

Abstract
Twelve pyridazinones (T1–T12) containing the (2-fluorophenyl) piperazine moiety were designed, synthesized, and evaluated for monoamine oxidase (MAO) -A and -B inhibitory activities. T6 was found to be the most potent MAO-B inhibitor with an IC50 value of 0.013 µM, followed by T3 (IC50 = 0.039 µM). Inhibitory potency for MAO-B was more enhanced by meta bromo substitution (T6) than by para bromo substitution (T7). For para substitution, inhibitory potencies for MAO-B were as follows: -Cl (T3) > -N(CH3)2 (T12) > -OCH3 (T9) > Br (T7) > F (T5) > -CH3 (T11) > -H (T1). T6 and T3 efficiently inhibited MAO-A with IC50 values of 1.57 and 4.19 µM and had the highest selectivity indices (SIs) for MAO-B (120.8 and 107.4, respectively). T3 and T6 were found to be reversible and competitive inhibitors of MAO-B with Ki values of 0.014 and 0.0071, respectively. Moreover, T6 was less toxic to healthy fibroblast cells (L929) than T3. Molecular docking simulations with MAO binding sites returned higher docking scores for T6 and T3 with MAO-B than with MAO-A. These results suggest that T3 and T6 are selective, reversible, and competitive inhibitors of MAO-B and should be considered lead candidates for the treatment of neurodegenerative disorders like Alzheimer’s disease.
Keywords: monoamine oxidase, pyridazinone, kinetics, reversibility, ADME, molecular docking
1. Introduction
Alzheimer’s disease (AD) is a fatal neurodegenerative disease characterized by progressive cognitive dysfunctions, an inability to perform daily activities, and by the various neuropsychiatric symptoms of late-stage AD [1]. AD is also associated with a reduction in the number of neurons in the limbic system, which manages memory processes [2]. Furthermore, the levels of many neurotransmitters, especially acetylcholine, are diminished in cerebral cortex in concert with neuronal loss, and the levels of other neurotransmitters, such as cholinergic, noncholinergic, serotonergic, dopaminergic, and neuropeptidergic neurotransmitters are altered in AD [3].
It has been reported that monoamine oxidase-B (MAO-B) activity is enhanced in AD, particularly in reactive astrocytes near β-amyloid (Aβ) plaques [4]. In a more recent study, it was reported that the MAO-B levels were elevated in astrocytes and in pyramidal neurons in the frontal cortex, hippocampus CA1 region, and entorhinal cortex [5]. MAO-B inhibitors can reduce neuronal damage by inhibiting oxidative deamination, and it is believed that MAO-B inhibition reduces oxidative stress and neurodegeneration, and thereby, delays AD progression [6,7,8]. Many scaffolds, such as chalcones, chromones, coumarins, thiazolidinones, isatins, and pyrazolines, have been reported to inhibit MAO-B selectively and to exhibit promising neuroprotective properties [9,10,11,12,13,14,15,16,17,18,19,20,21,22]. In addition, MAO-B inhibitors have been recently designed with two aryl or heteroaryl hydrophobic rings separated by short enamide, unsaturated ketone, multi-conjugated ketone, amides, or hydrazone spacers [23,24,25,26,27].
Pyridazinones are pyridazine-containing heterocycles with two adjacent ring nitrogen atoms and keto functionality. Several FDA-approved drugs with a pyridazinone nucleus, such as pimobendan, bemoradan, indolidan, and levosimendan, have been commercialized over the past 30 years as antihypertensive, and azanrinone and emorfazone are now marketed as cardiotonic and anti-inflammatory agents, respectively [28]. Recently, our group explored a novel class of pyridazinone hydrazides with pendant piperazine moieties and investigated their abilities to competitively and reversibly inhibit MAO-B [29].
Here, we report the MAO-A and MAO-B inhibitory effects of a series of novel hydrazone derivatives (T1–T12) bearing a phenyl ring with different substituents. We previously reported the anticancer activities of seven of these derivatives (T1–T7) [30]. The other five (T8–T12) were synthesized during the present study. Lead compounds were further examined by investigating enzyme inhibition kinetics, reversibility and cytotoxicity to normal cell lines. In addition, binding modes to MAO-A and MAO-B and interactions with their active sites were determined by molecular docking simulations. Structure-activity relationships were investigated by comparing in vitro enzyme inhibition and molecular docking results.
2. Results and Discussion
2.1. Chemistry
The synthetic routes used to produce the 12 derivatives are shown in Scheme 1. Compounds were synthesized by reacting commercial 3,6-dichloropyridazine with (2-fluorophenyl)piperazine in ethanol to afford 3-chloro-6-[4-(2-fluorophenyl)piperazine-1-yl]pyridazine. The physical and spectral properties of 3-chloro-6-[4-(2-fluorophenyl)piperazine-1-yl]pyridazine concurred with literature data [30]. Compound 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone was obtained by hydrolyzing 3-chloro-6-substituted pyridazine in hot glacial acetic acid. Ethyl 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetate was obtained by reacting 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone with ethyl bromoacetate in acetone in the presence of K2CO3. Then, 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetohydrazide was synthesized by condensing ethyl 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetate with hydrazine hydrate (yield 99%). The title compounds T1–T7, which contain benzalhydrazone, were obtained by condensing 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetohydrazide with substituted/non-substituted benzaldehydes, as we previously described [31]. The other five compounds (T8–12) were synthesized using the same methods and are reported for the first time. The molecular structures of these compounds were confirmed by 1H-NMR, 13C-NMR, and HRMS (Supplementary Materials). The molecular structures, yields, melting points, molecular weights, and molecular formulas of T1–T12 are provided in Table 1.
Scheme 1.

Synthetic pathway for the production of 6-(4-(2-fluorophenyl) piperazine-1-yl)-3(2H)-pyridazinone-2-acetyl-2-(substituted/non-substituted benzal)hydrazone derivatives (T1–T12).
Table 1.
Molecular structures, yields, melting points, molecular weights, and molecular formulae of T1-12.
