

Abstract
Unidirectional coherence transfer is highly efficient in intrinsically disordered proteins (IDPs). Their elevated ps-ns timescale dynamics ensures long transverse (T2) relaxation times allowing sophisticated coherence transfer pathway selection in comparison to folded proteins. 1Hα-detection ensures non-susceptibility to chemical exchange with the solvent and enables chemical shift assignment of consecutive proline residues, typically abundant in IDPs. However, many IDPs undergo a disorder-to-order transition upon interaction with their target protein, which leads to the loss of the favorable relaxation properties. Long coherence transfer routes now result in prohibitively large decrease in sensitivity. We introduce a novel 4D 1Hα-detected experiment HACANCOi, together with its 3D implementation, which warrant high sensitivity for the assignment of proline-rich regions in IDPs in complex with a globular protein. The experiment correlates 1Hαi, 13Cαi, 15Ni and spins by transferring the magnetization concomitantly from 13Cαi to 15Ni and . The B1 domain of protein G (GB1), and the enteropathogenic E. coli EspF in complex with human SNX9 SH3, serve as model systems to demonstrate the attainable sensitivity and successful sequential assignment.
Electronic supplementary material
The online version of this article (10.1007/s10858-020-00347-5) contains supplementary material, which is available to authorized users.
Keywords: E.coli, EspF, GB1, Intrinsically disordered protein, IDP, Resonance assignment, SNX9 SH3
Introduction
Intrinsically disordered proteins (IDPs), proteins with intrinsically disordered regions (IDRs) or modular proteins containing intrinsically disordered linkers (IDLs) have raised great interest in the scientific community in the last two decades. Not only because these proteins or regions have been tightly linked to disease related biology, but also because they have significantly influenced our perception of mechanistic structural biology in general. The fundamental difference between the classical folded protein kingdom and their disordered rivals, often referred to as the ‘dark proteome’ calls for different means to study them using NMR spectroscopic tools.
Whereas prohibitively fast transverse relaxation is the biggest hurdle in the structure determination of larger folded proteins and complexes, resonance overlap constitutes the major hindrance in the chemical shift assignment and hence structural characterization of disordered systems. Resonance overlap substantially limits the feasibility of the well-established amide proton (HN) detection based experiments which rely on Cα/Cβ shifts to obtain sequential connections, commonly used for the assignment of folded proteins (for reviews see e.g. Sattler et al. 1999; Permi and Annila 2004). Even though amide signals in a 1H, 15N HSQC of an IDP can show fair dispersion, residue type dependent 13Cα and 13Cβ clustering strongly reduces the applicability of these experiments. Carbonyl carbons, however, show greater dispersion thanks to their larger sensitivity to the type of neighboring residues in the primary sequence, and therefore better meet the demands of assignment (Yao et al. 1997; Mäntylahti et al. 2009; Bermel et al. 2012). Combination of 13C′ dispersion with the high sensitivity and robustness of the HN-detection based experiments, additionally exploiting their compatibility with the TROSY and BEST implementations (Solyom et al. 2013; Brutscher et al. 2015), has been a successful work-around scheme for the assignment of many disordered systems. These include experiments which exploit increase of dimensionality from conventional 3D to 4-7D spectra (Fiorito et al. 2006; Motackova et al. 2010; Nováček et al. 2011; Kazimierczuk et al. 2013; Brutscher et al. 2015; Yoshimura et al. 2015). Recently we and others have proposed HN-detected experiments which allow bridging the gaps in amino acid sequence fragments due to single prolines in PXP or XPX moieties (Liu and Yang 2003; Hellman et al. 2014; Tossavainen et al. 2020).
The HN/C′ approach is, however, less optimal for studies at neutral or alkali sample pH and/or proteins that contain a high percentage of prolines. To this end, two different approaches have been proposed; the direct 13C, especially 13C′, detection based experiments (Bermel et al. 2006a, 2009, 2012) and the 1Hα based detection (Mäntylahti et al. 2010, 2011). The direct detection of 13C′ spins instead of detection of HN renders the assignment non-susceptible to chemical exchange with the solvent at pH higher than 7. In addition, this approach enables the assignment of consecutive proline residues and is therefore suitable for studying proline rich IDRs/IDLs. However, to fully reclaim the advantages of 13C-detection, a probehead having 13C as the inner coil is required as an inherent sensitivity loss by a factor of eight, owing to the S/N = γH3/2/γC3/2 dependence on sensitivity, applies in comparison to 1H-detection. Showalter and co-workers have evaluated that sample concentration with the 13C-detection approach should not fall significantly below 500 μM when using three-dimensional 13C-detected experiments for chemical shift assignment (Bastidas et al. 2015).
Similar to its 13C-detected counterpart, the 1Hα-detection based assignment strategy is per se insensitive to chemical exchange with the solvent as 1Hαs are considered as non-labile protons irrespective of pH. Also, assignment of consecutive prolines can readily be accomplished, making these experiments attractive alternatives to 13C-detection based pulse schemes (Mäntylahti et al. 2010, 2011; Permi and Hellman 2012; Tossavainen et al. 2020). Moreover, unless a 13C-detection optimized probehead is used, these experiments offer superior sensitivity with respect to 13C-detected experiments (Wong et al. 2020), therefore enabling significant time savings. Previously we have successfully used a set of three 3D 1Hα-detected experiments, i.e. iHA(CA)NCO, HA(CA)CON and (HACA)CON(CA)HA in the assignment of several IDPs and proline-rich IDLs (Hellman et al. 2014; Aitio et al. 2012; Tossavainen et al. 2020). Extension to 4D, by utilizing 13Cα chemical shifts, facilitated the assignment of 161-residue BilRI, bacterial interleukin receptor I, from an opportunistic oral pathogen A. actinomycetemcomitans, half of whose amino acid sequence is composed of three residues only, that is alanines (23%), lysines (14%) and aspartic acids (13%) (Tossavainen et al. 2020).
Indeed, the unidirectional coherence transfer is highly efficient in IDPs. Thanks to their elevated ps-ns timescale dynamics IDPs have long transverse relaxation times (T2s) allowing more sophisticated coherence transfer pathway selection in comparison to globular proteins. In 1Hα-detected experiments this can further be boosted by measuring spectra at an elevated temperature, 35–45 °C, as temperature dependent exchange rate of labile amide protons with solvent is not a problem with 1Hα-detected experiments. The coherence transfer efficiency of the unidirectional experiments, however, drops dramatically in the case of folded proteins, or when an IDP or IDR/L folds upon binding when interacting with its target. In the latter case, favorable relaxation properties are partially lost at least for the residues directly and sometimes indirectly involved in binding. Under these conditions, the long coherence transfer routes become a liability resulting in prohibitively large losses in sensitivity in these experiments. In this manuscript, we introduce a novel 4D (3D) 1Hα-detected experiment HACANCOi, which warrants high sensitivity for the assignment of proline-rich regions in IDPs in complex with a globular protein.
