

Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a stress-induced cardiac channelopathy that has a high mortality in untreated patients. Our understanding has grown tremendously since CPVT was first described as a clinical syndrome in 1995. It is now established that the deadly arrhythmias are caused by unregulated ‘pathological’ calcium release from the sarcoplasmic reticulum (SR), the major calcium storage organelle in striated muscle. Important questions remain regarding the molecular mechanisms that are responsible for the pathological calcium release, regarding the tissue origin of the arrhythmic beats that initiate ventricular tachycardia, and regarding optimal therapeutic approaches. At present, mutations in six genes involved in SR calcium release have been identified as the genetic cause of CPVT: RYR2 (encoding ryanodine receptor calcium release channel), CASQ2 (encoding cardiac calsequestrin), TRDN (encoding triadin), CALM1, CALM2 and CALM3 (encoding identical calmodulin protein). Here, we review each CPVT subtype and how CPVT mutations alter protein function, RyR2 calcium release channel regulation, and cellular calcium handling. We then discuss research and hypotheses surrounding the tissue mechanisms underlying CPVT, such as the pathophysiological role of sinus node dysfunction in CPVT, and whether the arrhythmogenic beats originate from the conduction system or the ventricular working myocardium. Finally, we review the treatments that are available for patients with CPVT, their efficacy, and how therapy could be improved in the future.
Keywords: arrhythmia, calcium, heart excitation
Graphical Abstract
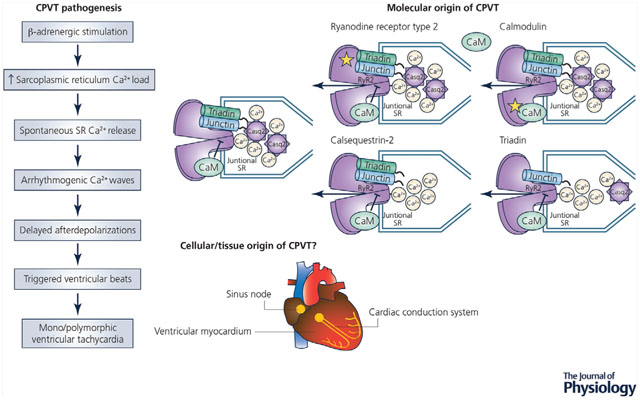
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a cardiac arrhythmia characterized by the presence of ventricular tachycardia in response to β-adrenergic receptor stimulation. Here, we describe the mechanistic progression from β-adrenergic stimulation to the formation of mono/polymorphic ventricular tachycardia. The goal of this review is to highlight the current molecular mechanisms that lead to CPVT followed by a discussion of the current hypotheses and research around the cellular/tissue origin of the arrhythmias.
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal, stress-induced cardiac channelopathy. First described in 1995, CPVT is characterized by polymorphic ventricular arrhythmias that are triggered by catecholamines released during exercise, stress or sudden emotion in individuals with structurally normal hearts (Leenhardt et al. 1995). Symptoms range from palpitations to cardiac arrest, with mortality rates between 30 and 50% in untreated individuals by age 40 (Pérez-Riera et al. 2018). Patients are normally diagnosed during early childhood but initial symptoms can occur in patients as old as 40 years of age (Pérez-Riera et al. 2018). CPVT is rare, with an estimated prevalence of 1:5000 to 1:10,000 depending on the population studied (Modell et al. 2012; Pérez-Riera et al. 2018). The true prevalence of CPVT is likely higher, since CPVT cases are frequently missed as most patients present with a normal resting electrocardiogram and structurally normal heart on cardiac workup (Imberti et al. 2016).
Human genetic studies have established that CPVT is caused by mutations in genes that encode proteins of the sarcoplasmic reticulum (SR) calcium release complex depicted in Fig. 1 (Swan et al. 1999; Lahat et al. 2001a,b). Supported mostly by experimental studies in mouse CPVT models (Cerrone et al. 2005; Knollmann et al. 2006; Rizzi et al. 2008; Uchinoumi et al. 2010), the current understanding of cellular CPVT pathophysiology is that catecholamines released during stress or exercise activate β-adrenergic receptor signalling, leading to a cellular chain reaction that culminates in pathological calcium release during diastole and calcium-triggered action potentials as described in Fig. 2.
Figure 1. The sarcoplasmic reticulum (SR) calcium release complex in cardiac muscle.
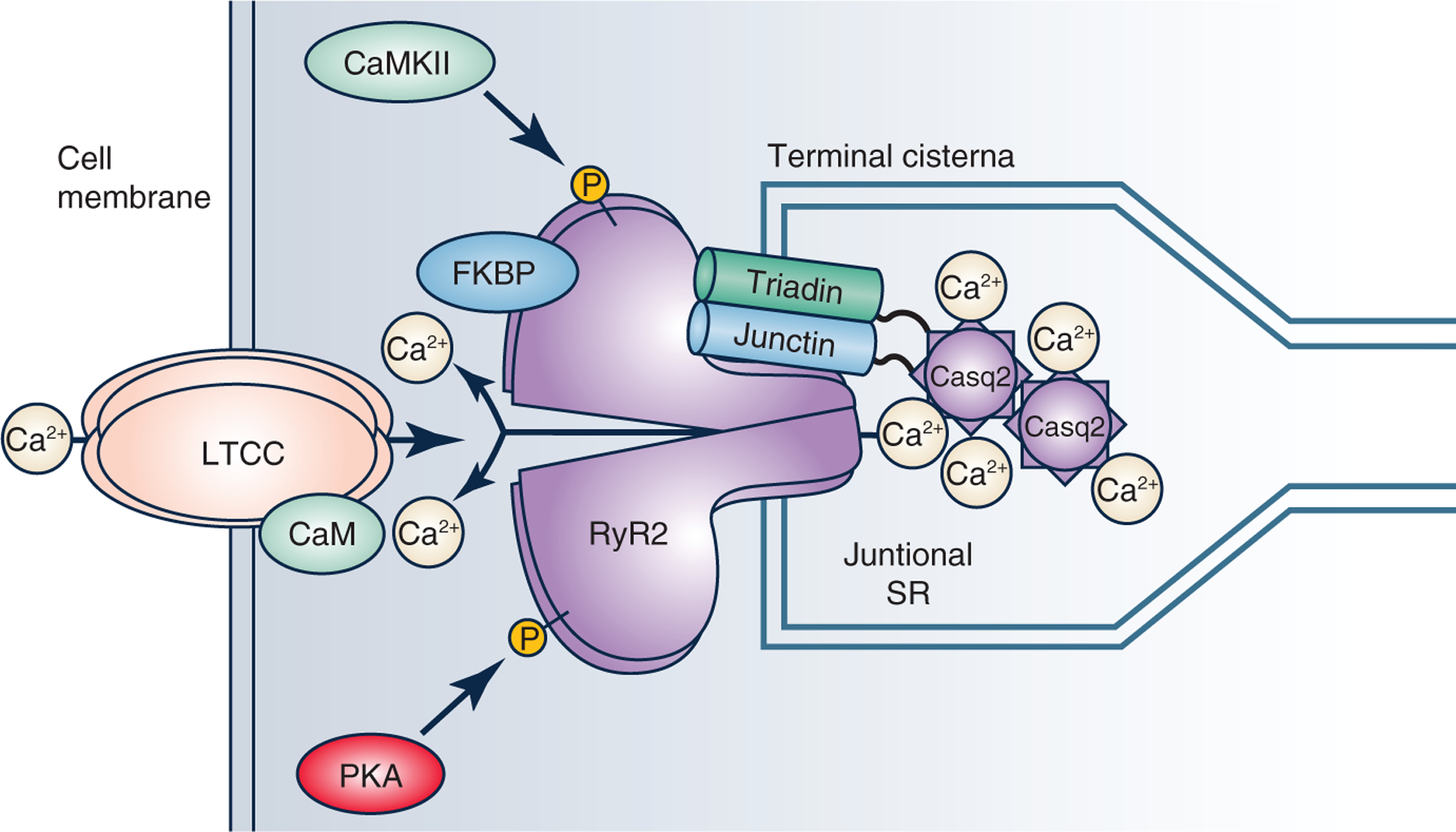
Pictured above are the proteins that are involved in the regulation of calcium release from the SR during excitation-contraction coupling. The ryanodine receptor type 2 (RyR2) is a large conductance calcium channel located in the junctional SR membrane that is gated by calcium influx via the L-type calcium channels (LTCC) in the cell membrane. RyR2 open probability is regulated by post-translational modifications (e.g. phosphorylation by calcium-calmodulin kinase II, CaMKII, protein kinase A, PKA), by cytosolic RyR2 binding proteins (calmodulin [CaM], immunophillins such as FK506 binding protein [FKBP]) and SR luminal proteins (calsequestrin [Casq2], triadin, junction). Casq2 forms polymers that are anchored to RyR2 and the junctional SR by triadin and junctin. CaM bound to LTCC mediates calcium-dependent inactivation of LTCC.
Figure 2. Cellular pathogenesis of catecholaminergic polymorphic ventricular tachycardia (CPVT).
