

Abstract
The production of hydrogen by water electrolysis benefits from the development of water oxidation catalysts. This development process can be aided by the postulation of design rules for catalytic systems. The analysis of the reactivity of molecular complexes can be complicated by their decomposition under catalytic conditions into nanoparticles that may also be active. Such a misinterpretation can lead to incorrect design rules. In this study, the nickel‐based water oxidation catalyst [NiII(meso‐L)](ClO4)2, which was previously thought to operate as a molecular catalyst, is found to decompose to form a NiOx layer in a pH 7.0 phosphate buffer under prolonged catalytic conditions, as indicated by controlled potential electrolysis, electrochemical quartz crystal microbalance, and X‐ray photoelectron spectroscopy measurements. Interestingly, the formed NiOx layer desorbs from the surface of the electrode under less anodic potentials. Therefore, no nickel species can be detected on the electrode after electrolysis. Catalyst decomposition is strongly dependent on the pH and buffer, as there is no indication of NiOx layer formation at pH 6.5 in phosphate buffer nor in a pH 7.0 acetate buffer. Under these conditions, the activity stems from a molecular species, but currents are much lower. This study demonstrates the importance of in situ characterization methods for catalyst decomposition and metal oxide layer formation, and previously proposed design elements for nickel‐based catalysts need to be revised.
Keywords: electrocatalysis, electrochemistry, nanoparticles, nickel, water splitting
Without a trace: Controlled potential electrolysis and electrochemical quartz microbalance techniques show that, under electrocatalytic water oxidation conditions, [NiII(meso‐L)](ClO4)2 decomposes to form an active NiOx layer in pH 7.0 phosphate buffer. This layer desorbs from the electrode at less anodic potentials, preventing ex situ detection. Catalyst decomposition is found to be highly buffer dependent.
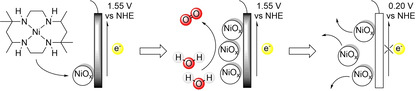
To combat climate change, the harvest of renewable energy is important to avoid greenhouse gas emissions through the use of fossil resources. Solar and wind power supply are steadily increasing, but due to their intermittency energy storage solutions are required. In this context the storage of renewable energy into chemical fuels, such as hydrogen, is important.[ 1 , 2 , 3 , 4 , 5 ] Hydrogen can be generated by water electrolysis using sustainable energy, and this reaction is therefore a topic of much recent interest.[ 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 ] Water electrolysis consists of two half reactions: proton reduction and water oxidation. Water oxidation requires two water molecules and the transfer of four electrons and four protons to produce oxygen, resulting in complicated reaction mechanisms.[ 19 , 20 , 21 , 22 , 23 , 24 ] Therefore, in water splitting catalysis, the water oxidation reaction is generally the limiting factor in terms of rate and energy losses. The development of novel water oxidation catalysts (WOCs) typically aims to improve these two properties of the catalyst, that is, to increase the activity and reduce the overpotential required for water oxidation catalysis. Molecular catalysts are generally more active per metal center and are easier to study at a higher level of detail than their heterogeneous counterparts. In addition, their ligand frameworks are relatively easy to modify, and with that the key properties of the catalyst can be more easily optimized. Interestingly, catalytic rates can be increased by orders of magnitude by modification of the ligand.[ 25 , 26 , 27 , 28 , 29 , 30 ] Knowledge about the mechanism by which a catalyst operates can lead to design rules, which facilitates catalyst development. Molecular ruthenium‐ and iridium‐based WOCs are currently state of the art in terms of activity and overpotential,[ 31 , 32 , 33 , 34 , 35 , 36 ] but recent research showed that also catalysts based on first row transition metals, like iron and nickel, have potential to be developed into proper catalysts for this reaction.[ 37 , 38 , 39 , 40 , 41 , 42 ] A downside of catalysts based on these more available and affordable metals, is that so far they generally have a lower activity. Their molecular complexes are generally also more susceptible to ligand degradation because the ligands bind less strongly to these metals and as a result nanoparticles are often formed.[ 43 , 44 ] Preventing ligand degradation is one of the challenges in the development of molecular catalysts to avoid the formation of metal nanoparticles.[ 45 , 46 , 47 , 48 ] These nanoparticles can also be active in water oxidation, complicating the analysis of the properties of molecular catalysts (Scheme 1).[ 49 , 50 , 51 ] More importantly, nanoparticle formation can lead to incorrect postulates for catalyst design as activity attributed to a complex could originate from metal nanoparticles. Detection of in situ‐generated nanoparticles can be very challenging. Hence, publications on molecular WOCs generally include multiple experiments to prove that the catalyst activity stems from a molecular species.[ 44 , 52 , 53 , 54 ] In the context of our ongoing research to explore novel first‐row metal‐based WOCs, [55] we were interested in developing novel nickel‐based catalysts. We aimed at applying previously developed design rules to optimize the catalyst, [56] but found that active NiOx layers are formed under certain conditions. Herein, we report the details of this study (Scheme 1).
Scheme 1.
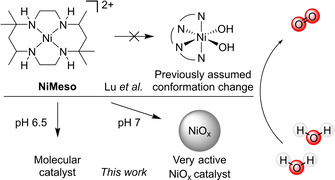
Water oxidation by NiMeso was previously proposed to be catalyzed by a molecular species that undergoes a conformational change. This study has shown that the catalyst in a pH 7.0 phosphate buffer decomposes in situ during prolonged electrolysis towards a NiOx layer, which is the main contributor to catalytic activity under these conditions. At open circuit voltage, the generated NiOx layer desorbs, which prevents ex situ detection. The catalyst does not show any signs of decomposition at pH 6.5.
The group of Lu presented the first electrochemically active molecular nickel‐based water oxidation catalyst [NiII(meso‐L)](ClO4)2 (NiMeso) based on the cyclam‐like meso ligand in 2014 (Scheme 1). [56] Since then, multiple ligand systems are published that allow for electrochemical water oxidation with molecular nickel complexes. [38] Examples include other cyclam‐based ligands,[ 57 , 58 , 59 , 60 ] porphyrin‐based systems, [61] pyridine‐based ligands,[ 62 , 63 , 64 , 65 , 66 ] and oxamidate‐type ligands.[ 67 , 68 ] As nickel oxide is also active in water oxidation, one should be careful when studying these catalysts to ensure that the activity stems from a molecular species.[ 10 , 69 ] There have indeed been several publications about nanoparticle formation during catalysis experiments with nickel‐based molecular catalysts.[ 68 , 70 , 71 ] The group of Najafpour showed the deposition of nickel oxide from a salen precursor under basic conditions. [70] After a thorough investigation, the formation of nickel oxide particles from a phthalocyanine complex was also observed. [71] Garrido‐Barros et al. investigated oxamidate‐type complexes and revealed that both the molecular complex and the NiOx layer, deposited after decomposition of the complex, were active in water oxidation. [68]
The NiMeso catalyst is known to function at a low overpotential under neutral conditions. The group of Lu describes that they observe an unusual pattern under prolonged water oxidation conditions: the current density steadily increases until a plateau is observed. [56] Current increase during controlled potential electrolysis (CPE) is often attributed to nanoparticle formation on the surface of the electrode,[ 72 , 73 , 74 ] however, no nickel deposition could be detected on the electrode using various techniques nor is the rinsed electrode active in catalysis. These two observations lead to the conclusion that the activity derives from a stable molecular catalyst. Interestingly, a current increase is not observed when the solution is stirred. Lu and co‐workers explained the unusual pattern in CPE by a conformational change of the ligand, going from a structure with two trans vacant sites to a structure with two cis vacant sites. The latter species is postulated to be the active catalyst and, as its concentration builds up during the CPE experiment, it was assumed to be responsible for the observed current increase. As a design rule for future nickel‐based WOCs they suggest the use of ligand frameworks that enforce two cis vacant sites. In follow‐up papers, they report two new nickel complexes based on ligands that enforce this coordination, however, these complexes operate at a higher overpotential and a lower activity than the original catalyst.[ 64 , 65 ] Interested in the principles of design rules for molecular catalysts, we investigate the nature of the catalytic nickel‐species in more detail using complementary electrochemical techniques to increase our understanding of the observed current increase.
CPE experiments under reported conditions (phosphate buffer, pH 7), [56] at various catalyst concentrations, show a plateau current at similar current densities of approximately 1.4‐1.5 mA cm−2 at 1.55 V vs. NHE for 0.5 mM, 1.0 mM, and 5.0 mM NiMeso (Figure 1a). For a molecular catalyst in solution the current density would be expected to increase linearly, which should lead to change of current density by a factor 10. Also, the time to reach the plateau current is influenced by the concentration of the nickel catalyst, as observed in approximately the first 100 s of the measurement, implying concentration dependent saturation behavior. The observed concentration independent plateau current clearly conflicts with the hypothesis that the nickel catalyst has to change conformation during CPE, as the plateau current should be linearly dependent on concentration in that scenario. Instead, these experiments do indicate the accumulation of an active species over time until a saturation point is reached. To gain more insight in the nature of the concentration of the nickel species, we performed electrochemical quartz crystal microbalance (EQCM) measurements. With these experiments the mass of the electrode during electrochemical measurements can be monitored, and this provides information on catalyst adsorption.[ 75 , 76 , 77 ] EQCM during 600 s of CPE under catalytic conditions demonstrates that the mass of the electrode steadily increases over time (Figure 1b). These observations indicate the adsorption of a species on the electrode surface upon applying an anodic potential. This increase in mass is considerably higher than a blank experiment with only buffer solution. The observed rate of mass accumulation during catalytic experiments depends on the catalyst concentration (Figure 1b). Whereas the current density doesn't increase much after 300 sec, the mass of the electrode continues to rise until the potential is set to 0.20 V vs. NHE after 600 sec, to reach final buildups of approximately 0.8 and 1.5 μg cm−2 for 0.5 and 1.0 mM nickel complex, respectively (Figure 1b). These observations, combined with the concentration dependent rate of plateau formation (Figure 1a), are a strong indication that the catalyst decomposes to form a NiOx species that adsorbs to the electrode with simultaneous current increase. Once the surface is saturated, the current plateaus, but the nickel species continues to adsorb. This interpretation is supported by the quantity of mass increase at the electrode, which agrees well with a few monolayers on a low surface area electrode (Figure 1b).[ 78 , 79 , 80 ] When the applied potential is set to 0.20 V vs. NHE (after 600 s) a quick desorption of all previously accumulated mass from the electrode surface is observed. The deposition and the desorption of the nickel material can be explained qualitatively by the Pourbaix diagram of nickel (see the Supporting Information, Figure S1). [81] At neutral pH and without anodic potential, the stable form of nickel is the soluble Ni2+. Under more oxidative potentials Ni3O4, Ni2O3 and NiO2 become thermodynamically more stable, and these forms of nickel are insoluble in aqueous buffer. Therefore, at the water oxidation conditions employed, the most stable state will be a solid nickel oxide species. When the potential is lowered, the nickel will be reduced to a Ni2+ species, which again is soluble in the medium.
Figure 1.
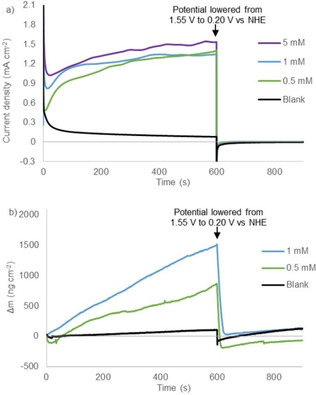
Controlled potential electrolysis measurements at 1.55 V vs. NHE for 600 s followed by 300 s at 0.20 V vs. NHE (a) and simultaneous electrochemical quartz microbalance measurements (b) of the buffer solution (black), 0.5 mM NiMeso (green), 1.0 mM NiMeso (blue) and 5.0 mM NiMeso (purple) in a 0.1 M pH 7.0 phosphate buffer using a gold working electrode.
An alternative explanation would be that the NiMeso catalyst, oxidized to a nickel(III) species formed at a potential of 0.87 V vs. NHE, containing two axial phosphate ligands ([NiIII(meso‐L)](HPO4)2) would form the active layer.[ 56 , 82 ] This complex is reported to be stable for hours. [58] To exclude that this molecular species with axial phosphate groups anchors to the surface, we performed CPE and EQCM experiments at a 1.20 V vs. NHE (Figure 2). At this potential, the nickel(III) species is formed, but the potential is too low for efficient catalysis. Under these conditions, no increase in electrode mass is observed, showing that these molecular species do not deposite on the surface at this potential. Instead, under catalytic conditions NiMeso decomposes to form likely a nickel oxide material, which explains the increase in activity.
Figure 2.
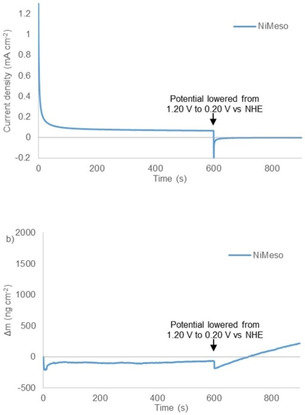
Controlled potential electrolysis measurements at 1.20 V vs. NHE (non‐catalytic conditions) for 600 s followed by 300 s at 0.2 V vs. NHE (a) and simultaneous electrochemical quartz microbalance measurements (b) of 1 mM NiMeso (blue) in a 0.1 M pH 7.0 phosphate buffer using a gold working electrode.
Similar deposition and desorption behavior was reported by Cho and Chang in 2017 for a nickel salt in phosphate buffer. [83] They showed the formation of a thin ‘Ni−Pi’ layer which consists of nickel oxide with incorporated phosphate anions. This layer was formed from a 1.0 mM solution of Ni(NO3)2 during cyclic voltammetry at high anodic potentials and desorbed from the surface when the potential was lowered below 0.90 V vs. Ag/AgCl. When we performed CPE/EQCM experiments of nickel salts at 1.55 V vs. NHE, we indeed observed the supposed ‘Ni−Pi’ layer formation up to 7.5 μg cm−2 after 600 s and a current density up to 2.4 mA cm−2 (Figure 3). In line with the results of Cho and Chang, this layer detaches upon lowering the potential. [83] This shows that the deposition formed from NiMeso has properties similar to a ‘Ni−Pi’ layer formed from bare nickel salts, giving a strong indication that the formed layer consists of the same material. Cho and Chang showed that the ‘Ni−Pi’ layer also forms under more basic conditions, at which it is stable upon lowering the potential, in accord with the Pourbaix diagram (Figure S1). [83] They characterized the nickel oxide species generated at pH 11 ex situ, showing an amorphous deposition with the incorporation of phosphate anions. The nickel deposit generated from the NiMeso complex likely consists of a similar amorphous nickel oxide material incorporating phosphate as the conditions of the experiments are very similar.
Figure 3.
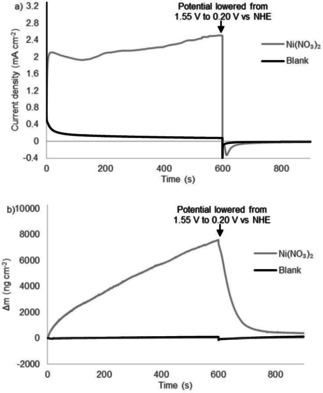
Controlled potential electrolysis measurements at 1.55 V vs. NHE for 600 s followed by 300 s at 0.20 V vs. NHE (a) and simultaneous electrochemical quartz microbalance measurements (b) of the buffer solution (black) and 1.0 mM Ni(NO3)2 (gray) in a 0.1 M pH 7.0 phosphate buffer using a gold working electrode.
To establish that the deposited material from the NiMeso complex is catalytically active, we performed several CPE experiments under various stirring conditions. An increase in current density to about 0.6 mA cm−2 is observed at 1.55 V vs. NHE under non‐stirring conditions using a fluorine doped tin oxide (FTO) electrode (Figure 4). In accordance with literature, [56] a lower current density of about 0.3 mA cm−2, that is constant in time, is observed under stirring conditions (green line in Figure 4). This lower observed current density, coupled with the absence of an increase in current, suggests that under stirring conditions the NiOx layer does not form and catalyst activity stems from a molecular species. We also performed an experiment without stirring, thus allowing for layer buildup, and started stirring after the current density stabilizes (purple line in Figure 4). Directly at the onset of stirring, the current density increases from 0.6 to 1.1 mA cm−2. This increase in current density upon stirring is a result of better mixing and is a strong indication that the deposited material is catalytically active. Importantly, the NiOx layer is stable upon stirring but does not form under stirring conditions. This can be explained by the fact that prior to NiOx layer formation, the molecular Ni‐complex has to decompose. Such a decomposition mechanism is apparently perturbed under stirring conditions, likely because the nickel complex resides for a relatively short time in the electrochemical double layer. Therefore, we conclude that the nickel complex slowly decomposes during non‐stirred CPE experiments and deposition of the formed nickel oxide layer on the electrode surface produces an active catalyst layer. To verify if the slow decomposition behavior of the nickel complex is induced by the ligand, we conduct a similar CPE experiment with a ligand‐free Ni(NO3)2 solution. Without stirring, the current density stabilizes after a few seconds to approximately 1.0 mA cm−2, while the current density with stirring increases to around 2.0 mA cm−2 in a few minutes (Figure S2). This suggests that if the nickel precursor is not supported by a ligand, which first has to be degraded by oxidation, the nickel salt can immediately form an active NiOx layer.
Figure 4.
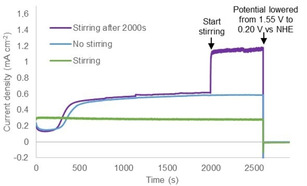
Controlled potential electrolysis measurements at 1.55 V vs. NHE for 2500 s followed by 300 s at 0.20 V vs. NHE of 1.0 mM NiMeso while stirring (800 rpm, green), while not stirring (blue), and while stirring after 2000s (800 rpm, purple) in a 0.1 M pH 7.0 phosphate buffer using a FTO working electrode.
To characterize the deposited film, we performed X‐ray photoelectron spectroscopy (XPS) measurements on the electrodes after CPE. When performing a 5 h CPE experiment while stirring the NiMeso solution, no nickel can be detected on the electrode after the electrolysis experiment. In contrast, in an identical experiment without stirring, during which layer formation is observed (see above), XPS measurements on the used electrode shows small signals indicating deposition of nickel on the electrode during the CPE experiment (Figure 5a,b; 0.44 % Ni content). The detected nickel content is minimal, in line with the rapid desorption observed during EQCM experiments under these conditions, and thus the signal‐to‐noise ratio is too low for detailed assignment. As NiOx layers are known to be stable at higher pH, [83] we performed similar 5 h CPE experiments in which the pH was rapidly tuned from 7 to 8.5 in the last seconds of CPE to prevent the desorption. Importantly, in a control experiment with continuous stirring and a similar change in pH in the final seconds, no nickel is detected on the electrode (Figure 5c), in line with our observations that no NiOx deposition occurs under stirring conditions. However, for an experiment with NiMeso without stirring during CPE, allowing for NiOx buildup, we observe clear XPS signals in the Ni 2p part of the spectrum, indicative of a NiOx layer (Figure 5d). The peak at 855.1 eV is associated to Ni 2p 3/2 with a satellite peak at 860.7 eV. The peak related to Ni 2p 1/2 appears at 872.6 eV and its satellite peak appears at 880.4 eV. These data are consistent with the formation of NiO layers on the electrode. [84] The total Ni content was determined to be 7.35 % for this sample. These XPS experiments confirm the formation of a NiOx layer from the molecular NiMeso species, in line with our EQCM and CPE experiments.
Figure 5.
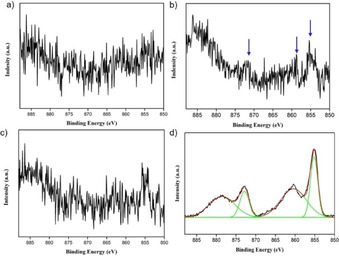
High‐resolution Ni 2p XPS spectra of the FTO electrode after 5 h of controlled potential electrolysis at 1.55 V vs. NHE of 1.0 mM NiMeso in a 0.1 M pH 7.0 phosphate buffer with stirring (a), without stirring (b), with stirring and pH tuning to 8.5 (c), and without stirring and with pH tuning to 8.5 (d).
Catalyst decomposition and water oxidation are known to be pH and buffer dependent. To study the sensitivity of the catalyst system to the reaction conditions, we also performed EQCM experiments in a pH 6.5 phosphate buffer and in a pH 7.0 acetate buffer. To our surprise, the CPE experiments in phosphate buffer at pH 6.5 show no current increase over time and EQCM measurements reveal almost no change in electrode mass, thereby suggesting that no NiOx layer is formed under these conditions (Figure 6). Under these conditions the water oxidation activity likely originates from the molecular catalyst. Thus, the stability of nickel‐based complexes appears to be very sensitive to pH. Notable is that the current density is significantly lower when no NiOx layer formation occurs. Likewise, no deposition is observed when performing the experiment in a pH 7.0 acetate buffer instead of phosphate (Figures S3 and S4). CPE/EQCM experiments with a solution of a nickel salt in a pH 7.0 acetate buffer do reveal increased current and electrode mass (Figures S5 and S6). This indicates that without a surrounding ligand, which protects the nickel metal, deposition of a NiOx layer can occur under these conditions. Buffer‐dependent nanoparticle formation has been observed for other metals like cerium. [85] We conclude that the stability of the molecular nickel complex is highly sensitive to pH and the type of buffer used.
Figure 6.
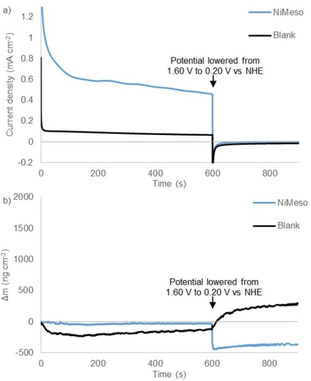
Controlled potential electrolysis measurements at 1.60 V vs. NHE for 600 s followed by 300 s at 0.20 V vs. NHE (a) and simultaneous electrochemical quartz microbalance measurements (b) of the buffer solution (black) and 1.0 mM NiMeso (blue) in a 0.1 M pH 6.5 phosphate buffer using a gold working electrode.
In conclusion, we have demonstrated on the basis of CPE, EQCM, and XPS experiments that a nickel complex with a cyclam‐based ligand (NiMeso) slowly decomposes under electrochemical water oxidation catalysis conditions in a pH 7.0 phosphate buffer, possibly initiated by ligand oxidation, forming a NiOx layer (Scheme 2). The NiOx layer desorbs at open circuit voltage, and thus remained previously undetected, [56] indicating that in situ experiments, like EQCM, are of vital importance to studying these (and likely other) systems. During CPE experiments of an unstirred solution of the molecular nickel complex, an increase in current density is observed until a concentration‐independent plateau is reached. EQCM shows an increase in electrode mass simultaneously with the increase in current density. The deposited material desorbs at less anodic potentials, as previously observed for nickel salts, which are known to form a ‘Ni−Pi’ layer during electrochemistry. The NiOx layer formed from NiMeso was confirmed by XPS experiments after a non‐stirred CPE experiment. In a CPE experiment with NiMeso but with a stirred solution, the current density did not increase over time. This suggests that the nickel complex slowly decomposes to form a NiOx layer. The formed NiOx layer is stable under catalytic conditions, but desorbs from the electrode at less anodic potentials, which can be understood from the Pourbaix diagram of nickel. As the species desorbs at open circuit voltage, there is no evidence of the layer formation after catalysis (Scheme 2). Therefore, it is not viable to attribute the absence of a current increase in consecutive cyclic voltammograms to the absence of metal oxide deposition (or nanoparticle formation). These results demonstrate the importance of in situ techniques for establishing the molecularity of homogeneous WOCs. Recently, the detection with EQCM of a transient cobalt deposit that is active in proton reduction was also reported, showing the broader applicability of the technique. [86]
Scheme 2.
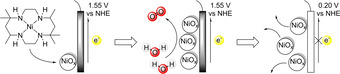
The NiMeso catalyst decomposes to NiOx under catalytic conditions in a pH 7.0 phosphate buffer. The formed NiOx layer desorbs from the surface at neutral potential.
Interestingly, the decomposition of the nickel catalyst and layer formation are pH and buffer dependent, as no layer formation is observed in a pH 6.5 phosphate buffer or in a pH 7.0 acetate buffer. Under these conditions, the molecular complex is likely active, indicating the importance of confirming molecularity even in very similar buffer systems. Most importantly, this work shows that the previously reported high activity at low overpotential ascribed to the molecular complex stems from decomposed deposited material and, as such, the design rules, reported to achieve highly active catalysts based on NiMeso, may not apply. Indeed, the necessity for two cis vacant sites, which was applied as design element for two nickel complexes, did not result in higher activity. [65]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organization for Scientific Research (NWO).
J. Hessels, F. Yu, R. J. Detz, J. N. H. Reek, ChemSusChem 2020, 13, 5625.
References
- 1. Lewis N. S., Nocera D. G., Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eckenhoff W. T., Eisenberg R., Dalton Trans. 2012, 41, 13004–13021. [DOI] [PubMed] [Google Scholar]
- 3. Dempsey J. L., Esswein A. J., Manke D. R., Rosenthal J., Soper J. D., Nocera D. G., Inorg. Chem. 2005, 44, 6879–6892. [DOI] [PubMed] [Google Scholar]
- 4. Gong J., Li C., Wasielewski M. R., Chem. Soc. Rev. 2019, 48, 1862–1864. [DOI] [PubMed] [Google Scholar]
- 5. Kim J. H., Hansora D., Sharma P., Jang J. W., Lee J. S., Chem. Soc. Rev. 2019, 48, 1908–1971. [DOI] [PubMed] [Google Scholar]
- 6. Detz R. J., Reek J. N. H., van der Zwaan B. C. C., Energy Environ. Sci. 2018, 11, 1653–1669. [Google Scholar]
- 7. Brudvig G. W., Reek J. N. H., Sakai K., Spiccia L., Sun L., ChemPlusChem 2016, 81, 1017–1019. [DOI] [PubMed] [Google Scholar]
- 8. Brudvig G. W., Campagna S., Chem. Soc. Rev. 2017, 46, 6085–6087. [DOI] [PubMed] [Google Scholar]
- 9. Suen N. T., Hung S. F., Quan Q., Zhang N., Xu Y. J., Chen H. M., Chem. Soc. Rev. 2017, 46, 337–365. [DOI] [PubMed] [Google Scholar]
- 10. Singh A., Spiccia L., Coord. Chem. Rev. 2013, 257, 2607–2622. [Google Scholar]
- 11. McCrory C. C. L., Jung S., Peters J. C., Jaramillo T. F., J. Am. Chem. Soc. 2013, 135, 16977–16987. [DOI] [PubMed] [Google Scholar]
- 12. Andreiadis E. S., Chavarot-Kerlidou M., Fontecave M., Artero V., Photochem. Photobiol. 2011, 87, 946–964. [DOI] [PubMed] [Google Scholar]
- 13. Zhang B., Sun L., Chem. Soc. Rev. 2019, 48, 2216–2264. [DOI] [PubMed] [Google Scholar]
- 14. Evans R. M., Siritanaratkul B., Megarity C. F., Pandey K., Esterle T. F., Badiani S., Armstrong F. A., Chem. Soc. Rev. 2019, 48, 2039–2052. [DOI] [PubMed] [Google Scholar]
- 15. Hetterscheid D. G. H., Reek J. N. H., Angew. Chem. Int. Ed. 2012, 51, 9740–9747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9878–9885; . [Google Scholar]
- 16. Kärkäs M. D., Verho O., Johnston E. V., Åkermark B., Chem. Rev. 2014, 114, 11863–12001. [DOI] [PubMed] [Google Scholar]
- 17. Blakemore J. D., Crabtree R. H., Brudvig G. W., Chem. Rev. 2015, 115, 12974–13005. [DOI] [PubMed] [Google Scholar]
- 18. He Y., Hamann T., Wang D., Chem. Soc. Rev. 2019, 48, 2182–2215. [DOI] [PubMed] [Google Scholar]
- 19. Meyer T. J., Sheridan M. V., Sherman B. D., Chem. Soc. Rev. 2017, 46, 6148–6169. [DOI] [PubMed] [Google Scholar]
- 20. Shaffer D. W., Xie Y., Concepcion J. J., Chem. Soc. Rev. 2017, 46, 6170–6193. [DOI] [PubMed] [Google Scholar]
- 21. Sala X., Maji S., Bofill R., García-Antón J., Escriche L., Llobet A., Acc. Chem. Res. 2014, 47, 504–516. [DOI] [PubMed] [Google Scholar]
- 22. Matheu R., Garrido-Barros P., Gil-Sepulcre M., Ertem M. Z., Sala X., Gimbert-Suriñach C., Llobet A., Nat. Rev. Chem. 2019, 3, 331–341. [Google Scholar]
- 23. Hessels J., Detz R. J., Koper M. T. M., Reek J. N. H., Chem. Eur. J. 2017, 23, 16413–16418. [DOI] [PubMed] [Google Scholar]
- 24. Wasylenko D. J., Palmer R. D., Berlinguette C. P., Chem. Commun. 2013, 49, 218–227. [DOI] [PubMed] [Google Scholar]
- 25. Helm M. L., Stewart M. P., Bullock R. M., DuBois M. R., DuBois D. L., Science 2011, 333, 863–866. [DOI] [PubMed] [Google Scholar]
- 26. Yu F., Poole D., Mathew S., Yan N., Hessels J., Orth N., Ivanović-Burmazović I., Reek J. N. H., Angew. Chem. Int. Ed. 2018, 57, 11247–11251; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11417–11421; . [Google Scholar]
- 27. Azcarate I., Constentin C., Robert M., Savéant J. M., J. Am. Chem. Soc. 2016, 138, 16639–16644. [DOI] [PubMed] [Google Scholar]
- 28. Pérez I. M., Engelmann X., Lee Y. M., Yoo M., Kumaran E., Farquhar E. R., Bill E., England J., Nam W., Swart M., Ray K., Angew. Chem. Int. Ed. 2017, 56, 14384–14388; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14576–14580. [Google Scholar]
- 29. Nakazono T., Sakai K., Dalton Trans. 2016, 45, 12649–12652. [DOI] [PubMed] [Google Scholar]
- 30. Codolà Z., Gamba I., Acuña-Parés F., Casadevall C., Clémancey N., Latour J. M., Luis J. M., Lorret-Fillol J., Costas M., J. Am. Chem. Soc. 2019, 141, 323–333. [DOI] [PubMed] [Google Scholar]
- 31. Duan L., Bozoglian F., Mandal S., Stewart B., Privalov T., Llobet A., Sun L., Nat. Chem. 2012, 4, 418–423. [DOI] [PubMed] [Google Scholar]
- 32. Kamdar J. M., Grotjahn D. B., Molecules 2019, 24, 494–517. [Google Scholar]
- 33. Thomsen J. M., Huang D. L., Crabtree R. H., Brudvig G. W., Dalton Trans. 2015, 44, 12452–12472. [DOI] [PubMed] [Google Scholar]
- 34. Sheehan S. W., Thomsen J. M., Hintermair U., Crabtree R. H., Brudvig G. W., Schmuttenmaer C. A., Nat. Commun. 2015, 6, 6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamamoto M., Föhlinger J., Petersson J., Hammarström L., Imahori H., Angew. Chem. Int. Ed. 2017, 56, 3329–3333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3377–3381. [Google Scholar]
- 36. Creus J., Matheu R., Peñafiel I., Moonshiram D., Blondeau P., Benet-Buchholz J., García-Antón J., Sala X., Godard C., Llobet A., Angew. Chem. Int. Ed. 2016, 55, 15382–15386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15608–15612. [Google Scholar]
- 37. Liu T., Zhang B., Sun L., Chem. Asian J. 2019, 14, 31–43. [DOI] [PubMed] [Google Scholar]
- 38. Wang J. W., Liu W. J., Zhong D. C., Lu T. B., Coord. Chem. Rev. 2018, 378, 237–261. [Google Scholar]
- 39. Parent A. R., Sakai K., ChemSusChem 2014, 7, 2070–2080. [DOI] [PubMed] [Google Scholar]
- 40. Nakazono T., Parent A. R., Sakai K., Chem. Commun. 2013, 49, 6325–6327. [DOI] [PubMed] [Google Scholar]
- 41. Najafpour M. M., Renger G., Holyńska M., Moghaddam A. N., Aro E. M., Carpentier R., Nishihara H., Eaton-Rye J. J., Shen J. R., Allakhverdiev S. I., Chem. Rev. 2016, 116, 2886–2936. [DOI] [PubMed] [Google Scholar]
- 42. Lorret-Fillol J., Codolà Z., Garcia-Bosch I., Gómez L., Pla J. J., Costas M., Nat. Chem. 2011, 3, 807–813. [DOI] [PubMed] [Google Scholar]
- 43. Daniel Q., Ambre R. B., Zhang B., Philippe B., Chen H., Li F., Fan K., Ahmadi S., Rensmo H., Sun L., ACS Catal. 2017, 7, 1143–1149. [Google Scholar]
- 44. Hong D., Mandal S., Yamada Y., Lee Y. M., Nam W., Llobet A., Fukuzumi S., Inorg. Chem. 2013, 52, 9522–9531. [DOI] [PubMed] [Google Scholar]
- 45. Wu X., Li F., Zhang B., Sun L., J. Photochem. Photobiol. C 2015, 25, 71–89. [Google Scholar]
- 46. Li J., Güttinger R., Moré R., Song F., Wan W., Patzke G. R., Chem. Soc. Rev. 2017, 46, 6124–6147. [DOI] [PubMed] [Google Scholar]
- 47. Artero V., Fontecave M., Chem. Soc. Rev. 2013, 42, 2338–2356. [DOI] [PubMed] [Google Scholar]
- 48. van Dijk B., Hofmann J. P., Hetterscheid D. G. H., Phys. Chem. Chem. Phys. 2018, 20, 19625–19634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Asraf M. A., Younus H. A., Yusubov M., Verpoort F., Catal. Sci. Technol. 2015, 5, 4901–4925. [Google Scholar]
- 50. Matheu R., Francas L., Chernev P., Ertem M. Z., Batista V., Haumann M., Sala X., Llobet A., ACS Catal. 2015, 5, 3422–3429. [Google Scholar]
- 51. Blakemore J. D., Mara M. W., Kushner-Lenhoff M. N., Schley N. D., Konezny S. J., Rivalta I., Negre C. F. A., Snoeberger R. C., Kokhan O., Huang J., Stickrath A., Tran L. A., Parr M. L., Chen L. X., Tiede D. M., Batista V. S., Crabtree R. H., Brudvig G. W., Inorg. Chem. 2013, 52, 1860–1871. [DOI] [PubMed] [Google Scholar]
- 52. Schley N. D., Blakemore J. D., Subbaiyan N. K., Incarvito C. D., D'Souza F., Crabtree R. H., Brudvig G. W., J. Am. Chem. Soc. 2011, 133, 10473–10481. [DOI] [PubMed] [Google Scholar]
- 53. Esarey S. L., Holland J. C., Bartlett B. M., Inorg. Chem. 2016, 55, 11040–11049. [DOI] [PubMed] [Google Scholar]
- 54. Singer Hobbs M., Sackville E. V., Smith A. J., Edler K. J., Hintermair U., ChemCatChem 2019, 11, 5313–5321. [Google Scholar]
- 55. Detz R. J., Abiri Z., Kluwer A. M., Reek J. N. H., ChemSusChem 2015, 8, 3057–3061. [DOI] [PubMed] [Google Scholar]
- 56. Zhang M., Zhang M. T., Hou C., Ke Z. F., Lu T. B., Angew. Chem. Int. Ed. 2014, 126, 13258–13264; [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 13042–13048. [DOI] [PubMed] [Google Scholar]
- 57. Chen G., Chen L., Ng S. M., Lau T. C., ChemSusChem 2014, 7, 127–134. [DOI] [PubMed] [Google Scholar]
- 58. Wang J. W., Hou C., Huang H. H., Liu W. J., Ke Z. F., Lu T. B., Catal. Sci. Technol. 2017, 7, 5585–5593. [Google Scholar]
- 59. Ariela B., Yaniv W., Dror S., Haya K., Yael A., Eric M., Dan M., Dalton Trans. 2017, 46, 10774–10779. [DOI] [PubMed] [Google Scholar]
- 60. Zhang L. H., Yu F., Shi Y., Li F., Li H., Chem. Commun. 2019, 55, 6122–6125. [DOI] [PubMed] [Google Scholar]
- 61. Han Y., Wu Y., Lai W., Cao R., Inorg. Chem. 2015, 54, 5604–5613. [DOI] [PubMed] [Google Scholar]
- 62. Wang L., Duan L., Ambre R. B., Daniel Q., Chen H., Sun J., Das B., Thapper A., Uhlig J., Dinér P., Sun L., J. Catal. 2016, 335, 72–78. [Google Scholar]
- 63. Wang D., Bruner C. O., Inorg. Chem. 2017, 56, 13638–13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang J. W., Zhang X. Q., Huang H. H., Lu T. B., ChemCatChem 2016, 8, 3287–3293. [Google Scholar]
- 65. Luo G. Y., Huang H. H., Wang J. W., Lu T. B., ChemSusChem 2016, 9, 485–491. [DOI] [PubMed] [Google Scholar]
- 66. Shen J., Wang M., He T., Jiang J., Hu M., Chem. Commun. 2018, 54, 9019–9022. [DOI] [PubMed] [Google Scholar]
- 67. Lin J., Kang P., Liang X., Ma B., Ding Y., Electrochim. Acta 2017, 258, 353–359. [Google Scholar]
- 68. Garrido-Barros P., Grau S., Drouet S., Benet-Buchholz J., Gimbert-Surinach C., Llobet A., ACS Catal. 2019, 9, 3936–3945. [Google Scholar]
- 69. Dincă M., Surendranath Y., Nocera D. G., Proc. Natl. Acad. Sci. USA 2010, 107, 10337–10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feizi H., Shiri F., Bagheri R., Singh J. P., Chae K. H., Song Z., Najafpour M. M., Catal. Sci. Technol. 2018, 8, 3954–3968. [Google Scholar]
- 71. Feizi H., Bagheri R., Jagličić Z., Singh J. P., Chae K. H., Song Z., Najafpour M. M., Dalton Trans. 2019, 48, 547–557. [DOI] [PubMed] [Google Scholar]
- 72. Lee K. J., McCarthy B. D., Dempsey J. L., Chem. Soc. Rev. 2019, 48, 2927–2945. [DOI] [PubMed] [Google Scholar]
- 73. Kanan M. W., Nocera D. G., Science 2008, 321, 1072–1075. [DOI] [PubMed] [Google Scholar]
- 74. Hetterscheid D. G. H., van der Ham C. J. M., Diaz-Morales O., Verhoeven M. W. G. M., Longo A., Banerjee D., Niemantsverdriet J. W., Reek J. N. H., Feiters M. C., Phys. Chem. Chem. Phys. 2016, 18, 10931–10940. [DOI] [PubMed] [Google Scholar]
- 75. Schley N. D., Blakemore J. D., Subbaiyan N. K., Incarvito C. D., D'Souza F., Crabtree R. H., Brudvig G. W., J. Am. Chem. Soc. 2011, 133, 10473–10481. [DOI] [PubMed] [Google Scholar]
- 76. Buttry D. A., Ward M. D., Chem. Rev. 1992, 92, 1355–1379. [Google Scholar]
- 77. Kottrup K. G., D'Agostini S., van Langevelde P. H., Siegler M. A., Hetterscheid D. G. H., ACS Catal. 2018, 8, 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang J., Vukmirović M. B., Sasaki K., Uribe F., Adžić R. R., J. Serb. Chem. Soc. 2005, 70, 513–525. [Google Scholar]
- 79. Inaba R., Khademhosseini A., Suzuki H., Fukuda J., Biomaterials 2009, 30, 3573–3579. [DOI] [PubMed] [Google Scholar]
- 80. Yu Y., Dubey M., Bernasek S. L., Dismukes G. C., Langmuir 2007, 23, 8257–8263. [DOI] [PubMed] [Google Scholar]
- 81. Ciesielczyk F., Bartczak P., Wieszczycka K., Siwińska-Stefańska K., Nowacka M., Jesionowski T., Adsorption 2013, 19, 423–434. [Google Scholar]
- 82. Zeigerson E., Bar I., Bernstein J., Kirschenbaum L. J., Meyerstein D., Inorg. Chem. 1982, 21, 73–80. [Google Scholar]
- 83. Cho S. K., Chang J., ACS Omega 2017, 2, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang N., Guo G., He B., Zhu J., Wu J., Qiu J., J. Alloys Compd. 2020, 838, 155578. [Google Scholar]
- 85. Singh S., Dosani T., Karakoti A. S., Kumar A., Seal S., Self W. T., Biomaterials 2011, 32, 6745–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sconyers D. J., Blakemore J. D., Chem. Commun. 2017, 53, 7286–7289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary