

Abstract
A series of substituted azothiophenes was prepared and investigated toward their isomerization behavior. Compared to azobenzene (AB), the presented compounds showed red‐shifted absorption and almost quantitative photoisomerization to their (Z) states. Furthermore, it was found that electron‐withdrawing substitution on the phenyl moiety increases, while electron‐donating substitution decreases the thermal half‐lives of the (Z)‐isomers due to higher or lower stabilization by a lone pair–π interaction. Additionally, computational analysis of the isomerization revealed that a pure singlet state transition state is unlikely in azothiophenes. A pathway via intersystem crossing to a triplet energy surface of lower energy than the singlet surface provided a better fit with experimental data of the (Z)→(E) isomerization. The insights gained in this study provide the necessary guidelines to design effective thiophenylazo‐photoswitches for applications in photopharmacology, material sciences, or solar energy harvesting applications.
Keywords: azothiophenes, heteroarylazobenzenes, photoswitches, weak noncovalent interactions
Switch it up! A series of substituted thiophenylazobenzenes was synthesized. The thermal (Z)→(E) isomerization rates can be tuned by attractive lone pair⋅⋅⋅π interactions, which stabilize the (Z) isomer: Lowering the electron density at the phenyl or increasing the electron density at the thiophenyl moieties increases the half‐lives up to two orders of magnitude. Computations support involvement of a triplet TS in the thermal (E)→(Z) isomerization process.
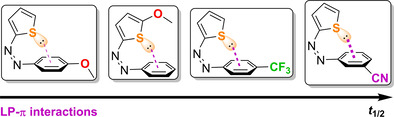
Introduction
The reversible photoisomerization properties of azobenzene (AB) have been investigated and applied in a vast number of studies since its discovery in 1937. [1] They have been used in various applications such as catalysis, [2] photobiology and photopharmacology, [3] information storage, [4] or solar energy storage systems. [5] The stable (E) isomer, featuring a strong π,π* absorption at 300–350 nm and a weak n,π* around 450 nm, depending on functional group substitution, can be photochemically converted with UV light (≈330 nm) to the metastable (Z) state in ≈80 %. (Z)‐AB on the other hand shows a strongly decreased π,π* absorption in the 350 nm range and a slightly increased absorption around 450 nm. Upon irradiation with visible light (≈450 nm) or thermal energy input, (Z)‐AB can be converted back to the (E) form. [6] The thermal half‐live of (Z)‐AB in solution is ≈2 days at room temperature and can be strongly altered by substitution on the phenyl rings. Introducing ortho‐fluorine groups for instance prolongs t 1/2 up to years, [7] while electronic push‐pull systems decrease the half‐lives to the sub‐second timescale. [8] Other parameters controlling the switching are the incorporation of AB in macrocycles. [9] Also subtle influences such as London dispersion and solvation have an effect.[ 10 , 11 ]
One goal in AB research is to achieve quantitative photoisomerization in both directions, red‐shifted and ideally separated absorption for (E) and (Z) states in the visible region for in vivo applications or solar energy harvesting, while maximizing the (Z) isomer half‐life to create an efficient two‐state photoswitch. In recent years, heteroarylazobenzenes (HetABs) have moved into the focus of interest, because they show highly interesting properties that even outperform those of the successfully and intensely studied ABs. [12] For example, HetABs with pyridine, [13] pyrazole, [14] tetrazole, [15] imidazole,[ 16 , 17 ] indole, [18] or thiazole [19] heterocycles allowed shifting of the π,π* bands up to 560 nm and showed almost quantitative switching to each photostationary state (PSS). Additionally, the thermal half‐lives of the (Z) isomers showed a broad variety, ranging from picoseconds [13] to multiple years. [20]
Following this approach, Fuchter and co‐workers presented a series of N‐heterocyclic ABs with different properties, controlled by systematic heteroaryl design (Figure 1).[ 15 , 20 ] In their studies, they established N‐methylpyrazole‐AB 1, which shows quantitative photoswitching in both directions, as well as very high thermal stability of the corresponding (Z) isomer with a half‐life of 1000 days. This long half‐live was rationalized by favourable CH⋅⋅⋅π interactions in the (Z) isomer, leading to an unprecedented T‐shaped geometry in azopyrrole 2. If the corresponding H is substituted by a methyl group, the (Z)‐T isomer cannot be formed, and a lower half‐live is observed in azopyrrole 3.[ 15 , 20 ] In a recent study, the groups of Wachtveitl, Dreuw and us reported 2‐thiophenyl‐AB 4 a (Figure 1). [21] The compound was found to also feature almost quantitative (E)→(Z) photoisomerization, excellent spectral separation of the (E) and (Z) isomers and extremely high quantum yields for both photoisomerization directions. Also, a T‐shaped geometry of the Z‐isomer was found to be the lowest energy conformation in this case, due to favourable lone pair (LP)⋅⋅⋅π interaction. Interestingly, 4 a showed a comparable half‐life of 2 h to azoyprrole 2 at room temperature.
Figure 1.
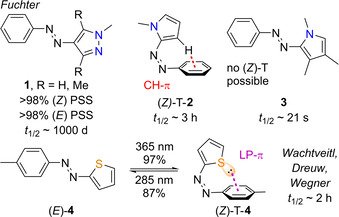
HetABs by Fuchter[ 15 , 20 ] (1–3) and Wegner, Dreuw, Wachtveitl (4). [21] Azopyrrole 2 as well as thiophenylazo 4 a form T‐shaped Z isomers, that are stabilized by CH⋅⋅⋅π and lone pair⋅⋅⋅π interactions, respectively. In contrast, azopyrrole 3 cannot adopt a T‐shaped (Z) isomer due to methyl substitution.
With these fundamental insights, the discovered LP⋅⋅⋅π interactions could now be used to tune the thermal isomerization behaviour of the azothiophenes. If the π‐electron density of the phenyl moiety is lowered, a stronger electron attraction of the sulfur LP, and thus a more stable (Z) isomer, should be the consequence. In opposite, if the π‐electron density is increased, less favourable electron interaction with the thiophenyl moiety should be the result. If this initial hypothesis is true, a higher stability of the unsubstituted (Z)‐T‐azothiophene isomer compared to the previously reported p‐methylphenyl compound 4 a is expected.
