

Abstract
Stem cell replacement offers a great potential for cardiac regenerative therapy. However, one of the critical barriers to stem cell therapy is a significant loss of transplanted stem cells from ischemia and inflammation in the host environment. Here, we tested the hypothesis that inhibition of the soluble epoxide hydrolase (sEH) enzyme using sEH inhibitors (sEHIs) to decrease inflammation and fibrosis in the host myocardium may increase the survival of the transplanted human induced pluripotent stem cell derived‐cardiomyocytes (hiPSC‐CMs) in a murine postmyocardial infarction model. A specific sEHI (1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea [TPPU]) and CRISPR/Cas9 gene editing were used to test the hypothesis. TPPU results in a significant increase in the retention of transplanted cells compared with cell treatment alone. The increase in the retention of hiPSC‐CMs translates into an improvement in the fractional shortening and a decrease in adverse remodeling. Mechanistically, we demonstrate a significant decrease in oxidative stress and apoptosis not only in transplanted hiPSC‐CMs but also in the host environment. CRISPR/Cas9‐mediated gene silencing of the sEH enzyme reduces cleaved caspase‐3 in hiPSC‐CMs challenged with angiotensin II, suggesting that knockdown of the sEH enzyme protects the hiPSC‐CMs from undergoing apoptosis. Our findings demonstrate that suppression of inflammation and fibrosis using an sEHI represents a promising adjuvant to cardiac stem cell‐based therapy. Very little is known regarding the role of this class of compounds in stem cell‐based therapy. There is consequently an enormous opportunity to uncover a potentially powerful class of compounds, which may be used effectively in the clinical setting.
Keywords: cardiac stem cell‐based therapy, CRISPR/Cas9, human induced pluripotent stem cell derived‐cardiomyocytes, myocardial infarction, soluble epoxide hydrolase inhibitors
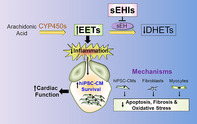
Stem cell replacement offers a great potential for cardiac regenerative therapy. However, one of the critical barriers to stem cell therapy is a significant loss of transplanted stem cells from ischemia and inflammation in the host environment. Here, we demonstrate that inhibition of the soluble epoxide hydrolase (sEH) enzyme to decrease inflammation and fibrosis in the host myocardium increases the survival of the transplanted human induced pluripotent stem cell derived‐cardiomyocytes (hiPSC‐CMs) in a murine postmyocardial infarction model.
Significance statement.
Stem cell replacement offers a great potential for cardiac regenerative therapy. However, there is a significant loss of transplanted stem cells from ischemia and inflammation in the host environment. This study demonstrates beneficial effects of inhibitors of soluble epoxide hydrolase (sEHI) in cell‐based therapy using human induced pluripotent stem cell‐derived cardiomyocytes in a preclinical model. sEHI results in a significant increase in the retention of transplanted stem cells and an improvement in cardiac function. Very little is known regarding this class of compounds in cell‐based therapy. There is consequently an opportunity to uncover a potentially powerful class of inhibitors, which may be used in clinical settings.
1. INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality in the United States. 1 , 2 , 3 , 4 Myocardial infarction (MI) results in a significant loss of more than a billion cardiomyocytes leading to a decrease in cardiac function. 5 Clinically, a delayed recovery of cardiac function may be observed with revascularization and medical therapy. However, a subset of patients do not recover their cardiac function despite optimal medical therapies and develop progressive adverse structural and electrical remodeling leading to heart failure (HF) with lethal consequences. Despite advances in therapy and management, HF remains a life‐threatening disease with a 5‐year mortality rate of 45% to 60%. 6 HF affects approximately 5.7 million people in the United States at an annual cost of nearly $30 billion, and is estimated to affect 9 million people at a cost of nearly $80 billion by 2030. 6 Currently, cardiac transplantation and left ventricular assist device are the only options for patients with end‐stage HF. However, due to limited donor hearts, nearly twice the number of patients are waiting for transplantation per year compared to the number of patients receiving cardiac transplantations. 6 Therefore, there is a compelling need to seek new options for patients suffering from HF.
Since adult cardiac myocytes are unable to proliferate sufficiently to replace the damaged tissue, stem cell therapy represents a promising approach for the treatment of HF, since it aims at generating new functional myocardium and inducing neoangiogenesis. To date, various stem cells have been used in clinical trials with each cell type presenting several advantages and limitations. 7 , 8 , 9 However, therapeutic strategies using cell‐based therapy have not produced full restorative functions. 10 , 11 A high rate of transplanted stem‐cell loss (90% within the first few days) has been observed due to inflammation in the host environment. 12 , 13 , 14 Therefore, therapeutic strategies for treating the damaged myocardium should focus on both replacing the myocardial tissue as well as decreasing inflammation.
One of the most conserved inflammatory responses is the activation of phospholipase A2 and the release of arachidonic acid, which is metabolized through the cyclooxygenase, lipoxygenase, and cytochrome P450 (CYP450) pathways. The action of CYP450 on arachidonic acid leads to the formation of epoxyeicosatrienoic acids (EETs), which have been shown to be anti‐inflammatory and pro‐fibrinolytic with several cardioprotective effects. It has been suggested that reduced levels of EETs contribute to CVDs. 15 However, EETs are further metabolized by the soluble epoxide hydrolase (sEH) enzyme to form corresponding diols, dihydroxyeicosatrienoic acids (DHETs) with diminished anti‐inflammatory activities. 15 Therefore, the cardioprotective activity of EETs can be enhanced by preventing the catalysis of EETs using inhibitors of the sEH enzyme (sEHIs). 16 Indeed, we have previously demonstrated that treatment with sEHIs results in decreased inflammation and cardiac fibrosis. 17 We have further shown that several sEHIs reduce ventricular myocyte hypertrophy, electrical remodeling and decrease both atrial and ventricular arrhythmia inducibility in several clinically relevant animal models. 17 , 18 , 19 , 20 , 21
We hypothesize that simultaneous reduction of inflammation in conjunction with stem cell transplantation would enhance stem cell survival and engraftment leading to an improvement in cardiac function. Hence in the present study, we used a two‐pronged approach to concurrently reduce inflammation and replace the lost myocardium using an anti‐inflammatory drug and human induced pluripotent stem cell‐derived cardiomyocytes (hiPSC‐CMs) in a preclinical model of murine post‐MI. We demonstrate that the dual treatment approach with sEHI and hiPSC‐CMs leads to a significant improvement in cardiac function as well as a decrease in fibrosis by limiting the pathological mitogen‐activated protein kinase (MAPK) signaling cascade, reactive oxygen species (ROS), and apoptosis in hiPSC‐CMs. Further examination of the host myocardial environment demonstrates the positive effects of sEHIs in significantly decreasing oxidative stress and apoptosis of cardiomyocytes and nonmyocyte cells (NMCs). Additionally, there is a restoration of the transient outward K+ current by sEHI in hiPSC‐CMs challenged with angiotensin II (ANG II), consistent with the prevention of electrical remodeling. Moreover, CRISPR/Cas9‐mediated gene silencing of the sEH enzyme reduces cleaved caspase‐3 in hiPSC‐CMs challenged with ANG II, suggesting that knockdown of the sEH enzyme protects the hiPSC‐CMs from undergoing apoptosis. Our study provides evidence that sEHI may represent an effective adjuvant therapy for cell transplantation since it addresses an unanticipated setback from inflammation‐mediated stem cell loss and fibrosis that may impede the success of the current cell‐based therapies.
2. METHODS
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996) and was approved by the University of California, Davis Institutional Animal Care and Use Committee. Detailed methods are presented in Supporting Information Materials and Methods.
2.1. Soluble epoxide hydrolase inhibitor
Selective and potent inhibitors (low nanomolar Ki's) for the rodent and human soluble epoxide hydrolase enzyme (sEH) were developed based on the catalytic mechanism and the x‐ray structure of the murine sEH (PDB access #: 1CQZ and 1CR6). 22 , 23 , 24 The potency, pharmacokinetics, and physiochemical properties of 11 different sEHIs were conducted. 1‐Trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea (TPPU, containing a piperidine ring) was found to have high inhibitory potency, drug‐like physicochemical properties, pharmacokinetics with high area under the curve values, a relatively longer half‐life and lower plasma protein binding properties than many previous compounds. 25 , 26 For these pharmacokinetic favorable characteristics, TPPU was chosen for this study. TPPU was found to be a low nanomolar inhibitor of the sEH enzyme and was previously tested against commercial drug targets and the NIH screen of drug targets; there were no hits at 10 micromolar (over 1000‐fold higher concentration than the effective concentration for the sEH enzyme). In addition, the x‐ray crystal structure of the sEH enzyme and the inhibitors has been solved. 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 There is no evidence to date of off‐target binding sites.
2.2. MI model in mice
MI was generated in 8‐ to 12‐week‐old NOD SCID gamma (NSG) as previously described. 17 Briefly, animals were anesthetized with ketamine 50 mg/kg and xylazine 2.5 mg/kg intraperitoneally. Intubation was performed perorally and mechanical ventilation was initiated. MI was performed using ligation of the left anterior descending coronary artery for 45 minutes. The chest was closed with 3‐0 Dexon rib sutures, 5‐0 Dexon II muscle sutures, and buried skin sutures. Negative plural pressure was re‐established via a temporary chest tube until spontaneous breathing occurred. Sham‐operated animals underwent the same procedure without tying the suture.
Echocardiograms using Vevo 2100 (FUJIFILM, Toronto, Canada) were performed 1 week after surgery after which mice were randomized into six different groups: Sham ± sEHI, MI ± sEHI, and MI + hiPSC‐CM ± sEHI. Treatment with sEHI included TPPU (15 mg/L) given in the drinking water for a period of 3 weeks. 35 The animals were followed for a period of 3 weeks at which time repeat echocardiograms were performed.
2.3. Magnetic resonance imaging
Imaging was performed using a Bruker BioSpec 7T horizontal bore system designed specifically for small animal imaging (Bruker Corporation, Billerica, Massachusetts). Mice were anesthetized with 1.5% isoflurane with oxygen in an induction chamber, placed in the magnetic resonance imaging (MRI) scanner, and were fitted with a nose cone connected to a vaporizer. Physiological parameters such as respiration and body temperature were monitored with a physiological monitoring unit that provides feedback on a warm air blower to keep the animal at normal body temperature during the scan. A retrospective gating technique to produce snapshots of the heart cycle was used and the data were collected using ParaVision 5.1 software (Bruker Corporation) and analyzed using Amira 6.0 software (Thermo Fisher Scientific, Waltham, Massachusetts).
2.4. Hemodynamic monitoring
Mice were anesthetized by intraperitoneal injection of 80 mg/kg of ketamine and 5 mg/kg of xylazine and maintained at 37°C. Recording of pressure and volume was performed by using Millar Pressure‐Volume System MPVS‐300 (Millar, Inc., Houston, Texas), Power Lab, and Lab Chart 6.0 software (AD Instruments, Colorado Springs, Colorado).
2.5. Bioluminescence imaging
Serial bioluminescence imaging (BLI) was performed on hiPSC‐CMs transplanted and TPPU treated mice on week 1, 2, 3, and 4 post‐MI with the use of the IVIS Spectrum (Perkin Elmer, Waltham, Massachusetts) imaging system using a thermoeletrically cooled back‐thinned, back‐illuminated CCD camera, with a 2048 × 2048 array of 13.5 micropixels and a 16‐bit digitizer. Mice were anesthetized with isoflurane and were injected intraperitoneally with d‐Luciferin (375 mg/kg body weight). Peak signals from a fixed region of interest were obtained and signals were quantified (photons.s‐1.cm‐2.sr‐1). 36
2.6. Human‐induced pluripotent stem cell‐derived cardiomyocytes
hiPSCs (19‐9‐7 T,WiCell, Madison, Wisconsin) were transduced with the puromycin gene under the α‐myosin heavy chain (MHC) promoter to enable enrichment of cardiomyocytes postdifferentiation and a double fusion construct of reporter genes; firefly luciferase (Fluc) for BLI and enhanced green fluorescent protein (GFP) to enable in vivo tracking of cell engraftment longitudinally. 36 hiPSCs were plated and differentiated for 30 days using a directed differentiation protocol. 37 hiPSC‐CMs enriched with puromycin and treated with tumor necrosis factor‐α (TNF‐α; 20 ng/mL for 20 minutes) or angiotensin II (ANG II, Sigma‐Aldrich; 1 μM for 24 hours) with or without TPPU (1 μM) were fixed and stained with antimyosin light chain‐2a (MLC2a), anti‐myosin light chain‐2v (MLC2v), and anti‐pERK1/2 antibodies (Cell Signaling Technology, Danvers, Massachusetts) before flow cytometric analyses as described above.
2.7. Measurement of plasma cytokine levels
Plasma cytokine levels from samples collected 3 weeks after sham or MI operation were analyzed using a Cytometric Bead Array kit and Cytometric Bead Array Analysis software following the manufacturer's protocol. 17
2.8. Statistical analysis
Data are presented as mean ± SEM. Statistical comparisons were analyzed by one‐way analysis of variance (ANOVA) followed by Bonferroni tests and Tukey's‐Kramer honest significant difference analyses for post hoc comparison. Statistical significance was considered to be achieved when P < .05.
3. RESULTS
3.1. Generation and characterization of hiPSC‐CMs for cell transplantation
hiPSCs were transduced with the puromycin gene under the α‐MHC promoter, to enable enrichment of cardiomyocytes postdifferentiation, as well as a double fusion construct of reporter genes (a kind gift from Dr Joseph Wu, Stanford University), containing firefly luciferase (Fluc) for BLI and enhanced GFP to enable in vivo tracking of cell engraftment longitudinally. 36 Transduced hiPSCs revealed high protein expression levels of pluripotency markers, including Oct3/4, SSEA4, TRA‐1‐60, and TRA‐1‐81 as assessed by flow cytometry (Figure S1A,B) and stained positive for pluripotency marker Oct3/4 when assessed by confocal immunofluorescence microscopy (Figure S1C). Positive selection of GFP+ hiPSCs was achieved by sorting the cells using florescent‐activated cell sorting (FACS; Figure 1A,B ). Next, GFP+ hiPSCs were differentiated into beating clusters of hiPSC‐CMs (Movie S1) using the small‐molecule based protocol and enriched with puromycin treatment with ~76% MHC positive cells as assessed by flow cytometry (Figure 1C). 38 Immunostaining of hiPSC‐CMs revealed the expression of MLC2v and MLC2a (Figure 1D). The use of these cells for transplantation and in vivo imaging was validated in vitro by the stable Fluc activity with a strong correlation between Fluc activity and cell number (R 2 = 0.985; Figure S2A,B).
FIGURE 1.
