentially (Del’Guidice, Latapy et al. 2015). ERK2 phosphorylates an unknown residue of GSK3β, increasing the efficiency of phosphorylation of other proteins, including FXR1 for proteasomal degradation (Del’Guidice, Latapy et al. 2015). This phosphorylation event is sensitive to ERK2 and GSK3β inhibitors, providing an opportunity to use these inhibitors for FXR protein-related diseases.
Sumoylation:
Sumoylation, a reversible PTM, involves the small ubiquitin-like modifier (SUMO) protein and is involved in many cellular signaling pathways (Hickey, Wilson et al. 2012). The enzymatic pathway uses the SUMO paralogs (~100 amino acids; ~11 kDa) and occurs in the covalent enzymatic conjugation of specific lysine residues of the substrate proteins (Hickey, Wilson et al. 2012). Sumoylation regulates a myriad of neurodevelopmental processes by preventing protein–protein interactions and/or by providing new binding sites for novel interacting proteins (Schorova and Martin 2016). A recent work by Khayachi and co-workers showed that FMRP is an active substrate of the SUMO pathway in neurons (Khayachi, Gwizdek et al. 2018). This group identified the sumoylation sites on FMRP that are induced by the activation of metabotropic glutamate receptors (mGlu5Rs). Modification of FMRP promotes the dissociation of FMRP–FMRP dimers and frees FMRP from the RNA granules that allowing local translation of the protein. FMRP-sumoylation is a major PTM that promotes FMRP-mediated neuronal function such as synaptic plasticity, neurotransmitter release, and neuronal network formation in mammalian brain.
Ubiquitinylation:
A recent publication established that post-translational modification of FXR1 leads to its ubiquitinylation in cancer cells (Qie, Majumder et al. 2017). In Head and Neck Squamous Cell Carcinoma (HNSCC) cell lines, the authors demonstrated that FXR1 is ubiquitinylated on an unknown C-terminal lysine residue by an E3 Ligase F-box only protein 4 (Fbxo4). The authors here made an important point regarding the regulation involving GSK3β that phosphorylates Fbxo4 to help degrade FXR1 in the cytoplasm. GSK3β mediated phosphorylation is necessary for Fbxo4 dimerization and activation and this event of Fbxo4-dependent ubiquitinylation leads to degradation of FXR1 by the proteasomal machinery. However, the data also suggest that FXR1 negatively regulates the translation of Fbxo4 by binding to the transcript. Thus, the authors propose the existence of a novel feedback loop, whereby FXR1 prevents translation of a target that would cause its degradation. This mechanism may partially account for the overexpression of FXR1 observed in HNSCC (Majumder, House et al. 2016, Qie, Majumder et al. 2017).
Role of FXR family proteins in Fragile X Syndrome
FMRP:
The indicators of Fragile X Syndrome (FXS) depend on various factors like sex, age, genetic and molecular variations, and environmental factors. Silencing of the FMR1 gene encoding FMRP (also known as synaptic functional regulator FMR1) is the predominant cause of FXS, the most common form of inherited intellectual disability. FXS is mostly caused by an expansion of CGG repeat sequence in the promoter region of FMR1 that leads to methylation and transcriptional, and translational/functional silencing of FMRP (Figure 2). Patients with >200 CGG repeats show a full FMRP mutation; they and those with between 45 and 54 CGG (intermediate) or between 55 and 200 CGG (premutation) repeats show an excessive transcription of FMR1 alleles and principally manifest two phenotypes, fragile X tremor ataxia syndrome (FXTAS) and fragile X primary ovarian insufficiency (FXPOI) (Tassone, Beilina et al. 2007)(Figure 2). Individuals with a less severe form of FXS have cells containing methylated or unmethylated FMRP alleles where a few copies of FMRP are produced. An early report highlighted patients who did not have the CGG repeats but instead had an FMR1 deletion that caused FXS (Gedeon, Baker et al. 1992) (Figure 2). Recent advances in high throughput targeted screening techniques have also identified individuals with a deletion or point mutation in FMRP (mutations in the coding region: S27X nonsense mutation, 1457insG, Arg138Gln, Gly266Glu, and Ile304Asn) rendering a dysfunctional or absent protein (Myrick, Hashimoto et al. 2015, Suhl and Warren 2015, Quartier, Poquet et al. 2017). A missense mutation within the amino-terminal domain (includes the nuclear localization signal) of FMRP, Arg138Gln, interrupts both calcium-activated potassium (BK) channel and chromatin binding to incur developmental delays in males (Collins, Bray et al. 2010, Alpatov, Lesch et al. 2014, Myrick, Hashimoto et al. 2015). A recent study showed that a specific in-frame deletion of exon 8, which encodes for the KH1 domain in FMR1, is enough to cause FXS-like phenotypes in rats (Golden, Breen et al. 2019). This deletion renders behavioral and genetic patterns within the medial prefrontal cortex (mPFC) of rats, which map to two weighted gene co-expression network modules that are conserved in the human frontal cortex and contains known FMRP targets (Golden, Breen et al. 2019). Besides, the mutation Ile304Asn in the KH2 domain of FMRP is found to be associated with a severe form of FXS, reported in a single patient (De Boulle, Verkerk et al. 1993). Although it is still unclear whether an impaired nucleic acidic recognition or a defective protein binding is the result of a single mutation that renders the FMRP dysfunction, an initial biophysical experiments show that the mutation somehow destabilizes the KH2 domain of FMRP (Di Marino, Achsel et al. 2014).
Figure 2: The CGG trinucleotide repeats at the 5′-UTR of FMR1cause FXS.
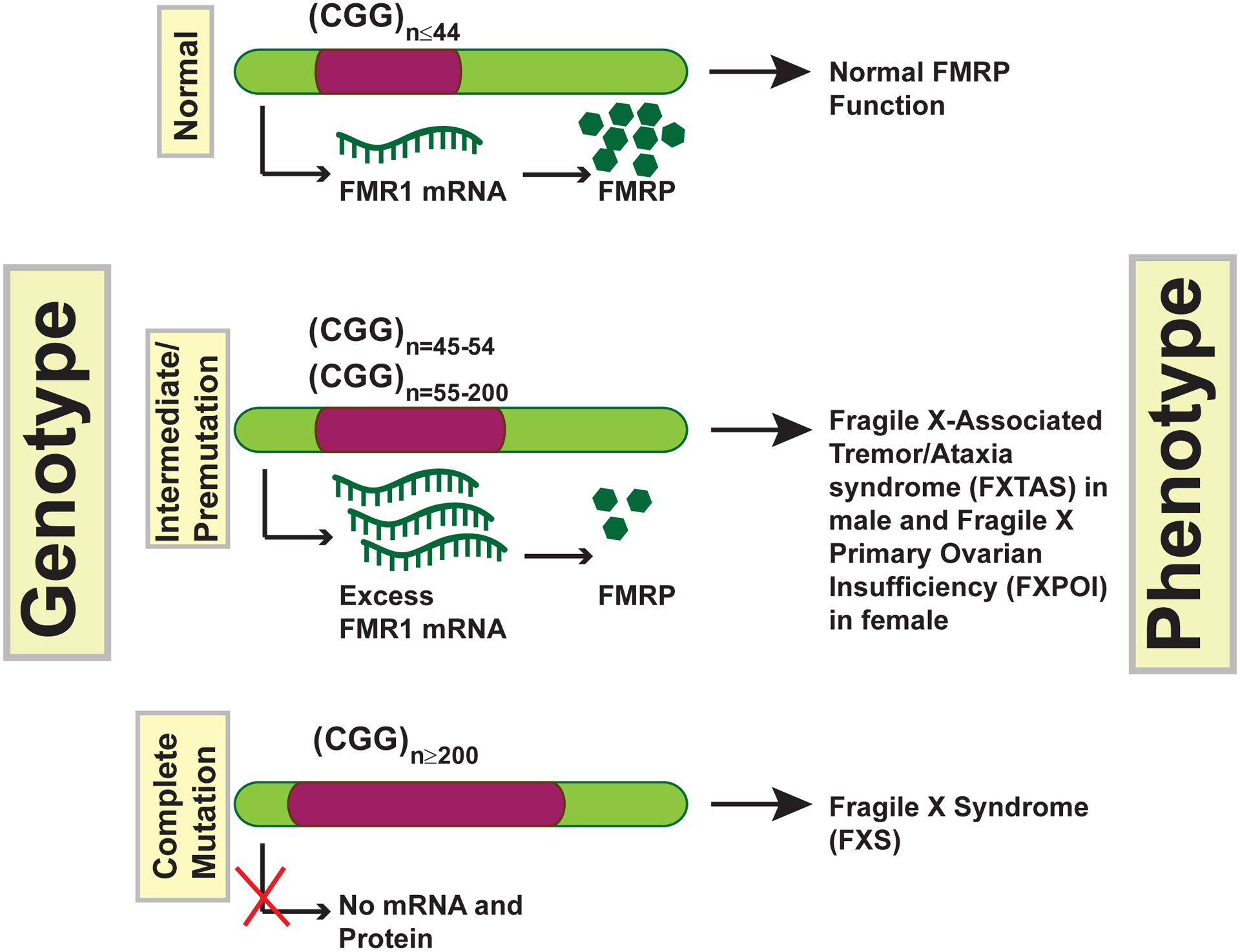
Mutations in the FMR1 gene can lead to several different diseases. Healthy individuals have less than 44 CGG repeats. FMR1 intermediate and premutation carriers can have between 45–54 and 55–200 repeats, respectively. In these cases, FMR1 mRNA is expressed at higher levels than in healthy individuals, although for obscure reasons, FMRP protein levels go down compared to normal due to unclear reasons. These carriers often have an increased chance of developing two disorders, FRAXA in males and FXPOI in females. In cases having more than 200 repeats, the “full mutation,” the FMR1 gene is hypermethylated and silenced, which is the primary cause of FXS.
