

Abstract
Motivation
Functional genomics experiments generate genomewide signal profiles that are dense information sources for annotating the regulatory elements. These profiles measure epigenetic activity at the nucleotide resolution and they exhibit distinctive patterns as they fluctuate along the genome. Most notable of these patterns are the valley patterns that are prevalently observed in assays such as ChIP Sequencing and bisulfite sequencing. The genomic positions of valleys pinpoint locations of cis-regulatory elements such as enhancers and insulators. Systematic identification of the valleys provides novel information for delineating the annotation of regulatory elements. Nevertheless, the valleys are not reported by majority of the analysis pipelines.
Results
We describe EpiSAFARI, a computational method for sensitive detection of valleys from diverse types of epigenetic profiles. EpiSAFARI employs a novel smoothing method for decreasing noise in signal profiles and accounts for technical factors such as sparse signals, mappability and nucleotide content. In performance comparisons, EpiSAFARI performs favorably in terms of accuracy. The histone modification valleys detected by EpiSAFARI exhibit high conservation, transcription factor binding and they are enriched in nascent transcription. In addition, the large clusters of histone valleys are found to be enriched at the promoters of the developmentally associated genes. Differential histone valleys exhibit concordance with differential DNase signal at cell line specific valleys. DNA methylation valleys exhibit elevated conservation and high transcription factor binding. Specifically, we observed enriched binding of transcription factors associated with chromatin structure around methyl-valleys.
Availability and implementation
EpiSAFARI is publicly available at https://github.com/harmancilab/EpiSAFARI.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Sequencing based functional genomics experiments (ENCODE Project Consortium, 2012; Hasin et al., 2017; Sharing Epigenomes Globally, 2018), such as chromatin immunoprecipitation sequencing (ChIP-Seq), are being widely used to study the regulatory processes underpinning phenotypic variation (Esteller, 2008; McVicker et al., 2013), such as PheWAS (Denny et al., 2016), HAWAS (Sun et al., 2016), and understanding epigenetic control of gene expression (Dong and Weng, 2013; Zhang and Reinberg, 2001). Most commonly, the data from these assays are summarized into signal profiles that represent epigenetic measurements at the signal nucleotide resolution, e.g. the fold-change signal or the read coverage at each nucleotide. Although the signal profiles can provide biological insight for comprehensive discovery and annotation of functional elements in the genome, most of the current analysis pipelines focus mainly on the identification of broad regions with signal enrichments, such as peaks (Harmanci et al., 2014; Rozowsky et al., 2009; Thomas et al., 2017; Zhang et al., 2008). In particular, there is much information that is encoded in the fluctuations of the signal profiles along the genome. Most notable of these fluctuations are the troughs, valleys, or canyons in the genomewide signal profiles. Troughs and valleys are generally used in the literature to refer to the punctate regions where signal profile exhibits ‘V’ shaped patterns such that a dip in the signal is observed between two summits (Sethi et al., 2018). Previous studies have generally focused on punctate valleys at the length scales of up to 5kbs. Canyons are generally used in the context of DNA methylation to refer to broad regions with depleted signals. The signal profile over canyons are similar to a broad ‘U’ shaped pattern with large basins (Jeong et al., 2014; Xie et al., 2013). A recent publication referred to these broad canyon domains as ‘nadirs’ (Jeong et al., 2017). Canyons have large spectrum of lengths from punctate (several kilobases) to very broad (upto megabases).
The valleys are commonly observed in the signal profiles generated from many epigenetic assays such as ChIP-Seq, DNA methylation (bisulfite sequencing) (Li et al., 2018; Xie et al., 2013), open chromatin measurement [DNase sequencing (Madrigal and Krajewski, 2012) and MNase-Seq] and replication timing sequencing (Audit et al., 2013; Dorschner et al., 2009). Among these, the replication timing valleys show the largest length scales (tens of megabases) and DNase valleys show the shortest length scales (At the order of 20 base pairs). In this study we are focusing on punctate valleys at the order of 100 base pairs to 5kbs. The main reason for this is that recent literature shows that the valleys at this length scale are enriched in cis-regulatory elements which can help enhance annotation of functional elements detected from functional genomics signals.
The valleys are important information bearing regions within the epigenetic signal profiles that can pinpoint the locations of cis-regulatory elements such as enhancers and insulators. Thus, the valleys can potentially enable researchers to anatomize and enhance the annotations of regulatory elements. The efficient and systematic identification of valleys can substantially increase the utility of functional genomics experiments. The information that are encoded in the valleys are currently left under-utilized because they are not reported explicitly by most of the analysis pipelines.
Here we present EpiSAFARI, which performs sensitive statistical detection of punctate valleys from the fluctuations in the genome-wide signal profiles. The core algorithm is based on spline smoothing of the epigenetic signal profiles followed by valley detection using the smoothed signal profile. EpiSAFARI can analyze sparse signals such as DNA methylation signals that are non-zero only at cytosine residues on the genome. EpiSAFARI integrates technical factors such as sequence context and mappability. These factors can impact the valleys by causing ‘non-biological valleys’ to manifest (Benjamini and Speed, 2012; Harmanci et al., 2014). Overall, EpiSAFARI performs favorably compared to other tools and can potentially help elucidate functional information about regulatory elements.