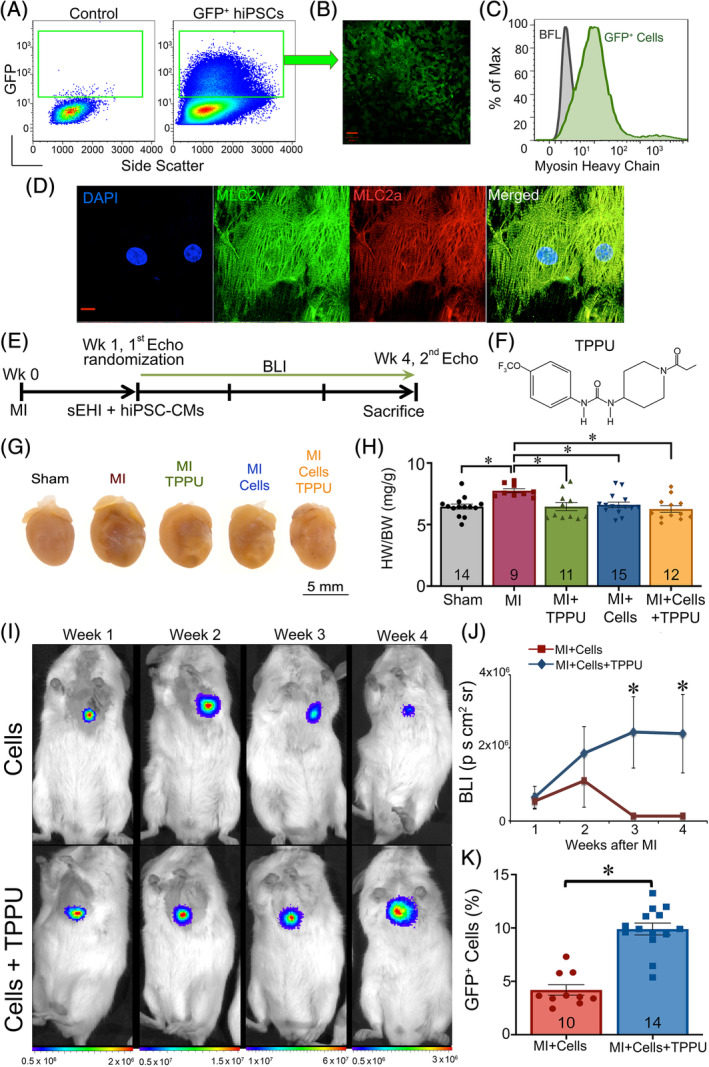
TPPU improves hiPSC‐CMs retention in a murine MI model: A, FACS for hiPSCs transduced with firefly luciferase (Fluc) and GFP. B, Confocal images of cultured FAC sorted GFP+ hiPSCs. Scale bar = 50 μM. C, Representative myosin heavy chain expression of hiPSC‐CMs at day 25 after differentiation assessed by flow cytometry. D, Confocal images showing expression of MLC2v (green) and MLC2a (red) in hiPSC‐CMs with cell nuclei stained with 4′,6‐diamidino‐2‐phenylindole (DAPI, blue). Scale bar = 20 μM. E, Schematic representation of the experimental protocol. F, Structure of the sEHI, TPPU used in our studies. Mice were randomized 1 week after MI to TPPU (15 mg/L) orally in drinking water or vehicle alone. Treatment was continued for 3 weeks. G, Photomicrographs of whole hearts from sham‐operated, MI, MI + TPPU, MI + Cell, and MI + Cell + TPPU mice. H, Summary data for heart weight/body weight (HW/BW) from the five groups of mice. I, Longitudinal in vivo BLI of hiPSC‐CMs transplanted into NSG mice at week 1, 2, 3, and 4 with (bottom panel) and without (top panel) sEHI treatment. J, Quantification of BLI signals from (E). K, Flow cytometric analyses of percentages of GFP+ hiPSC‐CMs after 4 weeks from mice receiving transplanted stem cells alone compared to transplanted cells plus sEHI treatment. Individual data points are shown together with the bar graphs in panels (H) and (K), with numbers shown within the bar graphs representing the numbers of animals for each group. Cells = hiPSC‐CMs. Data presented are Mean ± SEM, *P < .05 by ANOVA. BFL, background fluorescence; BLI, bioluminescence imaging; FACS, florescent‐activated cell sorting; GFP, green fluorescent protein; hiPSC‐CMs, human induced pluripotent stem cell derived‐cardiomyocytes; MI, myocardial infarction; sEHI, soluble epoxide hydrolase inhibitor; TPPU, 1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea; Wk, week
MI was generated in 8‐ to 12‐week‐old NOD‐scid gamma‐irradiated (NSG, Institute of Regenerative Cures, UC Davis) male and female mice using previously described techniques (Figure 1E). 17 One week after the surgery, mice were randomized into six experimental groups: Sham ± sEHI, MI ± sEHI, and MI + hiPSC‐CM ± sEHI. Transplantation of 1.5 × 106 hiPSC‐CMs into the border zones was performed using ultrasound‐guided (VisualSonics Vevo 2100, FUJIFILM, Toronto, Canada) injection (Figure S2C). Mice were randomized to receive either TPPU (15 mg/L; Figure 1F) orally in drinking water or vehicle alone for 3 weeks. 39 TPPU was selected as the sEHI of choice after evaluating the pharmacokinetic and physiological properties of 11 different sEHIs based on our prior publication. 17 , 25 , 26 Sham‐operated mice were also randomized to receive either TPPU or vehicle alone at week 1 for 3 weeks.
Whole heart images from the MI mice after 3‐week follow‐up exhibited cardiac dilatation (Figure 1G). As expected, hearts from the sham‐operated group showed no significant hypertrophy or dilatation. Summary data in Figure 1H illustrate a significant increase in the ratios of heart weight/body weight in the MI group compared with sham‐operated hearts. Treatment with TPPU, hiPSC‐CMs and both hiPSC‐CMs + TPPU resulted in a significant decrease in the heart weight and the heart weight/body weight ratios in the MI animals. The investigators were blinded to the treatment groups. A total of 98 NSG male and female animals were used. Eight animals died in the perioperative period, leaving a total of 90 mice in the study.
3.2. TPPU enhances the survival of hiPSC‐CMs
We next investigated the therapeutic potential of TPPU on the survival of hiPSC‐CMs after transplantation into the border zone in the post MI model. To test the engraftment of transplanted hiPSC‐CMs longitudinally, in vivo BLI (IVIS Xenogen Corp, Alameda, California) was performed. 36 BLI at 3 and 4 weeks demonstrated a significant increase in the survival of GFP+ cells in mice with TPPU treatment compared to the mice with cells only (Figure 1I,J; 2.3 × 106 ± 1 × 106 and 1.3 × 104 ± 5 × 104 photons/(s cm2 sr), respectively, at week 4). Interestingly, there was an increase in the BLI signal at week 2 and 3 post‐transplantation in the TPPU‐treated group attributing to the proliferation of transplanted cells. Quantification by flow cytometry showed significantly higher percentages of GFP+ hiPSC‐CMs in the TPPU treated mice (9.9% ± 0.5%) compared to the nontreated mice with hiPSC‐CMs (4.19% ± 0.4%; Figure 1K).
3.3. TPPU attenuates cardiac fibrosis post‐MI
We hypothesize that tissue injury from MI results in robust inflammatory responses leading to recruitment of fibroblasts that may inhibit cell engraftment. Hence, to examine the possible beneficial effect of sEHI in preventing cardiac fibrosis in hiPSC‐CMs transplantation, we quantified interstitial fibrosis in cardiac sections (5 μM) from corresponding areas in six groups of animals using Masson's Trichrome stain (Figure 2A) as we have previously described. 17 , 21 Analyses of the infarcted area at 4 weeks post‐MI were performed in a blinded fashion and demonstrated a significant decrease in the infarct size post‐MI after hiPSC‐CM transplantation, TPPU treatment, or the combination of hiPSC‐CM and TPPU, compared to MI alone (Figure 2A,B). There were no significant differences between the two sham‐operated groups as expected. The data suggest that treatment with TPPU and cells transplantation post‐MI prevents adverse cardiac remodeling in part by reducing infarct size and cardiac fibrosis.
FIGURE 2.