Materials and methods
Cloning, expression, and purification of SNX9 SH3 and EspF
The gene encoding the SH3 domain (residues 1–64) of human SNX9 (Sorting nexin 9) (UniProt Q9Y5X1) (SNX9 SH3) was cloned to pET15b vector (Novagen) into the NdeI and XhoI sites. The gene encoding residues 115–161 of EspF (UniProt B7UM88) (EspF) was cloned to pET15b vector with N-terminal GB1 fusion protein into the NdeI and XhoI sites. TEV protease (from Tobacco Etch Virus) cleavage site was added between GB1 fusion protein and EspF. Both protein constructs carried the N-terminal His-tag. All the genes were synthetic, obtained from GenScript Inc., USA.
Production of 15N and 13C labeled or unlabeled SNX9 SH3 or EspF proteins was carried out by transforming plasmids into the BL21(DE3) cells. The cells were grown in M9 minimal media, supplemented with 1 g/l of 15NH4Cl and 2 g/l 13C-D-glucose as the sole nitrogen or nitrogen and carbon source, respectively, or in LB media for obtaining unlabeled proteins. Cell culture was incubated at 37 °C and the temperature was decreased to 16 °C when OD of the cell culture reached 0.4 and protein production was induced with 1 mM IPTG when OD of the cell culture reached 0.6. The cells were further incubated at 16 °C for 16 h and collected by centrifugation. The cells were disrupted with sonication and the resulting supernatant was clarified by centrifugation with 30,000×g.
The clarified supernatant of His-Tagged SNX9 SH3 and His-Tagged GB1-EspF fusion protein was applied to the 1-mL His GraviTrap column (GE Healthcare) according to the manufacturer’s instructions. Imidazole was removed from eluted proteins by PD-10 (Ge Healthcare) before protease cleavage.
The His-Tag from the SNX9-SH3 was removed by thrombin protease and from the His-Tag-GB1-EspF fusion by TEV protease digestion. Digestion mixtures were applied to His GraviTrap column. Cleaved EspF and SNX9 SH3 eluted with flow-through, that were concentrated and applied into the Superdex75 (16/60) or Superdex30 (16/60) gel filtration column, respectively. Columns were equilibrated with NMR buffer (20 mM sodium phosphate pH 6.5, 50 mM NaCl). Fractions, containing purified proteins, were pooled and concentrated by Vivaspin2 (SartoriusStedim). All the gel filtrations were performed by using the ÄKTA Purifier FLPC purification system (GE Healthcare).
NMR spectroscopy
Two-dimensional iHA(CA)N(CO) and HA(CA)N(COi) experiments were measured at four different temperatures 5, 15, 25 and 35 °C using 2 mM 15N, 13C labeled B1 domain of protein G (GB1), dissolved in 95%/5% H2O/D2O sodium phosphate buffer, pH 5.5. Three/four-dimensional HA(CA)NCOi/HACANCOi as well as three-dimensonal HA(CA)CON experiment were acquired for a binary complex between 1.9 mM 15N, 13C labeled SNX9 SH3 and 1.5 mM 47-residue repeat of EspF and 2.5 mM unlabeled SNX9 SH3 and 1.5 mM 15N, 13C labeled EspF complex, respectively. All spectra were recorded on a Bruker AVANCE III HD 800 MHz spectrometer equipped with a TCI 1H/13C/15N cryoprobe. Traditional sampling was used for collecting two- and three-dimensional data whereas non-uniform sampling (NUS) with the NUS-sampler algorithm and random order sampling at density of 7% was employed for collecting four-dimensional data (Table 1). No sample point optimization, based on either T2 or J coupling values, was employed during the data collection. The NMR data were processed with Topspin 3.5 software package (Bruker Inc.) either using fast-Fourier transform or multi-dimensional decomposition (MDD) data processing for NUS data with non-recursive MDD and specifying constant-time dimension for t3. The reconstruction took roughly 2 h on a standard PC attached to the spectrometer. Motivation for using traditional sampling instead of NUS in 3D data was to warrant optimal S/N for the weakest, binding exchange broadened, cross peaks in the complex without sacrifying the resolution. Data analysis was accomplished using CcpNmr analysis v. 2.4.2 (Vranken et al. 2005). Chemical shifts and structural data of the SNX9-SH3 – EspF complex will be published elsewhere.
Table 1.
Data acquisition parameters
| Number of complex points (acquisition time) | |||||||
|---|---|---|---|---|---|---|---|
| Experiment | F1 (ms) | F2 (ms) | F3 (ms) | F4 (ms) | nD | Number of scans | Experimental time |
| iHA(CA)N(CO) | 200 (36.2) 15N | - | - | 256 (53.3) 1Hα | 2 | 32 | 1 h 20 m |
| HA(CA)N(CO)i | 200 (36.2) 12N | - | - | - | 2 | 32 | 1 h 20 m |
| HA(CA)NCOi | 64 (13.2) 13C’ | 80 (11.7) 15N | - | 256 (53.3) 1Hα | 3 | 16 | 22 h |
| HA(CA)CON | 80 (11.7) 15N | 64 (13.2) 13C’ | - | 256 (53.3) 1Hα | 3 | 8 | 11 h |
| HACANCOi | 80 (11.7) 15N | 64 (10.2) 15N | 48 (4.0) 13Cα | 256 (53.3) 1Hα | 4 | 8 | 23 h |
Results and discussion
Our group is actively working with IDPs harboring SH3 domain targeting short linear motifs (SLiMs). Classically SH3 domain interactions are mediated by proline-rich SLiMs. Due to the lack of an amide proton in the N-substituted proline residue, structural characterization of these epitopes using conventional HN-detected experiments is challenging. To overcome this limitation, we have employed a suite of 3D (or 4D) 1Hα-detected experiments for the assignment of these proline-rich SLiMs (Mäntylahti et al. 2010, 2011; Permi and Hellman 2012; Tossavainen et al. 2020). Although this approach has been very successful for assigning several IDPs and their complexes (Aitio et al. 2010, 2012; Hellman et al. 2014; Tossavainen et al. 2017; Thapa et al. 2020), the sensitivity of these experiments may become an issue upon complex formation, stemming from a longer magnetization transfer delay needed for the unidirectional coherence transfer pathway selection. To this end, we have designed a novel 4D (together with its 3D implementation) 1Hα-detected experiment, which offers significantly higher sensitivity in more slowly tumbling systems, e.g. IDPs in complex with structural domains, at the expense of data redundancy, that is exhibition of intraresidual and sequential cross peaks simultaneously in one experiment.
