

Germline variants when organized as genomic patterns are associated with cancer risk, oncogenic pathways, and clinical outcomes.
Abstract
There is an ongoing debate on the importance of genetic factors in cancer development, where gene-centered cancer predisposition seems to show that only 5 to 10% of the cancer cases are inheritable. By conducting a systematic analysis of germline genomes of 9712 cancer patients representing 22 common cancer types along with 16,670 noncancer individuals, we identified seven cancer-associated germline genomic patterns (CGGPs), which summarized trinucleotide mutational spectra of germline genomes. A few CGGPs were consistently enriched in the germline genomes of patients whose tumors had smoking signatures or correlated with oncogenesis- and genome instability–related mutations. Furthermore, subgroups defined by the CGGPs were significantly associated with distinct oncogenic pathways, tumor histological subtypes, and prognosis in 13 common cancer types, suggesting that germline genomic patterns enable to inform treatment and clinical outcomes. These results provided evidence that cancer risk and clinical outcomes could be encoded in germline genomes.
INTRODUCTION
The importance of genetic factors, unmodifiable random intrinsic DNA replication errors (“bad-luck”) (1, 2), and environmental factors (“environment-driven”) (3) in cancer development has been an ongoing debate. Sorting out the contribution importance of these factors to cancer development is critical to understand tumorigenesis, which can help in making treatment decisions and can direct in developing prevention strategies aimed at reducing cancer burden. At present, both bad-luck and environment-driven hypotheses are dominant and suggest that heredity plays a minimal role in tumorigenesis.
It is noted that both bad-luck and environment-driven hypotheses are cancer cell centered, which means that they only tackle the question from the cancer cell point of view but do not consider other aspects such as the host immune system. The contribution of genetic predisposition to cancer development and progression has been recognized for centuries and has not yet been widely investigated (4–6). Compared to somatic mutations in tumors, germline malignant variants face looser selection pressure and are inherited along with numerous passenger mutations (7). Systemic genome sequencing of normal tissues of cancer patients provides a considerable chance to study the germline genomic variants.
As the cancer-driving genetic germline variants distribute sparsely across genomes and are restricted to a small set of genes (7), investigations of germline genetic variants have been largely restricted to the known cancer driver genes including tumor suppressors and the ones closely related to DNA repair, oncogenic signaling pathways, and cell cycle (8, 9). For example, individuals diagnosed with colorectal cancer among the first-degree relatives who have Lynch syndrome (10) have been shown to carry DNA repair defects that disabled DNA damage resurrection in germ lines and therefore accumulated genetic alterations that led to colon cancer. Germline mutated Ras has been shown to be associated with developmental disorders (11) and cardiofaciocutaneous syndrome (12). Germline BRCA1/2 mutations are known to be directly associated with increased risks in multiple cancer types including breast and ovarian cancers (13–15). More recently, the systemic analysis of the associations between germline variants and cancer susceptibility has been performed (16). This analysis, on the one hand, supports the idea that the important cancer-related information is implicated in germline genomes, but on the other hand, each of the selected variants has a small penetrance for a small fraction (i.e., 1 to 2%) of the population only (i.e., depending on allele frequencies), indicating that individual germline variants could not be a sole informative descriptor of germline genomes. Consequently, although individual gene- or variant-centered studies have proven to be informative, so far, only a handful of associations between genes and cancer risk have been determined (17).
Thus, we took another approach and investigated whether a genomic pattern or a substantial, repeatedly occurring sequential profile in germline genomes could serve as a promising measurement for malignant genetic predisposition. To this end, we conducted a systematic analysis of the germline genomes of 9712 cancer patients representing 22 cancer types and 16,670 noncancer individuals and revealed seven cancer-related germline genomic patterns. Further investigations focusing on 7214 cancer patients of European ancestry highlighted that one of them (i.e., susceptibility genomic pattern for smoking or a conditional genetic risk factor) was significantly more enriched in the germline genomes of smoker patients than in those of nonsmoker patients. These results suggested that germline genomic patterns could provide an inspiring measurement for cancer susceptibility. Furthermore, germline subgroups defined by the patterns were significantly correlated to clinically meaningful differences in terms of cancer histological subtypes, oncogenic mechanisms, and survival outcomes in 10 common cancer types. These results implied that cancer genetic risk could be encoded in germline genomes in the form of not only genes such as BRCA1/2 but also genomic patterns. We speculated that further analysis of germline genomic patterns may unravel genetic mechanisms of diseases, where molecular mechanisms could be beyond the current gene-centered paradigm.
RESULTS
Genomic patterns in cancer germline genomes
We obtained 430,772,708 germline substitutions from the whole-exome sequencing data of 9712 cancer patients in The Cancer Genome Atlas (TCGA) (18), representing 22 cancer types and 46,998,783 somatic substitutions from their paired tumor genomes. Germline mutational catalog was generated by summarizing potential substitution profiles, where variations were incorporated with sequential sequence contexts. Meanwhile, whole-exome data of 16,670 noncancer individuals from three cohorts (see Materials and Methods) were merged to form a noncancer dataset, which served as the background. Blood cancers were not included for further analysis (see Materials and Methods). Cancers from 22 primary sites included adrenal gland (ACC), bladder (BLCA), bone marrow (LAML), brain (LGG and GBM), breast (BRCA), cervix (CESC), colon (COAD), eye (UVM), head and neck (HNSC), kidney (KIRP, KIRC, and KICH), liver (LIHC), lung (LUAD and LUSC), lymph node (DLBC), ovary (OV), pancreas (PAAD), bone, muscle, and fat (SARC), prostate (PRAD), skin (SKCM), stomach (STAD), testis (TGCT), thyroid (THCA), and uterus (UCEC).
Genetic variants aggregated in a germline genome could be viewed as accumulated outcomes of mutational processes that happened to its ancestral genomes and evolutionary genetic polymorphisms. A germline genomic pattern presents a pan-genome enrichment of recurring substitutions in a sequence context within germline genomes. Intuitively, a genomic pattern pertaining to cancer risk is most likely to display detectable enrichment in the germline genomes of cancer patients. Genomic patterns are buried in the high-dimensional genomic sequence features so that extracting the genomic patterns becomes a tedious and computationally intensive task by applying statistical methods. Nonnegative matrix factorization (NMF) enables interpretable feature extraction from high-dimensional data (19, 20) and has been used in extracting biological meaningful somatic mutational signatures in tumors (21–23). Therefore, we adapted this approach for germline mutational catalogs (Materials and Methods; figs. S1 and S2), demonstrated its stability, and then applied it to the germline mutational catalogs of the cancer patients and noncancer population (for background noise reduction). By doing so, we identified seven distinct genomic patterns [named cancer-associated germline genomic pattern (CGGP)] (Fig. 1 and data S1) along with their contributions that were represented by their weighing factors (data S2). Further analysis confirmed that none of the genomic patterns were sequencing artifacts (23). To test the robustness of the germline genomic patterns against the effects of potential sequencing artifacts, we have performed various validation tests using different sets of variants, subsets of samples, or conditions to reidentified CGGPs. Briefly, we have tried to (i) remove variants in repeats and outlier samples that are likely to be associated with batch effects according to previous studies (named as the DP20masked condition here) (24–26); (ii) remove the variants in low mappability regions (similar to the DP20masked condition); (iii) use alternative read depth thresholds (similar to the DP20masked condition); (iv) remove low-quality variants; (v) consider subsets of samples from different sequencing centers, whole-genome amplification protocol before sequencing, or exome capture array platforms; (vi) split the dataset by population ancestry [following the implication from Harris and Pritchard (27)]; (vii) use exon-defined strand-specific mutational profiles; and (viii) use germline variants called by Huang et al. (16). Detailed methods are available in Supplementary Materials and Methods. In general, we found that the genomic patterns were reproducible (with collapsed cosine similarities ≥ 0.95 and average cosine similarities ≥ 0.75 to the original germline genomic patterns) in these conditions (table S1). Besides, the seven CGGPs were not evenly stable across all conditions. CGGP_C, CGGP_D, CGGP_E, CGGP_F, and CGGP_G were stable for most of the conditions. By contrast, CGGP_A and CGGP_B were less stable, showing a cosine similarity of <0.75 in several conditions. Therefore, these two CGGPs should be interpreted with caution, and these two CGGPs could be further refined in the future when more cancer germline genomic data are available. Last, as the DP20masked condition might reduce the influence of repeat sequences and outlier samples, we also included the CGGP genomic pattern matrix and the genomic pattern’s weighing factor matrix of DP20masked condition for each TCGA sample available in data S1 and S2, respectively.