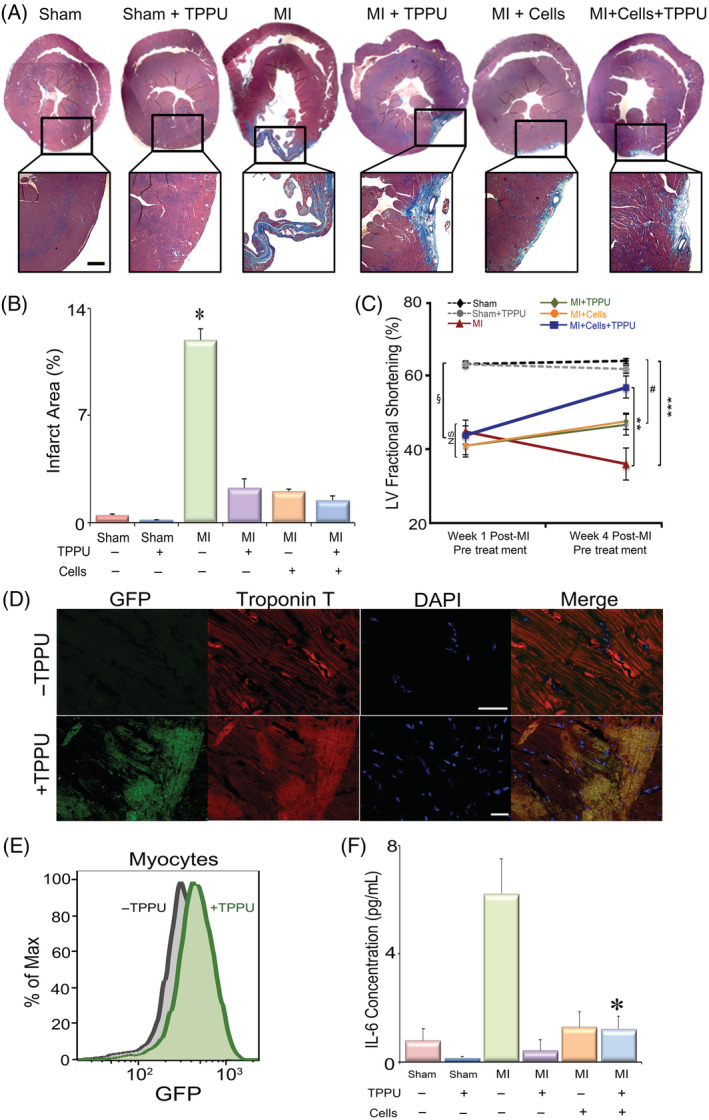
TPPU improves cardiac function and decreases fibrosis as assessed by echocardiography and immunohistochemistry: A, Cardiac sections stained with Masson's trichrome to demonstrate the amount of collagen deposition in the six groups of animals, Sham ± TPPU, MI ± TPPU, MI + Cells (hiPSC‐CM) ± TPPU. Scale bar = 200 μm. B, Quantification of the % infarct zone. n = 3 per group, *P < .05. C, FS data from M‐mode echocardiography pre‐ and post‐treatment in the six groups of mice. n = 6 to 15 per group. § P < .05 comparing Sham groups with MI groups before randomization to different treatment arms, **P < .05 comparing MI with MI + Cells + TPPU at week 4, # P < .05 comparing MI + Cells or MI + TPPU with Sham at week 4 and ***P < .05 comparing MI with Sham and Sham‐TPPU treated groups at week 4 by analysis of variance (ANOVA). D, Confocal immunofluorescence images of GFP+ hiPSC‐CMs expressing both GFP and cardiac troponin T from hearts treated with hiPSC‐CMs alone (top panel) or hiPSC‐CMs plus TPPU (bottom panel). Scale bar = 10 μM. E, Flow cytometric analyses showing the presence of GFP+ hiPSC‐CMs in hearts treated with or without TPPU. F, Serum concentration of cytokine, IL‐6. *P < .05 comparing MI with MI + Cells + TPPU at week 4 by ANOVA. n = 6 to 15 per group. Cells = hiPSC‐CMs. Mean ± SEM. FS, fractional shortening; GFP, green fluorescent protein; hiPSC‐CMs, human induced pluripotent stem cell derived‐cardiomyocytes; MI, myocardial infarction; TPPU, 1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea
3.4. Dual treatment significantly improves cardiac function
To assess the potential therapeutic benefits of TPPU treatment in cell‐based therapy after MI, we compared the functional recovery from six groups of animals. Functional analyses using echocardiography (Figure 2C) and MRI (Figure S3) were performed to evaluate the effect of TPPU in cell transplantation on chamber size and systolic function. Two‐dimensional and M‐mode echocardiography showed a significant improvement in left ventricular fractional shortening (FS) in the MI + hiPSC‐CM + TPPU group after 3 weeks of treatment (57% ± 3%) compared to before treatment (44% ± 2%, P < .05). As expected, there was a significant decrease in the FS in the MI alone group from week 1 (45% ± 3%) compared to week 4 (36% ± 4%), suggesting adverse remodeling. Moreover, all MI mice in the four different groups before randomization showed similar FS that was not significantly different among the four groups (P = NS), but significantly different from the two sham groups. Vehicle injection along with MI + TPPU (51% ± 3%) showed a comparable improvement in FS to the MI + TPPU group (47% ± 2%) suggesting no adverse effect from the vehicle injection (Figure S4A,B). Indeed, treatment with both sEHI and cells resulted in a significant improvement in FS compared to MI alone group (TPPU + Cells = 57% ± 2% compared to MI alone = 36% ± 4%) after 3 weeks of follow‐up. In addition, the MI + Cells + TPPU group was not statistically different from the Sham or Sham + TPPU groups (P = NS). In contrast, the MI only, MI + Cells, or MI + TPPU groups were significantly different from the Sham or Sham + TPPU groups (Figure 2C).
To better understand the mechanistic underpinning for the preserved left ventricular function postcell transplantation in the ischemic mice treated with and without TPPU treatment, we performed flow cytometric and histological analyses. Histologic analyses of cardiac sections via confocal immunofluorescence microscopic examination at week 4 post‐TPPU treatment demonstrated the presence of GFP+ hiPSC‐CMs expressing both GFP and cardiac troponin T (Figure 2D). However, the presence of GFP+ hiPSC‐CMs was not detected in sections without TPPU treatment, which is consistent with the decrease in BLI signals measure over the same period of time. The presence of GFP+ hiPSC‐CMs was further quantified by flow cytometry (Figure 2E) using antibodies directed against troponin T and GFP, demonstrating a significantly higher percentages of GFP+ hiPSC‐CMs in mice treated with dual therapy.
3.5. Treatment with TPPU results in a significant reduction in inflammatory cytokines
Following MI, a network of profibrotic factors including pro‐inflammatory cytokines are activated and are known to contribute toward inflammation in the clinical setting. 40 Our previous studies have documented the strong anti‐inflammatory activities of several sEHIs in preclinical models. 17 , 18 , 19 , 20 , 21 Here, our data demonstrated a significant increase in proinflammatory cytokine, interleukin 6 (IL‐6), in the MI mice (Figure 2F). Importantly, dual treatment with TPPU and hiPSC‐CMs significantly decreased the cytokine levels. An unbiased approach of metabolic profiling of oxylipids was also performed to document the target engagement by TPPU, demonstrating significant increases in EETs/DHETs ratios from arachidonic acid, EpODE/DiHODE ratios from α‐linolenic acid, and EpOME/DiHOME ratios from linoleic acid in the TPPU + Cell‐treated group compared with TPPU only and cell‐only groups (Figure S5).
3.6. Dual treatment improves cardiac contractility
Since FS is load‐dependent, we further performed hemodynamic monitoring comparing the five groups of animals to obtain the load‐independent indicator of cardiac contractility. Figure 3 shows representative recordings of left ventricular pressure (Figure 3A), volume (Figure 3B), and developed pressure (dP/dt; Figure 3C) in sham, MI, MI + TPPU, MI + hiPSC‐CMs, and MI + hiPSC‐CM + TPPU mice. To determine the end‐systolic P‐V relationship, we obtained a series of P‐V loops by altering the preload and derived the slope of the end systolic P‐V relationship (Ees), a load‐independent measure of cardiac contractility. The right and downward shift of the pressure‐volume (P‐V) loops in the MI mice indicates reduced end‐systolic pressure (ESP) and relatively larger end‐systolic and end‐diastolic volumes (Figure 3D) compared to the sham‐operated mice. The ESP (Figure 3E), maximum dP/dt (Figure 3G), and Ees (Figure 3H) were significantly reduced while the end‐systolic volume (Figure 3F) was significantly increased in the MI mice compared to sham‐operated mice. Importantly, Ees from the MI + TPPU + Cells significantly improved from to the MI alone group. In contrast, there were no significant differences among MI alone, MI + TPPU, or MI + hiPSC‐CMs groups (Figure 3H). We also demonstrate a significant improvement in the rate of relaxation of developed pressure (dP/dt min), an indicator of cardiac diastolic function, with TPPU‐, hiPSC‐CMs‐, or TPPU + hiPSC‐CMs‐treated groups compared to MI alone group (Figure S4C).
FIGURE 3.
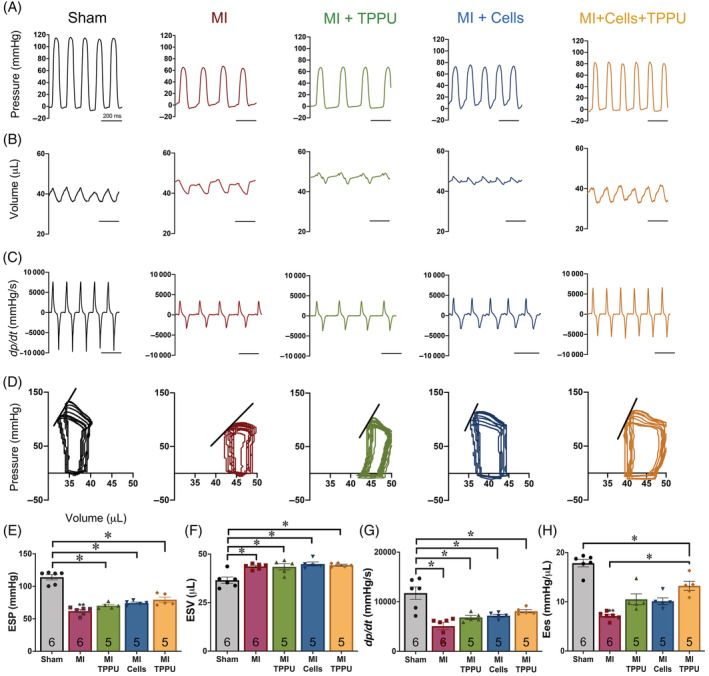
Hemodynamic monitoring demonstrates an improvement in contractility with hiPSC‐CMs and TPPU treatment: Recording traces of left ventricular (A) pressure, (B) volume, and (C) derivative of pressure with respect to time (dP/dt) from Sham‐operated, MI ± TPPU, and MI + hiPSC‐CMs ± TPPU mice. D, Pressure‐volume (P‐V) loop analyses. Pressure and volume have been calibrated. The volume calibration was performed using cuvette (P/N 910‐1049, Millar, Inc.) filled with fresh heparinized 37°C mouse blood. Summary data for (E) ESP, (F) ESV, (G) maximum dP/dt, and (H) slope for the end‐systolic P‐V relationship (Ees). Individual data points are presented together with bar graphs representing mean ± SEM. Number shown within the bar graphs represent the number of animals used. *P < .05 by analysis of variance (ANOVA). Cells = hiPSC‐CMs. ESP, end‐systolic pressure; ESV, end‐systolic volume; hiPSC‐CMs, human induced pluripotent stem cell derived‐cardiomyocytes; MI, myocardial infarction; TPPU, 1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea
3.7. Attenuated cardiac remodeling may be attributed to reduced oxidative stress and apoptosis
Mechanistically, we hypothesize that the increased cell survival and engraftment of transplanted hiPSC‐CMs with TPPU treatment, resulting in an improvement in cardiac function, may be due to a decrease in oxidative stress and apoptosis in the transplanted hiPSC‐CMs. Flow cytometric analyses were used to quantify the level of oxidative stress (production of ROS) in cardiomyocytes and NMCs as we have previously described. 21 Indeed, there was a significant decrease in ROS levels in hiPSC‐CMs isolated from the MI + hiPSC‐CM + TPPU group compared to the MI + hiPSC‐CM group without sEHI treatment (Figure 4A,B). Similarly, apoptosis quantified using single‐cell based flow cytometry in the transplanted hiPSC‐CM was significantly decreased with TPPU treatment (Figure 4C,D).