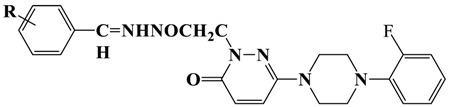
| |||||
|---|---|---|---|---|---|
| Compound No. | R | Yield (%) | P | MM | Molecular Formula |
| T1 | H | 79.90 | 226–228 | 434.48 | C23H23FN6O2 |
| T2 | 4-CH3 | 78.15 | 198–200 | 448.49 | C24H25FN6O2 |
| T3 | 2-Cl | 73.19 | 203–205 | 468.91 | C23H22ClFN6O2 |
| T4 | 4-Cl | 91.21 | 192–194 | 468.91 | C23H22ClFN6O2 |
| T5 | 4-Br | 85.69 | 209–211 | 513.36 | C23H22BrFN6O2 |
| T6 | 2-CH3O | 77.45 | 239–241 | 464.49 | C24H25FN6O3 |
| T7 | 4-N(CH3)2 | 68.77 | 237–239 | 477.53 | C25H28FN7O2 |
| T8 | 3-Br | 90.08 | 207–208 | 513.36 | C23H22BrFN6O2 |
| T9 | 2-F | 88.36 | 225–226 | 452.47 | C23H22 F2N6O2 |
| T10 | 4-F | 57.70 | 178–180 | 452.47 | C23H22 F2N6O2 |
| T11 | 4- OCH3 | 75.32 | 148–150 | 464.49 | C24H25FN6O3 |
| T12 | 2-CH3 | 72.21 | 244–245 | 448.49 | C24H25FN6O2 |
It was observed that these compounds were in two stereoisomeric forms by interpretation of NMR spectra. For example, T10 existed as two stereoisomeric forms in 1H-NMR spectrum (Figure 1A), indicating the resonance of methylene attached to the carbonyl group at 5.30 and 4.87 ppm (2H; s; CH2CO) and two pairs of singlet (one pair at 8.0 and 8.4 ppm and another one at 9.5 and 10.6 ppm) (Figure 1B). In addition, the carbons of pyridazinone C6, pyridazinone C4, and 2-fluorophenyl C5 were double in 13C-NMR spectrum at low MHz (Figure 1C). These pairs of signals clearly show that these compounds exist in two stereoisomeric forms, as other isomers described previously [32,33].
Figure 1.

Structures of two conformational isomers (A), and 1H-NMR (B) and 13C-NMR (C) spectra of T10.
2.2. MAO Inhibitory Activities
Of the 12 derivatives, six potently inhibited MAO-B with residual activities of <50% at 1.0 µM (Table 2). T6 most potently inhibited MAO-B, with an IC50 value of 0.013 µM, and was followed by T3 and T4 (IC50 = 0.039 and 0.099 µM, respectively). T2, T12, and T9 also potently inhibited MAO-B (IC50 = 0.10, 0.15, and 0.20 µM, respectively). Bromine at the meta position (T6) increased MAO-B inhibitory activity more so than at the para position (T7). Regarding substituents at the para position, MAO-B inhibitory activities increased in the order: –Cl (T3) > –N(CH3)2 (T12) > -OCH3 (T9) > -Br (T7) > -F (T5) > -CH3 (T11) > -H (T1). In addition, T6 efficiently inhibited MAO-A with an IC50 value of 1.57 µM, followed by T3 (IC50 = 4.19 µM) (Table 2), and T6 also had the highest selectivity index (SI) value (120.8) for MAO-B, followed by T3 (SI = 107.4).
Table 2.
Inhibitions of recombinant human MAO-A and MAO-B by T1 to T12 a.
| Compounds | Residual Activity (%) | IC50 (µM) | SI b | ||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
| (10 µM) | (1.0 µM) | ||||
| T1 | 96.6 ± 1.20 | 99.4 ± 0.85 | - | 7.68 ± 0.14 | - |
| T2 | 24.6 ± 2.86 | 4.22 ± 0.85 | 4.29 ± 0.12 | 0.10 ± 0.035 | 42.9 |
| T3 | 24.6 ± 0.97 | 3.01 ± 0.85 | 4.19 ± 0.27 | 0.039 ± 0.0028 | 107.4 |
| T4 | 75.2 ± 4.70 | 6.00 ± 0.94 | - | 0.099 ± 0.0069 | - |
| T5 | 84.0 ± 0.68 | 54.0 ± 2.83 | - | 1.68 ± 0.11 | - |
| T6 | 20.7 ± 0.62 | −15.3 ± 0.94 | 1.57 ± 0.80 | 0.013 ± 0.0016 | 120.8 |
| T7 | 25.4 ± 6.92 | 57.8 ± 9.58 | 4.43 ± 0.29 | 0.87 ± 0.056 | - |
| T8 | 73.3 ± 2.86 | 67.2 ± 9.58 | - | 2.67 ± 0.051 | - |
| T9 | 35.2 ± 0.56 | 2.60 ± 0.74 | 6.76 ± 0.46 | 0.20 ± 0.0017 | 33.8 |
| T10 | 33.7 ± 4.70 | 95.3 ± 6.70 | 7.35 ± 0.059 | 35.3 ± 0.39 | 0.21 |
| T11 | 70.4 ± 8.50 | 61.1 ± 2.98 | - | 6.86 ± 0.37 | - |
| T12 | 61.8 ± 1.65 | 17.9 ± 5.95 | - | 0.15 ± 0.021 | - |
| Toloxatone | 1.08 ± 0.025 | - | |||
| Lazabemide | - | 0.14 ± 0.011 | |||
| Clorgyline | 0.0070 ± 0.00070 | - | |||
| Pargyline | - | 0.030 ± 0.00083 | |||
a Results are expressed as the means ± standard errors of duplicate or triplicate experiments; b Selectivity index (SI) values shown are expressed as IC50 of MAO-A/IC50 of MAO-B; IC50 values for reference compounds were determined after preincubating them for 30 min with MAO-A or MAO-B.
The presence of different substituents on the phenyl hydrazone ring did not appreciably impact MAO selectivity, whereas the introduction of halogens, methoxy, methyl, or dimethylamino groups reduced MAO-B inhibition versus non-substituted hydrazone. The lead compounds T6 and T3, which had methoxy or chlorine groups at the ortho position of the phenyl system, were 11 and 3 times more potent, respectively, than the reference reversible MAO-B inhibitor lazabemide. Furthermore, T6 was twice as potent as the reference irreversible MAO-B inhibitor pargyline. Shifting methoxy group from the ortho to the para position of the phenyl ring (T11) dramatically reduced MAO-B inhibition (IC50 = 6.86 ± 0.37 µM) and shifting chlorine from the ortho to the para (T4) reduced MAO-B inhibition. These observations indicate that ortho is preferred over para in terms of MAO-B inhibition. Interestingly, recent studies have reported that chlorine and methoxy groups have considerable impacts on MAO-B inhibition [34,35].