Fig. 1. CGGPs deciphered from the germline genomes of cancer patients.
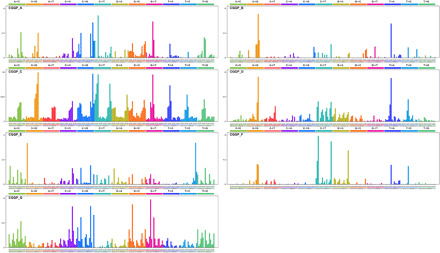
The profile of each CGGP is displayed in the order of 12 substitution subtypes: A>C, A>G, A>T, C>A, C>G, C>T, G>A, G>C, G>T, T>A, T>C, and T>G, which are also denoted in different colors. The characters in the bottom subtabs indicate the reference trinucleotides, and the substituted nucleotides in the central position are colored based on the 12 substitution subtypes.
Figure 1 illustrates the visualization of seven CGGPs. CGGP_A, CGGP_D, and CGGP_F were mainly characterized by an enrichment of transition mutations. CGGP_F was characterized by A>G and T>C variants, while CGGP_A and CGGP_D contained mainly C/T>T/C and G/A>A/G variants. Other context-dependent mutations were also revealed in these three patterns, and when combined with other CGGPs, CGGP_A and CGGP_D mainly contributed to lung, pancreas, and stomach cancers.
Signals of CGG>CAG and CCG>CTG were the traits of CGGP_B. When used in combination with CGGP_C or CGGP_E, CGGP_B was a potential contributor to the brain, breast, cervix, colon, rectum, kidney, lung, and stomach cancers (see below).
CGGP_C appeared to be a dominant pattern, with the strongest assigned signals and weights in samples among CGGPs. This pattern was mainly accounted for the transitions of A>G, G>A, C>T, and T>C with modest preferences for sequence contexts.
Conspicuous signals in CGGP_E led to the formation of a local triple nucleotide profile of repeated nucleotides, including CAC>CCC, GAG>GGG, ACA>AAA, GCG>GGG, CGC>CCC, TGT>TTT, CTC>CCC, and GTG>GGG. We found that CGGP_E was a genetic susceptibility germline genomic pattern for tobacco smoke, and the smokers whose germ line carried CGGP_E had elevated risks in 13 common cancer types. Although enrichment of CGGP_E would imply biologically meaningful information in certain cancer types, the combination of multiple CGGPs provides much more insights (see below).
CGGP_G was characterized in the preference of TA/GT>TG/AT and AC/TA>AT/CA mutations as well as modest overall assigned signals in other mutational profiles. Weights (i.e., representing the contribution of the pattern to a sample) for CGGP_G in germline genomes of cancer patients were significantly higher than those granted by noncancer individuals; the differences between them were also the most prominent in seven CGGPs (fig. S3). To examine whether the seven genomic patterns were cancer specific, we applied our modified NMF approach to the germline exome sequences of noncancer individuals (n = 16,670) and identified six genomic patterns. Patterns except CGGP_E were reproduced (cosine similarities were 0.99, 0.98, 1.00, 0.97, 0.93, and 1.00 for CGGP_ A, B, C, D, F, and G, respectively). We further extended this analysis to the germline dataset derived from the combination of the cancer patients and noncancer individuals (n = 9712 + 16,670) and found that all seven patterns were reproducible. These results implied that CGGP_E might be a pattern that was more enriched in cancer patients’ germline genomes than noncancer individuals.
To examine whether genomic patterns are different between Asian, African, and European ancestry patients, we tried to reproduce CGGPs in 7214 TCGA cancer patients of European ancestry. The resulting CGGPs had a high similarity with the original CGGPs (cosine similarity = 0.99; data S1). The algorithm for generating CGGPs requires several thousands of samples to obtain stable CGGPs; however, the sample sizes of the Asian ancestry (n = 593) and African ancestry (n = 898) patients in TCGA were not sufficient to obtain stable CGGPs. Therefore, we decided to focus on the patients of European ancestry in the following analyses.
CGGP_E was a susceptibility genomic pattern for tobacco smoke
A genetic predisposition could collaborate with exogenous cancer risk factors to drive tumorigenesis. While tobacco smoke is a well-recognized exogenous risk factor for inducing cancer, less than 15% of the smokers would end up developing lung cancer. Individual fates are affected not only by exposing to mutagens but also by their genetically determined sensitivity to mutagens. Thus, we determined whether any CGGP could be associated with tobacco smoking in cancer patients. We therefore examined the enrichment of each CGGP between the germ lines of smoker and nonsmoker patients.
Previously, the Catalogue of Somatic Mutations in Cancer (COSMIC) has identified 30 tumor somatic mutational signatures (http://cancer.sanger.ac.uk/cosmic/signatures), in which signatures 4 and 29 have been reported to be the somatic mutational imprints of tobacco smoking and tobacco chewing habit in tumors. It has been reported that at least 17 cancer types were linked to smoking based on the presence of signatures 4 and 29 in tumors associated with smoking (28). To examine the association between CGGPs with smoking, we partitioned the cancer patients into two groups: One was affected by tobacco smoking and the other was not, based on the presence of signatures 4 or 29 in tumors using the methods described previously (28). Comparative analyses of each of the CGGPs in the germline genomes between smokers and nonsmokers revealed that CGGP_E was significantly enriched in the smoking group across 13 common cancer types including lung (LUAD + LUSC, P = 8.53 × 10−2, ratio of means = 1.12; t test), brain (LGG + GBM, P = 1.05 × 10−12, ratio of means = 1.58; t test), prostate (P = 5.31 × 10−11, ratio of means = 2.68; t test), kidney (KIRP + KIRC + KICH, P = 5.08 × 10−5, ratio of means = 1.20; t test), breast (P = 5.85 × 10−4, ratio of means = 1.18; t test), stomach (P = 9.48 × 10−3, ratio of means = 1.23; t test), rectal (P = 4.95 × 10−2, ratio of means = 1.19; t test), thyroid (P = 3.43 × 10−2, ratio of means = 1.71; t test), and uterine (P = 5.48 × 10−2, ratio of means = 1.11; t test) cancers (Fig. 2A). Cancer types with insufficient sample sizes (i.e., less than 50) were not examined. A higher weight of CGGP_E in the germline genome had a significant positive correlation with the presence of smoking-related somatic mutational signature (COSMIC signature 4) in tumor genome as for the whole patient population (Fig. 2B).
Fig. 2. The relationship between germline genomic pattern E and tobacco mutagen sensitivity.
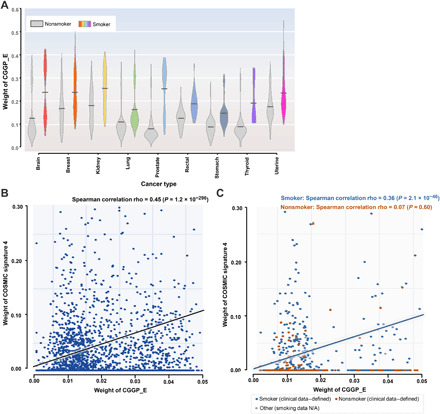
(A) Germline genomic pattern (CGGP_E) is highly enriched in smoker patients than in nonsmoker patients in nine cancer types (P < 0.1, t test). Gray violin boxes represent nonsmoker patients, while the boxes with other colors represent smoker patients. (B) Relative contributions (i.e., weights) of CGGP_E in germline genomes are significantly correlated to the possession of smoking-related somatic mutational signature 4 (COSMIC signature 4) in their paired tumor genomes. (C) Relative contributions (i.e., weights) of CGGP_E in germline genomes are positively correlated with COSMIC signature 4 in their paired tumors in the TCGA clinical information–defined smokers but not in the TCGA clinical information–defined nonsmokers. Notably, only cancer types with a sizable amount of clinical smoking information (e.g., HNSC, Lung, and PAAD) were considered. N/A, not applicable.