Herein, we present a fundamental study on substituted arylazothiophenes 4 with electron‐donating and withdrawing substituents on the phenyl, as well as on the thiophenyl moieties. Their synthesis, spectral properties and photoisomerization, as well as thermal (Z)→(E) isomerization kinetics have been studied. Furthermore, deeper insight into the thermal isomerization mechanism was obtained by high‐level computational analysis, which provides a solid basis for the design of novel thiophenyl‐AB photoswitches.
Results and Discussion
Synthesis
Initially, 4‐methylphenylazothiophene 4 a was successfully synthesized by lithiation of thiophene using nBuLi, followed by the addition to the corresponding diazonium tetrafluoroborate at low temperature.[ 17 , 21 ] However, these conditions failed for the preparation of the unsubstituted azothiophene (AT) 4 b. Therefore, a rather mild dithiophenylzinc reagent was prepared after treating 2‐bromothiophene (5 a) with Knochel's “turbo” Grignard reagent, [22] which could be coupled successfully with various substituted benzenediazonium tetrafluoroborates (6) to obtain substituted thiophenylazobenzenes 4 in moderate to good yields (Table 1).
Table 1.
Synthesis of thiophenylazobenzenes 4 b–l using dithiophenylzinc reagents and aryldiazonium tetrafluoroborates (6) as building blocks.
|
| ||||
|---|---|---|---|---|
|
Entry |
R1 |
R2 |
Product |
Isolated yield [%] |
|
1 |
H |
H |
4 b |
59 |
|
2 |
H |
4‐OMe |
4 c |
42 |
|
3 |
H |
4‐CN |
4 d |
70 |
|
4 |
H |
4‐CF3 |
4 e |
61 |
|
5 |
H |
3‐OMe |
4 f |
20[a] |
|
6 |
H |
3‐CN |
4 g |
60 |
|
7 |
OMe |
H |
4 h |
24 |
|
8 |
OMe |
4‐CN |
4 i |
48 |
|
9 |
Me |
4‐CN |
4 j |
70 |
|
10 |
Me |
4‐CF3 |
4 k |
54 |
|
11 |
CN |
H |
4 l |
0.3[b] |
[a] The 3‐methoxybenzenediazonium salt 6 f was added neat in small portions to the thiophenylzinc reagent at −60 °C due to its instability in solution. [b] The Grignard reagent was directly added to the diazonium suspension at −80 °C.
However, 5‐cyanothiophenylazobenzene (4 l) could not be obtained via this protocol. The corresponding dithiophenylzinc reagent did not react with the diazonium salt. At elevated temperatures only polymerization was observed. Hence, the cyanothiophenyl‐Grignard reagent was directly added to the diazonium suspension at low temperature to access the desired azobenzene 4 l in trace amounts (Table 1, entry 11). Again, mainly polymer formation was observed. Nevertheless, enough material for characterization and all following investigations could be acquired.
Spectroscopic properties
After the successful syntheses of thiophenylazobenzenes 4 b–l, the compounds were investigated using UV/Vis spectroscopy. All derivatives showed similar absorption spectra to the previously reported 4‐methylphenylazothiophene (4 a). [21] Compared to azobenzene, the arylazothiophenes exhibited significantly red‐shifted π,π* transitions, up to 414 nm in the case of 4 i (see Supporting Information). Furthermore, the n,π* transitions were found to overlap with the π,π* bands, resulting in small shoulders of the π,π* bands offsets. In general, the stronger the electronic push‐pull character of the system like 4 i, the more red‐shift was observed for the corresponding compounds.
Upon irradiation into the π,π* bands with light of corresponding wavelengths (Supporting Information), significant changes in the spectra were observed, indicating (E)→(Z) photoisomerization. In all compounds, except of 4 h,i, the π,π* intensity could be decreased to almost zero. The spectra of the resulting (Z) isomer showed new maxima in the 300 nm region separated from the initial (E) state. When irradiating the (Z) enriched states at 305 nm, mixtures of (E) and (Z) isomers were obtained. This behaviour can be rationalized by the absorption maximum of the (Z) isomers, which are located significantly below 305 nm. In this region the (E) and (Z) spectra of the compounds overlap. Thus, efficient (Z)→(E) photoisomerization was not possible with the available LEDs in our laboratory. In the case of 5‐cyanothiophenylazo 4 l, the back‐switching did not lead to a clear isosbestic point in the spectra. Despite the fact that repeated photoisomerization for 10 cycles showed only minor signs of decomposition, a reversible photoisomerization of 4 l cannot be guaranteed (Figure S3 e in Supporting Information). The spectroscopic and photoisomerization results are summarized in Table 2.
Table 2.
Irradiation wavelengths of the (E) isomers and photostationary state compositions for azothiophenes 4 a–l. For (Z)→(E) isomerization, all samples were irradiated at 305 nm.
|
Entry |
Compound |
(E)→(Z) irradiation wavelength [nm] |
(E)→(Z) PSS composition[a] [%] |
(Z)→(E) PSS composition[a] [%] |
||
|---|---|---|---|---|---|---|
|
|
|
|
(E) |
(Z) |
(E) |
(Z) |
|
1 |
4 b |
365 |
1 |
99 |
51 |
49 |
|
2 |
4 c |
385 |
6 |
94 |
64 |
36 |
|
3 |
4 d |
365 |
3 |
97 |
63 |
37 |
|
4 |
4 e |
365 |
2 |
98 |
59 |
41 |
|
5 |
4 f |
365 |
3 |
97 |
51 |
59 |
|
6 |
4 g |
365 |
<1 |
>99 |
57 |
43 |
|
7 |
4 h[ b] |
405 |
– |
– |
– |
– |
|
8 |
4 i [b] |
405 |
– |
– |
– |
– |
|
9 |
4 j |
385 |
5 |
95 |
59 |
41 |
|
10 |
4 k |
365 |
4 |
96 |
50 |
50 |
[a] Determined by HPLC at the corresponding isosbestic points of the spectra. [b] (Z) isomers could not be determined by HPLC analysis; the OMe‐substitution increases the overall basicity of these compounds facilitating the protonation by residual silanol groups of the column material leading to back‐switching.
Thermal isomerization kinetics
The thermal (Z)→(E) isomerization rates of all arylazothiophenes were determined by UV/Vis spectroscopy. Similar to London dispersion‐stabilized (Z)‐all‐meta‐alkyl ABs, [10] the LP⋅⋅⋅π interactions only affect the Z isomers due to spatial separation of the moieties in the transition state (TS) and the (E) isomer. Thus, higher or lower activation energies for the thermal isomerizations are the result, which provide indications for the stabilization energies due to LP⋅⋅⋅π interactions.