While present in most tissues, FMRP is highly expressed in brain and testis and plays a critical role in the translational control of several mRNAs in postsynaptic neurons, including the regulation of mGluRI (Darnell and Klann 2013, Bartley, O’Keefe et al. 2014, Bartley, O’Keefe et al. 2016). FMRP binds to the dendritic, small non-coding brain cytoplasmic RNA BC1 (Zalfa, Giorgi et al. 2003) where BC1 acts as a bridging molecule between FMRP and the substrate mRNAs to help FMRP acting as a translational repressor of specific mRNAs at synapses. Peptide mapping, molecular modelling and docking simulations also revealed that BC1 is 2′-O-methylated in the 5’-haripin region that is recognized by the second tudor domain of FMRP and interestingly this PTM affects this RNA-protein interaction (Lacoux, Di Marino et al. 2012). Other cellular functions of FMRP include a chromatin dependent DNA damage response (Alpatov, Lesch et al. 2014), RNA editing (Alpatov, Lesch et al. 2014, Shamay-Ramot, Khermesh et al. 2015), regulation of neuronal activity (Akins, Leblanc et al. 2012), and activation of the potassium channels KCNT1 (Brown, Kronengold et al. 2010). In an RNA- dependent or -independent manner RNA helicase MOV10 links FMRP with the miRNA pathway and this interaction promotes MOV10 association with mRNAs in brain cells (Muddashetty, Nalavadi et al. 2011). FMRP controls a plethora of mRNAs; however, only a few have been validated at the translational level to play a direct role in FXS pathobiology. A few of the FMRP targets are GABAA and GABAB receptor subunits, phosphatidylinositol 3-kinase enhancer (PIKE), matrix metalloproteinase 9 (MMP9), glycogen synthase kinase 3 (GSK3), amyloid precursor protein (APP) and diacylglycerol kinase-κ (DGKκ) (reviewed in detail in (Hagerman, Berry-Kravis et al. 2017)). Nonfunctional FMRP results in dysregulation of the DgKk-dependent mGluRI pathway, leading to impaired synaptic plasticity that involves aberrant mGluRI signaling (Tabet, Moutin et al. 2016). Additionally, mGluR5 plays a major role in the positive regulation of FMRP expression that linked to Alzheimer’s disease (Hamilton, Esseltine et al. 2014). Thus, proper FMRP function is vital for complex downstream signaling of NMDA, mGluR5, and BDNF receptors (Richter, Bassell et al. 2015).
In addition to FMRP-RNA interactions, a smaller number of studies have demonstrated associations between FMRP and other proteins. FMRP generally binds to other proteins through the Tudor domain at the N-terminus (Adams-Cioaba, Guo et al. 2010). However, the interaction between FMRP and CYFIP1 was found to involve the KH0 domain (Schenck, Bardoni et al. 2003, Abekhoukh and Bardoni 2014), and this interaction plays a direct role in FXS. A trimeric complex of the eIF4E-CYFIP1-FMRP proteins is known to regulate mRNA translation, but a failure of this complex due to lack of either FMRP or CYFIP1 can be directly linked with the onset of FXS (Napoli, Mercaldo et al. 2008). Notably, CYFIP1 is down-regulated in a group of patients with FXS who also have the Prader-Willi phenotype, and CYFIP1 has recently been connected to schizophrenia (Napoli, Mercaldo et al. 2008, De Rubeis, Pasciuto et al. 2013, Di Marino, Chillemi et al. 2015). Thus, proper FMRP function requires its interacting partner proteins to alleviate FXS.
Following the generation of the FMR1 knockout mouse in the early ‘90s, several studies have successfully applied it to validate various cellular and psychological traits found in human FXS patients. These mice show a behavioral phenotype similar to fragile X syndrome (Huber, Gallagher et al. 2002). FMRP is one of the proteins known to be synthesized in response to group 1 metabotropic glutamate receptor (mGluR) activation (Weiler and Greenough 1999). However, a report documenting increased translation dependent mGluRI signaling in the FMR1 knockout mouse brain (Huber, Gallagher et al. 2002) suggested FMRP’s role as a negative regulator of translation. The study further showed that intensified long-term depression (LTD) and/or mGluR function is a plausible cause of the behavioral phenotype in fragile X syndrome. In a separate study, the GABAergic signaling pathway was found to be downregulated in FXS and other neurodevelopmental disorders (Braat and Kooy 2015). Functional GABAergic signaling is vital for normal neural circuit function (Braat and Kooy 2015). Disturbance in the GABAergic interneurons, critical for regulating most of the excitatory neurons, can disrupt the excitatory/inhibitory balance (E/I balance) and, consequently, precipitate dysfunction in cognitive processes. A series of independent studies demonstrated the causative role that compromised GABAA receptor-mediated signaling plays in FXS in FMR1 knockout mice (Centonze, Rossi et al. 2008, Sabanov, Braat et al. 2017) (reviewed in (Hagerman, Berry-Kravis et al. 2017)).
Both the loss and gain of FMRP functions cause different variants of FXS, suggesting a delicate balance in the protein level is required for a proper neuronal purpose. Here, it is worth mentioning that carriers with a full mutation that leads to silencing of FMR1, do not suffer from ovarian dysfunction (Sherman, Curnow et al. 2014). Thus, the significant reduction of the FMR1 protein product, FMRP, does not appear to affect premature ovarian insufficiency (POI). Fragile X-associated POI (FXPOI), the loss of the ovarian hormonal function in a woman before or at the age of 40 years, is caused by an increase in the FMR1 levels that unexpectedly lowers protein levels leading to cellular toxicity (Barasoain, Barrenetxea et al. 2016). It is thought that FMR1 gain-of-function toxicity may account for FXPOI and the other premutation (PM)-associated disorder, fragile X-associated tremor/ataxia syndrome (FXTAS) (Tassone, Beilina et al. 2007, Sherman, Curnow et al. 2014). Although lower FMRP levels might contribute to neuronal dysfunction in FXTAS patients, it is unlikely to be the major cause of neurodegeneration in FXTAS (Renoux, Carducci et al. 2014). FXTAS presents with neuronal cell death that occurs due to the impaired development of nuclear lamina in the neurons harboring the FMRpolyG form is found (Sellier, Buijsen et al. 2017). Together these findings demonstrate that exquisite regulation of FMRP expression levels is critical for neuronal cell growth and development.
FXR1:
While to date there is no direct evidence linking FXR1 to FXS, in human neuroblastoma cells (SH-SY5Y), FXR1 is found to be targeted by a microRNA, miR-19b-3p, which plays a significant role in the molecular pathology of FXS (Ma, Tian et al. 2016). Additionally, FXR1 shares strong homology with FMRP (Zhang, O’Connor et al. 1995) and thus is considered an autism-associated protein. FXR1 expression can be abundant in the cerebrum, cerebellum, liver, kidney, testis, skeletal muscle, myocardium, and other tissues, especially in skeletal muscle and myocardium where FMRP is produced at significantly lower levels. FXR1 plays a vital role in healthy muscle development and has been implicated in Facioscapulohumeral muscular dystrophy (FSHD) (Ma, Tian et al. 2016). Interestingly, FXR1 forms heterodimer with FMRP, and these proteins have common mRNA targets (Tamanini, Bontekoe et al. 1999, Darnell, Fraser et al. 2009) and, in turn, may also play a key role in FXS. In addition to FMRP, FXR1 binds to other autism-associated proteins (Sakai, Shaw et al. 2011), and SNPs in FXR1 are often associated with severe autistic phenotypes FXR1 is also involved in bipolar disorder and schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics 2014, Liu, Bipolar Genome et al. 2016). A GWAS including a large cohort of patients with bipolar disorder accompanied by eating disorders (i.e., binge eating, purging, dietary restriction) revealed genetic variation within SOX2-OT with a secondary peak in the adjacent FXR1 gene on chromosome 3q26.33 (Liu, Bipolar Genome et al. 2016). The schizophrenia susceptibility gene, Disrupted-In-Schizophrenia 1 (Disc1), is a scaffolding protein that interacts with multiple pathways, including GSK3β (Mao, Ge et al. 2009). GSK3β hyper-phosphorylates FXR1, and their interaction is found to affect emotional stability in humans (Del’Guidice, Latapy et al. 2015). Moreover, the FXR1-GSK3β pathway controls the glutamatergic neurotransmission by regulating AMPA receptor subunits GluA1 and GluA2 with a vesicular glutamate transporter VGlut1 (Khlghatyan, Evstratova et al. 2018). Before this, Cook et al. showed that FXR1 binds to one of the AMPA receptor subunits, GluA2 mRNA through a conserved GU-rich element in its 5’UTR to downregulate its translation and the loss of FXR1 from the postnatal forebrain of mice enhanced expression of the AMPA receptor subunit GluA2 (Cook, Nuro et al. 2014). Unlike autism, schizophrenia is more directly linked to adult neurogenesis, and FXR1 appears to be an essential protein implicated in this disease. Uncovering the role of FXR1 in neurogenesis will undoubtedly open new avenues to better understand various neurological disorders and develop therapeutics for neurodegenerative diseases.
FXR2
As for FXR1, the function of FXR2, the other autosomal homolog of FMRP in FXS, is not well understood. One study showed that FMRP and FXR2 together regulate the significant scaffolding postsynaptic density protein, PSD95 (Fernandez, Li et al. 2015). These investigators suggested that FXR2 acts as a translational activator of PSD95, as the absence of FXR2P, reduced PSD95 mRNA translation in the hippocampus, ultimately affecting its fine-tuning during synaptic activity. Notably, FXR2 knockout mice show similar specific behavioral patterns as FMR1 knockout mice (Bontekoe, McIlwain et al. 2002, Spencer, Serysheva et al. 2006), and FMR1/FXR2 double knockout mice exhibit heightened behavioral phenotypes in locomotor and cognitive processes compared to single knockout or wild type. However, only FXR2 mutant mice show some distinct behavioral phenotypes suggesting a role of FXR2 in central nervous system function (Bontekoe, McIlwain et al. 2002). Further investigation is needed to fully reveal FXR2’s role in FXS and other neurological disorders.
FXR family proteins and their contribution to cancer
FMRP:
Although a significant body of work has illuminated the role of FMRP in FXS, only a handful of studies have produced evidence of its direct or indirect involvement of FMRP in cancer. There have been reports that FXS patients have decreased risk and exhibit a lower incidence of cancer (Schultz-Pedersen, Hasle et al. 2001, Luca, Averna et al. 2013), and one case study highlighted reduced glioblastoma invasion in a patient diagnosed with FXS (Kalkunte, Macarthur et al. 2007). In contrast, a high level of FMRP is linked to metastatic breast cancer, and overexpression of the protein in primary breast tumors induces lung metastasis (Luca, Averna et al. 2013). Additional findings indicate that FMR1 is overexpressed in hepatocellular carcinoma (Liu, Zhu et al. 2007). A recent report showed that a high level of FMRP expression correlates significantly with metastatic melanoma tissues, and that a reduction of FMRP in metastatic melanoma cell lines affects cell migration, invasion, and adhesion (Zalfa, Panasiti et al. 2017). These findings demonstrate that overexpressed FMRP can be linked to certain types of cancers.
As an RNA binding protein, FMRP binds to a variety of RNA targets while controlling their translation, transport, and stability (Bardoni, Abekhoukh et al. 2012). FMRP is known to bind to G-quadruplex (G4) motifs on RNA, a structure organized in repeated guanine tetrad units, on RNA through the RGG domain (Didiot, Tian et al. 2008, Maurin, Zongaro et al. 2014). The G-quadruplex is involved in regulating different steps of RNA metabolism; in particular, FMRP-RNA complexes regulate translation and additional post-transcriptional gene regulatory functions (Didiot, Tian et al. 2008, Maurin, Zongaro et al. 2014). RNA G4 structures have been shown to regulate the expression of genes implicated in the hallmarks of cancer, including TP53, VEGF, hTERT, TGFB2, and other essential oncogenes (Cammas and Millevoi 2017). These findings support the conclusion that FMRP is overexpressed in specific cancer and plays a significant role in tumor progression through altered FMRP-dependent G4 mRNA expression. The copper/zinc dismutase SOD1 is overexpressed in cancers and localized in the cytoplasm, the inter-membrane space of mitochondria, and the nucleus (Papa, Manfredi et al. 2014). Through its RGG motif, FMRP recognizes the SoSLIP RNA motif (Sod1 mRNA Stem Loops Interacting with FMRP) and binds Sod1 mRNA with a high affinity to activate its translation (Bechara, Didiot et al. 2009). A detailed review (Papa, Manfredi et al. 2014) compiles the emerging evidence for the role of SOD1 in multiple cancers and how it can be targeted for therapy. FMRP overexpression and positive regulation of Sod1 translation can directly or indirectly influence cancer progression.
FXR1:
Two seminal papers that define FXR1’s role in cancer drew attention in recent years (Qian, Hassanein et al. 2015, Majumder, House et al. 2016) (Figure 3). Qian et al. first showed that FXR1 could promote tumor progression in non-small cell lung cancer (NSCLC) by regulating two other oncogenes within the same chromosome 3q amplicon (Figre 3). The authors inferred that elevated FXR1 expression could be a candidate biomarker predictive of poor survival in NSCLC, opening avenues for a novel therapeutic target (Qian, Hassanein et al. 2015). Around the same time, Majumder et al. demonstrated that overexpressed FXR1 binds and destabilizes p21 to reduce p21 protein expression in oral cancer cells (Majumder, House et al. 2016). This group also showed that FXR1 binds and stabilizes TERC (telomerase RNA component) RNA in oral cancer cells (Majumder, House et al. 2016). These reports demonstrated the oncogenic role of FXR1, as reflected in its overexpression in various cancer cells and tissues. By using both in vivo and in vitro biochemical assays, the two groups established that FXR1 is overexpressed in lung, breast, and head and neck cancer compared to normal tissues and that the increased expression is critical for cancer cell growth and proliferation (Qian, Hassanein et al. 2015, Majumder, House et al. 2016) (Figure 3). Qian et al. identified the mechanisms by which FXR1 regulates the function of oncogenes like protein kinase C, iota, and epithelial cell transforming 2 by binding and forming a complex (Qian, Hassanein et al. 2015). High expression of FXR1 in head and neck cancer blocks cellular senescence (Majumder, House et al. 2016), an essential tumor-suppressing mechanism invoked to attenuate transformation-promoting responses to various cellular stresses like oxidative stress or reactive oxygen species (ROS) generation, telomere attrition, and oncogene activation. A permanent G1-phase cell cycle arrest, where cells remain metabolically active, can be a salient signature of senescence. Senescence plays a vital role in abating tumorigenesis (Braig, Lee et al. 2005, Chen, Trotman et al. 2005). By blocking this cellular function, FXR1 promotes head and neck cancer initiation and proliferation (Fernandez and Mallette 2016, Majumder, House et al. 2016). In subsequent work, others showed how FXR1 uses an ingenious feedback mechanism to downregulate its substrate Fbxo4 to bypass senescence and contribute to neoplastic progression in head and neck cancer (Qie, Majumder et al. 2017). Fbxo4, a tumor suppressor, is a component of an Skp1-Cul1-F-box E3 ligase (Qie, Majumder et al. 2017).