We further examined these associations in cancer smokers and nonsmokers defined by the clinical annotations. Among the TCGA samples, there were four cancer types (BLCA, HNSC, Lung, and PAAD) annotated with smoking information (28), representing 1667 smokers and 481 nonsmokers. We found that CGGP_E was significantly enriched in smokers than in nonsmokers (P = 4.50 × 10−26, ratio = 1.20; t test). When restricting the same analysis to individual cancer types, we found that similar results were obtained in lung cancer (P = 2.93 × 10−3, sample ratio = 179:906, ratio = 1.18; t test) and HNSC (P = 4.97 × 10−2, sample ratio = 208:393, ratio = 1.10; t test) but not in BLCA and PAAD. Notably, bladder tumors rarely contain signature 4 in tumor samples (28), and it may explain why we did not observe the enrichment of CGGP_E in smokers. It should be noted that some nonsmokers defined by clinical information could be passive smokers, with exposure to radon or air pollution or with self-reported mistakes. Furthermore, a positive association (Spearman correlation rho = 0.36, P = 2.1 × 10−46; Fig. 2C) between CGGP_E and signature 4 was found in clinically annotated smokers but not in nonsmokers. When restricted to specific cancer type, Spearman correlations between CGGP_E with signature 4 in lung and head and neck cancers were 0.13 (P = 1.2 × 10−4) and 0.44 (P < 2.2 × 10−16), respectively. The Spearman correlation in pancreatic cancer was not available because of the lack of enough smoking samples, while for TCGA clinical information–defined nonsmokers, no significant correlation was found for these cancer types. Together, the results were similar when smokers and nonsmokers were defined either by signatures 4 and 29 or by clinically annotated smoking information.
Next, we determined whether age and gender of patients affect the association between CGGP_E and signature 4. We found that CGGP_E and signature 4 were positively associated in both male (n = 1152, Spearman rho = 0.48, P < 2.2 × 10−16) and female (n = 515, Spearman rho = 0.21, P = 8.0 × 10−6) smokers with a similar association strength, suggesting that smoking-induced cancer risk did not differ appreciably between genders. These results agree with previous epidemiologic surveys of lung cancer risk and smoking (29, 30). Further, such positive associations were observed in both younger smokers (<66 years old at diagnosis, n = 726, Spearman rho = 0.46, P < 2.2 × 10−16) and older smokers (>65 years old at diagnosis, n = 864, Spearman rho = 0.19, P = 6.2 × 10−8), suggesting that for a person who had a higher contribution of CGGP_E in the germline genome, tobacco smoke did not substantially affect more for younger than older smokers to get cancer. However, we did not have the data about the starting smoking age for these patients. Thus, one should be cautious when interpreting these results.
We also examined the association between smoking quantity and CGGP_E and found no significant association between CGGP_E and pack-years of smoking. A positive correlation was reported between smoking quantity and signature 4 in lung and liver tumors but not in other cancer types (28). Together, these results suggested that CGGP_E might be a susceptible CGGP for smoking, i.e., smoking alone could not necessarily drive tumorigenesis unless CGGP_E was presented in the germline genome of a tobacco smoker. Furthermore, in general, a higher contribution of CGGP_E implied shorter survival (P = 0.01, log-rank test). More significant results were observed in four cancer types including adrenal gland (P = 1.1 × 10−4), bladder (P = 5.3 × 10−4), kidney (P = 6.2 × 10−3), and stomach cancers (P = 3.3 × 10−3). These results suggested that CGGP_E could have a quantitative effect on cancer development and prognosis. Unexpectedly, such a correlation was not observed in lung cancer (see Discussion). Further, we obtained similar results when assigning CGGP_E in a binary fashion to patients (see Materials and Methods).
To further understand the molecular mechanisms of CGGP_E’s impact on cancer development, we ranked patients on the basis of the contributions of CGGP_E in germ lines in descending order and selected patients from the upper quartile and the lower quartile to form two subgroups. Genes containing mutations that fitted CGGP_E were examined and compared between the two groups. Well-known cancer drivers, such as fibronectin type III (FN3), receptors of tyrosine kinases (RTKs), and genes encoding epidermal growth factor (EGF)–like domain proteins and insulin-like growth factor binding proteins, were significantly more frequently mutated in the upper quartile group [false discovery rate (FDR) < 0.05; data S3]. These results suggested that the presence of CGGP_E in germ lines might introduce significantly more mutations to RTKs and other cancer driver genes, which, in turn, could induce higher carcinogenesis risks for individuals when exposed to mutagenic agents. Thus, we speculated that, except tobacco smoke, CGGP_E might be a susceptibility genomic pattern for other mutagens. This hypothesis could be further tested in the future when related data become available.
These results provided a potential rationale for the long-established observation that less than 20% of heavy smokers would develop lung cancer in their lifetime. When extending the same analysis to other CGGPs to understand molecular mechanisms, we did not obtain biologically meaningful results. However, in specific tumor types, other CGGPs showed a casual positive correlation with smoking signatures [e.g., CGGP_G was significantly enriched in smoker patients of colorectal (P = 3.2 × 10−7), head and neck (P = 3.6 × 10−5), kidney (P = 9.1 × 10−18), lung (P = 9.3 × 10−19), prostate (P = 1.0 × 10−32), stomach (P = 2.9 × 10−15), and thyroid (P = 6.4 × 10−6) cancers], suggesting that latent mechanisms might still be revealed for the CGGPs.
CGGPs affected the somatic mutation of key oncogenic genes in tumors
To explore whether the CGGPs are associated with somatic mutations of key cancer drivers and family history of cancer, in each cancer type, we partitioned the patients into two subgroups, either those who carried or those who did not carry any mutation in a given gene, and examined whether the weighing factors of CGGPs differed between the subgroups (see Materials and Methods). Because of the sample size limitation, only the most frequently somatically mutated genes across all cancer types reported by TCGA were examined: TP53, PIK3CA, KMT2D, FAT4, ARID1A, PTEN, KMT2C, APC, KRAS, FAT1, ATRX, NF1, ZFHX3, IDH1, ATM, TRRAP, RNF213, AKAP9, and GRIN2A. Patients of each cancer type were partitioned based on whether a given gene was nonsynonymously mutated in their tumor tissues, and distributions of CGGP weighing factors were examined between such subgroups.
Our observations implied that CGGPs had impacts on which somatically mutated genes were to be selected in tumors (Fig. 3 and table S2). In 18 cancer types, we observed that higher weighing factors of certain CGGPs were associated with a higher somatic mutation frequency of the above genes (see Materials and Methods). Across six cancer types (LUSC, BLCA, COAD, OV, PAAD, and THCA), AKAP9 mutation was significantly associated with higher weighing factors of CGGP_C, D, or G. For BLCA, COAD, GBM, OV, and SKCM, KMT2D mutation was significantly associated with higher weighing factors of CGGP_A, CGGP_B, CGGP_B and F, CGGP_E, and CGGP_F and G. Some associations were observed in individual cancer types. For example, significant associations were observed between APC somatic mutations and CGGP_A in COAD and between ATM somatic mutation and CGGP_A in KIRC. These results suggested that germline genomic patterns could exert constraints on somatically mutated genes in tumors, and although CGGP_E was prominent in cancer patients, the combination of multiple germline genomic patterns might exert stronger constraints on selecting tumor somatic mutations. Therefore, CGGP combinations could serve as features for classifying cancer patients through thresholding or unsupervised clustering methods.
Fig. 3. Enrichment of germline CGGPs implied somatic mutations in oncogenic genes in paired tumors.

Elevated weighing factors of CGGPs are correlated with the higher prevalence of somatic mutations on the 18 most frequently mutated genes across all cancer types. The results for 18 common cancer (sub)types are summarized as a pie chart array. Each small pie chart in the plot is split to seven slots (starting from the top-right slot and going clockwise, CGGP_A to CGGP_G alphabetically). A gray slot indicates that no significant result is observed in corresponding cancer (sub)type and somatically mutated gene pair. An orange or a light blue slot indicates a positive (ratio of mean CGGP weights > 1, mutated samples versus nonmutated samples) or negative (ratio of mean CGGP weights < 1, mutated samples versus nonmutated samples) significant association (FDR < 0.25) of a CGGP and a somatically mutated gene in corresponding cancer (sub)type, respectively. A dark pink or a dark blue slot indicates a high confidence association (FDR < 0.25 and empirical P < 0.05 by 10,000 times of randomization tests) of a CGGP and a somatically mutated gene in corresponding cancer (sub)type, respectively.