FIGURE 4.
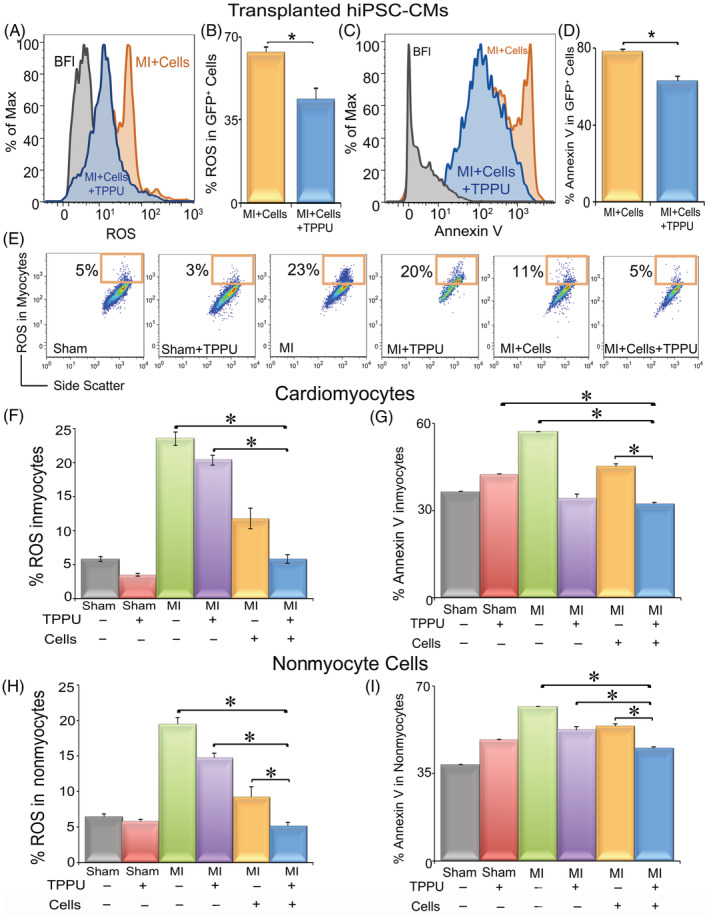
Flow cytometric analyses demonstrate a significant decrease in ROS and apoptosis in transplanted hiPSC‐CMs, host cardiomyocytes, and host nonmyocyte cells isolated from MI mice treated with both hiPSC‐CMs and TPPU: A, Flow cytometric analyses showing the median fluorescence intensity of ROS from transplanted hiPSC‐CMs. B, Summary data from (A) comparing cells isolated from mice receiving hiPSC‐CMs alone or hiPSC‐CMs and TPPU. C, Flow cytometric analyses showing the median fluorescence intensity of Annexin V indicating the degree of apoptosis in transplanted hiPSC‐CMs. D, Summary data from (C) comparing cells isolated from mice receiving hiPSC‐CMs alone or hiPSC‐CMs and TPPU. E, Flow cytometric analyses of ROS from host cardiomyocytes. x and y‐axes represent arbitrary units. F, Summary data from (E) comparing percentages of ROS in host cardiomyocytes isolated from six groups of mice. G, Summary data from flow cytometric analyses of Annexin V from host cardiomyocytes. H,I, Summary data from flow cytometric analyses of ROS and Annexin V from host NMCs, respectively. Mean ± SEM. n = 3 to 6 per group. *P < .05 by analysis of variance (ANOVA). Cells = hiPSC‐CMs. hiPSC‐CMs, human induced pluripotent stem cell derived‐cardiomyocytes; NMCs, nonmyocyte cells; ROS, reactive oxygen species; TPPU, 1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea
Additionally, the increased cell survival and engraftment of transplanted hiPSC‐CMs with TPPU treatment, resulting in an improvement in cardiac function, may be attributed to the cardioprotective mechanisms of TPPU on the host‐microenvironment. To test this, we measured ROS and apoptosis of host‐CMs as well as NMCs. There was a significant decrease in the levels of ROS and apoptosis of the host‐CMs (Figure 4E‐G) isolated from the hiPSC‐CM transplanted‐TPPU treated group compared to the MI, MI + hiPSC‐CMs, and MI + TPPU groups. Similarly, host NMCs isolated from the hiPSC‐CM transplanted‐TPPU treated group demonstrated a significant decrease in ROS and apoptosis compared to those isolated from MI, MI + hiPSC‐CM, and MI + TPPU groups (Figure 4H,I).
3.8. TPPU‐treatment decreases apoptosis and electrical remodeling in hiPSC‐CMs
To decipher the mechanisms of TPPU on hiPSC‐CMs, several types of in vitro analyses were performed. One of the robust responses to cardiac injury‐activated inflammation is the production of inflammatory cytokines, which activate multiple signaling pathways including MAPKs leading to oxidative stress and apoptosis. Therefore, we examined the members of the MAPK signaling cascade, extracellular signal‐regulated kinases (ERK1/2). 17 Differentiated hiPSC‐CMs (MLC2a+/MLC2v+) were stimulated using inflammatory cytokine TNF‐α (20 ng/mL for 20 minutes) with or without TPPU (1 μM). Analysis of stimulated hiPSC‐CMs demonstrated a significant increase in phosphorylated ERK1/2 (pERK1/2) compared with the control and control‐TPPU‐treated cells (Figure 5A,B). Treatment of TNF‐α stimulated hiPSC‐CMs with TPPU significantly decreased the pERK1/2 level (Figure 5A,B). Flow cytometric analyses enabled us to analyze the activity of pERK1/2 on a per‐cell basis by utilizing several markers to separate the hiPSC‐CMs from other cell types. Moreover, we have previously verified the flow cytometric analyses of pERK1/2 using Western blot analyses of FACS of specific cell population. 17
FIGURE 5.