Figure 3: FXR1 overexpression promotes cancer growth and metastasis.
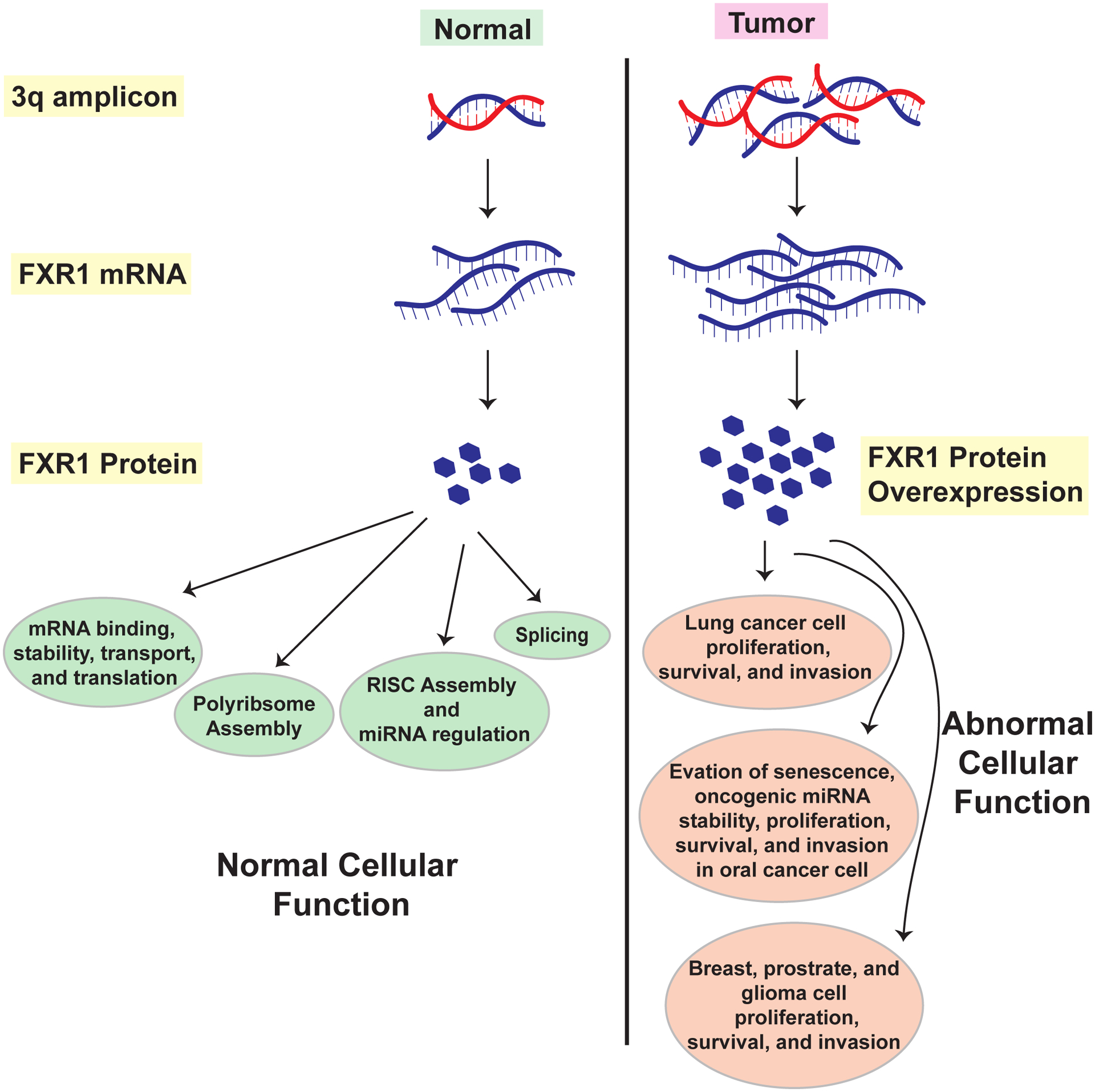
Chromosome 3q is often found to be amplified in several cancer types, including lung and head and neck. A high level of DNA followed by mRNA results in heightened the levels of FXR1 protein. Overexpressed FXR1 deregulates tumor suppressors and stabilize oncogenic miRNAs to help promote cancer progression.
A recent publication from our laboratory shows that FXR1 preferentially binds and regulates the levels of specific miRNAs in HNSCC, especially the oncogenic miRNA miR301a-3p (Majumder and Palanisamy 2020). Interestingly, miR301a-3p targets CDKN1A (p21) for degradation, which is also a target of FXR1 (Majumder, House et al. 2016, Majumder and Palanisamy 2020). FXR1 binds and maintains a steady level of miR301a-3p in HNSCC by protecting it from a 3’−5’ exonuclease, PNPT1 mediated decay, and consequently, miR301a-3p binds to the 3’-UTR of p21 mRNA for degradation (Majumder and Palanisamy 2020). FXR1 has been implicated in other cancers besides lung, breast, and head and neck. Cao et al. showed that FXR1 and lncRNA MIR17HG are upregulated in glioma tissues and cell lines (Cao, Zheng et al. 2019), in which FXR1 stabilizes the lncRNA. This study also found that the cells’ progression is inhibited by the downregulation of FXR1 or MIR17HG. Fan et al. reported that FXR1 knockdown inhibited cell proliferation in TP53/FXR2 co-deletion cancers and uncovered a novel role of FXR1 in gene transcription (Fan, Yue et al. 2017). Utilizing ChIP-MS and ChIP coupling followed by high-throughput sequencing (ChIP-seq), the study revealed the molecular mechanism that FXR1 employs to regulate cell proliferation. It also showed how FXR1 recruits transcription factor STAT1 or STAT3 to certain gene promoter regions, where it co-localizes with histone H3 lysine 4 trimethylation (H3K4me3) to regulate transcription (Fan, Yue et al. 2017). It is evident from these studies that high levels of FXR1 protein regulate transcription, post-transcription, and translation of several target genes while degrading certain tumor suppressors and activating oncogenes, to play a critical role in cancer growth and proliferation.
FXR2:
Compared to FXR1 and FMRP, little is known about the role of FXR2 in cancer. According to the human protein atlas, expression of FXR2 has low specificity in cancer; however, according to protein atlas, FXR2 over-expression seems favorable for patients’ survival in pancreatic cancer (Uhlen, Zhang et al. 2017). Gumireddy et al. showed that lncRNA Tre is upregulated in paired samples of primary breast cancer and lymph-node metastases (Gumireddy, Li et al. 2013), and that its expression stimulates tumor invasion. TreRNA forms a novel ribonucleoprotein (RNP) complex with RNA-binding proteins (hnRNP K, FXR1, and FXR2), PUF60 and SF3B3, and the complex is required for the RNA’s functions.
