

Abstract
A phenotypic high-throughput screen identified a benzamide small molecule with activity against small cell lung cancer cells. A “clickable” benzamide probe was designed that irreversibly bound a single 50 kDa cellular protein, identified by mass spectrometry as β-tubulin. Moreover, the anti-cancer potency of a series of benzamide analogs strongly correlated with probe competition, indicating that β-tubulin was the functional target. Additional evidence suggested that benzamides covalently modified Cys239 within the colchicine binding site. Consistent with this mechanism, benzamides impaired growth of microtubules formed with β-tubulin harboring Cys239, but not β3 tubulin encoding Ser239. We therefore designed an aldehyde-containing analog capable of trapping Ser239 in β3 tubulin, presumably as a hemiacetal. Using a forward genetics strategy, we identified benzamide-resistant cell lines harboring a Thr238Ala mutation in β-tubulin sufficient to induce compound resistance. The disclosed chemical probes are useful to identify other colchicine site binders, a frequent target of structurally diverse small molecules.
Introduction
The identification and development of new cancer therapies remain a challenge. Target-based screens have proven effective at identifying small molecule inhibitors for oncogenic kinases that are recurrently mutated in many cancers. However, for tumors without mutations in these kinases, phenotypic screens offer a powerful complementary approach in which small molecules with anti-cancer activity can be identified in high-throughput format. The establishment of preliminary structure–activity relationships around these initial HTS hits is essential to facilitate the development of chemical probes suitable for target identification. If the biological target lends itself to a more translational evaluation, then more sophisticated medicinal chemistry optimization is useful for initial in vivo proof of concept studies in animal models of human cancer.
Small cell lung cancer (SCLC) is an aggressive neuroendocrine (NE) malignancy, and a diagnosis of SCLC portends poor survival. Treatments for SCLC have remained largely unchanged, and cytotoxic chemotherapies remain the foundation of therapy for SCLC. The failure to improve outcomes in SCLC stems in part from a lack of recurrent mutations that encode chemically tractable proteins. We leveraged genetically defined cancer cell lines derived from a mouse model of SCLC to facilitate phenotypic screening for anti-cancer small molecules with activity against SCLC and other NE tumors.1−4 Here, we report the identification of a series of benzamides that exhibit anti-cancer activity to murine and human SCLC cell lines. We utilize a combination of medicinal chemistry, probe reagent development, forward genetics, and in vitro assays of microtubule dynamics to demonstrate that these small molecules covalently modify β-tubulin via nucleophilic aromatic substitution.
Results and Discussion
Identification and Structure–Activity Relationship Studies (SAR) of Benzamide SCLC Toxins
We identified a benzamide-phenyl piperazine from a phenotypic high-throughput chemical screen aimed at identifying small molecules with selective anti-cancer activity against a panel of mouse and human SCLC cell lines. Benzamide 1 was active against three independent murine SCLC (mSCLC) cell lines with an average EC50 of 3.21 ± 0.6 μM (Figure 1A,B). Moreover, three human SCLC cell lines, H889, H2107, and H128, exhibited sensitivity, suggesting conserved expression of the protein target of compound 1 in mouse and human cells (Supporting InformationFigure 1). A literature search identified a previous study reporting that benzamide 1 was toxic to yeast cells through inhibition of Sec14, a protein essential for membrane trafficking in the Golgi-endosomal system.5 However, less than 30% amino acid sequence conservation between yeast and mammalian Sec14 orthologs raised the possibility that compound 1 induced SCLC cell death through Sec14-independent mechanisms. We therefore initiated an unbiased medicinal chemistry and forward genetics approach to identify the target of benzamide 1.
Figure 1.

Structure–activity relationships of 4-chloro-3-nitrobenzamide SCLC toxins. (A) Table with half-maximal inhibitory concentrations (IC50) for benzamide analogs 1–15 against murine SCLC cell line 319N1. Standard error is indicated between brackets and is based on n = 2 technical replicates. (B) Dose–response curves of compound 1 against three murine SCLC cell lines. (C) Dose–response curves of compounds 1, 10, and 15 against murine SCLC cell line 319N1.
We synthesized analogs of compound 1 to understand structure–activity relationships that would enable the development of chemical probes for target identification. Systematic modification to the phenyl piperazine ring revealed that substitutions at the R1, R2, and R3 positions impacted toxicity (Figure 1A,C). The chlorine substituent at position R1 slightly improved potency (2), whereas ortho-fluorine analog 12 was 2-fold less active than parent benzamide 1. At the R2 position, bromine, chlorine, and cyano groups improved activity ∼5 to 7-fold (analogs 6–8), whereas meta-methyl and methoxy-substituted analogs 3 and 4 were only ∼3-fold more potent. Of the various 2,5-disubstituted combinations tested, analog 10 (R1 = Me and R2 = Cl) was the most potent benzamide with an EC50 of 182 nM. In contrast, modifications at the para-position (R3 = F, Me, and MeO; analogs 11 and 13–15) significantly impacted the cytotoxic activity within this series.