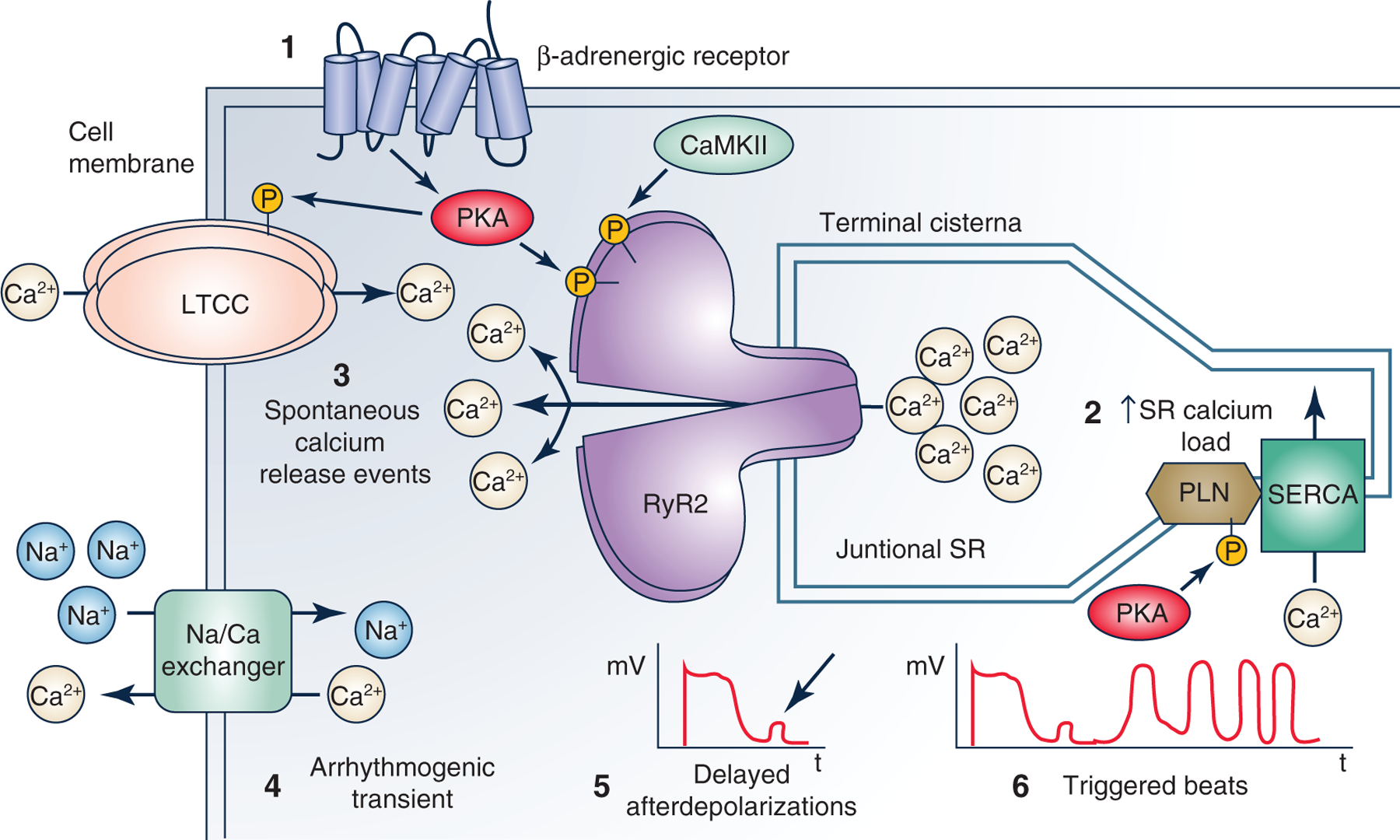
The cartoon illustrates cellular mechanisms underlying CPVT caused by the loss of calsequetrin. 1, catecholamines released during stress or exercise activates β-adrenergic receptor signalling, leading to cardiomyocyte calcium loading and enhanced sarcoplasmic reticulum (SR) Ca uptake. 2, the increased SR calcium load is a physiological response necessary for increasing cardiac output during the physiological fight or flight response (Bers, 2001). Normally, ventricular myocytes can handle the increased SR calcium load. 3, if a CPVT mutation is present, RyR2 SR calcium release channels open spontaneously during late diastole, causing unregulated ‘pathological’ SR calcium release termed ‘spontaneous calcium release events’ (SCR). 4, the rise in cytosolic calcium during the SCR activates the electrogenic sodium calcium exchanger, which generates an arrhythmogenic transient inward current. 5, this induces a cell membrane depolarization termed ‘delayed afterdepolarizations’ (DADs). 6, DADs are a well-established cellular mechanism that can then cause triggered beats that lead to ventricular arrhythmias (Priori & Corr, 1990).
Open questions remain as to how exactly mutations in CPVT genes cause functional alterations in the SR calcium release machinery that leads to pathological calcium release during diastole (molecular mechanisms). Even less well understood is how the altered cellular function causes CPVT at the whole-heart and in vivo level. It remains to be determined if and to what extent the dysfunction of the sinus node, the cardiac conduction system and the ventricular working myocardium contribute mechanistically to CPVT (tissue mechanisms). Here, we review the current understanding of the molecular and tissue mechanisms of CPVT to help consolidate the information and shed light on the areas where more work is needed. Our goal is to help advance our understanding of CPVT pathophysiology and its treatment.
Molecular mechanisms of CPVT
As of 2019, six different CPVT disease genes have been identified, which account for 60–75% of CPVT cases. The genetic cause of the remaining clinical CPVT cases is not yet known (Pérez-Riera et al. 2018; Roston et al. 2018b). All six CPVT genes (RYR2, CASQ2, TRDN, CALM1, CALM2, CALM3) encode proteins that are directly involved in regulating SR calcium release during excitation-contraction (EC) coupling (Fig. 1). Intheheart, electrical activation couples to mechanical force via the secondary messenger calcium (Bers, 2002). Membrane depolarization during the cardiac action potential opens L-type calcium channels (Fig. 1), which bring calcium into the cell. Calcium binds to and opens RyR2 located in the terminal cisternae of the SR, the junctional SR (Fig. 1), a process known as calcium-induced calcium release (CICR) (Fabiato, 1985). During systole, cytosolic calcium initiates myofilament contraction before being taken back up into the SR or pumped out into extracellular space during diastole. Proteins regulating SR calcium release can be categorized based on function and location. Proteins that are located within the SR lumen (e.g. calsequestrin, histidine-rich calcium-binding protein) affect the levels of free calcium present in the SR during the EC cycle. Proteins located within the junctional membrane of the SR (e.g. triadin, junctin) facilitate the interaction between calcium-handling proteins (e.g. calsequestrin and RyR2). Finally, there are a group of proteins that bind to the cytoplasmic surface of RyR2 (e.g. calmodulin, FK506 binding proteins) and regulate RyR2 sensitivity to cytoplasmic and SR luminal calcium levels. The following section reviews the proteins of interest in CPVT, their physiological role, and how mutations lead to CPVT.
Ryanodine receptor type 2
Gain-of-function mutations in the RYR2 gene are found in about 95% of patients with a genetically confirmed diagnosis of CPVT (Pérez-Riera et al. 2018) and are designated as CPVT type 1 (CPVT1). CPVT1 is autosomal-dominant and was first described in 1999 (Swan et al. 1999) before being mapped to RYR2 in 2001 (Priori et al. 2001). Since then, more than 200 gain-of-function variants in RYR2 have been discovered. Loss-of-function RYR2 variants also exist but are less common and associated with ventricular arrhythmia syndromes distinct from CPVT (Roston et al. 2017). RYR2 encodes the cardiac ryanodine receptor (RyR2), a 565 kD protein that forms a homotetrameric, high-conductance, cation-selective channel that releases calcium from the SR (Fig. 1) (Seidel et al. 2015). RyR2 interacts with many other proteins including calsequestrin 2 (Costello et al. 1986; Franzini-Armstrong et al. 1987), triadin (Guo et al. 1996), junctin (Zhang et al. 1997), calmodulin (Yamaguchi et al. 2003, 2007), junctophilin, and the immunophilins FKBP 12 and FKBP 12.6 (Jayaraman et al. 1992; Yano et al. 2009). FKBPs are thought to stabilize the closed conformation of RyR2 and prevent diastolic release of calcium from the SR (Wehrens et al. 2003). Currently, there are several hypotheses as to why mutations in RYR2 lead to CPVT, see Fig. 3 (Ikemoto & Yamamoto, 2002; Wehrens et al. 2003; Jiang et al. 2004; Liu et al. 2009). One theory is that mutations in RYR2 affect the ability of FKBP 12.6 to interact with RyR2, leading to dissociation of FKBP 12.6 and the opening of RyR2 during diastole (Wehrens et al. 2003). However, others have challenged this hypothesis (Xiao et al. 2007), and at the present time it seems unlikely that loss of FKBP 12.6 is responsible for CPVT. Rather, FKBPs still bind to mutant RyR2 but fail to inhibit them (Zhang et al. 2016). The most widely held hypothesis states that CPVT mutations sensitize RyR2 channels to SR luminal calcium, causing them to open at a lower intra-SR calcium concentration, termed store overload-induced calcium release (SOICR) (Jiang et al. 2004). According to the SOICR hypothesis, mutations in RYR2 decrease the threshold of SR calcium that is required to activate RyR2, leading to an increased probability of calcium leak and diastolic SR calcium release. A third hypothesis focuses on the interactions within the structure of RyR2. Normally, intramolecular interactions occur between the N-terminal and central domain of RyR2 monomers, termed ‘zipping’, which are critical to stabilizing the protein. When RyR2 is activated during the EC coupling cycle, the intramolecular interactions are weakened, ‘unzipping’ the domains, opening the channel, and causing the release of calcium. Mutations in RYR2 that occur within the interaction domains have been shown to cause a similar unzipping, which results in calcium leak (Ikemoto & Yamamoto, 2002; George et al. 2006; Sumitomo, 2016). While it is still debated which hypothesis is correct, common to all of them is that CPVT-linked RYR2 mutations increase the likelihood of spontaneous RyR2 openings and pathological calcium release during diastole.
Figure 3. Summary of current hypotheses for how RyR2 mutations could lead to catecholaminergic polymorphic ventricular tachycardia.
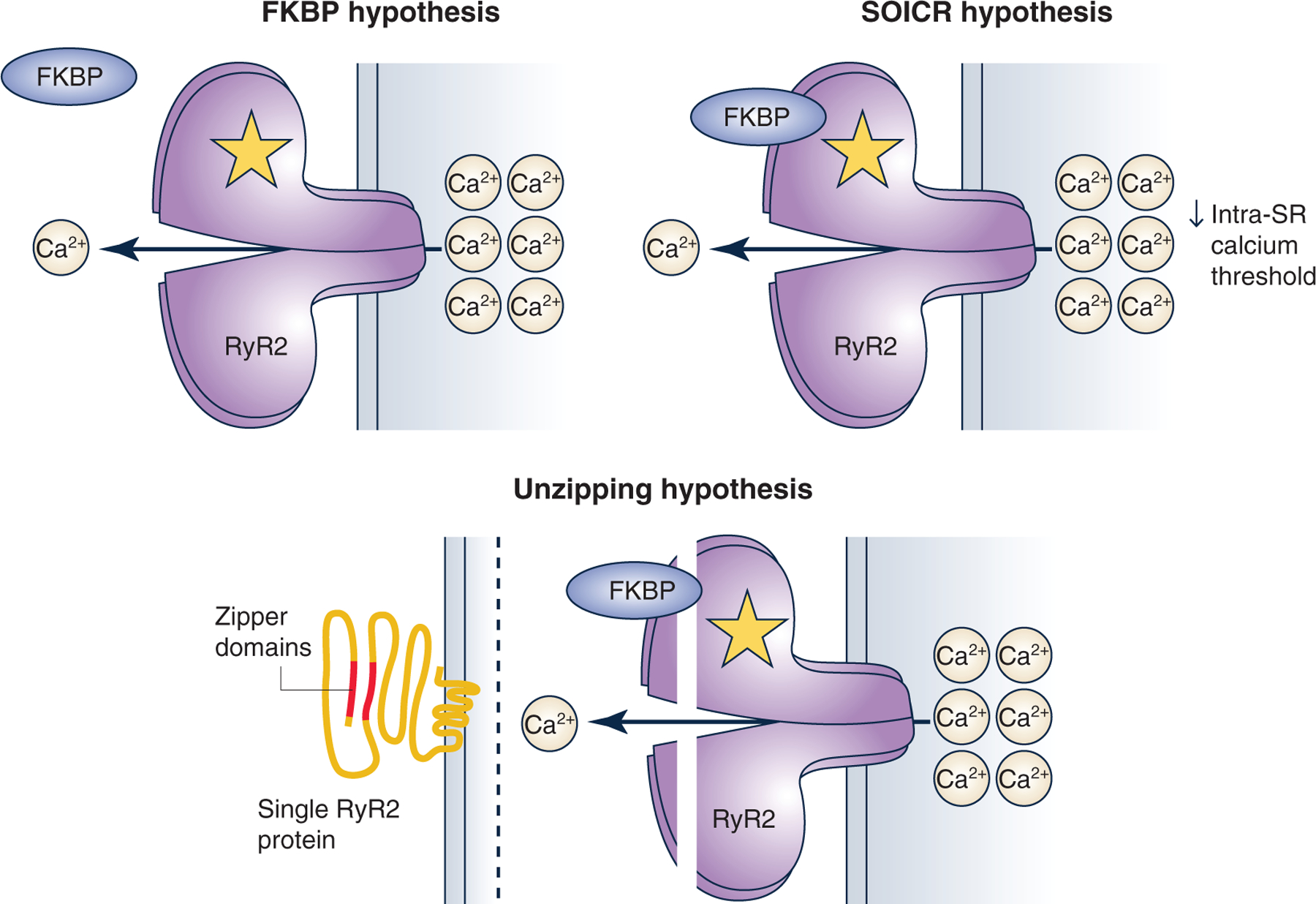
The first theory states that mutations prevent FKBP binding to RyR2. The second theory states that a mutation can lower the intra-sarcoplasmic reticulum (SR) calcium threshold needed RyR2 to open during diastole, termed ‘store overload-induced calcium release’ (SOICR). Finally, the ‘unzipping’ theory stems from the observation that the N-terminal and central domain of RyR2 interact with one another forming a tight seal. Mutations in RYR2 can affect the interaction and lead to an unzipping of the protein, making RyR2 more prone to open spontaneously.