The measured half‐lives at 20 °C are shown in Figure 2. The compounds were grouped in para‐phenyl‐substituted (red bars), meta‐phenyl (green), thiophene substituted (blue) and push‐pull (purple) substituted derivatives. While the parent, unsubstituted compound 4 b (grey bar) showed a t 1/2=7.06 h at 20 °C, the electron‐rich, para‐substituted derivatives 4 c and 4 a had shorter half‐lives, explainable by lower stabilization of the (Z)‐T isomers by less attractive LP⋅⋅⋅π interaction. Their aromatic π systems already possess high electron density, that diminishes attraction between the sulfur LP and the phenyl ring. In accordance to that, higher half‐lives for the electron‐deficient azothiophenes 4 d,e were observed due to the favourable stabilization of the (Z)‐T isomer. Additionally, the para‐substitution in azothiophenes 4 a,c,d could have a stabilizing effect on the TS, which relates to their generally lower half‐lives compared to the unsubstituted analogue 4 b.
Figure 2.
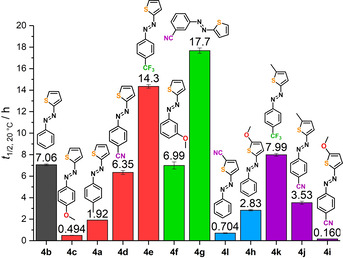
Half‐lives of azothiophenes 4 a–l at 20 °C in acetonitrile. Red bars: p‐phenyl substituted, green: m‐phenyl substituted, blue: thiophene substituted, purple: push‐pull‐systems. t 1/2 of 4 a was taken from ref. [21].
Analysis of the meta‐substituted derivatives, 3‐methoxy‐phenylazothiophene 4 f showed an almost equal half‐life compared to the parent compound 4 b, although bearing an electron‐donating methoxy group. However, the +M effect of the methoxy groups does not directly affect the N=N bond because of its meta position, and therefore the electronic nature of the transition state seems not to be influenced and thus leads to a similar half‐life to 4 b. Furthermore, the increased electron density in the phenyl moiety seems to destabilize the (Z)‐T isomer only slightly. Analysing compound 4 g, the influence of an electron‐withdrawing cyano‐group in meta position becomes dominant, resulting in the highest half‐live observed in this study for azothiophene 4 g.
Considering the half‐lives of the thiophene‐substituted derivatives 4 h,l (Figure 2, blue bars), the general trend of increasing the electron density at the donor, for example, the sulfur atom, rises the thermal isomerization half‐lives from electron poor 5‐cyanothiophenyl‐ (4 l, ≈0.7 h) to electron rich 5‐methoxythiophenyl‐azobenzene (4 h, ≈2.8 h). Again, substitution of the thiophene moiety in position 5 could stabilize the TS and thus lead to shorter half‐lives compared to the parent azothiophene 4 b. Interestingly, when the thiophene is substituted with electron donating and the phenyl moiety has an electron withdrawing group (4 i,j,k), the half‐life does not increase, but rather decreases. The push‐pull substitution pattern, which is known to lead to short half‐lives, [2] seems to outweigh the stabilizing LP⋅⋅⋅π interactions.
In order to get deeper insight into the thermal isomerization mechanism, the reaction rates were measured at four different temperatures in acetonitrile. An Exner‐plot [23] (Figure S6) indicated that all investigated azothiophenes 4 b–l followed the same thermal isomerization mechanism at the experimental temperature range (10–25 °C).
Computations
After experimental determination of the kinetic parameters, computational studies on the thermal isomerization energy pathway were performed on the DLPNO‐CCSD(T)/def2‐TZVP//PBE0‐D3(BJ)/def2‐TZVP level of theory, using the implicit SMD model for acetonitrile. [24] Two different conformations of the (E) and (Z) isomers are possible. [16] As shown for the unsubstituted azothiophene (4 b), a NNCS‐cis and a NNCS‐trans configuration exists for the (E) isomer (Figure 3, top), where the (E)‐cis isomer was found to be more stable by 2.7 kcal mol−1. A rotational scan revealed a barrier of ≈6 kcal mol−1, which ensures a very fast equilibration at room temperature, making the (E)‐cis the predominant form in ≈99 % according to the Boltzmann distribution.
Figure 3.
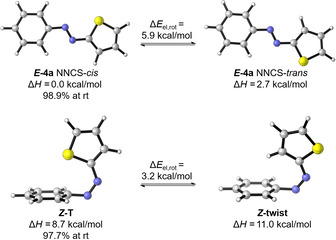
Electronic ground‐state isomers of azothiophene 4 b. Energies were computed on a DLPNO‐CCSD(T)/def2‐TZVP//PBE0‐D3(BJ)/def2‐TZVP level of theory.
Considering all possible (Z) isomers, the T‐shaped isomer was found to be 2.3 kcal mol−1 more stable than the twisted conformation (Figure 3, bottom). Again, a rotational scan revealed a barrier of 3.2 kcal mol−1 from (Z)‐twist to (Z)‐T, resulting in a very fast dynamic equilibrium in favour of 97.7 % of the (Z)‐T isomer at room temperature. Based on this conformational analysis, the (Z)‐T→(E)‐cis pathway should be the lowest in energy and therefore determine the kinetics of the thermal isomerization in azothiophenes.
Next, TS computations on the ground state revealed that four different TSs exist for the thermal (Z)→(E) interconversion (see Supporting Information). Intrinsic Reaction Coordinate (IRC) computations revealed that each (Z) isomer can interconvert via one specific TS to either the (E)‐cis or (E)‐trans isomer. As expected, the lowest‐lying TS was found to be the (Z)‐T→(E)‐cis pathway, proceeding in a mixed inversion‐rotation mechanism. However, a ΔG ≠=31.6 kcal mol−1 was calculated for the thermal isomerization of 4 b, which is significantly higher than the experimentally determined value of 23.2 kcal mol−1. Although the description of the thermal isomerization of azobenzenes is usually based on a pure S0 thermal isomerization energy pathway, detailed investigations revealed that a crossing of the S0 and T1 energy hypersurfaces during thermal isomerization can occur. Computations of such a pathway provides lower activation energies that are in good agreement with experimental values. [25] Here, it should be mentioned that DFT reproduces experimental activation energies for the azobenzene isomerization surprisingly well due to its general underestimation of TS energies. However, it does not describe the physical reality, which is not a pure S0 pathway.