Initial experiments had shown that the 4-chloro-3-nitro substitution pattern was an essential feature required for the ability of the benzamides to affect growth of the mSCLC cell line 319N1. For example, removing either of these substituents from the benzamide ring resulted in complete eradication of antiproliferative activity (see SI Table 1 and SI Figure 2, compounds 28 and 29). As shown in Figure 2A,B, we designed and evaluated several other analogs with alternate chloro/nitro substitution patterns (16–19), but all resulted in significantly less active benzamides when compared to 4-chloro-3-nitrobenzamide analog 10 (EC50 = 182 nM). These initial results, with the position of the chlorine substituent ortho and para to the mesomeric electron-withdrawing nitro and benzamide carbonyl in the most active analog 10, raised the possibility that nucleophilic aromatic substitution (SNAr) was important for the activity of these molecules. According to this model, we hypothesized that a nucleophilic amino acid side chain within a benzamide binding protein would displace the 4-chloro substituent via SNAr, resulting in a covalent bond to the target protein (Figure 2D).
Figure 2.

Structure–activity relationships of the 4-chloro-3-nitrobenzamide warhead reveal that benzamides bind covalently to p50 via aromatic nucleophilic substitution. (A) Table with half-maximal inhibitory concentrations (IC50) for benzamide analogs against murine SCLC line 319N1. Standard error is indicated between brackets and is based on n = 2 technical replicates. (B and C) Dose–response curves for analogs 10 and 16–23 against murine SCLC cell line 319N1. (D) Model for potential covalent cross-linking of the 4-chloro-3-nitrobenzamide warhead with binding protein via nucleophilic aromatic substitution. (E) Benzamide alkyne probe reagents 24 and 25 with half-maximal inhibitory concentrations (IC50) against murine SCLC line 319N1. Standard error is indicated between brackets and is based on n = 2 technical replicates. (F) SDS-PAGE gels of fluorescent azide dye-treated cell lysates obtained from 518T2 murine SCLC cells incubated with increasing concentrations of analogs 24 and 25.
To further evaluate this model, we generated analogs in which the chlorine leaving group was replaced with other leaving groups including fluorine, bromine, or iodine (Figure 2A,C). The activity of 20 (EC50 = 818 nM), 10 (EC50 = 182 nM), 21 (EC50 = 55 nM), and 22 (EC50 = 51 nM) positively correlated with the leaving group potential and the size of the halide substituent at position 4. Interestingly, analog 23 bearing 4-OCF3 was the most potent benzamide tested (EC50 = 20 nM).6 The impact of all these benzamide ring modifications was consistent with a nucleophilic aromatic substitution mediating a covalent compound–protein interaction.
Benzamides Bind Covalently to a Protein p50, Identified as β-Tubulin
We developed alkyne-modified chemical probes to visualize potential covalent small molecule protein complexes by SDS-PAGE via a copper-mediated click reaction with a fluorescent azide dye for in-gel detection.7−9 Based on the SAR described in Figure 1, we decided to introduce the alkyne “click” handle in the meta-position of the piperazinophenyl ring as a phenolic propargyl ether. Alkyne 24 harbored the benzamide SNAr warhead, whereas the corresponding alkyne analog 25 was substituted with a methyl group of similar van der Waals radius to chlorine but would not be expected to covalently bind to the target protein (Figure 2E). Consistent with this, methyl-substituted benzamide 25 was 10-fold less active compared to 24 (Figure 2E).
To visualize potential covalently modified proteins, murine SCLC cells were incubated with varying concentrations of either 24 or 25 (from 50 nM to 50 μM). Protein lysates were then subjected to the copper-mediated click reaction using a fluorescent azide dye (Alexa Fluor 532), and protein-probe complexes were visualized using an SDS-PAGE gel. A single dominant 50 kDa protein (hereafter referred as p50) was covalently modified by SNAr reactive probe 24, a band which was not observed in lysates incubated with 25 (Figure 2F and SI Figure 3).
Although the above experiments clearly demonstrated that the benzamide probe with an active SNAr warhead (24) was capable of forming an irreversible complex with p50, it remained to be determined whether this interaction was relevant to the anti-cancer activity of these compounds. To address this question in a quantitative fashion, we selected nine benzamide analogs that varied in potency between 51 nM and 4.36 μM in the cell-based phenotypic assay (cytotoxicity against mSCLC cell line 319N1) for a cell-based competition experiment with the clickable alkyne probe 24 in the probe-binding assay (Figure 3A and SI Figure 4). We identified a positive correlation between the cellular toxicity (IC50) of these nine analogs (see Figures 1A and 2A) and their respective ability to displace alkyne probe 24 (EC50) with an R2 value of 0.8895 (Figure 3B). These results were consistent with p50 binding being responsible for the anti-cancer activity of this series of benzamide analogs.