The 4D HACANCO experiment
The magnetization transfer pathway of the new experiment we dubbed HACANCOi is shown in Fig. 1a. In brief, magnetization is first transferred from 1Hα(i) to 13Cα(i) and then simultaneously to 13C’(i) and 15N(i), as well as to 15 N(i + 1). The proposed 4D HACANCOi experiment is shown in Fig. 1b, whereas the inset b′ shows the alternative implementation for the 3D HA(CA)NCOi scheme. The coherence flows through the 4D HACANCOi, and 3D HA(CA)NCOi, experiment are as follows (Eqs. 1, 2):
| 1 |
| 2 |
Fig. 1.

Description of the HACANCOi experiment. a Schematic presentation of magnetization transfer pathway during the proposed 4D HACANCOi experiment and 4D iHACANCO scheme (Mäntylahti et al. 2010; Tossavainen et al. 2020). Black arrows indicate the so-called out-and-back transfer pathway from 1Hα(i) to 13Cα(i) and further to 13C′(i) and 15N(i)/15N(i + 1). b 4D HACANCOi experiment to correlate 1Hα(i), 13Cα(i), 13C′(i) and 15N(i)/15N(i + 1) chemical shifts. Inset b′ 3D HA(CA)NCOi experiment to correlate 1Hα(i), 13C′(i) and 15N(i)/15N(i + 1) chemical shifts. Narrow and wide filled bars on 1H and 15N channels correspond to rectangular 90° and 180° pulses, respectively, applied with phase x unless otherwise stated. All 13C pulses are band-selective shaped pulses, denoted by filled narrow bars (90°) and filled and unfilled half ellipsoids (180°). Pulses denoted with unfilled bars are applied on-resonance. The 1H, 15N, 13C′, and 13Cα carrier positions are 4.7 (water), 121 (center of 15N spectral region), 174 ppm (center of 13C′ spectral region), and 54 ppm (center of 13Cα spectral region). The 13C carrier is initially set to the middle of 13C′ region (174 ppm), and shifted to 13Cα region (54 ppm) prior to 90° 15N pulse ϕ3. All band-selective 90° and 180° pulses for 13Cα (54 ppm) and 13C′ (174 ppm) have the shape of Q5 and Q3 (Emsley and Bodenhausen 1992) and duration of 240.0 μs and 192.0 μs at 800 MHz, respectively. The adiabatic 180° Chirp broadband inversion pulse, denoted with striped half ellipsoid in both 13C channels, for inverting 13Cα and 13C′ magnetization in the middle of t2 period had duration of 500 μs at 800 MHz (Böhlen and Bodenhausen 1993). The Waltz-65 sequence (Zhou et al. 2007) with strength of 4.17 kHz was employed to decouple 1H spins. The GARP (Shaka et al. 1985, 1987) with field strength of 4.55 kHz was used to decouple 13C during acquisition. Delay durations: τ = 1/(4JHC) ~ 1.7 ms; τ2 = 3.4 ms (optimized for non-glycine residues) or 2.2–2.6 ms (for observing both glycine and non-glycine residues); ε = duration of GH + field recovery ~ 1.2 ms; 2TC = 1/(2JCαC’) ~ 9.5 ms; 2TCAN ~ 28 ms. Maximum t3 is restrained t3,max < 2.0*(TCAN–τ2). Frequency discrimination in 13C′ and 15N dimensions is obtained using the States-TPPI protocol (Marion et al. 1989) applied to ϕ1 and ϕ2, respectively, whereas the quadrature detection in 13Cα dimension is obtained using the sensitivity-enhanced gradient selection (Kay et al. 1992; Schleucher et al. 1994). The echo and antiecho signals in 13Cα dimension are collected separately by inverting the sign of the GC gradient pulse together with the inversion of ψ, respectively. Phase cycling: ϕ1 = x, − x; ϕ2 = 2(x), 2(− x); ϕ3 = 4(y), 4(− y); ϕ4 = y; ψ = x; rec. = x, 2(− x), x, − x, 2(x), − x. Gradient strengths (% of max G/cm) and durations (ms): G1 = 17%, 0.234 ms; G2 = 40%, 1.0 ms; G3 = 60%, 1.0 ms; G4 = 25%, 1.0 ms; G5 = 35.7%, 0.234 ms; G6 = 7.1%, 0.234 ms; GC = 80.0%, 1.0 ms; GH = 20.1% 1.0 ms. The pulse sequences’ code and parameter file for Bruker Avance system are available from authors upon request
The experiments start with the 1Hα(i) → 13Cα(i) transfer, and the density operator immediately after the ϕ4 pulse is described as (time point a)
| 3 |
Next, the magnetization is simultaneously transferred to both 15N(i) and 15N(i + 1) spins as well as to the 13C′(i) spin using 13Cα–15N and 13Cα–13C′ INEPT schemes, while refocusing the antiphase coherence with respect to the 1Hα spin. The composite pulse decoupling is then applied to remove the scalar interaction between 1Hα and 13Cα spins during the long transfer from the 13Cα to 15N spin. The density operator at time point b is
| 4 |
where = sin(2π1JCαC’TC)cosm(2π1JCαCβTCAN)sin(2π1JCαHατ2)cosn−1(2π1JCαHατ2), = sin(2π1JNCαTCAN)cos(2π2JNCαTCAN) and = cos(2π1JNCαTCAN)sin(2π2JNCαTCAN) (m equals 0 for glycines and 1 for other residues, n is the number of protons attached to the α-carbon i.e. 1 for non-glycine residues and 2 for glycines). This is followed by labeling of the 13C’ chemical shift in t1 and 15 N chemical shift in t2. Thus, the relevant density operator after the ϕ3 at the time point c is
| 5 |
During the ensuing delay 2*TCAN, the desired coherence is refocused with respect to 15N and 13C′ spins and antiphase coherence with respect to the 1Hα is generated during τ2. In the 4D HACANCOi experiment, this period is used for labeling of the chemical shift of 13Cα in a constant-time manner. Therefore, the relevant density operator prior to the coherence order selective coherence transfer (COS-CT) step is (time point d)
| 6 |
For the 3D HA(CA)NCO the back-transfer step (inset b′ to Fig. 1) deviates slightly as no chemical shift of 13Cα is labeled during t3. Finally, COS-CT with gradient echo (Kay et al. 1992; Schleucher et al. 1994) is used to transfer the magnetization back to 1Hα and the signal of interest corresponds to the density operator at the time point e
| 7 |
Hence, the 4D HACANCOi experiment shows intraresidual cross peaks at ωHα(i), ωCα(i), ωN(i), ωC’(i), and sequential correlations at ωHα(i), ωCα(i), ωN(i+1), ωC’(i), frequencies, respectively. Analogously the 3D HA(CA)NCOi experiment exhibits intraresidual correlations at ωHα(i), ωN(i), ωC’(i), and sequential correlations at ωHα(i), ωN(i+1), ωC’(i) coordinates. Of note, given the frequency labeling of the 13Cα chemical shift during t3 takes place in a constant-time manner and employs sensitivity enhanced gradient echo during t3, there is no dimensionality-associated sensitivity loss between the 3D and 4D versions of the experiment.
To minimize adjustments of sample conditions and maximize availability of structural restraints from one sample, that is, HN-based distance restraints and 15N spin relaxation rates for instance, we prefer measuring the 1Hα-detected experiments in 95%/5% H2O/D2O. This demands a relatively robust water suppression as Hα spins often resonate very close to the residual water signal. To meet these requirements, the water flip-back approach together with the sensitivity-enhanced heteronuclear gradient-echo implementation is employed. Therefore, the bulk water magnetization is spin-locked throughout most of the experiment and residual transversal water magnetization components are dephased by the pulsed field gradients. In our hands this approach works sufficiently as shown for the first increment of the HACANCOi experiment from 15N,13C EspF: unlabeled SNX9-SH3 complex (Supplementary Fig. 1). Water suppression can be further optimized by adjusting the length of the gradient pulse G3 (1–4 ms) between the t1 and t2 evolution periods. Nevertheless, if the protein concentration is low, < 0.3 mM, use of pure D2O as a solvent is worth considering.
Coherence transfer efficiency of HACANCOi
The coherence transfer efficiency for the intraresidual and interresidual correlations in the 4D HACANCOi scheme can be calculated according to Eqs. 8 and 9:
| 8 |
| 9 |
Assuming a transverse relaxation time of 100 ms for the 13Cα spin in an IDP, the coherence transfer efficiency I for the first increment is 0.23 (0.08) for the intraresidual (sequential) pathway. For small (T2,Cα = 50 ms) and medium (T2,Cα = 30 ms) sized proteins, having residues in an α-helical conformation, the transfer efficiency becomes 0.13 (0.04) and 0.06 (0.02), respectively. In these calculations, we set 1JCαC’ = 53 Hz, 1JC’N = 15 Hz and 1JCαCβ = 35 Hz. In addition, 1JNCα and 2JNCα values in random coil (α-helical) conformation are assumed to be 10.6 (9.6) Hz and 7.5 (6.4) Hz, respectively (Delaglio et al. 1991). We have neglected one-bond 1JCαHα coupling and 1Hα transverse relaxation times in these calculations since the pulse sequences compared here and those previously shown, have a similar implementation for 1H–13C transfer. In order to maximize the coherence transfer efficiency for the intraresidual pathway, the delay 2TCAN should be set to 28 ms. If we calculate the theoretical coherence transfer efficiencies and compare the performance of the proposed 4D HACANCOi experiment with the 4D iHACANCO experiment (Tossavainen et al. 2020), we observe that for the assignment of IDPs, the two experiments offer similar sensitivity, IHACANCOi = 0.228 and IiHACANCO = 0.214, as can be seen in Fig. 2. However, the intraresidual 4D iHACANCO scheme lifts the data redundancy problem by displaying solely the intraresidual cross peaks (Mäntylahti et al. 2010; Tossavainen et al. 2020). Remarkably, for the assignment of small and medium sized globular proteins, in which the mobility of the polypeptide chain, and hence the transverse relaxation time, is dominated by the overall rotational tumbling, the proposed 4D HACANCOi experiment clearly outperforms the 4D iHACANCO. This is exemplified in Fig. 2, which shows that the transfer efficiency of the new 4D HACANCOi experiment is almost two-fold higher than that of the 4D iHACANCO experiment in small globular proteins. The difference is even more pronounced in the case of medium sized globular proteins, approaching a three times higher throughput for the new experiment.
Fig. 2.

Comparison of theoretical coherence transfer efficiencies for the HACANCOi and the intraresidual iHACANCO experiments. Calculations were carried out for three proteins having diverging T2 relaxation properties. The bars on the left hand side represent the coherence transfer efficiency for a typical IDP, having T2 relaxation times of 100, 200 and 200 ms for 13Cα, 13C′ and 15N, respectively. These values are significantly shorter for small and (medium sized) globular protein, i.e. 50 (30), 100 (60) and 100 (60) ms. For calculations, we assumed 1JCαC′ = 53 Hz, 1JC′N = 15 Hz and 1JCαCβ = 35 Hz. For an IDP, random coil conformation with 1JNCα = 10.6 Hz and 2JNCα = 7.5 Hz values is presumed. For small and medium sized structural proteins, α-helical conformation is assumed with 9.6 Hz and 6.4 Hz scalar interactions for one- and two-bond couplings between 13Cα and 15N, respectively (Delaglio et al. 1991)
Comparison of attainable sensitivities between the HACANCOi and iHACANCO experiments
For experimental verification of the coherence transfer efficiency in the new 4D HACANCOi experiment, we next compared the attainable intensities for the intraresidual cross peaks between the two two-dimensional experiments, i.e. the 2D implementation of the proposed HA(CA)N(COi) and the 2D iHA(CA)N(CO) pulse schemes (Mäntylahti et al. 2010). As can be recognized from Fig. 3, showing the per residue signal to noise ratio for the small B1 domain of protein G (GB1, molecular weight 6.6 kDa) between the two experiments, the sensitivity of the HACANCOi experiment is superior with respect to iHACANCO in all measured temperatures. On average, the largest gain in sensitivity is observed for the largest molecule, simulated with lowering the GB1 sample temperature to 5 ºC. The correlation times of GB1 at 5, 15, 25, and 35 °C are 7.5, 5.4, 4.1 and 3.2 ns, which further correspond to molecular weights of 13, 9, 7 and 5 kDa, respectively. The sensitivity gain in loop regions is somewhat more pronounced.
Fig. 3.

Comparison of sensitivities between the proposed HACANCOi and the intraresidual iHACANCO experiments. a Bar graphs of per residue signal to noise ratios in the two experiments at four temperatures. The horizontal axis shows the amino acid sequence. On top of the panels is shown the secondary structure as present in the 25 °C NMR solution structure of GB1 (PDB ID 2GB1, Gronenborn et al. 1991), arrows representing β-strands and the wave an α-helix. Measurements for the S/N ratios were carried out at four different temperatures, 5, 15, 25, and 35 °C. The ratios were measured from two-dimensional 1Hα, 15N correlation spectra of iHA(CA)N(CO) and HA(CA)N(CO)i experiments. For glycine two 1Hαs, if not degenerate, the average of the ratios is shown. Due to overlap with the residual water signal or with an interresidual peak some of the bars are missing from the graph. The bar of E56 is not shown due to the 90º phase difference in the signal of the C-terminal residue, and thereby meaningless comparison (Mäntylahti et al. 2010). b Expansions of GB1 iHA(CA)N(CO) and HA(CA)N(CO)i spectra at 5 and 35 °C. Peak assignment is shown in the 5 °C HA(CA)N(CO)i spectrum for both the i and i + 1 peaks. The base level of iHA(CA)N(CO) and HA(CA)N(CO)i experiments at each temperature is identical i.e. the peak intensities are directly comparable between the two experiments
The sensitivity enhancement scheme with the gradient echo warrants high quality water suppression, but is likely to become inefficient for high molecular weight complexes beyond 20 kDa. To this end, the conventional back-INEPT transfer from 13Cα to 1Hα might be an advantageous implementation (See Supplementary Material, Fig. S2).
Application of 3D HA(CA)NCOi and 4D HACANCOi pulse schemes for the assignment of EPEC EspF–SNX9 SH3 complex
To test the applicability of the novel 3D HA(CA)NCOi and 4D HACANCOi experiments, we employed them for the sequential assignment of the enteropathogenic E. coli (EPEC) EspF–SNX9 SH3 complex. SH3 domains are ubiquitous structural modules that typically recognize proline-rich linear motifs, harboured in IDPs/IDRs (Saksela and Permi 2012). This motif in our target, the second 47-residue repeat of EspF, contains five prolines in a stretch of seven residues. Figure 4 displays strip plots of 3D HA(CA)NCOi and 3D HA(CA)CON (Mäntylahti et al. 2010) spectra of this stretch collected from a sample composed of 15N, 13C labeled EspF bound to unlabeled SNX9 SH3. It exemplifies the robustness of the Hα-detection strategy for establishing sequential connectivities through proline-rich regions Indeed, the “sequential walk” through multiple consecutive proline residues, located at the EspF–SH3 binding interface, was successful with spectra measured from a sample dissolved in 95%/5% H2O/D2O. This is remarkable considering that three of the prolines’ Hα shifts nearly coincide with that of the residual water signal. Use of 1Hα, 13C’ and 15N chemical shifts warranted good dispersion of signals and full backbone resonance assignment of bound EPEC EspF was obtained. All expected intra- and interresidual peaks were present in the HA(CA)NCOi and HA(CA)CON spectra, respectively.
Fig. 4.

Representative strip plots from the proposed 3D HA(CA)NCOi (left panel, red contours) and 3D HA(CA)CON (right panel, blue contours) experiments from the proline-rich region 75RPAPPPP81 of 15N, 13C labeled EspF in complex with unlabeled SNX9 SH3. In the HA(CA)NCOi experiment strong intraresidual ωHα(i)–ωC′(i)–ωN(i) correlations are observed together with weaker (or absent) sequential ωHα(i)–ωC′(i)–ωN(i+1) correlations
For the assignment of 1H, 13C, 15N backbone resonances of SNX9 SH3, we prepared a reversibly labeled complex, that is 15N, 13C labeled SNX9 SH3 was bound to unlabeled EPEC EspF. In order to link the 13Cα chemical shifts directly to 1Hα resonances and to obtain additional signal dispersion without compromising the overall sensitivity, we employed the 4D HACANCOi experiment. Figure 5 shows an excerpt of 4D HACANCOi strip plots in which the intraresidual 15 N(i) and interresidual 15 N(i + 1) chemical shifts are correlated with their corresponding 1Hα(i), 13C′(i) and 13Cα(i) chemical shifts. This provides highly dispersed resonance correlation maps, but also readily offers 1Hα, 13Cα, and 13C’ chemical shift data, which are the most useful spins for calculating transiently populated secondary structures in IDPs as they are particularly sensitive to the ϕ/ψ angles of the protein backbone (Borcherds and Daughdrill 2018). Except for two residues whose Hα resonances are very close to that of residual water, all expected intra peaks were present in the 4D spectrum.
Fig. 5.
