

Abstract
Spermatozoa contain a repertoire of RNAs considered to be potential functional fertility biomarkers. In this study, the gene expression of human sperm subpopulations with high (F1) and low (F2) motility from healthy normozoospermic (N) and asthenozoospermic (A) individuals was evaluated using RNA microarray followed by functional genomic analysis of differentially expressed genes. Results from A–F1 versus N–F1, A–F2 versus N–F2, N–F1 versus N–F2, and A–F1 versus A–F2 comparisons showed a considerably larger set of downregulated genes in tests versus controls. Gene ontology (GO) analysis of A–F1 versus N–F1 identified 507 overrepresented biological processes (BPs), several of which are associated with sperm physiology. In addition, gene set enrichment analysis of the same contrast showed 110 BPs, 36 cellular components, and 31 molecular functions, several of which are involved in sperm motility. A leading-edge analysis of selected GO terms resulted in several downregulated genes encoding to dyneins and kinesins, both related to sperm physiology. Furthermore, the predicted activation state of asthenozoospermia was increased, while fertility, cell movement of sperm, and gametogenesis were decreased. Interestingly, several downregulated genes characteristic of the canonical pathway protein ubiquitination were involved in asthenozoospermia activation. Conversely, GO analysis of A–F2 versus N–F2 did not identify overrepresented BPs, although the gene set enrichment analysis detected six enriched BPs, one cellular component, and two molecular functions. Overall, the results show differences in gene transcription between sperm subpopulations from asthenozoospermic and normozoospermic semen samples and allowed the identification of gene sets relevant to sperm physiology and reproduction.
Keywords: asthenozoospermia, male infertility, microarray, sperm, transcriptome
INTRODUCTION
Spermatogenesis and sperm maturation are multifactorial processes that result in the production of differentiated spermatozoa with the ability to become capacitated and fertilize. In andrology laboratories, standard semen evaluation is the primary approach to assess sperm-fertilizing ability, by detecting anomalies in sperm concentration, motility, and morphology. However, it has a limited diagnostic power, and identification of novel markers of sperm function is increasingly demanded.1,2 To this end, several studies have characterized the presence of RNAs in mature spermatozoa,3,4 including comparisons between infertile and fertile men.5 The potential utility of sperm RNA as molecular fertility markers, mainly those consistently present in samples from fertile men, has been previously evaluated.6,7 Sperm RNA is a complex mixture of different types of RNA, some of which may represent a particular RNA population with functional roles after delivery into the oocyte.3,8 Besides this, some RNAs may also have physiological roles during the epigenetic reprogramming of sperm chromatin9 and sperm maturation and may therefore be involved in their ability to fertilize.3,10 However, recent studies indicate that a substantial portion of sperm mRNA is fragmented, though some transcripts are maintained intact with potential roles during sperm transit through the female reproductive tract, fertilization, and early embryogenesis.11,12,13
Asthenozoospermia, defined as a semen sample with a proportion of progressively motile spermatozoa below the lower WHO reference limit,14 is a major cause of male infertility, and several genes and molecular markers have been linked to this condition.15 This is relevant because assisted reproductive technologies require the isolation of progressively motile spermatozoa from those that are immotile and incapable of fertilizing.16 Indeed, only a small percentage of spermatozoa can achieve fertilization, and their isolation and full characterization represents a significant challenge in the study of male infertility. In this study, we compared the gene expression profiling of high (F1) and low (F2) sperm motility fractions isolated from the semen samples of asthenozoospermic and normozoospermic men by microarray technology. The aim was to identify transcripts that differ between sperm subpopulations and to determine their functional relevance in sperm physiology and reproduction.
MATERIALS AND METHODS
Semen samples and sperm analysis
This study was approved by the Human Ethical and Scientific Review Committees of the National Institute of Medical Sciences and Nutrition Salvador Zubirán, Mexico City, Mexico (reference number 1516, from 04/27/2015), and all participants gave written informed consent for the use of their semen in research. Human semen samples obtained by masturbation from normozoospermic (N) and asthenozoospermic (A) volunteers were collected in sterile containers and evaluated within 1 h after ejaculation according to the WHO reference guidelines.14 To isolate sperm subpopulations, liquefied semen samples were centrifuged at 800 g for 30 min through a 90%/60% discontinuous density gradient (Isolate; Irvine Scientific, Irvine, CA, USA). For each sample, two sperm subpopulations were recovered and named as A–F1/N–F1 (sperm pellets) and A–F2/N–F2 (90%/60% interphases). Sperm fractions were washed with phosphate-buffered saline; re-assessed for cell concentration, motility, and purity (visual absence of somatic cell contamination); and processed for RNA extractions. For analysis by microarray technology, F1 and F2 samples recovered from three A and three N semen donors were grouped into four study groups (three A–F1, three A–F2, three N–F1, and three N–F2), for a total of 12 microarrays.
RNA isolation and microarray hybridization
Total RNA from sperm subpopulations was extracted as previously described.7,17 Briefly, sperm samples were suspended in 1 ml of TRIzol® reagent (Invitrogen, Carlsbad, CA, USA), passed through 20G hypodermic needles, and separated by the addition of 0.2-ml chloroform. The RNA concentration was determined by using an ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and its quality was checked using an Agilent Bioanalyzer 2100 (Agilent Technologies Inc., Palo Alto, CA, USA) by loading samples onto RNA nano chips included in the RNA 6000 Nano kit (Agilent Technologies) for analysis with the Eukaryote Total RNA Nano Assay (version 2.6) and following sperm RNA quality criteria previously described.15 To confirm the absence of contamination by somatic cell RNA, reverse transcription polymerase chain reaction (RT-PCR) for CD45, c-kit, and E-cadherin was performed with primers indicated in Supplementary Table 1, and the resulting PCR amplicons were analyzed by gel electrophoresis.15,18
Supplementary Table 1.
Primer sequences used for polymerase chain reaction
| Gene | Accession number | Forward | Reverse |
|---|---|---|---|
| ACE | NM_000789.3 | AACTACCCGGAGGGCATAG | CCGGTCATATTCCTCCACAA |
| SLC25A1 | HM037273.1 | CCCCATGGAGACCATCAA | GTGAGGCCCTGGTACGTC |
| STRBP | NM_018387.4 | TCTAATTAGGGACGAATTGGAGA | TCCAATAAGTCCGGAGGATCT |
| MT-CYB | HM046248.1 | CAACAACCGCTATGTATTTCGT | GGTTTTTATGTACTACAGGTGGTCAA |
| CD-45 | NM_002838.3 | AGTCAAAGTTATTGTTATGCTGACAGA | TGCTTTCCTTCTCCCCAGTA |
| C-KIT | NM_000222.2 | ATGGCATGCAATGTGT | GGCAGTACAGAAGCAGAGCA |
| E-CAD | Z13009.1 | TCTACTGCATCACTGGCCAAGGAGCTG | AGCTTGAACCACCAGGGTATACGTAGG |
| SMCP | NM_030663.2 | TTTGGCTTCTGATAGTCATGGA | CACACATCTTCTGAACACTTGGA |
| RPL32 | NM_000994.3 | GAAGTTCCTGGTCCACAACG | GAGCGATCTCGGCACAGTA |
For microarray experiments, target cDNAs were prepared according to the Whole-Transcript PLUS (WT) Sense Target Labeling Protocol (Affymetrix, Inc., Santa Clara, CA, USA) as previously described.19,20 The cRNA products used as templates for second cycles of cDNA synthesis in the presence of dUTPs were fragmented by uracil-DNA glycosidase and apurin apirymidin endonuclease. The fragments (40–70 mer) were then biotin labeled with deoxynucleotide terminal addition reaction. Labeled single-stranded cDNAs were hybridized during 17 h at 45°C onto a GeneChip® Human Gene 2.0 ST Array (Affymetrix), containing 40 716 total RefSeq transcripts for 30 654 NM RefSeq well-established annotations for the measurement of protein coding and long intergenic noncoding RNA transcripts. Washing and staining with streptavidin-phycoerythrin were performed with the Affymetrix Fluidics Station 450 using the Affymetrix Staining Kit. Finally, the microarrays were scanned for fluorescence signals in a GeneChip Scanner 3000 7G (Affymetrix).
Data analysis
Microarray raw data were background corrected by the Robust Multiarray Average method21 and normalized by quantile normalization.22 The analyses involved four contrasts (A–F1 vs N–F1, A–F2 vs N–F2, N–F1 vs N–F2, and A–F1 vs A–F2), and the differentially expressed genes (DEGs) were determined by linear statistical models with arbitrary coefficients from the Bioconductor library limma.23,24 Multiple hypotheses were corrected by applying a false discovery rate (FDR). The up- and down-DEGs were selected on the bases of a fold-change |FC| >2 and P < 0.01. All analyses were performed by R software (version 3.2.1, 2015; The R Foundation for Statistical Computing, Vienna, Austria; https://www.r-project.org/).
Gene ontology analysis
Gene ontology (GO) was analyzed by Babelomics 5.0's FatiGO (http://babelomics.bioinfo.cipf.es) from GO terms generated by the list of DEGs, as previously described.25,26 The nomenclature of the enriched biological processes (BPs) included terms of the Gene Ontology Consortium.27 The adjusted P-values were calculated by an exact Fisher's test that evaluated the significant overrepresentation of functional terms in the list of DEGs with respect to the rest of the human genome after correcting for multiple tests (multiple hypotheses, one for each functional term) by FDR.28 A REVIGO (reduce + visualize Gene Ontology; http://revigo.irb.hr) analysis was done to identify and avoid redundancy of the most representative BPs, as well as for the construction of TreeMaps.29 Moreover, gene set enrichment analysis (GSEA; http://www.broadinstitute.org/gsea) was performed with an FDR <0.05 and normalized enrichment scores (NES) ≥2.0, and a leading-edge analysis was done to identify common gene subsets within the selected GO terms.