Single CGGPs and their combinations were associated with cancer types
In search of meaningful one or more CGGP combinations to distinguish cancer and noncancer genomes, we conducted statistical analyses of comprehensive combinations of the CGGPs (k = 1, 2, and 3). Germline genomic data of the noncancer population (n = 16,670) were further used, and statistical comparisons were conducted between patients of each cancer type and noncancer population (see Materials and Methods). We demonstrated that a set of single CGGPs or their combinations were significantly more prevalent in cancer patients compared to noncancer population, and germline groups defined by single CGGPs or their combinations (k = 1, 2, and 3) were significantly enriched in distinct cancer types (Fig. 4 and table S3). Genomic patterns CGGP_E, D, and G have more contributions to cancer samples than noncancer samples in most cancer types, whereas CGGP_A and F have smaller contributions to cancer samples than noncancer samples in most cancer types (Fig. 4). CGGP_B’s contributions were higher in some cancer types but smaller in some other cancer types than noncancer samples (Fig. 4). These results suggested that germline genomes of cancer and noncancer could have certain differences in genomic sequence arrangements; further, they have an implication for the germline pattern’s role in tissue-specific carcinogenesis. For example, patients with higher contributions of CGGP_E were more enriched in being diagnosed with 13 of 16 cancer types, such as LUAD, LUSC, SKCM, and STAD (FDR = 1.83 × 10−96, 1.04 × 10−80, 9.92 × 10−30, and 1.55 × 10−59, respectively, χ2 test). Patients with higher contributions of the CGGP_B + CGGP_E combination in their germ lines were more enriched in being diagnosed with BLCA and GBM (FDR = 4.11 × 10−9 and 4.51 × 10−12, respectively, χ2 test). GBM, BLCA, BRCA, STAD, SKCM, COAD, and THCA diagnoses were significantly enriched in patients with higher contributions of the CGGP_D + CGGP_E combination (FDR = 6.94 × 10−57, 8.56 × 10−53, 7.04 × 10−157, 4.87 × 10−67, 1.36 × 10−48, 1.89 × 10−61, and 1.51 × 10−76, respectively, χ2 test). Along this line, one tri-CGGP combination, CGGP_B + CGGP_D + CGGP_E, was enriched in GBM (FDR = 2.59 × 10−11, χ2 test). Furthermore, patients who carried one CGGP (CGGP_A/CGGP_D/CGGP_E/CGGP_F) in their germline genomes were significantly enriched with six COSMIC somatic mutational signatures in their tumor tissues compared to patients without the CGGPs (table S4). Patients who carried the CGGP_D + CGGP_E combination in their germline genomes were significantly enriched with six COSMIC signatures in their corresponding tumor tissues in comparison with patients without this CGGP combination, implying that germline genomic patterns had the potential to shape or select somatic mutational processes during tumorigenesis. In summary, these results suggested that CGGPs or their combinations could be associated with triggering of endogenous mutations in tumors and could shape the carcinogenesis and tumor proliferation process.
Fig. 4. Differential contribution of CGGPs in the germline genomes between cancer and noncancer samples.
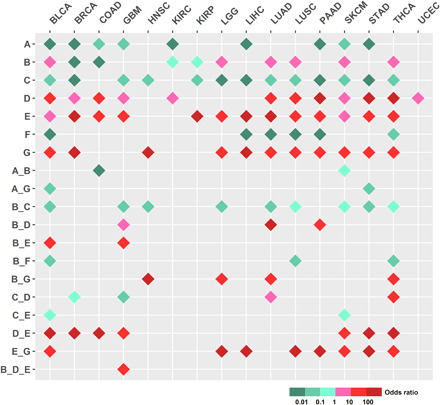
One CGGP or CGGP combinations that had less than 50 samples were excluded. χ2 test (FDR < 0.25) was used to test whether cancer and noncancer samples could be distinguished by CGGPs or CGGP combinations.
We also examined the differential contributions of CGGPs or CGGP combinations between cancer types and subtypes. As shown in table S5, CGGP_A was more enriched in LIHC than in LUAD (FDR = 0.01, odds ratio = 2.45) and BRCA (FDR = 0.03, odds ratio = 2.14). Further, the CGGP_A + CGGP_E combination was more enriched in LUSC than in LUAD (FDR = 0.02, odds ratio = 4.61), BRCA (FDR = 4.57 × 10−3, odds ratio = 3.80), and UCEC (FDR = 0.03, odds ratio = 3.48). The enrichments of certain CGGP (or CGGP combination) in GBM, OV, TGCT, LIHC, LUAD, and BRCA in comparison with other cancer types were also observed. CGGPs also differentially contributed to cancer subtypes of breast and lung cancers (table S5). For example, either single CGGP_A, B, C, D, and G or the combinations of CGGP_A + CGGP_G, CGGP_B + CGGP_C, CGGP_B + CGGP_F, and CGGP_C + CGGP_G were significantly different between the basal and luminal breast cancer subtypes. Similarly, the CGGP_A + CGGP_E combination could distinguish LUAD and LUSC lung cancer subtypes. These results suggested that CGGPs could distinguish cancer types and subtypes.
CGGP-defined germline subgroups were associated with distinct tumor histological subtypes, oncogenic pathways, and prognosis
We next explored whether CGGPs could classify germline genomes into subgroups for a given cancer type. To do so, we clustered the patients of each cancer type (cancer types with insufficient sample sizes were excluded for the analysis) based on CGGP contribution profiles in germline genomes. The partitions were conducted using unsupervised hierarchical clustering. For most cancer types, the CGGPs were able to classify germ lines into stable subgroups; by applying dimension reduction methods such as principal components analysis (PCA) ahead of hierarchical clustering, we obtained similar results. CGGP-defined germline subgroups exhibited correlations with cancer histological subtypes in four cancer types: brain, lung, kidney, and stomach cancers. Cancer types that lacked histology diagnosis information were not analyzed. Brain cancer patients were clustered into three germline subgroups. Subgroup 1 was enriched with GBM samples, while subgroup 3 was enriched with less-aggressive astrocytoma samples (Fig. 5A). Subgroup 2 was also enriched with GBM samples but, unlike subgroup 1, was in favor of CGGP_A rather than CGGP_E (subgroup 1 + 2 n = 289, subgroup 2 n = 520, P = 1.00 × 10−4 for GBM, χ2 test). GBM-enriched germline subgroups had significantly shorter survival than the other subgroup (P = 4.00 × 10−5 and 7.00 × 10−16; log-rank test). Likewise, three germline subgroups were found in kidney cancer patients. Two of the subgroups were enriched with clear cell renal carcinoma; the other was dominated by KIRP and KICH (P = 2.40 × 10−2, χ2 test). Significant survival differences were also observed between the subgroups (subgroup 1 versus 2: P = 7.00 × 10−2, subgroup 1 versus 3: P = 4.00 × 10−3; log-rank test). Furthermore, three germline subgroups of lung cancer patients were significantly enriched with adenocarcinoma and squamous cell carcinoma samples (subgroup 1 n = 124, subgroup 2 + 3 n = 603, P = 1.60 × 10−3, χ2 test). These results suggested that CGGPs in the germline genomes of these three tissues could determine which histological subtypes of their tumors could be developed and the patient prognosis as well.
Fig. 5. Associations between CGGP-defined subgroups with cancer histological subtypes, oncogenic pathways, and clinical outcomes.
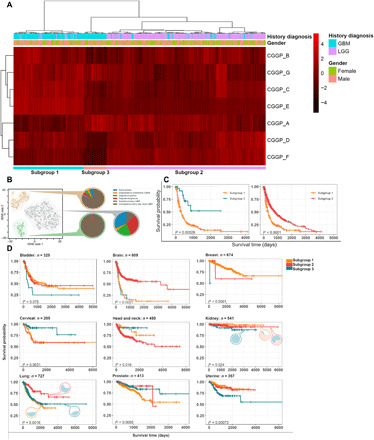
(A) Brain tumor patients were partitioned into subgroups with distinct clinical and biological traits through unsupervised hierarchical clustering of the distribution profile of the genomic patterns in patients’ germ lines. (B) Visualization of tSNE illustrated the subgroups identified in A1. (C) Significant survival differences between the brain subgroups. (D) Significant survival differences between germline subgroups in bladder, breast, cervical, head and neck, kidney, lung, prostate, and uterine cancers. Cancer types with insufficient sample size or without histological diagnosis information were excluded from analyses.
In nine common cancer types (bladder, brain, breast, cervical, head and neck, kidney, lung, prostate, and uterine cancers), CGGP-defined germline subgroups were significantly associated with prognosis (log-rank test, P = 7.50 × 10−2, 1.00 × 10−4, 1.00 × 10−4, 3.10 × 10−3, 1.60 × 10−3, 2.40 × 10−2, 1.60 × 10−3, 9.50 × 10−3, and 7.30 × 10−4, respectively; for cancer types with more than two subgroups, the most significant P value was reported) (Fig. 5B). These results suggested that for these nine common cancer types, clinical outcomes were partially determined by the CGGPs in germline genomes.