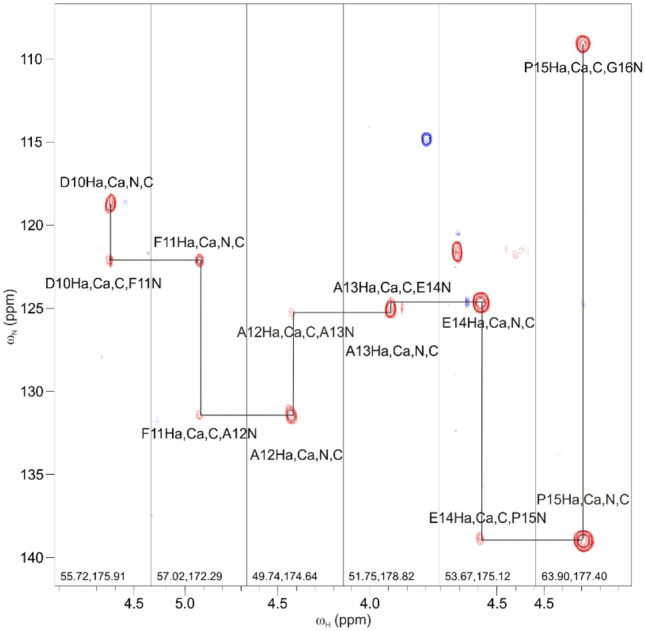
Strip plots of the proposed 4D HACANCOi experiment. Spectra display the “sequential walk” through amino acid residue stretch 10DFAAEP15 in 15N, 13C labeled SNX9 SH3 in complex with unlabeled EPEC EspF. This stretch forms a part of the EPEC EspF binding site on SNX9 SH3. Both intraresidual 1Hα(i), 13Cα(i), 13C′(i), 15N(i) and sequential 1Hα(i), 13Cα(i), 13C’(i), 15N(i + 1) correlations are observed, allowing sequential assignment of 1Hα, 13Cα, 13C′ and 15N backbone resonances. Although in the example given here, 4D HACANCOi displayed both intra- and interresidual correlations, usually a complementary 4D HACACON experiment (Tossavainen et al. 2020) is needed to confirm interresidual connectivities
Assignment of disordered proteins confronts the well-established HN-detected triple-resonance approaches due to clustering of 13Cα/13Cβ chemical shifts by residue type, susceptibility of HN resonances to chemical exchange broadening at alkali or neutral pH, and also high abundance of prolines in IDPs (Mäntylahti et al. 2010). To circumvent problems associated with the HN-detection on IDPs, the 13C-detection strategy has been employed successfully. It does not suffer from the exchange broadening of signals and yet allows assignment of prolines. Like HN-detected experiments they often apply the ‘CON’ strategy for connecting neighboring residues with highly degenerate chemical shifts (Pantoja-Uceda and Santoro 2014; Sahu et al. 2014; Brutscher et al. 2015; Chaves-Arquero et al. 2018). Another salient feature of the 13C-detected strategy is the absence of the disturbing residual water signal, i.e. no special care needs to be undertaken for water handling in these experiments. Further improvements in resolution and sensitivity stem from the fact that two- and three-bond 13C–13C couplings are much smaller in size than the corresponding 1H–1H couplings. The caveat of this strategy is the inherently lower sensitivity of 13C-detection, by a factor of 8, with respect to 1H-detection. This can be partially compensated (by a factor of ca. 2) by using a triple-resonance probehead with 13C as the inner coil. We have proposed using 1Hα-detection as an alternative to the NH- or 13C-detection strategies (Mäntylahti et al. 2010, 2011). Indeed, 1Hα-detection offers the best of both worlds, higher sensitivity of 1H-detection in general and the possibility to work on IDPs under alkali conditions and/or abundant proline content. Thus far our assignment strategy has followed solely unidirectional coherence transfer routes. That is, active purging of redundant information between spectra using so-called intraresidual (Brutscher 2002; Permi 2002) or sequential coherence transfer was employed in the 3D and 4D iHACANCO, HACACON and (HACA)CONCAHA experiments (Mäntylahti et al. 2010, 2011; Tossavainen et al. 2020). In this paper, we have introduced the novel HACANCOi experiment that offers superior sensitivity for the assignment of IDPs which establish a slowly dissociating complex with their binding partner thus rendering their relaxation properties less favorable towards more selective pulse sequences. The new experiment utilizes a superior 15N–13C′ correlation map to maximize chemical shift dispersion in challenging IDP/IDRs (Yao et al. 1997; Mäntylahti et al. 2009; Bermel et al. 2012), although both i and i + 1 correlations are visible in the 15N dimension. The coherence transfer efficiency is clearly superior to the corresponding intraresidual only iHACANCO experiment (Mäntylahti et al. 2010; Tossavainen et al. 2020) even for the smallest globular protein or complex. Recently, Wong et al. proposed a suite of Hα− detected experiments, including e.g., haCONHA and haNCOHA that yield Hα(i), C′(i), N(i + 1), and Hα(i + 1), C′(i), N(i + 1) (as well as Hα(i), C′(i), N(i + 1)) correlations, respectively (Wong et al. 2020). They found haNCONHA to be on average 3.5 times more sensitive than the 13C-detected counterpart, haCON. Our proposed HACANCOi experiment, based on the calculation shown above, has 1.9 and 2.8 times higher theoretical S/N for small and medium sized proteins/complexes, respectively, compared to that of haNCOHA. This stems from the fact that in HACANCOi, magnetization is transferred simultaneously from the 13Cα(i) spin to 13C′(i) and to 15N(i)/15N(i + 1) spins during the delay 2TCAN. The experiment is then conceptually similar to the HN-detected HNCO,CA TROSY experiment proposed by Konrat et al. (1999) for the assignment of high-molecular weight proteins.
In case of high molecular weight complexes beyond 20 kDa, the sensitivity-enhanced Rance-Kay scheme may become inefficient in comparison to the conventional back-INEPT transfer. To this end, the version of the pulse sequence without the sensitivity-enhanced gradient selection shown in the Supplementary material should be employed (Fig. S2). In such a case, it may be necessary to dissolve the sample in pure D2O instead of H2O/D2O for sufficient water suppression. Distinguishing glycine residues from other residues is not as straightforward as in the iHACANCO experiment since contains a cosm(2π2JCαCβTCAN) term, where 2TCAN is set to 28 ms. Therefore, separation of glycines based on their 180° difference in sign is not possible with the proposed experiment due to dependence. However, extending the first τ2 delay to 4.4 ms yields a 180° phase inversion for glycines, if deemed necessary (Mäntylahti et al. 2009).
It is noteworthy that the proposed HACANCOi experiment as well as the iHACANCO and HACACON (Mäntylahti et al. 2010; Tossavainen et al. 2020) schemes are optimal for proline assignment from the coherence transfer efficiency point of view. The magnetization is transferred direcly from the 13Cα(i) spin to the 15N(i) spin of proline. Indeed, being an N-substituted residue, the 15N spin of proline exhibits 1JNCα and 1JNCδ couplings of similar size and hence the straight-through 1H → 13C → 15 N → 13C → 1H type of experiments, such as (HACA)CONCAH (Mäntylahti et al. 2011; Yao et al. 2014; Tossavainen et al. 2020) are less sensitive than the out-and-back (1H ← → 13C ← → 15N) type experiments shown here.