Ingenuity pathway analysis
Selected bio-functions associated with sperm physiology and reproduction were analyzed by using ingenuity pathway analysis (IPA; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). Bio-functions were considered relevant when an absolute z-score >2 and P < 0.01 were fulfilled. Furthermore, from a hypothesis-driven approach to the DEGs in A–F1 versus N–F1 comparison on sperm physiology bio-functions, a molecule activity prediction (MAP) by IPA was analyzed as previously described.19,30,31
Quantitative real-time PCR
For microarray validation of DEGs, 1-μg total RNA from each sperm sample was reversely transcribed by using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. Quantitative real-time PCR (qPCR) was performed on a Light Cycler® 2.0 Detection System with a TaqMan PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA). Primers and probes for PCR amplifications were designed with the online software from Universal Probe Library Assay Design Center of Roche (https://lifescience.roche.com/en_mx/brands/universal-probe-library.html#assay-design-center), and each pair of primers was submitted to the National Center for Biotechnology Information BLAST search to ensure specificity for the target mRNA. The oligonucleotide sequences used for gene amplification are shown in Supplementary Table 1. The comparative CT method32 was used to quantify the expression of target genes after normalizing against ribosomal protein L32 (RPL32)as the housekeeping gene.33,34
Statistical analyses
NCSS software (NCSS, Kaysville, UT, USA) was used for data analysis. From the homogeneity and distribution of the data, either Student's t- test or nonparametric Mann–Whitney U test was performed to compare differences between the A and N control samples. P ≤ 0.05 was considered statistically significant.
RESULTS
Sperm characteristics
The analysis of A semen samples showed significantly lower progressive motility than N samples. Moreover, the comparison of sperm subpopulations showed a lower percentage of progressive motility in A–F1 than N–F1, but there was no statistical difference between A–F2 and N–F2 (Supplementary Table 2).
Supplementary Table 2.
Semen parameters of samples used for microarray analysis
| Parameter | Normozoospermic (n=3) | Asthenozoospermic (n=3) |
|---|---|---|
| Age | 27.67±2.08 | 34.67±4.51 |
| Volume (mL) | 3.60±1.38 | 4.80±1.92 |
| Concentration (1×106 Spz ml−1) | 84.84±37.24 | 46.00±15.71 |
| Total sperm count (×106) | 331.27±118.21 | 238.4±175.69 |
| Morphology (% of normal forms) | ≥30 | ≥30 |
| Semen progressive motility (%) | 77.00±7.51 | 19.33±8.08* |
| F1 progressive motility | 76.00±6.96 | 40.50±23.57 |
| F2 progressive motility | 24.00±12.68** | 27.75±20.98** |
Data expressed as mean±s.d. Comparisons between groups were determined by Student’s t-test. *P≤0.05 versus normozoospermic; **P≤0.05 versus F1 progressive motility in each group. Spz: spermatozoa; s.d: standard deviation
RNA sample quality
Sperm RNA samples' integrity and purity were evaluated by microelectropherograms, with controls of RNA from peripheral blood mononuclear cells (PBMC). RNA isolated from PBMC showed two peaks corresponding to 18S and 28S rRNA that were absent from sperm RNA (Supplementary Figure 1a (1.2MB, tif) ). Moreover, no amplicons were observed when the RNA from sperm subpopulations was analyzed for specific somatic RNA markers (Supplementary Figure 1b (1.2MB, tif) ).
Differential gene expression in sperm subpopulations and GO analysis
The unsupervised hierarchical clustering of A–F1/N–F1 (Figure 1a) and A–F2/N–F2 (Figure 1b) revealed two sample groups in each contrast. Analysis of A–F1 versus N–F1 resulted in 1863 DEGs, comprising 116 (6.2%) up- and 1747 (93.8%) down-regulated genes. Conversely, the A–F2 versus N–F2 contrast identified 207 DEGs, including 17 (8.2%) up- and 190 (91.8%) down-regulated genes. However, intragroup subpopulation comparisons analyzed as N–F1 versus N–F2 and A–F1 versus A–F2 resulted in 61 (7 up- and 54 down-regulated genes) and 908 DEGs (42 up- and 866 down-regulated genes), respectively. The corresponding heatmaps are shown in Supplementary Figure 2 (1.2MB, tif) . Moreover, the top up- and down-regulated genes from each of the four contrasts are summarized in Supplementary Table 3–6. The directionality of three down- and one up-regulated DEG from A–F1 versus N–F1 was confirmed by qPCR (Supplementary Table 7).
Figure 1.

Gene clustering of the microarray data showing pair-wise comparisons. (a) A–F1 versus N–F1; (b) A–F2 versus N–F2. Heatmaps show one sample for each column and one gene for each horizontal line. Color indicates gene expression value intensities (z-score), where red signifies upregulation, green downregulation, and black unchanged. A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility; A-F2: asthenozoospermic sperm subpopulation with low motility; N-F2: normozoospermic sperm subpopulation with low motility.
Supplementary Table 3.
Top up- and down-differentially expressed genes in A–F1 versus N–F1
| Gene name | Gene symbol | FC | P |
|---|---|---|---|
| Overexpressed in A–F1 | |||
| Mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1 | MT-ND1 | 4.60 | 1.07E-03 |
| MicroRNA 933 | MIR933 | 4.28 | 3.39E-03 |
| MicroRNA 657 | MIR657 | 4.27 | 5.39E-03 |
| Mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 4L | MT-ND4L | 4.18 | 3.37E-03 |
| Mitochondrially encoded cytochrome b | MT-CYB | 4.11 | 1.94E-03 |
| MicroRNA 4461 | MIR4461 | 4.08 | 4.99E-04 |
| Mitochondrially encoded cytochrome C oxidase I | MT-CO1 | 3.92 | 2.82E-03 |
| Long intergenic nonprotein coding RNA 863 | LINC00863 | 3.72 | 6.49E-03 |
| microRNA 4748 | MIR4748 | 3.55 | 4.33E-03 |
| Mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 5 | MT-ND5 | 3.40 | 7.18E-03 |
| Underexpressed in A–F1 | |||
| ADAM metallopeptidase domain 3A (pseudogene) | ADAM3A | −13.24 | 2.61E-03 |
| Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 4 | APOBEC4 | −7.99 | 2.21E-05 |
| Small nucleolar RNA, C/D box 71 | SNORD71 | −7.52 | 9.17E-04 |
| StAR-related lipid transfer domain containing 6 | STARD6 | −7.49 | 1.22E-06 |
| Cilia- and flagella-associated protein 206 | CFAP206 | −7.21 | 5.46E-04 |
| Leucine-rich repeat containing 9 | LRRC9 | −7.07 | 8.62E-05 |
| Glyceronephosphate O-acyltransferase | GNPAT | −6.49 | 6.41E-06 |
| Long intergenic nonprotein coding RNA 1364 | LINC01364 | −6.32 | 1.87E-05 |
| RORA antisense RNA 2 | RORA-AS2 | −6.06 | 1.09E-04 |
| Arginine vasopressin-induced 1 | AVIP1 | −6.06 | 6.55E-05 |
Negative values mean downregulation. Gene names and symbols assigned after identity verification in the HUGO Gene Nomenclature Committee (https://www.genenames.org) or the NCBI Gene resource (https://www.ncbi.nlm.nih.gov/gene). DEGs: differentially expressed genes; FC: fold change; NADH: nicotinamide adenine dinucleotide
Supplementary Table 6.
Top up- and down-differentially expressed genes in A–F1 versus A–F2
| Gene name | Gene symbol | FC | P |
|---|---|---|---|
| Overexpressed in A-F1 | |||
| MicroRNA 1250 | MIR1250 | 3.21 | 1.18E-03 |
| MicroRNA 924 | MIR924 | 2.94 | 6.70E-04 |
| MicroRNA 3135b | MIR3135B | 2.89 | 1.89E-03 |
| MicroRNA 4686 | MIR4686 | 2.63 | 2.18E-04 |
| Olfactory receptor family 2 subfamily L member 1 pseudogene | OR2L1P | 2.61 | 5.40E-03 |
| POU class 4 homeobox 3 | POU4F3 | 2.55 | 8.19E-03 |
| MicroRNA 1301 | MIR1301 | 2.45 | 3.34E-03 |
| Long intergenic nonprotein coding RNA 701 | LINC00701 | 2.42 | 3.69E-04 |
| MicroRNA 3116-2 | MIR3116-2 | 2.39 | 1.97E-04 |
| MicroRNA 4294 | MIR4294 | 2.37 | 6.29E-03 |
| Underexpressed in A–F1 | |||
| Serglycin | SRGN | −23.90 | 4.87E-04 |
| Nicotinamide phosphoribosyltransferase | NAMPT | −22.46 | 3.36E-03 |
| Major histocompatibility complex, class II, DR alpha | HLA-DRA | −20.18 | 2.42E-03 |
| Small nucleolar RNA, C/D box 71 | SNORD71 | −18.73 | 2.91E-05 |
| Metastasis associated lung adenocarcinoma transcript 1 | MALAT1 | −15.52 | 3.05E-05 |
| Small nucleolar RNA, C/D box 116-15 | SNOR116-15 | −15.03 | 1.09E-05 |
| Lysosomal protein transmembrane 5 | LAPTM5 | −13.97 | 2.80E-04 |
| Fc fragment of IgE receptor Ig | FCER1G | −13.86 | 1.29E-03 |
| TYRO protein tyrosine kinase-binding protein | TYROBP | −13.83 | 1.68E-03 |
| Small nucleolar RNA, C/D box 116-24 | SNORD116-24 | −13.69 | 2.78E-05 |
Negative values mean downregulation. Gene names and symbols assigned after identity verification in the HUGO Gene Nomenclature Committee (https://www.genenames.org) or the NCBI Gene resource (https://www.ncbi.nlm.nih.gov/gene). DEGs: differentially expressed genes; FC: fold change
Supplementary Table 7.
Validation of selected differentially expressed genes identified by microarray analysis using quantitative real-time polymerase chain reaction
| Gene symbol | Gene name | A–F1 versus N–F1 (FC) | |
|---|---|---|---|
| Microarray | qPCR* | ||
| MT-CYB | Mitochondrially encoded cytochrome b | 4.11 | 2.79 |
| SLC25A1 | Plasma membrane citrate carrier | −2.55 | −3.49 |
| ACE | Angiotensin I-converting enzyme | −2.17 | −1.79 |
| STRBP | Spermatid perinuclear RNA-binding protein | −2.11 | −2.90 |
Negative values mean downregulation. n≥3. *P≤0.05 for gene targets in A–F1 versus N–F1 by qPCR. DEGs: differentially expressed genes; qPCR: quantitative real-time polymerase chain reaction; FC: fold change
Supplementary Table 4.