2.3. Kinetics of MAO-B Inhibitions
Lineweaver-Burk plots and secondary plots obtained for MAO-B inhibition showed that T3 and T6 were competitive inhibitors (Figure 2A,C), with Ki values of 0.014 ± 0.0014 and 0.0071 ± 0.0033 µM, respectively (Figure 2B,D). These results suggest that T3 and T6 are competitive inhibitors of MAO-B.
Figure 2.

Lineweaver-Burk plots for MAO-B inhibition by T3 or T6 (A,C), and respective secondary plots (B,D) of slopes vs. inhibitor concentrations.
2.4. Reversibility Studies
Reversibility studies were performed using a dialysis-based method. Dialysis recovered MAO-B inhibition by T3 from 22.2% (AU) to 79.4% (AD) (Figure 3). T6 was also recovered from 36.9% (AU) to 79.0% (AD) (Figure 3). These degrees of recovery were similar to that observed for lazabemide (the reversible reference MAO-B inhibitor) (from 36.9% to 83.0%). However, inhibition by pargyline (irreversible MAO-B inhibitor) was not recovered. These experiments showed that inhibitions of MAO-B by T3 and T6 were recovered by dialysis to the reversible reference level, thus showing that T3 and T6 reversibly inhibit MAO-B.
Figure 3.

Recoveries of MAO-B inhibitions by T3 and T6 by dialysis.
2.5. Cytotoxic Effects of the Active Pyridazinone Derivatives
The cytotoxic effects of T3 and T6 were investigated using L929 cells (a healthy fibroblast cell line). T3 caused complete cell death at 50 and 100 µM), but no significant death at 1, 10, or 20 µM (Figure 4). T6 did not have a cytotoxic effect at any dose (Figure 4). The IC50 values of T3 and T6 were 27.05 and 120.6 µM, respectively. These results suggested that T6 is a more appropriate drug candidate [30].
Figure 4.

L929 cell viabilities after treatments with T3 (A) or T6 (B). nc, normal control. The experiments were performed with three sets at each concentration.
2.6. Molecular Docking Studies
Docking simulations were used to investigate molecular interactions and binding modes of T3 or T6 with MAO-A or MAO-B. Simulations docking scores are provided in Table 3. The ortho-fluorine phenyl rings of T3 and T6 interacted with F352 of MAO-A by π-π contact and by hydrophobic interaction with Y444, Y407, and the FAD of MAO-A (Figure 5). Moreover, the pyridazinone ring of T6 was well oriented towards F208 of MAO-A, and established another hydrophobic interaction. The ortho positioned fluorine on the phenyl ring of T3 or T6 faced a hydrophobic cage consisting of FAD, Y435, and Y398 of MAO-B, similar to MAO-A binding mode (Figure 6). Pyridazinone rings participated in hydrophobic interactions with the MAO-B selective residue Y326, and the hydrazone moieties of T3 and T6 formed hydrogen bonds with the E84 side-chain and the T201 backbone of MAO-B.
Table 3.
Docking score values for T3 and T6 interactions with MAO-A or MAO-B.
| Compounds | Docking Score (kcal/mol) | |
|---|---|---|
| MAO-A | MAO-B | |
| T3 | −8.20 | −9.45 |
| T6 | −8.43 | −9.56 |
Figure 5.

Top scored poses of T3 (A, green sticks) and T6 (B, magenta sticks) towards MAO-A. Green lines show π-π interactions. Water molecules are rendered as cyan spheres.
Figure 6.

Top scored poses of T3 (A, green sticks) and T6 (B, magenta sticks) towards MAO-B. Red arrows indicate hydrogen bonds. Water molecules are rendered as cyan spheres.
Computational studies of T3 or T6 interactions with MAO-A or MAO-B provided satisfactory explanations for the experimental biological data obtained. Docking scores of T3 and T6 agreed with IC50 experimental values, which indicated T6 is better than T3. Regarding binding modes, the ortho-fluorine phenyl rings of T3 and T6 face the hydrophobic cages of MAO-A and MAO-B. Interestingly, the main difference between MAO-A and MAO-B interactions centers on the presence of two hydrogen bonds between the hydrazone moieties of T3 and T6 and E84 and T201 of MAO-B, due to no hydrogen bond with their corresponding sites V93 and V210 in MAO-A, thus supporting that T3 and T6 are selective MAO-B inhibitors.
2.7. ADME Prediction
The physicochemical parameters and likenesses of T3 and T6 were further investigated by BOILED-Egg construction and using the SwissADME online platform. These online programs determine whether compounds are suitable for oral administration based on the bioavailability radar panel containing physicochemical properties such as flexibility, insolubility, lipophilicity, saturation, size, and polarity, and provide rapid appraisals of the drug-likenesses of drug candidates. The BOILED-Egg constructions of T3 and T6 predicted that both are well absorbed in the GI tract but in outside of blood-brain barrier (BBB) permeation range (Figure 7). T3 and T6 were shown to lie within the pink regions (optimal ranges) (Figure 8). Bioavailability radar predicted both compounds had acceptable pharmacokinetics and satisfied drug-likeness rules. These results suggest that T3 and T6 should be considered potential drug candidates.
Figure 7.

The BOILED-Egg construction of T3 and T6.
Figure 8.

The bioavailability radar of T3 (A) and T6 (B).
3. Materials and Methods
3.1. Chemical Studies
All chemicals used in this study were purchased from Aldrich, Fluka AG, or E. Merck. 3-Chloro-6-substituted pyridazines, 3-chloro-6-(4-(2-fluorophenyl)piperazine-1-yl)pyridazine, 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone, ethyl 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl acetate, and 6-(4-(2-fluorophenyl) piperazine-1-yl)-3(2H)-pyridazinone-2-yl acetohydrazide were synthesized as previously described [20,21,22,23,24]. The purities of these compounds were checked by TLC using Merck Kieselgel F254 plates. Melting points were determined using an Electrothermal 9200 (Cole-Parmer Instrument Co. Vernon Hills, IL, USA), and IR spectra were recorded on a Perkin Elmer Spectrometer using the ATR technique. 1H-NMR and 13C-NMR spectra were recorded on a Brucker Avonce Ultrashield FT-NMR spectrometer in DMSO at 600 and 75 MHz, respectively. Mass spectra (HRMS) were obtained using a combined Waters Acquity Ultra Performance Liquid Chromatography Micromass and an LCT Premier XE UPLC/MS TOFF spectrophotometer (Waters Corp, Milford, MA, USA) in ESI+ and ESI− modes.