In order to investigate if a S0‐T1‐S0 crossing could also be possible in the azothiophene system, the singlet and triplet isomerization pathways were computed on the TPSSh/def2‐TZVP level of theory (Figure 4). [26] Two minima were located on the T1 pathway, one with a NNCS‐cis and one with a NNCS‐trans‐like geometry. Both structures showed a CNNC dihedral angle of ≈108° and an energy difference of ΔH=1.7 kcal mol−1. A transition state was found for the interconversion with a free activation enthalpy of 9.6 kcal mol−1 to the one and 11.3 kcal mol−1 to the opposite direction. IRC computations of the transition state revealed a torsional movement around the CNNC dihedral angle for the interconversion of the two triplet minima. As an approximation, the IRC curves for the S0 and T1 states were superimposed and are shown in Figure 4. It becomes obvious that the T1 crosses the S0 pathway twice during thermal isomerization, which is a hint that the thermal isomerization of azothiophenes 4 might not occur via a pure singlet state. Upon isomerization, a triplet state seems to become energetically favourable when the N=N bond is extremely bent. After a second crossing, the isomerization follows the S0 pathway to a shoulder which can be assigned to the N=N‐(E) conformation with the phenyl‐ and thiophenyl‐rings still in an orthogonal orientation to each other. Finally, both aromatic rings rotate in opposite direction until the (E)‐cis minimum is reached.
Figure 4.
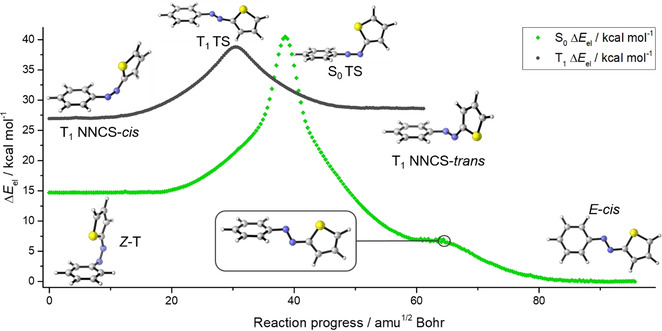
S0 and T1 isomerization pathways of azothiophene 4 b (TPSSh/def2‐TZVP). The T1 path crosses the S0 path twice, which is a hint that the thermal isomerization of azothiophenes does not exclusively occur via a pure S0 pathway.
Conclusions
In summary, a series of substituted arylazothiophenes 4 was synthesized. In general, the investigated azothiophenes showed red‐shifted absorption spectra compared to azobenzene. They were found to almost quantitatively isomerize to the (Z) states upon irradiation. Kinetic measurements showed that the electronic nature of the π systems strongly influences the thermal half‐lives, where electron‐withdrawing substitution on the phenyl moiety generally prolongs, and electron‐donating groups shorten the half‐life. Furthermore, increasing electron density at the thiophene scaffold also led to higher half‐lives. These findings can be explained with stronger or weaker LP⋅⋅⋅π interactions of the sulfur LP with the phenyl π‐system, providing different stabilization for the corresponding (Z)‐T isomers. Since such an interaction is geometrically neither in the transition state nor the (E) isomer possible, the energetic gap between the (E) and (Z)‐T isomers influence the thermal isomerization rate significantly. Additionally, a computational study on the isomerization mechanism revealed that a pure singlet state mechanism is not likely. An approximate overlay of both the S0 and T1 isomerization IRC curves revealed two crossing points of the pathways, indicating that the thermal isomerization of azothiophenes does not solely occur on the S0 energy surface.
The insights gained in this research provide essential parameters for the design of new heterocyclic photoswitches for applications ranging from photobiology, material sciences to data and energy storage devices.
Experimental Section
General procedure A for the synthesis of azothiophenes: A dry Schlenk‐tube under nitrogen atmosphere was charged with iPrMgCl⋅LiCl (1.3 mol L−1 in THF, 4.0 mmol, 2.0 equiv.) and neat 2‐bromothiophene (3.0 mmol, 1.5 equiv.) was added dropwise at rt. After stirring for 45 min at rt, the solution was diluted with dry THF (2 mL), cooled to −20 °C and ZnBr2 in dry THF (1.6 mmol, 0.8 equiv.) was added. After warming to rt, the solution was stirred for 30 min and was then added dropwise to a suspension of the corresponding diazonium tetrafluoroborate (2.0 mmol, 1.0 equiv.) in a mixture of dry NMP and dry THF (1:1, 4 mL) at −40 °C. After complete addition, the suspension was allowed to warm to −20 °C and was stirred at this temperature for 2 h. After quenching with sat. aq. NH4Cl (5 mL) and water (5 mL), the mixture was diluted and extracted with Et2O (3x10 mL), the combined organic phases were dried over MgSO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography to yield the products as red to orange solids.
General procedure B for the synthesis of azothiophenes: A dry Schlenk‐tube under nitrogen atmosphere was charged with a solution of the corresponding 2‐bromothiophene 5 (3.0 mmol, 1.5 equiv.) in dry THF (2 mL). Then, iPrMgCl⋅LiCl (1.3 mol L−1 in THF, 4.0 mmol, 2.0 equiv.) was added dropwise at rt. The reaction was stirred until GC‐MS showed full conversion of the bromothiophene at rt. After cooling to −20 °C, ZnBr2 in dry THF (1.6 mmol, 0.80 equiv.) was added and the suspension was allowed to warm to rt and was stirred for 30 min. The thiophenylzinc solution was then added dropwise to a suspension of the corresponding diazonium tetrafluoroborate (2.0 mmol, 1.0 equiv.) in a mixture of dry NMP and dry THF (1:1, 4 mL) at −40 °C. After complete addition, the suspension was allowed to warm to −20 °C and was stirred at this temperature for 2 h. After quenching with sat. aq. NH4Cl (5 mL) and water (5 mL), the mixture was diluted and extracted with Et2O or DCM (3x 10 mL), the combined organic phases were dried over MgSO4, filtered and concentrated. The residue was purified by silica gel flash column chromatography to yield the products as red to orange solids.
Thiophenylazobenzene (4 b): Procedure A: iPrMgCl⋅LiCl (3.2 mL, 4.2 mmol, 2.1 equiv.), 2‐bromothiophene (0.30 mL, 3.0 mmol, 1.5 equiv.), ZnBr2 in dry THF (1.0 mol L−1, 1.6 mL, 1.6 mmol, 0.8 equiv.), benzenediazonium tetrafluoroborate (386 mg, 2.01 mmol, 1.00 equiv.), chromatography: cyclohexane/EtOAc; 100:1, 75:1, 50:1, orange to red solid (222 mg, 59 %). 1H NMR (400 MHz, CDCl3) δ 7.90–7.84 (m, 2 H), 7.81 (dd, J=3.9, 1.3 Hz, 1 H), 7.54–7.39 (m, 4 H), 7.17 ppm (dd, J=5.4, 3.8 Hz, 2 H); 13C NMR (101 MHz, CDCl3) δ 160.5, 152.2, 131.9, 130.9, 129.2, 128.6, 127.6, 123.0 ppm; HRMS (ESI): m/z for C10H8N2SNa+; calcd 211.0300, found 211.0302; m.p. 72 °C.
4‐Methoxyphenylazothiophene (4 c): Procedure B: iPrMgCl⋅LiCl (3.1 mL, 4.0 mmol, 2.0 equiv.), 2‐bromothiophene (0.30 mL, 3.0 mmol, 1.5 equiv.) in dry THF (2 mL), 2 h stirring at rt, ZnBr2 in dry THF (2.0 mL, 2.0 mmol, 1.0 equiv.), 4‐methoxybenzenediazonium tetrafluoroborate (445 mg, 2.00 mmol, 1.00 equiv.), DCM extraction, chromatography: cyclohexane/EtOAc; 7:1, red solid (182 mg, 42 %). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J=9.0 Hz, 2 H), 7.72 (dd, J=3.8, 1.3 Hz, 1 H), 7.35 (dd, J=5.3, 1.3 Hz, 1 H), 7.14 (dd, J=5.4, 3.8 Hz, 1 H), 6.99 (d, J=9.0 Hz, 2 H), 3.88 ppm (s, 3 H); 13C NMR (101 MHz, CDCl3) δ 162.1, 160.8, 146.5, 130.4, 127.6, 127.4, 124.8, 114.4, 55.7 ppm; HRMS (ESI): m/z for C11H10N2OSNa+; calcd 241.0406, found 241.0404; m.p. 85 °C.
4‐Cyanophenylazothiophene (4 d): Procedure B: iPrMgCl⋅LiCl (2.8 mL, 3.6 mmol, 1.8 equiv.), 2‐bromothiophene (0.30 mL, 3.0 mmol, 1.5 equiv.) in dry THF (2 mL), 1.5 h stirring at rt, ZnBr2 in dry THF (2.0 mL, 2.0 mmol, 1.0 equiv.), 4‐cyanobenzenediazonium tetrafluoroborate (441 mg, 2.01 mmol, 1.00 equiv.), DCM extraction, chromatography: cyclohexane/EtOAc; 10:1, red solid (299 mg, 70 %). 1H NMR (400 MHz, CDCl3) δ 7.98–7.87 (m, 3 H), 7.81–7.74 (m, 2 H), 7.52 (dd, J=5.4, 1.3 Hz, 1 H), 7.21 ppm (dd, J=5.4, 3.9 Hz, 1 H); 13C NMR (101 MHz, CDCl3) δ 160.1, 154.3, 134.2, 133.3, 130.8, 128.1, 123.4, 118.6, 113.7 ppm; HRMS (ESI): m/z for C11H7N3SNa+; calcd 236.0253, found 236.0251; m.p. 160 °C (decomposition).
4‐(Trifluoromethyl)phenylazothiophene (4 e): Procedure A: iPrMgCl⋅LiCl (3.2 mL, 4.2 mmol, 2.1 equiv.), 2‐bromothiophene (0.30 mL, 3.0 mmol, 1.5 equiv.), ZnBr2 in dry THF (1.6 mL, 1.6 mmol, 0.80 equiv.), 4‐(trifluoromethyl)benzenediazonium tetrafluoroborate (528 mg, 2.03 mmol, 1.00 equiv.), chromatography: cyclohexane/EtOAc; 30:1, orange solid (314 mg, 61 %). 1H NMR (400 MHz, CDCl3) δ 7.99–7.91 (m, 2 H), 7.88 (dd, J=3.8, 1.3 Hz, 1 H), 7.75 (d, J=8.3 Hz, 2 H), 7.49 (dd, J=5.4, 1.3 Hz, 1 H), 7.20 ppm (dd, J=5.4, 3.9 Hz, 1 H); 13C NMR (101 MHz, CDCl3) δ 160.0, 154.0, 133.4, 131.9 (q, J=32.6 Hz), 129.9, 127.8, 126.3 (q, J=3.8 Hz), 125.3, 123.0 ppm; 19F NMR (377 MHz, CDCl3) δ−62.5 ppm; HRMS (ESI): m/z for C12H9N2F3SNa+; calcd 250.0409, found 250.0406; m.p. 114 °C.
3‐Methoxyphenylazothiophene (4 f): A dry Schlenk‐tube under nitrogen‐atmosphere was charged with iPrMgCl⋅LiCl (1.3 mol L−1 in THF, 5.0 mL, 6.5 mmol, 1.3 equiv.) and 2‐bromothiophene (0.50 mL, 5.2 mmol, 1.0 equiv.) was added dropwise at rt. After stirring for 45 min, the solution was diluted with dry THF (4 mL), cooled to −20 °C and ZnBr2 in dry THF (1.0 mol L−1, 2.8 mL, 2.8 mmol, 0.54 equiv.) was added dropwise. After complete addition, the mixture was warmed to rt, stirred for 30 min, diluted with dry THF (12 mL) and cooled to −60 °C afterwards. At this temperature, 3‐methoxybenzenediazonium tetrafluoroborate (1.34 g, 6.03 mmol, 1.17 equiv.) was added in small portions over a period of 30 min. The deep red suspension was stirred at −60 °C for 4 h. At this temperature, the reaction was quenched by the dropwise addition of half‐saturated aq. NH4Cl (10 mL) and was then slowly warmed to rt and diluted with Et2O (25 mL) and water (10 mL). After phase separation, the aqueous phase was extracted with Et2O (3×20 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by two consecutive flash column chromatographies (cyclohexane/EtOAc; 1:1, then cyclohexane/EtOAc; 10:1) to yield a red oil, which slowly crystallized to a red solid after solvent evaporation while cooling to rt (225 mg, 20 %).1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J=3.9, 1.3 Hz, 1 H), 7.50 (ddd, J=7.8, 1.7, 1.0 Hz, 1 H), 7.45–7.36 (m, 3 H), 7.17 (dd, J=5.4, 3.8 Hz, 1 H), 7.01 (ddd, J=8.1, 2.5, 1.0 Hz, 1 H), 3.89 ppm (s, 3 H); 13C NMR (101 MHz, CDCl3) δ 160.4, 160.4, 153.5, 131.9, 129.9, 128.7, 127.6, 117.9, 117.1, 105.9, 55.6 ppm; HRMS (ESI): m/z for C11H11N2OS+; calcd 219.0857, found 219.0589; m.p. 63 °C.
3‐Cyanophenylazothiophene (4 g): Procedure A: iPrMgCl⋅LiCl (3.2 mL, 4.2 mmol, 2.1 equiv.), 2‐bromothiophene (0.30 mL, 3.0 mmol, 1.5 equiv.), ZnBr2 in dry THF (1.6 mL, 1.6 mmol, 0.80 equiv.), 3‐cyanobenzenediazonium tetrafluoroborate (437 mg, 2.01 mmol, 1.00 equiv), chromatography: cyclohexane/EtOAc; 5:1, orange solid (258 mg, 60 %). 1H NMR (400 MHz, CDCl3) δ 8.14 (t, J=1.8 Hz, 1 H), 8.08 (ddd, J=8.0, 2.0, 1.2 Hz, 1 H), 7.88 (dd, J=3.9, 1.3 Hz, 1 H), 7.70 (dt, J=7.7, 1.4 Hz, 1 H), 7.60 (t, J=7.8 Hz, 1 H), 7.50 (dd, J=5.3, 1.3 Hz, 1 H), 7.20 ppm (dd, J=5.4, 3.9 Hz, 1 H); 13C NMR (101 MHz, CDCl3) δ 159.9, 152.2, 133.8, 133.5, 130.3, 130.2, 128.0, 127.4, 126.1, 118.3, 113.5 ppm; HRMS (ESI): m/z for C11H7N3SNa+; calcd 236.0253, found 236.0256; m.p. 126 °C.
5‐Methoxythiophenylazobenzene (4 h): Procedure B: iPrMgCl⋅LiCl (2.6 mL, 3.4 mmol, 2.0 equiv.), 5‐bromo‐2‐methoxythiophene (0.31 mL, 2.6 mmol, 1.5 equiv.) in dry THF (1.7 mL), stirred for 30 min, ZnBr2 in dry THF (1.7 mL, 1.7 mmol, 1.0 equiv.), benzenediazonium tetrafluoroborate (329 mg, 1.7 mmol, 1.00 equiv.) in dry THF/NMP, 1:1 (3.4 mL), stirred at −20 °C for 3.25 h, DCM extraction, two consecutive chromatographies: cyclohexane/EtOAc; 5:1, a red oil (87 mg, 24 %). 1H NMR (400 MHz, CDCl3) δ 7.81–7.75 (m, 2 H), 7.54 (d, J=4.4 Hz, 1 H), 7.49–7.42 (m, 2 H), 7.41–7.34 (m, 1 H), 6.33 (d, J=4.4 Hz, 1 H), 3.97 ppm (s, 3 H); 13C NMR (101 MHz, CDCl3) δ 171.1, 152.3, 147.3, 132.5, 129.8, 129.1, 122.5, 105.3, 60.0 ppm; HRMS (ESI): m/z for C11H11N2S+; calcd 219.0587, found 219.0587.
4‐Cyanophenyl‐5‐methoxythiophenyldiazene (4 i): Procedure B: iPrMgCl⋅LiCl (0.85 mL, 1.1 mmol, 2.0 equiv.), 2‐bromo‐5‐methoxythiophene (0.10 mL, 0.82 mmol, 1.0 equiv.) in dry THF (0.5 mL), stirred for 2.5 h, ZnBr2 in dry THF (0.45 mL, 0.45 mmol, 0.80 equiv.) stirred for 70 min, 4‐cyanobenzenediazonium tetrafluoroborate (136 mg, 0.564 mmol, 1.00 equiv.) in dry THF/NMP, 1:1 (1 mL), stirred at −20 °C for 1 h and at rt for another hour, chromatography: cyclohexane/EtOAc; 5:1, dark red solid (66 mg, 48 %). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J=8.7 Hz, 2 H), 7.72 (d, J=8.8 Hz, 2 H), 7.64 (d, J=4.5 Hz, 1 H), 6.40 (d, J=4.5 Hz, 1 H), 4.01 ppm (s, 3 H); 13C NMR (101 MHz, CDCl3) δ 173.6, 154.7, 146.8, 135.6, 133.2, 122.9, 119.0, 112.3, 106.6, 60.3 ppm; HRMS (ESI): m/z for C12H9N3OSNa+; calcd 266.0358, found 266.0359; m.p. 164 °C.
4‐Cyanophenyl‐5‐methylthiophenyldiazene (4 j): Procedure B: iPrMgCl⋅LiCl (3.0 mL, 3.9 mmol, 1.9 equiv.), 2‐bromo‐5‐methylthiophene (0.36 mL, 3.0 mmol, 1.5 equiv.) in dry THF (2.0 mL), stirred at rt for 45 min, ZnBr2 in dry THF (1.6 mL, 1.6 mmol, 0.78 equiv.), 4‐cyanobenzenediazonium tetrafluoroborate (445 mg, 2.05 mmol, 1.00 equiv.) in dry THF/NMP, 4:1 (10 mL) at −50 °C. Stirred at −30 °C for 2 h, chromatography: cyclohexane/EtOAc/DCM; 7.5:1:1, dark red solid (325 mg, 70 %). 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J=8.6 Hz, 2 H), 7.75 (d, J=8.6 Hz, 2 H), 7.70 (d, J=3.9 Hz, 1 H), 6.97–6.84 (m, 1 H), 2.56 ppm (d, J=1.0 Hz, 3 H); 13C NMR (101 MHz, CDCl3) δ 157.8, 154.5, 147.5, 135.2, 133.2, 126.9, 123.2, 118.8, 113.1, 17.0 ppm; HRMS (ESI): m/z for C12H9N3SNa+; calcd 250.0409, found 250.0384; m.p. 146 °C.