Conclusions
We have introduced a new NMR experiment, HACANCOi, using 1Hα-detection for the assignment of backbone 1Hαi, 13Cαi, 15Ni and 13C′i resonances in 15N, 13C labeled proteins. In comparison to the established intraresidual iHACANCO experiment (Mäntylahti et al. 2010; Tossavainen et al. 2020), the novel experiment yields two cross peaks per residue and hence suboptimal resolution, but offers superior sensitivity which may become a limiting factor in the assignment of IDPs forming a complex with a more slowly tumbling structural protein. As we have demonstrated here, the new experiment thrives in molecular systems established by a disordered region bound to a globular domain in which the attainable transverse relaxation rates resemble those of structural proteins rather than IDPs with highly elevated dynamics. The new experiment further extends the possibilities of employing the 1Hα-detection approach to systems enriched with proline residues and/or studied under alkali conditions where detection of amide protons becomes difficult.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work is supported by the grant from the Academy of Finland (Grant Number 288235 to PP).
Funding
Open access funding provided by University of Jyväskylä (JYU).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aitio O, Hellman M, Kazlauskas A, Vingadassalom DF, Leong JM, Saksela K, Permi P. Recognition of tandem PxxP motifs as a unique Src homology 3-binding mode triggers pathogen-driven actin assembly. Proc Natl Acad Sci. 2010;107:21743–21748. doi: 10.1073/pnas.1010243107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitio O, Hellman M, Skehan B, Kesti T, Leong JM, Saksela K, Permi P. Enterohaemorrhagic Escherichia coli exploits a tryptophan switch to hijack host f-actin assembly. Structure. 2012;20:1692–1703. doi: 10.1016/j.str.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastidas M, Gibbs EB, Sahu D, Showalter SA. A primer for carbon-detected NMR applications to intrinsically disordered proteins in solution. Concepts Magn Reson. 2015;44:54–66. doi: 10.1002/cmr.a.21327. [DOI] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R. 13C-detected protonless NMR spectroscopy of proteins in solution. Prog Nucl Magn Reson Spectrosc. 2006;48:25–45. doi: 10.1016/j.pnmrs.2005.09.002. [DOI] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Kümmerle R, Pierrattelli R. Novel 13C direct detection experiments, including extension to the third dimension, to perform the complete assignment of proteins. J Magn Reson. 2006;178:56–64. doi: 10.1016/j.jmr.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Pierattelli R. Speeding up (13)C direct detection biomolecular NMR spectroscopy. J Am Chem Soc. 2009;131:15339–15345. doi: 10.1021/ja9058525. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Gonnelli L, Koźmiński W, Piai A, Pierattelli R, Stanek J. Speeding up sequence specific assignment of IDPs. J Biomol NMR. 2012;53:293–301. doi: 10.1007/s10858-012-9639-0. [DOI] [PubMed] [Google Scholar]
- Böhlen J-M, Bodenhausen G. Experimental aspects of chirp NMR spectroscopy. J Magn Reson Ser A. 1993;102:293–301. doi: 10.1006/jmra.1993.1107. [DOI] [Google Scholar]
- Borcherds WM, Daughdrill GW. Using NMR chemical shifts to determine residue-specific secondary structure populations for intrinsically disordered proteins. Meth Enzymol. 2018;611:101–136. doi: 10.1016/bs.mie.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutscher B. Intraresidue HNCA and COHNCA experiments for protein backbone resonance assignment. J Magn Reson. 2002;156:155–159. doi: 10.1006/jmre.2002.2546. [DOI] [PubMed] [Google Scholar]
- Brutscher B, Felli IC, Gil-Caballero S, Hosek T, Kummerle R, Piai A, Pietarelli R, Solyom Z. NMR methods for the study of intrinsically disordered proteins structure, dynamics, and interactions: general overview and practical guidelines. Adv Exp Med Biol. 2015;870:49–122. doi: 10.1007/978-3-319-20164-1_3. [DOI] [PubMed] [Google Scholar]
- Chaves-Arquero B, Pantoja-Uceda D, Roque A, Ponte J, Suau P, Jimenez MA. A CON-based NMR assignment strategy for pro-rich intrinsically disordered proteins with low signal dispersion: the C-terminal domain of histone H1.0 as a case study. J Biomol NMR. 2018;72:139–148. doi: 10.1007/s10858-018-0213-2. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Torchia DA, Bax A. Measurement of 15N–13C J couplings in staphylococcal nuclease. J Biomol NMR. 1991;1:439–446. doi: 10.1007/bf02192865. [DOI] [PubMed] [Google Scholar]
- Emsley L, Bodenhausen G. Optimization of shaped selective pulses for NMR using a quaternion description of their overall propagators. J Magn Reson. 1992;97:135–148. doi: 10.1016/0022-2364(92)90242-Y. [DOI] [Google Scholar]
- Fiorito F, Hiller S, Wider G, Wüthrich K. Automated resonance assignment of proteins: 6D APSY-NMR. J Biomol NMR. 2006;35:27–37. doi: 10.1007/s10858-006-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, Clore GM. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1999;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- Hellman M, Piirainen H, Jaakola V, Permi P. Bridge over troubled proline: assignment of intrinsically disordered proteins using (HCA)CON(CAN)H and (HCA)N(CA)CO(N)H experiments concomitantly with HNCO and i(HCA)CO(CA)NH. J Biomol NMR. 2014;58:49–60. doi: 10.1007/s10858-013-9804-0. [DOI] [PubMed] [Google Scholar]
- Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. doi: 10.1021/ja00052a088. [DOI] [Google Scholar]
- Kazimierczuk K, Stanek J, Zawadzka-Kazimierczuk A, Koźmiński W. High-dimensional NMR spectra for structural studies of biomolecules. ChemPhysChem. 2013;14:3015–3025. doi: 10.1002/cphc.201300277. [DOI] [PubMed] [Google Scholar]
- Konrat R, Yang D, Kay LE. A 4D TROSY-based pulse scheme for correlating 1HNi, 15Ni, 13Cαi, 13C’i-1 chemical shifts in high molecular weight, 15N, 13C, 2H labeled proteins. J Biomol NMR. 1999;15:309–313. doi: 10.1023/A:1008310617047. [DOI] [PubMed] [Google Scholar]
