

Summary
Classically, follicle-stimulating hormone receptor (FSHR)-driven cAMP-mediated signaling boosts human ovarian follicle growth and oocyte maturation. However, contradicting in vitro data suggest a different view on physiological significance of FSHR-mediated cAMP signaling. We found that the G-protein-coupled estrogen receptor (GPER) heteromerizes with FSHR, reprogramming cAMP/death signals into proliferative stimuli fundamental for sustaining oocyte survival. In human granulosa cells, survival signals are missing at high FSHR:GPER ratio, which negatively impacts follicle maturation and strongly correlates with preferential Gαs protein/cAMP-pathway coupling and FSH responsiveness of patients undergoing controlled ovarian stimulation. In contrast, FSHR/GPER heteromers triggered anti-apoptotic/proliferative FSH signaling delivered via the Gβγ dimer, whereas impairment of heteromer formation or GPER knockdown enhanced the FSH-dependent cell death and steroidogenesis. Therefore, our findings indicate how oocyte maturation depends on the capability of GPER to shape FSHR selective signals, indicating hormone receptor heteromers may be a marker of cell proliferation.
Subject Areas: Molecular Biology, Female Reproductive Endocrinology, Endocrine Regulation
Graphical Abstract
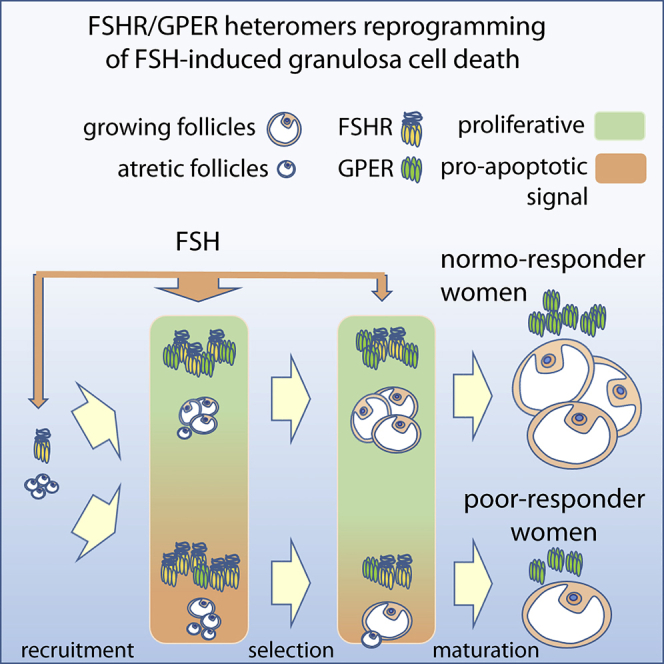
Highlights
-
•
G-protein-coupled estrogen receptor (GPER) interacts with FSH receptor (FSHR)
-
•
FSHR/GPER heteromers reprogram FSH-induced death signals to proliferative stimuli
-
•
Anti-apoptotic signaling of heteromers is via a GPER-Gαs inhibitory complex and Gβγ
-
•
Heteromer formation impacts follicle maturation and FSH responses of IVF patients
Molecular Biology; Female Reproductive Endocrinology; Endocrine Regulation
Introduction
Ovarian follicular growth and dominance in women of reproductive age is a physiological example of how a tightly regulated equilibrium between active cell proliferation and apoptosis results in the selection of a single dominant follicle at the expense of all others. Key players of this game are sex hormones, follitropin (FSH) and 17β-estradiol (E2), which stimulate cell viability and proliferative signals in the gonads and certain tumor cells (Correia et al., 2015; Lizneva et al., 2019). Sex-hormone receptors are druggable targets in fertility and cancer treatment to control cell death and survival.
The FSH-receptor (FSHR) stimulates Gαs protein-dependent cAMP/PKA activation, resulting in cAMP-response element binding protein (CREB) phosphorylation and steroidogenic activity, necessary to produce estrogens that, in turn, are well-known stimulators of growth (Casarini and Crépieux, 2019). However, a pro-apoptotic role of FSH has also been proposed (Amsterdam et al., 1998, 2003), and, intriguingly, prolonged FSHR overexpression (Casarini et al., 2016) or accumulation of high intracellular cAMP levels (Aharoni et al., 1995; Yoshida et al., 2000) are a prerequisite for both steroid synthesis and cell death (Breckwoldt et al., 1996). The FSH-related pro-apoptotic activity occurs when high FSHR expression is induced (Casarini et al., 2016), providing a plausible reason why no consistent steroidogenic cell lines permanently overexpressing the FSHR exist so far (Casarini et al., 2018; Revankar et al., 2004). Other FSHR functions have been reported to be mediated by Gαi and Gαq proteins, the Gβγ dimer (Gloaguen et al., 2011; Ulloa-Aguirre et al., 2018), and other molecules inducing proliferative signals under low FSHR density in the cell membrane (Tranchant et al., 2011). FSHR-mediated activation of protein kinase B (AKT) occurs downstream of G protein activation (Gonzalez-Robayna et al., 2000; Sayers and Hanyaloglu, 2018) and results in anti-apoptotic and proliferative activity in FSHR-expressing ovarian (Rossi et al., 2017) and cancer (Chen et al., 2016) cells.
Estrogens activate two nuclear receptors, ERα and ERβ, and a membrane G-protein-coupled receptor (GPCR), named GPER, mediating rapid E2-induced intracellular responses such as calcium ion (Ca2+ mobilization (Revankar et al., 2005; Thomas et al., 2005)) and found in ovarian tissues throughout the follicular phase (Heublein et al., 2012). Although GPER is coupled to Gαs, it is unable to trigger intracellular cAMP accumulation in response to E2 in a number of cell models (Broselid et al., 2014). This is due to the interaction of GPER with both the membrane-associated guanylate kinases (MAGUK) and protein-kinase-A-anchoring protein 5 (AKAP5), constitutively inhibiting cAMP in a Gαi/o-independent manner (Broselid et al., 2014). However, opposite data demonstrating that the receptor may trigger cAMP activation were also provided in certain cell models (Filardo et al., 2007; Thomas et al., 2005), suggesting the existence of cell- or culture-specific mechanisms regulating GPER mode of action. GPER mediates survival/proliferation signals through phosphoinositide 3 (PI3K)/phospho-AKT (pAKT) and phospho-extracellular-regulated kinases 1 and 2 (pERK1/2) activation (Gonzalez de Valdivia et al., 2017), as well as Gβγ-dependent mechanisms upregulating proto-oncogenes (Maggiolini et al., 2004).
Ovarian granulosa cells express both FSHR and GPER (Prossnitz and Maggiolini, 2009) modulating a network of proliferative signals (Heublein et al., 2013; Pavlik et al., 2011) fundamental for regulating gametogenesis (Heublein et al., 2012). Consistent with its impact on cell growth, FSHR expression was found in pathological contexts characterized by uncontrolled cell proliferation, such as tumors (Choi et al., 2004) or endometriosis (Ponikwicka-Tyszko et al., 2016), suggesting that it could be a target for still outstanding anti-cancer therapies (Perales-Puchalt et al., 2019). Similarly, GPER expression has been described in several tumor cells (Filardo, 2018), including breast, endometrium, and ovary (Barton et al., 2018), where it may cooperate with FSHR in inducing uncontrolled cell proliferation (Heublein et al., 2013; Li et al., 2007).
Physiologically, high FSHR expression occurs transitorily in the ovarian granulosa cells, decreasing once a single dominant follicle is selected (Jeppesen et al., 2012). We reasoned that the opposing nature of FSHR activity, proliferative and pro-apoptotic, may rely on the cooperation with other factors modulating FSHR signaling. In particular, as described for other structurally similar GPCRs (Guitart et al., 2019; Ji et al., 2004; Jonas et al., 2018; Rivero-Müller et al., 2010), FSHR signaling may be regulated by forming heteromers with other receptors (Casarini et al., 2018). Here, we demonstrate that GPER and FSHR interactions reprogram FSHR-related death into life signals, upregulating the viability of FSHR-/GPER-expressing cells including human ovarian granulosa cells. Together, our data provide a mechanistic model for dominant follicle selection in humans and a novel therapeutic target for poor FSH-responder women undergoing controlled ovarian stimulation.
Results
FSH and cAMP Pro-apoptotic Potential Is Associated with FSHR Expression Levels and Is Counteracted by E2
We first evaluated the FSHR-related potential of activating steroidogenic and pro-apoptotic signals, likely associated with cAMP production (Aharoni et al., 1995; Casarini and Crépieux, 2019). Intracellular accumulation of cAMP depends on FSHR-Gαs protein coupling, which constitutively increases together with receptor expression levels (Tubio et al., 2010). This was demonstrated by employing Gαs- and FSHR-tagged BRET biosensors, which resulted in left-shifted BRET saturation curves at the maximal concentration of receptor-encoding plasmid used to transfect HEK293 (HEK293/FSHR) cells. This indicates a greater receptor-G protein association affinity is achieved when FSHR is highly expressed (Figures 1A and S1). The receptor expression-dependent association with Gαs was not observed with other G proteins reported to be FSHR intracellular interactors, such as Gαi and Gαq (Figures 1B–1E, S2 and S3). Although Gαs likely competes with other interactors for binding a low number of FSHRs, the potential of decreasing cell viability (Figure 1F) and of mediating both basal and FSH-induced cAMP activation was FSHR concentration dependent (Figure 1G). Increases in intracellular cAMP is deleterious for the viability of FSHR-expressing cells (Casarini et al., 2016). Indeed, treatment of either HEK293/FSHR or human primary granulosa cells with 8-br-cAMP induces reduction of cell viability in a concentration-dependent manner (Figures 1H and 1I). Interestingly, the cAMP-analog-induced decrease of granulosa cell viability was inhibited by 50 pg/ml E2 (Figure 1I), suggesting possible crosstalk between opposing gonadotropin- and estrogen-mediated intracellular actions. Indeed, in the absence of E2, FSH treatment induced a decrease of HEK293/FSHR cell viability (Figures 1J and 1K).