Calsequestrin 2
The second most common cause of CPVT is mutations in the CASQ2 gene, termed CPVT2 (Lahat et al. 2001a,b). Calsequestrin was first associated with CPVT when missense mutations were discovered in a family with autosomal recessive CPVT in 2001 and nonsense mutations in a similar family in 2002 (Postma et al. 2002). Calsequestrin-linked CPVT was thought to be an autosomal recessive disease only (Postma et al. 2002; de la Fuente et al. 2008; Kirchhefer et al. 2010), but in 2016 a report of a novel CASQ2 mutation (K180R) in a family with autosomal-dominant inheritance of CPVT was published (Gray et al. 2016). Calsequestrin is a calcium-binding protein located in the terminal cisternae that form the junctional SR (Fig. 1). Calsequestrin is a high-capacity, low-affinity calcium-binding protein that binds 40–50 calcium ions through its 60–70 negatively charged amino acid residues (Yano & Zarain-Herzberg, 1994). While it is known that calsequestrin forms homo-polymers in a calcium-dependent manner (Park et al. 2003), not much is known about the structure of the polymers. A recent report utilized very low pH (3.5) conditions to crystalize a novel structure for the calsequestrin polymer filament (Fig. 4) (Titus et al. 2019). The filaments are an assembly of calsequestrin dimers that interact with one another to form three different thioredoxin helices that come together to form the filament (Fig. 4a). Two of the three domains form a double helix at the core of the filament, while the final domain creates an outer ‘collar’ and winds around the inner double helix (Fig. 4b). The new structure was then used to identify novel potential calcium-binding sites. The analysis found calcium-binding sites that bridged both intra- and inter-dimer interfaces of calsequestrin, suggesting that calcium is involved in the ability for calsequestrin to form dimers and those dimers to form subsequent filaments (Titus et al. 2019). Functionally, calsequestrin regulates the amount of calcium released from the SR during EC coupling by buffering intra-SR calcium (Zhang et al. 1997; Bers, 2002).
Figure 4. The new cardiac calsequestrin filament (Titus et al. 2019, with permission).
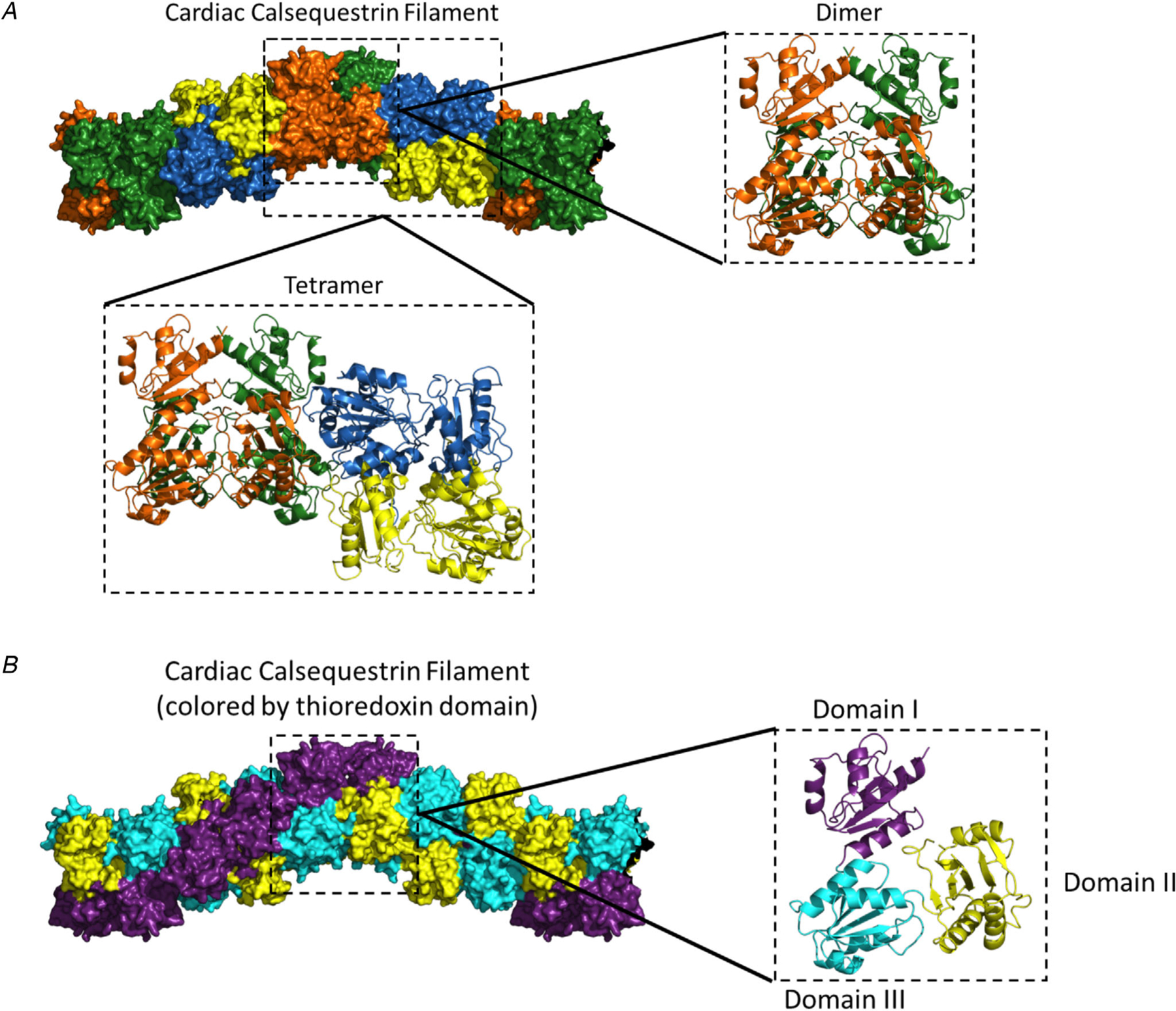
Pictured above is the putative structure of the cardiac calsequestrin filament. a, putative calsequestrin filament including its dimeric and tetrameric assembly. b, the filament exhibits a helical structure at the domain level. For simplicity, calsequestrin monomers are coloured by thioredoxin domain (domain I, purple; domain II, yellow; domain III, cyan). The filament is formed by an inner thioredoxin double helix (domains II and III) with an outer thioredoxin single helix (domain I) wrapped around the double helical core. Right side: The monomers are translated but remain in their dimer-forming orientation.
To regulate calcium release, calsequestrin is anchored to RyR2 by two proteins, triadin and junctin. It has been suggested that the interaction between calsequestrin and RyR2 may contribute to the refractory period of calcium release that occurs after each physiological CICR, but the mechanism is still not well understood (Györke et al. 2009; Katz et al. 2009; Liu et al. 2009). Studies using skeletal calsequestrin (Casq1, not expressed in the heart) showed changes in diffusional mobility of Casq1 after depletion of SR calcium (Manno et al. 2017). If cardiac calsequestrin (Casq2) acts in a similar manner in the heart, the findings from Casq1 would provide a plausible mechanism for how calsequestrin is regulating calcium release and termination. Hence, the simplest explanation of why mutations in calsequestrin cause CPVT is that they impair calsequestrin’s ability to buffer calcium in the SR, resulting in a much faster rise of intra-SR free calcium concentration close to RyR2 release channels. Although intra-SR calcium kinetics have not yet been measured experimentally in calsequestrin CPVT models, there is extensive evidence that absence of calsequestrin in mice leads to hyperactive RyR2 channels, impaired calcium-release termination, a shortened calcium release refractory period, and enhanced spontaneous release of calcium (Knollmann et al. 2006; Chopra et al. 2007; Kryshtal et al. 2015). While impaired calcium buffering is the generally accepted mechanism for calsequestrin nonsense mutations that result in loss of calsequestrin, missense mutations (e.g. R33Q) may alter calsequestrin interaction with RyR2 in addition to reducing calcium buffering (Terentyev et al. 2006). For autosomal-dominant CPVT2, the current hypothesis is that calsequestrin mutations affect the ability for polymerization to occur. Two autosomal-dominant mutations (K180R and S173I) were unable to form polymers in a turbidity assay. Interestingly, the defect for the K180R mutation only occurred in the presence of magnesium. While the exact mechanism is unknown, magnesium may affect the ability of calsequestrin to form filaments and could be involved in the pathogenesis of the K180R mutation (Titus et al. 2019). More work is needed to understand the physiological role of calsequestrin in the EC coupling cycle, to determine the prevalence of autosomal-dominant calsequestrin mutations, and to understand how they cause CPVT.
Calmodulin
Three different genes - CALM1, CALM2, CALM3 - encode identical calmodulin proteins and are located on chromosomes 14q32, 2p21 and 19q13, respectively. Mutations in each of the three calmodulin genes have been linked to three distinct genetic arrhythmia disorders: CPVT (Nyegaard et al. 2012; Yin et al. 2014; Gomez-Hurtado et al. 2016), idiopathic ventricular fibrillation (Marsman et al. 2014), and long-QT syndrome (Jiménez-Jáimez et al. 2016). Structurally, calmodulin has an alpha-helical structure and contains four classical calcium-binding EF hand motifs, two on each N- and C-terminal lobe. The motifs can bind to one calcium ion each with the N-terminal lobe having a lower affinity for calcium (Linse et al. 1991; VanScyoc et al. 2002). When calcium binds to calmodulin, the protein will undergo a conformational change that exposes the hydrophobic patches in each lobe. The patches contain a large amount of methionine residue that utilizes a central linker between the two lobes to interact with a large number of protein targets (Yamniuk & Vogel, 2004). Within the EC coupling machinery, calmodulin binds to RyR2 and the L-type calcium channel (LTCC) (Fig. 1). The binding of calmodulin to RyR2 inhibits calcium release from the SR during diastole (Yamaguchi et al. 2003, 2007). Calcium binding to calmodulin inactivates LTCC channels (Peterson et al. 1999; Zühlke et al. 1999), a process termed calcium-dependent inactivation. In addition to RyR2 and LTCC, calmodulin regulates other membrane ion channels important for the cardiac action potential (e.g. Nav 1.5, KCNQ1). Mutations in the three CALM genes have a wide range of effects, which may explain the diverse arrhythmia phenotypes associated with calmodulin mutations. For example, calmodulin mutants linked to CPVT either fail to inhibit or even activate RyR2 (Hwang et al. 2014; Gomez-Hurtado et al. 2016). CPVT calmodulin mutants tend to bind to RyR2 with higher affinity than wild-type calmodulin (Hwang et al. 2014; Gomez-Hurtado et al. 2016), which can explain their autosomal-dominant mode of action. On the other hand, calmodulin mutants associated with LQTS do not affect RyR2, but rather impair LTCC inactivation, leading to a profound action potential lengthening and QT prolongation (Limpitikul et al. 2014). Due to the many calmodulin targets within the cell, calmodulin mutations may also cause ventricular arrhythmias by other mechanisms, but so far none have been definitively confirmed.