3.2. Synthesis of Intermediates
3.2.1. Synthesis of 3-chloro-6-[4-(2-fluorophenyl)piperazine-1-yl]pyridazine (1)
A solution containing 0.01 mol of 3,6-dichloropyridazine and 0.01 mol of (2-fluorophenyl)piperazine in 15 mL of ethanol was stirred under reflux at 120 °C for 6 h with the cooler adjusted to 15 °C as previously described [36]. The reaction mix was poured into ice water, and the precipitate obtained by filtration was purified by crystallization from ethanol. Melting points of non-original compounds were in accord with previously reported values [36].
3.2.2. Synthesis of 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone (2)
A solution of 0.05 mol of 3-chloro-6-[4-(2-fluorophenyl)piperazine-1-yl]pyridazine in 30 mL glacial acetic acid was refluxed for 6 h. The acetic acid was then removed under reduced pressure, and the residue obtained was dissolved in water and extracted with chloroform. The organic phase was dried over sodium sulfate and evaporated under reduced pressure, and the residue was purified by recrystallization from ethanol. Melting points of non-original compounds were in accord with previously reported values [36].
3.2.3. Synthesis of ethyl 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetate (3)
A mixture of 0.01 mol 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone, 0.02 mol ethyl bromoacetate, and 0.02 mol potassium carbonate in acetone (40 mL) was refluxed overnight. After the mixture had cooled, organic salts were filtered off, the solvent was evaporated, and the residue was purified by recrystallization from n-hexane to give the esters. Melting points of non-original compounds were in accord with previously reported values [36].
3.2.4. Synthesis of 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetohydrazide (4)
Hydrazine hydrate (99%, 3 mL) was added to the solution of 0.01 mol ethyl 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetate in 25 mL methanol and stirred for 3 h at room temperature. The precipitate obtained was filtered off, washed with water, dried, and recrystallized from ethanol. Melting points of non-original compounds were in accord with previously reported values [36].
3.2.5. General Procedure for the Synthesis of T1–T12
A solution containing 0.01 mol 6-(4-(2-fluorophenyl)piperazine-1-yl)-3(2H)-pyridazinone-2-yl-acetohydrazide and 0.01 mol substituted/non-substituted benzaldehyde was stirred in ethanol (15 mL), refluxed for 6 h, and poured into ice water. The precipitate was filtered, dried, and crystallized from methanol:water (8:2, v/v) [34]. All spectral data of the five new compounds (T8–T12) were in accord with assigned structures.
3.2.6. 6-[4-(2-Fluorophenyl)piperazine-1-yl]-3(2H)-pyridazinone-2-acetyl-2-(3-bromobenzal)hydrazon (T8)
White crystals (methanol/water); yield 90.08%; m.p. 207–208 °C; 1H-NMR (DMSO-d6, 300 MHz): δ 3.18 (4H; t; CH2N; b+b′), 3.45 (4H; t; CH2N; a+a′), 5.31 (2H; s; CH2CO), 6.96 (1H; d; J = 4.14 Hz, pyridazinone H5), 6.97–7.87 (9H; m; phenyl protons, pyridazinone H4), 8.58 (1H; s; -N=CH-) and 10.80 (1H; s; -NH-N). 13C-NMR (DMSO-d6, 300 MHz), δ 46.82 (2C, CH2-N; b+b′), 50.05 (2C, CH2-N; a+a′), 53.14 (1C; -N-CH2-C=O), 115.13 (1C; =CH), 119.10 (1C; pyridazinone C5), 122.96 (1C; 2-fluorophenyl C4), 124.55 (1C; 3-bromophenyl C5), 125.90 (1C; 2-fluorophenyl C5), 126.05 (1C; 3-bromophenyl C2), 129.72 (1C; 3-bromophenyl C6), 130.26 (1C; pyridazinone C4), 131.08 (1C; 2-fluorophenyl C6), 134.22 (1C; 3-bromophenyl C3), 142.99 (1C; 2-fluorophenyl C3), 147.39 (1C; 3-bromophenyl C1), 149.13 (1C; 2-fluorophenyl C1), 154.40 (1C; 3-bromophenyl C4), 156.56 (1C; pyridazinone C6), 158.53 (1C; 2-fluorophenyl C2), 161.02 (1C; CH2-N-C=O), 168.75 (1C; pyridazinone C3); C23H22BrFN6O2 MS (ESI+) calculated: 513.3714, found: m/e 513.10526 (M+; 100.0%).
3.2.7. 6-[4-(2-Fluorophenyl)piperazine-1-yl]-3(2H)-pyridazinone-2-acetyl-2-(2-fluorobenzal)hydrazon (T9)
White crystals (methanol/water); yield 88.36%; m.p. 225–226 °C; 1H-NMR (DMSO-d6, 300 MHz): δ; 3.19 (4H; t; CH2N; b+b′), 3.47 (4H; t; CH2N; a+a′), 5.31 (2H; s; CH2CO), 6.94 (1H; d; J = 4.15 Hz, pyridazinone H5), 6.95–7.49 (9H; m; phenyl protons, pyridazinone H4), 8.96 (1H; s; -N=CH-) and 10.69 (1H; s; -NH-N). 13C-NMR (DMSO-d6, 300 MHz), 46.63 (2C, CH2N; b+b′), 50.07 (2C, CH2N; a+a′), 53.29 (1C; -N-CH2-C=O), 115.83 (1C; =CH), 116.14 (1C; pyridazinone C5), 119.06 (1C; 2-fluorophenyl C6′), 122.90 (1C; 2-fluorophenyl C5′), 124.51 (1C; 2-fluorophenyl C4′), 125.84 (1C; 2-fluorophenyl C5), 129.07 (1C; 2-fluorophenyl C4), 129.15 (1C; pyridazinone C4), 129.62 (1C; 2-fluorophenyl C6), 129.70 (1C; 2-fluorophenyl C3), 129.78 (1C; 2-fluorophenyl C3′), 131.09 (1C; 2-fluorophenyl C1), 143.43 (1C; 2-fluorophenyl C1′), 147.80 (1C; 2-fluorophenyl C2′), 149.09 (1C; pyridazinone C6), 154.54 (1C; 2-fluorophenyl C2), 158.79 (1C; CH2-N-C=O) and 168.73 (1C; pyridazinone C3); C23H22F2N6O2 MS (ESI+) calculated: 452.47, found: 453.18594 m/e (M+; 100.0%).