4‐(Trifluoromethyl)phenyl‐5‐methylthiophenyldiazene (4 k): Procedure B: iPrMgCl⋅LiCl (2.8 mL, 3.6 mmol, 2.2 equiv.), 2‐bromo‐5‐methylthiophene (0.36 mL, 3.0 mmol, 1.8 equiv.) in dry THF (2.0 mL), a stirred at rt for 1 h, ZnBr2 in dry THF (1.6 mL, 1.6 mmol, 1.0 equiv.), 4‐(trifluoromethyl)benzene‐diazonium tetrafluoroborate (442 mg, 1.68 mmol, 1.00 equiv.), chromatography: n‐pentane/Et2O; 50:1), red crystalline solid (244 mg, 54 %). 1H NMR (400 MHz, CDCl3) δ 7.93–7.86 (m, 2 H), 7.77–7.65 (m, 3 H), 6.92–6.86 (m, 1 H), 2.56 ppm (s, 3 H); 19F NMR (377 MHz, CDCl3) δ−62.5 ppm; 13C NMR (101 MHz, CDCl3) δ 157.9, 154.3, 146.5, 134.4, 131.4 (q, J=32.3 Hz), 126.6, 126.4 (q, J=3.8 Hz), 125.5, 122.9, 16.9 ppm; HRMS (ESI): m/z for C12H9F3N2SNa+; calcd 293.0331, found 293.0330; m.p. 106 °C.
5‐Cyanothiophenyl‐phenyldiazene (4 l): To a solution of 2‐bromo‐5‐cyanothiophene (599 mg, 3.19 mmol, 1.04 equiv.) in dry THF (2 mL) under a nitrogen‐atmosphere at −60 °C, iPrMgCl⋅LiCl (1.3 mol L−1 in THF, 3.2 mL, 4.2 mmol, 1.4 equiv.) was added dropwise. After complete addition, the red solution was stirred at −60 °C for 50 min and was then added dropwise to a suspension of benzenediazonium tetrafluoroborate (590 mg, 3.07 mmol, 1.00 equiv.) in dry THF (4 mL) at −80 °C. The deep red solution was stirred at this temperature for 30 min and was allowed to warm to rt and stirred for 1.5 h. After diluting with Et2O (5 mL) and quenching the reaction with sat. aq. NH4Cl (5 mL) and water (5 mL), the phases were separated and the aqueous phase was extracted with Et2O (10 mL). After drying over MgSO4, filtration and evaporation of the solvents under reduced pressure, the residue was purified by two consecutive flash column chromatographies (SiO2, cyclohexane/EtOAc; 10:1) to yield the product as a red solid (2 mg, 0.3 %). 1H NMR (400 MHz, CDCl3) δ 7.93–7.87 (m, 1 H), 7.77 (d, J=4.1 Hz, 1 H), 7.65 (d, J=4.1 Hz, 0 H), 7.57–7.49 ppm (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 164.6, 151.7, 137.3, 132.7, 129.9, 129.5, 123.7, 114.5, 111.0 ppm; HRMS (ESI): m/z for C11H7N3SNa+; calcd 236.0253, found 236.0250. Melting point was not determined due to limited amount of substance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG; WE 5601/6‐1) is gratefully acknowledged. We thank Dr. Urs Gellrich and Tizian Müller for fruitful discussions. Open access funding enabled and organized by Projekt DEAL.
A. H. Heindl, H. A. Wegner, Chem. Eur. J. 2020, 26, 13730.
References
- 1. Hartley G. S., Nature 1937, 140, 281. [Google Scholar]
- 2.
- 2a. Stoll R. S., Hecht S., Angew. Chem. Int. Ed. 2010, 49, 5054–5075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5176–5200; [Google Scholar]
- 2b. Ueno A., Takahashi K., Osa T., J. Chem. Soc. Chem. Commun. 1980, 837–838; [Google Scholar]
- 2c. Cacciapaglia R., Di Stefano S., Mandolini L., J. Am. Chem. Soc. 2003, 125, 2224–2227; [DOI] [PubMed] [Google Scholar]
- 2d. Niedek D., Erb F. R., Topp C., Seitz A., Wende R. C., Eckhardt A. K., Kind J., Herold D., Thiele C. M., Schreiner P. R., J. Org. Chem. 2020, 85, 1835–1846. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Beharry A. A., Woolley G. A., Chem. Soc. Rev. 2011, 40, 4422–4437; [DOI] [PubMed] [Google Scholar]
- 3b. van der Berg J. P., Velema W. A., Szymanski W., Driessen A. J. M., Feringa B. L., Chem. Sci. 2015, 6, 3593–3598; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Bellotto S., Chen S., Rentero Rebollo I., Wegner H. A., Heinis C., J. Am. Chem. Soc. 2014, 136, 5880–5883; [DOI] [PubMed] [Google Scholar]
- 3d. Polosukhina A., Litt J., Tochitsky I., Nemargut J., Sychev Y., de Kouchkovsky I., Huang T., Borges K., Trauner D., van Gelder R. N., Kramer R. H., Neuron 2012, 75, 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Liu Z. F., Hashimoto K., Fujishima A., Nature 1990, 347, 658–660; [Google Scholar]
- 4b. Ikeda T., Tsutsumi O., Science 1995, 268, 1873–1875. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Han G. G. D., Li H., Grossman J. C., Nat. Commun. 2017, 8, 1446; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Cho E. N., Zhitomirsky D., Han G. G. D., Liu Y., Grossman J. C., ACS Appl. Mater. Interfaces 2017, 9, 8679–8687; [DOI] [PubMed] [Google Scholar]
- 5c. Jiang Y., Huang J., Feng W., Zhao X., Wang T., Li C., Luo W., Sol. Energ. Mater. Sol. Cells 2019, 193, 198–205; [Google Scholar]
- 5d. Dong L., Feng Y., Wang L., Feng W., Chem. Soc. Rev. 2018, 47, 7339–7368. [DOI] [PubMed] [Google Scholar]
- 6. Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 7. Knie C., Utecht M., Zhao F., Kulla H., Kovalenko S., Brouwer A. M., Saalfrank P., Hecht S., Bléger D., Chem. Eur. J. 2014, 20, 16492–16501. [DOI] [PubMed] [Google Scholar]
- 8. Whitten D. G., Wildes P. D., Pacifici J. G., Irick G., J. Am. Chem. Soc. 1971, 93, 2004–2008. [Google Scholar]
- 9.