Conclusion and future perspective:
Over the last few decades, instead of pursuing in depth understanding of the molecular mechanisms underlying these diseases, inordinate efforts have been to enumerating and characterizing the symptoms of FXS and several other neurological disorders involving FXR family proteins, instead of in depth understanding of the molecular mechanism of the disease. Although various therapeutic approaches and specific FXS treatments and various therapeutic approaches are being developed and tested on FXS animal models and patients, delving into the molecular basis of the disease is of utmost necessity. For example, recent efforts demonstrate that demethylation of the CGG repeats in post-mitotic FXS neurons reactivates FMRP, restoring the protein’s function (Liu, Wu et al. 2018). This novel molecular finding establishes that demethylation of the CGG expansion is sufficient to promote FMRP function, suggesting a potential therapeutic strategy for FXS. Transcriptional reactivation of FMR1 might provide an alternative strategy to achieve the treatment options for FXS. This approach has been well-reviewed (Tabolacci, Palumbo et al. 2016), wherein the authors suggested that a better understanding of the molecular basis of FXS pathophysiology is critical to providing new therapeutic options based on such transcriptional activation of proteins.
The FXR family members share similar structures, especially in their N-terminal and central domains, the regions that are mainly involved in protein-protein interactions and functions (Ramos, Hollingworth et al. 2006). Although, to date FMRP is mainly linked to FXS, and FXR1 to cancer, their specific structural and functional differences which can account for that which might also play a role in their differential expression and divergent regulatory networks. For example, the ability of FXR1 to repress GluA2 synthesis is unique to FXR1 and is not a property of FXR2 or FMRP. Ultimately, the loss of FXR1P-mediated GluA2 repression heightens activity-dependent synaptic delivery of GluA2, increasing its incorporation at potentiated synapses. Association between the Gsk3β-FXR1 pathway and glutamatergic signaling also suggests how it may contribute to emotional regulation in response to mood stabilizers, or in illnesses like mood disorders and schizophrenia. Thus, FXR1 has a critical role in limiting synaptic plasticity and memory storage in the brain and fulfills an unexpected and dissimilar function among Fragile X proteins in these processes.
While considerable advances have been made in FXS, understanding how FXR protein family members are modulated, and how they regulate a number of essential genes in cancer, is still at a very preliminary stage. In multiple cancers, among the three proteins, FXR1 tends to show a higher expression and have a distinct gene regulatory network including tumor suppressor genes, miRNAs, and lncRNA. Recent works has illuminated the expression of specific FXR1 isoforms and how certain isoforms are included, excluded, or skipped during transcription (McClure and Palanisamy 2019). Further studies on FMRP and FXR2 will be needed to understand their role in oncogenesis.
In this review we have recapitulated the biological, biochemical, structural, and functional characteristics of the FXR family of proteins, with a focus on the structural basis of their RNA-binding specificities, their roles in fragile X syndrome and cancer, as well as the prospects for appropriate therapeutic strategies. Much remains to discovered regarding the significance of differential expression of the proteins in various tissues and how these affects human disease. These questions will be answered by uncovering details about their expression at co- and post-transcriptional levels, along with the effects of post-transcriptional and post-translational modifications under various conditions. High-impact future studies will explore the domains of FXR family proteins and their post-translational modifications, including methylation, ubiquitinylation, phosphorylation, sumoylation, and acetylation, and how the relevant RNA biology-related functions play out in fragile X and cancer models. FXR family protein functions have been the object of increasing study and scrutiny over the past thirty years. However, their co-existence, recognition of novel RNA substrates, PTM changes related to protein-protein interactions, and functions, whether deleterious or beneficial in terms of human disease, require further investigation.
Footnotes
Conflict of interest statement:
The authors declare no conflict of interest for the study.
References:
- Abekhoukh S and Bardoni B (2014). “CYFIP family proteins between autism and intellectual disability: links with Fragile X syndrome.” Front Cell Neurosci 8: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams-Cioaba MA, Guo Y, Bian C, Amaya MF, Lam R, Wasney GA, Vedadi M, Xu C and Min J (2010). “Structural studies of the tandem Tudor domains of fragile X mental retardation related proteins FXR1 and FXR2.” PLoS One 5(11): e13559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinolfi S, Bagni C, Musco G, Gibson T, Mazzarella L and Pastore A (1999). “Dissecting FMR1, the protein responsible for fragile X syndrome, in its structural and functional domains.” Rna 5(9): 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akins MR, Leblanc HF, Stackpole EE, Chyung E and Fallon JR (2012). “Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions.” J Comp Neurol 520(16): 3687–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stutzer A, Armache KJ, Simon MD, Xu C, Ali M, Murn J, Prisic S, Kutateladze TG, Vakoc CR, Min J, Kingston RE, Fischle W, Warren ST, Page DC and Shi Y (2014). “A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response.” Cell 157(4): 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker CE, de Diego Otero Y, Bontekoe C, Raghoe P, Luteijn T, Hoogeveen AT, Oostra BA and Willemsen R (2000). “Immunocytochemical and biochemical characterization of FMRP, FXR1P, and FXR2P in the mouse.” Exp Cell Res 258(1): 162–170. [DOI] [PubMed] [Google Scholar]
- Barasoain M, Barrenetxea G, Huerta I, Telez M, Criado B and Arrieta I (2016). “Study of the Genetic Etiology of Primary Ovarian Insufficiency: FMR1 Gene.” Genes (Basel) 7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni B, Abekhoukh S, Zongaro S and Melko M (2012). “Intellectual disabilities, neuronal posttranscriptional RNA metabolism, and RNA-binding proteins: three actors for a complex scenario.” Prog Brain Res 197: 29–51. [DOI] [PubMed] [Google Scholar]
- Bardoni B, Sittler A, Shen Y and Mandel JL (1997). “Analysis of domains affecting intracellular localization of the FMRP protein.” Neurobiol Dis 4(5): 329–336. [DOI] [PubMed] [Google Scholar]
- Bartley CM, O’Keefe RA, Blice-Baum A, Mihailescu MR, Gong X, Miyares L, Karaca E and Bordey A (2016). “Mammalian FMRP S499 Is Phosphorylated by CK2 and Promotes Secondary Phosphorylation of FMRP.” eNeuro 3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley CM, O’Keefe RA and Bordey A (2014). “FMRP S499 is phosphorylated independent of mTORC1-S6K1 activity.” PLoS One 9(5): e96956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H and Bardoni B (2009). “A novel function for fragile X mental retardation protein in translational activation.” PLoS Biol 7(1): e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell E, Zhang X and Ceman S (2010). “Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA.” Human Molecular Genetics 19(7): 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontekoe CJ, McIlwain KL, Nieuwenhuizen IM, Yuva-Paylor LA, Nellis A, Willemsen R, Fang Z, Kirkpatrick L, Bakker CE, McAninch R, Cheng NC, Merriweather M, Hoogeveen AT, Nelson D, Paylor R and Oostra BA (2002). “Knockout mouse model for Fxr2: a model for mental retardation.” Hum Mol Genet 11(5): 487–498. [DOI] [PubMed] [Google Scholar]
- Borreca A, Gironi K, Amadoro G and Ammassari-Teule M (2016). “Opposite Dysregulation of Fragile-X Mental Retardation Protein and Heteronuclear Ribonucleoprotein C Protein Associates with Enhanced APP Translation in Alzheimer Disease.” Mol Neurobiol 53(5): 3227–3234. [DOI] [PubMed] [Google Scholar]