Top up- and down-differentially expressed genes in A–F2 versus N–F2
| Gene name | Gene symbol | FC | P |
|---|---|---|---|
| Overexpressed in A–F2 | |||
| Small nucleolar RNA, C/D box 32B | SNORD32B | 4.72 | 8.96E-03 |
| Mucin 21, cell surface associated | MUC21 | 4.50 | 6.33E-03 |
| Cytochrome P450 family 2 subfamily B member 7, pseudogene | CYP2B7P | 3.75 | 8.14E-04 |
| Small nucleolar RNA, C/D box 8 | SNORD8 | 3.35 | 7.09E-03 |
| Glia maturation factor gamma | GMFG | 3.33 | 5.76E-03 |
| Small nucleolar RNA, H/ACA box 7B | SNORA7B | 2.80 | 5.94E-03 |
| Small nucleolar RNA, C/D box 104 | SNORD104 | 2.42 | 3.29E-03 |
| Triggering receptor expressed on myeloid cells 2 | TREM2 | 2.34 | 4.36E-03 |
| Small nucleolar RNA, C/D box 94 | SNORD94 | 2.26 | 3.47E-03 |
| Histone cluster 1 H2B family member a | HIST1H2BA | 2.22 | 5.69E-04 |
| Underexpressed in A–F2 | |||
| VPS54 subunit of GARP complex | VPS54 | 3.85 | 1.19E-03 |
| CPEB2 divergent transcript | CPEB2-AS1 | 3.51 | 3.30E-04 |
| Long intergenic nonprotein coding RNA 1364 | LINC01364 | 3.50 | 6.06E-03 |
| Apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 4 | APOBEC4 | 3.34 | 1.36E-05 |
| Long intergenic nonprotein coding RNA 2372 | LINC02372 | 3.21 | 3.61E-05 |
| B4GALT4 antisense RNA 1 | B4GALT4-AS1 | 3.21 | 1.16E-03 |
| Adhesion G protein-coupled receptor L4 | ADGRL4 | 3.16 | 5.73E-04 |
| Glyceronephosphate O-acyltransferase | GNPAT | 3.11 | 2.83E-04 |
| LIF receptor subunit alpha | LIFR | 3.08 | 1.69E-03 |
| Long intergenic nonprotein coding RNA 1797 | LINC01797 | 3.05 | 6.60E-04 |
Negative values mean downregulation. Gene names and symbols assigned after identity verification in the HUGO Gene Nomenclature Committee (https://www.genenames.org) or the NCBI Gene resource (https://www.ncbi.nlm.nih.gov/gene). DEGs: differentially expressed genes; FC: fold change
Supplementary Table 5.
Top up- and down-differentially expressed genes in N–F1 versus N–F2
| Gene name | Gene symbol | FC | P |
|---|---|---|---|
| Overexpressed in N–F1 | |||
| Coiled-coil domain containing 13 | CCDC13 | 2.49 | 7.27E-03 |
| HSFY1 pseudogene 1 | HSFY1P1 | 2.47 | 1.59E-03 |
| RNA, U6atac small nuclear (U12-dependent splicing) | RNU6ATAC | 2.32 | 3.70E-03 |
| Cbp/p300 interacting transactivator with Glu/Asp rich carboxy-terminal domain 4 | CITED4 | 2.31 | 8.60E-03 |
| Myosin heavy chain 16 pseudogene | MYH16 | 2.28 | 6.08E-03 |
| Family with sequence similarity 230 member C | FAM230C | 2.10 | 2.98E-03 |
| Long intergenic nonprotein coding RNA 355 | LINC00355 | 2.06 | 4.95E-03 |
| Underexpressed in N–F1 | |||
| Nicotinamide phosphoribosyltransferase | NAMPT | −15.82 | 2.04E-03 |
| Serglycin | SRGN | −13.61 | 2.18E-03 |
| Secreted phosphoprotein 1 | SPP1 | −13.51 | 1.12E-03 |
| Lysosomal protein transmembrane 5 | LAPTM5 | −10.91 | 3.40E-03 |
| Metallothionein 1X | MT1X | −6.64 | 1.07E-03 |
| Cathepsin D | CTSD | −6.30 | 1.35E-03 |
| Mitochondrially encoded cytochrome c oxidase I | COX1 | −5.94 | 6.56E-03 |
| C-X-C motif chemokine receptor 4 | CXCR4 | −4.33 | 4.91E-03 |
| Mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 2 | MT-ND2 | −4.23 | 8.50E-03 |
| Hypoxia-inducible factor 1 subunit alpha | HIF1A | −4.18 | 7.57E-03 |
Negative values mean downregulation. Gene names and symbols assigned after identity verification in the HUGO Gene Nomenclature Committee (https://www.genenames.org) or the NCBI Gene resource (https://www.ncbi.nlm.nih.gov/gene). DEGs: differentially expressed genes; FC: fold change
GO analysis identified 507 BPs overrepresented in A–F1 versus N–F1. The ten BPs with the highest statistical significance (log10 P > -9.9) are shown in Table 1. BPs involved in reproduction, spermatid differentiation, and cell movement were significantly overrepresented. Most genes within each BP were downregulated. A REVIGO TreeMap was constructed to summarize the findings of the GO analysis (Supplementary Figure 3 (1.2MB, tif) ). On the other hand, no BP was overrepresented in the A–F2 versus N–F2 comparison.
Table 1.
Top ten biological processes identified from 507 gene ontology terms summarized by REVIGO in A–F1 versus N–F1
| GO ID | Description | Log10 P value | Genes |
|---|---|---|---|
| 0022412 | Cellular process involved in reproduction in multicellular organism | −31.29 | ↓DDX20, ↓CEP57, ↓PCSK4, ↓KDM3A, ↓CFTR, ↓MTOR, ↓PRKDC, ↓STK11, ↓ZPBP2, ↓PDILT,
↓PDE3A, ↓SRPK1, ↓SPINK2, ↓SPAM1, ↓TTLL5, ↓HORMAD1, ↓NEURL1, ↓KLHL10, ↓OCA2, ↓CATSPERD, ↓TGFBR1, ↓TSSK2, ↓PIWIL1, ↓DZIP1, ↓NUP210L, ↓NPHP1, ↓AGFG1, ↓AFF4, ↓TMF1, ↓SERPINA5, ↓GALNTL5, ↓TRIP13, ↓MOV10L1, ↓PSME4, ↓SYCP1, ↓RNF17, ↓IFT88, ↓DIAPH 2, ↓STRBP, ↓ADAM2, ↓CASC5, ↓CRISP1, ↓SMAD5, ↓CABYR |
| 0048515 | Spermatid differentiation | −27.95 | ↓CEP57, ↓PCSK4, ↓KDM3A, ↓CFTR, ↓STK11, ↓ZPBP2, ↓PDILT, ↓SRPK1, ↓SPINK2, ↓TTLL5,
↓NEURL1, ↓KLHL10, ↓OCA2, ↓CATSPERD, ↓TSSK2, ↓PIWIL1, ↓NUP210L, ↓NPHP1, ↓AGFG1, ↓AFF4, ↓TMF1, ↓GALNTL5, ↓TRIP13, ↓PSME4, ↓SYCP1, ↓RNF17, ↓IFT88, ↓STRBP, ↓CASC5, ↓CABYR |
| 0007286 | Spermatid development | −27.37 | ↓CEP57, ↓PCSK4, ↓KDM3A, ↓CFTR, ↓STK11, ↓ZPBP2, ↓PDILT, ↓SRPK1, ↓SPINK2, ↓TTLL5,
↓NEURL1, ↓KLHL10, ↓OCA2, ↓CATSPERD, ↓TSSK2, ↓PIWIL1, ↓NUP210L, ↓AGFG1, ↓AFF4, ↓TMF1, ↓GALNTL5, ↓TRIP13, ↓PSME4, ↓SYCP1, ↓RNF17, ↓IFT88, ↓STRBP, ↓CASC5, ↓CABYR |
| 0060271 | Cilium assembly | −19.88 | ↓KIAA0586, ↓C2CD3, ↓ZMYND10, ↓RFX3, ↓NEK1, ↓FBF1, ↓ARL13B, ↓TTLL5, ↓LRRC6,
c ↓NEURL1, ↓CELSR2, ↓ATP6V0D1, ↓DNAAF1, ↓DZIP1, ↓TMEM67, ↓TTBK2, ↓AHI1, ↓PCM1, ↓ABLIM1, ↓IFT74, ↓IFT80, ↓IFT81, ↓IFT88, ↓TRAF3IP1, ↓TCTN2, ↓FOPNL, ↓CEP164, ↓ACTR2, ↓RPGRIP1L, ↓KIF3A, ↓KIF24, ↓KIF27 |
| 0007018 | Microtubule-based movement | −19.24 | ↓RHOT1, ↓KIF21B, ↓KIF16B, ↓KIFAP3, ↓NDE1, ↓RFX3, ↓NEFM, ↓ARMC4, ↓CDC42, ↓MAP1S,
↓TTLL6, ↓LRRC6, ↓CELSR2, ↓DNAAF1, ↓KTN1, ↓RAB1A, ↓DNAH9, ↓DNAH3, ↓DNAH1, ↓DNAH7, ↓DNAH8, ↓DNAH12, ↓DNAH14, ↓PCM1, ↓IFT74, ↓IFT81, ↓TRAF3IP1, ↓AP3D1, ↓KIFC3, ↓ACTR3, ↓ACTR2, ↓KIF3A, ↓KIF24, ↓KIF27, ↓KIF2C, ↓KIF2A, ↓CABYR |
| 0009566 | Fertilization | −18.44 | ↓PCSK4, ↓TUBGCP3, ↓CLGN, ↓ZPBP2, ↓CCT3, ↓CCT4, ↓SPINK2, ↓SPAM1, ↓TTLL5, ↓KLHL10,
↓CATSPERD, ↓CATSPERG, ↓ATP8B3, ↓TDRD9, ↓ARSA, ↓PLCZ1, ↓WDR48, ↓PLCD4, ↓ADAM21, ↓SPACA3, ↓SERPINA5, ↓TRIM36, ↓MAEL, ↓SYCP2, ↓TEX15, ↓ADAM2, ↓UBE3A, ↓CRISP1, ↓CACNA1H |
| 0000209 | Protein polyubiquitination | −12.58 | ↓WWP2, ↓HACE1, ↓ITCH, ↓BTRC, ↓CDC27, ↓UBE2G1, ↓UBE2R2, ↓SMURF1, ↓DTL, ↓HUWE1,
↓DZIP3, ↓TPP2, ↓AMFR, ↓TRIP12, ↓RNF216, ↓PSMB1, ↓PSMD3, ↓PSMD6, ↓PSMD8, ↓PSME4, ↓PSMF1, ↓RBCK1, ↓RNF167, ↓RNF111, ↓UBE2K, ↓UBE3A, ↓UBE3C, ↓SHARPIN, ↓RC3H2 |
| 0001701 | In utero embryonic development | −12.51 | ↓MED1, ↓C2CD3, ↓MKL2, ↓EPB41L5, ↓FLVCR1, ↓TAPT1, ↓HORMAD1, ↓SOX6, ↓EPN1, ↓STIL,
↓PRKCSH, ↓BRK1, ↓NCOA1, ↓NCOA6, ↓BPTF, ↓TGFBR1, ↓WDTC1, ↓COL4A3BP, ↓FUT8, ↓SP2, ↓EPAS1, ↓BIRC6, ↓UBR3, ↓SEC24D, ↓DUSP3, ↓RPGRIP1L, ↓SMAD4, ↓SMAD2, ↓KIF3A, ↓INTS1 |