- Liu X, Yang D. HN(CA)N and HN(COCA)N experiments for assignment of large disordered proteins. J Biomol NMR. 2003;57:83–89. doi: 10.1007/s10858-013-9783-1. [DOI] [PubMed] [Google Scholar]
- Mäntylahti S, Tossavainen H, Hellman M, Permi P. An intraresidual i(HCA)CO(CA)NH experiment for the assignment of main-chain resonances in 15N, 13C labeled proteins. J Biomol NMR. 2009;45:301–310. doi: 10.1007/s10858-009-9373-4. [DOI] [PubMed] [Google Scholar]
- Mäntylahti S, Aitio O, Hellman M, Permi P. HA-detected experiments for the backbone assignment of intrinsically disordered proteins. J Biomol NMR. 2010;47:171–181. doi: 10.1007/s10858-010-9421-0. [DOI] [PubMed] [Google Scholar]
- Mäntylahti S, Hellman M, Permi P. Extension of the HA-detection based approach: (HCA)CON(CA)H and (HCA)NCO(CA)H experiments for the main-chain assignment of intrinsically disordered proteins. J Biomol NMR. 2011;49:99–109. doi: 10.1007/s10858-011-9470-z. [DOI] [PubMed] [Google Scholar]
- Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR-spectra without phase cycling – application to the study of hydrogen-exchange in proteins. J Magn Reson. 1989;85:393–399. doi: 10.1016/0022-2364(89)90152-2. [DOI] [Google Scholar]
- Motáčková V, Nováček J, Zawadzka-Kazimierczuk A, Kazimierczuk K, Zídek L, Sanderová H, Krásný L, Koźmiński W, Sklenář V. Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J Biomol NMR. 2010;48:169–177. doi: 10.1007/s10858-010-9447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nováček J, Zawadzka-Kazimierczuk A, Papoušková V, Zídek L, Sanderová H, Krásný L, Koźmiński W, Sklenář V. 5D 13C-detected experiments for backbone assignment of unstructured proteins with a very low signal dispersion. J Biomol NMR. 2011;50:1–11. doi: 10.1007/s10858-011-9496-2. [DOI] [PubMed] [Google Scholar]
- Pantoja-Uceda D, Santoro J. New 13C-detected experiments for the assignment of intrinsically disordered proteins. J Biomol NMR. 2014;59:43–50. doi: 10.1007/s10858-014-9827-1. [DOI] [PubMed] [Google Scholar]
- Permi P. Intraresidual HNCA: An experiment for correlating only intraresidual backbone resonances. J Biomol NMR. 2002;23:201–209. doi: 10.1023/A:1019819514298. [DOI] [PubMed] [Google Scholar]
- Permi P, Annila A. Coherence transfer in proteins. Prog Nucl Magn Reson Spectrosc. 2004;44:97–137. doi: 10.1016/j.pnmrs.2003.12.001. [DOI] [Google Scholar]
- Permi P, Hellman M. Alpha proton detection based backbone assignment of intrinsically disordered proteins. Methods Mol Biol. 2012;895:211–226. doi: 10.1007/978-1-61779-927-3_15. [DOI] [PubMed] [Google Scholar]
- Sahu D, Bastidas M, Showalter S. Generating NMR chemical shift assignments of intrinsically disordered proteins using carbon-detect NMR methods. Anal Biochem. 2014;449:17–25. doi: 10.1016/j.ab.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, Permi P. SH3 domain ligand binding: what’s the consensus and where’s the specificity? FEBS Lett. 2012;586:2609–2614. doi: 10.1016/j.febslet.2012.04.042. [DOI] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc. 1999;34:93–202. doi: 10.1016/S0079-6565(98)00025-9. [DOI] [Google Scholar]
- Schleucher J, Schwendinger M, Sattler M, Schmidt P, Schedletzky O, Glaser SJ, Sørensen OW, Griesinger C. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J Biomol NMR. 1994;4:301–306. doi: 10.1007/BF00175254. [DOI] [PubMed] [Google Scholar]
- Shaka AJ, Keeler J. Broadband spin decoupling in isotropic liquids. Prog Nucl Magn Reson Spectrosc. 1987;19:47–129. doi: 10.1016/0079-6565(87)80008-0. [DOI] [Google Scholar]
- Shaka AJ, Barker PB, Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson. 1985;64:547–552. doi: 10.1016/0022-2364(85)90122-2. [DOI] [Google Scholar]
- Solyom Z, Schwarten M, Geist L, Konrat R, Willbold D, Brutscher B. BEST-TROSY experiments for time-efficient sequential resonance assignment of large disordered proteins. J Biomol NMR. 2013;55:311–321. doi: 10.1007/s10858-013-9715-0. [DOI] [PubMed] [Google Scholar]
- Thapa CJ, Haataja T, Pentikäinen U, Permi P. 1H, 13C and 15N NMR chemical shift assignments of cAMP-regulated phosphoprotein-19 and -16 (ARPP-19 and ARPP-16) Biomol NMR Assign. 2020 doi: 10.1007/s12104-020-09951-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tossavainen H, Hellman M, Vainonen JP, Kangasjärvi J, Permi P. 1H, 13C and 15N NMR chemical shift assignments of A.thaliana RCD1 RST. Biomol NMR Assign. 2017;11:207–210. doi: 10.1007/s12104-017-9749-4. [DOI] [PubMed] [Google Scholar]
- Tossavainen H, Salovaara S, Hellman M, Ihalin R, Permi P. Dispersion from Cα and NH: 4D experiments for backbone resonance assignment of intrinsically disordered proteins. J Biomol NMR. 2020;74:147–159. doi: 10.1007/s10858-020-00299-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wong LE, Kim TH, Muhandiram DR, Forman-Kay JD, Kay LE. NMR experiments for studies of dilute and condensed protein phases: application to the phase-separating protein CAPRIN1. J Am Chem Soc. 2020;142:2471–2489. doi: 10.1021/jacs.9b12208. [DOI] [PubMed] [Google Scholar]
- Yao J, Dyson HJ, Wright PE. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997;419:285–289. doi: 10.1016/s0014-5793(97)01474-9. [DOI] [PubMed] [Google Scholar]
- Yao X, Becker S, Zweckstetter M. A six-dimensional alpha proton detection-based APSY experiment for backbone assignment of intrinsically disordered proteins. J Biomol NMR. 2014;60:231–240. doi: 10.1007/s10858-014-9872-9. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Kulminskaya N, Mulder FAA. Easy and unambigous sequential assignments of intrinsically disordered proteins by correlating the backbone 15N and 13C’ chemical shifts of multiple contiguous residues in highly resolved 3D spectra. J Biomol NMR. 2015;61:109–121. doi: 10.1007/s10858-014-9890-7. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Kümmerle R, Qiu X, Redwine D, Cong R, Taha A, Baugh D, Winniford B. A new decoupling method for accurate quantification of polyethylene copolymer composition and triad sequence distribution with 13C NMR. J Magn Reson. 2007;187:225–233. doi: 10.1016/j.jmr.2007.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.