Triadin
Triadin is a trans SR membrane protein that forms a complex with RyR2, calsequestrin and junctin to create the SR calcium release unit (Jones et al. 1995; Kobayashi & Jones, 1999). Triadin localizes to the junctional SR membrane in both cardiac and skeletal muscle (Fig. 1). Triadin has a single membrane-spanning domain, a short N-terminal segment located in the cytoplasm, a long C-terminal tail that projects intraluminally and is highly basic and charged, and a long run of charged amino acid residues called ‘KEKE’ association motifs that promote protein-protein interactions (Knudson et al. 1993; Jones et al. 1995; Guo et al. 1996; Zhang et al. 1997). Triadin is thought to act mainly as a scaffolding protein to help anchor calsequestrin to RyR2 near the junctional SR membrane. The anchoring properties of triadin are important for maintaining the ultrastructure of the terminal cisterna. When triadin is knocked out in mice (Chopra et al. 2009), protein levels of RyR2, calsequestrin and junctin were all reduced despite normal RNA expression. Consistent with the role of triadin as the primary anchoring protein that retains calsequestrin in the terminal cisterna, calsequestrin can be detected in the free SR of triadin knockout myocytes (Chopra et al. 2009). Loss of triadin also resulted in profound structural remodelling of the terminal cisterna, with an approximately 50% reduction of the junctional SR-t-tubule interface. As a result of the structural remodelling, coupling efficiency between LTCC and RyR2 was impaired and LTCC calcium-dependent inactivation reduced. Due to the impaired LTCC inactivation, triadin loss results in a gain-of-function effect on LTCC, increased calcium current and prolonged cardiac action potential. The ensuing cellular and SR calcium overload causes an increase in spontaneous calcium release events especially during catecholaminergic stimulation (Chopra et al. 2009). Other roles of triadin have been proposed, such as the ability of overexpressed triadin to enhance SR calcium release by directly affecting the RyR2 channel, but the work was conducted in cultured myocytes and needs to be confirmed at the single channel level (Terentyev et al. 2005). The first triadin mutation linked to CPVT was discovered in 2012 and another was found in 2015 (Roux-Buisson et al. 2012; Rooryck et al. 2015). Triadin mutations are thought to result in decreased levels of the protein. For example, the triadin-T59R mutation renders the triadin protein unstable and leads to enhanced degradation (Roux-Buisson et al. 2012). More recently, triadin mutations were also found in patients suffering from long-QT. The mutations caused a frameshift in the TRDN gene leading to a syndrome termed ‘triadin knockout syndrome’ (Altmann et al. 2015). Patients suffered from multiple episodes of exertion-induced syncope and cardiac arrest in early childhood. Other symptoms include mild to moderate muscle weakness. Further electrocardiographic workup demonstrated extensive T-wave inversion and QT prolongation. Normally, patients with similar findings would be classified as having long-QT syndrome. It has been proposed that patients with triadin mutations that decrease the levels of triadin should be diagnosed with ‘triadin knockout syndrome’ (Altmann et al. 2015), which is essentially an overlap syndrome with features of both CPVT and long-QT.
Other candidate genes for CPVT
Although not yet identified, mutations in other genes that are integral to SR calcium handling could potentially cause CPVT and should be screened for in patients carrying a clinical diagnosis of CPVT. Junctin, encoded by the gene aspartyl-beta-hydroxylase (ASPH), is a junctional SR protein (Fig. 1) that interacts with triadin and RyR2 (Jones et al. 1995; Kobayashi & Jones, 1999). Like triadin, junctin functions as a scaffold for calsequestrin and is involved in the polymerization of calsequestrin as calcium increases in the SR (Lee et al. 2012). Other CPVT candidates are proteins that regulate RyR2 calcium sensitivity such as FKBP 12 and FKBP 12.6 (Fig. 1). Both proteins are expressed in cardiac myocytes and are thought to promote the closed state of the channel. FKBP 12 and 12.6 bind to the same region of RyR2 which suggests that the competition between them is important for the functional outcome of RyR2 (Gonano & Jones, 2017). Another candidate is histidine-rich calcium (HRC) binding protein, a SR luminal calcium-binding buffering that is upregulated when calsequestrin is knocked out (Murphy et al. 2011). Increased HRC may activate RyR2 calcium release channels and contribute to CPVT (Liu et al. 2015).
Tissue mechanisms underlying CPVT
It is generally agreed upon that CPVT mutations render the RyR2 calcium release channels hyperactive, generating pathological calcium release during diastole. What remain unclear are the tissue mechanisms of CPVT. For example, a key question is the mechanistic contribution of low sinus heart rates, since sinus node dysfunction is a hallmark of CPVT in patients and animal models (Faggioni et al. 2014b; Miyata et al. 2018). Another key question is whether the arrhythmic trigger that causes the ventricular arrhythmias originates from ventricular cardiomyocytes in the working myocardium or from specialized cells of the cardiac conduction system, the Purkinje cells. Theoretical considerations and modelling studies favour the ventricular Purkinje network as the primary cellular source of CPVT (Xie et al. 2010) but conclusive experimental evidence is lacking.
Pathophysiological role of sinus node dysfunction in CPVT
Sinus node dysfunction and bradycardia are well-documented phenotypes of CPVT in humans and in mouse models of CPVT (Faggioni et al. 2014b; Miyata et al. 2018). The mechanisms of how CPVT mutations cause sinus node dysfunction are thought to be multifactorial. For example, one theory is that the loss of calsequestrin causes a regional microfibrosis in the sinus node, which would alter the generation and propagation of an electrical impulse through the node (Glukhov et al. 2015). It is hypothesized that the microfibrosis could form from apoptosis and fibrogenesis due to an overload of diastolic calcium (Swaminathan et al. 2011). The increase in calcium levels could also trigger the activation of CaMKII, which has been shown to be involved in the upregulation of genes that could promote structural remodelling (Huke & Knollmann, 2011). Understanding how sinus node dysfunction originates is important as sinus node chronotropic incompetence has been shown to be a risk predictor for ventricular arrhythmia in children and young adults with CPVT (Franciosi et al. 2019). A likely explanation is that low sinus rates prolong the diastolic interval, allowing the spontaneous SR calcium release to occur before CICR during the next action potential can empty the SR and reset the SR calcium clock. Our group tested this hypothesis experimentally at the cellular level and in vivo using the Casq2 KO mouse CPVT model (Faggioni et al. 2013). We found that increasing the pacing rate reduced the likelihood of spontaneous calcium release and triggered beats in isolated cardiomyocytes. In vivo, artificially raising resting heart rates with atropine or by overdrive pacing prevented CPVT (Faggioni et al. 2013). To study the role of the cardiac conduction system in CPVT in more detail, our group recently developed two novel mouse models where the CASQ2 gene can be either inactivated (i.e. conditional KO) or activated (i.e. conditional rescue) by Cre-mediated recombination (Flores et al. 2018). Thus, we were able to modulate CASQ2 gene expression in a tissue and temporal manner by crossing our new mouse models with mice that carry Cre controlled by tissue selective and/or inducible promotors. For example, using the HCN4KiT-Cre system (Hoesl et al. 2008), we were able to re-express Casq2 in the SA node of adult Casq2 KO mice, which accelerated sinus heart rates and prevented CPVT (Flores et al. 2018), as illustrated in Fig. 5. Hence, dysfunction of the sinoatrial node and the ensuing slow sinus heart rates may independently contribute to arrhythmia risk in CPVT.
Figure 5. Cardiac conduction system (CSS) targeted CASQ2 gene deletion or rescue.
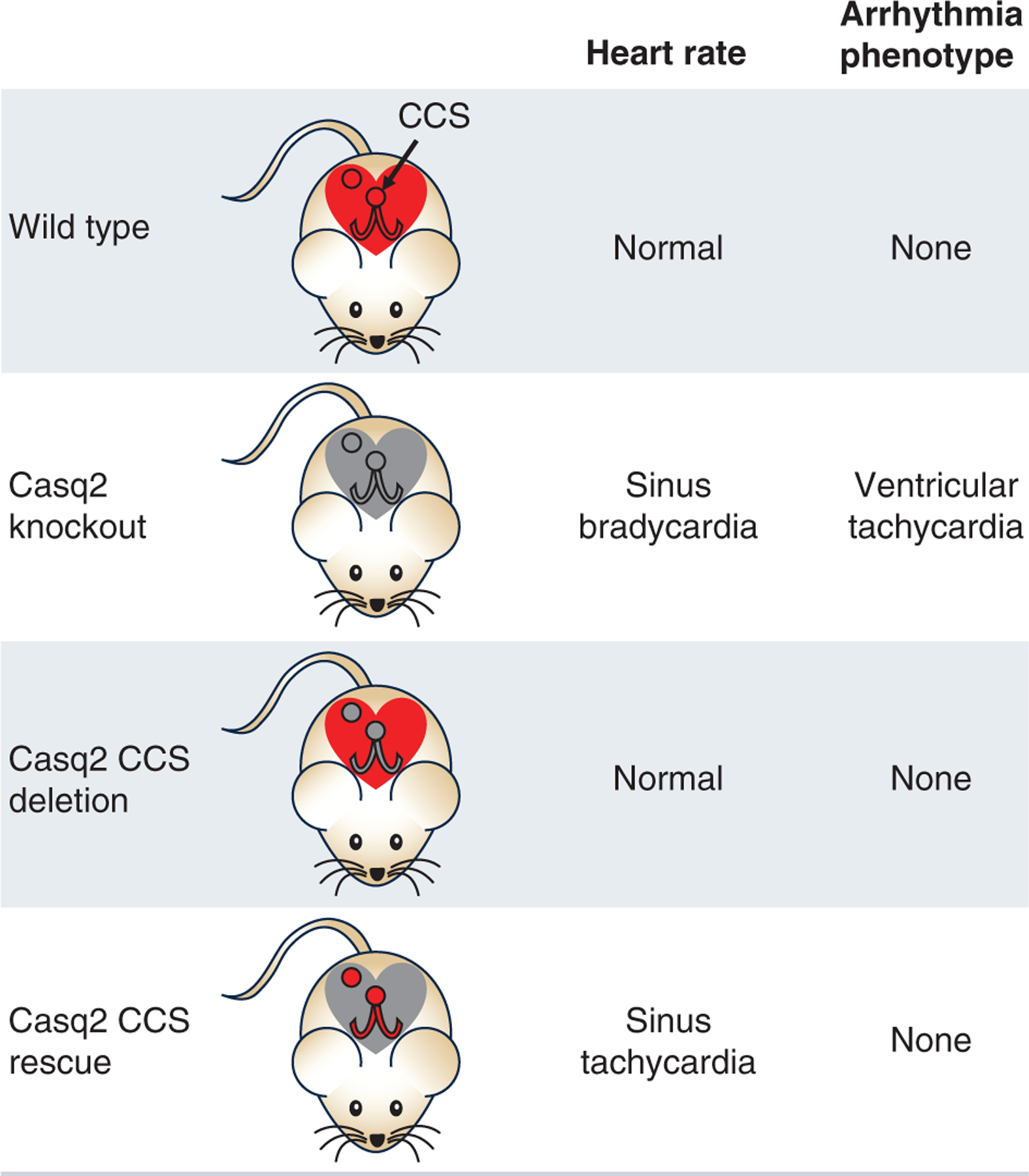
Cartoon showing the heart rate and ventricular arrhythmia phenotype of mice with conditional deletion or rescue of calsequestrin in the CCS. Red colour indicates functional CASQ2.