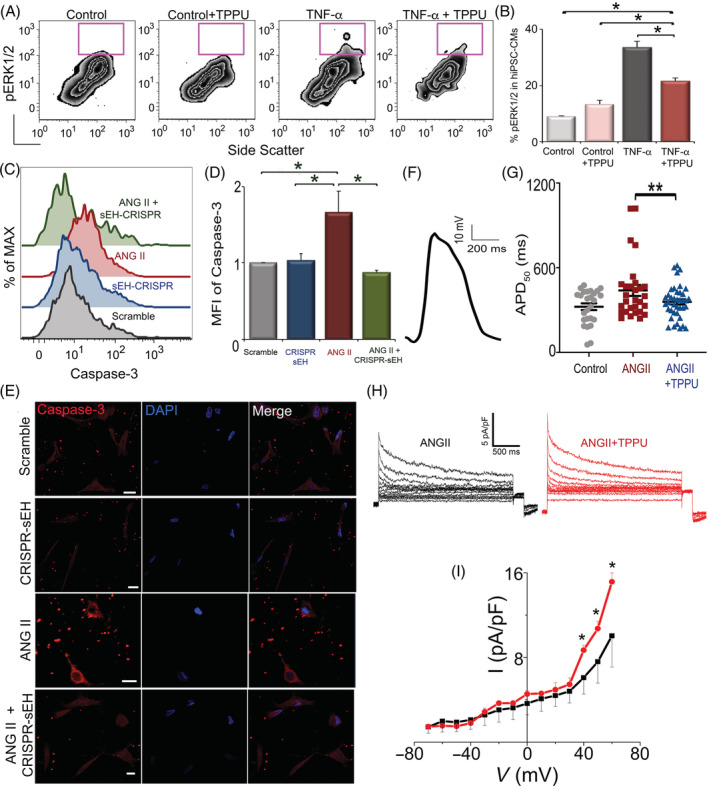
Treatment with TPPU results in a significant reduction in pERK1/2 and recovery of Ito and APD50 in hiPSC‐CMs in vitro: A, Flow cytometric analyses of pERK1/2 signal from four different groups of hiPSC‐CMs (no treatment, treated with TPPU, treated with TNF‐α, or treated with TNF‐α and TPPU). x and y‐axes represent arbitrary units. B, Summary data from (A). C, Flow cytometric analysis of hiPSC‐CMs treated with Scramble, CRISPR‐sEH, ANG II, and ANGII+CRISPR‐sEH. D, Summary data from (C). E, Immunofluorescence images showing an increase in cleaved caspase‐3 in angiotensin II (ANG II, 1 μM for 24 hours) treated hiPSC‐CMs, compared with the scramble, sEH‐CRISPR, and sEH‐CRISPR‐ANG II‐treated cells. F, Representative optical recordings of action potential from hiPSC‐CMs. G, APD50 from control, hiPSC‐CMs treated with ANG II alone or ANG II and TPPU. n = 30 to 100 cells per group of biological repeats from three experiments. H, Transient outward K+ current recordings from hiPSC‐CMs and (I) the corresponding current‐voltage (I‐V) relationship. Representative results are shown. Mean ± SEM. *P < .05 by analysis of variance (ANOVA), **P < .05 by t test. APD50, action potential duration at 50% repolarization; hiPSC‐CMs, human induced pluripotent stem cell derived‐cardiomyocytes; TNF‐α, tumor necrosis factor‐α; TPPU, 1‐trifluoromethoxyphenyl‐3‐(1‐propionylpiperidine‐4‐yl)urea
3.9. CRISPR/Cas9‐mediated gene silencing of the sEH enzyme reduces cleaved caspase‐3 in hiPSC‐CMs challenged with ANG II
Next, in order to demonstrate that the improvement in stem cell transplantation is directly due to the inhibition of the sEH enzyme and not secondary to the off‐target effects, we performed genome editing to silence the sEH enzyme in hiPSC‐CMs using CRISPR/Cas9. Apoptosis was assessed using cleaved caspase‐3, a critical process during apoptosis. We first demonstrated the expression of the sEH enzyme in hiPSC‐CMs using flow cytometry (Figure S5). Flow cytometry and immunofluorescence microscopy showed a significant increase in cleaved caspase‐3 in ANG II (1 μM for 24 hours)‐treated hiPSC‐CMs compared to hiPSC‐CMs with gene silencing of the sEH enzyme treated with ANG II. Two different control groups (hiPSC‐CMs treated with scramble sequences or hiPSC‐CMs with gene silencing of the sEH enzyme without ANG II treatment) were used (Figure 5C‐E).
3.10. Reduction in electrical remodeling by TPPU treatment in hiPSC‐CMs challenged with ANG II
Finally, to evaluate the effect of TPPU on electrical remodeling of hiPSC‐CMs, we challenged the hiPSC‐CMs with ANG II to assess the change in electrophysiological properties using optical and patch‐clamp recordings. Action potentials of hiPSC‐CMs stimulated with ANG II were recorded optically. Treatment with TPPU prevented the increase of the action potential duration at 50% repolarization (APD50) in hiPSC‐CMs challenged with ANG II. APD50 in the TPPU‐treated group were similar to control group (Figure 5F,G). Consistently, hiPSC‐CMs activated with ANG II demonstrated a significant downregulation of the transient outward K+ current assayed using patch‐clamp recordings from single hiPSC‐CMs. Treatment with TPPU prevented the downregulation of the transient outward K+ current (Figure 5H,I). Collectively, the results support that treatment with TPPU leads to an improvement in hiPSC‐CMs including a decrease in oxidative stress, apoptosis, and electrical remodeling.
4. DISCUSSION
Adult cardiac myocytes are unable to proliferate sufficiently to replace the damaged tissue; therefore, stem cell therapy represents a promising approach for the treatment of HF, since it is centered on the premise of regenerating new functional myocardium and inducing neoangiogenesis. In the current study, we demonstrate the therapeutic efficacy of sEHI in hiPSC‐CM cell transplantation in a preclinical model. Treatment with sEHI results in a significant decrease in apoptosis and oxidative stress in transplanted stem cells, leading to a significant increase in the retention of hiPSC‐CMs. In addition, there is an improvement in the host cardiac milieu with sEHI treatment as demonstrated by a significant decrease in ROS and apoptosis in the host cardiomyocytes and nonmyocytes, making it more conducive to the survival of transplanted hiPSC‐CMs. Finally, the dual therapy with sEHI and cardiac stem cell transplantation results in a concomitant decrease in fibrosis and a significant improvement in cardiac function, assayed using both noninvasive and invasive in vivo analyses. Mechanistically, treatment with sEHI results in a decrease in ERK1/2 activation and electrical remodeling in hiPSC‐CMs. Our data provide evidence that reduction of inflammation by increasing the biological activities of EETs can improve stem cell engraftment by optimizing the host microenvironment and the transplanted cells. Mitigation of inflammation using sEHI in cardiac stem cell transplantation may represent an effective therapy post‐MI since it addresses the effects of oxidative stress and the associated inflammation‐mediated fibrosis that may impede the success of the current cell‐based therapies.
4.1. Current challenges in cardiac cell‐based therapy
Stem cells possess the capacity for sustained proliferation, cardiac differentiation as well as trophic functions which enables myocardial repair. To date, various stem cells have been used in clinical trials with each cell type presenting several advantages and limitations. 7 , 8 Since the original description of iPSCs, the field has greatly expanded. Importantly, a large numbers of studies have refined the techniques for efficient directed‐differentiation of iPSCs into cardiomyocytes. 38 , 41 In addition, multiple studies have provided evidence for the utilities of iPSC‐CMs in cardiac transplantation in animal models. 7 However, therapeutic strategies using cell‐based therapy to combat ischemic cardiomyopathy have not produced full restorative functions. 10 , 11 One of the main challenges of cardiac stem cell therapy is the survival and retention of transplanted cells in the hostile milieu. A high rate of transplanted stem‐cell loss, ~90% within the first few days has been observed due to ischemic environment and inflammation. 42 , 43 For transplanted cells to mechanical and electrically couple with native cardiomyocytes to ultimately contract synchronously, they would first need to survive the initial robust inflammatory response associated with cardiac tissue injury.
4.2. Detrimental effects of inflammatory mediators and oxidative stress
Robust inflammatory mediators are one of the prime causes of cell death. 42 , 43 , 44 The recruitment of inflammatory cells such as neutrophils and monocytes into the infarcted heart tissue causes the production of inflammatory cytokines such as TNF‐α, IL‐6, IL‐1, and chemokines, which further secrete proteolytic enzymes and ROS. 43 , 44 , 45 These mediators are severely cytotoxic and compromise survival of transplanted cells. 42 IL‐1 is elevated in infarcted myocardium and facilitates apoptosis of cardiomyocytes and adverse remodeling. 46 Cell death further increases inflammatory responses leading to attenuation of cardiac function. In response to inflammation, stem cells produce ROS and also activate mitochondrial and endoplasmic reticulum stress and death receptor pathways eliciting apoptosis. 47 Inflammation activates ERK1/2 leading to fibrosis and apoptosis, thereby reduces cell engraftment. ERK1/2 members of the MAPK superfamily are known to regulate several critical processes including stem cell differentiation, proliferation, and survival. Therefore, it is critical that the transplanted stem cells can overcome the harsh inflammatory microenvironment and survive in the injured myocardium for significant repairs to occur.