| 0035036 | Sperm–egg recognition | −11.52 | ↓PCSK4, ↓CLGN, ↓ZPBP2, ↓CCT3, ↓CCT4, ↓SPAM1, ↓CATSPERD, ↓CATSPERG, ↓ATP8B3, ↓ARSA,
↓ADAM21, ↓SPACA3, ↓ADAM2, ↓CRISP1 |
| 0007030 | Golgi organization | −9.90 | ↓NPLOC4, ↓HACE1, ↓BLZF1, ↓BCAS3, ↓TMED5, ↓GOLGA5, ↓ARFGEF1, ↓PLEKHM2, ↓CDC42,
↓USP6NL, ↓ATL3, ↓DYM, ↓GCC2, ↓RAB1A, ↓KIFC3 |
Arrows (↓) show gene expression directionality. The biological processes summarized by REVIGO (http://revigo.irb.hr) were generated with FatiGO (http://babelomics.bioinfo.cipf.es) using the list of 1863 DEGs. GO ID: gene ontology identification code. A-F1: asthenozoospermic sperm subpopulation with high motility; N-F1: normozoospermic sperm subpopulation with high motility
Gene set enrichment analysis
Leading-edge analysis of the 110 BPs identified in the A-F1 versusN-F1 comparison showed a set of genes, including dynein axonemal intermediate chain 1 (DNAI1), dynein axonemal heavy chain 1 (DNAH1) and 7 (DNAH7), leucine-rich repeat containing 6 (LRRC6), tetratricopeptide repeat domain 26 (TTC26), sperm-associated antigen 16 (SPAG16), CF transmembrane conductance regulator (CFTR), ubiquitin conjugating enzyme E2 B (UBE2B), and zona pellucida binding protein 2 (ZPBP2), that overlapped with several physiologically relevant BPs (with NES ≥2 and FDR <0.05) (Figure 2a). Most of the enriched BPs identified were involved in spermatogenesis and sperm physiology, including fertilization, sperm motility, axonemal dynein complex assembly, male gamete generation, cilium morphogenesis, and microtubule-based movement, with the last two showing the highest NES (Supplementary Table 8). Furthermore, 36 cellular components (CC) were identified in the same contrast (Supplementary Table 9), which highlights a subset of genes comprising the kinesin family members 2A (KIF2A), 2B (KIF2B), and 2C (KIF2C); intraflagellar transport 81 (IFT81); NME/NM23 family member 8 (TXNDC3); DNAH8, DNAH1, and DNAH7; A-kinase anchoring protein 3 (AKAP3) and 4 (AKAP4) involved in motile cilium, dynein complex, axonemal part, microtubule-associated complex, sperm flagellum, and kinesin complex (Figure 2b). Finally, 31 molecular functions (MFs) were also identified (Supplementary Table 10), where the kinesin family members 3B (KIF3B), C3 (KIFC3), 16 (KIF16), and 20 (KIF20), KIF2A, KIF2C,KIF2B, DNAH1, DNAH7, DNAH8, DNAH6, and DNAH9 genes overlapped with the MFs' ATPase activity signal and microtubule motor activity. Moreover, the genes KIF16, KIF3B, KIFC3, and KIF20 also overlapped with the MF ATP-dependent microtubule motor activity (Figure 2c).
Figure 2.
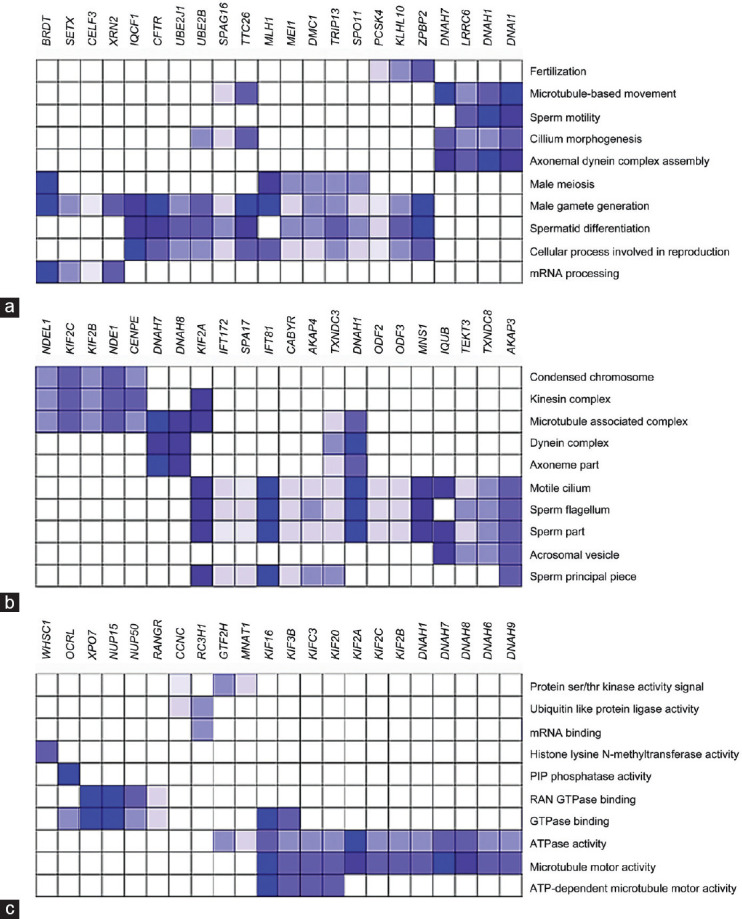
Leading-edge analysis of the A–F1 versus N–F1. (a) Biological processes; (b) cellular components; (c) molecular functions. Columns: genes within the core enrichment of the shown GO terms; rows: selected GO terms from the gene set enrichment analysis (Supplementary Table 8–10). Gene expression values are represented in colors, and the range of colors (from light to dark blue) shows the level of expression values (from low to lowest) (http://www.broadinstitute.org/gsea). GO: gene ontology; A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.
Supplementary Table 8.
Biological processes identified by gene set enrichment analysis in A–F1 versus N–F1 contrast
| Biological processes | NES | FDR |
|---|---|---|
| Cilium morphogenesis | −2.77 | 0.00 |
| Microtubule-based movement | −2.76 | 0.00 |
| Cilium organization | −2.74 | 0.00 |
| Microtubule-based process | −2.70 | 0.00 |
| Male gamete generation | −2.60 | 0.00 |
| Chromosome segregation | −2.60 | 0.00 |
| Cellular component assembly involved in morphogenesis | −2.59 | 0.00 |
| Microtubule organizing center organization | −2.59 | 0.00 |
| Spermatid differentiation | −2.55 | 0.00 |
| Microtubule cytoskeleton organization | −2.54 | 0.00 |
| Organelle fission | −2.53 | 0.00 |
| Meiotic cell cycle | −2.53 | 0.00 |
| Meiotic cell cycle process | −2.52 | 0.00 |
| Meiotic chromosome segregation | −2.51 | 0.00 |
| Cell projection assembly | −2.51 | 0.00 |
| Nuclear chromosome segregation | −2.50 | 0.00 |
| Nuclear export | −2.47 | 0.00 |
| Meiosis I | −2.46 | 0.00 |
| Male meiosis | −2.45 | 0.00 |
| Centrosome cycle | −2.45 | 0.00 |
| Golgi vesicle transport | −2.43 | 0.00 |
| Protein modification by small protein removal | −2.42 | 0.00 |
| RNA localization | −2.40 | 0.00 |
| Regulation of cellular response to heat | −2.40 | 0.00 |
| Organelle assembly | −2.40 | 0.00 |
| Golgi to vacuole transport | −2.39 | 0.00 |
| Golgi organization | −2.39 | 0.00 |
| Reciprocal DNA recombination | −2.35 | 0.00 |
| Cytoskeleton-dependent intracellular transport | −2.34 | 0.00 |
| Cell cycle G2 M phase transition | −2.33 | 0.00 |
| Sperm motility | −2.33 | 0.00 |
| Axonemal dynein complex assembly | −2.33 | 0.00 |
| Mitotic nuclear division | −2.32 | 0.00 |
| Protein ubiquitination involved in ubiquitin-dependent protein catabolic process | −2.31 | 0.00 |
| Centrosome duplication | −2.29 | 0.00 |
| Establishment of localization by movement along microtubule | −2.29 | 0.00 |
| Sister chromatid cohesion | −2.29 | 0.00 |
| Ribonucleoprotein complex localization | −2.28 | 0.00 |
| Sperm–egg recognition | −2.26 | 0.00 |
| tRNA transport | −2.26 | 0.00 |
| Post Golgi vesicle-mediated transport | −2.26 | 0.00 |
| Intra Golgi vesicle-mediated transport | −2.26 | 0.00 |
| Sister chromatid segregation | −2.25 | 0.00 |
| Multiorganism localization | −2.25 | 0.00 |
| Cilium movement | −2.25 | 0.00 |
| DNA synthesis involved in DNA repair | −2.25 | 0.00 |
| Mitotic spindle organization | −2.25 | 0.00 |
| Germ cell development | −2.24 | 0.00 |
| Nucleobase-containing compound transport | −2.24 | 0.00 |
| Nucleus organization | −2.24 | 0.00 |
| Nuclear envelope organization | −2.24 | 0.00 |
| Chromosome separation | −2.23 | 0.00 |
| Membrane disassembly | −2.23 | 0.00 |
| Protein transport along microtubule | −2.22 | 0.00 |
| DNA repair | −2.21 | 0.00 |
| Homologous chromosome segregation | −2.21 | 0.00 |
| Retrograde transport vesicle recycling within Golgi | −2.21 | 0.00 |
| Protein localization to Golgi apparatus | −2.20 | 0.00 |
| Fertilization | −2.20 | 0.00 |
| Synapsis | −2.20 | 0.00 |
| Chromosome organization involved in meiotic cell cycle | −2.19 | 0.00 |
| Golgi to endosome transport | −2.19 | 0.00 |
| Cytoplasmic microtubule organization | −2.19 | 0.00 |
| Spindle assembly | −2.19 | 0.00 |
| Peptidyl lysine modification | −2.18 | 0.00 |
| Cellular process involved in reproduction in multicellular organism | −2.18 | 0.00 |
| Protein polyubiquitination | −2.18 | 0.00 |
| Mitotic spindle assembly | −2.16 | 0.00 |
| Protein localization to vacuole | −2.16 | 0.00 |
| Microtubule bundle formation | −2.15 | 0.00 |
| Epithelial cilium movement | −2.14 | 0.00 |
| Cell–cell recognition | −2.14 | 0.00 |
| ER to Golgi vesicle-mediated transport | −2.14 | 0.00 |