- Braat S and Kooy RF (2015). “The GABAA Receptor as a Therapeutic Target for Neurodevelopmental Disorders.” Neuron 86(5): 1119–1130. [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T and Schmitt CA (2005). “Oncogene-induced senescence as an initial barrier in lymphoma development.” Nature 436(7051): 660–665. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D and Kaczmarek LK (2010). “Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack.” Nat Neurosci 13(7): 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD and Warren ST (1998). “Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein.” J Biol Chem 273(25): 15521–15527. [DOI] [PubMed] [Google Scholar]
- Bureau A, Beaulieu JM, Paccalet T, Chagnon YC and Maziade M (2017). “The interaction of GSK3B and FXR1 genotypes may influence the mania and depression dimensions in mood disorders.” J Affect Disord 213: 172–177. [DOI] [PubMed] [Google Scholar]
- Cammas A and Millevoi S (2017). “RNA G-quadruplexes: emerging mechanisms in disease.” Nucleic Acids Res 45(4): 1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Gao R, Yu C, Chen L and Feng Y (2019). “The RNA-binding protein FXR1 modulates prostate cancer progression by regulating FBXO4.” Funct Integr Genomics 19(3): 487–496. [DOI] [PubMed] [Google Scholar]
- Cao S, Zheng J, Liu X, Liu Y, Ruan X, Ma J, Liu L, Wang D, Yang C, Cai H, Li Z, Feng Z and Xue Y (2019). “FXR1 promotes the malignant biological behavior of glioma cells via stabilizing MIR17HG.” J Exp Clin Cancer Res 38(1): 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, De Chiara V, Musella A, Prosperetti C, Calabresi P, Bernardi G and Bagni C (2008). “Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome.” Biol Psychiatry 63(10): 963–973. [DOI] [PubMed] [Google Scholar]
- Cheever A and Ceman S (2009). “Phosphorylation of FMRP inhibits association with Dicer.” RNA 15(3): 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C and Pandolfi PP (2005). “Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis.” Nature 436(7051): 725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME and Warren ST (2010). “Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males.” Am J Med Genet A 152A(10): 2512–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D, Nuro E, Jones EV, Altimimi HF, Farmer WT, Gandin V, Hanna E, Zong R, Barbon A, Nelson DL, Topisirovic I, Rochford J, Stellwagen D, Beique JC and Murai KK (2014). “FXR1P limits long-term memory, long-lasting synaptic potentiation, and de novo GluA2 translation.” Cell Rep 9(4): 1402–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D, Sanchez-Carbente Mdel R, Lachance C, Radzioch D, Tremblay S, Khandjian EW, DesGroseillers L and Murai KK (2011). “Fragile X related protein 1 clusters with ribosomes and messenger RNAs at a subset of dendritic spines in the mouse hippocampus.” PLoS One 6(10): e26120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Annessa I, Cicconardi F and Di Marino D (2019). “Handling FMRP and its molecular partners: Structural insights into Fragile X Syndrome.” Prog Biophys Mol Biol 141: 3–14. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Fraser CE, Mostovetsky O and Darnell RB (2009). “Discrimination of common and unique RNA-binding activities among Fragile X mental retardation protein paralogs.” Hum Mol Genet 18(17): 3164–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST and Darnell RB (2001). “Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function.” Cell 107(4): 489–499. [DOI] [PubMed] [Google Scholar]
- Darnell JC and Klann E (2013). “The translation of translational control by FMRP: therapeutic targets for FXS.” Nat Neurosci 16(11): 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA and Willems PJ (1993). “A point mutation in the FMR-1 gene associated with fragile X mental retardation.” Nat Genet 3(1): 31–35. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Pasciuto E, Li KW, Fernandez E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, Grant SG, Poujol C, Choquet D, Achsel T, Posthuma D, Smit AB and Bagni C (2013). “CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation.” Neuron 79(6): 1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del’Guidice T, Latapy C, Rampino A, Khlghatyan J, Lemasson M, Gelao B, Quarto T, Rizzo G, Barbeau A, Lamarre C, Bertolino A, Blasi G and Beaulieu JM (2015). “FXR1P is a GSK3beta substrate regulating mood and emotion processing.” Proc Natl Acad Sci U S A 112(33): E4610–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marino D, Achsel T, Lacoux C, Falconi M and Bagni C (2014). “Molecular dynamics simulations show how the FMRP Ile304Asn mutation destabilizes the KH2 domain structure and affects its function.” J Biomol Struct Dyn 32(3): 337–350. [DOI] [PubMed] [Google Scholar]
- Di Marino D, Chillemi G, De Rubeis S, Tramontano A, Achsel T and Bagni C (2015). “MD and Docking Studies Reveal That the Functional Switch of CYFIP1 is Mediated by a Butterfly-like Motion.” J Chem Theory Comput 11(7): 3401–3410. [DOI] [PubMed] [Google Scholar]
- Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL and Moine H (2008). “The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer.” Nucleic Acids Res 36(15): 4902–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhanskaya N, Merz G, Aletta JM and Denman RB (2006). “Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP.” J Cell Sci 119(Pt 9): 1933–1946. [DOI] [PubMed] [Google Scholar]
- Dube M, Huot ME and Khandjian EW (2000). “Muscle specific fragile X related protein 1 isoforms are sequestered in the nucleus of undifferentiated myoblast.” BMC Genet 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart DE, Malter HE, Feng Y and Warren ST (1996). “The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals.” Hum Mol Genet 5(8): 1083–1091. [DOI] [PubMed] [Google Scholar]
- Fan Y, Yue J, Xiao M, Han-Zhang H, Wang YV, Ma C, Deng Z, Li Y, Yu Y, Wang X, Niu S, Hua Y, Weng Z, Atadja P, Li E and Xiang B (2017). “FXR1 regulates transcription and is required for growth of human cancer cells with TP53/FXR2 homozygous deletion.” Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH and Folsom TD (2015). “GABA receptor subunit distribution and FMRP-mGluR5 signaling abnormalities in the cerebellum of subjects with schizophrenia, mood disorders, and autism.” Schizophr Res 167(1–3): 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Kneeland RE, Liesch SB and Folsom TD (2010). “Fragile X mental retardation protein levels are decreased in major psychiatric disorders.” Schizophr Res 124(1–3): 246–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, Li KW, Rajan N, De Rubeis S, Fiers M, Smit AB, Achsel T and Bagni C (2015). “FXR2P Exerts a Positive Translational Control and Is Required for the Activity-Dependent Increase of PSD95 Expression.” J Neurosci 35(25): 9402–9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E and Mallette FA (2016). “The Rise of FXR1: Escaping Cellular Senescence in Head and Neck Squamous Cell Carcinoma.” PLoS Genet 12(11): e1006344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsom TD, Thuras PD and Fatemi SH (2015). “Protein expression of targets of the FMRP regulon is altered in brains of subjects with schizophrenia and mood disorders.” Schizophr Res 165(2–3): 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridell RA, Benson RE, Hua J, Bogerd HP and Cullen BR (1996). “A nuclear role for the Fragile X mental retardation protein.” Embo j 15(19): 5408–5414. [PMC free article] [PubMed] [Google Scholar]
- Gedeon AK, Baker E, Robinson H, Partington MW, Gross B, Manca A, Korn B, Poustka A, Yu S, Sutherland GR and et al. (1992). “Fragile X syndrome without CCG amplification has an FMR1 deletion.” Nat Genet 1(5): 341–344. [DOI] [PubMed] [Google Scholar]