Figure 3.

Binding of benzamides to the β-tubulin colchicine site is responsible for toxicity in SCLC cells. (A) SDS-PAGE gels of fluorescent azide dye-treated cell lysates obtained from 518T2 murine SCLC cells incubated with alkyne probe 24 (5 μM) after pre-treatment with increasing concentrations of compounds 1–4, 6, 10, and 20–22. (B) Correlation among nine benzamide analogs for toxicity (IC50) and p50 binding (EC50). (C) Silver-stained SDS-PAGE gel of pull-down samples treated with probe 24 or probe 25. (D) Fold change of β5 tubulin peptide abundance in sample 24 normalized to sample 25. (E) SDS-PAGE gels of fluorescent azide dye-treated cell lysates obtained from 518T2 murine SCLC cells incubated with alkyne probe 24 (5 μM) after pre-treatment with increasing concentrations of colchicine or vincristine.
To enable identification of p50 using chemical probe–protein precipitation and mass spectrometry-based proteomics, the cross-linking experiment was repeated on a larger scale. After incubating cells with covalent alkyne probe 24 or with non-covalent probe 25, lysates were prepared and clicked to a biotin azide (instead of a fluorescent azide). The resulting biotin–protein–probe complexes were precipitated with streptavidin beads followed by boiling the beads. Proteins in the eluate were analyzed by SDS-PAGE and visualized using silver staining, confirming that p50 was pulled down in the sample treated with covalent probe 24 but not in the sample with compound 25 (Figure 3C). Shotgun LC–MS/MS analysis identified β-tubulin as the only protein enriched in the probe 24 treated sample (SI Table 2), of which β-tubulin isoform 5 (Tubb5; β5 tubulin) was the most enriched (12.95-fold) of the various isoforms.
Benzamides Bind β-Tubulin in the Colchicine Binding Pocket and Alter Microtubule Dynamics
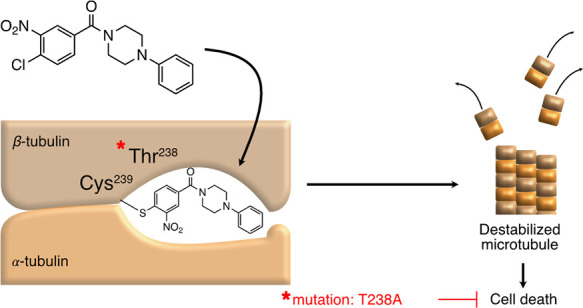
Notably, when focusing on the peptide counts for Tubb5 (β5 tubulin), we found that there was one single peptide (aa217-241) that was depleted from the probe-only treated sample (Figure 3D). We hypothesized that this peptide was depleted from the MS dataset due to a mass shift resulting from a covalent modification with the probe. The 217-241 peptide contained a single cysteine, raising the possibility that this residue acted as the nucleophile in the SNAr reaction with the probe. There have been at least two other small molecules reported to covalently modify β-tubulin at Cys239. T0070907 was a compound initially identified as an inhibitor of PPARγ via covalent modification of Cys313.10 Subsequently, T0070907 was shown to also induce tubulin degradation11,12 followed by a report by Yang et al. showing covalent modification of Cys239 of β-tubulin via displacement of the chlorine para to the nitro group in the 2-Cl-5-nitrobenzamide moiety of T0070907.13 Another compound, T138067 was also shown to bind covalently to β-tubulin through Cys239 by displacement of the para-fluorine in the pentafluorophenylsulfonamide warhead of T138067.14,15
Colchicine is a natural product that binds β-tubulin in a pocket located near the αβ-tubulin heterodimer interface. Cys239 lies within the colchicine binding pocket of β-tubulin and has been shown to be important for colchicine activity.16 Because our data suggested that benzamides interact with Cys239 within the colchicine binding pocket, we performed a competition experiment between covalent benzamide probe 24 and colchicine. As depicted in Figure 3E, colchicine was able to displace binding of alkyne probe 24 (5 μM) to p50 in a dose-dependent manner with an EC50 of 2.8 μM. Also, no such probe competition was observed with vincristine, a microtubule-destabilizing agent that acts at a site distinct from the colchicine site (Figure 3E). This data provided additional evidence that benzamides bound β-tubulin within the colchicine binding pocket.
Colchicine and other drugs that target β-tubulin impair microtubule dynamics and cause cell cycle arrest at the G2/M transition due to the impairment of chromosome segregation during mitosis.17 As expected, cell cycle analysis indicated that mSCLC cells treated with 300 nM colchicine or benzamide analog 21 (IC50 = 69 nM) for 24 h induced G2/M cell cycle arrest (Figure 4A).