These results implied that CGGP profiles in germ lines could also have influences on the oncogenic pathways in tumor tissues. In each of the above nine cancer types, we selected the differentially expressed genes between subgroups and performed functional enrichment analysis to examine the affected oncogenic pathways (see Materials and Methods). The main differences between subgroups 1 and 2 of brain cancer patients were ubiquitin and protein degradation functions and metabolism pathways (Table 1). Notably, subgroups 1 and 2 were both mainly consisted of GBM patients but were diverse in carrier status of germline patterns. Genes related to the ubiquitin and protein degradation functions and metabolism pathways were differentially expressed in tumor tissues between the subgroups of at least six cancer types. Differential expression of the genes in the biological processes related to cell cycle and mitochondria between the subgroups was observed in lung cancer (Table 1).
Table 1. Significant differences in the gene expression profile of the corresponding tumor tissues between the CGGP-defined subgroups.
| Cancer type |
CGGP-defined subgroups |
Function term | P | FDR |
| Bladder | 2 versus 3 | Pathways and metabolism | 9.80 × 10−13 | 1.81 × 10−9 |
| Bladder | 2 versus 3 | Ubiquitin and protein degradation |
6.20 × 10−6 | 0.01 |
| Bladder | 1 versus 3 | Pathways and metabolism | 8.31 × 10−9 | 1.54 × 10−5 |
| Brain | 1 versus2 | Ubiquitin and protein degradation |
7.87 × 10−6 | 0.01 |
| Brain | 1 versus2 | Pathways and metabolism | 1.13 × 10−5 | 0.02 |
| Cervical | 1 versus 3 | Pathways and metabolism | 9.80 × 10−13 | 1.81 × 10−9 |
| Cervical | 1 versus 3 | Ubiquitin and protein degradation |
9.03 × 10−6 | 0.02 |
| Cervical | 2 versus 3 | Pathways and metabolism | 8.31 × 10−9 | 1.54 × 10−5 |
| Kidney | 3 versus 1 | Pathways and metabolism | 9.80 × 10−13 | 1.81 × 10−9 |
| Kidney | 3 versus 1 | Ubiquitin and protein degradation |
6.20 × 10−6 | 0.01 |
| Lung | 2 versus 1 | Pathways and metabolism | 8.12 × 10−17 | 2.00 × 10−13 |
| Lung | 2 versus 1 | Ubiquitin and protein degradation |
7.18 × 10−12 | 1.29 × 10−8 |
| Lung | 2 versus 1 | Mitochondria | 1.69 × 10−8 | 3.04 × 10−5 |
| Lung | 2 versus 1 | Cell cycle | 4.70 × 10−8 | 8.42 × 10−5 |
| Lung | 3 versus 1 | Ubiquitin and protein degradation |
2.14 × 10−7 | 3.97 × 10−4 |
| Uterine | 3 versus 1 | Pathways and metabolism | 9.80 × 10−13 | 1.81 × 10−13 |
| Uterine | 3 versus 1 | Ubiquitin and protein degradation |
2.04 × 10−5 | 0.04 |
| Uterine | 2 versus 1 | Pathways and metabolism | 8.31 × 10−9 | 1.54 × 10−5 |
Next, we examined whether normal tissues had different gene expression programs between the CGGP-defined subgroups. With a limited number of normal tissue expression profiles available in TCGA, only cancer types with sufficient overall and subgroup-wise sample size, i.e., breast (n = 76), kidney (n = 122), and lung (n = 95) cancers, were selected for this analysis. We identified differentially expressed genes in the normal tissues between the subgroups of breast, kidney, and lung cancers. Genes were functionally clustered and annotated using the same method described above. Gene Ontology (GO) terms of biological processes, molecular functions, and pathways showed significant differences between subgroups (table S6). For instance, differentially expressed genes between subgroups 1 and 2 of breast cancer were enriched in the following biological processes and functions: cell cycle (FDR = 2.0 × 10−19), mitosis (FDR = 3.3 × 10−18), mitotic nuclear division (FDR = 3.4 × 10−14), cell division (FDR = 1.1 × 10−13), and centromere (FDR = 2.4 × 10−9). The modulated genes between subgroups 2 and 3 of breast cancer have enriched functions, and pathways that are related to ribosome and mRNA splicing differed (FDR < 10−10). The above results implied that the impacts CGGPs imposed on patients’ tumor tissues were prominent but indirect. For example, divergence of adenosine triphosphate (ATP)–related metabolism between kidney cancer’s normal tissue subgroups 1 and 3 transformed to differences in immune response functions in tumors. This was in accordance with the fact that carcinogenesis diseases are influenced by both genetics and environmental factors. Together, these results implied that contributions of CGGPs in patients’ germline genomes could play an important role in affecting gene regulation programs in normal tissues and are implicitly associated with oncogenic pathways in tumor progression and metastasis.
DISCUSSION
In this study, germline genomes paired with tumor genomes of 7214 cancer patients of European ancestry and germline genomes of 16,670 noncancer individuals were analyzed to reveal enriched sequential context–dependent variant profiles that could be associated with cancer risk, tumorigenesis, and clinical outcomes. CGGPs provide an aspiring method to examine the impact of germline genomes on cancer development, latent molecular mechanisms, and clinical outcomes. The interaction of genetic and environmental factors often leads to tumorigenesis. We showed that individuals with CGGP_E could be sensitive to tobacco smoke for developing 13 cancer types. Thus, for an individual who has a higher weight of CGGP_E in his/her germ line, it could be possible to reduce the risk of cancer development by actively avoiding exposures to tobacco smoke. Unexpectedly, although a higher contribution of CGGP_E in germ lines conferred a poor prognosis in various cancer types, such an association was not observed in lung cancer patients. One of the explanations could be that tobacco mutagens are directly exposed to the lung so that tobacco mutagens could overpower the conditional genetic factors for lung cancer development and metastasis. Thus, the clinical outcomes of lung cancer patients could be mainly driven by tobacco smoking. This hypothesis agrees with the fact that more than 85% of lung cancer patients are smokers. On the other hand, for other cancer types such as bladder cancer, the tissues are indirectly exposed to tobacco mutagens so that germline CGGP_E could play a more important role in tumorigenesis and outcomes.
Distinct combinations of CGGPs in germ lines were associated with instinct cancer types, suggesting that germline genetic architecture with different fractions of the CGGPs has an impact on tumorigenesis in a tissue-specific manner and has also constraints on many aspects of tumorigenesis and metastasis. For example, the germline subgroups defined by CGGPs were significantly associated with tumor histological subtypes, mechanisms of carcinogenesis, and prognosis for at least 13 common cancer types, and the strengths of CGGPs in germline genomes were associated with key somatic cancer drivers in paired tumors. We also showed that CGGPs and their combinations in patients’ germ lines had constraints on key somatic mutated genes and mutational processes (COSMIC somatic signatures) in paired tumors. It seems that they are mainly associated with higher genome instability. Germline genetic architecture could also affect the gene regulatory programs in normal tissues. For example, we demonstrated that gene expression programs of the normal tissues between the CGGP-defined subgroups in at least three cancers were notably modulated and enriched in cell cycle or other general biological processes that could contribute to cancer progression and metastasis. These results suggest that congenital germline variants encode some subtle causalities of tumorigenesis and metastasis. The quantitative analysis of germline variants provides intriguing depictions of genomic features, which are associated with genetic diseases. We speculate that other genetic diseases could also be affected by their own specific germline genomic patterns. Furthermore, clustering analysis of germline genomes using the genomic patterns puts forward a novel notion of population-scale genomic clustering, whereas previous efforts focus on representing general geographic subgroups through gene-based evolutionary features (31, 32).
To quantify germline-defined intrinsic cancer risk, however, it would require larger cohorts that can represent the general population and a more sophisticated evaluating approach for the weighing factors. In this study, we focused on the proof of concept, i.e., proving the existence of germline genomic patterns and their impacts on somatic mutational events and clinical outcomes. Stricter criteria were chosen when we needed to translate consecutive weighing factors (in forms of float numbers) to binary CGGP assignments for these purposes. In theory, it is possible to assume a given individual to have a higher or lower risk of cancer by using the CGGPs, but to accurately quantify intrinsic cancer risks, one needs more appropriate criteria. For example, in our stricter rules, the boundary of confidence intervals was set to the point of 95% confidence, but it is widely acknowledged that the lifetime risk of cancer is around 30 to 40% in the general population. This issue cannot be resolved simply by relaxing the boundary to 30%, because we have shown that CGGPs are better considered in groups instead of independent individual patterns, and therefore, the intervals should be reconciled to this idea. Further investigations are needed to estimate intrinsic cancer risks in the future.