Figure 1.

Excessive FSHR Expression Levels Negatively Impact HEK293 Cell Viability
(A) Association of FSHR to the Gαs protein increases with higher receptor expression levels was assessed in HEK293 cells co-expressing the FSHR/rluc- (donor) and the Gαs protein/venus-tagged BRET biosensor. Data were interpolated by non-linear regression and compared by Kruskal Wallis test and Dunn's post-test (100 and 200 ng/well of FSHR-encoding plasmid versus the 25 ng/well condition; p = 0.0014; mean ± SEM; n = 5).
(B–E) Comparison of constitutive FSHR coupling to Gαs, Gαq, and Gαi with increasing receptor expression levels, in HEK293 cells transfected using 25 (B), 50 (C), 100 (D), and 200 (E) ng/well of FSHR/rluc-encoding plasmid. Receptor level-dependent increase in affinity occurs with Gαs but not with Gαq and Gαi proteins (Kruskal Wallis test and Dunn's post-test; p < 0.0001; mean ± SEM; n = 5).
(F) Transfected HEK293 cell viability increases at low FSHR-encoding plasmid amounts, whereas decreases at high plasmid concentration. Data were represented by box and whiskers plots (∗ = significantly different versus coupled mock-transfected sample; Kruskal Wallis test and Dunn's post-test; p < 0.05; n = 6)
(G) Basal and FSH-induced intracellular cAMP levels with increasing FSHR expression levels. Data are represented as mean ± SEM and interpolated by linear regression (n = 4).
(H) Colorimetric assay reveals the relationship between increasing intracellular cAMP concentration and decreasing of HEK293/FSHR cell viability. Cells were treated with increasing concentrations of 8-br-cAMP or 200 mM forskolin (∗ = significantly different versus 0.0 8-br-cAMP-treated samples; Kruskal Wallis test and Dunn's post-test; p = 0.0019).
(I) In human primary granulosa lutein cells, 50 pg/ml E2 co-treatment inhibits the decrease in cell viability induced by the cAMP analog (∗ = significantly different versus 0.0 8-br-cAMP-treated samples; Kruskal Wallis test and Dunn's post-test; p < 0.05; n = 8).
(J) Cell viability is lower in HEK293/FSHR transfected cells stimulated with FSH compared with vehicle (Mann-Whitney's U-test; p = 0.0003; n = 8).
(K) Summary: FSHR coupling to the Gαs protein/cAMP-pathway, occurring at high receptor expression levels and in the absence of E2, decreases cell viability. This action is counteracted by estrogen.
FSHR and GPER Form Heteromeric Complexes at the Cell Membrane
In order to define the mechanism linking FSH and GPER/estrogen-mediated intracellular networks, we employed distinct approaches to examine the formation of possible heterodimers/oligomers involving FSHR and GPER at the cell membrane. Investigation at the atomic level relied on a protein-protein docking-based approach, the FiPD-based approach (Casciari et al., 2006; Fanelli et al., 2013) applied to the structural models of the two receptors. The two independent docking runs by using FSHR as a target and GPER as a probe and vice versa converged on the same predicted architecture of the heterodimer (Figure 2A), ranked among the best 10 out of 4,000 solutions, and were characterized by good membrane topology. Remarkably, when using FSHR as a target, the predicted docking solution was the best in score (i.e. rank #1) out of 4,000, belonged to the most populated solution cluster, and showed a good membrane topology (MemTop) score (0.578) (see Methods). The FSHR-GPER interface in the predicted heterodimer is characterized by contacts between H6 and H7 from FSHR and H7 and H6 from GPER, respectively (Figure 2A).
Figure 2.

The FSHR Forms Heteromers with GPER
(A) Predicted structural model of the heterodimer between FSHR (green) and GPER (violet) seen in directions perpendicular (left) and parallel (right) to the bundle main axis. In this dimer, H6 of FSHR interacts with H7 of GPER and H6 of GPER interacts with H7 of FSHR.
(B) Western blotting for FSHR and GPER transient expression and co-expression in HEK293 cells, using validated receptor-specific antibodies (Figure S6). β-ACTIN was used as loading control.
(C–E) Representative confocal microscopy image of tagged-GPER and FSHR co-localization by immunofluorescence, in HEK293 cells transiently transfected with FSHR and GPER. A specific primary antibody was used for GPER followed by a TRITC-labelled secondary antibody, whereas nuclei were blue-stained by DAPI. FSHR was visualized by the venus tag. Bar = 25 μm.
(F) Formation of FSHR/rluc- and GPER/venus-tagged heteromers in transfected HEK293 cells. BRET ratio values resulting from molecular interactions were represented as mean ± SEM. Specific association is indicated by data interpolation using non-linear regression, which results in BRET saturation curve (n = 4).
(G and H) FSHR-GPER associations at the single-molecule level were visualized and quantitated by photo-activated localization microscopy with photo-activatable dyes (PD-PALM) in HEK293 cells.
(I) Representative reconstructed PD-PALM images of detected FLAG-GPER and HA-FSHR molecule at the plasma membrane. Images are reconstructed from 2-2 μm2 areas after x-y coordinate localization using QuickPALM followed by a 50 nm radius neighborhood analysis of receptor molecules. Scale bar represents 0.3 μm.
(J) Quantitative analysis of hetero-, homo-, and monomeric forms of FSHR and GPER when concomitantly expressed in HEK 293, using dual channel PD-PALM; mean ± SEM, n = 5.
(K) Quantitative evaluation of the types of FSHR and GPER homomers. Data are expressed as percentage of total receptor forms, including monomers; mean ± SEM, n = 10.
(L) Quantitative analysis of heteromeric assemblies between FSHR and GPER using dual channel PD-PALM reveals diverse heterodimeric complexes; mean ± SEM, n = 8 cells.
(M) Analysis of individual protomer composition within heterotrimers and heterotetramers demonstrates the preferential occurrence of FSHR protomers versus GPER protomers within these individual multimers; mean ± SEM, n = 8.
Western blotting and immunofluorescent staining of HEK293/FSHR-GPER cells confirmed expression of both untagged receptors in cell lysates (Figure 2B) and their co-localization at the cell surface (Figures 2C–2E). No signals were detected in GPER- and FSHR-negative cells (Figure 2E). The physical interaction between the two receptors was demonstrated by BRET. In transfected HEK293 cells, transiently expressing both FSHR-rluc and venus-tagged GPER (GPER/rluc) biosensors, a BRET saturation curve was observed with increasing acceptor concentration, indicating specific interactions between the two receptors (Figures 2F, S4, and S5). Further evidence of FSHR-GPER heteromer assembly at the plasma membrane was provided by total internal reflection microscopy (TIRF-M) and photo-activated localization microscopy with photoactivatable dyes (PD-PALM) (Figures 2G–2I), a super-resolution imaging approach we have previously employed to quantitate protomer composition within asymmetric heteromer complexes between LHCGR mutants and between FSHR and LHCGR at the plasma membrane (Jonas et al., 2015, 2018). HEK 293 cells expressing HA-tagged FSHR and FLAG-tagged GPER were labeled with CAGE500-conjugated anti-HA and CAGE552-conjugated anti-FLAG antibodies and fixed for PD-PALM with TIRF-M imaging (Figures 2G–2I). Both FSHR and GPER existed as pre-existing monomers, homomers, and heteromers (Figure 2J). Although both receptors formed a similar percentage of monomers, within the associated populations FSHR exhibited a greater number of homomers over heteromers, whereas conversely the predominant associated form for GPER was heteromers with FSHR. Furthermore, FSHR exhibited a range of homomeric complexes (from dimers, to a range of oligomers) similar to our previous reports with LHCGR (Jonas et al., 2015), whereas at a low level compared with all associations, the GPER-GPER complexes that were detected were primarily dimeric (Figure 2K). GPER-FSHR heteromers at the plasma membrane were also observed in cells co-expressing these receptors, consistent with the BRET data. The heteromer population consisted of heterodimers and a range of hetero-oligomeric complexes (Figure 2L). As we have previously demonstrated that receptor complexes >5 protomers are density dependent, whereas low-order oligomers are density independent (Jonas et al., 2015), we focused further analysis of the protomer identity within individual low-order hetero-oligomeric complexes. This revealed such complexes favored formation of FSHR dominant hetero-oligomers (Figure 2M), consistent with the “preference” for this receptor to exhibit diverse oligomeric forms, and also indicates potential asymmetry in these low-order hetero-oligomers. Overall, these data demonstrate that FSHR and GPER preform into distinct heteromeric assemblies at the plasma membrane and that GPER associations are preferentially formed with FSHR than with itself.
FSH Stimulation of FSHR-GPER Heteromers Promotes Cell Viability via Inhibition of cAMP Signaling
A potential functional association of GPER with FSHR in modulating cell viability was next investigated. HEK293/GPER expressing cells displayed a rapid intracellular Ca2+ increase (Figure 3A) occurring immediately upon 50 pg/mL estradiol addition, demonstrating expression of functional GPER (Revankar et al., 2005). BRET measurements also revealed that GPER can couple to Gαs (Figure 3B), although the receptor is unable to mediate cAMP production upon ligand binding (Figure 3C), consistent with previous reports demonstrating the association of GPER with a cAMP/PKA inhibitory complex assembled by MAGUK and AKAP5 proteins (Broselid et al., 2014). Intracellular cAMP levels were maximally stimulated upon FSH treatment of HEK293/FSHR cells but, interestingly, not in HEK293 cells co-expressing FSHR and GPER (Figure 3C), nor in GPER-expressing cells (Figures 3C and S7). These data indicate that the presence of GPER inhibits the increase of intracellular cAMP, independent of the presence or absence of E2, in FSHR-expressing cells. This effect is specifically targeted to FSHR, because LH treatment of LHCGR-GPER co-expressing HEK293 cells induces an increase in intracellular cAMP to similar levels as in the absence of GPER (Figure S8). Moreover, GPER-dependent inhibition of FSH-induced cAMP signaling occurs even under blockade of the nuclear estrogen receptor by fulvestrant (Figure 3D), thus excluding its involvement. The ability of GPER to inhibit FSH-mediated cAMP signaling was confirmed by measurement of FSH-induced CREB phosphorylation, a downstream cAMP-dependent event (Figure 3E).
Figure 3.

FSHR-GPER Crosstalk Promotes FSH-Stimulated Cell Viability via Gβγ Dimers
(A) GPER transiently expressed in HEK 293 cells simulates E2-induced intracellular Ca2+ increase (Kruskal-Wallis test; p < 0.0001; n = 8; mean ± SEM). Signals were captured by BRET over 150 s, in the presence of the calcium-biosensor. PBS (vehicle)-treated cells provided basal levels. Cells lysed by Triton X- were positive controls and only a 43-s time window is representatively shown. Compounds were added at the 21-s time point.
(B) GPER/rluc-coupling to the Gαs protein/venus-tagged was demonstrated by BRET. Values are mean ± SEM, and the logarithmic curve was obtained after interpolation using non-linear regression (n = 8).
(C) 10-nM FSH induced cAMP increase in HEK293 cells expressing either one or both FSHR and GPER. 50 pg/ml E2 were added as indicated, whereas mock-transfected and forskolin-treated cells served as basal and positive control, respectively. cAMP values are indicated as induced BRET changes over vehicle-treated mocks (∗ = significantly different versus FSH-treated mocks; two-way ANOVA with Dunnett's correction for multiple tests; p < 0.0001; n = 5; mean ± SEM).
(D) Evaluation of 10-nM FSH-induced intracellular cAMP increase, in the presence and in the absence of the nuclear estrogen receptor blockade by fulvestrant. ∗ = significantly different versus FSH-treated mocks; two-way ANOVA with Dunnett's correction for multiple tests; p < 0.0001; n = 5; means ± SEM.
(E) Representative Western blotting analysis of the cAMP-dependent pCREB activation, in HEK293 cells expressing either one or both FSHR and GPER. 50 pg/ml E2 were added where indicated and total ERK served as normalizer.
(F) Decrease of BRET signal indicating a reduced FSHR/rluc-Gαs protein/venus interaction along with increasing untagged GPER expression levels (n = 3; mean ± SEM). The receptor-tyrosine kinase colony-stimulating factor-1 receptor (cFMS) was the negative control.
(G) Proposed model of the FSHR/cAMP signaling blockade by the GPER-associated inhibitory machinery. The MAGUK/AKAP5 complex might physically interact with the Gαs protein coupled to FSHR upon heteromer formation, resulting in cAMP signaling blockade. At this point we hypothesized that FSH-dependent intracellular signals modulating cell viability could be activated by the βγ dimer.
(H) Evaluation of Gαs protein-dependence of pCREB and βγ dimer-dependence of 15-min pAKT activation in HEK293 cells expressing either one or both FSHR and GPER, respectively. The βγ dimer inhibitor gallein was used where indicated and total ERK was the normalizer.
(I) Semi-quantification of the 15-min pAKT signal obtained by western blotting in HEK293/FSHR-GPER cells treated by FSH (∗ = significantly different versus gallein-treated cells; Mann-Whitney's U-test; p = 0.002; n = 6; see Figure S9 for semi-quantification of pAKT signals).
(J) Decreasing of 10 nM FSH-induced cell viability in FSHR-GPER-co-expressing HEK293 cells due to βγ dimer-blockade by gallein. Results were compared with cell viability data from gallein-untreated cells and HEK293/FSHR cells of Figure 1J (cells transfected by 1 × 103 ng/well FSHR-encoding ± 1 × 103 ng/well GPER-encoding plasmid; ∗ = different versus mock; Kruskal-Wallis test; p < 0.0001; n = 8; mean ± SEM).
To determine if FSHR and Gαs basal coupling was altered in cells co-expressing GPER, FSHR-Gαs coupling was measured via BRET. Increasing levels of GPER decreased BRET signals between FSHR and Gαs (Figures 3F and S8). Although it is unknown whether the decay of the BRET signal corresponds to an uncoupling or structural rearrangement of the FSHR-Gαs complex, it is indicative of a physical perturbation of such complex by GPER. We hypothesized that the GPER/MAGUK/AKAP5 complex associates with FSHR upon heteromer formation perturbing FSHR-Gαs pre-coupling. This disables the ability of FSH to increase cAMP, while not affecting Gβγ-dependent signaling by FSH (Figure 3G). This hypothesis was explored by measuring Gβγ-dependent AKT phosphorylation by FSHR-GPER heteromers, in response to FSH (Figure 3H). Acute FSH treatment (15 min) did not induce pAKT activation in HEK293/FSHR cells, which requires chronic FSH stimulation to detect activation (Nechamen et al., 2007). In contrast, pAKT activation was increased in FSH-treated HEK293/FSHR-GPER cells and was inhibited by the selective Gβγ inhibitor gallein (Figure 3I). GPER also increased cell viability following FSH treatment in FSHR-GPER cells (Figure 3J), compared with HEK293/FSHR cells (Figure 1J), which critically was also inhibited by gallein. Taken together, these results demonstrate that GPER is capable of downregulating FSHR/Gαs/cAMP to increase cell viability via a Gβγ-dependent mechanism.
Disruption of FSHR-GPER Heteromer Formation Results in FSH-Induced Decrease in Cell Viability
To demonstrate that in cells co-expressing FSHR and GPER, FSH/FSHR-mediated increase in cAMP and cell viability was directly due to FSHR-GPER heteromer formation, a mutant GPER (GPER(mut)) was created that would not form heteromers with FSHR. To this purpose, the predicted structural model of the heterodimer was exploited to drive site-directed mutagenesis. All the H6 and H7 non-glycine and non-alanine amino acids facing FSHR were replaced by alanines, leading to a GPER form mutated in fifteen positions distributed along the whole length of the two helices, the last amino acid being the beginning of H8 (Figure 4A). GPER(mut) was still able to activate calcium signaling (Figure 4B) but was unable to form heteromers with FSHR as demonstrated by the lack of a saturated BRET signal (Figure 4C). GPER(mut) was expressed at the plasma membrane in cells co-expressing FSHR as detected by PD-PALM (Figures 4D–4F) and was expressed at similar levels to wild-type (WT) GPER (Figures 4G and S10). A dramatic reduction in the number of heteromeric associations occurred (from 40.3% ± 3.1% heteromers for WT GPER to 14.6% ± 3.4% for GPER(mut), Figures 2J and 4H)), while the homomer population which even with WT GPER was not altered (Figure 4H). Unexpectedly, the organization of GPER homomers resulted in an increase in the proportion of homodimers (Figure 4I), compared with WT GPER (Figure 2K), possibly suggesting that distinct homomer interface with a distinct affinity to the heteromer interface and/or mutation of H6-7 results in enhanced stabilization of a homodimer association. Critically, quantitative analysis of individual heteromer complexes revealed that while all forms were decreased, it was primarily formation of the hetero-oligomers that was impeded through mutation of GPER (Figure 4I). In cells co-expressing GPER(mut) and FSHR, 10-nM FSH treatment induced increases in intracellular cAMP (Figure 4J) and decreased cell viability when compared with FSH-treated cells co-expressing wild-type GPER and FSHR (Figure 4K). Therefore, inhibition of FSHR-mediated cAMP signaling and increased cell viability is due to a physical interaction between FSHR and GPER (Figure 4L).
Figure 4.

Decrease of Cell Viability by Disruption of FSHR-GPER Heteromers
(A) Side view, in a direction perpendicular to the bundle main axis, of the GPER structural model. The receptor regions are colored as follows: H1, H2, H3, H4, H5, H6, H7, and H8 are, respectively, blue, orange, green, pink, yellow, aquamarine, violet, and red; I1 and E1 are slate; I2 and E2 are gray; and I3 and E3 are magenta. The receptor amino acids participating in the interface with FSHR and subjected to alanine replacement are represented as spheres centered on the Cα-atom. They include Q255, R259, L262, L266, V267, V270, and V277 in H6, L304, T305, I308, L319, I323, and F326, and L327 in H7, and E329 in H8. To obtain a GPER(mut) molecule unable to form heteromers, interacting residues indicated in the box were changed to alanine by de novo DNA synthesis.
(B) Demonstration of GPER(mut) functionality by BRET using the calcium-biosensor, in transiently transfected HEK293 cells. The mutant receptor mediates E2-induced intracellular Ca2+ increase compared with vehicle, over 150 s (two-way ANOVA; p < 0.0001; n = 8; mean ± SEM). Compounds were injected at the 21-s time point.
(C) FSHR/rluc- and GPER(mut)/venus-tagged proteins do not form heteromers. BRET ratio values resulting from molecular interactions are represented as mean ± SEM, together with data from non-mutant GPER (Figure 2F). Specific association is indicated by data interpolation using linear regression (n = 4).
(D–I) Confirmation of GPER(mut) membrane localization and lack of heteromerization with FSHR.
(D–F) Representative reconstructed PD-PALM images and heatmap of associations following localization and neighborhood analysis. Scale bar = 0.3 μm
(G) Similar number of GPER and GPER(mut) receptors were expressed at the cell membrane (p = 0.06; Mann-Whitney's U-test).
(H) Quantitative analysis of hetero-, homo-, and monomeric forms of GPER(mut) when co-expressed with FSHR in HEK293, using dual channel PD-PALM (∗ = significantly different versus monomers; unpaired t test; p < 0.0001; mean ± SEM, n = 19).
(I) Quantitative evaluation of the types of GPER(mut) homomers and of heteromeric assemblies with FSHR as a percentage of all receptor forms, including monomers. Mean ± SEM, n = 8 cells.
(J) cAMP increase induced by 10 nM FSH, in HEK293 cells expressing either one or both FSHR and GPER(mut). Mock-transfected and forskolin-treated cells served as basal and positive control, respectively. cAMP values were measured by ELISA and indicated pmol/mL (∗ = significantly different versus FSH-treated mocks; two-way ANOVA with Tukey's correction for multiple tests; p ≤ 0.0106; n = 8; mean ± SEM).
(K) Comparison of HEK293/FSHR-GPER (Figure 3J) and HEK293/FSHR-GPER(mut) cell viability, under treatment with 10 nM FSH (∗ = different versus HEK293/FSHR-GPER; Mann-Whitney's U-test; p = 0.0003; n = 8; means ± SEM).
(L) Proposed model showing the inability of GPER(mut) to inhibit the activation of FSH-stimulated cAMP production with ensuing decrease of cell viability due to disruption of heteromerization.
GPER Requires the MAGUK/AKAP5 Complex to Inhibit FSHR-Mediated Decrease in Cell Viability
The role of known GPER-linked machinery (Broselid et al., 2014) in inhibiting FSHR-mediated cAMP response was evaluated in HEK293 cells via a genome editing approach. AKAP5 was knocked-out in HEK 293 cells by CRISPR/Cas9 (AKAP5-KO HEK293 cells) (Figures 5A, S11, and S12). In these cells, GPER still exhibited heteromer formation with FSHR, as measured by BRET (Figure 5B). However, FSH treatment of AKAP5-KO HEK293 cells, co-expressing GPER and FSHR, induced intracellular cAMP generation (Figure 5C), demonstrating AKAP5 is essential in exerting GPER-dependent inhibition of cAMP production occurring via FSHR. Consistent with the cAMP data, the GPER rescue of cell viability requires AKAP5 (Figure 5D), strengthening the role of this inhibitory machinery in counteracting FSHR activity by GPER. Therefore, one of the molecular mechanisms regulating FSHR-activation of Gαs protein-dependent signals requires the association of GPER and AKAP5, as cells expressing FSHR/GPER, but lacking AKAP5, are able to generate cAMP and thus reduce FSHR-dependent cell viability (Figure 5E).
Figure 5.

Cell Viability Decreases upon Disrupting the GPER-Associated MAGUK/AKAP5 Molecular Inhibitory Complex
(A) Model depicting the development of the AKAP5-KO HEK293 cell line by CRISPR/Cas9. The absence of AKAP5 leads to disruption of the GPER-associated inhibitory machinery without impairing FSHR-GPER heteromer formation.
(B) FSHR/rluc-GPER/venus-tagged heteromer formation evaluated by BRET, in AKAP5-KO HEK293 cells. Values are expressed as mean ± SEM and interpolated by non-linear regression (n = 4).
(C) Intracellular cAMP increase following 10 nM FSH-treatment of AKAP5-KO HEK293 cells, transiently expressing FSHR and/or GPER. Fifty pg/ml E2 were added where indicated and signals acquired by BRET. ∗ = significantly different versus FSH-treated mocks; two-way ANOVA with Tukey's post-hoc test; p < 0.0001; n = 6; means ± SEM.
(D) Viability of FSHR- and/or GPER-expressing AKAP5-KO HEK293 cells in the presence and in the absence of 10 nM FSH (∗ = different versus mock; two-way ANOVA with Dunnett's post-hoc test; p < 0.0001; n = 8; means ± SEM).
(E) Model describing the failure of the inhibition of FSHR/Gαs protein signaling and cell viability decrease, in the absence of AKAP5.
FSHR and GPER Co-Expression Is Linked to Ovarian Follicle Maturation: In Vivo, Pharmacological Relevance in an Assisted Reproduction Setting
To investigate if FSHR-GPER co-expression may have physiological and clinical relevance, the levels of these receptors at mRNA and protein were evaluated in granulosa cells collected from ovarian follicular fluids of women undergoing FSH stimulation for assisted reproduction techniques (ART). The presence of both FSHR and GPER protein in granulosa cells was demonstrated by immunostaining of human ovarian follicle tissue sections (Figures 6A and 6B) following antibody validation (Figure S6). This was confirmed by Western blotting of human granulosa and transfected HEK293 cell lysates, in the presence and in the absence of GPER mRNA depletion by siRNA (Figure 6C), where the anti-FSHR or anti-GPER antibodies produced a signal in cells expressing only these receptors (Figure 6C).
Figure 6.

Link Between FSHR-GPER Co-expression and Follicular Growth Response to FSH
(A and B) Representative determination of FSHR and GPER expression in granulosa cells at the antral follicular stage, by immunohistochemistry. Ovarian sections were treated by specific anti-FSHR or -GPER primary antibodies, over hematoxylin background staining (bar = 200 μm).
(C) Western blotting demonstrating the presence of both FSHR and GPER in primary human granulosa cell lysates and the inhibition of expression of GPER by 48-h siRNA. HEK293 transiently transfected with FSHR- and GPER-encoding plasmids were used as controls. The efficacy of GPER siRNA is shown and compared with control siRNA (mock)-treated granulosa cells. β-ACTIN was used as a loading control.
(D) Correlation between FSHR and GPER gene expression levels in granulosa cells collected from donor normo- (n = 61) and poor-responder (n = 30) women undergoing FSH stimulation for assisted reproduction. Each patient is represented by a point, and mRNA levels were measured by real-time PCR, normalized over the RPS7 housekeeping gene and interpolated by linear regression. Immunohistochemistry and uncropped western blotting for antibody validation are provided as supplemental materials (Figure S6).
(E–J) Representative images of GPER and FSHR co-localization in 48-h mock- (E-G) and GPER siRNA-treated (H-J) human granulosa cells, detected by immunofluorescence. Specific primary antibody was used for FSHR and GPER binding, as well as TRITC- (E, H) and FITC-labelled (F, I) secondary antibodies, respectively. Nuclei (blue) were stained by DAPI (G, J). Bar = 25 μm.
(K) Intracellular cAMP levels measured in control and GPER siRNA-treated granulosa cells by ELISA, in the presence or absence of 10 nM FSH. Data are represented by box and whiskers plots (∗ = different versus vehicle/mock-treated cells; two-way ANOVA and Sidak's multiple comparisons test; p ≤ 0.0074; n = 8).
(L) Granulosa cell viability after 48-h treatment with control/GPER siRNA. Effects of 10-nM FSH were also assessed 24 h before measurements (∗ = different versus vehicle/mock-treated cells; Kruskal-Wallis with Dunn's correction for multiple tests; p = 0.0002; n = 12).
(M) Evaluation of procaspase 3 cleavage in granulosa cells under 48-h GPER depletion by siRNA. 10-nM FSH was added 24 h before analysis, as indicated, while total ERK was the loading control.
(N) Progesterone levels measured in media of control and GPER siRNA-treated granulosa cells, maintained 24 h in the presence or in the absence of 10 nM FSH, by immunoassay. (∗ = different versus vehicle/mock-treated cells; two-way ANOVA and Fisher's test; p ≤ 0.001; n = 6).
(O) Model describing the FSHR/Gαs protein-dependent activation of the steroidogenic/apoptotic pathway, under GPER depletion by siRNA.
(P) Correlation between oocyte number and the ratio between E2 serum levels and cumulative FSH dose of normo- (n = 61) and poor-responder (n = 30) women. Patients are represented by points, and data were interpolated using linear regression.
FSHR/GPER mRNA expression levels were matched with clinical parameters indicative of in vivo proliferation. A group of 91 women were subdivided into two groups; “normo-responders” were defined as women providing >4 oocytes upon controlled ovarian stimulation, whereas “poor responders” as women providing ≤4 oocytes (Ferraretti et al., 2011; Polyzos and Sunkara, 2015). Granulosa cells from donor women were cultured for one week to enable cells to recover following gonadotropin hyperstimulation during ART procedure and mRNA levels of FSHR and GPER quantified, plotted in an X-Y graph, and interpolated by non-linear regression. In normo-responder women, FSHR expression increased linearly together with GPER transcripts (Figures 6D, S13, and S14). The functional importance of such a ratio is supported by the finding that in FSH poor responders, low oocyte yield is correlated to higher FSHR over GPER expression, suggesting this may impact cell survival.
The role of GPER in counteracting intracellular death signals through crosstalk with FSHR was confirmed using human primary granulosa cells, where the effects of FSH were evaluated in the presence and absence of GPER via siRNA (Figures 6C and 6E–6J). These data suggest FSHR-GPER interaction occurring also in hGLC, as confirmed by proximity ligation assay (Figure S15). Interestingly, GPER-depleted granulosa cells exhibited both increased basal and FSH-induced cAMP production, compared with mock-treated cells (Figure 6K), and similar results were obtained in AKAP5 siRNA-treated cells (Figure S16). Moreover, 10-nM FSH treatment reduced cell viability following GPER depletion (Figure 6L) and induced procaspase 3 cleavage as detected by Western blotting (Figure 6M), suggesting the protective role of GPER from gonadotropin-induced cell death. In these experiments, all samples were maintained under LHCGR depletion via siRNA, as LHCGR has been reported to negatively regulate cAMP signaling from FSHR via LHCGR/FSHR heteromers (Feng et al., 2013). As increases in intracellular cAMP (Figure 6K) is known to drive steroid synthesis in granulosa cells, the amount of progesterone secreted in FSH-treated cells was also significantly increased in human granulosa cells depleted of GPER (Figure 6N), thus supporting previously proposed links between steroidogenic and pro-apoptotic pathways in ovarian cells (Amsterdam et al., 2003; Breckwoldt et al., 1996).
These data strongly support a functional requirement of FSHR-GPER associations in the ovary. As we have shown that proportional amounts of both receptor transcripts correlate with high oocyte yield (Figure 6D), in FSH poor responders, low oocyte number is correlated to higher FSHR over GPER expression, presumably resulting in deleterious FSHR/cAMP-dependent effects for cell survival (Figure 6O). The concept is further assessed by plotting biochemical parameters against the number of oocytes achieving maturation after controlled ovarian stimulation procedures (Figure 6P). The ratio between estradiol levels and cumulative FSH dose, assumed to be indicative of the steroid capability to inhibit the selective pressure via activation of proliferative and anti-apoptotic signals in growing oocytes, are correlated in normo-responder women, whereas they are not in poor responders who have lower levels of GPER (Figure 6P). These data further support our findings in human granulosa cells, whereby the reduction of GPER results in enhanced FSH-mediated steroidogenesis but increased loss of cell viability and subsequent decrease in oocyte yield in these patients.
Discussion
This work demonstrates that GPER heteromerization with FSHR shifts the preferential signal transduction from cAMP to pAKT activation, reducing cAMP-dependent apoptosis and favoring cell survival. Our results demonstrate novel aspects of FSHR function, by extending the number of transmembrane partners of FSHR to steroid hormone GPCRs. We also identify the mechanism underpinning GPER-FSHR heteromer inhibition of cAMP signaling is via the GPER-associated MAGUK-AKAP5 protein complex. Critically, our data demonstrate important implications for ovarian physiology and FSH use in fertility treatment, identifying for the first time that FSHR-GPER heteromer represent a druggable target to regulate cell death and survival and improve fertility outcomes in poor FSH responders.
The ability of FSHR-GPER to form heteromers was demonstrated complementarily using different approaches and, interestingly, revealed that GPER formed minimal homomeric associations and preferential formation with FSHR in complexes containing a higher number of FSHR than GPER molecules. In that respect, as FSH is thought to bind and activate FSHR trimers (Jiang et al., 2014), our data could support a role for these asymmetric lower order hetero-oligomers in regulating cell viability. This inhibitory role of GPER on cAMP is specific to FSHR, because GPER had no effect on LHCGR-mediated cAMP, reflecting structural differences between FSHR and LHCGR, in line with the different and specific physiological role of LH (Casarini et al., 2012, 2017). Moreover, it is worth of note that LHCGR-mediated cAMP increase is not linked to cell death, likely due to anti-apoptotic pathways simultaneously activated by the ligand (Casarini et al., 2012, 2016, 2017). Upon FSHR and GPER co-expression, disruption or structural rearrangements of the FSHR-Gαs protein interaction occurs. These rearrangements are GPER dependent and negatively impact the FSH-induced cAMP production via the GPER-related anchoring complex AKAP5 (Broselid et al., 2014). Data were confirmed in FSHR-GPER co-expressing human primary granulosa cells, where siRNA knockdown of native GPER expression enhanced the FSHR-mediated cAMP steroidogenic pathway (Broselid et al., 2014) and negatively impacted cell viability. Although our data support the lack of E2-induced cAMP increase via GPER, opposite findings were also obtained (Filardo et al., 2007; Thomas et al., 2005). We could speculate that GPER-mediated cAMP increase could depend on specific conditions, such as the cell type, MAGUK/AKAP5 expression levels, or the type of estrogen binding the receptor. Intact FSHR-GPER complexes responded to FSH treatment by stimulating acute βγ-dependent pAKT activation, which has been associated with cell migration and proliferative events in several cell models (Kamal et al., 2014; Matoba et al., 2018; Ouelaa-Benslama et al., 2012; Surve et al., 2014), and were proposed as a target for tumor-suppressing therapies (Cantley and Neel, 1999). We found this FSHR-associated pAKT activation occurs relatively rapidly upon cell treatment by FSH, in contrast to what has been previously described (Gloaguen et al., 2011). Overall, our findings fulfill the recently proposed criteria for demonstrating functional GPCR heteromers in native tissue: (a) receptor co-localization/interaction, (b) exhibition of distinct functional properties by heteromers compared with protomers, and (c) loss of heteromer-specific properties upon heteromer disruption (Gomes et al., 2016); in our study this was via knockdown of GPER in primary human granulosa cells; equivalent findings were observed with the GPER(mut) that identified TM6-7 as the heteromer interface that when mutated could specifically disrupt interactions with FSHR. To date, these criteria have only been fulfilled by a small subset of family A GPCR heteromers (Gomes et al., 2016). Intriguingly, GPER(mut) homodimers were more prevalent compared with WT GPER, perhaps suggesting a distinct homodimer interface that exhibits a distinct affinity compared with the heteromer TM6-7 interface with FSHR, and/or the GPER(mut) perhaps stabilizes a receptor conformation that results in more stable homomer associations. It is important to note, however, that this mutation, even with altered homomeric associations, did not alter the ability of GPER to activate estradiol-induced calcium signaling.
Although proliferative and anti-apoptotic signals are preferentially activated at relatively low FSHR levels (Tranchant et al., 2011), the steroidogenic/pro-apoptotic pathway is stimulated in the presence of increasing receptor number. This is due to concentration-dependent variations of FSHR association affinity with its different intracellular effectors, as shown for other GPCRs (Bates et al., 2006), which is similar across different G proteins at low receptor expression levels, whereas preferential coupling to Gαs protein and activation of cAMP signaling occurs at high receptor expression levels (Tranchant et al., 2011). In addition, the findings in this study suggest this is likely due to a higher level of FSHR monomers/homomers over FSHR-GPER heteromers. A greater proportion of FSHR only complexes would thus program the cell fate toward FSHR/cAMP-dependent death, a finding well described in the literature (Aharoni et al., 1995; Amsterdam et al., 1998; Breckwoldt et al., 1996; Casarini et al., 2016; Maillet et al., 2002; Sasson et al., 2003; Sirotkin et al., 2008, 2018; Tajima et al., 2002) but neglected for a long-time due to the lack of evidence on the pro-apoptotic action of FSH in vivo. Indeed, FSH action is commonly associated with proliferative events, follicular growth being the physiological example, and as suggested by the presence of FSHR in pathological contexts, characterized by uncontrolled cell growth, such as cancer (Choi et al., 2004) or endometriosis (Ponikwicka-Tyszko et al., 2016), which indicated FSHR as a target of anti-cancer drugs (Perales-Puchalt et al., 2019).
The proliferative role of FSH is well known, and indeed this hormone is used as a drug for inducing controlled ovarian stimulation and in the clinical setting of infertility treatment (Behre, 2019; Santi et al., 2018). On the other hand, the physiological FSH action is directed to estrogen biosynthesis, enhancing cell growth in vivo (Wallach et al., 1996), and it is not surprising that both hormones are placed within proliferative contexts. We suggest that the formation of FSHR-GPER heteromers may be involved in the endocrine regulation of ovarian physiology (Hillier, 1994), providing a molecular mechanism explaining why one follicle becomes dominant although it is exposed to the same hormonal milieu of other follicles becoming atretic. Accordingly, FSHR-GPER heteromers would support dominance and further growth, whereas the lack/insufficiency of GPER expression, and thus GPER-FSHR heteromers, would direct FSH action toward apoptosis in the follicles, which become atretic. Conversely, GPER KO mice feature pathological conditions such as altered glucose and lipid metabolism (Sharma and Prossnitz, 2016) and tumorigenesis (Marjon et al., 2014), without any specific reproductive phenotype (Prossnitz and Hathaway, 2015). However, the mouse is a multiovulatory species, and species-specific mechanisms underlying the endocrine regulation of multi- versus mono-ovulation are different (Driancourt et al., 1991; Webb et al., 2016). Whether FSHR-GPER heteromerization results from the evolution in mono-ovulatory mammals is a topic of future studies.
The ability of these heteromers to modulate opposing apoptotic and proliferative pathways could have implications in hormone-dependent cancers. A number of pro-apoptotic or anti-proliferative actions have been previously associated with FSHR function (Casarini and Crépieux, 2019; Casarini et al., 2016), especially in conditions of high receptor expression levels, similar to other GPCRs (Revankar et al., 2004). Our data support a mechanism by which FSHR-GPER heteromeric complexes occur and function in certain receptor-expressing tumor cells (Heublein et al., 2013), inhibiting the pro-apoptotic, cAMP pathway and enhancing the activation of proliferative signals by FSH, thereby upregulating tumor growth. A similar mechanism was previously described for other GPCRs (Moreno et al., 2014; Rozenfeld et al., 2011). Patients affected by ovarian carcinoma co-expressing FSHR and GPER have lower prognosis than those with cancer cells expressing FSHR or GPER alone (Heublein et al., 2013). FSHR-GPER heteromerization should be investigated in tumor tissues expressing both receptors, thus representing a potential target for specific drug design. The structural model of the FSHR-GPER interaction predicted and supported by the ability to disrupt the proposed interface at H6-7 may provide a rationale for future structure-based drug design/discovery.
In terms of clinical applications, our study provides evidence for the potential application of these results in improving outcomes in assisted reproduction and infertility treatment. The possibility to boost follicular growth and maturation in women who are poor responders to ovarian stimulation with FSH (e.g. due to advanced age) is a current challenge in reproductive medicine. Novel biologicals could be designed to specifically and transitorily favor FSHR-GPER oligomerization and, thereby, follicular growth and rescue. Indeed, our data suggest that women who are poor responders to FSH stimulation in an assisted reproduction program exhibit low expression of GPER that is not correlated with FSHR expression, possibly reflecting reduced ability to form heteromers and thus an insufficient pro-proliferative FSH action in such conditions.
In summary, we have demonstrated that GPER shifts the pro-apoptotic effects of high FSHR expression levels toward upregulation of cell viability. This occurs via formation of heteromeric complexes capable of inhibiting the cAMP pathway but stimulating pAKT, deviating FSHR coupling from Gαs to Gβγ. Our data provide a novel and promising target for improving both infertility treatment and cancer therapy, through the development of drugs favoring or inhibiting FSHR-GPER heteromer function, respectively.
Limitations of the Study
Although gene expression data in granulosa-luteal cells from women undergoing ART were provided, this is an in vitro-based study and receptor-receptor heteromer formation would need further evaluation in ovarian follicles of different developmental stages.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Livio Casarini: livio.casarini@unimore.it.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Data that support the findings of this study are available from the Lead Contact on reasonable request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by the Italian Ministry of University and Research (MIUR). M.S. is an LE STUDIUM RESEARCH FELLOW, Loire Valley Institute for Advanced Studies, Orléans & Tours, France, - INRA - Center Val de Loire, 37380 Nouzilly, France, receiving funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 665790. We would like to thank Dr Andreas Bruckbauer at the Facility for Imaging of Light Microscopy (FILM), Imperial College London, for technical support with PALM. A.C.H. was supported by grants from the BBSRC (BB/1008004/1) and Genesis Research Trust, N.S.S is supported by an Imperial College London President's Scholarship. Grant “Departments of Excellence Program” from MIUR to the Department of Biomedical, Metabolic and Neural Sciences (University of Modena and Reggio Emilia). Polish National Science Centre (NCN) grants: DEC-2015/17/B/NZ1/01777, DEC-2017/25/B/NZ4/02364.
Author Contributions
LC designed the study, managed experiments, performed data analysis and interpretation, and wrote the manuscript. CL, EP, SL, and LR performed BRET and western blotting experiments and data analysis. SS, and BM performed BRET and gene expression analysis. SM did immunostainings. CA performed gene expression analysis. NSS have applied the PALM method. JC created the CRISP/Cas9-modified cells. GB, FP, ALM, and MTV provided scientific support, primary cells and tissues, and manuscript editing. ML and GO provided scientific support, data interpretation, and manuscript editing. FGK was involved in the management of immunostainings and manuscript editing. FF did bioinformatics analyzes, data interpretation, and manuscript editing. ARM managed CRISPR/Cas9 experiments, supported data analysis, and did manuscript editing. ACH supported experiments and study design, provided data interpretation, scientific support, and manuscript writing. MS provided study and scientific management, data interpretation, and manuscript writing.
Declaration of Interests
ML and GO are Merck Serono SpA employees without any conflict of interest.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101812.
Supplemental Information
References
- Aharoni D., Dantes A., Oren M., Amsterdam A. cAMP-mediated signals as determinants for apoptosis in primary granulosa cells. Exp. Cell Res. 1995;218:271–282. doi: 10.1006/excr.1995.1156. [DOI] [PubMed] [Google Scholar]
- Amsterdam A., Dantes A., Hosokawa K., Schere-Levy C.P., Kotsuji F., Aharoni D. Steroid regulation during apoptosis of ovarian follicular cells. Steroids. 1998;63:314–318. doi: 10.1016/s0039-128x(98)00016-6. [DOI] [PubMed] [Google Scholar]
- Amsterdam A., Sasson R., Keren-Tal I., Aharoni D., Dantes A., Rimon E., Land A., Cohen T., Dor Y., Hirsh L. Alternative pathways of ovarian apoptosis: death for life. Biochem. Pharmacol. 2003;66:1355–1362. doi: 10.1016/s0006-2952(03)00485-4. [DOI] [PubMed] [Google Scholar]
- Barton M., Filardo E.J., Lolait S.J., Thomas P., Maggiolini M., Prossnitz E.R. Twenty years of the G protein-coupled estrogen receptor GPER: historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018;176:4–15. doi: 10.1016/j.jsbmb.2017.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates B., Zhang L., Nawoschik S., Kodangattil S., Tseng E., Kopsco D., Kramer A., Shan Q., Taylor N., Johnson J. Characterization of Gpr101 expression and G-protein coupling selectivity. Brain Res. 2006;1087:1–14. doi: 10.1016/j.brainres.2006.02.123. [DOI] [PubMed] [Google Scholar]
- Behre H.M. Clinical use of FSH in male infertility. Front. Endocrinol. (Lausanne). 2019;10:322. doi: 10.3389/fendo.2019.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckwoldt M., Selvaraj N., Aharoni D., Barash A., Segal I., Insler V., Amsterdam A. Expression of Ad4-BP/cytochrome P450 side chain cleavage enzyme and induction of cell death in long-term cultures of human granulosa cells. Mol. Hum. Reprod. 1996;2:391–400. doi: 10.1093/molehr/2.6.391. [DOI] [PubMed] [Google Scholar]
- Broselid S., Berg K.A., Chavera T.A., Kahn R., Clarke W.P., Olde B., Leeb-Lundberg L.M.F. G protein-coupled receptor 30 (GPR30) forms a plasma membrane complex with membrane-associated guanylate kinases (MAGUKs) and protein kinase A-anchoring protein 5 (AKAP5) that constitutively inhibits cAMP production. J. Biol. Chem. 2014;289:22117–22127. doi: 10.1074/jbc.M114.566893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley L.C., Neel B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarini L., Crépieux P. Molecular mechanisms of action of FSH. Front. Endocrinol. (Lausanne). 2019;10:305. doi: 10.3389/fendo.2019.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarini L., Lispi M., Longobardi S., Milosa F., La Marca A., Tagliasacchi D., Pignatti E., Simoni M. LH and hCG action on the same receptor results in quantitatively and qualitatively different intracellular signalling. PLoS One. 2012;7:e46682. doi: 10.1371/journal.pone.0046682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarini L., Reiter E., Simoni M. β-arrestins regulate gonadotropin receptor-mediated cell proliferation and apoptosis by controlling different FSHR or LHCGR intracellular signaling in the hGL5 cell line. Mol. Cell. Endocrinol. 2016;437:11–21. doi: 10.1016/j.mce.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Casarini L., Riccetti L., De Pascali F., Gilioli L., Marino M., Vecchi E., Morini D., Nicoli A., La Sala G.B., Simoni M. Estrogen modulates specific life and death signals induced by LH and hCG in human primary granulosa cells in vitro. Int. J. Mol. Sci. 2017;18:926. doi: 10.3390/ijms18050926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarini L., Santi D., Simoni M., Potì F. “Spare” luteinizing hormone receptors: facts and fiction. Trends Endocrinol. Metab. 2018;29:208–217. doi: 10.1016/j.tem.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Casciari D., Seeber M., Fanelli F. Quaternary structure predictions of transmembrane proteins starting from the monomer: a docking-based approach. BMC Bioinformatics. 2006;7:340. doi: 10.1186/1471-2105-7-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Bai M., Ning C., Xie B., Zhang J., Liao H., Xiong J., Tao X., Yan D., Xi X. Gankyrin facilitates follicle-stimulating hormone-driven ovarian cancer cell proliferation through the PI3K/AKT/HIF-1α/cyclin D1 pathway. Oncogene. 2016;35:2506–2517. doi: 10.1038/onc.2015.316. [DOI] [PubMed] [Google Scholar]
- Choi J.-H., Choi K.-C., Auersperg N., Leung P.C.K. Overexpression of follicle-stimulating hormone receptor activates oncogenic pathways in preneoplastic ovarian surface epithelial cells. J. Clin. Endocrinol. Metab. 2004;89:5508–5516. doi: 10.1210/jc.2004-0044. [DOI] [PubMed] [Google Scholar]
- Correia S., Cardoso H.J., Cavaco J.E., Socorro S. Oestrogens as apoptosis regulators in mammalian testis: angels or devils? Expert Rev. Mol. Med. 2015;17:e2. doi: 10.1017/erm.2014.25. [DOI] [PubMed] [Google Scholar]
- Driancourt M.A., Webb R., Fry R.C. Does follicular dominance occur in ewes? J. Reprod. Fertil. 1991;93:63–70. doi: 10.1530/jrf.0.0930063. [DOI] [PubMed] [Google Scholar]
- Fanelli F., Seeber M., Felline A., Casciari D., Raimondi F. Quaternary structure predictions and structural communication features of GPCR dimers. Prog. Mol. Biol. Transl. Sci. 2013;117:105–142. doi: 10.1016/B978-0-12-386931-9.00005-2. [DOI] [PubMed] [Google Scholar]
- Feng X., Zhang M., Guan R., Segaloff D.L. Heterodimerization between the lutropin and follitropin receptors is associated with an attenuation of hormone-dependent signaling. Endocrinology. 2013;154:3925–3930. doi: 10.1210/en.2013-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraretti A.P., La Marca A., Fauser B.C.J.M., Tarlatzis B., Nargund G., Gianaroli L., ESHRE working group on Poor Ovarian Response Definition ESHRE consensus on the definition of “poor response” to ovarian stimulation for in vitro fertilization: the Bologna criteria. Hum. Reprod. 2011;26:1616–1624. doi: 10.1093/humrep/der092. [DOI] [PubMed] [Google Scholar]
- Filardo E.J. A role for G-protein coupled estrogen receptor (GPER) in estrogen-induced carcinogenesis: dysregulated glandular homeostasis, survival and metastasis. J. Steroid Biochem. Mol. Biol. 2018;176:38–48. doi: 10.1016/j.jsbmb.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Filardo E., Quinn J., Pang Y., Graeber C., Shaw S., Dong J., Thomas P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148:3236–3245. doi: 10.1210/en.2006-1605. [DOI] [PubMed] [Google Scholar]
- Gloaguen P., Crépieux P., Heitzler D., Poupon A., Reiter E. Mapping the follicle-stimulating hormone-induced signaling networks. Front. Endocrinol. (Lausanne). 2011;2:45. doi: 10.3389/fendo.2011.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I., Ayoub M.A., Fujita W., Jaeger W.C., Pfleger K.D.G., Devi L.A. G protein-coupled receptor heteromers. Annu. Rev. Pharmacol. Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Robayna I.J., Falender A.E., Ochsner S., Firestone G.L., Richards J.S. Follicle-Stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-lnduced kinase (Sgk): evidence for A kinase-independent signaling by FSH in granulosa cells. Mol. Endocrinol. 2000;14:1283–1300. doi: 10.1210/mend.14.8.0500. [DOI] [PubMed] [Google Scholar]
- Gonzalez de Valdivia E., Broselid S., Kahn R., Olde B., Leeb-Lundberg L.M.F. G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J. Biol. Chem. 2017;292:9932–9943. doi: 10.1074/jbc.M116.765875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X., Moreno E., Rea W., Sánchez-Soto M., Cai N.-S., Quiroz C., Kumar V., Bourque L., Cortés A., Canela E.I. Biased G protein-independent signaling of dopamine D1-D3 receptor heteromers in the nucleus accumbens. Mol. Neurobiol. 2019;56:6756–6769. doi: 10.1007/s12035-019-1564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heublein S., Lenhard M., Vrekoussis T., Schoepfer J., Kuhn C., Friese K., Makrigiannakis A., Mayr D., Jeschke U. The G-protein-coupled estrogen receptor (GPER) is expressed in normal human ovaries and is upregulated in ovarian endometriosis and pelvic inflammatory disease involving the ovary. Reprod. Sci. 2012;19:1197–1204. doi: 10.1177/1933719112446085. [DOI] [PubMed] [Google Scholar]
- Heublein S., Mayr D., Vrekoussis T., Friese K., Hofmann S.S., Jeschke U., Lenhard M. The G-protein coupled estrogen receptor (GPER/GPR30) is a gonadotropin receptor dependent positive prognosticator in ovarian carcinoma patients. PLoS One. 2013;8:e71791. doi: 10.1371/journal.pone.0071791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier S.G. Current concepts of the roles of follicle stimulating hormone and luteinizing hormone in folliculogenesis. Hum. Reprod. 1994;9:188–191. doi: 10.1093/oxfordjournals.humrep.a138480. [DOI] [PubMed] [Google Scholar]
- Jeppesen J.V., Kristensen S.G., Nielsen M.E., Humaidan P., Dal Canto M., Fadini R., Schmidt K.T., Ernst E., Yding Andersen C. LH-receptor gene expression in human granulosa and cumulus cells from antral and preovulatory follicles. J. Clin. Endocrinol. Metab. 2012;97:E1524–E1531. doi: 10.1210/jc.2012-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji I., Lee C., Jeoung M., Koo Y., Sievert G.A., Ji T.H. Trans-activation of mutant follicle-stimulating hormone receptors selectively generates only one of two hormone signals. Mol. Endocrinol. 2004;18:968–978. doi: 10.1210/me.2003-0443. [DOI] [PubMed] [Google Scholar]
- Jiang X., Fischer D., Chen X., McKenna S.D., Liu H., Sriraman V., Yu H.N., Goutopoulos A., Arkinstall S., He X. Evidence for follicle-stimulating hormone receptor as a functional trimer. J. Biol. Chem. 2014;289:14273–14282. doi: 10.1074/jbc.M114.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas K.C., Fanelli F., Huhtaniemi I.T., Hanyaloglu A.C. Single molecule analysis of functionally asymmetric G protein-coupled receptor (GPCR) oligomers reveals diverse spatial and structural assemblies. J. Biol. Chem. 2015;290:3875–3892. doi: 10.1074/jbc.M114.622498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas K.C., Chen S., Virta M., Mora J., Franks S., Huhtaniemi I., Hanyaloglu A.C. Temporal reprogramming of calcium signalling via crosstalk of gonadotrophin receptors that associate as functionally asymmetric heteromers. Sci. Rep. 2018;8:2239. doi: 10.1038/s41598-018-20722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal F.A., Mickelsen D.M., Wegman K.M., Travers J.G., Moalem J., Hammes S.R., Smrcka A.V., Blaxall B.C. Simultaneous adrenal and cardiac g-protein-coupled receptor-gβγ inhibition halts heart failure progression. J. Am. Coll. Cardiol. 2014;63:2549–2557. doi: 10.1016/j.jacc.2014.02.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ganta S., Cheng C., Craig R., Ganta R.R., Freeman L.C. FSH stimulates ovarian cancer cell growth by action on growth factor variant receptor. Mol. Cell. Endocrinol. 2007;267:26–37. doi: 10.1016/j.mce.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizneva D., Rahimova A., Kim S.-M., Atabiekov I., Javaid S., Alamoush B., Taneja C., Khan A., Sun L., Azziz R. FSH beyond fertility. Front. Endocrinol. (Lausanne). 2019;10:136. doi: 10.3389/fendo.2019.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggiolini M., Vivacqua A., Fasanella G., Recchia A.G., Sisci D., Pezzi V., Montanaro D., Musti A.M., Picard D., Andò S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004;279:27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- Maillet G., Bréard E., Benhaïm A., Leymarie P., Féral C. Hormonal regulation of apoptosis in rabbit granulosa cells in vitro: evaluation by flow cytometric detection of plasma membrane phosphatidylserine externalization. Reproduction. 2002;123:243–251. doi: 10.1530/rep.0.1230243. [DOI] [PubMed] [Google Scholar]
- Marjon N.A., Hu C., Hathaway H.J., Prossnitz E.R. G protein-coupled estrogen receptor regulates mammary tumorigenesis and metastasis. Mol. Cancer Res. 2014;12:1644–1654. doi: 10.1158/1541-7786.MCR-14-0128-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba A., Matsuyama N., Shibata S., Masaki E., Emala C.W., Mizuta K. The free fatty acid receptor 1 promotes airway smooth muscle cell proliferation through MEK/ERK and PI3K/Akt signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018;314:L333–L348. doi: 10.1152/ajplung.00129.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E., Andradas C., Medrano M., Caffarel M.M., Pérez-Gómez E., Blasco-Benito S., Gómez-Cañas M., Pazos M.R., Irving A.J., Lluís C. Targeting CB2-GPR55 receptor heteromers modulates cancer cell signaling. J. Biol. Chem. 2014;289:21960–21972. doi: 10.1074/jbc.M114.561761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechamen C.A., Thomas R.M., Dias J.A. APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol. Cell. Endocrinol. 2007;260–262:93–99. doi: 10.1016/j.mce.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouelaa-Benslama R., De Wever O., Hendrix A., Sabbah M., Lambein K., Land D., Prévost G., Bracke M., Hung M.-C., Larsen A.K. Identification of a GαGβγ, AKT and PKCα signalome associated with invasive growth in two genetic models of human breast cancer cell epithelial-to-mesenchymal transition. Int. J. Oncol. 2012;41:189–200. doi: 10.3892/ijo.2012.1457. [DOI] [PubMed] [Google Scholar]
- Pavlik R., Wypior G., Hecht S., Papadopoulos P., Kupka M., Thaler C., Wiest I., Pestka A., Friese K., Jeschke U. Induction of G protein-coupled estrogen receptor (GPER) and nuclear steroid hormone receptors by gonadotropins in human granulosa cells. Histochem. Cell Biol. 2011;136:289–299. doi: 10.1007/s00418-011-0846-7. [DOI] [PubMed] [Google Scholar]
- Perales-Puchalt A., Wojtak K., Duperret E.K., Yang X., Slager A.M., Yan J., Muthumani K., Montaner L.J., Weiner D.B. Engineered DNA vaccination against follicle-stimulating hormone receptor delays ovarian cancer progression in animal models. Mol. Ther. 2019;27:314–325. doi: 10.1016/j.ymthe.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyzos N.P., Sunkara S.K. Sub-optimal responders following controlled ovarian stimulation: an overlooked group? Hum. Reprod. 2015;30:2005–2008. doi: 10.1093/humrep/dev149. [DOI] [PubMed] [Google Scholar]
- Ponikwicka-Tyszko D., Chrusciel M., Stelmaszewska J., Bernaczyk P., Sztachelska M., Sidorkiewicz I., Doroszko M., Tomaszewski J., Tapanainen J.S., Huhtaniemi I. Functional expression of FSH receptor in endometriotic lesions. J. Clin. Endocrinol. Metab. 2016;101:2905–2914. doi: 10.1210/jc.2016-1014. [DOI] [PubMed] [Google Scholar]
- Prossnitz E.R., Hathaway H.J. What have we learned about GPER function in physiology and disease from knockout mice? J. Steroid Biochem. Mol. Biol. 2015;153:114–126. doi: 10.1016/j.jsbmb.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz E.R., Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009;308:32–38. doi: 10.1016/j.mce.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar C.M., Vines C.M., Cimino D.F., Prossnitz E.R. Arrestins block G protein-coupled receptor-mediated apoptosis. J. Biol. Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- Revankar C.M., Cimino D.F., Sklar L.A., Arterburn J.B., Prossnitz E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Rivero-Müller A., Chou Y.-Y., Ji I., Lajic S., Hanyaloglu A.C., Jonas K., Rahman N., Ji T.H., Huhtaniemi I. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc. Natl. Acad. Sci. U. S. A. 2010;107:2319–2324. doi: 10.1073/pnas.0906695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi V., Lispi M., Longobardi S., Mattei M., Di Rella F., Salustri A., De Felici M., Klinger F.G. LH prevents cisplatin-induced apoptosis in oocytes and preserves female fertility in mouse. Cell Death Differ. 2017;24:72–82. doi: 10.1038/cdd.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenfeld R., Gupta A., Gagnidze K., Lim M.P., Gomes I., Lee-Ramos D., Nieto N., Devi L.A. AT1R-CB₁R heteromerization reveals a new mechanism for the pathogenic properties of angiotensin II. EMBO J. 2011;30:2350–2363. doi: 10.1038/emboj.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi D., Potì F., Simoni M., Casarini L. Pharmacogenetics of G-protein-coupled receptors variants: FSH receptor and infertility treatment. Best Pract. Res. Clin. Endocrinol. Metab. 2018;32:189–200. doi: 10.1016/j.beem.2018.01.001. [DOI] [PubMed] [Google Scholar]
- Sasson R., Dantes A., Tajima K., Amsterdam A. Novel genes modulated by FSH in normal and immortalized FSH-responsive cells: new insights into the mechanism of FSH action. FASEB J. 2003;17:1256–1266. doi: 10.1096/fj.02-0740com. [DOI] [PubMed] [Google Scholar]
- Sayers N., Hanyaloglu A.C. Intracellular follicle-stimulating hormone receptor trafficking and signaling. Front. Endocrinol. (Lausanne). 2018;9:653. doi: 10.3389/fendo.2018.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G., Prossnitz E.R. GPER/GPR30 knockout mice: effects of GPER on metabolism. Methods Mol. Biol. 2016;1366:489–502. doi: 10.1007/978-1-4939-3127-9_38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirotkin A.V., Benco A., Tandlmajerova A., Vasícek D., Kotwica J., Darlak K., Valenzuela F. Transcription factor p53 can regulate proliferation, apoptosis and secretory activity of luteinizing porcine ovarian granulosa cell cultured with and without ghrelin and FSH. Reproduction. 2008;136:611–618. doi: 10.1530/REP-08-0229. [DOI] [PubMed] [Google Scholar]
- Sirotkin A.V., Ben O A., Tandlmajerová A., Lauková M., Vaší Ek D., Laurin Ik J., Kornhauser J., Alwasel S., Harrath A.H. cAMP response element-binding protein 1 controls porcine ovarian cell proliferation, apoptosis, and FSH and insulin-like growth factor 1 response. Reprod. Fertil. Dev. 2018;30:1145–1153. doi: 10.1071/RD17508. [DOI] [PubMed] [Google Scholar]
- Surve C.R., Lehmann D., Smrcka A.V. A chemical biology approach demonstrates G protein βγ subunits are sufficient to mediate directional neutrophil chemotaxis. J. Biol. Chem. 2014;289:17791–17801. doi: 10.1074/jbc.M114.576827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima K., Hosokawa K., Yoshida Y., Dantes A., Sasson R., Kotsuji F., Amsterdam A. Establishment of FSH-responsive cell lines by transfection of pre-ovulatory human granulosa cells with mutated p53 (p53val135) and Ha-ras genes. Mol. Hum. Reprod. 2002;8:48–57. doi: 10.1093/molehr/8.1.48. [DOI] [PubMed] [Google Scholar]
- Thomas P., Pang Y., Filardo E.J., Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- Tranchant T., Durand G., Gauthier C., Crépieux P., Ulloa-Aguirre A., Royère D., Reiter E. Preferential β-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol. Cell. Endocrinol. 2011;331:109–118. doi: 10.1016/j.mce.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Tubio M.R., Fernandez N., Fitzsimons C.P., Copsel S., Santiago S., Shayo C., Davio C., Monczor F. Expression of a G Protein-coupled Receptor (GPCR) leads to attenuation of signaling by other GPCRs. J. Biol. Chem. 2010;285:14990–14998. doi: 10.1074/jbc.M109.099689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Aguirre A., Reiter E., Crépieux P. FSH receptor signaling: complexity of interactions and signal diversity. Endocrinology. 2018;159:3020–3035. doi: 10.1210/en.2018-00452. [DOI] [PubMed] [Google Scholar]
- Wallach E.E., Shoham Z., Schachter M. Estrogen biosynthesis—regulation, action, remote effects, and value of monitoring in ovarian stimulation cycles. Fertil. Steril. 1996;65:687–701. doi: 10.1016/s0015-0282(16)58197-7. [DOI] [PubMed] [Google Scholar]
- Webb R., Buratini J., Hernandez-Medrano J.H., Gutierrez C.G., Campbell B.K., Webb R., Buratini J., Hernandez-Medrano J.H., Gutierrez C.G., Campbell B.K. Follicle development and selection: past, present and future. Anim. Reprod. 2016;13:234–249. [Google Scholar]
- Yoshida Y., Hosokawa K., Dantes A., Tajima K., Kotsuji F., Amsterdam A. Theophylline and cisplatin synergize in down regulation of BCL-2 induction of apoptosis in human granulosa cells transformed by a mutated p53 (p53 val135) and Ha-ras oncogene. Int. J. Oncol. 2000;17:227–235. doi: 10.3892/ijo.17.2.227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available from the Lead Contact on reasonable request.