Figure 4.

Benzamide analog 21 inhibits microtubule growth and induces G2/M cell cycle arrest. (A) Cell cycle analysis by flow cytometry of 518T2 cells treated with DMSO, analog 21 (300 nM), or colchicine (300 nM) for 24 h. (B) Quantification of the growth rate of microtubules using brain tubulin from samples treated with DMSO (33 microtubules quantified) and samples treated with analog 21 (21 microtubules quantified); n = 1 trial.
Microtubules are dynamic structures in the cell that undergo phases of growth and shrinkage in a phenomenon called dynamic instability.18 Drugs that bind the colchicine binding pocket of β-tubulin alter these dynamics and destabilize microtubule fibers.19 We therefore performed in vitro microtubule formation assays to assess the impact of benzamide analog 21 on microtubule dynamics. Compared to control, the growth of bovine brain microtubules was reduced by approximately 80% in the presence of analog 21 even at 10:1 (tubulin/compound) ratio (Figure 4B and SI Figure 5A).
Targeting the β3 Tubulin Isoform with Aldehyde-Modified Benzamides
A common mechanism of resistance to taxanes, the most commonly used microtubule-targeting agent for cancer therapy, is the overexpression of β3 tubulin (encoded by TUBB3) in which Arg277 replaces Ser277 in the M loop, greatly reducing the binding of taxane agents.20,21 Therefore, agents that target a site distinct from where taxanes bind could have translational potential. Given that the benzamides described herein target the colchicine site, we considered whether these might be active against β3 tubulin overexpressing cancers.19,22 However, the isoform switch to β3 tubulin indirectly impacts the colchicine binding pocket because TUBB3 encodes serine at position 239, rather than cysteine. Given that benzamides covalently modified β-tubulin potentially via Cys239 (Figure 3D), their binding affinity for the Ser239 containing β3 isoform could be dramatically impaired. Indeed, our proteomic datasets indicated that β3 tubulin was enriched only 3-fold compared to the average 9.2-fold enrichment for the other isoforms of β-tubulin (β2A, β2B, β4A, β4B, and β5), which harbor a cysteine at position 239 (SI Table 2). Consistent with this observation, benzamide analog 21 (up to 10 μM) failed to impair the in vitro microtubule growth rate of tubulin preparations using human recombinant β3 tubulin (Figure 5B and SI Figure 5B),23 whereas this same compound (at 1 μM) dramatically affected the growth of brain tubulin containing primarily the Cys239 containing β-tubulin isoforms (Figure 4B).
Figure 5.

Formyl-substituted benzamide analog 26 enables the inhibition of the Ser239-containing β3 tubulin isoform. (A) Model for hemiacetal formation of benzaldehyde analog 26 with serine or cysteine residues. (B) Quantification of the growth rate of β3 tubulin microtubules treated with DMSO (n = 131 microtubules quantified over three trials) or analog 21 (n = 126 microtubules quantified over three trials). (C) Dose–response curve of compound 26 in murine SCLC cell line 319N1. (D) Quantification of the growth rate of microtubules using β3 tubulin treated with DMSO (n = 107 microtubules quantified over two trials) or analog 26 at 1 μM (n = 119 microtubules quantified over two trials), 5 μM (n = 102 microtubules quantified over two trials), and 10 μM (n = 71 microtubules quantified over two trials). (E) Quantification of the growth rate of microtubules using brain tubulin from samples treated with DMSO (n = 32 microtubules quantified over one trial) or analog 26 (n = 36 microtubules quantified over one trial).
We therefore set out to design an analog that would engage serine 239 in hemiacetal formation with an appropriately placed aldehyde. Importantly, it was anticipated that such an analog would retain the capacity to capture Cys239 isoforms in the form of a hemithioacetal instead of via SNAr (Figure 5A). Gratifyingly, replacing the halide substituent in the benzamide ring with a formyl group led to analog 26 that retained potent anti-cancer activity against 319N1 mSCLC cells (EC50 = 127 nM) (Figure 5C). Consistent with our hypothesis, benzaldehyde 26 dose-dependently altered the dynamics of microtubules composed exclusively of β3 tubulin in a dose-dependent manner (Figure 5D). Microtubule growth rate was reduced from 0.83 μm/min in the DMSO control to 0.67 μm/min with analog 26 (10:1 ratio β3 tubulin/27). Further growth rate reduction was achieved at higher analog 26 concentrations, reaching 0.31 μm/min at 10 μM (Figure 5D and SI Figure 5B). Importantly, benzaldehyde 26 also potently reduced the growth of microtubules in preparations of bovine brain tubulin, which is composed primarily of Cys239-containing β-tubulin isoforms (Figure 5E and SI Figure 5C). In conclusion, aldehyde analogs such as 26 target both Cys239 and Ser239 isoforms, presumably via hemiacetal/hemithioacetal formation, which lays out a potential chemical strategy to target cancers that acquire resistance through β3 tubulin overexpression.