Purkinje cells as the cellular source of CPVT
Theoretical considerations and modelling studies favour the ventricular Purkinje network as the primary cellular source of CPVT because of a favourable source-sink relationship (Xie et al. 2010). There are also key differences between Purkinje cells and ventricular myocytes that could explain why Purkinje cells are more likely to generate arrhythmias. Purkinje cells have a much lower number of T-tubules compared with ventricular cardiomyocytes and hence more corbular SR (Sommer & Johnson, 1968). The differences in organelle structure result in a calcium activation process that is unique to Purkinje cells, termed ‘reverse mode-EC coupling’. The calcium release units within the centre of Purkinje fibres do not respond to voltage but instead are activated by calcium waves (Stuyvers et al. 2005). Experimental evidence to support the proposition that CPVT originates from the Purkinje system stems from a mouse model with a heterozygous mutation in RYR2(RYR2/RYR2R4496C) (Cerrone et al. 2005). Optical mapping of whole mouse hearts demonstrated that breakthrough patterns from ventricular tachycardias may originate from the His-Purkinje network in both ventricles (Cerrone et al. 2007). To confirm that the arrhythmias were generated from the Purkinje fibres, chemical ablation (Lugol’s solution) of the right ventricular endocardial cavity was performed. Mice treated with Lugol’s had conversion of bi-directional VT into monomorphic VT, suggesting that endocardial Purkinje fibre ablation prevented the development of arrhythmias from the right ventricle (Cerrone et al. 2007). Follow-up studies in single cells found that RYR2/RYR2R4496C Purkinje cells developed spontaneous calcium release events and triggered beats at a significantly higher rate than ventricular myocytes (Herron et al. 2010; Kang et al. 2010).
The experimental studies in the RYR2/RYR2R4496C mice had suggested that alternating beats of bi-directional VT originated from the right and left bundle branches, but the mechanism by which the triggered activity generated an alternating pattern was not addressed (Cerrone et al. 2007). Using a simulated two-dimensional anatomic model of rabbit ventricles with a simplified His-Purkinje system, a computer modelling study attempted to gain a better understanding of the mechanism underlying bi-directional ventricular tachycardia, which is a hallmark of CPVT (Leenhardt et al. 1995). Based on the results of the modelling, the authors proposed that a ‘ping-pong’ mechanism, called reciprocating bigeminy, could account for the bi-directional VT pattern that was seen in CPVT (Baher et al. 2011). The ping-pong mechanism is based on the findings that above a certain threshold heart rate, a delayed afterdepolarization (DAD) triggers an action potential that initiates ventricular bigeminy, and that the threshold heart rate for bigeminy varies at different locations in the heart (Baher et al. 2011). The findings from the model suggested that DAD-triggered arrhythmias could cause ventricular bigeminy when a single site in the His-Purkinje system or the ventricular myocardium developed a DAD-triggered beat following a sinus beat. Bi-directional VT occurred when a second site developed ventricular bigeminy and reciprocally activated the first site by the ping-pong mechanism. Bi-directional VT could degenerate into polymorphic VT when the increase in heart rate recruited additional sites in the His-Purkinje to develop bigeminy (Baher et al. 2011). The data from the RYR2R4496C model suggests that the His-Purkinje system could serve as both the source of initiation and the propagation of ventricular arrhythmias.
While the above evidence suggests Purkinje cells in the conduction system as the source for CPVT, direct experimental evidence was lacking until recently, when Flores et al. (2018) tried to establish causation by knocking out calsequestrin only in the conduction system using the HCN4KiT-Cre recombinase system. Cre expression is driven by the promoter of the HCN4 gene, which is expressed in the sinus node and the cardiac conduction system but not in the ventricular working myocardium. Surprisingly, the deletion of calsequestrin in the Purkinje network did not produce a CPVT phenotype (Fig. 5) (Flores et al. 2018). Since deletion of calsequestrin is an established molecular mechanism of CPVT, the results of the study suggest that Purkinje cells in the conduction system may not be capable of generating CPVT on their own. Rather, ventricular myocytes may be the cellular culprit responsible for CPVT, which is discussed next.
Ventricular cardiomyocytes as the cellular source of CPVT
Based on experimental studies of cells isolated from the ventricular myocardium of CPVT mouse models, ventricular cardiomyocytes are clearly capable of generating spontaneous calcium release, DADs, and spontaneous action potentials in response to catecholaminergic stimulation, making them candidates to be the origin of ventricular arrhythmias in CPVT (Knollmann et al. 2006; Liu et al. 2006; Cerrone et al. 2007). Other studies support this. For example, human cardiomyocyte models of CPVT generated from patient-specific induced pluripotent stem cells (iPSCs) also exhibit DADs and action potentials triggered by spontaneous calcium release (Novak et al. 2012, 2015). Drug efficacy in isolated ventricular cardiomyocytes can predict anti-arrhythmic efficacy in mouse models and in humans with CPVT (Knollmann, 2011; Batiste et al. 2019). Furthermore, CPVT patients frequently present with atrial tachycardia or atrial fibrillation that can occur prior to or during their ventricular tachycardia (Leenhardt et al. 1995). Up to 74% of patients with CPVT experience supraventricular tachycardias at slower heart rates than the ventricular arrhythmias (Sumitomo et al. 2007; Cerrone et al. 2009; Sy et al. 2011). Based on optical mapping data from a calsequestrin null mouse model, the atrial tachyarrhythmias are driven by spontaneous calcium release events in atrial myocardium that cause DADs and atrial triggered beats (Faggioni et al. 2014a), supporting the suggestion that that calcium-triggered tachyarrhythmias can originate outside the specialized conduction system. Finally, in clinical studies of CPVT patients, 60–70% of ventricular ectopy originated from the right and left ventricular outflow tracts (Sumitomo et al. 2003; Sy et al. 2011). Although there is no universal agreement on where to find Purkinje fibres in the human ventricle, researchers have suggested that the outflow tract has little or no Purkinje fibres present (Shimizu, 2009). However, a recent anatomical study of the human heart identified what appear to be specialized conducting cells in the right ventricular outflow tract. The specialized cells are thought to be the ramifications of the right bundle branch that do not extend past the pulmonary valves, but more studies are needed to understand the nature of these cells. Of note, some of the arrhythmia foci identified within the right ventricular outflow tract were at anatomical locations distinct from those of the specialized conduction cells (De Almeida et al. 2020). Together, the human data could indicate that ventricular cardiomyocytes are capable of triggering CPVT, at least in portions of the left or right outflow tract. More research with tissue-targeted genetic models such as those in Fig. 5 is needed to determine the tissue origin of CPVT, and to help understand how ventricular cardiomyocytes can overcome the sink–source mismatch to trigger CPVT.
Treatment of CPVT patients
For CPVT patients, there are several therapeutic options recommended by consensus guidelines (Priori et al. 2013). First-line therapy is beta-adrenergic receptor blockers (Class I recommendation). If patients on maximally tolerated beta-blocker therapy continue to have syncope or recurrent sustained VT, treatment should be intensified with combination medical therapy (e.g. adding flecainide, Class IIA) or left cardiac sympathetic denervation (Class IIB). There is no role for an implantable cardiac defibrillator (ICD) as standalone therapy, nor programmed electrical stimulation in CPVT (Class III).
Beta-adrenergic receptor inhibitors (beta-blockers)
Beta-blockers are the first-line drug therapy to treat CPVT since CPVT is triggered by beta-adrenergic stimulation. Based on clinical studies (Hayashi et al. 2009; Leren et al. 2016), the most effective beta-blocker is nadolol, although it is not known why nadolol is superior to other beta-blockers. Although beta-blockers are recommended as a first-line therapy, they are not completely protective: over 30% of patients experienced events during an eight-year follow-up period (van der Werf et al. 2012).
Flecainide and propafenone
The Class 1C anti-arrhythmic drugs flecainide and propafenone both prevent exercise-induced VT in mice and humans with CPVT (Watanabe et al. 2009; Hwang et al. 2011). At the cellular level, their efficacy can be attributed to their dual inhibition of sodium and RyR2 calcium release channels, resulting in a profound suppression of spontaneous calcium release and triggered beats. Since the initial report in 2009, five clinical studies including a randomized placebo-controlled trial (Kannankeril et al. 2017) have investigated flecainide therapy in CPVT patients that were experiencing exercise-induced ventricular arrhythmia despite therapy with beta-blockers. After the initiation of flecainide, approximately 80% of patients had either a partial or a complete suppression of exercise-induced VT (van der Werf et al. 2011; Khoury et al. 2013; Watanabe et al. 2013; Kannankeril et al. 2017; Wangüemert Pérez et al. 2018). Propafenone, another Class 1C anti-arrhythmic drug, has also been used to treat CPVT (Marx et al. 2019). Similar to flecainide, propafenone inhibits RyR2 single channels (Hwang et al. 2011) and inhibits arrhythmogenic calcium waves in CPVT cardiomyocytes (Savio-Galimberti & Knollmann, 2015). While both anti-arrhythmic drugs are effective clinically, additional studies are still needed to gain a better understanding of the role of sodium channel versus RyR2 inhibition, and the potential as a first-line therapy (Behere & Weindling, 2016).
Calcium channel blockers
Although effective in cell and animal models of CPVT (Katz et al. 2010; Alcalai et al. 2011), clinically LTCC blockers by and large provide only transient or partial benefit in CPVT patients already treated with beta-blockers (Swan et al. 2005; Rosso et al. 2007; Katz et al. 2010). Both beta-blockers and flecainide are superior at preventing VT. Calcium channel blockers could be considered for patients that are unable to take flecainide or in refractory cases of CPVT.
Cardiac sympathetic denervation
Left or bilateral cardiac sympathetic denervation reduces catecholamine signalling in the heart by preventing the release of norepinephrine from sympathetic nerve terminals. It was first reported to be successful for CPVT in 2008 when three patients that were refractory to their medications became symptom free after the procedure (Wilde et al. 2008). A larger follow-up study found a reduction of major cardiac events from 86% to 21% (De Ferrari et al. 2015). The most recent guidelines recommend sympathetic denervation as an option for patients that are still experiencing symptoms on maximum beta-blocker therapy (Class IIB).