- Golden CEM, Breen MS, Koro L, Sonar S, Niblo K, Browne A, Burlant N, Di Marino D, De Rubeis S, Baxter MG, Buxbaum JD and Harony-Nicolas H (2019). “Deletion of the KH1 Domain of Fmr1 Leads to Transcriptional Alterations and Attentional Deficits in Rats.” Cereb Cortex 29(5): 2228–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumireddy K, Li A, Yan J, Setoyama T, Johannes GJ, Orom UA, Tchou J, Liu Q, Zhang L, Speicher DW, Calin GA and Huang Q (2013). “Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step.” EMBO J 32(20): 2672–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM and Zhao X (2012). “Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome.” Hum Mol Genet 21(3): 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL and Hagerman PJ (2017). “Fragile X syndrome.” Nat Rev Dis Primers 3: 17065. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Esseltine JL, DeVries RA, Cregan SP and Ferguson SS (2014). “Metabotropic glutamate receptor 5 knockout reduces cognitive impairment and pathogenesis in a mouse model of Alzheimer’s disease.” Mol Brain 7: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey CM, Wilson NR and Hochstrasser M (2012). “Function and regulation of SUMO proteases.” Nat Rev Mol Cell Biol 13(12): 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST and Bear MF (2002). “Altered synaptic plasticity in a mouse model of fragile X mental retardation.” Proc Natl Acad Sci U S A 99(11): 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YS, Kim SS, Kim MS, Yoo NJ and Lee SH (2017). “Frameshift Mutation of FXR1 Encoding a RNA-Binding Protein in Gastric and Colorectal Cancers with Microsatellite Instability.” Pathol Oncol Res 23(2): 453–454. [DOI] [PubMed] [Google Scholar]
- Kalkunte R, Macarthur D and Morton R (2007). “Glioblastoma in a boy with fragile X: an unusual case of neuroprotection.” Arch Dis Child 92(9): 795–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaytor MD and Orr HT (2002). “The GSK3 beta signaling cascade and neurodegenerative disease.” Curr Opin Neurobiol 12(3): 275–278. [DOI] [PubMed] [Google Scholar]
- Khandjian EW, Corbin F, Woerly S and Rousseau F (1996). “The fragile X mental retardation protein is associated with ribosomes.” Nat Genet 12(1): 91–93. [DOI] [PubMed] [Google Scholar]
- Khayachi A, Gwizdek C, Poupon G, Alcor D, Chafai M, Casse F, Maurin T, Prieto M, Folci A, De Graeve F, Castagnola S, Gautier R, Schorova L, Loriol C, Pronot M, Besse F, Brau F, Deval E, Bardoni B and Martin S (2018). “Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation.” Nat Commun 9(1): 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khlghatyan J, Evstratova A, Chamberland S, Marakhovskaia A, Bahremand A, Toth K and Beaulieu JM (2018). “Mental Illnesses-Associated Fxr1 and Its Negative Regulator Gsk3beta Are Modulators of Anxiety and Glutamatergic Neurotransmission.” Front Mol Neurosci 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, McIlwain KA and Nelson DL (2001). “Comparative genomic sequence analysis of the FXR gene family: FMR1, FXR1, and FXR2.” Genomics 78(3): 169–177. [DOI] [PubMed] [Google Scholar]
- Lacoux C, Di Marino D, Boyl PP, Zalfa F, Yan B, Ciotti MT, Falconi M, Urlaub H, Achsel T, Mougin A, Caizergues-Ferrer M and Bagni C (2012). “BC1-FMRP interaction is modulated by 2’-O-methylation: RNA-binding activity of the tudor domain and translational regulation at synapses.” Nucleic Acids Res 40(9): 4086–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bipolar Genome S, Kelsoe JR and Greenwood TA (2016). “A genome-wide association study of bipolar disorder with comorbid eating disorder replicates the SOX2-OT region.” J Affect Disord 189: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, Li Y, Vershkov D, Cacace A, Young RA and Jaenisch R (2018). “Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene.” Cell 172(5): 979–992 e976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhu X, Zhu J, Liao S, Tang Q, Liu K, Guan X, Zhang J and Feng Z (2007). “Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray.” Oncol Rep 18(4): 943–951. [PubMed] [Google Scholar]
- Luca R, Averna M, Zalfa F, Vecchi M, Bianchi F, La Fata G, Del Nonno F, Nardacci R, Bianchi M, Nuciforo P, Munck S, Parrella P, Moura R, Signori E, Alston R, Kuchnio A, Farace MG, Fazio VM, Piacentini M, De Strooper B, Achsel T, Neri G, Neven P, Evans DG, Carmeliet P, Mazzone M and Bagni C (2013). “The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation.” EMBO Mol Med 5(10): 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Tian S, He S, Chen Q, Wang Z, Xiao X, Fu L and Lei X (2016). “The mechanism of action of FXR1P-related miR-19b-3p in SH-SY5Y.” Gene 588(1): 62–68. [DOI] [PubMed] [Google Scholar]
- Majumder M, House R, Palanisamy N, Qie S, Day TA, Neskey D, Diehl JA and Palanisamy V (2016). “RNA-Binding Protein FXR1 Regulates p21 and TERC RNA to Bypass p53-Mediated Cellular Senescence in OSCC.” PLoS Genet 12(9): e1006306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M and Palanisamy V (2020). “RNA binding protein FXR1-miR301a-3p axis contributes to p21WAF1 degradation in oral cancer.” PLoS Genet 16(1): e1008580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ and Tsai LH (2009). “Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling.” Cell 136(6): 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin T, Zongaro S and Bardoni B (2014). “Fragile X Syndrome: from molecular pathology to therapy.” Neurosci Biobehav Rev 46 Pt 2: 242–255. [DOI] [PubMed] [Google Scholar]
- McClure JJ and Palanisamy V (2019). “Muscle-Specific FXR1 Isoforms in Squamous Cell Cancer.” Trends Cancer 5(2): 82–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Nalavadi VC, Gross C, Yao X, Xing L, Laur O, Warren ST and Bassell GJ (2011). “Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling.” Mol Cell 42(5): 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrick LK, Hashimoto H, Cheng X and Warren ST (2015). “Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain.” Hum Mol Genet 24(6): 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, Witke W, Costa-Mattioli M, Sonenberg N, Achsel T and Bagni C (2008). “The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP.” Cell 134(6): 1042–1054. [DOI] [PubMed] [Google Scholar]
- Nordio L, Marques AT, Lecchi C, Luciano AM, Stefanello D and Giudice C (2018). “Immunohistochemical Expression of FXR1 in Canine Normal Tissues and Melanomas.” J Histochem Cytochem 66(8): 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Mittler G and Mann M (2004). “Identifying and quantifying in vivo methylation sites by heavy methyl SILAC.” Nat Methods 1(2): 119–126. [DOI] [PubMed] [Google Scholar]
- Papa L, Manfredi G and Germain D (2014). “SOD1, an unexpected novel target for cancer therapy.” Genes Cancer 5(1–2): 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps HM, Pierce JM, Murphy AJ, Correa H, Qian J, Massion PP and Lovvorn HN 3rd (2019). “FXR1 expression domain in Wilms tumor.” J Pediatr Surg 54(6): 1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Hassanein M, Hoeksema MD, Harris BK, Zou Y, Chen H, Lu P, Eisenberg R, Wang J, Espinosa A, Ji X, Harris FT, Rahman SM and Massion PP (2015). “The RNA binding protein FXR1 is a new driver in the 3q26–29 amplicon and predicts poor prognosis in human cancers.” Proc Natl Acad Sci U S A 112(11): 3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qie S, Majumder M, Mackiewicz K, Howley BV, Peterson YK, Howe PH, Palanisamy V and Diehl JA (2017). “Fbxo4-mediated degradation of Fxr1 suppresses tumorigenesis in head and neck squamous cell carcinoma.” Nat Commun 8(1): 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartier A, Poquet H, Gilbert-Dussardier B, Rossi M, Casteleyn AS, Portes VD, Feger C, Nourisson E, Kuentz P, Redin C, Thevenon J, Mosca-Boidron AL, Callier P, Muller J, Lesca G, Huet F, Geoffroy V, El Chehadeh S, Jung M, Trojak B, Le Gras S, Lehalle D, Jost B, Maury S, Masurel A, Edery P, Thauvin-Robinet C, Gerard B, Mandel JL, Faivre L and Piton A (2017). “Intragenic FMR1 disease-causing variants: a significant mutational mechanism leading to Fragile-X syndrome.” Eur J Hum Genet 25(4): 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A, Hollingworth D, Adinolfi S, Castets M, Kelly G, Frenkiel TA, Bardoni B and Pastore A (2006). “The structure of the N-terminal domain of the fragile X mental retardation protein: a platform for protein-protein interaction.” Structure 14(1): 21–31. [DOI] [PubMed] [Google Scholar]
- Renoux AJ, Carducci NM, Ahmady AA and Todd PK (2014). “Fragile X mental retardation protein expression in Alzheimer’s disease.” Front Genet 5: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ and Klann E (2015). “Dysregulation and restoration of translational homeostasis in fragile X syndrome.” Nat Rev Neurosci 16(10): 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzene M, Di Maira G, Tosoni K and Pinna LA (2010). “Assessment of CK2 constitutive activity in cancer cells.” Methods Enzymol 484: 495–514. [DOI] [PubMed] [Google Scholar]
- Sabanov V, Braat S, D’Andrea L, Willemsen R, Zeidler S, Rooms L, Bagni C, Kooy RF and Balschun D (2017). “Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice.” Neuropharmacology 116: 71–81. [DOI] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE and Zoghbi HY (2011). “Protein interactome reveals converging molecular pathways among autism disorders.” Sci Transl Med 3(86): 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Say E, Tay HG, Zhao ZS, Baskaran Y, Li R, Lim L and Manser E (2010). “A functional requirement for PAK1 binding to the KH(2) domain of the fragile X protein-related FXR1.” Mol Cell 38(2): 236–249. [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C and Moine H (2001). “The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif.” Embo j 20(17): 4803–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL and Giangrande A (2003). “CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein.” Neuron 38(6): 887–898. [DOI] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Moro A, Bagni C and Mandel JL (2001). “A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P.” Proc Natl Acad Sci U S A 98(15): 8844–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). “Biological insights from 108 schizophrenia-associated genetic loci.” Nature 511(7510): 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorova L and Martin S (2016). “Sumoylation in Synaptic Function and Dysfunction.” Front Synaptic Neurosci 8: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Pedersen S, Hasle H, Olsen JH and Friedrich U (2001). “Evidence of decreased risk of cancer in individuals with fragile X.” Am J Med Genet 103(3): 226–230. [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A, Champy MF, Sorg T, Pavlovic G, Wattenhofer-Donze M, Birling MC, Oulad-Abdelghani M, Eberling P, Ruffenach F, Joint M, Anheim M, Martinez-Cerdeno V, Tassone F, Willemsen R, Hukema RK, Viville S, Martinat C, Todd PK and Charlet-Berguerand N (2017). “Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome.” Neuron 93(2): 331–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamay-Ramot A, Khermesh K, Porath HT, Barak M, Pinto Y, Wachtel C, Zilberberg A, Lerer-Goldshtein T, Efroni S, Levanon EY and Appelbaum L (2015). “Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish.” PLoS Genet 11(12): e1005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SL, Curnow EC, Easley CA, Jin P, Hukema RK, Tejada MI, Willemsen R and Usdin K (2014). “Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI).” J Neurodev Disord 6(1): 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Choi M, Siomi MC, Nussbaum RL and Dreyfuss G (1994). “Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome.” Cell 77(1): 33–39. [DOI] [PubMed] [Google Scholar]
- Siomi H, Siomi MC, Nussbaum RL and Dreyfuss G (1993). “The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein.” Cell 74(2): 291–298. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Higashijima K, Ishizuka A and Siomi H (2002). “Casein kinase II phosphorylates the fragile X mental retardation protein and modulates its biological properties.” Mol Cell Biol 22(24): 8438–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Zhang Y, Siomi H and Dreyfuss G (1996). “Specific sequences in the fragile X syndrome protein FMR1 and the FXR proteins mediate their binding to 60S ribosomal subunits and the interactions among them.” Mol Cell Biol 16(7): 3825–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjekloca L, Konarev PV, Eccleston J, Taylor IA, Svergun DI and Pastore A (2009). “A study of the ultrastructure of fragile-X-related proteins.” Biochem J 419(2): 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL and Paylor R (2006). “Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins.” Hum Mol Genet 15(12): 1984–1994. [DOI] [PubMed] [Google Scholar]
- St-Denis N, Gabriel M, Turowec JP, Gloor GB, Li SS, Gingras AC and Litchfield DW (2015). “Systematic investigation of hierarchical phosphorylation by protein kinase CK2.” J Proteomics 118: 49–62. [DOI] [PubMed] [Google Scholar]
- Stetler A, Winograd C, Sayegh J, Cheever A, Patton E, Zhang X, Clarke S and Ceman S (2006). “Identification and characterization of the methyl arginines in the fragile X mental retardation protein Fmrp.” Hum Mol Genet 15(1): 87–96. [DOI] [PubMed] [Google Scholar]
- Suhl JA and Warren ST (2015). “Single-Nucleotide Mutations in FMR1 Reveal Novel Functions and Regulatory Mechanisms of the Fragile X Syndrome Protein FMRP.” J Exp Neurosci 9(Suppl 2): 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YJ, Conti J, Currie JR, Brown WT and Denman RB (2000). “RNAs that interact with the fragile X syndrome RNA binding protein FMRP.” Biochem Biophys Res Commun 275(3): 973–980. [DOI] [PubMed] [Google Scholar]
- Tabet R, Moutin E, Becker JA, Heintz D, Fouillen L, Flatter E, Krezel W, Alunni V, Koebel P, Dembele D, Tassone F, Bardoni B, Mandel JL, Vitale N, Muller D, Le Merrer J and Moine H (2016). “Fragile X Mental Retardation Protein (FMRP) controls diacylglycerol kinase activity in neurons.” Proc Natl Acad Sci U S A 113(26): E3619–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabolacci E, Palumbo F, Nobile V and Neri G (2016). “Transcriptional Reactivation of the FMR1 Gene. A Possible Approach to the Treatment of the Fragile X Syndrome.” Genes (Basel) 7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata A, Matsumoto N and Kato T (2017). “Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci.” Nat Commun 8: 14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamanini F, Bontekoe C, Bakker CE, van Unen L, Anar B, Willemsen R, Yoshida M, Galjaard H, Oostra BA and Hoogeveen AT (1999). “Different targets for the fragile X-related proteins revealed by their distinct nuclear localizations.” Hum Mol Genet 8(5): 863–869. [DOI] [PubMed] [Google Scholar]
- Tamanini F, Kirkpatrick LL, Schonkeren J, van Unen L, Bontekoe C, Bakker C, Nelson DL, Galjaard H, Oostra BA and Hoogeveen AT (2000). “The fragile X-related proteins FXR1P and FXR2P contain a functional nucleolar-targeting signal equivalent to the HIV-1 regulatory proteins.” Hum Mol Genet 9(10): 1487–1493. [DOI] [PubMed] [Google Scholar]
- Tamanini F, Meijer N, Verheij C, Willems PJ, Galjaard H, Oostra BA and Hoogeveen AT (1996). “FMRP is associated to the ribosomes via RNA.” Hum Mol Genet 5(6): 809–813. [DOI] [PubMed] [Google Scholar]
- Tassone F, Beilina A, Carosi C, Albertosi S, Bagni C, Li L, Glover K, Bentley D and Hagerman PJ (2007). “Elevated FMR1 mRNA in premutation carriers is due to increased transcription.” RNA 13(4): 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R, Pallesen J, Daugaard TF, Borglum AD and Nielsen AL (2013). “Genome wide assessment of mRNA in astrocyte protrusions by direct RNA sequencing reveals mRNA localization for the intermediate filament protein nestin.” Glia 61(11): 1922–1937. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A and Ponten F (2017). “A pathology atlas of the human cancer transcriptome.” Science 357(6352). [DOI] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT and Oostra BA (1993). “Characterization and localization of the FMR-1 gene product associated with fragile X syndrome.” Nature 363(6431): 722–724. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP and et al. (1991). “Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome.” Cell 65(5): 905–914. [DOI] [PubMed] [Google Scholar]
- Weiler IJ and Greenough WT (1999). “Synaptic synthesis of the Fragile X protein: possible involvement in synapse maturation and elimination.” Am J Med Genet 83(4): 248–252. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B and Bagni C (2003). “The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses.” Cell 112(3): 317–327. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Panasiti V, Carotti S, Zingariello M, Perrone G, Sancillo L, Pacini L, Luciani F, Roberti V, D’Amico S, Coppola R, Abate SO, Rana RA, De Luca A, Fiers M, Melocchi V, Bianchi F, Farace MG, Achsel T, Marine JC, Morini S and Bagni C (2017). “The fragile X mental retardation protein regulates tumor invasiveness-related pathways in melanoma cells.” Cell Death Dis 8(11): e3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA and Wu JQ (2014). “An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex.” J Neurosci 34(36): 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, O’Connor JP, Siomi MC, Srinivasan S, Dutra A, Nussbaum RL and Dreyfuss G (1995). “The fragile X mental retardation syndrome protein interacts with novel homologs FXR1 and FXR2.” Embo j 14(21): 5358–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]