Competition with Benzamide Probe 24 and Forward Genetics Identify Another HTS Hit That Binds β-Tubulin at the Colchicine Binding Site
Given that microtubules are frequently targeted by small molecules, by design or unintended,16,24,25 we contemplated that benzamide probe 24 might be useful to identify other toxins that act through binding at the β-tubulin colchicine site. When we examined our mSCLC HTS hit list, a chloronitrobenzenesulfonamidobenzofuran (27, IC50 = 2.8 μM, Figure 6A,B) was flagged as containing structural features essential for nucleophilic aromatic substitution. As shown in Figure 6C, sulfonamide 27 displaced probe 24 from p50 in a dose-responsive manner. This result is consistent with the notion that microtubules are the mechanistic target of compound 27, presumably via covalent capture of Cys239 β-tubulin isoforms.
Figure 6.

Tubb5T238A compound-resistant allele identified by forward genetics is sufficient to confer benzamide resistance in murine SCLC cells. (A) Structure of compound 27. (B) Dose–response curve of compounds 1 and 27 against a murine SCLC cell line 319N1. Standard error is indicated between brackets and is based on n = 2 technical replicates. (C) SDS-PAGE gels of fluorescent azide dye-treated cell lysates obtained from 518T2 murine SCLC cells incubated with alkyne probe 24 (5 μM) after pre-treatment with increasing concentrations of compound 27. (D and E) Dose–response curves of compounds 27 and 10 in parental Msh2-deficient 319N1-M2KO cell line and resistant clones A–E. (F) Table with half-maximal inhibitory concentrations (IC50) for benzamide analogs 10 and 27 against the parental 319N1-M2KO cell line and five resistant clones A–E. (G) Tubb5 is one of the only 22 genes recurrently mutated in all five resistant clones. (H) Crystal violet staining of murine SCLC (518T2) cells edited in Tubb5 harboring the T238A mutation (top). Binary images from crystal violet images enable clearer representation of alive cells in the Tubb5 (T238A) sample (bottom). (I) Dose–response curves of analog 21 in 518T2 cells and 518T2-Tubb5T238A cells.
Simultaneous to our efforts to develop probe reagents for biochemical target identification studies, we utilized a forward genetics target identification strategy that we recently reported.26 Deletion of the DNA mismatch repair protein MSH2 using CRISPR-Cas9 is sufficient to induce hypermutation in human and murine cancer cell lines. The increased mutation frequency facilitates the emergence of clones that harbor compound-resistant alleles in the target of small molecule cytotoxins. We therefore generated a barcoded (20-nucleotide barcode) clonal mSCLC cell line (319N1-M2KO) using a lentiviral vector encoding blasticidin resistance. We successfully generated and validated independent barcoded clones resistant to sulfonamide 27. We performed compound selections using 27 at five concentrations close to the lethal dose, as determined by 1 week of compound exposure (EC1001wk). Following 2 weeks of compound selection, compound-resistant colonies emerged and were expanded. To confirm in vitro resistance, we determined the IC50 for five independent 319N1-M2KO clones (A–E) and confirmed that all were resistant to 27 (IC50 > 2-fold change, Figure 6D,F). Interestingly, all five clones were cross-resistant to benzamide 10, suggesting a shared molecular target or resistance mechanism (IC50 > 2.5-fold-change, Figure 6E,F). The same clones were not cross-resistant to etoposide, suggesting that general resistance mechanisms were not responsible for the emergence of these clones in response to selection with sulfonamide 27 (SI Figure 6).
To identify recurrently mutated genes across multiple resistant clones, all five sulfonamide 27 resistant clones were subjected to exome sequencing. We identified 22 genes mutated in five out of five clones (Figure 6G). Among these was a common Tubb5 mutation (712A > G) that resulted in a T238A (Thr > Ala) amino acid substitution immediately adjacent to the Cys239. It was of interest that the T238A mutation had previously been identified in yeast tubulin (TUB2) as a resistance mutation to benomyl, a microtubule inhibitor that also binds at the colchicine binding site.27,28 To our knowledge, this mutation had not been previously identified in mammalian cells, but this nonetheless raised the possibility that T238A mediated resistance to the benzamide and sulfonamide compound reported here.