3.2.8. 6-[4-(2-Fluorophenyl)piperazine-1-yl]-3(2H)-pyridazinone-2-acetyl-2-(4-florobenzal)hydrazon (T10)
White crystals (methanol/water); yield 57.70%; m.p. 178–180 °C; 1H-NMR (DMSO-d6, 300 MHz): δ; 3.17 (4H; t; CH2N; b+b′), 3.46 (4H; t; CH2N; a+a′), 5.30 (2H; s; CH2CO), 6.84 (1H; d; J = 4.15 Hz, pyridazinone H5), 6.85–7.83 (9H; m; phenyl protons, pyridazinone H4), 8.08 (1H; s; -N=CH-) and 10.68 (1H; s; -NH-N). 13C-NMR (DMSO-d6, 300 MHz), 46.84 (2C, CH2N; b+b′), 50.07 (2C, CH2N; a+a′), 53.29 (1C; -N-CH2-C=O), 115.83 (1C; =CH), 116.05 (1C; pyridazinone C5), 116.35 (1C; 4-fluorophenyl C6), 122.90 (1C; 4-fluorophenyl C5), 124.51 (1C; 4-fluorophenyl C4), 125.84 (1C; 2-fluorophenyl C5), 129.07 (1C; 2-fluorophenyl C4), 129.62 (1C; pyridazinone C4), 129.70 (1C; 2-fluorophenyl C6), 130.68 (1C; 2-fluorophenyl C3), 139.71 (1C; 4-fluorophenyl C3), 147.80 (1C; 2-fluorophenyl C1), 149.09 (1C; 4-fluorophenyl C1), 154.54 (1C; 4-fluorophenyl C2), 158.79 (1C; pyridazinone C6), 162.52 (1C; 2-fluorophenyl C2), 165.16 (1C; CH2-N-C=O) and 168.73 (1C; pyridazinone C3); C23H22F2N6O2 MS (ESI+) calculated: 452.47, found: 453.18583 m/e (M+; 100.0%).
3.2.9. 6-[4-(2-Fluorophenyl)piperazine-1-yl]-3(2H)-pyridazinone-2-acetyl-2-(4-methoxybenzal)hydrazon (T11)
White crystals (methanol/water); yield 75.32%; m.p. 148–150 °C. 1H-NMR (DMSO-d6, 300 MHz): δ 3.17 (4H; t; CH2N; b+b′), 3.45 (4H; t; CH2N; a+a′), 3.85 (3H; s; CH3O), 5.30 (2H; s; CH2CO), 6.88 (1H; d; J = 4.15 Hz, pyridazinone H5), 6.89–7.79 (9H; m; phenyl protons, pyridazinone H4), 8.64 (1H; s; -N=CH-) and 10.55 (1H; s; -NH-N). 13C-NMR (DMSO-d6, 300 MHz), 46.83 (2C, CH2N; b+b′), 50.07 (2C, CH2N; a+a′), 53.03 (1C; -N-CH2-C=O), 55.38 (1C; -OCH3), 114.07 (1C; =CH), 116.12 (1C; pyridazinone C5), 119.07 (1C; 4-methoxyphenyl C6), 124.50 (1C; 4-methoxyphenyl C5), 125.77 (1C; 4-methoxyphenyl C4), 126.30 (1C; 2-fluorophenyl C5), 128.82 (1C; 2-fluorophenyl C4), 129.43 (1C; pyridazinone C4), 130.14 (1C; 2-fluorophenyl C6), 131.10 (1C; 2-fluorophenyl C3), 139.63 (1C; 4-methoxyphenyl C3), 144.42 (1C; 2-fluorophenyl C1), 149.04 (1C; 4-methoxyphenyl C1), 154.39 (1C; 4-methoxyphenyl C2), 156.83 (1C; pyridazinone C6), 158.82 (1C; 2-fluorophenyl C2), 161.35 (1C; CH2-N-C=O) and 168.56 (1C; pyridazinone C3); C24H25FN6O3 MS (ESI+) calculated: 464.49, found: 465.20506 m/e (M+; 100.0%).
3.2.10. 6-[4-(2-Fluorophenyl)piperazine-1-yl]-3(2H)-pyridazinone-2-acetyl-2-(2-methylbenzal)hydrazon (T12)
White crystals (methanol/water); yield 72.21%; m.p. 244–245 °C; 1H-NMR (DMSO-d6, 300 MHz): δ 2.50 (3H; s; -CH3), 3.17 (4H; t; CH2N; b+b′), 3.45 (4H; t; CH2N; a+a′), 5.30 (2H; s; CH2CO), 6.92 (1H; d; J = 4.19 Hz, pyridazinone H5), 6.92–7.77 (9H; m; phenyl protons, pyridazinone H4), 8.37 (1H; s; -N=CH-) and 10.58 (1H; s; -NH-N). 13C-NMR (DMSO-d6, 300 MHz), δ 20.16 (1C; -CH3), 46.69 (2C; CH2-N; b+b′), 50.10 (2C, CH2-N; a+a′), 53.36 (1C; -N-CH2-C=O), 116.13 (1C; =CH), 119.07 (1C; pyridazinone C5), 122.87 (1C; 4-methylphenyl C6), 124.54 (1C; 2-methylphenyl C2), 125.78 (1C; 2-methylphenyl C3), 126.24 (1C; 4-methylphenyl C5), 127.47 (1C; 4-methylphenyl C4), 129.96 (1C; pyridazinone C4), 130.85 (1C; 2-fluorophenyl C3), 131.48 (1C; 2-fluorophenyl C4), 137.13 (1C; 2-fluorophenyl C5), 143.70 (1C; 2-methylphenyl C1), 147.49 (1C; 2-fluorophenyl C6), 149.71 (1C; 2-fluorophenyl C1), 156.98 (1C; 2-fluorophenyl C2), 158.81 (1C; pyridazinone C6) 163.96 (1C; CH2-N-C=O) and 168.57 (1C; pyridazinone C3); C24H25FN6O2 MS (ESI+) calculated: 448.49, found: 449.21034 m/e (M+; 100.0%).
3.3. Enzyme Assays
Recombinant human MAO-A and MAO-B inhibitory activities of were continuously assayed as described previously [37], using kynuramine (0.06 mM) and benzylamine (0.3 mM), respectively. Substrate concentrations were 1.5 × and 2.3 × Km, respectively, and their Km values were 0.041 mM and 0.13 mM, respectively. MAO-A and MAO-B inhibitory activities were measured after preincubating enzymes and inhibitors for 15 min. Enzymes and chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA).