Implantable cardiac defibrillator
The 2013 guidelines recommended (Class I) that a patient with CPVT has an ICD implanted if the patient has survived a cardiac arrest or if a patient has syncope/documented sustained VT despite optimal medical management and/or left cardiac sympathetic denervation(Al-Khatib et al. 2018). A recent meta-analysis reviewed 53 studies containing 1429 CPVT patients, 35% of whom had an ICD implanted (Roston et al. 2018a). During follow-up, 40% of patients received at least one appropriate shock, 21% of patients received at least one inappropriate shock, and electrical storm (three or more sustained episodes of ventricular tachycardia, ventricular fibrillation, or appropriate shocks from an ICD within 24 hours) occurred in 20% of patients. Seven patients died despite ICD placement, with four deaths associated with electrical storm. The effectiveness of the shocks was also assessed. Some 99% of shocks for ventricular tachycardia failed despite being appropriate whereas 94% of shocks for ventricular fibrillation were successful (Roston et al. 2018a). Thus, the efficacy of ICD shocks in CPVT appears dependent on the arrhythmia mechanism - effective for ventricular fibrillation but ineffective for VT. (Miyake et al. 2013) A recent multicenter study of 136 CPVT patients who presented with cardiac arrest showed no survival benefit associated with ICD implant (van der Werf et al. 2019). These data indicate that ICD therapy can be both ineffective and proarrhythmic, and can cause serious medical complications and psychological burden, especially in paediatric populations. Hence, ICD implantation is controversial and of questionable utility for CPVT patients.
Possible future CPVT therapies
One promising approach is to target the sinus node dysfunction that is characteristic of CPVT. As proof of concept, artificially raising heart rates with atropine, by atrial overdrive pacing or by re-expressing Casq2 in the sinoatrial node prevented catecholamine-induced ventricular arrhythmia in the Casq2 KO mouse CPVT model (Faggioni et al. 2013; Flores et al. 2018). Observational patient data also suggests the efficacy of raising sinus heart rates as a novel therapeutic approach that can prevent exercise-induced VT (Faggioni et al. 2013), which we recently confirmed in a small open-label clinical trial, where raising sinus heart rates with atropine prevented or reduced exercise-induced ventricular ectopy in six CPVT patients (Kannankeril et al. 2019).
Another approach that has been tested successfully in CPVT mouse models is gene therapy via adeno-associated viral (AAV) vectors. Multiple studies have seen beneficial effects from delivering various calcium handling proteins such as calsequestrin (Denegri et al. 2014; Kurtzwald-Josefson et al. 2017; Cacheux et al. 2019) and an engineered calmodulin that inhibits RyR2 calcium release (Liu et al. 2018). AAV was also successfully used for in vivo CRISPR/Cas9-mediated gene editing of an RYR2 CPVT mutation in mice (Pan et al. 2018). As gene therapy continues to improve, the above studies demonstrate the power that a targeted approach could have on arrhythmic diseases, and potentially prevent patients from needing any pharmacological or surgical interventions.
More selective small molecule inhibitors of RyR2-mediated calcium release could provide a better tolerated and effective CPVT therapy. Dantrolene, a drug used clinically to treat hyperactive skeletal muscle calcium release, decreased exercise-induced ventricular ectopy in some but not all CPVT patients (Penttinen et al. 2015). Our group recently reported the discovery of ent-verticilide, a cyclic depsipeptide that has nanomolar potency, is selective for RyR2 over RyR1, and exhibits anti-arrhythmic efficacy in vivo (Batiste et al. 2019). Another promising drug target is the calmodulin-dependent serine-threonine protein kinase II (CaMKII) (Fig. 1). RyR2 phosphorylation by CaMKII increases RyR2 calcium leak. CaMKII inhibition with KN-93 (Liu et al. 2011) or with AAV-mediated delivery of a CaMKII peptide inhibitor (Bezzerides et al. 2019) was effective in suppressing arrhythmias in a murine model of CPVT. Finally, Kifuensine, an inhibitor of mannosidase-I, was used to successfully rescue expression of calsequestrin and reduce CPVT occurrence in triadin-KO mice by preventing proteasomal degradation of misfolded proteins (Cacheux et al. 2019).
Conclusion
The goal of this topical review was to update the field on the current molecular and tissue mechanisms of CPVT and highlight therapeutic approaches. At a molecular level, six genes that affect calcium handling have been found to cause CPVT: RYR2, CASQ2, TRDN, CALM1, CALM2 and CALM3. More work is needed to understand exactly how mutations disrupt protein function and cause pathological calcium release at the cellular level. At a tissue level, current research suggests that sinus node dysfunction contributes mechanistically to the development of exercise-induced ventricular ectopy and could be targeted therapeutically by increasing the sinus heart rate. Future studies are needed to definitively answer whether the ectopic beats originate in the ventricular conduction system (i.e. Purkinje cells) or in the ventricular working myocardium. Insights from studying the molecular and tissue mechanisms extend beyond CPVT, because RyR2 hyperactivity and abnormal calcium handling is also a common feature of structural heart diseases such as ischaemic cardiomyopathy or non-ischaemic heart failure, which are the leading cause of sudden death in the developed world. Hence, CPVT can serve as an important paradigm for studying calcium-related arrhythmia mechanisms and developing novel therapeutics that prevent ventricular arrhythmia and sudden death.
Acknowledgements
The authors would like to thank Erron Titus for giving us permission to use part of a figure he published on a pre-print server on the novel structure of the calsequestrin-2 filament, Fig. 5 of the review.
Funding
MJW is supported in part by the National Heart, Lung, and Blood Institute (NHLBI): F30-HL145917. BCK is supported in part by the NHLBI: R35-HL144980, the Leducq Foundation: 18CVD05, and the American Heart Association (AHA): 19SFRN34830019.
Biographies

Matthew Wleklinski is an MD/PhD student interested in studying how mutations in calcium handling proteins lead to cardiac arrhythmias and sudden death. His current work focuses on the protein calsequestrin and its role in catecholaminergic polymorphic ventricular tachycardia.

Dr Bjorn Knollmann, MD, PhD, is the director of the Vanderbilt Center for Arrhythmia Research and Therapeutics (VanCART) and the Ion Channel and Transporter Training Program at Vanderbilt University, Nashville, TN, USA. He also serves as a Professor in the Departments of Medicine, Pharmacology, and Clinical Pharmacology and was recently named the William Stokes Endowed Chair in Experimental Therapeutics. His primary research interests are finding new mechanisms and treatments for heart rhythm disorders. Work in his laboratory investigates molecular arrhythmia mechanisms involving alterations in the functioning of myofilaments and calcium release channels. In addition, the lab is developing and testing new anti-arrhythmic therapies in animal models and in humans.
Footnotes
This review was presented at the symposium ‘2018 Gordon Research Conference on Cardiac Regulatory Mechanisms’, which took place at Colby-Sawyer College, New London, NH, USA, 3–8 June 2018.
Competing interests
The authors of this review have no conflicts of interest to disclose.
References
- Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, Gillis AM, Granger CB, Hammill SC, Hlatky MA, Joglar JA, Kay GN, Matlock DD, Myerburg RJ & Page RL (2018). 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary. Circulation 138, e210–e271. [DOI] [PubMed] [Google Scholar]
- Alcalai R, Wakimoto H, Arad M, Planer D, Konno T, Wang L, Seidman JG, Seidman CE & Berul CI (2011). Prevention of ventricular arrhythmia and calcium dysregulation in a catecholaminergic polymorphic ventricular tachycardia mouse model carrying calsequestrin-2 mutation. J Cardiovasc Electrophysiol 22, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann HM, Tester DJ, Will ML, Middha S, Evans JM, Eckloff BW & Ackerman MJ (2015). Homozygous/Compound Heterozygous Triadin Mutations Associated With Autosomal-Recessive Long-QT Syndrome and Pediatric Sudden Cardiac Arrest: Elucidation of the Triadin Knockout Syndrome. Circulation 131, 2051–2060. [DOI] [PubMed] [Google Scholar]
- Baher AA, Uy M, Xie F, Garfinkel A, Qu Z & Weiss JN (2011). Bidirectional ventricular tachycardia: ping pong in the His-Purkinje system. Heart Rhythm 8, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiste SM, Blackwell DJ, Kim K, Kryshtal DO, Gomez-Hurtado N, Rebbeck RT, Cornea RL, Johnston JN & Knollmann BC (2019). Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic. Proc Natl Acad Sci U S A 116, 4810–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behere SP & Weindling SN (2016). Catecholaminergic polymorphic ventricular tachycardia: An exciting new era. Ann Pediatr Cardiol 9, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2001). Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers, Dordrecht; Boston. [Google Scholar]
- Bers DM (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Bezzerides VJ, Caballero A, Wang S, Ai Y, Hylind RJ, Lu F, Heims-Waldron DA, Chambers KD, Zhang D, Abrams DJ & Pu WT (2019). Gene Therapy for Catecholaminergic Polymorphic Ventricular Tachycardia by Inhibition of Ca. Circulation 140, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacheux M, Fauconnier J, Thireau J, Osseni A, Brocard J, Roux-Buisson N, Fauré J, Lacampagne A & Marty I (2019). Interplay between Triadin and Calsequestrin in the Pathogenesis of CPVT in the Mouse. Mol Ther. [DOI] [PMC free article] [PubMed]
- Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C & Priori SG (2005). Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res 96, e77–82. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Napolitano C & Priori SG (2009). Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm 6, 1652–1659. [DOI] [PubMed] [Google Scholar]
- Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG & Jalife J (2007). Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 101, 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C & Knollmann BC (2007). Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2 and trigger ventricular arrhythmias in mice. Circ Res 101, 617–626. [DOI] [PubMed] [Google Scholar]
- Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C & Knollmann BC (2009). Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A 106, 7636–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello B, Chadwick C, Saito A, Chu A, Maurer A & Fleischer S (1986). Characterization of the junctional face membrane from terminal cisternae of sarcoplasmic reticulum. J Cell Biol 103, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Almeida MC, Stephenson RS, Anderson RH, Benvenuti LA, Loukas M & Aiello VD (2020). Human subpulmonary infundibulum has an endocardial network of specialized conducting cardiomyocytes. Heart Rhythm 17, 123–130. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Dusi V, Spazzolini C, Bos JM, Abrams DJ, Berul CI, Crotti L, Davis AM, Eldar M, Kharlap M, Khoury A, Krahn AD, Leenhardt A, Moir CR, Odero A, Olde Nordkamp L, Paul T, Rosés I Noguer F, Shkolnikova M, Till J, Wilde AA, Ackerman MJ & Schwartz PJ (2015). Clinical Management of Catecholaminergic Polymorphic Ventricular Tachycardia: The Role of Left Cardiac Sympathetic Denervation. Circulation 131, 2185–2193. [DOI] [PubMed] [Google Scholar]
- de la Fuente S, Van Langen IM, Postma AV, Bikker H & Meijer A (2008). A case of catecholaminergic polymorphic ventricular tachycardia caused by two calsequestrin 2 mutations. Pacing Clin Electrophysiol 31, 916–919. [DOI] [PubMed] [Google Scholar]
- Denegri M, Bongianino R, Lodola F, Boncompagni S, De Giusti VC, Avelino-Cruz JE, Liu N, Persampieri S, Curcio A, Esposito F, Pietrangelo L, Marty I, Villani L, Moyaho A, Baiardi P, Auricchio A, Protasi F, Napolitano C & Priori SG (2014). Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation 129, 2673–2681. [DOI] [PubMed] [Google Scholar]