2 Materials and methods
2.1 EpiSAFARI algorithm
The input to EpiSAFARI is the signal profile or the mapped reads (Fig. 1). First, EpiSAFARI smooths the signal profile. To smooth the signal, EpiSAFARI divides genome into non-overlapping windows of length base pairs and applies spline-based smoothing to the raw signal profile in each window (Fig. 1, step 2, Supplementary Fig. S1). Although other smoothing approaches have been proposed (Harmanci et al., 2014; Knijnenburg et al., 2014), basis spline curves are advantageous because they do not rely on a model, they are flexible and they guarantee a continuous smoothed signal (Unser et al., 1993).
Fig. 1.

Illustration of the steps in EpiSAFARI algorithm. The input can be one of read coverage signal (bedGraph, wig, bigwig formatted) or mapped reads (sam formatted). The read coverage signal profile, , is smoothed using spline-based fitting in the second step. Smoothing computes the minimum least square fit of using the spline basis functions (). The smoothed signal () is plotted with lightly colored original signal to illustrate the effect of smoothing. In third step, valleys are detected. Red and green dashed lines indicate the minima (dip) and maxima locations (summits), respectively. A valley is defined as a dip surrounded by two summits. The blow-up illustrates the summit positions ( and ) and the dip of a valley () and the signal level at these locations denoted by , and . Next, the valleys are filtered with respect to distance between dip-to-summit distance () and the ratio between signal level at the summits to the dip (). The valleys that are removed are illustrated with grey shaded lines. In step 4, hill score, mappability and nucleotide content are assigned. In this step, multi-mappability profile and genome sequence are used as input. In the final step, statistical significance for the valleys are assigned. For each valley, the signal enrichment is estimated using binomial test for comparing the signal within base pair vicinity of the summits compared to the signal around base vicinity of the dip. The output from EpiSAFARI are the smoothed signal profiles (bedGraph formatted) and the valleys (bed formatted)
The spline curves are defined by a set of ‘knots’ and the degree of the polynomial (Supplementary Figs S1 and S2). The knots represent the positions where the polynomials meet such that the derivative of the curves are continuous up to the selected degree of the spline functions (Supplementary Fig. S1). While knot positions affect the accuracy of spline-based smoothing, general knot selection is a complex and open problem (Foley and Nielson, 1989). We compared 3 different knot selection procedures using different knot numbers and observed that uniform knot selection performs comparably in terms of accuracy to the random knot selection, and derivative-based knot selection procedures (Supplementary Methods, Supplementary Fig. S3). Since EpiSAFARI smooths signal with non-overlapping windows, the smoothing is performed for each window separately. We observed that uniform knot selection biases the detected valley dips periodically on the windows. When derivative or random knot selection is used, this bias is removed (Supplementary Fig. S3i–k). However, we observed slight enrichment of valley dips at the ends of the windows. These biases are removed when overlapping windows are used for spline smoothing (Supplementary Fig. S3l). For this, EpiSAFARI slides the windows with a stepping length that is smaller than window length. This way, each position is covered by multiple windows.
The degree of the splines represents the degree of the polynomials that make up the basis curves. By default, EpiSAFARI uses splines of degree 5 with 7 knots. We observed that increasing (or decreasing) the spline degree or knot numbers may decrease valley detection accuracy because they may cause underfitting or overfitting in the smoothing process (See Supplementary Methods). The spline degree and knot number parameters tune the complexity of smoothing and they can be changed by the user. After basis function generation, a linear minimum square error fit of the signal to the spline basis functions is computed.
| (1) |
| (2) |
where represents the vector that contains the original signal profile in the current window, denotes the set of spline basis functions and denotes the error minimizing weights. In order to decrease computational complexity, is formed by using the signal levels at points of interest in each window. The points of interest are selected as the positions where signal changes value. For sparse signals (e.g. WGBS-based DNA methylation), the points of interest are chosen such that only locations with non-zero signal values are selected. As the smoothing does not make any assumptions on the underlying signal, EpiSAFARI is applicable for analysis of the signal profiles generated diverse set of assays including microarray-based assays (Schumacher et al., 2006). The window length parameter, , determines the number of points of interest and can impact accuracy (Supplementary Methods, Supplementary Fig. S4a–c).
After smoothing, EpiSAFARI evaluates the maximum error among the points of interest. If the maximum error is higher than an anticipated error, EpiSAFARI increases the knot number and the spline degrees and re-iterates signal smoothing with the updated parameters. After all the windows on a chromosome are processed, they are concatenated to form the final smoothed profile. To ensure continuity of the signal, EpiSAFARI filters the concatenated signal profile using a median filter (of length base pairs) that can be changed by the user. By default, EpiSAFARI sets equal to 50 (Supplementary Methods, Supplementary Fig. S4g).
2.2 Valley detection
After smoothing the signal profile, next step is detection of the local extrema, i.e. local minima and maxima. The local extrema are identified as the genomic coordinates where derivative changes sign:
| (3) |
| (4) |
| (5) |
where denotes the smoothed signal value at genomic position. In (3) ad (4), and denotes the set of minima and maxima coordinates, respectively.
The valleys are defined by a local minimum (i.e. dip) and two nearby maxima (i.e. summits) located upstream and downstream the dip (Fig. 1, step 3). The candidate valleys satisfy following constraints:
| (6) |
where denotes the set of valleys, which are triplets of genomic positions . and denote the summit coordinates and denotes the position of valley’s dip between and such that maximum (minimum) distance from and to are bounded by () parameter. This ensures that the summits are not very far from (or near to) the dip. In addition, the ratio of the signal levels at both summits to the dip are bounded below by to ensure that there is difference between the signal levels at summits and signal at the dip. We set for sensitive detection of valleys in the experiments (Supplementary Methods, Supplementary Fig. S4f).
2.3 Assignment of hill scores
For each valley, EpiSAFARI computes a quality score for the two ‘hills’ on each valley (Fig. 1). A hill is the genomic region between the dip and the (left or right) summits. A good hill shows a monotonic increase between the dip and the summits (Supplementary Fig. S5a). To measure this, EpiSAFARI computes the fraction of positions in the left and right hill where the signal is increasing (i.e. going up-hill) while moving away from the dip to the summit. Hill score is computed as
| (7) |
where denotes the hill score and is an indicator function:
| (8) |
For a good valley, the hill score is close to 1.0, indicating that both hills to the left and right of the dip exhibit a monotonically increasing signal while moving away from the dip. If there are any segments with a down-hill trend, the hill score decreases (Step 4 in Fig. 1). Using a high hill score cutoff increases the topological quality of valleys but may adversely impact sensitivity of the valley detection (Supplementary Methods). On the other hand, decreasing the cutoff causes the reported valleys to overlap with each other (Supplementary Fig. S5b). We observed that the valleys with high qualities are separated in the distribution of the hill scores at the very high end of the distribution (Supplementary Methods, Supplementary Figs S5e–g and S6). We therefore use high hill score cutoff (0.90) to report only the valleys with high topological quality.
The mappability of valleys is very important to distinguish valleys caused by low mappability versus the real valleys caused by biological signal fluctuation. For this, EpiSAFARI uses the precomputed multi-mappability signal (Harmanci et al., 2014) profile. For each valley, EpiSAFARI computes the average and the maximum of the multi-mappability signal. In general, high multi-mappability corresponds to a low mappable region and these regions are filtered out in the downstream analysis.
2.4 Assignment of statistical significance
The next step is assignment of statistical significance to the detected valleys (Fig. 1). By statistical significance, we refer to how significant the depletion of the signal at the dip is compared to the signal levels at the summits. Thus, valleys with low P-value correspond to deep valleys. The assigned P-values are used to sort the valleys while performing enrichment analysis.
For a valley at , EpiSAFARI first computes the signal around the vicinity of the dip and the summits using
| (9) |
| (10) |
| (11) |
where , , denote the average signal in the base pair (100 base pairs by default) vicinity of the summits , and the dip (Supplementary Fig. S7). Next, EpiSAFARI computes the binomial P-value of enrichment of signal around summits compared to the dip:
| (12) |
| (13) |
where number of combinations for selecting items within items:
| (14) |
In order to assign the final P-value to the valley, we combine the P-values that are assigned to enrichment of the signal at the two summits. This process corresponds to combining the null models that are used to assign the two P-values for the observed summit-to-dip signal enrichment. We first use intersection of the null models as the joint null model (Supplementary Fig. S7a). Assuming that the left and right hills are independent, this corresponds to the direct multiplication of the P-values:
| (15) |
denotes the P-value computed by intersection-based combination of the P-values assigned to observed summit-to-dip signal enrichment (Supplementary Methods, Supplementary Figs S7 and S8).
After the P-values are assigned, the false discovery rate at which each valley would be deemed significant is estimated using Benjamini-Hochberg procedure (Benjamini, 2010).
The valleys that EpiSAFARI detected may overlap with each other although we generally observed that the overlap between detected valleys tends to be very small. To ensure that a non-redundant set of minima are reported, EpiSAFARI filters out the valleys whose dips are close to each other by selecting the most significant valley (i.e. lowest P-value) around local minima positions. Finally, EpiSAFARI annotates valleys with respect to genes and transcription factor binding peaks. We created a GFF file from the transcription factor binding peak regions from ENCODE project (ENCODE Project Consortium, 2012), which contains the transcription factor peaks that are identified by 690 ChIP-Seq experiments performed on cell lines and uniformly processed by the ENCODE Project. The smoothed signal profiles can be used for visualizing the signal (Supplementary Fig. S2).
An important factor about detecting valleys is the required sequencing depth. For analyzing the required read depth, we used a high depth H3K4me3 ChIP-Sequencing data from NA12878 sample (Kasowski et al., 2013) and identified valleys. We next computed the increase in the number of valleys with increasing read depth and the increase in the fraction of identified functional elements (Supplementary Fig. S4i and j). We found that beyond 35-40 million reads, the valley detection does not provide substantial additional information.
3 Results
3.1 Performance benchmarking
We first focused on comparing the valleys detected by EpiSAFARI with the existing tools. While several studies have focused on analysis of valleys in different contexts, we found that PARE (Pundhir et al., 2016) is available with implementation that can be used for comparison. In the comparison, we used the H3K4me3 histone modification ChIP-Seq data for NA12878 individual from the ENCODE Project (ENCODE Project Consortium, 2012). In general, H3K4me3 modification marks the promoters of the active genes. We have focused specifically on this modification because firstly it is a well-characterized mark and secondly PARE algorithm is tuned for analysis of this mark so that we are fair in comparison of the tools. We downloaded the two replicates that are available and pooled the reads from the replicates. PARE algorithm is run with default settings except that we extended the search space to 2000 base pairs (-v option) and we relaxed the FDR cutoff to 0.1 (-t option). For EpiSAFARI, we set the FDR cutoff to 0.05, filtered out the valleys with hill scores lower than 0.90. In general, we observed that EpiSAFARI identifies many more valleys compared to PARE. To make the comparison fair, we sorted the EpiSAFARI valleys with respect to increasing FDR (i.e. more significant first) and we sorted the PARE valleys with respect to decreasing score (i.e. higher score first) assigned by the algorithm. We then focused on the top 2000 valleys.
Currently, ChIP-Seq datasets are analyzed primarily in terms of peaks. To include the peaks in the comparison, we identified the peaks for the H3K4me3 data using MUSIC (Harmanci et al., 2014) and extracted the 200 base pair vicinity of the summits and used the summit regions in comparison with the valleys. Since we do not have a set of valleys that can directly serve as ground truth, we used different hypotheses to evaluate whether the identified valleys are biologically meaningful and used these to assess performance of methods.
We first focused on comparison of transcription factor binding activity around the valleys. We hypothesized that the real valleys must be enriched in transcription factor binding. ENCODE project supplies a large number of ChIP-Seq experiments and uniformly processed peak calls for many transcription factors for NA12878 sample. We pooled the available peaks calls from the 90 ChIP-Seq experiments for NA12878 sample. We then evaluated the fraction of top valleys that overlap with a transcription factor peak. In order to correct for the valley lengths reported by the methods, we used the valleys that are reported by PARE as they are and we used only the 200 base pair vicinity of the valley dips (i.e. dip location ± 100 base pairs) reported by EpiSAFARI. Figure 2a shows the fraction of top valleys (and summits) that overlap with a transcription factor peak while the number of top peaks is increased (x-axis). We observed that more than 90% of the top EpiSAFARI valleys that we evaluated overlap with a peak. The fraction of overlap decreases slowly as we increase the number of top valleys. PARE valleys, in contrast, show a fairly low overlap with a transcription factor peak (starting at 50%) and the overlap fraction increases as the number of top valleys is increased. Around 90% of the peak summits contain a transcription factor binding. This result indicates that the valleys (and the scores) detected by EpiSAFARI represent a better representative set of transcription factor activity compared to the other methods.
Fig. 2.
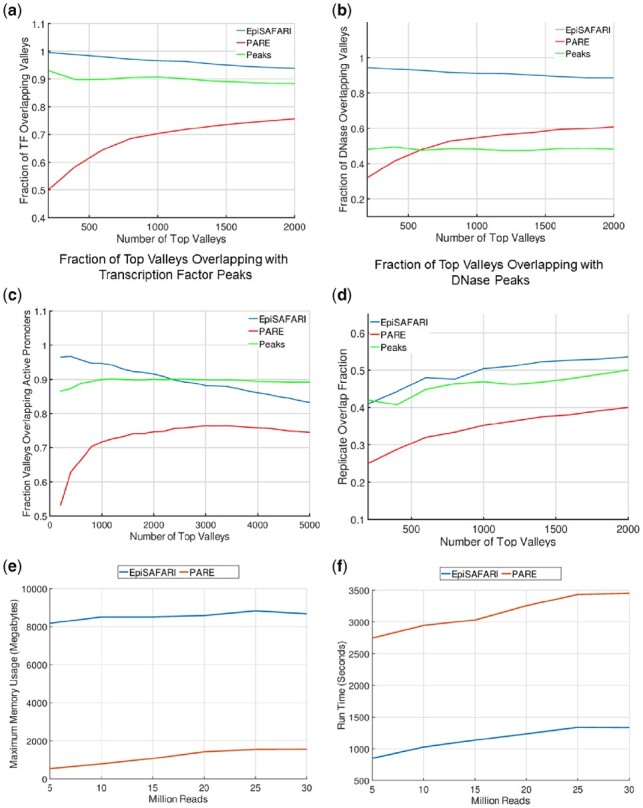
Comparisons. Comparison of the top H3K4me3 valleys in NA12878 sample as detected by EpiSAFARI (blue), by PARE (red) and peak summit regions detected by MUSIC (green). (a) The fraction of top valleys and summits that overlap with a transcription factor peak. X-axis shows the number of top valleys (summits) and y-axis shows the fraction of valleys that overlap with a transcription factor peak. (b) The fraction of top valleys that overlap with a DNase peak. (c) The fraction of top valleys (summits) that overlap with an active promoter. (d) Overlap fraction of the of valleys detected from the two replicates of GM12878 H3K4me3 dataset. (e) The memory requirements of EpiSAFARI and PARE with increasing read depth. X-axis shows the total number of reads and y-axis shows the required maximum main memory in gigabytes. (f) The time requirements of EpiSAFARI and PARE. X-axis shows the total number of reads and y-axis shows the required wall time in seconds
Another hypothesis about the valleys is that they are enriched in terms of open chromatin. To measure this, we downloaded the peaks of the DNase-1 hypersensitive site sequencing (DNase-Seq) data from the ENCODE project for NA12878 sample. These peaks represent the experimentally detected locations of genomic positions for accessible DNA. Similar to the previous comparison, we overlapped the top valleys detected by EpiSAFARI and PARE with the DNase peaks. Figure 2b shows the fraction of top valleys that overlap with a DNase peak. Similar to previous analysis, we used the 200 bp vicinity of the valley dip for EpiSAFARI valleys for this comparison. We observed that EpiSAFARI valleys show a much higher overlap fraction to the DNase peaks compared to PARE and peak summits. In addition, PARE valleys also show an increasing overlap fraction with decreasing score while EpiSAFARI valleys show a slowly decreasing overlap fraction with decreasing significance. This result indicates that EpiSAFARI valleys are better representatives of the accessible DNA positions compared to the valleys detected by PARE. The peak summits exhibit around 50% overlap with the DNase peaks.
We hypothesized that the top valleys of the H3K4me3 modification must be enriched in the active gene promoters. To identify the active gene promoters, we used the transcript expression quantifications from the ENCODE project for NA12878. We first identified the transcripts whose reported expression levels in terms of reads per kilobase per million mapped reads (RPKM) are higher than 0.05. We next extracted the 1000 base pair vicinity of the transcription start site of each transcript. These constitute the set of active promoters. We then overlapped the valleys with the active promoters and computed the fraction of top valleys that overlap with active promoters. Figure 2c shows the overlap fraction of top valleys with active promoters. EpiSAFARI shows a fairly high overlap (higher than 90%) at the top valleys and decreases as the number of top valleys decrease. For the top 2000 valleys, the overlap is always higher than 80%. The top valleys identified by PARE show 50% overlap with active promoters and the overlap increases as the number of top valleys is increased. The peak summits show around 90% overlap with the active promoter regions. This result shows that EpiSAFARI valleys capture the active promoter information better than PARE while detecting valleys. In comparison to peak summits, EpiSAFARI valleys exhibit higher accuracy for top 2000 valleys. We also compared the reproducibility of the valleys (and summits) identified by the methods. For this, we randomly divided the reads for NA12878 H3K4me3 to generate 2 replicates. We identified the valleys using EpiSAFARI and PARE and the peaks using MUSIC. We finally compared the replicates and computed the average consistency among identified valleys and summits. Figure 4d shows the replicate overlap for the top valleys and peak summits. All the methods show low replicate consistency for the top elements and consistency increases up to 50% as top element number is increased. In comparison, EpiSAFARI valleys (and peak summits) exhibit higher replicate consistency compared to PARE valleys. We also compared the run time and main memory requirements of EpiSAFARI and PARE (Fig. 2e and f). We found that PARE uses less memory than EpiSAFARI and EpiSAFARI has lower run time requirements.
Fig. 4.

Conservation and Transcription around Histone Valleys. (a) Characterization of the methyl-valleys. (a) Conservation around the 20 000 base pair vicinity of the dips of methyl-valleys as reported by EpiSAFARI. All valleys (blue), valleys that do not overlap with promoters (red) and random regions (green) are shown. (b) The fraction of the top methyl-valleys that overlap with a H1hESC transcription factor binding peaks. X-axis shows the top number of valleys and y-axis shows the fraction of valleys that overlap with at least one transcription factor peak. (c) Heatmap of the transcription factor binding on the top 1000 methyl-valleys. Each row is a methyl-valley and each column is a transcription factor. Within a row, white color indicates an overlap with the valley corresponding to the row and the peaks of the transcription factor that is corresponding to the column. Red indicates no overlap between the transcription factor’s peaks and the valley. The rows and columns are sorted by hierarchical clustering
3.2 Histone valleys are enriched in functional activity
We next computed the average conservation on the valleys identified by EpiSAFARI. To measure the conservation around the valleys, we aggregated the PhyloP conservation score (Kuhn et al., 2013) around the 20 000 base pair vicinity of the reported dips of the valleys (Fig. 3a). There is a substantial increase in the average conservation signal around the valley dip and conservation decreases with increasing distance to the dip. We also found that the valleys that do not overlap with H3K4me3 peaks (or with promoters) show increased conservation compared to random regions (Fig. 3a). These valleys potentially represent the novel elements that EpiSAFARI identified that could have been missed by peak callers.
Fig. 3.
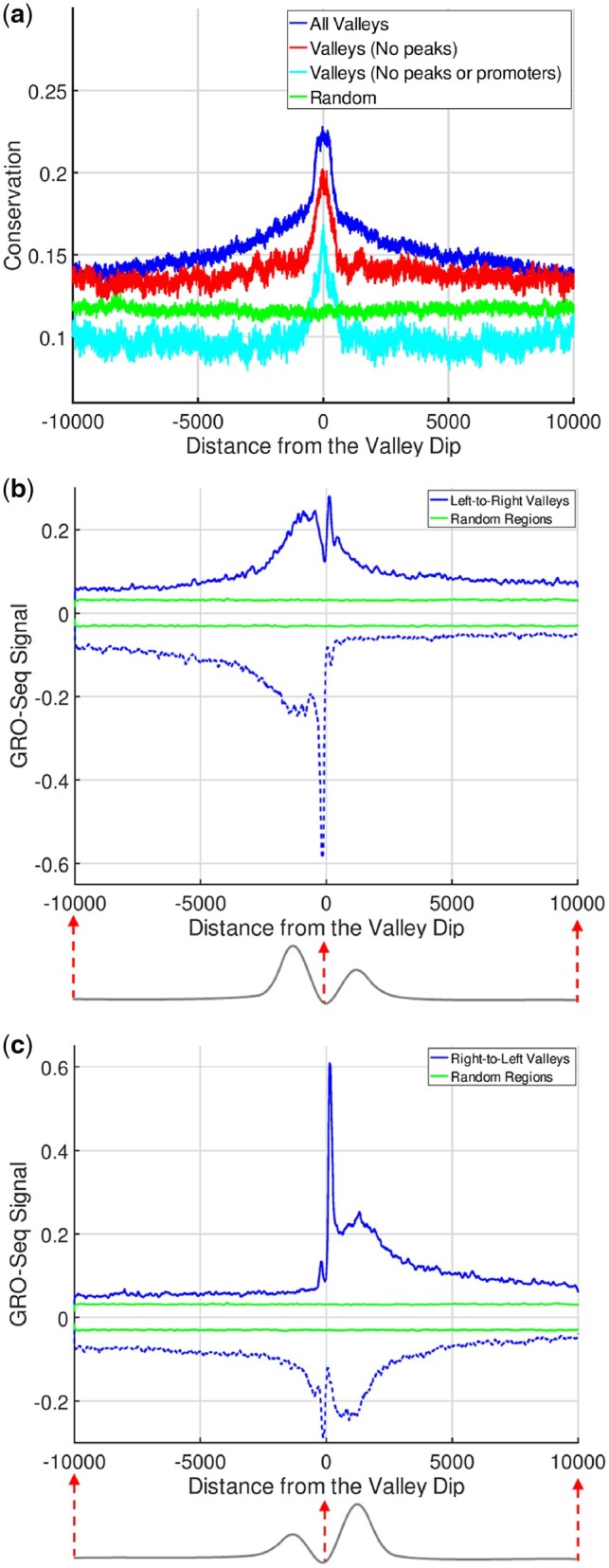
Conservation and Transcription around Histone Valleys. (a) Average conservation within 20 000 base pairs of the valley dips. X-axis shows the distance from the dip and y-axis shows the average PhyloP conservation score. The conservation around all valleys (blue), valleys that do not overlap with any H3K4me3 peaks (red), valleys that do not overlap with neither promoters nor peaks (cyan) and randomized regions (green) are shown. (b) The aggregation of GRO-Seq signal within 20 000 base pairs of left-to-right valleys (blue) and random regions (green). The bottom illustration points out the fact that left summit is taller than right summit. (c) The aggregation of GRO-Seq signal within 20 000 base pairs of right-to-left valleys where right summits are taller than left summits
We also hypothesized that the valleys as detected by EpiSAFARI may contain cis-regulatory elements such as promoters and enhancers (Supplementary Fig. S9). One line of evidence for existence of these elements is the nascent transcription at the valleys. To study this, we used the global run-on sequencing (GRO-Seq) data for NA12878 sample (See Data Availability). GRO-Seq data represent the genome-wide measurement of nascent transcription, i.e. RNA that has just been transcribed (or being transcribed) at each location in the genome (Core et al., 2008). We observed that there is an increased GRO-Seq signal on both positive and negative strands around the dip of the valley (Supplementary Fig. S10a and b).
An important observation about the valleys is that valleys may show asymmetry with respect to the signal levels at the left and right summit positions and this relates to transcriptional activity (Kundaje et al., 2012). In order to study the relation between valley shape asymmetry and transcriptional activity around the valleys, we divided the valleys into two groups. First group, we call left-to-right valleys (Bottom illustration in Fig. 3b), have higher signal on the left summit compared to right summit. Second group, right-to-left valleys, (Bottom illustration in Fig. 3c) contains higher signal on the right summit compared to the left summit. Figure 3b and c shows the average GRO-Seq signal around the 20 000 base pairs vicinity of the valley dips for left-to-right and right-to-left valleys, respectively. For left-to-right valleys, there is a sharp peak on the negative strand signal to the left of the dip position. The positive signal, while still high, does not show a corresponding sharp peak. In other words, left-to-right valleys are enriched in terms of negatively oriented nascent transcription. Similar pattern is seen for the right-to-left valleys (Fig. 3c) albeit on the positive strand. Overall, these results provide supporting evidence that the valleys detected by EpiSAFARI contain genomic elements of potential functional role. Furthermore, the valleys’ asymmetry can delineate the directionality of transcriptional activity around them.
An important downstream analysis is comparison of valleys from two samples. We studied the ‘differential valley detection’ to identify the valleys that are specific to samples under different conditions (Supplementary Fig. S13a). To compare the valleys from two samples (e.g. cell lines with different treatments) and to identify valleys specific to first sample, we first pool the valleys identified in the two samples. We next compute the difference profile by subtracting the signal profile for second sample from that of the first sample. This profile quantifies enrichment of signal at the valleys when sample 1 is compared to sample 2. Finally, we compute the significance of each of the pooled valleys using the difference profile (Supplementary Material). We compared the valleys from NA12878 and K562 cell lines, and identified the NA12878 and K562 specific valleys. To characterize the sample specific valleys, we analyzed the DNase signal from both cell lines and computed the fold-change at the cell line specific valleys. This analysis revealed that cell line specific valleys show significantly increased DNase signal (Supplementary Fig. S13b and c) compared to all valleys.
3.3 Methyl-valleys are enriched in chromatin structure associated transcription factor binding
As another application of EpiSAFARI, we next focused on analysis of the valleys in the DNA methylation signal of H1 embryonic stem cell line (H1hESC) measured by the whole genome bisulfite sequencing (WGBS) data from Roadmap Epigenome Project. The valleys in DNA methylation signals have been shown to contribute to important biological phenomena (Jeong et al., 2014; Li et al., 2018). We first downloaded the processed the WGBS signal profile from Roadmap Epigenome Project (Romanoski et al., 2015). This signal profile measures the fraction of methylated versus non-methylated cytosine residues at the CpG di-nucleotides. We identified valleys in DNA methylation signal, i.e. methyl-valleys, using sparse mode of EpiSAFARI. In valley detection, we set , and excluded the valleys that contain less than 20 CpG di-nucleotides as these may correspond to valleys with very sparse signals. While smoothing DNA methylation signals, is used (Supplementary Methods, Supplementary Fig. S4k–n).
The methyl-valleys generally show elevated conservation (Fig. 4a) compared to their surroundings. We overlapped the valleys with the transcription factor ChIP-Seq peaks for H1hESC cell line. The top methyl-valleys (including valleys that do not overlap promoters) are highly enriched in terms of transcription factor binding peaks (Fig. 4b). We next identified the fraction of transcription factors whose peaks overlap with the top 1000 valleys (Supplementary Fig. S11). Large fraction of methyl-valleys overlaps with transcription factors such as CTCF (60%), Rad21 (50%) and Znf143 (50%), which are known regulators of three-dimensional chromatin structure (Fig. 4c). A similar result has been reported in another study (Lin et al., 2017) where the authors show that chromatin structure associated transcription factors are enriched in undermethylated regions. We also used EpiSAFARI to detect the methyl-valleys for NA12878 cell line using a publicly available dataset (See Data Availability). While we did observe that the top methyl-valleys are enriched in transcription factor binding (Supplementary Fig. S12a), we did not observe the enrichment of the chromatin structure associated transcription factors (Supplementary Fig. 12b). These results provide evidence that the stratification of the transcription factor binding on the valleys can provide biological insight in epigenetic data analysis.
3.4 Supervalleys are enriched at promoters of developmental genes in embryonic stem cell line
It was previously shown that the broad domains of histone modification enrichments may represent super-enhancer regions (Pott and Lieb, 2015), which are associated with important biological phenomena such as cancer initiation. In addition, several studies showed that the broad H3K4me3 peaks are enriched around genes with certain biological roles (Benayoun et al., 2014; Dincer et al., 2015). We overlapped the H3K4me3 valleys for H1hESC cell line with the promoters and we identified the top 500 genes whose promoters overlap with highest number of valleys (Fig. 5a), which we refer to as supervalleys. Interestingly, these genes are significantly related to development, differentiation and DNA binding (Fig. 5b). Consequently, the clustering of the valleys with respect to proximity in genomic coordinates may be an indicator of elements with important biological functions. When we performed the supervalley analysis using H3K4me3 valleys of NA12878 cell line, we did not find any significant functional category.
Fig. 5.

Functional enrichment of the genes whose promoters overlap with supervalleys. (a) Illustration of a simple valley and a H3K4me3 supervalley. The simple valley consists of a double-peak pattern with a valley in it. The supervalleys contain many consecutive valleys that are clustered within a small genomic distance. Block diagram below illustrates the detection of supervalleys on promoters. The valleys are assigned to gene promoters such that a promoter is defined as the 20 kb vicinity of the transcription start site (TSS). The genes whose promoters overlap with the largest number of valleys are identified. The top 500 genes are used in gene ontology enrichment. (b) The most significant 10 GO terms (x-axis) that are detected from ontology enrichment as sorted with respect to enrichment false discovery rate (y-axis)
4. Discussion
We presented EpiSAFARI as a new method for detecting the valleys in epigenetic signal profiles. One of the main challenges related to the valley-centric analysis of epigenetic data is concretizing the definition ‘valley calling’ process. EpiSAFARI treats the valleys to be dips in the signal that are between two summits but this definition could potentially be revised to ensure that the valleys represent the functionally most meaningful regions. Another limitation that we have faced is defining quality metrics for the detected valleys. The hill score aims to measure the valley quality but we observed that it may be affected to a certain extent by the signal smoothing parameters. More robust measures of valley quality can elucidate the valley quality. Another challenge is defining statistical models for valley calling. Although we evaluated several statistical models that evaluate the significance of the valleys, the definition of statistical significance of valleys should be studied in more detail.
Several previous methods have utilized valleys in different contexts. These methods rely on smoothing of signal using kernel-based approaches [such as Gaussian (Knijnenburg et al., 2014) and wavelet filtering (Audit et al., 2013)] or modeling of the read clusters (such as PARE). In comparison, EpiSAFARI is advantageous to these methods for two reasons. Firstly, EpiSAFARI incorporates mappability and nucleotide content in filtering of the detected valleys. As we have demonstrated, these factors may create false positive valleys. Secondly, the kernel smoothing-based methods may fail to smooth sparse signals (such as DNA methylation) because smoothing of sparse signals will introduce many false positive valleys. On the other hand, EpiSAFARI computes an interpolation of the sparse signal to efficiently build a continuous smoothing of the sparse signals. Thus, the spline-based modeling of EpiSAFARI separates it from previous methods for modeling of both continuous and sparse signal profiles. EpiSAFARI utilizes a parametric spline-based strategy to smooth the signal before detection of the valleys. We studied extensively the impact of the parameters on valley detection accuracy. While knot selection may slightly bias results, these can be mitigated by using overlapping windows.
Supplementary Material
Acknowledgements
None.
Funding
This work was supported by National Institutes of Health of The United States grant 5R01NS079715 to VG.
Conflict of Interest: none declared.
Contributor Information
Arif Harmanci, School of Biomedical Informatics, Center for Precision Health, USA.
Akdes Serin Harmanci, School of Biomedical Informatics, Center for Systems Medicine, University of Texas Health Science Center, Houston, TX 77030, USA.
Jyothishmathi Swaminathan, Department of Pediatrics, USA.
Vidya Gopalakrishnan, Department of Pediatrics, USA; Department of Molecular and Cellular Oncology, USA; Brain Tumor Center, USA; Center for Cancer Epigenetics, University of Texas, M.D. Anderson Cancer Center, Houston, TX 77030, USA; M.D. Anderson UTHealth Graduate School of Biomedical Sciences, Houston, TX 77030, USA.
References
- Audit B. et al. (2013) Multiscale analysis of genome-wide replication timing profiles using a wavelet-based signal-processing algorithm. Nat. Protoc., 8, 98–110. [DOI] [PubMed] [Google Scholar]
- Benayoun B.A. et al. (2014) H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell, 158, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. (2010) Discovering the false discovery rate. J. R. Stat. Soc. Ser. B, 72, 405–416. [Google Scholar]
- Benjamini Y., Speed T.P. (2012) Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res., 40, e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D. et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L.J. et al. (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science, 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny J.C. et al. (2016) Phenome-wide association studies as a tool to advance precision medicine. Annu. Rev. Genomics Hum. Genet., 17, 353–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincer A. et al. (2015) Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic landscapes in the human brain. Transl. Psychiatry, 5, e679.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Weng Z. (2013) The correlation between histone modifications and gene expression. Epigenomics, 5, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner M.O. et al. (2009) Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA, 107, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. (2008) Epigenetics in Cancer. N. Engl. J. Med., 358, 1148–1159. [DOI] [PubMed] [Google Scholar]
- Foley T.A., Nielson G.M. (1989) Knot selection for parametric spline interpolation. In: Mathematical Methods in Computer Aided Geometric Design, pp. 261–271.
- Harmanci A. et al. (2014) MUSIC: identification of enriched regions in ChIP-Seq experiments using a mappability-corrected multiscale signal processing framework. Genome Biol., 15, 474.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y. et al. (2017) Multi-omics approaches to disease. Genome Biol., 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M. et al. (2017) A Cell type-specific Class of Chromatin Loops Anchored at Large DNA Methylation Nadirs. bioRxiv, 212928.
- Jeong M. et al. (2014) Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet., 46, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M. et al. (2013) Extensive variation in chromatin states across humans. Science (New York, NY), 342, 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg T.A. et al. (2014) Multiscale representation of genomic signals. Nat. Methods, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R.M. et al. (2013) The UCSC genome browser and associated tools. Brief. Bioinform., 14, 144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A. et al. (2012) Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res., 22, 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2018) Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol., 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. et al. (2017) Sparse conserved under-methylated CpGs are associated with high-order chromatin structure. Genome Biol., 18, 163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigal P., Krajewski P. (2012) Current bioinformatic approaches to identify DNase I hypersensitive sites and genomic footprints from DNase-seq data. Front. Genet., 3, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G. et al. (2013) Identification of genetic variants that affect histone modifications in human cells. Science (New York, NY), 342, 747–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott S., Lieb J.D. (2015) What are super-enhancers? Nat. Genet., 47, 8–12. [DOI] [PubMed] [Google Scholar]
- Pundhir S. et al. (2016) Peak-valley-peak pattern of histone modifications delineates active regulatory elements and their directionality. Nucleic Acids Res., 44, 4037–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanoski C.E. et al. (2015) Epigenomics: roadmap for regulation. Nature, 518, 314–316. [DOI] [PubMed] [Google Scholar]
- Rozowsky J. et al. (2009) PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol., 27, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher A. et al. (2006) Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res., 34, 528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi A. et al. (2018) A cross-organism framework for supervised enhancer prediction with epigenetic pattern recognition and targeted validation. bioRxiv, 385237. [DOI] [PMC free article] [PubMed]
- Sharing Epigenomes Globally (2018) Editorial. Nat. Methods, 15, 151. [Google Scholar]
- Sun W. et al. (2016) Histone acetylome-wide association study of autism spectrum disorder. Cell, 167, 1385–1397.e11. [DOI] [PubMed] [Google Scholar]
- Thomas R. et al. (2017) Features that define the best ChIP-seq peak calling algorithms. Brief. Bioinform, 18, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unser M. et al. (1993) B-spline signal processing. I. Theory. IEEE Trans. Signal Process, 41, 821–833. [Google Scholar]
- Xie W. et al. (2013) Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell, 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol., 9, R137.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Reinberg D. (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.