Here, we have demonstrated that heredity plays an important role in cancer causation. It has been reported that certain COSMIC signatures in tumors are significantly associated with tumor driver gene mutations (33). We reported that germline genomic patterns were significantly associated with COSMIC signatures. Therefore, it is possible that germline genomic patterns are associated with somatic mutational signatures and then associated with tumor driver mutations. For example, we found that CGGP_G in HNSC was associated with somatic mutations of PIK3CA and with COSMIC signature 2. Poulos et al. (33) found that COSMIC signature 2 was also significantly associated with PIK3CA mutation in HNSC. These results implied the causality of CGGP_G for PIK3CA mutation in HNSC tumors. These examples provide an argument against the bad-luck hypothesis, which proposes that tumorigenesis is a purely random process of DNA replication errors and heredity plays a minimal role in tumorigenesis. However, tumorigenesis is a complex process. In the future, it is important to develop a framework to systematically dissect out the contributors of either bad-luck, genetic, or environmental factors or certain combinations of these factors. Last, this study provides a conceptual advance in cancer genomics, where the CGGP is beyond the traditional cancer risk genes. Genomic patterns could be an unnoticed type of genomic “dark matter” encoded in germline genomes to influence cancer development and progression. They could also provide another dimension of genomic regulation for cancer development, progression, and metastasis. Moreover, further investigation of germline genomic patterns could uncover genetic mechanisms of diseases that are beyond the gene-centered molecular mechanisms.
MATERIALS AND METHODS
Cancers from 22 primary sites were included in this study. Note that although data derived from bone marrow, lymph node, and thymus were collected and integrated into the mutational catalogs (see below), only solid tumor types were analyzed.
Data and germline variant calling
To obtain germline variants of cancer patients, BAM files of the whole-exome sequencing of normal samples were collected from TCGA cohorts hosted at Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/). The samples represent a nonredundant set of 9712 individuals of 22 cancer types. Variant calling tool HaplotypeCaller from the GenomeAnalysisToolkit (GATK) (version 3.8-0-ge9d806836; java -XX:ParallelGCThreads = 4 -jar /thepath/GenomeAnalysisTK.jar -T HaplotypeCaller -nct 4 -R /thepath/GRCh38.d1.vd1.fa -I /thepath/bamfile.bam --genotyping_mode DISCOVERY -stand_call_conf 30 -o /thepath/bamfile.vcf) was used. We tested the HaplotypeCaller jointed calling mode and single sample calling mode by calling variants from chromosome 1 of randomly selected 1534 individuals from the 9712 individuals and found that the mutational catalogs derived from the jointed calling mode and those from the single sample calling mode highly resembled each other (fig. S4A). Similarly, switching to Mutect2 also did not affect the reproducibility (fig. S4B). Although samples representing blood cancers were included in the mutational catalog and the discovery of germline genomic patterns, corresponding individuals and cohort were not further analyzed because of the concern that tumor cells might contaminate the peripheral blood sample to the point undistinguishable for allele frequency (AF) thresholding method. Considering the robustness of our methodology (Supplementary Materials and Methods), removing these samples in the initial stages would not substantially change the results reported in this study. To obtain the tumor somatic variants for the 9712 patients, we retrieved their variants, which were called by VarScan 2 and provided by TCGA (current release at GDC, v12.0; Supplementary Materials and Methods) for COSMIC analysis to coordinate with the configuration of the previous study (28). As for oncogene analysis, the recently updated MC3 somatic mutation dataset (v0.2.8, controlled version) was adopted (34).
Control dataset or the noncancer dataset consisted of noncancer individuals collected from three cohorts: (i) Swedish Schizophrenia Population-Based Case-control Exome Sequencing (dbGaP ID: phs000473.v2.p2), representing 12,380 individuals of age 18 to 65. This cohort was also used by Genovese et al. (35) for investigating germline and hematopoiesis-derived somatic mutations. (ii) Myocardial Infarction Genetics Exome Sequencing Consortium (dbGaP ID: phs001000.v1.p1) collected the whole-exome sequencing data of 2322 noncancer individuals. (iii) Myocardial Infarction Genetics Exome Sequencing Consortium: Ottawa Heart Study (dbGaP ID: phs000806.v1.p1) contains whole-exome sequencing data of 1968 noncancer individuals. No individual was shared by the three cohorts. We used the samples labeled as the healthy control in these cohorts.
Criteria for determining germline variants
Only variants with read depth (DP) no less than 20 were retained for further inspection. Theoretically, germline homozygous and heterozygous variants would have variant allele frequency (VAF) signals residing around 1.0 and 0.5, respectively, with subtle influences introduced by the systemic sequencing and variant calling bias. Among the variants derived from peripheral blood of cancer patients, we observed a visible subset with VAF ranging from 0.2 to 0.3, implying that putative somatic mutations generated from clonal hematopoiesis existed across the population (fig. S2). To avoid picking up somatic mutation contaminations that emerged from clonal hematopoiesis, which was a readily detectable process (36, 37) in elderly people (>65 years old, 10%) and young population (1%), we developed a method that was similar to that described by Genovese et al. (35); specifically, we inputted VAFs of all variants to the Gaussian mixture model (GMM) with default configuration to determine the “soft” threshold (i.e., VAF intervals) for the high-confidence germline variants. In the TCGA cohort, the GMM component revealed that the 95% confidence interval of VAFs for heterozygous variants was 0.422 to 0.540. This interval was close to that reported previously (35). The interval of the noncancer dataset was calculated independently from patient germ line and yielded a similar scope (0.420 to 0.537). The criterion of VAF > 0.9 was imposed for the homozygous variants, as we observed that very few variants fell into the interval of 0.5 to 1.0 and the GMM model could not fit well around 0.9 to 1.0.
Discovering of genomic patterns using the NMF method
We termed the “trinucleotide profile” of a variant as the trinucleotide form by the immediate 5′ sequential context of the variant (i.e., the previous nucleotide), the variant itself, and the immediate 3′ sequential context (i.e., the next nucleotide). Previous studies in tumor somatic mutational signatures tended to focus on the transcribed strand only because of strand bias. We, however, decided to include both the transcribed strand and the nontranscribed strand to achieve presumably better signal capturing. In this study, substitutions have 12 possibilities, and each of the two context slots has four choices, which build up 192 potential trinucleotide profiles. For each population (i.e., cancer germ lines, cancer somatic mutations, and noncancer germ lines), the corresponding mutational catalog was a two-dimensional matrix of shape (192, number_of_samples), where each row was filled with nonnegative integers that counted the number of presences of the given profile within a sample.
NMF factorizes a matrix Vi × j into two smaller matrices, Wi × k and Hk × j. We modified the method developed previously (22) by introducing a hyperparameter that encouraged the exploration of distinct patterns through penalties addressed by the non-sparseness of pattern matrix. The rationale behind this was that germline genome, by nature, carried much more variants compared to somatic mutations in tumor genome, and the variants were less informative than the de novo mutations in tumors. In other words, somatic mutations in tumors were more closely associated with tumorigenesis, whereas germline variants were, by definition, inherited from parents and were “noisier” and not directly associated with specific biological processes or disease in most cases. The number of germline genomic patterns was determined using the silhouette method, which was previously implemented for extracting tumor mutational signatures (23). The silhouette score (i.e., the stability of the solved genomic pattern) would gradually decay as more germline genomic patterns were extracted (k = 2 to k = 7) and then drop drastically from k = 7 to k = 8, k = 8 to k = 9, and so on, where the score decreased from more than 0.5 to 0.2 or less. In addition, in k > 7 setups, the extra patterns would closely resemble previously known patterns (i.e., the ones found by k = 7 setup; cosine similarity > 0.9995). Therefore, k = 7 was the last point before overfitting. We tested the robustness of this NMF approach in several ways, where we showed that the approach was able to tolerate random noise. For example, randomly adding or removing up to 30% noise to the mutational catalog allowed the reproduction of original germline genomic patterns (see Supplementary Materials and Methods for details; the related source code is available as data S4). Germline genomic patterns, once made available, could be assigned ubiquitously to given germline genomes by converging the NMF model without updating the pattern matrix (Wixk). In other words, new individuals could be easily evaluated by the germline genomic patterns.
Distinguishing CGGPs between cancer types and noncancer population
To identify differential contributions of single CGGPs or their combinations between cancer types and noncancer samples, sample-wise normalization was first applied to the mutational catalog matrix (i.e., dividing each value of 192 mutational types of one sample by the sum of all 192 mutational types of this sample, ensuring that the sum of mutational catalogs for any sample is equal to 1) to alleviate the technical batch effect between TCGA and noncancer cohorts. The contribution weights of cancer and noncancer samples were resolved by taking the normalized mutational catalog matrix and the normalized CGGP matrix as inputs. Last, for each CGGP, samples were sorted in descending order of weighing factors, and those in the top 25% and the bottom 25% were selected to form two subgroups. χ2 test was applied to test whether there was an enrichment of a CGGP in the top 25% subgroup versus the bottom 25% subgroup.
Clustering and visualization of the CGGP-defined patient subgroups
For clustering germline genomes using CGGPs, the weighing factor matrix was treated as the feature matrix representing the samples, where each vector of length seven describes its corresponding germline genome. The samples were then clustered via hierarchical clustering, where the cluster detection followed the regular tree cutting principles. Similar results were obtained with or without PCA before clustering. tSNE (t-distributed stochastic neighbor embedding) (38) is a dimension reduction and visualization algorithm that has been applied to various fields including genome-wide association study (39) and protein similarity comparisons (40). This algorithm provides meaningful visualization of latent clusters inside high-dimensional data, which could serve as an aid to view hierarchical clustering results more intuitively.
Examining of CGGPs’ impact on COSMIC somatic signatures and important somatic mutations in oncogenic genes
We assigned COSMIC somatic signatures using the method and criteria reported in (21, 22) (Supplementary Materials and Methods). For each CGGP, patients of each cancer type were sorted in descending order of weighing factors, and individuals in the top 25% and bottom 25% were selected to form two subgroups. Similarly, for each COSMIC somatic signature, patients of each cancer type were sorted in descending order of weights, and individuals in the top 25% and bottom 25% were selected to form two subgroups. Then, the functional associations of the CGGP subgroups with the COSMIC signature subgroups could be further compared by using statistical methods. Here, χ2 test was used to examine whether higher weights of one CGGP in germ line would imply significantly more possession of a COSMIC somatic signature. When performing the correlation analysis between CGGP and COSMIC signature, the data points were first smoothed by fitting the LOESS model and the correlation was estimated by the Spearman correlation coefficient. We also performed sample-wise normalization to COSMIC contribution weighing matrix before analysis.
To examine the association between CGGPs and somatically mutated genes, for a given gene, patients of each cancer type were partitioned into two subgroups based on their somatic mutation status of the gene (presence/absence of mutation). If the mutated gene group contained more than nine samples, we conducted the following analyses. We conducted t test for each CGGP between the two groups to test whether higher/lower weights (i.e., ratio of mean CGGP weights of >1 or <1) of one CGGP could be associated with a somatically mutated gene. P values were FDR-corrected among each cancer type. Because a trio (i.e., a gene-CGGP pair with a ratio of mean CGGP weights of >1 or a gene-CGGP pair with a ratio of mean CGGP weights of <1) could be significant (FDR < 0.25) in multiple cancer types, we conducted a randomization test by reshuffling the P values (n = 10,000 times) to calculate whether the probability of a trio had FDR < 0.25 in two or more cancer types. An empirical P value of <0.05 from the randomization test was set to be significant.
Functional enrichment analysis of differentially expressed genes between the CGGP-defined cancer subgroups
The same method was used for germline data and somatic data. For individuals in a given tumor type and sample type (i.e., somatic or germ line), we obtained their gene expression profiles (raw counts) from the GDC repository. Multiple count values of the same gene were averaged. We ranked all probed genes based on their level of differential expression, measured by t test, between the two germline-defined subgroups. Then, we selected either the most significant 3000 genes or all genes that had P values less than 1.0 × 10−3. Raw counts and unadjusted P values were used because our purpose here was to rank genes, instead of a formal differential expression analysis. We fed the picked genes to the functional annotation tool of DAVID 6.8 (https://david.ncifcrf.gov/) for functional annotation and clustering.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Alberta Innovates—Health Solutions Translational Chair Program (to E.W.), the Canada Foundation for Innovation (to E.W.), the Canadian Institutes of Health Research (to E.W.), the Natural Sciences and Engineering Research Council (to E.W.), the Natural Science Foundation of China (81670462, 81970440, and 81921001 to Q.C.; 62062032 to X.L.), Peking University Basic Research Program (BMU2020JC001 to Q.C.), PKU-Baidu Fund (2019BD014 to Q.C.), and Chinese Government Scholarship Fund (no. 201908360052 to X.L.). Author contributions: E.W. and Q.C. conveyed the conceptual idea. E.W., Q.C., X.F., J.L., and B.L. designed the study. X.F., X.X., and M.A. performed data preparation. X.F., X.X., Y.Z., and X.L. conducted coding and mutational signature extraction. X.X., X.F., and Y.Z. contributed to downstream analysis. Y.Z., X.L., and D.L. contributed to the examination of algorithms and distributed computing setup and feature selection methodologies. All the authors discussed the data interpretation and proofread and corrected the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eaba4905/DC1
REFERENCES AND NOTES
- 1.Tomasetti C., Li L., Vogelstein B., Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasetti C., Vogelstein B., Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu S., Powers S., Zhu W., Hannun Y. A., Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman R., Iwai J., Genetic predisposition and stress-induced hypertension. Science 193, 161–162 (1976). [DOI] [PubMed] [Google Scholar]
- 5.McPherson K., Steel C. M., Dixon J. M., ABC of breast diseases—Breast cancer-epidemiology, risk factors, and genetics. BMJ 321, 624–628 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakagawa H., Liyanarachchi S., Davuluri R. V., Auer H., Martin E. W. Jr., de la Chapelle A., Frankel W. L., Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 23, 7366–7377 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Bignell G. R., Greenman C. D., Davies H., Butler A. P., Edkins S., Andrews J. M., Buck G., Chen L., Beare D., Latimer C., Widaa S., Hinton J., Fahey C., Fu B., Swamy S., Dalgliesh G. L., Teh B. T., Deloukas P., Yang F., Campbell P. J., Futreal P. A., Stratton M. R., Signatures of mutation and selection in the cancer genome. Nature 463, 893–898 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farmer H., Cabe N. M., Lord C. J., Tutt A. N. J., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M. B., Jackson S. P., Smith G. C. M., Ashworth A., Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Rahman N., Realizing the promise of cancer predisposition genes. Nature 505, 302–308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasen H. F., Watson P., Mecklin J. P., Lynch H. T., New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116, 1453–1456 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Schubbert S., Zenker M., Rowe S. L., Böll S., Klein C., Bollag G., van der Burgt I., Musante L., Kalscheuer V., Wehner L.-E., Nguyen H., West B., Zhang K. Y. J., Sistermans E., Rauch A., Niemeyer C. M., Shannon K., Kratz C. P., Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 38, 331–336 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Niihori T., Aoki Y., Narumi Y., Neri G., Cavé H., Verloes A., Okamoto N., Hennekam R. C. M., Gillessen-Kaesbach G., Wieczorek D., Kavamura M. I., Kurosawa K., Ohashi H., Wilson L., Heron D., Bonneau D., Corona G., Kaname T., Naritomi K., Baumann C., Matsumoto N., Kato K., Kure S., Matsubara Y., Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38, 294–296 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Antoniou A., Pharoah P. D. P., Narod S., Risch H. A., Eyfjord J. E., Hopper J. L., Loman N., Olsson H., Johannsson O., Borg A., Pasini B., Radice P., Manoukian S., Eccles D. M., Tang N., Olah E., Anton-Culver H., Warner E., Lubinski J., Gronwald J., Gorski B., Tulinius H., Thorlacius S., Eerola H., Nevanlinna H., Syrjäkoski K., Kallioniemi O.-P., Thompson D., Evans C., Peto J., Lalloo F., Evans D. G., Easton D. F., Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 72, 1117–1130 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gayther S. A., Russell P., Harrington P., Antoniou A. C., Easton D. F., Ponder B. A., The contribution of germline BRCA1 and BRCA2 mutations to familial ovarian cancer: No evidence for other ovarian cancer–susceptibility genes. Am. J. Hum. Genet. 65, 1021–1029 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell K. N., Wubbenhorst B., D’Andrea K., Garman B., Long J. M., Powers J., Rathbun K., Stopfer J. E., Zhu J., Bradbury A. R., Simon M. S., DeMichele A., Domchek S. M., Nathanson K. L., Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet. Med. 17, 630–638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang K.-L., Mashl R. J., Wu Y., Ritter D. I., Wang J., Oh C., Paczkowska M., Reynolds S., Wyczalkowski M. A., Oak N., Scott A. D., Krassowski M., Cherniack A. D., Houlahan K. E., Jayasinghe R., Wang L.-B., Zhou D. C., Liu D., Cao S., Kim Y. W., Koire A., McMichael J. F., Hucthagowder V., Kim T.-B., Hahn A., Wang C., McLellan M. D., Al-Mulla F., Johnson K. J., Network C. G. A. R., Lichtarge O., Boutros P. C., Raphael B., Lazar A. J., Zhang W., Wendl M. C., Govindan R., Jain S., Wheeler D., Kulkarni S., Dipersio J. F., Reimand J., Meric-Bernstam F., Chen K., Shmulevich I., Plon S. E., Chen F., Ding L., Pathogenic germline variants in 10,389 adult cancers. Cell 173, 355–370.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fletcher O., Houlston R. S., Architecture of inherited susceptibility to common cancer. Nat. Rev. Cancer 10, 353–361 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Grossman R. L., Heath A. P., Ferretti V., Varmus H. E., Lowy D. R., Kibbe W. A., Staudt L. M., Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry M. W., Browne M., Langville A. N., Pauca V. P., Plemmons R. J., Algorithms and applications for approximate nonnegative matrix factorization. Comput. Stat. Data Anal. 52, 155–173 (2007). [Google Scholar]
- 20.Lee D. D., Seung H. S., Algorithms for non-negative matrix factorization. Adv. Neural Inf. Process. Syst. 13, 556–562 (2001). [Google Scholar]
- 21.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Aparicio S. A. J. R., Behjati S., Biankin A. V., Bignell G. R., Bolli N., Borg A., Børresen-Dale A.-L., Boyault S., Burkhardt B., Butler A. P., Caldas C., Davies H. R., Desmedt C., Eils R., Eyfjörd J. E., Foekens J. A., Greaves M., Hosoda F., Hutter B., Ilicic T., Imbeaud S., Imielinski M., Jäger N., Jones D. T. W., Jones D., Knappskog S., Kool M., Lakhani S. R., López-Otín C., Martin S., Munshi N. C., Nakamura H., Northcott P. A., Pajic M., Papaemmanuil E., Paradiso A., Pearson J. V., Puente X. S., Raine K., Ramakrishna M., Richardson A. L., Richter J., Rosenstiel P., Schlesner M., Schumacher T. N., Span P. N., Teague J. W., Totoki Y., Tutt A. N. J., Valdés-Mas R., van Buuren M. M., van ‘t Veer L., Vincent-Salomon A., Waddell N., Yates L. R.; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain, Zucman-Rossi J., Futreal P. A., Dermott U. M., Lichter P., Meyerson M., Grimmond S. M., Siebert R., Campo E., Shibata T., Pfister S. M., Campbell P. J., Stratton M. R., Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov L. B., Nik-Zainal S., Wedge D. C., Campbell P. J., Stratton M. R., Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 3, 246–259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L. A., Menzies A., Martin S., Leung K., Chen L., Leroy C., Ramakrishna M., Rance R., Lau K. W., Mudie L. J., Varela I., McBride D. J., Bignell G. R., Cooke S. L., Shlien A., Gamble J., Whitmore I., Maddison M., Tarpey P. S., Davies H. R., Papaemmanuil E., Stephens P. J., Laren S. M., Butler A. P., Teague J. W., Jönsson G., Garber J. E., Silver D., Miron P., Fatima A., Boyault S., Langerød A., Tutt A., Martens J. W. M., Aparicio S. A. J. R., Borg Å., Salomon A. V., Thomas G., Børresen-Dale A.-L., Richardson A. L., Neuberger M. S., Futreal P. A., Campbell P. J., Stratton M. R.; Breast Cancer Working Group of the International Cancer Genome Consortium , Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasnic R., Brandes N., Zuk O., Linial M., Substantial batch effects in TCGA exome sequences undermine pan-cancer analysis of germline variants. BMC Cancer 19, 783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckley A. R., Standish K. A., Bhutani K., Ideker T., Lasken R. S., Carter H., Harismendy O., Schork N. J., Pan-cancer analysis reveals technical artifacts in TCGA germline variant calls. BMC Genomics 18, 458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koire A., Katsonis P., Lichtarge O., Repurposing germline exomes of the cancer genome atlas demands a cautious approach and sample-specific variant filtering. Pac. Symp. Biocomput. 21, 207–218 (2016). [PMC free article] [PubMed] [Google Scholar]
- 27.Harris K., Pritchard J. K., Rapid evolution of the human mutation spectrum. eLife 6, e24284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandrov L. B., Ju Y. S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T., Campbell P. J., Vineis P., Phillips D. H., Stratton M. R., Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freedman N. D., Leitzmann M. F., Hollenbeck A. R., Schatzkin A., Abnet C. C., Cigarette smoking and subsequent risk of lung cancer in men and women: Analysis of a prospective cohort study. Lancet Oncol. 9, 649–656 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Keeffe L. M., Taylor G., Huxley R. R., Mitchell P., Woodward M., Peters S. A. E., Smoking as a risk factor for lung cancer in women and men: A systematic review and meta-analysis. BMJ Open 8, e021611 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg N. A., Pritchard J. K., Weber J. L., Cann H. M., Kidd K. K., Zhivotovsky L. A., Feldman M. W., Genetic structure of human populations. Science 298, 2381–2385 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Tishkoff S. A., Reed F. A., Friedlaender F. R., Ehret C., Ranciaro A., Froment A., Hirbo J. B., Awomoyi A. A., Bodo J.-M., Doumbo O., Ibrahim M., Juma A. T., Kotze M. J., Lema G., Moore J. H., Mortensen H., Nyambo T. B., Omar S. A., Powell K., Pretorius G. S., Smith M. W., Thera M. A., Wambebe C., Weber J. L., Williams S. M., The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulos R. C., Wong Y. T., Ryan R., Pang H., Wong J. W. H., Analysis of 7,815 cancer exomes reveals associations between mutational processes and somatic driver mutations. PLOS Genet. 14, e1007779 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellrott K., Bailey M. H., Saksena G., Covington K. R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K. E., Lellan M. M., Sofia H. J., Hutter C., Getz G., Wheeler D., Ding L.; MC3 Working Group; Genome Atlas Research Network , Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genovese G., Kähler A. K., Handsaker R. E., Lindberg J., Rose S. A., Bakhoum S. F., Chambert K., Mick E., Neale B. M., Fromer M., Purcell S. M., Svantesson O., Landén M., Höglund M., Lehmann S., Gabriel S. B., Moran J. L., Lander E. S., Sullivan P. F., Sklar P., Grönberg H., Hultman C. M., McCarroll S. A., Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P. V., Mar B. G., Lindsley R. C., Mermel C. H., Burtt N., Chavez A., Higgins J. M., Moltchanov V., Kuo F. C., Kluk M. J., Henderson B., Kinnunen L., Koistinen H. A., Ladenvall C., Getz G., Correa A., Banahan B. F., Gabriel S., Kathiresan S., Stringham H. M., McCarthy M. I., Boehnke M., Tuomilehto J., Haiman C., Groop L., Atzmon G., Wilson J. G., Neuberg D., Altshuler D., Ebert B. L., Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie M., Lu C., Wang J., McLellan M. D., Johnson K. J., Wendl M. C., McMichael J. F., Schmidt H. K., Yellapantula V., Miller C. A., Ozenberger B. A., Welch J. S., Link D. C., Walter M. J., Mardis E. R., Dipersio J. F., Chen F., Wilson R. K., Ley T. J., Ding L., Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Maaten L., Hinton G., Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008). [Google Scholar]
- 39.Mejia-Roa E., Tabas-Madrid D., Setoain J., Garcia C., Tirado F., Pascual-Montano A., NMF-mGPU: Non-negative matrix factorization on multi-GPU systems. BMC Bioinformatics 16, 43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ray S., Bandyopadhyay S., A NMF based approach for integrating multiple data sources to predict HIV-1–human PPIs. BMC Bioinformatics 17, 121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eaba4905/DC1