Because multiple recurrent mutations existed in each clone, we sought to confirm that the T238A mutation was sufficient to confer resistance to benzamides. We engineered the T238A mutation into 518T2 mSCLC cells using CRISPR/Cas9. After transfection with Cas9 protein-sgRNA complexes targeting Tubb5 with an oligonucleotide repair template encoding the T238A mutation and a sham control, we selected cells with benzamide 21 at 400 nM for 2 weeks. To visualize compound resistant clones, plates were stained with crystal violet. Emerging clones were observed under the Tubb5T238A condition, whereas no clones were visible under the control condition (Figure 6H). Compound resistance to analog 21 was confirmed in 518T2Tubb5T238A cells compared to parental 518T2 cells (Figure 6I). Thus, introduction of a single T238A mutation in Tubb5 in murine SCLC cell lines was sufficient to endow resistance to the benzamides (10 and 21) and arylsulfonamide 27. This result established a causal relationship between the mutation and compound resistance.
Chemistry
Compounds 1, 7, 11, 12, and 27–29 were commercially available. As shown in Scheme 1, reaction of 4-chloro-3-nitrobenzoyl chloride with a collection of arylpiperazines I in the presence of triethylamine delivered benzamide analogs 2–6, 8–10, and 13–15. Analogs 16–23 and 26 were obtained via condensation of benzoic acid derivatives II with arylpiperazines I mediated by EDC as the dehydrating agent and DMAP as the catalyst. Benzamide analogs 24 and 25 were obtained from the corresponding phenolic benzamide intermediates via an additional propargylation with propargyl bromide and potassium carbonate in dimethylformamide.
Scheme 1. Synthesis of Benzamides.

Reagents and conditions: (a) NEt3, THF, rt; (b) (for 24 and 25) propargyl bromide, K2CO3, DMF; (c) EDC·HCl, DMAP, DMF, rt.
Conclusions
Here, we disclosed a multipronged approach to establish the mode of action of benzamides with anti-cancer activity to SCLC. Through medicinal chemistry, probe reagent development, and biochemical pull-down methods, we established that these benzamides target β-tubulin through the colchicine binding pocket using a nucleophilic aromatic substitution to covalently modify cysteine 239. Consistent with this mechanism of action, we demonstrated that these cytotoxins impair microtubule dynamics in vitro and induce cell cycle arrest at the G2/M transition. In addition, β-tubulin isoforms (including β3 tubulin) that encode serine at position 239 exhibited intrinsic resistance to the benzamide compounds as a result of the reduced nucleophilicity of serine compared to cysteine. We reported a strategy to engage serine-bearing β-tubulin isoforms by developing a benzaldehyde analog that targets serine 239 through hemiacetal formation. Forward genetics screening with a cell line harboring engineered DNA mismatch repair deficiency identified a compound-resistant allele in threonine 238 (Thr238Ala) that was sufficient to encode resistance to anti-cancer toxins bearing the chloronitrobenzene SNAr warhead.
β-tubulin is a well-validated therapeutic target of several classes of anti-cancer natural products and small molecules. However, β-tubulin is also increasingly identified as an unwanted off-target of small molecules purported to act on other protein targets. Here, we demonstrated the potential of our cell-based probe competition binding assay and expect that the disclosed benzamide probes will prove useful in drug discovery and development to identify compounds with the capacity, by design or not, to bind β-tubulin.
Experimental Section
General Methods for Chemistry
Unless otherwise specified, all commercially available reagents were used as received. Compound 1 was purchased from ChemBridge (#5348909). Compounds 7, 11, and 28–29 were purchased from ChemDiv (#Y031-8281, #Y031-8282, #Y030-0278, and #Y031-8034). Compound 27 was purchased from Princeton BioMolecular Research (#OSSK_101609). Compound 12 was purchased from Enamine (#Z30011284). All reactions using dried solvents were carried out under an atmosphere of argon in flame-dried glassware with magnetic stirring. A dry solvent was dispensed from a solvent purification system that passes the solvent through two columns of dry neutral alumina. Silica gel chromatographic purifications were performed by flash chromatography with a silica gel (Sigma, grade 60, 230–400 mesh) packed in glass columns (the eluting solvent was determined by thin-layer chromatography, TLC), or with an Isco Combiflash system using Redisep Rf Flash columns with size ranging from 4 to 80 g. Analytical TLC was performed on glass plates coated with a 0.25 mm silica gel using UV or by iodide or KMnO4 staining for visualization. Routine 1H and proton-decoupled 13C NMR spectra were obtained on an Agilent 400 MHz NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) from low to high field relative to the residual solvent. Multiplicities are given as s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), and m (multiplet). HRMS data were obtained from the Shimadzu Center for Advanced Analytical Chemistry (SCAAC) at U.T. Arlington. All purchased and synthetic compounds exhibited between 95 and 99% purity as determined by LC–MS analysis performed on an Agilent 1290 HPLC system using an Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm; Agilent) that was coupled to an Agilent 6130 mass spectrometer run in ESI mode in both positive and negative ionization with a scan range of 100–1100 m/z. Liquid chromatography was carried out at a flow rate of 0.5 mL/min at 20 °C with a 5 μL injection volume using the gradient elution with aqueous acetonitrile containing 0.1% formic acid. The gradient was adjusted based on the different polarities of different compounds.