Infarction associated hypoxia increases oxidative stress in the injured myocardium. ROS is known to influence critical cellular processes including gene expression of inflammatory cytokines and growth factors, metabolic alterations, calcium handling, ionic fluxes, and apoptosis. 48 ROS‐induced apoptotic factors from transplanted cells further increase oxidative stress in the myocardium. Apoptosis is induced by both intrinsic and extrinsic factors including inflammatory cytokines. Indeed, we demonstrate a significant decrease in ROS and apoptosis not only in transplanted hiPSC‐CMs but also in the host environment including cardiomyocytes and NMCs with sEHI treatment.
Another critical barrier for stem cell engraftment is the recruitment of fibroblasts, which form physical barriers at the site of injury and inhibit cell engraftment. Here, we directly demonstrate the beneficial effect of sEHI on the reduction of cardiac fibrosis and adverse remodeling consistent with our previous study. 17
4.3. Current strategies to improve cardiac cell‐based therapy
To combat the challenge of stem cell death in the injured myocardium immediately after transplantation, several pro‐survival strategies, which either activate endogenous cellular survival mechanisms or inhibit major cell death pathways have been used. 12 , 49 , 50 Indeed, studies have shown that inhibitors of inflammation increase cell survival by many fold. 43 Specifically, various strategies have been used to improve transplanted cell survival including expression of IL‐1 inhibitor and treatment with superoxide dismutase to reduce inflammation. 43 Our previous data demonstrate that sEHIs are potent anti‐inflammatory agents that significantly decrease the systemic levels of cytokines and chemokines. 17 , 18 , 19 , 51 , 52 , 53 , 54 , 55 , 56 , 57 In the present study, we demonstrate for the first time the broader use sEHIs in reducing inflammation to promote transplanted hiPSC‐CMs survival for cardiac regenerative therapy post‐MI. Moreover, the beneficial effects of sEHIs is relevant since we have previously demonstrated the presence of sEH enzyme in cardiomyocytes as well as hiPSC‐CMs. 21
Finally, we show that treatment of ANG II‐ and TNF‐α‐activated hiPSC‐CMs with sEHI leads to a decrease in the activation of ERK1/2 and electrical remodeling. Taken together, our data suggest that conditioning the hiPSC‐CMs with sEHI may help the cells to better survive the harsh conditions in the ischemic myocardium.
4.4. Perspectives and future directions
It is well documented that paracrine factors secreted or induced by transplanted stem cells contribute significantly to the improvement in cardiac function. Our findings (Figures 1 and 2) are consistent with previous studies which demonstrate that despite the scarcity of survival stem cells 3 to 4 weeks after transplantation, there are still significant benefit in the improvements in cardiac function, possibly from the paracrine signals such as growth factors, chemokines, and exosomes. 58 These paracrine signals may activate multiple targets including attenuation of inflammation, fibrosis, and apoptosis, which act synergistically to prevent structural and electrical remodeling.
There are other mechanisms apart from the generation of new myocardium and paracrine factors that may contribute to the beneficial impact, including mechanical stabilization, stimulation of neovascularization, and cell fusion, which remain be investigated. Future studies using RNA sequencing analyses to determine the gene pathways altered by sEHI treatment would be of great interest.
The selective and potent inhibitor for the sEH enzymes were developed based on the catalytic mechanism and the x‐ray structure of the murine sEH (PDB access #: 1CQZ and 1CR6). In addition, the x‐ray crystal structure of the sEH enzyme and the inhibitors has been solved. 27 , 30 , 31 , 32 , 33 , 34 There is no evidence to date of off‐target binding sites. 22 , 23 , 24 Furthermore, we have taken advantage of CRISPR/Cas9‐mediated gene silencing of the sEH enzyme to test our hypothesis. Finally, very little is known regarding the role of this class of compounds in cell‐based therapy. There is consequently an enormous opportunity to uncover a potentially powerful class of inhibitors, which may be used effectively in the clinical setting. Our study transcends the suppression of inflammation and scar formation in cardiac cell‐based therapy. Since the CYP450 pathway is evolutionarily preserved, the inhibitors have the potential to be utilized in other inflammatory‐related diseases.
CONFLICT OF INTEREST
J.Y. declared leadership position with EicOsis. D.K.L. declared advisory role with Novoheart Ltd. B.D.H., and N.C. declared intellectual property rights on a UC patent on the technology. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
P.S.: conception and study design, data acquisition, data analysis and interpretation, manuscript writing, review, and revision; N.C.: conception and study design, data analysis and interpretation, manuscript writing, review, and revision; P.N.T., J.H.L., X.‐D.Z., V.T., D.J.R.: data acquisition, data analysis and interpretation; J.Y., L.R., N.L., K.S.L., C.E.N., S.Y., S.G., J.N.Y.: data acquisition; D.K.L., E.N.Y., B.D.H.: data analysis and interpretation.
Supporting information
Appendix S1: Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank Dr Joseph Wu (Stanford University) for the double fusion construct of reporter genes (firefly luciferase and enhanced green fluorescent protein). Supported by American Heart Association (AHA) Career Development award (18CDA34110060) and Harold S. Geneen Charitable Trust Award (P. S.); Postdoctoral Fellowship from National Institutes of Health (NIH) T32 Training Grant in Basic & Translational Cardiovascular Science (NIH T32 HL086350) and NIH F32 HL149288 Postdoctoral Research Fellowship (P. N. T.); NIH K99R00 ES024806 (K. S. L.); the National Institute of Environmental Health Sciences (NIEHS) R35 ES030443 and NIEHS Superfund Research Program P42ES004699 (B. D. H.); NIH P01 AG051443 and NIH R01 DC015135 (E. N. Y.), AHA Beginning Grant‐in‐Aid 14BGIA18870087 and NIH R56 HL138392 (X.‐D. Z.); NIH R01 HL085727, NIH R01 HL085844, NIH R01 HL137228, NIH S10 RR033106, Research Award from the Rosenfeld Heart Foundation, U.S. Department of Veterans Affairs Merit Review Grants I01 BX000576, and I01 CX001490 (N. C.). N. C. is the holder of the Roger Tatarian Endowed Professorship in Cardiovascular Medicine and a part‐time staff physician at VA Northern California Health Care System, Mather, California.
Sirish P, Thai PN, Lee JH, et al. Suppression of inflammation and fibrosis using soluble epoxide hydrolase inhibitors enhances cardiac stem cell‐based therapy. STEM CELLS Transl Med. 2020;9:1570–1584. 10.1002/sctm.20-0143
Padmini Sirish and Phung N. Thai contributed equally to the work.
Funding information U.S. Department of Veterans Affairs Merit Review Grant, Grant/Award Numbers: I01 CX001490, I01 BX000576; Rosenfeld Foundation; American Heart Association Beginning Grant‐in‐Aid, Grant/Award Number: 14BGIA18870087; National Institute of Environmental Health Sciences River Award, Grant/Award Number: IR35 ES0443; National Institute of Environmental Health Sciences Superfund Program, Grant/Award Number: P42ES004699; National Institute of Environmental Health Sciences (NIEHS), Grant/Award Number: R35 ES030443; National Institutes of Health, Grant/Award Numbers: S10 RR033106, R01 HL137228, R01 HL085844, R01 HL085727, R56 HL138392, R01 DC015135, P01 AG051443, K99R00 ES024806; NIH F32 HL149288 Postdoctoral Research Fellowship; NIH T32 Training Grant in Basic & Translational Cardiovascular Science, Grant/Award Number: NIH T32 HL086350; Harold S. Geneen Charitable Trust Award; American Heart Association Career Development award, Grant/Award Number: 18CDA34110060
Contributor Information
Padmini Sirish, Email: psirish@ucdavis.edu.
Nipavan Chiamvimonvat, Email: nchiamvimonvat@ucdavis.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available within the paper or can be obtained from the corresponding author upon request.