- 9a. Reuter R., Wegner H. A., Chem. Commun. 2011, 47, 12267–12276; [DOI] [PubMed] [Google Scholar]
- 9b. Siewertsen R., Neumann H., Buchheim-Stehn B., Herges R., Näther C., Renth F., Temps F., J. Am. Chem. Soc. 2009, 131, 15594–15595; [DOI] [PubMed] [Google Scholar]
- 9c. Schweighauser L., Wegner H. A., Chem. Commun. 2013, 49, 4397–4399; [DOI] [PubMed] [Google Scholar]
- 9d. Schweighauser L., Häussinger D., Neuburger M., Wegner H. A., Org. Biomol. Chem. 2014, 12, 3371–3379; [DOI] [PubMed] [Google Scholar]
- 9e. Slavov C., Yang C., Heindl A. H., Stauch T., Wegner H. A., Dreuw A., Wachtveitl J., J. Phys. Chem. Lett. 2018, 9, 4776–4781; [DOI] [PubMed] [Google Scholar]
- 9f. Heindl A. H., Becker J., Wegner H. A., Chem. Sci. 2019, 10, 7418–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schweighauser L., Strauss M. A., Bellotto S., Wegner H. A., Angew. Chem. Int. Ed. 2015, 54, 13436–13439; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13636–13639. [Google Scholar]
- 11. Strauss M. A., Wegner H. A., Angew. Chem. Int. Ed. 2019, 58, 18552–18556; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18724–18729. [Google Scholar]
- 12. Crespi S., Simeth N. A., König B., Nat. Rev. Chem. 2019, 3, 133–146. [Google Scholar]
- 13. Garcia-Amorós J., Nonell S., Velasco D., Chem. Commun. 2012, 48, 3421–3423. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Zhang Z.-Y., He Y., Zhou Y., Yu C., Han L., Li T., Chemistry 2019, 25, 13402–13410; [DOI] [PubMed] [Google Scholar]
- 14b. Wang Y.-T., Liu X.-Y., Cui G., Fang W.-H., Thiel W., Angew. Chem. Int. Ed. 2016, 55, 14009–14013; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14215–14219; [Google Scholar]
- 14c. Stricker L., Böckmann M., Kirse T. M., Doltsinis N. L., Ravoo B. J., Chemistry 2018, 24, 8639–8647; [DOI] [PubMed] [Google Scholar]
- 14d. Gibson R. S. L., Calbo J., Fuchter M. J., ChemPhotoChem 2019, 3, 372–377; [Google Scholar]
- 14e. Devi S., Saraswat M., Grewal S., Venkataramani S., J. Org. Chem. 2018, 83, 4307–4322. [DOI] [PubMed] [Google Scholar]
- 15. Calbo J., Weston C. E., White A. J. P., Rzepa H. S., Contreras-García J., Fuchter M. J., J. Am. Chem. Soc. 2017, 139, 1261–1274. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Otsuki J., Suwa K., Sarker K. K., Sinha C., J. Phys. Chem. A 2007, 111, 1403–1409; [DOI] [PubMed] [Google Scholar]
- 16b. Otsuki J., Suwa K., Narutaki K., Sinha C., Yoshikawa I., Araki K., J. Phys. Chem. A 2005, 109, 8064–8069. [DOI] [PubMed] [Google Scholar]
- 17. Wendler T., Schütt C., Näther C., Herges R., J. Org. Chem. 2012, 77, 3284–3287. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Crespi S., Simeth N. A., Bellisario A., Fagnoni M., König B., J. Phys. Chem. A 2019, 123, 1814–1823; [DOI] [PubMed] [Google Scholar]
- 18b. Simeth N. A., Crespi S., Fagnoni M., König B., J. Am. Chem. Soc. 2018, 140, 2940–2946. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Garcia-Amorós J., Maerz B., Reig M., Cuadrado A., Blancafort L., Samoylova E., Velasco D., Chem. Eur. J. 2019, 25, 7726–7732; [DOI] [PubMed] [Google Scholar]
- 19b. Garcia-Amorós J., Castro M. C. R., Coelho P., Raposo M. M. M., Velasco D., Chem. Commun. 2013, 49, 11427–11429. [DOI] [PubMed] [Google Scholar]
- 20. Weston C. E., Richardson R. D., Haycock P. R., White A. J. P., Fuchter M. J., J. Am. Chem. Soc. 2014, 136, 11878–11881. [DOI] [PubMed] [Google Scholar]
- 21. Slavov C., Yang C., Heindl A. H., Wegner H. A., Dreuw A., Wachtveitl J., Angew. Chem. Int. Ed. 2020, 59, 380–387; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 388–395. [Google Scholar]
- 22.
- 22a. Piller F. M., Appukkuttan P., Gavryushin A., Helm M., Knochel P., Angew. Chem. Int. Ed. 2008, 47, 6802–6806; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6907–6911; [Google Scholar]
- 22b. Haag B., Peng Z., Knochel P., Org. Lett. 2009, 11, 4270–4273. [DOI] [PubMed] [Google Scholar]
- 23. Exner O. in Progress in Physical Organic Chemistry, Vol. 10 (Eds.: Streitwieser, Jr. A., Taft R. W.), Wiley, Hoboken, 1973, pp. 411–482. [Google Scholar]
- 24.
- 24a. Riplinger C., Sandhoefer B., Hansen A., Neese F., J. Chem. Phys. 2013, 139, 134101; [DOI] [PubMed] [Google Scholar]
- 24b. Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158–6170; [Google Scholar]
- 24c. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868; [DOI] [PubMed] [Google Scholar]
- 24d. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 24e. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 24f. Marenich A. V., Cramer C. J., Truhlar D. G., J. Phys. Chem. B 2009, 113, 6378–6396; [DOI] [PubMed] [Google Scholar]
- 24g. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Cembran A., Bernardi F., Garavelli M., Gagliardi L., Orlandi G., J. Am. Chem. Soc. 2004, 126, 3234–3243; [DOI] [PubMed] [Google Scholar]
- 25b. Gagliardi L., Orlandi G., Bernardi F., Cembran A., Garavelli M., Theor. Chem. Acc. 2004, 111, 363–372. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Staroverov V. N., Scuseria G. E., Tao J., Perdew J. P., J. Chem. Phys. 2003, 119, 12129–12137; [Google Scholar]
- 26b. Tao J., Perdew J. P., Staroverov V. N., Scuseria G. E., Phys. Rev. Lett. 2003, 91, 146401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary