

There is a Blood Commentary on this article in this issue.
Key Points
Bone marrow stromal cells from myeloma patients downregulate CD38 expression on myeloma cells through activation of the JAK-STAT3 pathway.
JAK inhibitor ruxolitinib upregulates CD38 expression on myeloma cells and enhances daratumumab-mediated cytotoxicity.
Abstract
Anti-CD38 monoclonal antibody (MoAb) treatments including daratumumab (DARA) are effective therapies for both newly diagnosed and relapsed multiple myeloma (MM). In this study, we examined the soluble factors that modulate CD38 expression and are associated with sensitivity to DARA-mediated antibody-dependent cellular cytotoxicity (ADCC) in the bone marrow (BM) microenvironment. Importantly, primary BM stromal cell (BMSC) culture supernatant (BMSC-sup) and interleukin-6 (IL-6) downregulated CD38 expression and reduced DARA-mediated ADCC. Both cytokine profiling of the BMSC-sup and genome-scale clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) knockout screening in MM cell lines identified and validated the JAK-STAT3 signaling pathway mediating CD38 downregulation, whereas the JAK-STAT1 pathway mediated CD38 upregulation. STAT3 knockdown abrogated BMSC-sup– and IL-6–induced CD38 downregulation on MM cell lines. We also confirmed that STAT3 and CD38 is negatively correlated in primary MM cells. To assess potential clinical relevance, pharmacological inhibition of the JAK-STAT pathway on BMSC-sup–induced CD38 downregulation was further examined. JAK inhibitor ruxolitinib inhibited STAT3 phosphorylation in MM cell lines, upregulated CD38 expression in MM cell lines and primary patient MM cells, and augmented DARA-mediated ADCC against MM cell lines. Taken together, our results suggest that CD38 expression on MM cells in the BM microenvironment is regulated by both STAT1 (positively) and STAT3 (negatively), and that inhibition of the JAK-STAT3 pathway represents a novel therapeutic option to enhance CD38 expression and anti-CD38 MoAb-mediated MM cytotoxicity.
Visual Abstract

Introduction
The treatment strategies for multiple myeloma (MM) incorporating proteasome inhibitor (PI) and immunomodulatory drug (IMiD) combinations have remarkably improved patient outcome. More recently, immune therapies have further transformed the treatment paradigm in MM.1 Specifically, monoclonal antibodies (MoAbs) directed against CD38 (daratumumab [DARA]) and SLAMF7 (elotuzumab) have already been approved by US Food and Drug Administration (FDA), and several other MoAbs directed against these and other antigens are under evaluation.2 Most recently, combination treatments incorporating MoAbs, IMiDs, and PIs are achieving remarkable response rates in both newly diagnosed and relapsed MM.3-7 Importantly, however, intrinsic or acquired resistance to therapy underlies relapse of disease in many patients.8 This resistance may be due to MM-intrinsic factors and/or accessory cells/cytokines mediating immunosuppression and drug resistance in the bone marrow milieu.
CD38 is a type II transmembrane glycoprotein that functions as an adhesion molecule and an ectoenzyme associated with calcium mobilization from the endothelial reticulum. Hematological cells including natural killer cells, T cells, and B cells, as well as nonhematological tissues such as prostatic epithelial cells and airway-striated muscle cells, express CD38.9-12 Importantly, CD38 is highly expressed on malignant plasma cells from MM patients, and anti-CD38 MoAbs including DARA, isatuximab (SAR650984), and MOR202 trigger MM cytotoxicity through induction of complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC),13 antibody-dependent cellular phagocytosis,14 and/or programmed cell death through Fcγ receptor–mediated cross-linking.15 Although DARA monotherapy remarkably achieves 30% responses in relapsed and refractory MM,16,17 resistance mechanisms are underlying in the majority of patients. As noted, DARA in combination with IMiDs or PIs achieves high extent, frequency, and depth of response in patients with relapsed and newly diagnosed MM; however, relapse of disease is commonly observed. All-trans retinoic acid (ATRA) or histone deacetylase inhibitors are reported strategies to enhance DARA-mediated cytotoxicity via upregulation of CD38 expression.18,19 Type I and II interferons also upregulate CD38 expression in various hematopoietic cell types.20-24 IMiDs have also been reported to upregulate CD38 expression and prime MM cells for DARA-mediated cytotoxicity through degradation of CD38 gene repressors, IKZF1 and IKZF3.25
Our and other studies have shown that bone marrow (BM) stromal cells (BMSCs) produce cytokines that mediate MM cell proliferation, survival, drug resistance, and migration in the BM microenvironment.26,27 Specifically, interleukin-6 (IL-6) plays a central role in MM cell proliferation and survival by activating Ras-dependent MAPK, phosphatidylinositol-3 kinase (PI3K)–protein kinase B (Akt), and Janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathways.28,29 IL-6 activates not only STAT3, but also to a lesser extent STAT1; aberrant STAT3 activation is oncogenic, whereas STAT1 activation triggered by interferon-γ (IFN-γ) stimulation has tumor-suppressor functions.20 To date, however, the biologic impact of the tumor-promoting, immunosuppressive MM BM microenvironment on sensitivity vs resistance to CD38-targeting immunotherapies has not yet been characterized.
In this study, we identify mechanisms in the BM milieu modulating CD38 expression on MM cells and sensitivity to CD38-targeted therapies. Importantly, our studies suggest that therapeutically targeting JAK-STAT3 with ruxolitinib may enhance sensitivity of tumor cells to CD38-targeted therapies in MM.
Methods
Cell lines
RPMI 8226, MM.1S, NCI-H929, and HEK293T cells were purchased from American Type Culture Collection (ATCC; Manassas, VA). MOLP-8 was purchased from DSMZ (Braunschweig, Germany). All MM cell lines (RPMI 8226, MM.1S, NCI-H929, and MOLP-8) were cultured in 5% CO2 at 37°C in RPMI 1640 medium containing 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO), 2 μM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA). HEK293T cells were cultured in Dulbecco modified Eagle medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cell lines were tested and authenticated by short tandem repeat DNA fingerprinting analysis (Molecular Diagnostic Laboratory, Dana-Farber Cancer Institute [DFCI], Boston, MA), and used within 3 months after thawing. Cell lines were also regularly tested for mycoplasma contamination using the MycoAlert mycoplasma detection kit (Lonza, Basel, Switzerland).
Primary patient MM cells and BMSCs
Primary patient MM cells were obtained from BM aspirates of MM patients, with written informed consent according to the Declaration of Helsinki. All procedures were performed using a protocol approved by the Institutional Review Board of the DFCI. Mononuclear cells were separated by using Ficoll-Paque PLUS (GE Healthcare Bio-Sciences, Pittsburgh, PA). Primary MM cells were further purified by CD138+ selection using anti-CD138 magnetic-activated cell separation microbeads (Miltenyi Biotec, San Diego, CA). BMSCs were cultured for 72 hours in the same culture medium as MM cells. Culture supernatants were then harvested, passed through 0.2-μm filters, mixed with 50% of control culture medium, and used as BMSC culture supernatant (BMSC-sup).
To analyze the correlation of STAT3 and CD38 expression in primary MM cells, we have used RNA-sequencing (RNAseq) data from 319 newly diagnosed MM patients who were enrolled in the Intergroupe Francophone du Myélome (IFM)/DFCI 2009 study.30,31 RNAseq samples were quantified using Salmon RNAseq quantification, and were normalized using Deseq2. Pearson correlation between STAT3 and CD38 expression levels after normalization was calculated using R and ggplot2.
Reagents and compounds
Recombinant human IL-6, tumor necrosis factor-α, macrophage inflammatory protein-1α (MIP-1α), stromal cell–derived factor-1α (SDF-1α), IL-1β, IL-8, insulin-like growth factor 1 (IGF-1), transforming growth factor-β (TGF-β), oncostatin-M (OSM), leukemia-inhibitory factor (LIF), IL-10, IL-2, IFN-β, and IFN-γ were purchased from R&D Systems (Minneapolis, MN). Ruxolitinib, ATRA, and panobinostat were purchased from Selleck Chemicals (Houston, TX), dissolved in dimethyl sulfoxide as stock solutions, and stored in a −20°C freezer. DARA and control immunoglobulin G1 humanized MoAb directed against human respiratory syncytial virus (felvizumab) were obtained from the DFCI pharmacy and from Novus Biologicals (Centennial, CO), respectively.
Flow cytometry
The MoAbs in supplemental Table 1 (available on the Blood Web site) were used for flow cytometric analyses. CD38 mean fluorescence intensity (MFI) was measured by BD FACSCanto and FlowJo_V10. Relative CD38 MFI was normalized to MFI of control cells. Primary MM cells were defined as the CD38+/CD138+ fraction of patient BM mononuclear cells. Isotype-matched MoAb-stained cells were used as negative controls.
MoAbs
All MoAbs in supplemental Table 1 were used according to the manufacturer’s recommendations/protocols.
qRT-PCR
Total RNA was isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany). Complementary DNA was then synthesized from 1 μg of total RNA with oligo(dT) primers using the Superscript III First-strand Synthesis kit (Thermo Fisher Scientific, Waltham, MA). Quantitative real-time polymerase chain reaction (PCR; qRT-PCR) was carried out using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and the 7500 Real-Time PCR system (Applied Biosystems). The relative amount of each transcript was determined using the relative standard curve method. The values were normalized to invariant control GAPDH expression. Specific primers for each gene are listed in supplemental Table 2.
Immunoblotting
Genome-scale CRISPR-Cas9 library screening
Library amplification, lentivirus production, cell transduction, DNA sequencing, as well as data processing and initial analysis, were performed as in our previous studies.34,35 Briefly, the clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) GeCKOv2 library containing 6 unique single-guide RNAs (sgRNAs) against each of 19 050 genes and 4 sgRNAs against each of 1864 microRNAs were infected into RPMI 8226 cells with high expression of CD38. Cells infected with the library were cultured in the presence of IL-6 (1 ng/mL).
Lentiviral production and infection
HEK 293T packaging cells were seeded into 6-well plates (8 × 105 cells per well) 1 day prior to transfection. Five hundred nanograms of lenti-CRISPRv2, EF.STAT3C.Ubc.GFP, or EF.STAT3DN.Ubc.GFP were transfected into HEK 293T cells, together with 500 ng of pCMV-dR8.2 dvpr and 100 ng of pCMV VSV-G for lentivirus packaging using TransIT-LT1 Transfection reagent (Mirus Bio LLC, Madison, WI). After 16 hours of transfection, cells were re-fed with fresh medium containing 30% (vol/vol) FBS to increase viral titer. After 24 hours, medium containing virus was harvested, passed through 0.45-μm filters, and used for infection. MM cells were spinoculated using ultracentrifugation at 25 000 rpm at 4°C for 2 hours with crude viral supernatant in the presence of 8 μg/mL polybrene, and then incubated in 5% CO2 at 37°C for 5 hours. After culture in fresh medium for 24 hours, infected cells were selected with puromycin dihydrochloride (1 μg/mL) for at least 2 days or G418 disulfate (600 μg/mL) for at least 6 days, followed by subsequent assays. All plasmid vectors used in this study are listed in supplemental Table 3.
CRISPR-Cas9 knockout of the STAT3 gene
We designed the sgRNA-targeting STAT3 gene by using the MIT CRISPR tool (supplemental Table 4). Oligonucleotides were annealed and subcloned into lenti-CRISPRv2 vectors. The sgRNA vectors were then transfected into HEK 293T cells, as described in the previous section.
siRNA transfection
Small-interfering RNAs (siRNAs) against STAT3 and STAT1 were purchased from Dharmacon (GE Healthcare Life Sciences, Marlborough, MA). The Amaxa transfection system (Thermo Fisher Scientific) was used for siRNA transfection into MM cells, as in our prior studies.36
BLI-based ADCC assay using luciferase-transfected cells
Luciferase-transfected RPMI 8226 cells were generated using murine stem cell virus internal ribosome entry site (MSCV IRES) luciferase plasmid. Cells were cocultured for 24 hours in 96-well U-bottom plates with natural killer cells from healthy donors at an effector-to-target ratio of 6:1, in the presence of either 1 μg/mL human IgG1 control antibody (felvizumab) or DARA. Beetle luciferin of the One-Glo EX Luciferase Assay System (Promega, Madison, WI) was then added, and survival of RPMI 8226 cells was determined by bioluminescence imaging (BLI). Specific lysis of target cells was calculated using the following formula: % lysis = [1 − (mean BLI signal in the presence of DARA/mean BLI signal in the presence of control antibody)] × 100%.
Statistical analysis
Experiments were performed at least 3 times, and biological triplicates were used in each experiment unless otherwise specified. Statistical significance was determined by the Student t test (not significant [NS or N]; *P < .05; **P < .01; ***P < .001). Error bars represent standard deviation.
Results
BMSC supernatant triggers reduction of CD38 expression on MM cells
We first examined the effect of the BM microenvironment on CD38 expression in MM cells. We cocultured MM cell lines RPMI 8226 and MOLP-8 with BMSCs for 5 days, and noted decreased CD38 expression in both MM cell lines (Figure 1A). Moreover, CD38 expression was lower on patient MM cells when they are cocultured with patient BM mononuclear cells (BMMCs) compared with MM cells without BMMCs (P = .050; Figure 1B).
Figure 1.
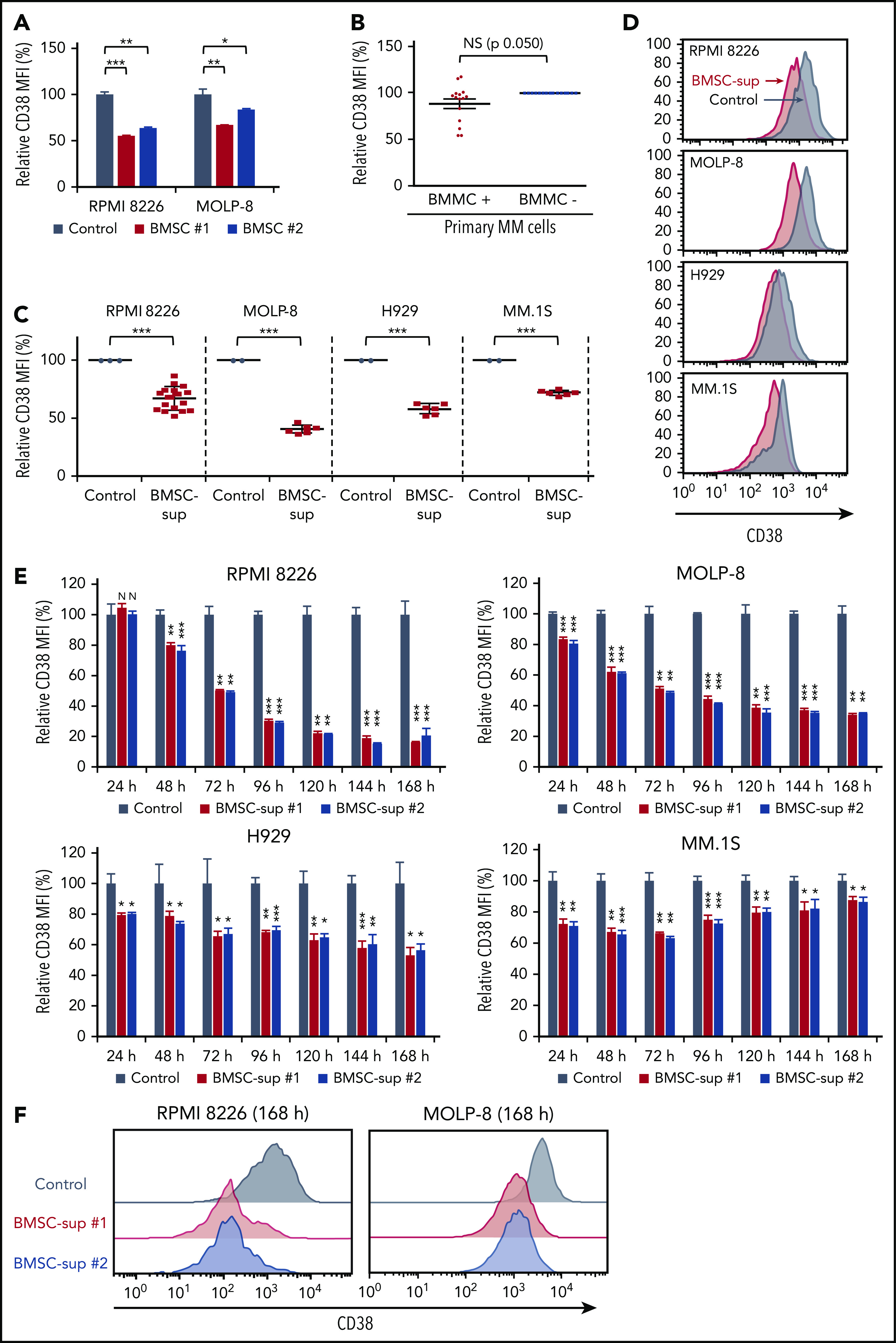
BMSCs and BMSC-sup reduce CD38 expression on MM cells. (A) RPMI 8226 and MOLP-8 cells were cocultured with or without 2 different bone marrow stromal cells (BMSC #1 or #2) for 5 days. After incubation, CD38 expression was measured by flow cytometry, and relative CD38 MFI was calculated in comparison with MFI of control. (B) CD138+ primary MM cells from 15 MM patients were cocultured with or without autologous BMMCs for 5 days. After incubation, CD38 expression on 7-AAD− and CD138+ MM cells was measured by flow cytometry, and relative CD38 MFI was calculated in comparison with MFI of MM cells cultured without BMMCs. (C) RPMI 8226, MOLP-8, H929, and MM.1S cells were cultured with control culture medium (control, n = 3 for RPMI 8226, and n = 2 for the other cell lines) or BMSC-sup from different MM patients (n = 17 for RPMI 8226, and n = 6 for the other cell lines) for 72 hours. CD38 expression on 7-AAD− cells was measured by flow cytometry, and relative CD38 MFI was calculated in comparison with MFI of control. (D) Representative histograms of CD38 expression of panel C. (E) Relative CD38 MFIs were measured after MM cells were cultured with control medium or 2 different BMSC-sup (BMSC-sup #1 or #2) for 24 to 168 hours. (F) Histograms of CD38 expression in RPMI 8226 and MOLP-8 cells cultured with control medium, BMSC-sup #1 or #2 for 168 hours. Data are shown as mean plus or minus standard error of the mean. *P < .05; **P < .01; ***P < .001.
To determine whether the decreased CD38 expression on MM cells was mediated by soluble factors secreted by BMSCs, we next cultured MM cell lines (RPMI 8226, MOLP-8, H929, and MM.1S) in the presence of BMSC-sup and evaluated CD38 expression by flow cytometry. As observed in MM-BMSC cocultures, BMSC-sup triggered significant reduction of CD38 expression on all MM cell lines, with MFI reduction ratios vs control cells of 32.6%, 59.2%, 41.6%, and 27.5% on RPMI 8226, MOLP-8, H929, and MM.1S cells, respectively (Figure 1C-D). To examine whether the effect of BMSC-sup on CD38 expression was time-dependent, we next cultured MM cells with BMSC-sups for 1 to 7 days, and noted that CD38 expression was downregulated in a time-dependent fashion in RPMI 8226, MOLP-8, and H929 cells, but not in MM.1S cells (Figure 1E-F). These results showed that soluble factors from BMSCs suppress CD38 expression on MM cells.
IL-6 downregulates CD38 expression on MM cells
To identify those soluble factors inducing decreased CD38 expression on MM cells, we evaluated the effect of MM-relevant (IL-6, MIP-1α, SDF-1α, IL-1β, IL-8, IGF-1, TGF-β, OSM, and LIF) and immune response–related (IL-10, IL-2, IFNβ, and IFN-γ) cytokines in the BM microenvironment on modulation of CD38 expression on MM cell lines. Importantly, IL-6 triggered the most significant decrease of CD38 expression, assessed by flow cytometric analysis (Figure 2A; supplemental Tables 5 and 6). Conversely, previous reports have demonstrated that IFN-β and IFN-γ upregulate CD38 expression in different cell types,20-24 and we here observed that both IFN-β and IFN-γ increased CD38 expression on MM cell lines (IFN-β > IFN-γ; Figure 2A; supplemental Tables 5 and 6; supplemental Figure 1A-B). Our study further showed that CD38 reduction on MM cell lines was triggered by IL-6 in a dose-dependent fashion (Figure 2B). qRT-PCR demonstrated that CD38 messenger RNA (mRNA) level in RPMI 8226 cells was also markedly downregulated after 12-hour treatment with BMSC-sup or IL-6 treatment (Figure 2C). IL-6 also significantly induced downregulation of CD38 mRNA level in primary patient MM cells (Figure 2D). These results suggest that IL-6 has a major role in BMSC-sup–mediated CD38 downregulation on MM cell lines and patient cells.
Figure 2.
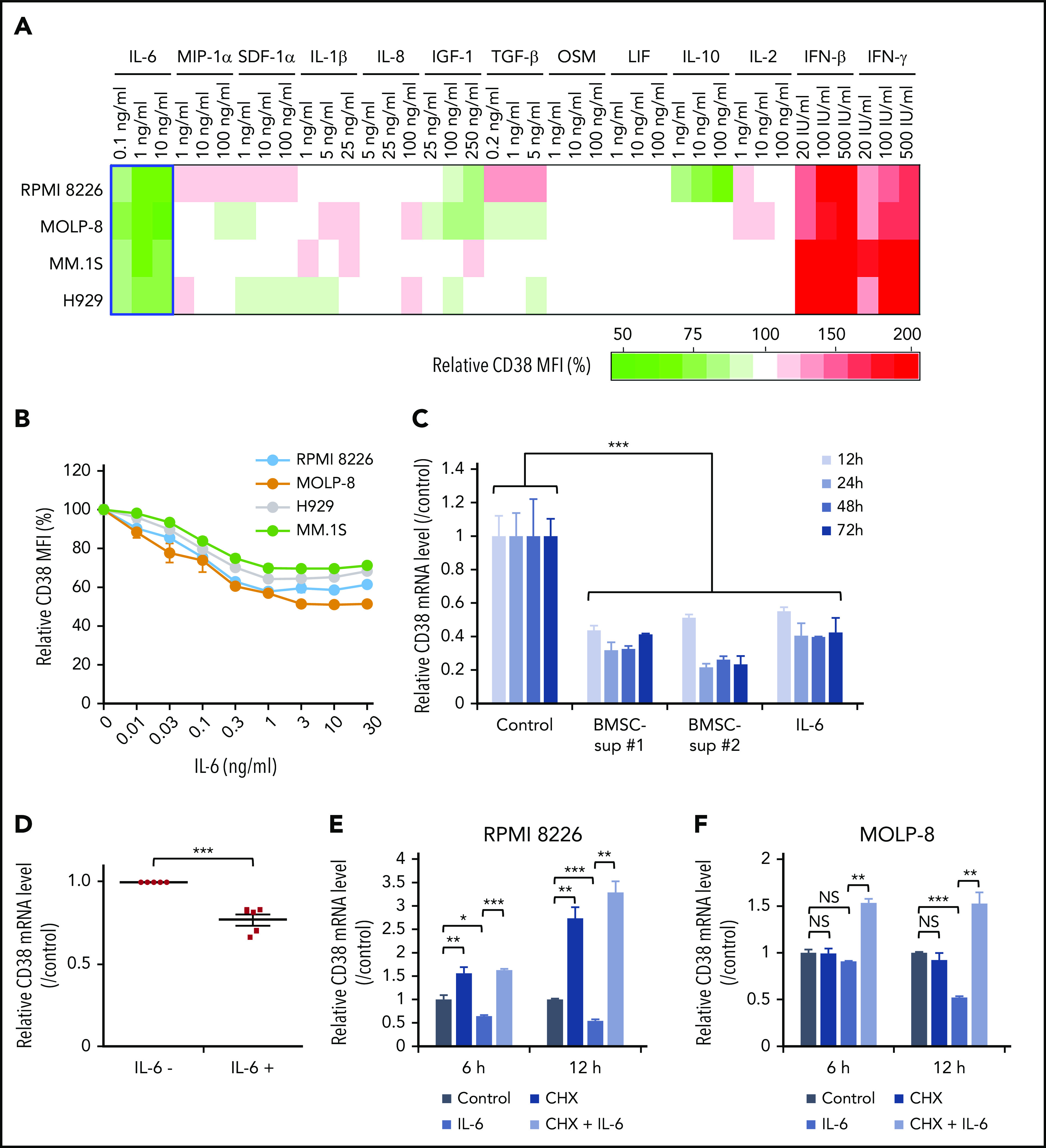
Cytokine profiling shows that IL-6 is the major soluble factor downregulating CD38 expression on MM cells. (A) Four MM cell lines were cultured with control culture medium or culture medium containing the indicated cytokines (IL-6, MIP-1α, SDF-1α, IL-1β, IL-8, IGF-1, TGF-β, OSM, LIF, IL-10, IL-2, IFN-β, IFN-γ) for 72 hours. CD38 expression was measured by flow cytometry after 72-hour incubation, and relative CD38 MFI was calculated in comparison with MFI of control. Heat map was created based on relative CD38 MFI as indicated in supplemental Table 5. (B) Four MM cell lines were treated for 72 hours with IL-6 (0.01-30 ng/mL), and CD38 expression was measured by flow cytometry. (C) RPMI 8226 cells were cultured with control culture media, 2 different BMSC-sup (BMSC-sup #1 or #2), or IL-6 (5 ng/mL) for 12, 24, 48, or 72 hours, and CD38 mRNA level was measured by qRT-PCR. (D) CD138+ primary MM cells separated from BMMCs of 5 MM patients using CD138 magnetic-activated cell separation beads were cultured with or without IL-6 (5 ng/mL), and the CD38 mRNA level was measured by qRT-PCR. (E-F) RPMI 8226 (E) and MOLP-8 (F) cells were cultured with control culture medium or IL-6 (5 ng/mL), in the absence or presence of CHX (10 µg/mL) for 6 hours or 12 hours, and the CD38 mRNA level was measured by qRT-PCR. Data are shown as mean plus or minus standard error of the mean. *P < .05; **P < .01; ***P < .001.
We next examined the molecular mechanisms whereby IL-6 or BMSC-sup downregulate CD38 expression on MM cells. We first evaluated the CD38 mRNA level in MM cell lines cultured with IL-6, in the presence or absence of protein synthesis inhibitor cycloheximide (CHX). Importantly, CHX completely abrogated CD38 downregulation induced by IL-6 in both RPMI 8226 and MOLP-8 cells (Figure 2E-F). These results suggest that protein synthesis triggered by IL-6–induced signaling is associated with downregulation of CD38 mRNA and protein expression.
Genome-scale CRISPR-Cas9 library screening identified STAT3 as a mediator of IL-6–induced CD38 downregulation
Because IL-6 activates signaling pathways including JAK-STATs, MEK–extracellular signal-regulated kinase, and PI3K-Akt to promote MM cell proliferation, survival, and migration,26,37 we next carried out unbiased and genome-scale CRISPR-Cas9 library screening to identify key molecules mediating IL-6–induced CD38 downregulation. Although IL-6 triggered downregulation of CD38 expression on the majority of RPMI 8226 cells, a small cell fraction maintained relatively high CD38 expression (CD38-high population). We therefore sorted those top 5% cells with CD38-high expression and compared these cells with nonsorted population (control cells). Using this approach, genes maintaining CD38 expression are enriched in the CD38-high population (Figure 3A).
Figure 3.
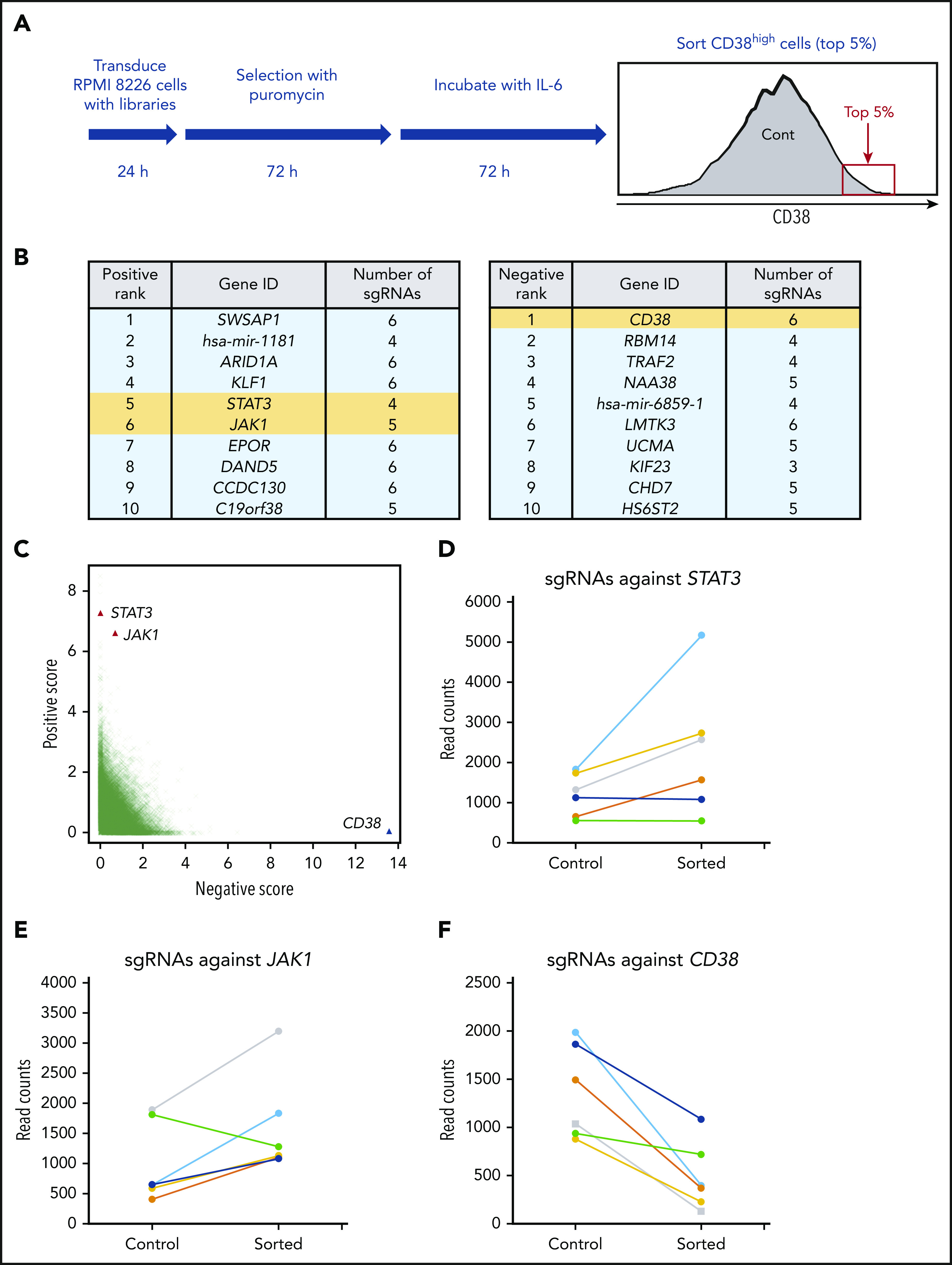
Genome-scale CRISPR-Cas9 knockout screening identifies STAT3 as a repressor of CD38. (A) Flowchart of genome-scale CRIPSR-Cas9 screening in RPMI 8226 cells. (B) List of top 10 positive (left panel) and negative (right panel) ranked genes; positive represents sgRNAs enriched in sorted cells compared with control cells, and negative the opposite. STAT3, JAK1, and CD38 are highlighted in yellow. (C) Enrichment of specific sgRNAs in CD38-high fraction. Each dot specifies 1 sgRNA. (D-F) Read counts of sgRNAs against STAT3 (D), JAK1 (E), and CD38 (F) in control and sorted cells.
Deep sequencing of embedded sgRNA barcodes showed coverage of at least 100 reads for 98.2% of sgRNAs in control cells (supplemental Figure 2A- B). Total read counts in control and sorted cells were similar, as shown in supplemental Figure 3C. The gene rank based on P value identified STAT3 and JAK1 as highly positively enriched genes, identifying these genes as potential mediators of IL-6–induced CD38 downregulation (Figure 3B left panel; Figure 3C). Conversely and as expected, CD38 was at the top of negatively ranked genes (Figure 3B right panel; Figure 3C). By analyses of read counts of 6 individual sgRNAs, 4 sgRNAs against STAT3 and 5 sgRNAs against JAK1 were increased in sorted cells (Figure 3, panels D and E, respectively); in contrast, read counts of all 6 sgRNAs against CD38 were decreased (Figure 3F). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the top 200 positive genes revealed significant enrichment of the JAK-STAT pathway (supplemental Table 7). Gene ontology enrichment analysis also showed 15 gene ontology terms, including positive regulation of gene expression and negative regulation of transcription from the RNA polymerase II promoter, as potential regulated factors that downregulate CD38 expression (supplemental Table 8). Importantly, we performed qRT-PCR validation of top-ranked genes identified in RPMI 8226 and MOLP-8 cells treated with IL-6, and observed that only STAT3 was upregulated by IL-6 (supplemental Figure 3A-B).
STAT3 negatively regulates CD38 expression
Because the JAK-STAT pathway was identified as a modulator of CD38 expression, we next examined the impact of STAT3 knockdown on CD38 expression in RPMI 8226 and MOLP-8 cells, in the presence or absence of BMSC-sup or IL-6. As we have shown previously herein, CD38 expression was significantly downregulated by BMSC-sup or IL-6 in control cells (sgNT#1), which was completely abrogated in STAT3 knockdown cells. Of note, STAT3 knockdown (sgSTAT#1 and #2) cells showed even higher baseline CD38 expression than in control (sgNT#1) cells (Figure 4A-B; supplemental Figure 4A-B). To further confirm STAT3-induced CD38 downregulation, we also transfected STAT3 siRNA into RPMI 8226 cells. Consistent with sgRNA results, STAT3 knockdown by siRNA also blocked CD38 downregulation induced by BMSC-sup or IL-6 (Figure 4C-D). Conversely, overexpression of constitutively active STAT3 triggered CD38 downregulation at both the protein and mRNA levels (Figure 4E-G).
Figure 4.
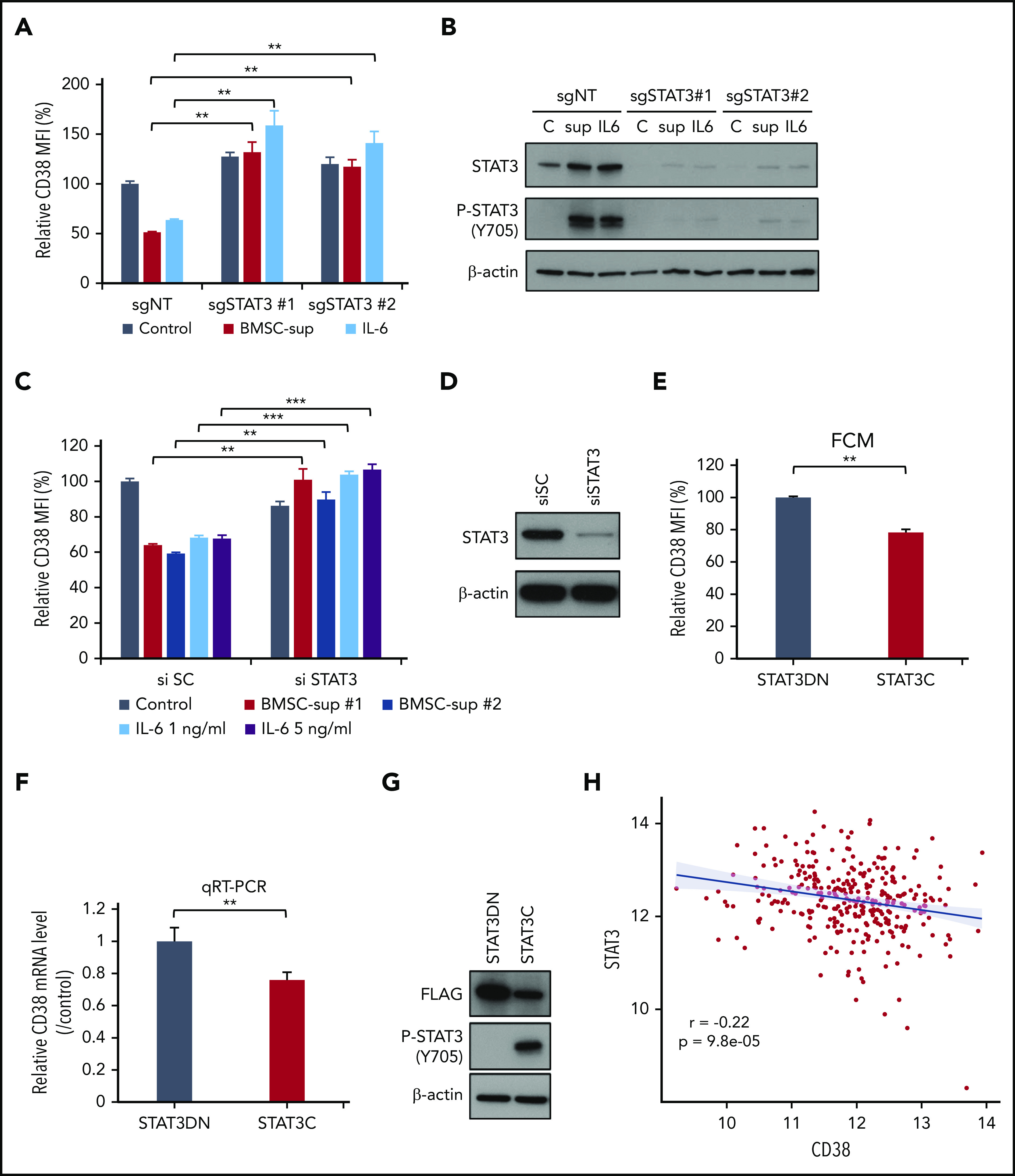
STAT3 negatively regulates CD38 expression. (A-B) RPMI 8226 cells were infected with single-guide (sg) nontarget control (sgNT), sgSTAT3 #1 or #2 expressing lentiviral vector, and cultured with control culture medium (Control), BMSC-sup, or IL-6 (1 ng/mL) for 72 hours. (C-D) RPMI 8226 cells were transfected with scrambled siRNA (si SC) or STAT3 siRNA (si STAT3). These cells were cultured with control culture medium, BMSC-sup, or IL-6 (1 or 5 ng/mL) for 48 hours. After incubation, CD38 expression was measured by flow cytometry (A, C). STAT3 downregulation was confirmed by immunoblotting (B, D). (E-G) RPMI 8226 cells were transfected with the dominant-negative STAT3 (STAT3DN) or constitutively active STAT3 (STAT3C) construct. CD38 protein expression (E) and mRNA level (F) were assessed by flow cytometry (FCM) and qRT-PCR, respectively. (G) Phosphorylated STAT3 (P-STAT3) expression was confirmed by immunoblotting. Relative CD38 MFI was calculated in comparison with MFI of control. Data are shown as mean plus or minus standard error of the mean. *P < .05; **P < .01; ***P < .001. (H) CD38 (x-axis) and STAT3 (y-axis) expression are negatively correlated (r, −0.22; P, 9.8e-05) at diagnosis in 319 newly diagnosed MM patient samples from the IFM/DFCI 2009 study. C, control culture medium; sup, BMSC-sup.
To assess clinical relevance, we further analyzed correlation of STAT3 and CD38 expression in primary MM cells using RNAseq data from 319 newly diagnosed MM patients. We confirmed a significant inverse correlation between STAT3 and CD38 expression in this data set (Figure 4H). Taken together, these data show that STAT3 negatively regulates CD38 expression in MM cell lines and patient cells.
STAT3 knockout enhances STAT1 in response to IL-6 and contributes to CD38 upregulation in the BM microenvironment
Previous studies have demonstrated that both IFN-β and IFN-γ upregulate CD38 via STAT1 activation.38 It has also been shown that IL-6 activates not only STAT3, but also STAT1.39 These studies, coupled with our results, suggest that CD38 is positively and negatively regulated by STAT1 and STAT3, respectively. Therefore, we next examined the impact of IL-6 on STAT1 and STAT3 activation in MM cells. IL-6 treatment of 6 hours induced potent phosphorylation of both STAT3 and STAT1 in both RPMI 8226 and MOLP-8 cells, whereas IFN-β and IFN-γ triggered only STAT1 phosphorylation (Figure 5A-B). We next evaluated the impact of STAT1 vs STAT3 activation on CD38 expression in MM cells. After establishing STAT3 stable knockout RPMI 8226 cells, we transiently knocked down STAT1 using STAT1 siRNA, in the presence of BMSC-sup or IL-6 (Figure 5C-D). Both BMSC-sup and IL-6 downregulated CD38 expression in STAT3 wild-type cells, which was not altered by STAT1 knockdown. In contrast, both BMSC-sup and IL-6 significantly upregulated CD38 expression in STAT3 knockout cells, which was abrogated by STAT1 knockdown (Figure 5C). Moreover, STAT3 knockout strongly enhanced STAT1 expression in response to IL-6 (Figure 5D), indicating that STAT3 negatively regulates STAT1 expression. Taken together, these results strongly suggest that CD38 on MM cells is positively and negatively regulated by STAT1 and STAT3, respectively. However, importantly, STAT1-induced CD38 upregulation is not able to overcome STAT3-induced CD38 downregulation.
Figure 5.
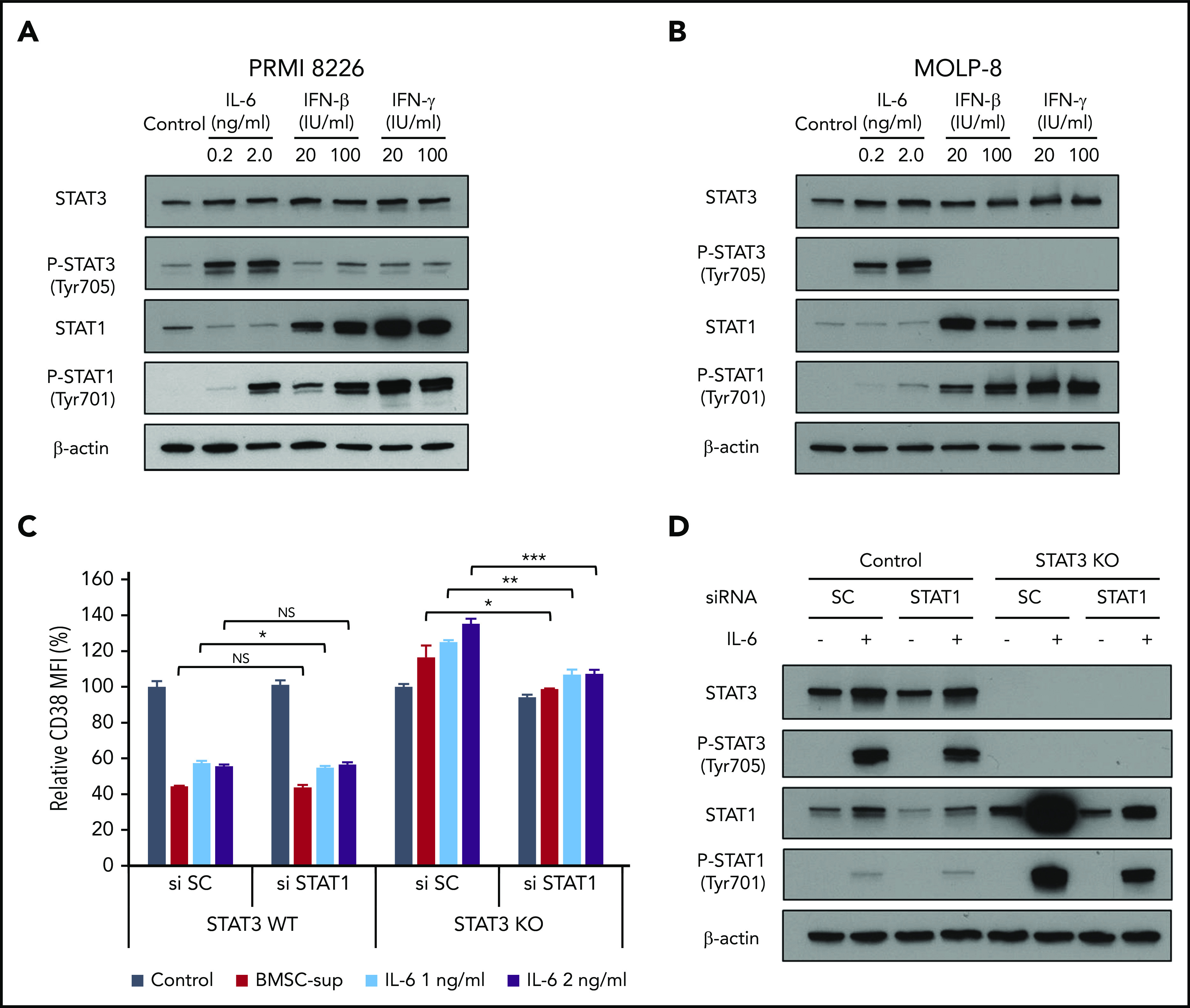
Regulation of CD38 by STAT1 and STAT3. (A-B) RPMI 8226 (A) and MOLP-8 (B) cells were cultured with IL-6, IFN-β, or IFN-γ for 6 hours, and then subjected to immunoblotting using indicated antibodies. (C) STAT3 stable knockout (KO) RPMI 8226 cells were transfected with scrambled siRNA (si SC) or STAT1 siRNA (si STAT1), and then cultured with BMSC-sup or IL-6 (1 or 2 ng/mL) for 72 hours. CD38 expression was measured by flow cytometry. (D) STAT3 stable knockout RPMI 8226 cells were transfected with scrambled (SC) siRNA or STAT1 siRNA, cultured in the absence or presence of IL-6 (2 ng/mL) for 6 hours, and then subjected to immunoblotting using indicated antibodies. Relative CD38 MFI was calculated in comparison with MFI of control. Data are shown as mean plus or minus standard error of the mean. *P < .05; **P < .01; ***P < .001.
Ruxolitinib suppresses STAT3 activation and enhances DARA-mediated ADCC against MM cell lines
Although MM cells express CD38, our data suggest that CD38 is downregulated by soluble factors (ie, IL-6) activating the JAK-STAT3 pathway in the BM microenvironment. We therefore hypothesized that inhibition of JAK-STAT3 signaling could upregulate CD38 expression, thereby enhancing DARA-mediated MM cytotoxicity. We first showed that JAK1/2 inhibitor ruxolitinib significantly decreased phosphorylation of both STAT1 and STAT3 induced by BMSC-sup or IL-6 (Figure 6A) in a dose-dependent fashion (Figure 6B). These results further confirm STAT3 as a negative regulator of CD38 expression. We next evaluated the effect of ruxolitinib on ADCC against MM cells induced by DARA in the presence of BMSC-sup. Consistent with CD38 upregulation on MM cells, ruxolitinib also abrogated the BMSC-sup–induced reduction of DARA-mediated ADCC (Figure 6C). These results suggest that the JAK1/2 inhibitor can be used to enhance DARA-mediated ADCC against MM cells in the BM microenvironment.
Figure 6.
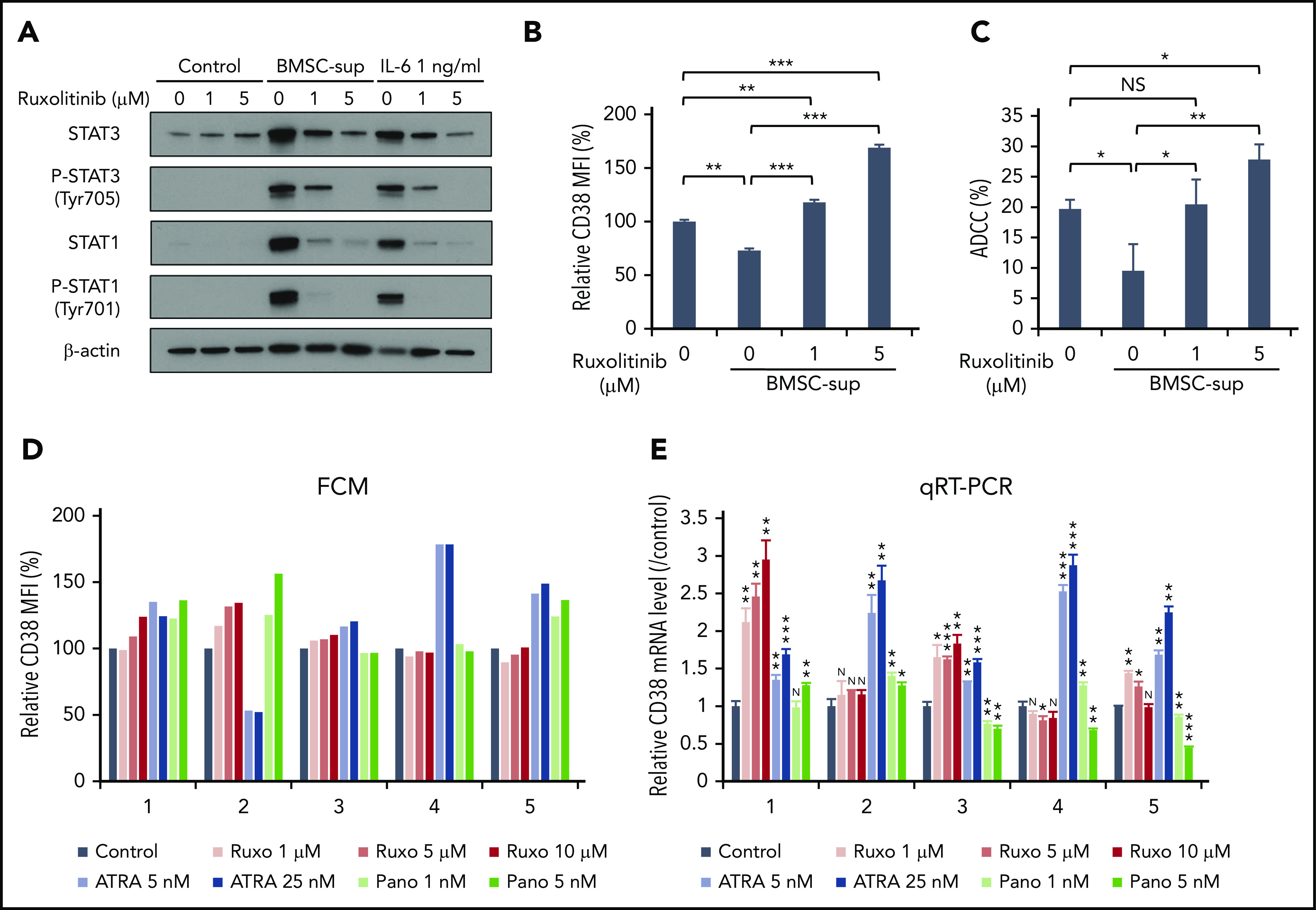
JAK inhibitor ruxolitinib upregulates CD38 expression and enhances DARA-mediated ADCC against MM cells. (A) RPMI 8226 cells were treated with solvent control or ruxolitinib (Ruxo; 1, 5 µM), in the absence or presence of BMSC-sup or IL-6 (1 ng/mL) for 48 hours. Whole-cell lysates were subjected to immunoblotting using indicated antibodies. (B-C) RPMI 8226 cells were treated with solvent control or ruxolitinib (1, 5 µM), in the absence or presence of BMSC-sup for 72 hours, and then subjected to flow cytometric analysis for CD38 expression (B) and DARA-mediated ADCC assay (C). (D) BMMCs of 5 MM patients were treated with solvent control, ruxolitinib (1, 5, 10 µM), ATRA (5, 25 nM), or panobinostat (Pano; 1, 5 nM) for 120 hours. After incubation, CD38 expression on 7-AAD− and CD138+ MM cells was measured by flow cytometry. (E) CD138+ MM cells separated from BMMCs of 5 MM patients were treated with solvent control, ruxolitinib (1, 5, 10 µM), ATRA (5, 25 nM), or panobinostat (1, 5 nM) for 120 hours. After incubation, relative CD38 mRNA level was assessed by qRT-PCR. Relative CD38 MFI was calculated in comparison with MFI of control. Data are shown as mean plus or minus standard error of the mean. *P < .05; **P < .01; ***P < .001.
CD38 expression on primary MM cells is differentially altered by ruxolitinib, ATRA, or panobinostat
ATRA has been reported to upregulates CD38 on MM cells,18 and a clinical trial of DARA in combination with ATRA is currently ongoing. Panobinostat has also been shown to upregulate CD38 on MM cells19; in our prior studies we showed that histone deacetylase inhibitors including panobinostat inhibited STAT3 activation.40 We therefore next compared the effects of ruxolitinib, ATRA, and panobinostat on CD38 cell-surface expression and CD38 mRNA in primary patient MM cells. BMMCs from 5 MM patients were cultured for 5 days with ruxolitinib (1, 5, 10 µM), ATRA (5, 25 nM), or panobinostat (1, 5 nM). Ruxolitinib triggered upregulation of CD38 MFI (Figure 6D) and CD38 mRNA (Figure 6E) in 3 and 4 MM patient samples, respectively. These results suggest that JAK1/2 inhibition may modulate CD38 mRNA and protein on patient MM cells.
Discussion
The BM microenvironment plays a crucial role in MM pathogenesis by promoting MM cell growth, survival, and drug resistance. Accessory cells and cytokines within the BM milieu modulate expression of signaling pathways within MM cells, as well as expression of cell-surface antigens and adhesion molecules. We therefore here hypothesized that CD38 expression on MM cells may be modulated in the BM milieu. Indeed, we observed that CD38 expression is significantly downregulated in the presence of BMSCs or BMSC-sup. Because BMSC-sup contains a number of cytokines and chemokines, we first focused on MM-relevant cytokines. Among these soluble factors, IL-6 showed the most potent downregulatory effect on CD38 expression in MM cell lines. Interestingly, other gp130 family cytokines, such as OSM or LIF, did not modulate CD38 expression even at high concentrations (100 ng/mL). On the other hand, IFN-β and IFN-γ markedly enhanced CD38 expression, whereas IGF-1 did not alter CD38 expression. These results suggest that CD38 expression on MM cells is downregulated primarily by IL-6 in the BM microenvironment.
IL-6 is able to activate multiple intracellular signaling cascades including the MEK–extracellular signal-regulated kinase, PI3K-Akt, and JAK-STAT pathways. To identify the molecule and/or signaling pathways mediating CD38 downregulation triggered by IL-6 treatment, we used genome-scale CRISPR-Cas9 knockout screening. Importantly, STAT3 was identified as the most potent negative regulator of CD38 expression, consistent with known activities of IL-6 as a potent activator of STAT3. Because STAT3 is a transcription factor that commonly positively regulates target genes, we hypothesized that STAT3 may indirectly inhibit CD38 via induction of a repressor of CD38. We therefore next examined the effect of IL-6 on CD38 in the presence of a translation inhibitor CHX, which completely abrogated IL-6–induced CD38 downregulation. Taken together, these results suggest that new protein synthesis is required for IL-6–triggered, STAT3 activation–mediated CD38 downregulation on MM cells.
To further validate the biologic impact of STAT3 on CD38 downregulation, we performed STAT3 knockdown and overexpression experiments. STAT3 knockdown/knockout significantly inhibited BMSC-sup– or IL-6–induced CD38 downregulation; conversely, transfection of constitutively active STAT3 downregulated CD38 expression. Moreover, we also analyzed the correlation of STAT3 and CD38 expression in primary patient MM cells, and confirmed a significant inverse correlation between STAT3 and CD38. These data further identify and confirm that STAT3 activity plays a crucial role in CD38 downregulation in MM cells.
Previous studies have shown that IFN-β and IFN-γ upregulate CD38 expression,20,24 and that IL-6 also activates STAT1. In MM, STAT3 can be oncogenic when aberrantly activated, whereas STAT1, a transcription factor formed after IFN-γ stimulation, has tumor-suppressor functions.41 Specifically, STAT-deficient cells have revealed the existence of reciprocal regulatory mechanisms by STAT3 and STAT1.22 Moreover, in mouse embryo fibroblasts with deficient STAT3, IL-6 mediates an IFN-γ–like response including prolonged STAT1 activation and the induction of multiple IFN-γ–inducible genes.42 Therefore, 1 of the functions of STAT3 in response to IL-6 is downregulation of STAT1 activity. Indeed, our study shows that knockout and/or knockdown of STAT3 upregulates CD38 expression on MM cell lines in the presence of IL-6; in contrast, knockdown of STAT1 in STAT3 stable knockout cells suppressed this CD38 upregulation. These results suggest that STAT3 negatively regulates STAT1. Taken together, our results show that STAT3 is a dominant factor modulating CD38 expression on MM cells in the BM microenvironment: even though either BMSC-sup or IL-6 treatment activates both STAT1 (positive regulator) and STAT3 (negative regulator) of CD38, the dominant phenotype is CD38 downregulation on MM cells.
We next asked whether JAK-STAT3 inhibition enhanced CD38 expression and DARA-mediated MM cell line cytotoxicity in the BM microenvironment. For this purpose, we used ruxolitinib, which acts by binding to the cytokine receptor and preventing JAK association, thereby decreasing its activity.43 As expected, ruxolitinib inhibited phosphorylation of both STAT1 and STAT3 induced by BMSC-sup. Importantly, ruxolitinib in a dose dependent fashion abrogated both BMSC-sup–induced CD38 downregulation on MM cells and enhanced DARA-mediated ADCC against MM cells.
Because ATRA and panobinostat have been reported to upregulate CD38 expression on MM cells, we here compared the effect of ATRA, panobinostat, and ruxolitinib on CD38 MFI and CD38 mRNA levels in primary patient MM cells. Each of these agents triggered upregulation of CD38 cell-surface expression and CD38 mRNA levels in a fraction of MM patient samples. An ongoing clinical trial is evaluating daratumumab in combination with ATRA (clinicaltrials.gov #NCT02751255).44 The variable effect of ruxolitinib observed in patient MM cells may be due to ruxolitinib’s inhibition of both STAT3 and STAT1 because we showed that STAT1 positively regulates, whereas STAT3 negatively regulates, CD38 expression. Importantly, our MM cell line knockdown experiments indicate that selective STAT3 inhibitors would be more effective at upregulating CD38 on MM cells, but no selective STAT3 inhibitors are currently available for clinical evaluation. The clinical significance of ruxolitinib modulating CD38 will be determined in future clinical trials, analogous to ongoing clinical trials evaluating combination ATRA with daratumumab anti-CD38 MoAb therapy.44
In conclusion, our results provide the rationale for novel combination treatment strategies utilizing CD38-targeting immunotherapy together with JAK-STAT pathway inhibitors to abrogate CD38 downregulation on MM cells, and thereby enhance sensitivity to CD38-targeted ADCC against MM in the BM milieu.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health Specialized Program of Research Excellence grant P50100707 (K.C.A.) and National Cancer Institute grants R01-CA050947 (K.C.A.) and R01-CA178264 (T.H. and K.C.A.), and by the Sheldon and Miriam Medical Research Foundation (K.C.A.).
K.C.A. is an American Cancer Society Clinical Research Professor.
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE153710).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.O., T.H., and K.C.A. developed the concept; D.O., J.L., H.O., and T.H. designed the experiments; D.O., J.L., and K.K. performed experiments; D.O., J.L., H.O., K.K., Y.-T.T., S.A., K.A., and T.H. provided technical support, advice, and supervision; D.O., J.L., M.K.S., and T.H. analyzed data; D.O. and Y.-T.T. provided clinical samples; and D.O., T.H., and K.C.A. wrote the manuscript.
Conflict-of-interest disclosure: K.C.A. serves on advisory boards to Celgene, Millennium-Takeda, Janssen, Sanofi-Aventis, Bristol Myers Squibb, Gilead, Precision Biosciences, and Tolero, and is a scientific founder of OncoPep and C4 Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Kenneth C. Anderson, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02215; e-mail: kenneth_anderson@dfci.harvard.edu.
REFERENCES
- 1.Kumar SK, Anderson KC. Immune therapies in multiple myeloma. Clin Cancer Res. 2016;22(22):5453-5460. [DOI] [PubMed] [Google Scholar]
- 2.Iftikhar A, Hassan H, Iftikhar N, et al. Investigational monoclonal antibodies in the treatment of multiple myeloma: a systematic review of agents under clinical development. Antibodies (Basel). 2019;8(2):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Facon T, Kumar S, Plesner T, et al. ; MAIA Trial Investigators . Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29-38. [DOI] [PubMed] [Google Scholar]
- 5.Mateos MV, Dimopoulos MA, Cavo M, et al. ; ALCYONE Trial Investigators . Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528. [DOI] [PubMed] [Google Scholar]
- 6.Dimopoulos MA, Oriol A, Nahi H, et al. ; POLLUX Investigators . Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331. [DOI] [PubMed] [Google Scholar]
- 7.Palumbo A, Chanan-Khan A, Weisel K, et al. ; CASTOR Investigators . Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766. [DOI] [PubMed] [Google Scholar]
- 8.de Mel S, Lim SH, Tung ML, Chng WJ. Implications of heterogeneity in multiple myeloma. BioMed Res Int. 2014;2014:232546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deaglio S, Mehta K, Malavasi F. Human CD38: a (r)evolutionary story of enzymes and receptors. Leuk Res. 2001;25(1):1-12. [DOI] [PubMed] [Google Scholar]
- 10.Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowell PD, Goldstein AS. Functional evidence that progenitor cells near sites of inflammation are precursors for aggressive prostate cancer. Mol Cell Oncol. 2017;4(2):e1279723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotlikoff MI, Kannan MS, Solway J, et al. Methodologic advancements in the study of airway smooth muscle. J Allergy Clin Immunol. 2004;114(suppl 2):S18-S31. [DOI] [PubMed] [Google Scholar]
- 13.de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840-1848. [DOI] [PubMed] [Google Scholar]
- 14.Overdijk MB, Verploegen S, Bögels M, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 2015;7(2):311-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Overdijk MB, Jansen JH, Nederend M, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcγ receptor-mediated cross-linking. J Immunol. 2016;197(3):807-813. [DOI] [PubMed] [Google Scholar]
- 16.Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207-1219. [DOI] [PubMed] [Google Scholar]
- 17.Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387(10027):1551-1560. [DOI] [PubMed] [Google Scholar]
- 18.Nijhof IS, Groen RW, Lokhorst HM, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015;29(10):2039-2049. [DOI] [PubMed] [Google Scholar]
- 19.García-Guerrero E, Gogishvili T, Danhof S, et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood. 2017;129(25):3386-3388. [DOI] [PubMed] [Google Scholar]
- 20.Bauvois B, Durant L, Laboureau J, et al. Upregulation of CD38 gene expression in leukemic B cells by interferon types I and II. J Interferon Cytokine Res. 1999;19(9):1059-1066. [DOI] [PubMed] [Google Scholar]
- 21.Mihara K, Yoshida T, Ishida S, et al. All-trans retinoic acid and interferon-α increase CD38 expression on adult T-cell leukemia cells and sensitize them to T cells bearing anti-CD38 chimeric antigen receptors. Blood Cancer J. 2016;6(5):e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musso T, Deaglio S, Franco L, et al. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J Leukoc Biol. 2001;69(4):605-612. [PubMed] [Google Scholar]
- 23.Bürgler S, Gimeno A, Parente-Ribes A, et al. Chronic lymphocytic leukemia cells express CD38 in response to Th1 cell-derived IFN-γ by a T-bet-dependent mechanism. J Immunol. 2015;194(2):827-835. [DOI] [PubMed] [Google Scholar]
- 24.Bat-Erdene A, Nakamura S, Oda A, et al. Class 1 HDAC and HDAC6 inhibition inversely regulates CD38 induction in myeloma cells via interferon-α and ATRA. Br J Haematol. 2019;185(5):969-974. [DOI] [PubMed] [Google Scholar]
- 25.Fedele PL, Willis SN, Liao Y, et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. 2018;132(20):2166-2178. [DOI] [PubMed] [Google Scholar]
- 26.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585-598. [DOI] [PubMed] [Google Scholar]
- 27.Kawano Y, Moschetta M, Manier S, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160-172. [DOI] [PubMed] [Google Scholar]
- 28.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86(4):1243-1254. [PubMed] [Google Scholar]
- 29.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374(Pt 1):1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Attal M, Lauwers-Cances V, Hulin C, et al. ; IFM 2009 Study . Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376(14):1311-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samur MK, Minvielle S, Gulla A, et al. Long intergenic non-coding RNAs have an independent impact on survival in multiple myeloma. Leukemia. 2018;32(12):2626-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hideshima T, Catley L, Yasui H, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107(10):4053-4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hideshima T, Neri P, Tassone P, et al. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin Cancer Res. 2006;12(19):5887-5894. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Song T, Zhou W, et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia. 2019;33(1):171-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joung J, Konermann S, Gootenberg JS, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening [published correction appears in Nat Protoc. 2019;14(7):2259]. Nat Protoc. 2017;12(4):828-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sagawa M, Ohguchi H, Harada T, et al. Ribonucleotide Reductase Catalytic Subunit M1 (RRM1) as a novel therapeutic target in multiple myeloma. Clin Cancer Res. 2017;23(17):5225-5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer. 2002;2(12):927-937. [DOI] [PubMed] [Google Scholar]
- 38.Stephanou A, Latchman DS. STAT-1: a novel regulator of apoptosis. Int J Exp Pathol. 2003;84(6):239-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13(2):211-217. [DOI] [PubMed] [Google Scholar]
- 40.Hideshima T, Cottini F, Ohguchi H, et al. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015;5(5):e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAK-STAT. 2012;1(2):65-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa-Pereira AP, Tininini S, Strobl B, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci USA. 2002;99(12):8043-8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-798. [DOI] [PubMed] [Google Scholar]
- 44.Frerichs KA, Minnema MC, Levin MD, et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma; results of the phase 1/2 Dara/ATRA study [abstract]. Blood. 2019;134(13). Abstract 653. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.