REFERENCES
- 1. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561‐1566. [DOI] [PubMed] [Google Scholar]
- 2. Ho KK, Pinsky JL, Kannel WB, et al. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993;22:6A‐13A. [DOI] [PubMed] [Google Scholar]
- 3. Dominguez LJ, Parrinello G, Amato P, Licata G. Trends of congestive heart failure epidemiology: contrast with clinical trial results. Cardiologia. 1999;44:801‐808. [PubMed] [Google Scholar]
- 4. Cohn J, Archibald D, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure: results of a veterans administration cooperative study. N Engl J Med. 1986;314:1547‐1552. [DOI] [PubMed] [Google Scholar]
- 5. Gerbin KA, Murry CE. The winding road to regenerating the human heart. Cardiovasc Pathol. 2015;24:133‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics‐2016 update: a report from the American Heart Association. Circulation. 2016;133:e38‐e360. [DOI] [PubMed] [Google Scholar]
- 7. Lalit PA, Hei DJ, Raval AN, Kamp TJ. Induced pluripotent stem cells for post‐myocardial infarction repair: remarkable opportunities and challenges. Circ Res. 2014;114:1328‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kai C, hD W. Clinical applications of stem cells for the heart. Circ Res. 2005;96:151‐163. [DOI] [PubMed] [Google Scholar]
- 9. Park SJ, Kim RY, Park BW, et al. Dual stem cell therapy synergistically improves cardiac function and vascular regeneration following myocardial infarction. Nat Commun. 2019;10:3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dimmeler S, Zeiher AM. Cell therapy of acute myocardial infarction: open questions. Cardiology. 2009;113:155‐160. [DOI] [PubMed] [Google Scholar]
- 11. Dimmeler S, Losordo D. Stem cells review series: an introduction. Circ Res. 2011;109:907‐909. [DOI] [PubMed] [Google Scholar]
- 12. Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro‐survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015‐1024. [DOI] [PubMed] [Google Scholar]
- 13. Chen IY, Wu JC. Cardiovascular molecular imaging: focus on clinical translation. Circulation. 2011;123:425‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang WY, Ebert AD, Narula J, Wu JC. Imaging cardiac stem cell therapy: translations to human clinical studies. J Cardiovasc Transl Res. 2011;4:514‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55‐90. [DOI] [PubMed] [Google Scholar]
- 16. Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311‐333. [DOI] [PubMed] [Google Scholar]
- 17. Sirish P, Li N, Liu JY, et al. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci USA. 2013;110:5618‐5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiamvimonvat N, Ho CM, Tsai HJ, Hammock BD. The soluble epoxide hydrolase as a pharmaceutical target for hypertension. J Cardiovasc Pharmacol. 2007;50:225‐237. [DOI] [PubMed] [Google Scholar]
- 19. Li N, Liu JY, Qiu H, et al. Use of metabolomic profiling in the study of arachidonic acid metabolism in cardiovascular disease. Congest Heart Fail. 2011;17:42‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu L, Timofeyev V, Li N, et al. Alpha‐actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+‐activated K+ channel (SK2 channel). Proc Natl Acad Sci USA. 2009;106:18402‐18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sirish P, Li N, Timofeyev V, et al. Molecular mechanisms and new treatment paradigm for atrial fibrillation. Circ Arrhythm Electrophysiol. 2016;9:e003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW. Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J Biol Chem. 2000;275:15265‐15270. [DOI] [PubMed] [Google Scholar]
- 23. Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc Natl Acad Sci USA. 1999;96:10637‐10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morisseau C, Goodrow MH, Dowdy D, et al. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA. 1999;96:8849‐8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ulu A, Appt S, Morisseau C, et al. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol. 2012;165:1401‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu JY, Lin YP, Qiu H, et al. Substituted phenyl groups improve the pharmacokinetic profile and anti‐inflammatory effect of urea‐based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci. 2013;48:619‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43:4716‐4723. [DOI] [PubMed] [Google Scholar]
- 28. Gomez GA, Morisseau C, Hammock BD, Christianson DW. Human soluble epoxide hydrolase: structural basis of inhibition by 4‐(3‐cyclohexylureido)‐carboxylic acids. Protein Sci. 2006;15:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garriga M, Caballero J. Insights into the structure of urea‐like compounds as inhibitors of the juvenile hormone epoxide hydrolase (JHEH) of the tobacco hornworm Manduca sexta: analysis of the binding modes and structure‐activity relationships of the inhibitors by docking and CoMFA calculations. Chemosphere. 2011;82:1604‐1613. [DOI] [PubMed] [Google Scholar]
- 30. Xing L, McDonald JJ, Kolodziej SA, et al. Discovery of potent inhibitors of soluble epoxide hydrolase by combinatorial library design and structure‐based virtual screening. J Med Chem. 2011;54:1211‐1222. [DOI] [PubMed] [Google Scholar]
- 31. Moser D, Achenbach J, Klingler FM, Estel la B, Hahn S, Proschak E. Evaluation of structure‐derived pharmacophore of soluble epoxide hydrolase inhibitors by virtual screening. Bioorg Med Chem Lett. 2012;22:6762‐6765. [DOI] [PubMed] [Google Scholar]
- 32. Amano Y, Yamaguchi T, Tanabe E. Structural insights into binding of inhibitors to soluble epoxide hydrolase gained by fragment screening and X‐ray crystallography. Bioorg Med Chem. 2014;22:2427‐2434. [DOI] [PubMed] [Google Scholar]
- 33. Takai K, Chiyo N, Nakajima T, et al. Three‐dimensional rational approach to the discovery of potent substituted cyclopropyl urea soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2015;25:1705‐1708. [DOI] [PubMed] [Google Scholar]
- 34. Amano Y, Tanabe E, Yamaguchi T. Identification of N‐ethylmethylamine as a novel scaffold for inhibitors of soluble epoxide hydrolase by crystallographic fragment screening. Bioorg Med Chem. 2015;23:2310‐2317. [DOI] [PubMed] [Google Scholar]
- 35. Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee AS, Wu JC. Imaging of embryonic stem cell migration in vivo. Methods Mol Biol. 2011;750:101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lian X, Zhang J, Azarin SM, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta‐catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lian X, Hsiao C, Wilson G, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E1848‐E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singla DK, Hacker TA, Ma L, et al. Transplantation of embryonic stem cells into the infarcted mouse heart: formation of multiple cell types. J Mol Cell Cardiol. 2006;40:195‐200. [DOI] [PubMed] [Google Scholar]
- 40. Suthahar N, Meijers WC, Sillje HHW, et al. From inflammation to fibrosis‐molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep. 2017;14:235‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 42. Der Sarkissian S, Levesque T, Noiseux N. Optimizing stem cells for cardiac repair: current status and new frontiers in regenerative cardiology. World J Stem Cells. 2017;9:9‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Don CW, Murry CE. Improving survival and efficacy of pluripotent stem cell‐derived cardiac grafts. J Cell Mol Med. 2013;17:1355‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi RZ, Li QP. Improving outcome of transplanted mesenchymal stem cells for ischemic heart disease. Biochem Biophys Res Commun. 2008;376:247‐250. [DOI] [PubMed] [Google Scholar]
- 45. Aurora AB, Olson EN. Immune modulation of stem cells and regeneration. Cell Stem Cell. 2014;15:14‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maekawa Y, Mizue N, Chan A, et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin‐1 receptor‐associated kinase‐4 signaling: a regulator of bone marrow‐derived dendritic cells. Circulation. 2009;120:1401‐1414. [DOI] [PubMed] [Google Scholar]
- 47. Rodrigues M, Turner O, Stolz D, Griffith LG, Wells A. Production of reactive oxygen species by multipotent stromal cells/mesenchymal stem cells upon exposure to fas ligand. Cell Transplant. 2012;21:2171‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye L, Chang YH, Xiong Q, et al. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell‐derived cardiovascular cells. Cell Stem Cell. 2014;15:750‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robey TE, Saiget MK, Reinecke H, Murry CE. Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol. 2008;45:567‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ai D, Pang W, Li N, et al. Soluble epoxide hydrolase plays an essential role in angiotensin II‐induced cardiac hypertrophy. Proc Natl Acad Sci USA. 2009;106:564‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Harris TR, Li N, Chiamvimonvat N, et al. The potential of soluble epoxide hydrolase inhibition in the treatment of cardiac hypertrophy. Congest Heart Fail. 2008;14:219‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li N, Liu JY, Timofeyev V, et al. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: insight gained using metabolomic approaches. J Mol Cell Cardiol. 2009;47:835‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu JY, Li N, Yang J, et al. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib‐mediated cardiovascular events. Proc Natl Acad Sci USA. 2010;107:17017‐17022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu JY, Yang J, Inceoglu B, et al. Inhibition of soluble epoxide hydrolase enhances the anti‐inflammatory effects of aspirin and 5‐lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol. 2010;79:880‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Qiu H, Li N, Liu JY, Harris TR, Hammock BD, Chiamvimonvat N. Soluble epoxide hydrolase inhibitors and heart failure. Cardiovasc Ther. 2011;29:99‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu D, Li N, He Y, et al. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:18733‐18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chimenti I, Smith RR, Li TS, et al. Relative roles of direct regeneration versus paracrine effects of human cardiosphere‐derived cells transplanted into infarcted mice. Circ Res. 2010;106:971‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Supporting information
Data Availability Statement
The data that support the findings of this study are available within the paper or can be obtained from the corresponding author upon request.