3.4. Enzyme Inhibitions and Kinetics
The inhibitory activities of the twelve compounds against MAO-A and MAO-B were first investigated at an inhibitor concentration of 10 µM. For the compounds showing residual activities of <50%, IC50 values for MAO-A and MAO-B inhibitions were determined. Kinetic experiments were carried out at five substrate and three inhibitor concentrations [38].
3.5. Analysis of Inhibitor Reversibilities
The reversibilities of T3 and T6 inhibitions were investigated by dialysis after preincubating them with MAO-B for 30 min, as previously described [39]. The concentrations used were T3 0.080 µM, T6 0.020 µM, lazabemide (reference reversible MAO-B inhibitor) 0.30 µM, and pargyline (reference irreversible MAO-B inhibitor) 0.060 µM. Reversibilities were determined using the relative activities of undialyzed (AU) and dialyzed (AD) samples [40].
3.6. Cytotoxicity
An MTS (5-(3-carboxymethoxyphenyl)-2-(4,5-dimethyl-thiazolyl)-3-(4-sulfophenyl)) tetrazolium assay was used to evaluate the cytotoxic activities of T3 and T6 by measuring formazan absorbance. MTS is converted to formazan by mitochondria in metabolically active cells via NAD(P)H-dependent dehydrogenase enzymes. L929 cells (a mouse fibroblast cell line) were used to evaluate the cytotoxicities. Briefly, cells (5 × 104 cells/well) were seeded into 96-well plates in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin, and incubated for 24 h. After supernatants were removed, the cells were treated with the compounds dissolved in DMSO (at 1, 10, 20, 50, or 100 µM) in triplicate for 48 h at 37 °C in a 5% CO2 atmosphere. MTS reagent (Uptima, Interchim, Montluçon, France) was then added to each well and incubated for 3 h. Absorbances were measured at 490 nm using a microplate reader (BioTek, Winooski, VT, USA), and cell viabilities were calculated as percentage of untreated controls [30].
3.7. Molecular Docking Studies
T3 and T6 were docked using X-ray solved structures of MAO-A and MAO-B obtained from the Protein Data Bank (PDB) by retrieving entries 2Z5X and 2V5Z, respectively [41,42]. Protein targets were prepared using the Protein Preparation Wizard Schrödinger Suite 2018-4 (New York, NY, USA) for refining and optimizing crystal structures after correcting for protonation states and adding missing hydrogen atoms [43,44]. Nine water molecules within MAO-A and eight water molecules within MAO-B were not deleted for docking simulations, as was previously performed [45]. To generate all possible tautomers and protonation states at physiological pH, T3 and T6 were treated using the LigPrep tool [46]. The obtained files were used for docking simulations by employing Grid-based Ligand Docking with Energetics (GLIDE) [47]. Enclosing boxes were automatically centered on the related cognate ligands, T3 and T6. The default force Field OPLS_20058 and the standard precision (SP) docking protocol with default settings were employed for the in silico analyses. To confirm the validity of docking studies, re-docking simulations were performed on T3 and T6 at their binding sites. The cognate ligands are flagged as HRM and SAG for MAO-A and MAO-B, respectively, and moved back to the original positions with Root Mean Square Deviations (RMSD) accounting for all heavy atoms equal to 0.760 Å and 0.395 Å for HRM and SAG, respectively [48].
3.8. ADME Prediction
ADME parameters were predicted in silico to determine a range of physical-chemical properties, lipophilicity, water solubility, and pharmacokinetic data, using the login-free website http://www.swissadme.ch/ [49].
4. Conclusions
The present work describes the synthesis of twelve pyridazinones (T1–T12) containing the 2-fluorophenyl substituted piperazine moiety. All synthesized compounds were evaluated for MAO-A and MAO-B inhibitory activities. Most of the compounds were selective MAO-B inhibitors, and T3 and T6 were found to be selective, competitive, and reversible MAO-B inhibitors. T3 and T6 potently and reversibly inhibited MAO-B in the sub-micromolar range, and were relatively non-toxic to healthy fibroblasts. We concluded that T3 and T6 offer new developmental leads for the treatment of neurodegenerative disorders.
Supplementary Materials
1H- and 13C-NMR spectral data are available in the Supplementary Materials.
Author Contributions
Conceptualization: Z.Ö., B.M. and H.K.; biological activity: J.M.O.; kinetics: J.M.O.; docking analysis: N.G., O.N.; synthesis: M.C., Z.Ö.; data curation: B.M., J.M.O.; writing—original draft preparation: B.M., S.E.B., M.U., M.A.A., A.M.; writing—review and editing: B.M. and K.H.; supervision: H.K.; funding acquisition: H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Republic of Korea government (NRF-2019R1A2C1088967) (to H.K.).
Conflicts of Interest
The authors have no conflict of interest to declare.
Sample Availability: Samples of the compounds are available from the authors.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Harilal S., Jose J., Parambi D.G.T., Kumar R., Mathew G.E., Uddin M.S., Kim H., Mathew B. Advancements in nanotherapeutics for Alzheimer’s disease: Current perspectives. J. Pharm. Pharmacol. 2019;71:1370–1383. doi: 10.1111/jphp.13132. [DOI] [PubMed] [Google Scholar]
- 2.Geldenhuys W.J., Darvesh A.S. Pharmacotherapy of Alzheimer’s disease: Current and future trends. Expert. Rev. Neurother. 2015;5:3–5. doi: 10.1586/14737175.2015.990884. [DOI] [PubMed] [Google Scholar]
- 3.Chiu P.Y., Wei C.Y. Donepezil in the one-year treatment of dementia with Lewy bodies and Alzheimer’s disease. J. Neurol. Sci. 2017;81:22. doi: 10.1016/j.jns.2017.08.913. [DOI] [Google Scholar]
- 4.Mathew B., Parambi D.G.T., Mathew G.E., Uddin M.S., Inasu S.T., Kim H., Marathakam A., Unnikrishnan M.K., Carradori S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019;352:e1900177. doi: 10.1002/ardp.201900177. [DOI] [PubMed] [Google Scholar]
- 5.Kim D., Baik S.H., Kang S., Cho S.W., Bae J., Cha M.Y., Sailor M.J., Jung I.M., Ahn K.H. Close correlation of monoamine oxidase activity with progress of Alzheimer’s disease in mice, observed by in vivo two-photon imaging. ACS Cent. Sci. 2016;2:967–975. doi: 10.1021/acscentsci.6b00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar B., Prakash V., Kumar V. A perspective on monoamine oxidase enzyme as drug target: Challenges and opportunities. Curr. Drug Targets. 2017;18:87–97. doi: 10.2174/1389450117666151209123402. [DOI] [PubMed] [Google Scholar]
- 7.Finberg J.P.M., Rabey J.M. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front. Pharmacol. 2016;7:340. doi: 10.3389/fphar.2016.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guglielmi P., Mathew B., Secci D., Carradori S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020:112650. doi: 10.1016/j.ejmech.2020.112650. [DOI] [PubMed] [Google Scholar]
- 9.Carradori S., Silvestri R. New frontiers in selective human MAO-B inhibitors. J. Med. Chem. 2015;58:6717–6732. doi: 10.1021/jm501690r. [DOI] [PubMed] [Google Scholar]
- 10.Tripathi R.K.P., Ayyannan S.R. Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update. Med. Res. Rev. 2019;39:1603–1706. doi: 10.1002/med.21561. [DOI] [PubMed] [Google Scholar]
- 11.Guglielmi P., Carradori S., Ammazzalorso A., Secci D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019;14:995–1035. doi: 10.1080/17460441.2019.1637415. [DOI] [PubMed] [Google Scholar]
- 12.Kumar B., Sheetal S., Mantha A.K., Kumar V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016;6:42660–42683. doi: 10.1039/C6RA00302H. [DOI] [Google Scholar]
- 13.Mathew B., Haridas A., Suresh J., Mathew G.E., Ucar G., Jayaprakash V. Monoamine oxidase inhibitory actions of chalcones. A mini review. Cent. Nerv. Syst. Agents Med. Chem. 2016;16:120–136. doi: 10.2174/1871524915666151002124443. [DOI] [PubMed] [Google Scholar]
- 14.Choi J.W., Jang B.K., Cho N., Park J.H., Yeon S.K., Ju E.J., Lee Y.S., Han G., Pae A.M., Kim D.J., et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg. Med. Chem. 2015;23:6486–6496. doi: 10.1016/j.bmc.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Mathew B., Mathew G.E., Petzer J.P., Petzer A. Structural exploration of synthetic chromones as selective MAO-B inhibitors. A Mini Review. Comb. Chem. High Throughput Screen. 2017;20:522–532. doi: 10.2174/1386207320666170227155517. [DOI] [PubMed] [Google Scholar]
- 16.Gaspar A., Silva T., Yáñez M., Viña D., Orallo F., Ortuso F., Uriarte E., Alcaro S., Borges F. Chromone, a privileged scaffold for the development of monoamine oxidase inhibition. J. Med. Chem. 2011;54:5165–5173. doi: 10.1021/jm2004267. [DOI] [PubMed] [Google Scholar]
- 17.Reis J., Cagide F., Chavarria D., Silva T.B., Fernandes C., Gaspar A., Uriarte E., Remiao F., Alcaro S., Ortuso F., et al. Discovery of new chemical entities of old targets. Insight on the lead optimization of chromones based monoamine oxidase B inhibitors. J. Med. Chem. 2016;59:5879–5893. doi: 10.1021/acs.jmedchem.6b00527. [DOI] [PubMed] [Google Scholar]
- 18.Badavath V.N., Baysal I., Ucar G., Sinha B.N., Jayaprakash V. Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis. ACS Med. Chem. Lett. 2016;7:56–61. doi: 10.1021/acsmedchemlett.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Secci D., Bolasco A., Chimenti P., Carradori S. The state of the art of pyrazole derivatives as monoamine oxidase inhibitors and antidepressant/anticonvulsant agents. Curr. Med. Chem. 2011;18:5114–5144. doi: 10.2174/092986711797636090. [DOI] [PubMed] [Google Scholar]
- 20.Mathew B., Suresh J., Anbazhagan S., Mathew G.E. Pyrazoline. A promising scaffold for the inhibition of monoamine oxidase. Cent. Nerv. Syst. Agents Med. Chem. 2013;13:195–206. doi: 10.2174/1871524914666140129122632. [DOI] [PubMed] [Google Scholar]
- 21.Matos M.J., Vina D., Vazquez-Rodriquez S., Uriarte E., Santana L. Focusing on new monoamine oxidase inhibitors. Differently substituted coumarins as an interesting scaffold. Curr. Top. Med. Chem. 2012;12:2210–2233. doi: 10.2174/156802612805220002. [DOI] [PubMed] [Google Scholar]
- 22.Guglielmi P., Secci D., Petzer A., Bagetta D., Chimenti P., Rotondi G., Ferrante C., Recinella L., Leone S., Alcaro S., et al. Benzo[b]tiophen-3-ol derivatives as effective inhibitors of human monoamine oxidase: Design, synthesis, and biological activity. J. Enzym. Inhib. Med. Chem. 2019;34:1511–1525. doi: 10.1080/14756366.2019.1653864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh J.M., Rangarajan T.M., Chaudhary R., Singh R.P., Singh M., Singh R.P., Tondo A.R., Gambacorta N., Nicolotti O., Mathew B., et al. Novel class of chalcone oxime ethers as potent monoamine oxidase-B and acetylcholinesterase inhibitors. Molecules. 2020;25:2356. doi: 10.3390/molecules25102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maliyakkal N., Eom B.H., Heo J.H., Almoyad M.A.A., Parambi D.G.T., Gambacorta N., Nicolotti O., Beeran A.A., Kim H., Mathew B. A new potent and selective monoamine oxidase-B inhibitor with extended conjugation in chalcone framework. 1-[4-(morpholin-4-yl)phenyl]-5-phenylpenta-2,4-dien-1-one. Chem. Med. Chem. 2020;15:1629–1633. doi: 10.1002/cmdc.202000305. [DOI] [PubMed] [Google Scholar]