| Axoneme assembly | −2.13 | 0.00 |
| Protein complex localization | −2.13 | 0.00 |
| DNA biosynthetic process | −2.13 | 0.00 |
| Cell division | −2.12 | 0.00 |
| Gene silencing by RNA | −2.11 | 0.00 |
| Regulation of chromosome segregation | −2.11 | 0.00 |
| Binding of sperm to zona pellucida | −2.09 | 0.00 |
| Protein sumoylation | −2.09 | 0.00 |
| Androgen receptor signaling pathway | −2.09 | 0.00 |
| Cell cycle phase transition | −2.08 | 0.00 |
| Spindle checkpoint | −2.08 | 0.00 |
| Protein localization to centrosome | −2.07 | 0.00 |
| Negative regulation of mRNA metabolic process | −2.07 | 0.00 |
| Intracellular steroid hormone receptor signaling pathway | −2.07 | 0.00 |
| Retrograde vesicle-mediated transport Golgi to ER | −2.06 | 0.00 |
| Regulation of organelle assembly | −2.06 | 0.00 |
| Single fertilization | −2.06 | 0.00 |
| Chromatin modification | −2.05 | 0.00 |
| Establishment of protein localization to vacuole | −2.05 | 0.00 |
| Endomembrane system organization | −2.05 | 0.00 |
| Postreplication repair | −2.04 | 0.00 |
| Male meiosis I | −2.04 | 0.00 |
| Nucleotide excision repair | −2.04 | 0.00 |
| Double-strand break repair | −2.03 | 0.00 |
| NLS-bearing protein import into nucleus | −2.03 | 0.00 |
| Regulation of proteasomal ubiquitin-dependent protein catabolic process | −2.02 | 0.00 |
| Organelle transport along microtubule | −2.02 | 0.00 |
| mRNA processing | −2.02 | 0.00 |
| DNA recombination | −2.02 | 0.00 |
| Vesicle coating | −2.02 | 0.00 |
| Regulation of microtubule-based process | −2.02 | 0.00 |
| RNA secondary structure unwinding | −2.02 | 0.00 |
| Regulation of proteasomal protein catabolic process | −2.01 | 0.00 |
| Protein export from nucleus | −2.01 | 0.00 |
| Negative regulation of protein complex disassembly | −2.01 | 0.00 |
| Myelin assembly | −2.01 | 0.00 |
| Gene silencing | −2.00 | 0.00 |
The 110 biological processes showed in this table were selected on the basis of NES ≥2.0 and FDR <0.05. The molecular signature database (v6.2) searched was biological processes using GSEA approach (http://www.broadinstitute.org/gsea). The ten biological processes in bold text were included in the leading-edge analysis. NES: normalized enrichment score; FDR: false discovery rate; GSEA: gene set enrichment analysis
Supplementary Table 9.
Cellular components identified by gene set enrichment analysis in A–F1 versus N–F1 contrast
| Cellular components | NES | FDR |
|---|---|---|
| Centrosome | −2.71 | 0.00 |
| Motile cilium | −2.61 | 0.00 |
| Cilium | −2.59 | 0.00 |
| Microtubule organizing center part | −2.58 | 0.00 |
| Ciliary basal body | −2.56 | 0.00 |
| Nuclear pore | −2.56 | 0.00 |
| Centriole | −2.54 | 0.00 |
| Ciliary part | −2.54 | 0.00 |
| Kinesin complex | −2.52 | 0.00 |
| Ciliary plasm | −2.50 | 0.00 |
| Microtubule-associated complex | −2.47 | 0.00 |
| Sperm part | −2.37 | 0.00 |
| Intraciliary transport particle | −2.36 | 0.00 |
| Kinetochore | −2.35 | 0.00 |
| Dynein complex | −2.34 | 0.00 |
| Ciliary tip | −2.33 | 0.00 |
| Spindle | −2.28 | 0.00 |
| Axoneme part | −2.25 | 0.00 |
| Microtubule | −2.24 | 0.00 |
| Chromosome centromeric region | −2.23 | 0.00 |
| Sperm flagellum | −2.22 | 0.00 |
| Condensed chromosome centromeric region | −2.21 | 0.00 |
| Condensed chromosome | −2.19 | 0.00 |
| Spindle pole | −2.13 | 0.00 |
| Pericentriolar material | −2.12 | 0.00 |
| Intraciliary transport particle B | −2.10 | 0.00 |
| Inclusion body | −2.09 | 0.00 |
| Nuclear envelope | −2.07 | 0.00 |
| Chromosomal region | −2.05 | 0.00 |
| Acrosomal vesicle | −2.04 | 0.00 |
| DNA repair complex | −2.04 | 0.00 |
| Acetyltransferase complex | −2.02 | 0.00 |
| DNA-directed RNA polymerase II holoenzyme | −2.01 | 0.00 |
| Sperm’s principal piece | −2.01 | 0.00 |
| Ribonucleoprotein granule | −2.00 | 0.00 |
| PML body | −2.00 | 0.00 |
The 36 cellular components showed in this table were selected on the basis of NES ≥2.0 and FDR <0.05. The molecular signature database (v6.2) searched was cellular components using GSEA approach (http://www.broadinstitute.org/gsea). The ten cellular components in bold text were included in the leading-edge analysis. NES: normalized enrichment score; FDR: false discovery rate; GSEA: gene set enrichment analysis
Supplementary Table 10.
Molecular functions identified by gene set enrichment analysis in A–F1 versus N–F1 contrast
| Molecular functions | NES | FDR |
|---|---|---|
| Microtubule motor activity | −2.82 | 0.00 |
| Motor activity | −2.47 | 0.00 |
| Ligase activity | −2.39 | 0.00 |
| Helicase activity | −2.36 | 0.00 |
| Nuclear localization sequence binding | −2.35 | 0.00 |
| RAN GTPase binding | −2.33 | 0.00 |
| Ubiquitin-like protein-specific protease activity | −2.32 | 0.00 |
| Signal sequence binding | −2.30 | 0.00 |
| Tubulin binding | −2.30 | 0.00 |
| Protein serine threonine kinase activity | −2.29 | 0.00 |
| Thiol-dependent ubiquitin-specific protease activity | −2.28 | 0.00 |
| Ubiquitin-like protein ligase activity | −2.26 | 0.00 |
| ATPase activity | −2.25 | 0.00 |
| Microtubule binding | −2.24 | 0.00 |
| Nucleocytoplasmic transporter activity | −2.23 | 0.00 |
| Translation initiation factor activity | −2.23 | 0.00 |
| Ubiquitin-like protein binding | −2.18 | 0.00 |
| Translation factor activity RNA binding | −2.16 | 0.00 |
| DNA-dependent ATPase activity | −2.14 | 0.00 |
| Peptide N acetyltransferase activity | −2.10 | 0.00 |
| Purine NTP-dependent helicase activity | −2.10 | 0.00 |
| Ubiquitin-like protein transferase activity | −2.09 | 0.00 |
| Protein transporter activity | −2.08 | 0.00 |
| mRNA binding | −2.05 | 0.00 |
| RNA helicase activity | −2.04 | 0.00 |
| DNA-directed DNA polymerase activity | −2.03 | 0.00 |
| Lysine acetylated histone binding | −2.03 | 0.00 |
| Histone lysine N methyltransferase activity | −2.03 | 0.00 |
| GTPase binding | −2.03 | 0.00 |
| Phosphatidylinositol phosphate phosphatase activity | −2.01 | 0.00 |
| ATP-dependent microtubule motor activity | −2.01 | 0.00 |
The 31 molecular functions showed in this table were selected on the basis of NES ≥2.0 and FDR <0.05. The molecular signature database (v6.2) searched was molecular functions using GSEA approach (http://www.broadinstitute.org/gsea). The ten molecular functions in bold text were included in the leading-edge analysis. NES: normalized enrichment score; FDR: false discovery rate
The core enrichment of microtubule-based movement showed that genes KIF2A, KIF2B, KIF2C, DNAH1, DNAH7, and DNAH8 (Figure 3) were also identified in the main CCs and MFs (Figure 2b and 2c). Conversely, A–F2 versus N–F2 showed the enrichment of only six BPs (highlighting cilium morphogenesis and cilium organization; (Supplementary Table 11), one CC (cilium), and two MFs (ubiquitin-like protein-specific protease activity and RAN GTPase binding).
Figure 3.
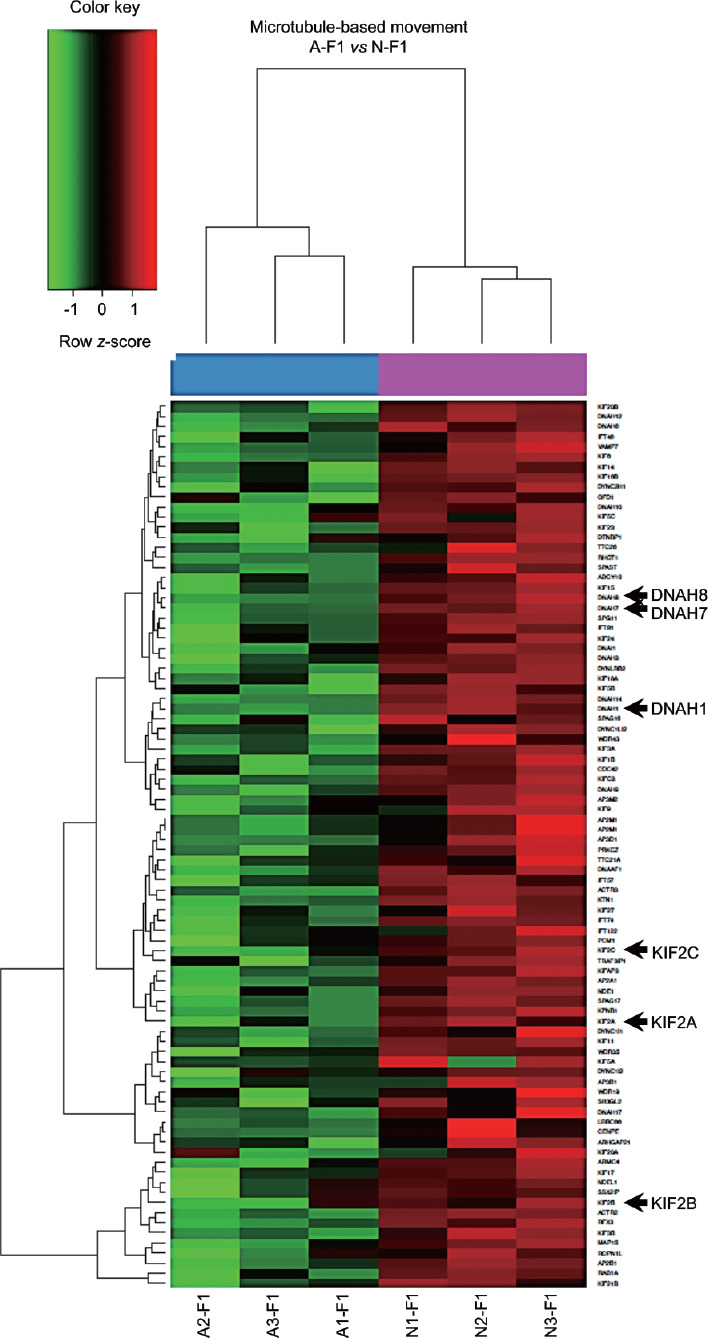
Heatmap of the core enrichment of the biological process microtubule-based movement. Columns correspond to sperm samples analyzed, whereas lines represent genes within the core enrichment. Arrows indicate genes downregulated in A–F1 that were also identified in the leading-edge analysis of biological processes, cellular components, and molecular functions, as shown in Figure 2. Color indicates gene expression value intensities (z-score), where red signifies upregulation, green downregulation, and black unchanged. A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.
Supplementary Table 11.
Biological processes identified by gene set enrichment analysis in A–F2 versus N–F2 contrast
| Biological processes | NES | FDR |
|---|---|---|
| Cilium morphogenesis | −2.17 | 0.01 |
| Cilium organization | −2.13 | 0.01 |
| Peroxisome organization | −2.09 | 0.02 |
| Cellular component assembly involved in morphogenesis | −2.09 | 0.02 |
| Protein modification by small protein removal | −2.08 | 0.01 |
| Chromosome separation | −2.03 | 0.03 |
The six biological processes showed in this table were selected on the basis of NES ≥2.0 and FDR <0.05. The molecular signature database (v6.2) searched was biological processes using GSEA approach (http://www.broadinstitute.org/gsea). NES: normalized enrichment score; FDR: false discovery rate
Predicted activation state of bio-functions
IPA core analysis of the list of DEGs from the A–F1 versus N–F1 comparison showed that the predicted activation state of several bio-functions involved in reproduction, either increased or decreased (Table 2). Sperm disorders, asthenozoospermia, oligozoospermia, and infertility were predicted to be increased. In addition, fertility was predicted to be decreased, along with microtubule dynamics, organization of cytoskeleton, formation of cilia, and cell movement of sperm.
Table 2.
Bio-functions predicted activation state in the A–F1 versus N–F1 comparison by ingenuity pathway analysis core analysis
| Bio-functions | Activation z-score | P | Predicted activation state |
|---|---|---|---|
| Sperm disorder | 6.97 | 1.07×10−5 | Increased |
| Asthenozoospermia | 4.47 | 1.00×10−4 | Increased |
| Oligozoospermia | 4.36 | 4.24×10−3 | Increased |
| Cell death of male germ cells | 2.24 | 6.53×10−3 | Increased |
| Infertility | 2.21 | 5.12×10−4 | Increased |
| Depolymerization of microtubules | 2.16 | 7.01×10−3 | Increased |
| Degeneration of embryonic tissue | 2.16 | 8.31×10−3 | Increased |
| Fertility | −6.44 | 6.20×10−4 | Decreased |
| Microtubule dynamics | −6.04 | 1.85×10−7 | Decreased |
| Organization of cytoplasm | −5.85 | 2.08×10−8 | Decreased |
| Organization of cytoskeleton | −5.85 | 1.26×10−6 | Decreased |
| Repair of DNA | −4.53 | 1.22×10−3 | Decreased |
| Formation of cilia | −3.55 | 3.41×10−12 | Decreased |
| Transport of vesicles | −2.65 | 2.34×10−3 | Decreased |
| Transport of protein | −2.56 | 1.69×10−4 | Decreased |
| Cell movement of sperm | −2.53 | 1.73×10−4 | Decreased |
| Gametogenesis | −2.17 | 2.17×10−13 | Decreased |
A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility; IPA: ingenuity pathway analysis
Molecule activity prediction of bio-functions
The MAP analysis by IPA of DEGs from the A–F1 versus N–F1 comparison predicted bio-function fertility to be inhibited, on the basis of downregulation of 13 genes where Angiotensin-converting enzyme (ACE) and ADAM Metallopeptidase Domain 2 (ADAM2) are involved in the canonical pathway (CP) axonal guidance signaling (Figure 4a). Similarly, MAP analyses of the bio-function capacitation, acrosome reaction, and binding of sperm were also predicted to be inhibited (Supplementary Figure 4 (1.2MB, tif) ). Conversely, asthenozoospermia was predicted to be activated on the basis of the downregulation of 21 genes including Ubiquitin Conjugating Enzyme E2 J1 (UBE2J1), Heat Shock Protein Family A 4 Like (HSPA4L), and DnaJ Heat Shock Protein Family (Hsp40) A1 (DNAJA1), which areall involved in the CP protein ubiquitination pathway (Figure 4b).
Figure 4.
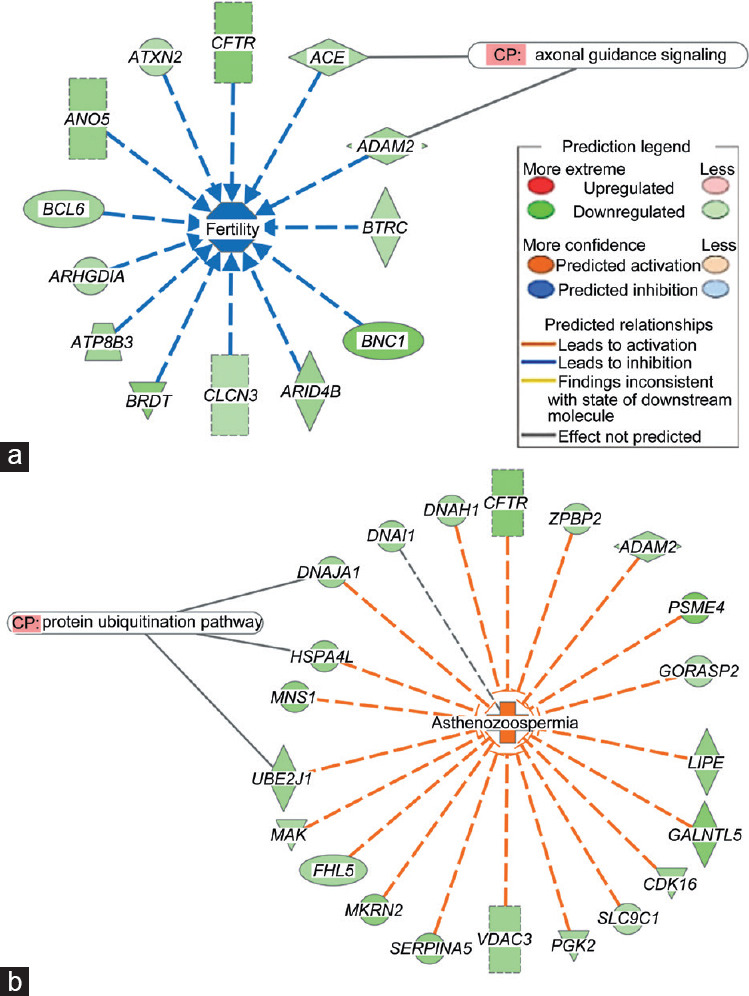
Molecular activity prediction by IPA from the DEGs in A–F1 versus N–F1 contrast. (a) Fertility (inhibited); (b) asthenozoospermia (activated). Colors indicate predicted relationships of gene expression levels and bio-functions, and color intensities reflect the degree of gene expression or bio-function activity. IPA: ingenuity pathway analysis (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/); DEGs: differentially expressed genes; A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility; CP: canonical pathway.
DISCUSSION
Approximately 13% of couples worldwide seek medical advice for fertility purposes. Unexplained infertility is commonly treated with expensive and possibly unneeded high-complexity technologies. The recurrent use of in-vitro fertilization and intracytoplasmic sperm injection in assisted reproductive technologies is a consequence of the inadequacy of current male fertility diagnostic methods.35,36 Therefore, new approaches to studying spermatozoa at the cellular and molecular level are still required. In this regard, the characterization of sperm RNA profiling has become a promising strategy to reveal the mechanisms leading to altered sperm parameter values. The study of sperm physiology using high-throughput measurement technology such as microarray analysis has previously revealed several biomarkers of human sperm function.37,38 This approach may help to increase our understanding of the basic molecular mechanisms underlying the pathogenesis of male infertility.
In natural conception, a small fraction of the spermatozoa in an ejaculate reaches the fertilization site. Sperm selection takes places in the female genital tract, where sperm motility is a key feature to reach the oocyte. Hence, analysis of sperm subpopulations based on differential motility may represent a valuable model for studying sperm physiology and reproduction. In 2012, Jodar et al.15 performed the first study of human sperm transcriptome of asthenozoospermic semen samples using microarray analysis and showed a differential expression pattern between the F1 of patients and controls. We also found a transcriptomic profile in A–F1 different from that of N–F1 samples, but decided further to compare sperm subpopulations with different motilities obtained from density gradients. As expected, F2 subpopulations presented lower motility values than the corresponding F1 from A and N samples, but there were considerably fewer DEGs in A–F2 versus N–F2 than A–F1 versus N–F1, which indicates that F2 from A and N samples had similar expression profiles. Moreover, these data indicate that genomic transcriptional differences between the A and N samples reside mainly in the highly motile sperm subpopulation. However, when the internal contrasts were analyzed, N–F1 versus N–F2 showed considerably fewer differences in DEGs than A–F1 versus A–F2, which suggests that normozoospermic sperm subpopulations are more homogeneous than asthenozoospermic. Furthermore, overlapping of DEGs from N–F1 versus N–F2 and A–F1 versus A–F2 evidenced the downregulation of nicotinamide phosphoribosyltransferase, serglycin, and lysosomal protein transmembrane 5, but these transcripts did not show differential expression in A–F1 versus N–F1 or A–F2 versus N–F2, which suggests that their overexpression in F2 fractions may be associated with sperm's reduced motility. Similarly, both A–F1 versus N–F1 and A–F2 versus N–F2 contrasts identified the downregulation in A fractions of apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 4, glyceronephosphate O-acyltransferase, and long intergenic nonprotein coding RNA 1364, indicating that the underexpression of these three transcripts could be related to asthenozoospermia and therefore may be used as potential candidates for future investigations.
As the A–F1 versus N–F1 contrast resulted in a higher number of DEGs, a deeper analysis showed that among the more upregulated RNAs in A–F1, several of the mitochondrial origin (including mitochondrially encoded nicotinamide adenine dinucleotide reduced [NADH]:ubiquinone oxidoreductase core subunits) and microRNAs were highlighted. The differential expression of microRNAs in asthenozoospermia has been described previously,39,40 and upregulation of microRNA 27b seems to be specifically involved in reduced cysteine-rich secretory protein 2 expression in asthenozoospermia.41 All of these indicate that microRNAs may play an important role in the manifestations of this pathology.
As expected, microtubule-based movement was among the most overrepresented BP, and a core enrichment analysis of this BP revealed several genes involved in sperm motility that were also identified by the leading-edge GSEA. This analysis also identified KIF2A, KIF2B, KIF2C, DNAH1, DNAH7, and DNAH8 as consistently downregulated in A–F1, all of them being members of the axonemal dynein and kinesin families, which play fundamental roles in the mammalian sperm flagellum.42 Previous investigations indicate that other members of the dynein and kinesin gene families are also differentially expressed in asthenozoospermia.5 DNAH1 mutations have formerly been associated with sperm defects and infertility.43,44,45 Furthermore, naturally occurring mutations in some other genes identified in the present investigation, such as CFTR and LRRC6, have also been associated with several pathologies that involve morphological anomalies of the flagellum, resulting in asthenozoospermia and male infertility.46,47
Protein polyubiquitination was identified as a top BP, and the canonical pathway protein ubiquitination was also associated with asthenozoospermia, which suggests that proper regulation of protein degradation in mature spermatozoa is fundamental for obtaining healthy normozoospermic spermatozoa. Indeed, previous investigations have demonstrated that increased sperm ubiquitination is inversely associated with sperm motility48 and that there is a positive correlation between ubiquitinated spermatozoa and the percentage of spermatozoa with abnormal chromatin.49
In addition, data mining analysis of the A–F1 versus N–F1 comparison identified specific GO terms associated with activation of the bio-function asthenozoospermia and with the inhibition of fertility, acrosome reaction, capacitation, and sperm binding. Previously, GO analyses of different pathological sperm profiles have shown several GO terms consistent with alterations in morphology, motility, and sperm count5,50,51,52 that were also detected in the present study, such as spermatid differentiation and development and repair of DNA. This suggests that asthenozoospermia has common mechanisms with some sperm bio-functions essential for fertilization, and should therefore be seen as a complex pathology not only involving a compromised cell motility, but also other cellular malfunctions, jeopardizing the proper delivery of DNA/RNA cargo to the oocyte. This hypothesis may have implications for the management of asthenozoospermic patients in reproductive assisted facilities. However, whether the RNAs herein identified are of relevance in sperm capacitation, cell fusion, or early stages of embryo development requires further investigation.
Finally, owing to the high level of downregulated mRNAs and upregulated microRNAs observed in spermatozoa from asthenozoospermic patients, we suggest that an environmental origin via epigenetic genomic markers could be considered among the causes of semen abnormalities. In conclusion, our analysis of sperm RNA fractions from asthenozoospermic patients identified a number of gene sets significantly associated with male infertility due to low sperm motility. These results provide new ideas for further studies on male infertility diagnosis and treatment and for the development of strategies for research and development in male contraception.
AUTHOR CONTRIBUTIONS
PCC, SLA, FL, and MC designed the experimental study. PCC, EBC, and HSGM selected and processed the sperm samples. PCC, MC, and DB acquired and interpreted experimental results. SLA and CRE performed the bioinformatics analyses. PCC, SLA, CRE, FL, and MC analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declare no competing interests.
Purity and quality controls of sperm RNA samples. (a) Agilent bioanalyzer analysis of RNA from sperm and control somatic cells (PBMCs). In PBMCs, the majority of the RNA corresponds to rRNA (18s and 28S), while RNA from sperm samples are rRNA depleted; (b) Electrophoresis analysis of RT-PCR products corresponding to specific markers for leukocyte (CD45), germ cells (c-kit), epithelial cells (E-cad), and spermatozoa (SMCP). F1: sperm subpopulation F1; F2: sperm subpopulation F2; L: human leukocytes; P: epithelial cells; T: trophoblast explants; C: negative control; PBMCs: peripheral blood mononuclear cells; SMCP: sperm mitochondrial-associated cysteine-rich protein; RT-PCR: real-time polymerase chain reaction.
Gene clustering of the microarray data of N–F1 versus N–F2 and A–F1 versus A–F2 contrasts. Heatmaps show one sample for column and one gene for each horizontal line. Color indicates gene expression value intensities (z-score). Red: upregulation; Green: downregulation; Black: no change. N–F1: normozoospermic sperm subpopulation with high motility; N-F2: normozoospermic sperm subpopulation with low motility; A-F1: asthenozoospermic sperm subpopulation with high motility; A-F2: asthenozoospermic sperm subpopulation with low motility.
REVIGO TreeMap summarizing gene ontology biological process categories overrepresented in A–F1 versus N–F1. FatiGO was used to identify Gene Ontology Biological Processes overrepresented among transcripts more highly expressed. Categories with P-values below 0.01 were used to generate the treemap, colored by functional category. The box size is proportional to the P-value of each category. A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.
Molecule activity prediction of three sperm bio-functions using the DEG in A–F1 versus N–F1 contrast. (a) Capacitation of sperm; (b) acrosome reaction of sperm; (c) binding of sperm. Colors indicate predicted relationships of gene expression levels and bio-functions; color intensities reflect the gene expression degree or bio-function activity (http://ingenuity.com). A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.
ACKNOWLEDGMENTS
We thank Elisabeth Raab MD, MPH (University of Southern California, Los Angeles, CA, USA), and Paolo Rinaudo MD, PhD (University of California San Francisco, San Francisco, CA, USA), for the critical revision of the manuscript. Financial support was provided by a grant from Cathedra Salvador Zubirán (National Autonomous University of Mexico and National Institute of Medical Sciences and Nutrition Salvador Zubirán, Mexico City, Mexico) and funds from the Department of Reproduction Biology of the National Institute of Medical Sciences and Nutrition Salvador Zubirán, Patronage of the National Institute of Medical Sciences and Nutrition Salvador Zubirán, FunSaEd A.C. (Mexico), and Tambre Foundation (Madrid, Spain).
Supplementary Information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Sakkas D, Ramalingam M, Garrido N, Barratt CL. Sperm selection in natural conception: what can we learn from mother nature to improve assisted reproduction outcomes? Hum Reprod Update. 2015;21:711–26. doi: 10.1093/humupd/dmv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sousa AP, Amaral A, Baptista M, Tavares R, Caballero Campo P, et al. Not all sperm are equal: functional mitochondria characterize a subpopulation of human sperm with better fertilization potential. PLoS One. 2011;6:e18112. doi: 10.1371/journal.pone.0018112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jodar M, Selvaraju S, Sendler E, Diamond MP, Krawetz SA, et al. The presence, role and clinical use of spermatozoal RNAs. Hum Reprod Update. 2013;19:604–24. doi: 10.1093/humupd/dmt031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrido N, Garcia-Herrero S, Meseguer M. Assessment of sperm using mRNA microarray technology. Fertil Steril. 2013;99:1008–22. doi: 10.1016/j.fertnstert.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Bansal SK, Gupta N, Sankhwar SN, Rajender S. Differential genes expression between fertile and infertile spermatozoa revealed by transcriptome analysis. PLoS One. 2015;10:e0127007. doi: 10.1371/journal.pone.0127007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonache S, Mata A, Ramos MD, Bassas L, Larriba S. Sperm gene expression profile is related to pregnancy rate after insemination and is predictive of low fecundity in normozoospermic men. Hum Reprod. 2012;27:1556–67. doi: 10.1093/humrep/des074. [DOI] [PubMed] [Google Scholar]
- 7.Ostermeier GC, Dix DJ, Miller D, Khatri P, Krawetz SA. Spermatozoal RNA profiles of normal fertile men. Lancet. 2002;360:772–7. doi: 10.1016/S0140-6736(02)09899-9. [DOI] [PubMed] [Google Scholar]
- 8.Boerke A, Dieleman SJ, Gadella BM. A possible role for sperm RNA in early embryo development. Theriogenology. 2007;68:S147–55. doi: 10.1016/j.theriogenology.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 9.Hamatani T. Human spermatozoal RNAs. Fertil Steril. 2012;97:275–81. doi: 10.1016/j.fertnstert.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Li RH, Ran MX, Zhang Y, Liang K, et al. High throughput small RNA and transcriptome sequencing reveal capacitation-related microRNAs and mRNA in boar sperm. BMC Genomics. 2018;19:736. doi: 10.1186/s12864-018-5132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson GD, Mackie P, Jodar M, Moskovtsev S, Krawetz SA. Chromatin and extracellular vesicle associated sperm RNAs. Nucleic Acids Res. 2015;43:6847–59. doi: 10.1093/nar/gkv591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selvaraju S, Parthipan S, Somashekar L, Kolte AP, Binsila BK, et al. Occurrence and functional significance of the transcriptome in bovine (Bos taurus) spermatozoa. Sci Rep. 2017;7:42392. doi: 10.1038/srep42392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sendler E, Johnson GD, Mao S, Goodrich RJ, Diamond MP, et al. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Res. 2013;41:4104–17. doi: 10.1093/nar/gkt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen. Geneva: World Health Organization Press; 2010. [Google Scholar]
- 15.Jodar M, Kalko S, Castillo J, Ballesca JL, Oliva R. Differential RNAs in the sperm cells of asthenozoospermic patients. Hum Reprod. 2012;27:1431–8. doi: 10.1093/humrep/des021. [DOI] [PubMed] [Google Scholar]
- 16.Henkel R. Sperm preparation: state-of-the-art-physiological aspects and application of advanced sperm preparation methods. Asian J Androl. 2012;14:260–9. doi: 10.1038/aja.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caballero-Campo P, Lin W, Simbulan R, Liu X, Feuer S, et al. Advanced paternal age affects sperm count and anogenital distance in mouse offspring. Reprod Sci. 2018;25:515–22. doi: 10.1177/1933719118759441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambard S, Galeraud-Denis I, Martin G, Levy R, Chocat A, et al. Analysis and significance of mRNA in human ejaculated sperm from normozoospermic donors: relationship to sperm motility and capacitation. Mol Hum Reprod. 2004;10:535–41. doi: 10.1093/molehr/gah064. [DOI] [PubMed] [Google Scholar]
- 19.Lira-Albarran S, Durand M, Larrea-Schiavon MF, Gonzalez L, Barrera D, et al. Ulipristal acetate administration at mid-cycle changes gene expression profiling of endometrial biopsies taken during the receptive period of the human menstrual cycle. Mol Cell Endocrinol. 2017;447:1–11. doi: 10.1016/j.mce.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 20.Lira-Albarran S, Larrea-Schiavon MF, Gonzalez L, Durand M, Rangel C, et al. The effects of levonorgestrel on FSH-stimulated primary rat granulosa cell cultures through gene expression profiling are associated to hormone and folliculogenesis processes. Mol Cell Endocrinol. 2017;439:337–45. doi: 10.1016/j.mce.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 21.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 23.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 25.Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–80. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- 26.Alonso R, Salavert F, Garcia-Garcia F, Carbonell-Caballero J, Bleda M, et al. Babelomics 5.0: functional interpretation for new generations of genomic data. Nucleic Acids Res. 2015;43:W117–21. doi: 10.1093/nar/gkv384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colquhoun D. An investigation of the false discovery rate and the misinterpretation of p-values. R Soc Open Sci. 2014;1:140216. doi: 10.1098/rsos.140216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lira-Albarran S, Durand M, Barrera D, Vega C, Becerra RG, et al. A single preovulatory administration of ulipristal acetate affects the decidualization process of the human endometrium during the receptive period of the menstrual cycle. Mol Cell Endocrinol. 2018;476:70–8. doi: 10.1016/j.mce.2018.04.010. [DOI] [PubMed] [Google Scholar]
- 31.Lira-Albarran S, Vega CC, Durand M, Rangel C, Larrea F. Functional genomic analysis of the human receptive endometrium transcriptome upon administration of mifepristone at the time of follicle rupture. Mol Cell Endocrinol. 2019;485:88–96. doi: 10.1016/j.mce.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Schefe JH, Lehmann KE, Buschmann IR, Unger T, Funke-Kaiser H. Quantitative real-time RT-PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J Mol Med (Berl) 2006;84:901–10. doi: 10.1007/s00109-006-0097-6. [DOI] [PubMed] [Google Scholar]
- 33.Barragan M, Martinez A, Llonch S, Pujol A, Vernaeve V, et al. Effect of ribonucleic acid (RNA) isolation methods on putative reference genes messenger RNA abundance in human spermatozoa. Andrology. 2015;3:797–804. doi: 10.1111/andr.12053. [DOI] [PubMed] [Google Scholar]
- 34.Perez-Rico A, Crespo F, Sanmartin ML, De Santiago A, Vega-Pla JL. Determining ACTB, ATP5B and RPL32 as optimal reference genes for quantitative RT-PCR studies of cryopreserved stallion semen. Anim Reprod Sci. 2014;149:204–11. doi: 10.1016/j.anireprosci.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Feuer S, Rinaudo P. From embryos to adults: a DOHaD perspective onin vitro fertilization and other assisted reproductive technologies. Healthcare (Basel) 2016;4:51. doi: 10.3390/healthcare4030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dyer S, Chambers GM, de Mouzon J, Nygren KG, Zegers-Hochschild F, et al. International committee for monitoring assisted reproductive technologies world report: assisted reproductive technology 2008, 2009 and 2010. Hum Reprod. 2016;31:1588–609. doi: 10.1093/humrep/dew082. [DOI] [PubMed] [Google Scholar]
- 37.Zhang T, Wu J, Liao C, Ni Z, Zheng J, et al. System analysis of teratozoospermia mRNA profile based on integrated bioinformatics tools. Mol Med Rep. 2018;18:1297–304. doi: 10.3892/mmr.2018.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Zhang P, Song D, Xiong S, Zhang H, et al. Expression profiles and characteristics of human lncRNA in normal and asthenozoospermia sperm. Biol Reprod. 2018;100:982–93. doi: 10.1093/biolre/ioy253. [DOI] [PubMed] [Google Scholar]
- 39.Abu-Halima M, Hammadeh M, Schmitt J, Leidinger P, Keller A, et al. Altered microRNA expression profiles of human spermatozoa in patients with different spermatogenic impairments. Fertil Steril. 2013;99:1249–55e16. doi: 10.1016/j.fertnstert.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 40.Salas-Huetos A, Blanco J, Vidal F, Godo A, Grossmann M, et al. Spermatozoa from patients with seminal alterations exhibit a differential micro-ribonucleic acid profile. Fertil Steril. 2015;104:591–601. doi: 10.1016/j.fertnstert.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 41.Zhou JH, Zhou QZ, Lyu XM, Zhu T, Chen ZJ, et al. The expression of cysteine-rich secretory protein 2 (CRISP2) and its specific regulator miR-27b in the spermatozoa of patients with asthenozoospermia. Biol Reprod. 2015;92:28. doi: 10.1095/biolreprod.114.124487. [DOI] [PubMed] [Google Scholar]
- 42.Lindemann CB, Lesich KA. Functional anatomy of the mammalian sperm flagellum. Cytoskeleton. 2016;73:652–69. doi: 10.1002/cm.21338. [DOI] [PubMed] [Google Scholar]
- 43.Amiri-Yekta A, Coutton C, Kherraf ZE, Karaouzene T, Le Tanno P, et al. Whole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutations. Hum Reprod. 2016;31:2872–80. doi: 10.1093/humrep/dew262. [DOI] [PubMed] [Google Scholar]
- 44.Sha Y, Yang X, Mei L, Ji Z, Wang X, et al. DNAH1 gene mutations and their potential association with dysplasia of the sperm fibrous sheath and infertility in the Han Chinese population. Fertil Steril. 2017;107:1312–8e2. doi: 10.1016/j.fertnstert.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 45.Ben Khelifa M, Coutton C, Zouari R, Karaouzene T, Rendu J, et al. Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet. 2014;94:95–104. doi: 10.1016/j.ajhg.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li CY, Jiang LY, Chen WY, Li K, Sheng HQ, et al. CFTR is essential for sperm fertilizing capacity and is correlated with sperm quality in humans. Hum Reprod. 2010;25:317–27. doi: 10.1093/humrep/dep406. [DOI] [PubMed] [Google Scholar]
- 47.Kott E, Duquesnoy P, Copin B, Legendre M, Dastot-Le Moal F, et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet. 2012;91:958–64. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutovsky P, Hauser R, Sutovsky M. Increased levels of sperm ubiquitin correlate with semen quality in men from an andrology laboratory clinic population. Hum Reprod. 2004;19:628–38. doi: 10.1093/humrep/deh131. [DOI] [PubMed] [Google Scholar]
- 49.Hodjat M, Akhondi MA, Al-Hasani S, Mobaraki M, Sadeghi MR. Increased sperm ubiquitination correlates with abnormal chromatin integrity. Reprod Biomed Online. 2008;17:324–30. doi: 10.1016/s1472-6483(10)60215-5. [DOI] [PubMed] [Google Scholar]
- 50.Fu G, Wei Y, Wang X, Yu L. Identification of candidate causal genes and their associated pathogenic mechanisms underlying teratozoospermia based on the spermatozoa transcript profiles. Andrologia. 2016;48:576–83. doi: 10.1111/and.12484. [DOI] [PubMed] [Google Scholar]
- 51.Liu XX, Cai L, Liu FJ. An in silico analysis of human sperm genes associated with asthenozoospermia and its implication in male infertility. Medicine (Baltimore) 2018;97:e13338. doi: 10.1097/MD.0000000000013338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montjean D, De La Grange P, Gentien D, Rapinat A, Belloc S, et al. Sperm transcriptome profiling in oligozoospermia. J Assist Reprod Genet. 2012;29:3–10. doi: 10.1007/s10815-011-9644-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Purity and quality controls of sperm RNA samples. (a) Agilent bioanalyzer analysis of RNA from sperm and control somatic cells (PBMCs). In PBMCs, the majority of the RNA corresponds to rRNA (18s and 28S), while RNA from sperm samples are rRNA depleted; (b) Electrophoresis analysis of RT-PCR products corresponding to specific markers for leukocyte (CD45), germ cells (c-kit), epithelial cells (E-cad), and spermatozoa (SMCP). F1: sperm subpopulation F1; F2: sperm subpopulation F2; L: human leukocytes; P: epithelial cells; T: trophoblast explants; C: negative control; PBMCs: peripheral blood mononuclear cells; SMCP: sperm mitochondrial-associated cysteine-rich protein; RT-PCR: real-time polymerase chain reaction.
Gene clustering of the microarray data of N–F1 versus N–F2 and A–F1 versus A–F2 contrasts. Heatmaps show one sample for column and one gene for each horizontal line. Color indicates gene expression value intensities (z-score). Red: upregulation; Green: downregulation; Black: no change. N–F1: normozoospermic sperm subpopulation with high motility; N-F2: normozoospermic sperm subpopulation with low motility; A-F1: asthenozoospermic sperm subpopulation with high motility; A-F2: asthenozoospermic sperm subpopulation with low motility.
REVIGO TreeMap summarizing gene ontology biological process categories overrepresented in A–F1 versus N–F1. FatiGO was used to identify Gene Ontology Biological Processes overrepresented among transcripts more highly expressed. Categories with P-values below 0.01 were used to generate the treemap, colored by functional category. The box size is proportional to the P-value of each category. A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.
Molecule activity prediction of three sperm bio-functions using the DEG in A–F1 versus N–F1 contrast. (a) Capacitation of sperm; (b) acrosome reaction of sperm; (c) binding of sperm. Colors indicate predicted relationships of gene expression levels and bio-functions; color intensities reflect the gene expression degree or bio-function activity (http://ingenuity.com). A–F1: asthenozoospermic sperm subpopulation with high motility; N–F1: normozoospermic sperm subpopulation with high motility.