- Fabiato A (1985). Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol 85, 247–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggioni M, Hwang HS, van der Werf C, Nederend I, Kannankeril PJ, Wilde AA & Knollmann BC (2013). Accelerated sinus rhythm prevents catecholaminergic polymorphic ventricular tachycardia in mice and in patients. Circ Res 112, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D & Knollmann BC (2014a). Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ Arrhythm Electrophysiol 7, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggioni M, van der Werf C & Knollmann BC (2014b). Sinus node dysfunction in catecholaminergic polymorphic ventricular tachycardia: risk factor and potential therapeutic target? Trends Cardiovasc Med 24, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores DJ, Duong T, Brandenberger LO, Mitra A, Shirali A, Johnson JC, Springer D, Noguchi A, Yu ZX, Ebert SN, Ludwig A, Knollmann BC, Levin MD & Pfeifer K (2018). Conditional ablation and conditional rescue models for Casq2 elucidate the role of development and of cell-type specific expression of Casq2 in the CPVT2 phenotype. Hum Mol Genet 27, 1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciosi S, Roston TM, Perry FKG, Knollmann BC, Kannankeril PJ & Sanatani S (2019). Chronotropic incompetence as a risk predictor in children and young adults with catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol 30, 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Kenney LJ & Varriano-Marston E (1987). The structure of calsequestrin in triads of vertebrate skeletal muscle: a deep-etch study. J Cell Biol 105, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CH, Jundi H, Walters N, Thomas NL, West RR & Lai FA (2006). Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res 98, 88–97. [DOI] [PubMed] [Google Scholar]
- Glukhov AV, Kalyanasundaram A, Lou Q, Hage LT, Hansen BJ, Belevych AE, Mohler PJ, Knollmann BC, Periasamy M, Györke S & Fedorov VV (2015). Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex1. Eur Heart J 36, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Hurtado N, Boczek NJ, Kryshtal DO, Johnson CN, Sun J, Nitu FR, Cornea RL, Chazin WJ, Calvert ML, Tester DJ, Ackerman MJ & Knollmann BC (2016). Novel CPVT-Associated Calmodulin Mutation in CALM3 (CALM3-A103V) Activates Arrhythmogenic Ca Waves and Sparks. Circ Arrhythm Electrophysiol 9, pii: e004161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonano LA & Jones PP (2017). FK506-binding proteins 12 and 12.6 (FKBPs) as regulators of cardiac Ryanodine Receptors: Insights from new functional and structural knowledge. Channels (Austin) 11, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray B, Bagnall RD, Lam L, Ingles J, Turner C, Haan E, Davis A, Yang PC, Clancy CE, Sy RW & Semsarian C (2016). A novel heterozygous mutation in cardiac calsequestrin causes autosomal dominant catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 13, 1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Jorgensen AO, Jones LR & Campbell KP (1996). Biochemical characterization and molecular cloning of cardiac triadin. J Biol Chem 271, 458–465. [DOI] [PubMed] [Google Scholar]
- Györke S, Stevens SC & Terentyev D (2009). Cardiac calsequestrin: quest inside the SR. J Physiol 587, 3091–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Denjoy I, Extramiana F, Maltret A, Buisson NR, Lupoglazoff JM, Klug D, Takatsuki S, Villain E, Kamblock J, Messali A, Guicheney P, Lunardi J & Leenhardt A (2009). Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 119, 2426–2434. [DOI] [PubMed] [Google Scholar]
- Herron TJ, Milstein ML, Anumonwo J, Priori SG & Jalife J (2010). Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 7, 1122–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesl E, Stieber J, Herrmann S, Feil S, Tybl E, Hofmann F, Feil R & Ludwig A (2008). Tamoxifen-inducible gene deletion in the cardiac conduction system. J Mol Cell Cardiol 45, 62–69. [DOI] [PubMed] [Google Scholar]
- Huke S & Knollmann BC (2011). Oxidized CaMKII: a “heart stopper” for the sinus node? J Clin Invest 121, 2975–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, Yin H & Knollmann BC (2011). Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 4, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL, Cornea RL, Bers DM & Knollmann BC (2014). Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res 114, 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto N & Yamamoto T (2002). Regulation of calcium release by interdomain interaction within ryanodine receptors. Front Biosci 7, d671–683. [DOI] [PubMed] [Google Scholar]
- Imberti JF, Underwood K, Mazzanti A & Priori SG (2016). Clinical Challenges in Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ 25, 777–783. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P & Marks AR (1992). FK506 binding protein associated with the calcium release channel (ryanodine receptor). J Biol Chem 267, 9474–9477. [PubMed] [Google Scholar]
- Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H & Chen SR (2004). RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A 101, 13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Jáimez J, Palomino Doza J, Ortega Á, Macías-Ruiz R, Perin F, Rodríguez-Vázquez del Rey MM, Ortiz-Genga M, Monserrat L, Barriales-Villa R, Blanca E, Álvarez M & Tercedor L (2016). Calmodulin 2 Mutation N98S Is Associated with Unexplained Cardiac Arrest in Infants Due to Low Clinical Penetrance Electrical Disorders. PLoS One 11, e0153851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LR, Zhang L, Sanborn K, Jorgensen AO & Kelley J (1995). Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem 270, 30787–30796. [DOI] [PubMed] [Google Scholar]
- Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG & Fishman GI (2010). Purkinje cells from RyR2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res 107, 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannankeril PJ, Moore JP, Cerrone M, Priori SG, Kertesz NJ, Ro PS, Batra AS, Kaufman ES, Fairbrother DL, Saarel EV, Etheridge SP, Kanter RJ, Carboni MP, Dzurik MV, Fountain D, Chen H, Ely EW, Roden DM & Knollmann BC (2017). Efficacy of Flecainide in the Treatment of Catecholaminergic Polymorphic Ventricular Tachycardia: A Randomized Clinical Trial. JAMA Cardiol 2, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannankeril PJ, Shoemaker MB, Gayle KA, Fountain D, Roden DM & Knollmann BC (2019). Atropine-Induced Sinus Tachycardia Protects Against Exercise-Induced Ventricular Arrhythmias in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. Europace. [DOI] [PMC free article] [PubMed]
- Katz G, Arad M & Eldar M (2009). Catecholaminergic polymorphic ventricular tachycardia from bedside to bench and beyond. Curr Probl Cardiol 34, 9–43. [DOI] [PubMed] [Google Scholar]
- Katz G, Khoury A, Kurtzwald E, Hochhauser E, Porat E, Shainberg A, Seidman JG, Seidman CE, Lorber A, Eldar M & Arad M (2010). Optimizing catecholaminergic polymorphic ventricular tachycardia therapy in calsequestrin-mutant mice. Heart Rhythm 7, 1676–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury A, Marai I, Suleiman M, Blich M, Lorber A, Gepstein L & Boulos M (2013). Flecainide therapy suppresses exercise-induced ventricular arrhythmias in patients with CASQ2-associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 10, 1671–1675. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Wehrmeister D, Postma AV, Pohlentz G, Mormann M, Kucerova D, Müller FU, Schmitz W, Schulze-Bahr E, Wilde AA & Neumann J (2010). The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J Mol Cell Cardiol 49, 95–105. [DOI] [PubMed] [Google Scholar]
- Knollmann BC (2011). Carvedilol tweaks calcium release to ease arrhythmias. Nat Med 17, 923–924. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C & Pfeifer K (2006). Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116, 2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Stang KK, Moomaw CR, Slaughter CA & Campbell KP (1993). Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin). J Biol Chem 268, 12646–12654. [PubMed] [Google Scholar]
- Kobayashi YM & Jones LR (1999). Identification of triadin 1 as the predominant triadin isoform expressed in mammalian myocardium. J Biol Chem 274, 28660–28668. [DOI] [PubMed] [Google Scholar]
- Kryshtal DO, Gryshchenko O, Gomez-Hurtado N & Knollmann BC (2015). Impaired calcium-calmodulin-dependent inactivation of Cav1.2 contributes to loss of sarcoplasmic reticulum calcium release refractoriness in mice lacking calsequestrin 2. J Mol Cell Cardiol 82, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzwald-Josefson E, Yadin D, Harun-Khun S, Waldman M, Aravot D, Shainberg A, Eldar M, Hochhauser E & Arad M (2017). Viral delivered gene therapy to treat catecholaminergic polymorphic ventricular tachycardia (CPVT2) in mouse models. Heart Rhythm 14, 1053–1060. [DOI] [PubMed] [Google Scholar]
- Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B & Pras E (2001a). Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation 103, 2822–2827. [DOI] [PubMed] [Google Scholar]
- Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D & Eldar M (2001b). A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 69, 1378–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Maeng JS, Choi JY, Lee YR, Hwang CY, Park SS, Park HK, Chung BH, Lee SG, Kim YS, Jeon H, Eom SH, Kang C, Kim DH & Kwon KS (2012). Role of Junctin protein interactions in cellular dynamics of calsequestrin polymer upon calcium perturbation. J Biol Chem 287, 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD & Coumel P (1995). Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91, 1512–1519. [DOI] [PubMed] [Google Scholar]
- Leren IS, Saberniak J, Majid E, Haland TF, Edvardsen T & Haugaa KH (2016). Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 13, 433–440. [DOI] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL & Yue DT (2014). Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol 74, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S, Helmersson A & Forsén S (1991). Calcium binding to calmodulin and its globular domains. J Biol Chem 266, 8050–8054. [PubMed] [Google Scholar]
- Liu B, Ho HT, Brunello L, Unudurthi SD, Lou Q, Belevych AE, Qian L, Kim DH, Cho C, Janssen PM, Hund TJ, Knollmann BC, Kranias EG & Györke S (2015). Ablation of HRC alleviates cardiac arrhythmia and improves abnormal Ca handling in CASQ2 knockout mice prone to CPVT. Cardiovasc Res 108, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Walton SD, Ho HT, Belevych AE, Tikunova SB, Bonilla I, Shettigar V, Knollmann BC, Priori SG, Volpe P, Radwański PB, Davis JP & Györke S (2018). Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J Am Heart Assoc 7 pii: e008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA & Priori SG (2006). Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 99, 292–298. [DOI] [PubMed] [Google Scholar]
- Liu N, Rizzi N, Boveri L & Priori SG (2009). Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J Mol Cell Cardiol 46, 149–159. [DOI] [PubMed] [Google Scholar]
- Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA & Priori SG (2011). Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/−) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol 50, 214–222. [DOI] [PubMed] [Google Scholar]
- Manno C, Figueroa LC, Gillespie D, Fitts R, Kang C, Franzini-Armstrong C & Rios E (2017). Calsequestrin depolymerizes when calcium is depleted in the sarcoplasmic reticulum of working muscle. Proc Natl Acad Sci U S A 114, E638–E647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman RF, Barc J, Beekman L, Alders M, Dooijes D, van den Wijngaard A, Ratbi I, Sefiani A, Bhuiyan ZA, Wilde AA & Bezzina CR (2014). A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol 63, 259–266. [DOI] [PubMed] [Google Scholar]
- Marx A, Lange B, Nalenz C, Hoffmann B, Rostock T & Konrad T (2019). A 35-year effective treatment of catecholaminergic polymorphic ventricular tachycardia with propafenone. HeartRhythm Case Rep 5, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake CY, Webster G, Czosek RJ, Kantoch MJ, Dubin AM, Avasarala K & Atallah J (2013). Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol, 6, 579–587. [DOI] [PubMed] [Google Scholar]
- Miyata K, Ohno S, Itoh H & Horie M (2018). Bradycardia Is a Specific Phenotype of Catecholaminergic Polymorphic Ventricular Tachycardia Induced by RYR2 Mutations. Intern Med 57, 1813–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell SM, Bradley DJ & Lehmann MH (2012). Genetic testing for long QT syndrome and the category of cardiac ion channelopathies. PLoS Curr, 4, e4f9995f9969e9996c9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RM, Mollica JP, Beard NA, Knollmann BC & Lamb GD (2011). Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+-binding protein changes in CSQ2 knockout mice. Am J Physiol Heart Circ Physiol 300, H595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Barad L, Lorber A, Gherghiceanu M, Reiter I, Eisen B, Eldor L, Itskovitz-Eldor J, Eldar M, Arad M & Binah O (2015). Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J Cell Mol Med 19, 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Barad L, Zeevi-Levin N, Shick R, Shtrichman R, Lorber A, Itskovitz-Eldor J & Binah O (2012). Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to β-adrenergic stimulation. J Cell Mol Med 16, 468–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M & Børglum AD (2012). Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet 91, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Philippen L, Lahiri SK, Lee C, Park SH, Word TA, Li N, Jarrett KE, Gupta R, Reynolds JO, Lin J, Bao G, Lagor WR & Wehrens XHT (2018). In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Res 123, 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Wu S, Dunker AK & Kang C (2003). Polymerization of calsequestrin. Implications for Ca2+ regulation. J Biol Chem 278, 16176–16182. [DOI] [PubMed] [Google Scholar]
- Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K & Aalto-Setälä K (2015). Correction: Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS One 10, e0134746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP & Yue DT (1999). Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 22, 549–558. [DOI] [PubMed] [Google Scholar]
- Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA & Guicheney P (2002). Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res 91, e21–26. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V & Danieli GA (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200. [DOI] [PubMed] [Google Scholar]
- Priori SG & Corr PB (1990). Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol-Heart C Physiol 258, H1796–H1805. [DOI] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C-E, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G & Tracy C (2013) HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 10, 1932–1963. [DOI] [PubMed] [Google Scholar]
- Pérez-Riera AR, Barbosa-Barros R, de Rezende Barbosa MPC, Daminello-Raimundo R, de Lucca AA & de Abreu LC (2018). Catecholaminergic polymorphic ventricular tachycardia, an update. Ann Noninvasive Electrocardiol 23, e12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P & Priori SG (2008). Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res 103, 298–306. [DOI] [PubMed] [Google Scholar]
- Rooryck C, Kyndt F, Bozon D, Roux-Buisson N, Sacher F, Probst V & Thambo JB (2015). New Family With Catecholaminergic Polymorphic Ventricular Tachycardia Linked to the Triadin Gene. J Cardiovasc Electrophysiol 26, 1146–1150. [DOI] [PubMed] [Google Scholar]
- Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, Belhassen B & Viskin S (2007). Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 4, 1149–1154. [DOI] [PubMed] [Google Scholar]
- Roston TM, Jones K, Hawkins NM, Bos JM, Schwartz PJ, Perry F, Ackerman MJ, Laksman ZWM, Kaul P, Lieve KVV, Atallah J, Krahn AD & Sanatani S (2018a). Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: A systematic review. Heart Rhythm 15, 1791–1799. [DOI] [PubMed] [Google Scholar]
- Roston TM, Sanatani S & Chen SR (2017). Suppression-of-function mutations in the cardiac ryanodine receptor: Emerging evidence for a novel arrhythmia syndrome? Heart Rhythm 14, 108–109. [DOI] [PubMed] [Google Scholar]
- Roston TM, Yuchi Z, Kannankeril PJ, Hathaway J, Vinocur JM, Etheridge SP, Potts JE, Maginot KR, Salerno JC, Cohen MI, Hamilton RM, Pflaumer A, Mohammed S, Kimlicka L, Kanter RJ, LaPage MJ, Collins KK, Gebauer RA, Temple JD, Batra AS, Erickson C, Miszczak-Knecht M, Kubuš P, Bar-Cohen Y, Kantoch M, Thomas VC, Hessling G, Anderson C, Young ML, Choi SHJ, Cabrera Ortega M, Lau YR, Johnsrude CL, Fournier A, Van Petegem F & Sanatani S (2018b). The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace 20, 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, Fauconnier J, Brocard J, Denjoy I, Durand P, Guicheney P, Kyndt F, Leenhardt A, Le Marec H, Lucet V, Mabo P, Probst V, Monnier N, Ray PF, Santoni E, Trémeaux P, Lacampagne A, Fauré J, Lunardi J & Marty I (2012). Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet 21, 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio-Galimberti E & Knollmann BC (2015). Channel Activity of Cardiac Ryanodine Receptors (RyR2) Determines Potency and Efficacy of Flecainide and R-Propafenone against Arrhythmogenic Calcium Waves in Ventricular Cardiomyocytes. PLoS One 10, e0131179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel M, Lai FA & Zissimopoulos S (2015). Structural and functional interactions within ryanodine receptor. Biochem Soc Trans 43, 377–383. [DOI] [PubMed] [Google Scholar]
- Shimizu W (2009). Arrhythmias originating from the right ventricular outflow tract: how to distinguish “malignant” from “benign”? Heart Rhythm 6, 1507–1511. [DOI] [PubMed] [Google Scholar]
- Sommer JR & Johnson EA (1968). Cardiac muscle. A comparative study of Purkinje fibers and ventricular fibers. J Cell Biol 36, 497–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuyvers BD, Dun W, Matkovich S, Sorrentino V, Boyden PA & ter Keurs HE (2005). Ca2+ sparks and waves in canine purkinje cells: a triple layered system of Ca2+ activation. Circ Res 97, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo N (2016). Current topics in catecholaminergic polymorphic ventricular tachycardia. J Arrhythm 32, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo N, Harada K, Nagashima M, Yasuda T, Nakamura Y, Aragaki Y, Saito A, Kurosaki K, Jouo K, Koujiro M, Konishi S, Matsuoka S, Oono T, Hayakawa S, Miura M, Ushinohama H, Shibata T & Niimura I (2003). Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumitomo N, Sakurada H, Taniguchi K, Matsumura M, Abe O, Miyashita M, Kanamaru H, Karasawa K, Ayusawa M, Fukamizu S, Nagaoka I, Horie M, Harada K & Hiraoka M (2007). Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J 71, 1606–1609. [DOI] [PubMed] [Google Scholar]
- Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ & Anderson ME (2011). Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest 121, 3277–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan H, Laitinen P, Kontula K & Toivonen L (2005). Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol 16, 162–166. [DOI] [PubMed] [Google Scholar]
- Swan H, Piippo K, Viitasalo M, Heikkilä P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K & Toivonen L (1999). Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol 34, 2035–2042. [DOI] [PubMed] [Google Scholar]
- Sy RW, Gollob MH, Klein GJ, Yee R, Skanes AC, Gula LJ, Leong-Sit P, Gow RM, Green MS, Birnie DH & Krahn AD (2011). Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 8, 864–871. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC & Gyorke S (2005). Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res 96, 651–658. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG & Gyorke S (2006). Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res 98, 1151–1158. [DOI] [PubMed] [Google Scholar]
- Titus EW, Deiter FH, Shi C, Wojciak J, Scheinman M, Jura N & Deo RC (2019). The structure of a calsequestrin filament reveals mechanisms of familial arrhythmia. bioRxiv, 672303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N & Matsuzaki M (2010). Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res 106, 1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf C, Kannankeril PJ, Sacher F, Krahn AD, Viskin S, Leenhardt A, Shimizu W, Sumitomo N, Fish FA, Bhuiyan ZA, Willems AR, van der Veen MJ, Watanabe H, Laborderie J, Haïssaguerre M, Knollmann BC & Wilde AA (2011). Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 57, 2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf C, Lieve KV, Bos JM, Lane CM, Denjoy I, Roses-Noguer F, Aiba T, Wada Y, Ingles J, Leren IS, Rudic B, Schwartz PJ, Maltret A, Sacher F, Skinner JR, Krahn AD, Roston TM, Tfelt-Hansen J, Swan H, Robyns T, Ohno S, Roberts JD, van den Berg MP, Kammeraad JA, Probst V, Kannankeril PJ, Blom NA, Behr ER, Borggrefe M, Haugaa KH, Semsarian C, Horie M, Shimizu W, Till JA, Leenhardt A, Ackerman MJ & Wilde AA (2019). Implantable cardioverter-defibrillators in previously undiagnosed patients with catecholaminergic polymorphic ventricular tachycardia resuscitated from sudden cardiac arrest. Eur Heart J 40, 2953–2961. [DOI] [PubMed] [Google Scholar]
- van der Werf C, Zwinderman AH & Wilde AA (2012). Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace 14, 175–183. [DOI] [PubMed] [Google Scholar]
- VanScyoc WS, Sorensen BR, Rusinova E, Laws WR, Ross JB & Shea MA (2002). Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys J 83, 2767–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangüemert Pérez F, Hernández Afonso JS, Groba Marco MDV, Caballero Dorta E, Álvarez Acosta L, Campuzano Larrea O, Pérez G, Brugada Terradellas J & Brugada Terradellas R (2018). Flecainide Reduces Ventricular Arrhythmias in Patients With Genotype RyR2-positive Catecholaminergic Polymorphic Ventricular Tachycardia. Rev Esp Cardiol (Engl Ed) 71, 185–191. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA & Knollmann BC (2009). Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15, 380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, Rosso R, Bhuiyan ZA, Bikker H, Kannankeril PJ, Horie M, Minamino T, Viskin S, Knollmann BC, Till J & Wilde AA (2013). Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 10, 542–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ & Marks AR (2003). FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113, 829–840. [DOI] [PubMed] [Google Scholar]
- Wilde AA, Bhuiyan ZA, Crotti L, Facchini M, De Ferrari GM, Paul T, Ferrandi C, Koolbergen DR, Odero A & Schwartz PJ (2008). Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med 358, 2024–2029. [DOI] [PubMed] [Google Scholar]
- Xiao J, Tian X, Jones PP, Bolstad J, Kong H, Wang R, Zhang L, Duff HJ, Gillis AM, Fleischer S, Kotlikoff M, Copello JA & Chen SR (2007). Removal of FKBP12.6 does not alter the conductance and activation of the cardiac ryanodine receptor or the susceptibility to stress-induced ventricular arrhythmias. J Biol Chem 282, 34828–34838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z & Weiss JN (2010). So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 99, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Takahashi N, Xu L, Smithies O & Meissner G (2007). Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest 117, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Xu L, Pasek DA, Evans KE & Meissner G (2003). Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor). J Biol Chem 278, 23480–23486. [DOI] [PubMed] [Google Scholar]
- Yamniuk AP & Vogel HJ (2004). Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol Biotechnol 27, 33–57. [DOI] [PubMed] [Google Scholar]
- Yano K & Zarain-Herzberg A (1994). Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem 135, 61–70. [DOI] [PubMed] [Google Scholar]
- Yano M, Yamamoto T, Kobayashi S & Matsuzaki M (2009). Role of ryanodine receptor as a Ca2(+) regulatory center in normal and failing hearts. J Cardiol 53, 1–7. [DOI] [PubMed] [Google Scholar]
- Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, George AL & Satin J (2014). Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc 3, e000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Waddell HM, Wu E, Dholakia J, Okolo CA, McLay JC & Jones PP (2016). FKBPs facilitate the termination of spontaneous Ca2+ release in wild-type RyR2 but not CPVT mutant RyR2. Biochem J 473, 2049–2060. [DOI] [PubMed] [Google Scholar]
- Zhang L, Kelley J, Schmeisser G, Kobayashi YM & Jones LR (1997). Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem 272, 23389–23397. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW & Reuter H (1999). Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399, 159–162. [DOI] [PubMed] [Google Scholar]