General Procedure A for the Preparation of Benzamides from 4-Chloro-3-nitrobenzoyl Chloride and Arylpiperazines I
A solution of 4-chloro-3-nitrobenzoyl chloride, arylpiperazine I, and triethylamine in THF was stirred at room temperature for 16 h. The concentration of the reaction mixture followed by flash chromatography (gradient of 0–50% EtOAc in hexane) gave the corresponding benzamide.
General Procedure B for the Preparation of Benzamides from Substituted Benzoic Acids II and Arylpiperazines I
To a solution of arylpiperazine I, benzoic acid derivative II, and DMAP in DMF was added EDC·HCl at rt. The resulting reaction mixture was stirred for 16 h. Flash chromatography (gradient of 0–40% EtOAc in hexane) gave the corresponding benzamide.
(4-Chloro-3-nitrophenyl)(4-(2-chlorophenyl)piperazin-1-yl)methanone (2)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (75.0 mg, 0.34 mmol), 1-(2-chlorophenyl)piperazine HCl salt (104.0 mg, 0.44 mmol), and Et3N (0.90 mL, 6.45 mmol) in THF (4 mL). Yield 89 mg (68%) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 1.6 Hz, 1H), 7.66–7.55 (m, 2H), 7.36 (dd, J = 8.2, 1.5 Hz, 1H), 7.26–7.16 (m, 1H), 7.05–6.96 (m, 2H), 3.94 (bs, 2H), 3.60 (bs, 2H), 3.22–2.92 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 166.8, 148.3, 147.7, 135.5, 132.3, 131.8, 130.75, 128.9, 128.51, 127.79, 124.66, 124.57, 120.62, 51.58, 50.92, 48.15, 42.7; HRMS calculated for C17H15Cl2N3O3 [M + H]+ 380.0563, found 380.0541.
(4-Chloro-3-nitrophenyl)(4-(3-methoxyphenyl)piperazin-1-yl)methanone (3)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (106.0 mg, 0.48 mmol), 1-(3-methoxyphenyl)piperazine (251.0 mg, 1.31 mmol), and Et3N (0.30 mL, 2.15 mmol) in THF (4 mL). Yield 146 mg (80%) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 1.7 Hz, 1H), 7.64–7.55 (m, 2H), 7.17 (t, J = 8.0 Hz, 1H), 6.56–6.47 (m, 1H), 6.47–6.41 (m, 2H), 3.88 (s, 2H), 3.76 (s, 3H), 3.57 (d, J = 8.9 Hz, 2H), 3.32–3.01 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 166.65, 160.6, 152.0, 147.7, 135.3, 132.3, 131.9, 130.0, 128.5, 124.7, 109.4, 105.5, 103.3, 55.2, 49.7, 49.3, 47.6, 42.4; HRMS calculated for C18H18ClN3O4 [M + H]+ 376.1059, found 376.1042.
(4-Chloro-3-nitrophenyl)(4-(m-tolyl)piperazin-1-yl)methanone (4)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (75.0 mg, 0.34 mmol), 1-(3-methylphenyl)piperazine (124.0 mg, 0.70 mmol), and Et3N (0.30 mL, 2.15 mmol) in THF (4 mL). Yield 110 mg (89%) as light-yellow gummy solid. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 1.7 Hz, 1H), 7.66–7.57 (m, 2H), 7.17 (t, J = 7.9 Hz, 1H), 6.78–6.70 (m, 3H), 3.91 (bs, 2H), 3.58 (bs, 2H), 3.19 (m, 4H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.7, 150.7, 147.8, 139.1, 135.4, 132.3, 131.9, 129.15, 128.6, 124.7, 121.85, 117.75, 114.0, 49.9, 47.8, 42.5, 21.75; HRMS calculated for C18H18ClN3O3 [M + H]+ 360.1109, found 360.1090.
(4-Chloro-3-nitrophenyl)(4-(2,5-dimethylphenyl)piperazin-1-yl)methanone (5)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (75.0 mg, 0.34 mmol), 1-(2,5-dimethylphenyl)piperazine (122.0 mg, 0.64 mmol), and Et3N (0.60 mL, 4.30 mmol) in THF (4 mL). Yield 122 mg (96%) as a light-yellow thick oil. 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 1.5 Hz, 1H), 7.63 (d, J = 1.2 Hz, 2H), 7.09 (d, J = 7.6 Hz, 1H), 6.88–6.79 (m, 2H), 4.04–3.81 (m, 2H), 3.59 (bs, 2H), 3.03–2.82 (m, 4H), 2.31 (s, 3H), 2.28 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.8, 150.35, 147.8, 136.4, 135.6, 132.3, 131.9, 131.1, 129.4, 128.45, 124.7, 124.65, 120.1, 52.1, 51.7, 48.5, 43.05, 21.2, 17.4; HRMS calculated for C19H20ClN3O3 [M + H]+ 374.1266, found 374.1242.