- 25.Hagenow J., Hagenow S., Grau K., Khanfar M., Hefke L., Proschak E., Stark H. Reversible small molecule inhibitors of MAO A and MAO B with anilide motifs. Drug Des. Dev. Ther. 2020;14:371–393. doi: 10.2147/DDDT.S236586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kavully F.S., Oh J.M., Dev S., Kaipakasseri S., Palakkathondi A., Vengamthodi A., Azeez R.F.A., Tondo A.R., Nicolotti O., Kim H., et al. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020;72:916–926. doi: 10.1111/jphp.13264. [DOI] [PubMed] [Google Scholar]
- 27.Turan-Zitouni G., Hussein W., Saglık B.N., Tabbi A., Korkut B. Design, synthesis and biological evaluation of novel N-pyridyl-hydrazone derivatives as potential monoamine oxidase (MAO) inhibitors. Molecules. 2018;23:113–124. doi: 10.3390/molecules23010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubey S., Bhosle P.A. Pyridazinone: An important element of pharmacophore possessing broad spectrum of activity. Med. Chem. Res. 2015;24:3579–3598. doi: 10.1007/s00044-015-1398-5. [DOI] [Google Scholar]
- 29.Özdemir Z., Alagöz M.A., Uslu H., Karakurt A., Erikci A., Ucar G., Uysa M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020;72:692–704. doi: 10.1007/s43440-020-00070-w. [DOI] [PubMed] [Google Scholar]
- 30.Özdemir Z., Başak-Türkmen N., Ayhan İ., Çiftçi O., Uysal M. Synthesis of new 6-[4-(2-fluorophenylpiperazine-1-yl)]-3(2h)-pyridazinone-2-acethyl-2-(substitutedbenzal)hydrazine derivatives and evaluation of their cytotoxic effects in liver and colon cancer cell lines. Pharm. Chem. J. 2019;52:923–929. doi: 10.1007/s11094-019-01927-y. [DOI] [Google Scholar]
- 31.Özdemir Z., Utku S., Mathew B., Carradori S., Orlando G., Simone S.D., Alagöz M.A., Özçelik A.B., Uysal M., Claudio Ferrante F. Synthesis and biological evaluation of new 3(2H)-pyridazinone derivatives as non-toxic anti-cancer compounds against colon carcinoma cells. J. Enzym. Inhib. Med. Chem. 2020;35:1100–1109. doi: 10.1080/14756366.2020.1755670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vergara S., Carda M., Agut R., Yepes L.M., Velez I.D., Robledo S.M., Galeano W.C. Synthesis, antiprotozoal activity and cytotoxicity in U-937 macrophages of triclosan–hydrazone hybrids. Med. Chem. Res. 2017;26:3262–3273. doi: 10.1007/s00044-017-2019-2. [DOI] [Google Scholar]
- 33.Govindasami T., Pandey A., Palanivelu N., Pandey A. Synthesis, Characterization and antibacterial activity of biologically important vanillin related hydrazone derivatives. Int. J. Org. Chem. 2011;1:71–77. doi: 10.4236/ijoc.2011.13012. [DOI] [Google Scholar]
- 34.Mathew B., Mathew G.E., Ucar G., Joy M., Nafna E.K., Suresh J. Monoamine oxidase inhibitory activity of methoxy-substituted chalcones. Int. J. Biol. Macromol. 2017;104:1321–1329. doi: 10.1016/j.ijbiomac.2017.05.162. [DOI] [PubMed] [Google Scholar]
- 35.Mathew B., Ucar G., Mathew G.E., Mathew S., Purapurath P.K., Moolayil F., Mohan S., Gupta S.V. Monoamine oxidase inhibitory activity: Methyl- versus chloro-chalcone derivatives. Chem. Med. Chem. 2016;11:2649–2655. doi: 10.1002/cmdc.201600497. [DOI] [PubMed] [Google Scholar]
- 36.Özçelik A.B., Özdemir Z., Sari S., Utku S., Uysal M. A new series of pyridazinone derivatives as cholinesterases inhibitors: Synthesis, in vitro activity and molecular modeling studies. Pharmacol. Rep. 2019;71:1253–1263. doi: 10.1016/j.pharep.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Mathew B., Baek S.C., Parambi D.G.T., Lee J.P., Joy M., Annie Rilda P.R., Randev R.V., Nithyamol P., Vijayan V., Inasu S.T., et al. Selected aryl thiosemicarbazones as a new class of multi-targeted monoamine oxidase inhibitors. Med. Chem. Comm. 2018;9:1871–1881. doi: 10.1039/C8MD00399H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H.W., Ryu H.W., Kang M.G., Park D., Oh S.R., Kim H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016;26:4714–4719. doi: 10.1016/j.bmcl.2016.08.044. [DOI] [PubMed] [Google Scholar]
- 39.Baek S.C., Lee H.W., Ryu H.W., Kang M.G., Park D., Kim S.H., Cho M.L., Oh S.R., Kim H. Selective inhibition of monoamine oxidase A by hispidol. Bioorg. Med. Chem. Lett. 2018;15:584–588. doi: 10.1016/j.bmcl.2018.01.049. [DOI] [PubMed] [Google Scholar]
- 40.Parambi D.G.T., Oh J.M., Baek S.C., Lee J.P., Tondo A.R., Nicolotti O., Kim H., Mathew B. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019;93:103335. doi: 10.1016/j.bioorg.2019.103335. [DOI] [PubMed] [Google Scholar]
- 41.Son S.Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binda C., Wang J., Pisani L., Caccia C., Carotti A., Salvati P., Edmondson D.E., Mattevi A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007;50:5848–5852. doi: 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- 43.Schrödinger Release 2018-2 . Prime. Schrödinger, LLC; New York, NY, USA: 2018. [Google Scholar]
- 44.Schrödinger Release 2018-2 . Glide. Schrödinger, LLC; New York, NY, USA: 2018. [Google Scholar]
- 45.Mangiatordi G.F., Alberga D., Pisani L., Gadaleta D., Trisciuzzi D., Farina R., Carotti A., Lattanzi G., Catto M., Nicolotti O. A rational approach to elucidate human monoamine oxidase molecular selectivity. Eur. J. Pharm. Sci. 2017;101:90–99. doi: 10.1016/j.ejps.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 46.LigPrep. Schrödinger, LLC; New York, NY, USA: 2018. [Google Scholar]
- 47.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 48.Reeta, Baek S.C., Lee J.P., Rangarajan T.M., Ayushee, Singh R.P., Singh M., Mangiatordi G.F., Nicolotti O., Kim H., et al. Ethyl acetohydroxamate incorporated chalcones: Unveiling a novel class of chalcones for multitarget monoamine oxidase-B inhibitors against Alzheimer’s disease. CNS Neurol. Disord. Drug Targets. 2019;8:643–654. doi: 10.2174/1871527318666190906101326. [DOI] [PubMed] [Google Scholar]
- 49.Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.