(4-(3-Bromophenyl)piperazin-1-yl)(4-chloro-3-nitrophenyl)methanone (6)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (49.0 mg, 0.22 mmol), 1-(3-bromophenyl)piperazine (55.0 mg, 0.23 mmol), and Et3N (0.10 mL, 0.72 mmol) in THF (2 mL). Yield 81 mg (92%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 1.8 Hz, 1H), 7.66–7.56 (m, 2H), 7.12 (t, J = 8.0 Hz, 1H), 7.06–6.98 (m, 2H), 6.82 (ddd, J = 8.3, 2.3, 1.0 Hz, 1H), 3.74 (m, 9 Hz, 4H), 3.38–3.03 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 166.7, 151.8, 147.8, 135.1, 132.4, 131.8, 130.6, 128.7, 124.7, 123.5, 123.3, 119.55, 115.2, 49.2, 47.5, 42.3; HRMS calculated for C17H15BrClN3O3 [M + H]+ 424.0058, found 424.0030.
3-(4-(4-Chloro-3-nitrobenzoyl)piperazin-1-yl)benzonitrile (8)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (100.0 mg, 0.45 mmol), 1-(3-cyanophenyl)piperazine (172.0 mg, 0.92 mmol), and Et3N (0.3 mL, 2.15 mmol) in THF (4 mL). Yield 135 mg (80%) as a light-yellow gummy solid. 1H NMR (400 MHz, CDCl3) δ 7.94 (t, J = 1.2 Hz, 1H), 7.59 (d, J = 1.5 Hz, 2H), 7.37–7.26 (m, 1H), 7.14–7.05 (m, 3H), 4.08–3.47 (m, 4H), 3.22 (bs, 4H); 13C NMR (101 MHz, CDCl3) δ 166.7, 150.7, 147.7, 135.1, 132.4, 131.9, 130.15, 128.6, 124.7, 123.55, 120.6, 119.1, 119.05, 113.0, 48.6, 47.3, 42.1; HRMS calculated for C18H15ClN4O3 [M + H]+ 371.0905, found 371.0882.
(4-Chloro-3-nitrophenyl)(4-(2,5-dichlorophenyl)piperazin-1-yl)methanone (9)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (65.0 mg, 0.30 mmol), 1-(2,5-dichlorophenyl)piperazine (103.0 mg, 0.34 mmol), and Et3N (0.30 mL, 2.20 mmol) in THF (3 mL). Yield 26 mg (21%) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 1.8 Hz, 1H), 7.67–7.57 (m, 2H), 7.30 (d, J = 8.4 Hz, 1H), 7.05–6.95 (m, 2H), 3.95 (bs, 2H), 3.61 (bs, 2H), 3.25–2.91 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 166.9, 149.3, 147.9, 135.4, 133.4, 132.5, 131.9, 131.6, 128.8, 127.3, 124.75, 124.6, 121.2, 51.5, 50.9, 48.1, 42.7; HRMS calculated for C17H15Cl3N3O [M + H]+ 414.0174, found 414.0155.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(4-chloro-3-nitrophenyl)methanone (10)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (77.0 mg, 0.35 mmol), 1-(5-chloro-2-methylphenyl)piperazine (121.0 mg, 0.57 mmol), and Et3N (0.30 mL, 2.15 mmol) in THF (4 mL). Yield 140 mg (68%) as a light-yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 1.6 Hz, 1H), 7.61 (d, J = 2.0 Hz, 2H), 7.09 (d, J = 8.0 Hz, 1H), 7.02–6.90 (m, 2H), 3.90 (bs, 2H), 3.57 (bs, 2H), 3.10–2.71 (m, 4H), 2.26 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.8, 151.5, 147.7, 135.4, 132.3, 132.2, 131.85, 131.8, 131.0, 128.5, 124.6, 123.9, 119.8, 51.7, 51.5, 48.2, 42.8, 17.4; HRMS calculated for C18H17Cl2N3O3 [M + Na]+ 416.0539, found 416.0512.
(4-Chloro-3-nitrophenyl)(4-(2,4-difluorophenyl)piperazin-1-yl)methanone (13)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (75.0 mg, 0.34 mmol), 1-(2,4-difluorophenyl)piperazine (111.0 mg, 0.56 mmol), and Et3N (0.60 mL, 4.30 mmol) in THF (4 mL). Yield 92 mg (71%) as a light-yellow gummy solid. 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 1.7 Hz, 1H), 7.68–7.55 (m, 2H), 6.89 (td, J = 9.9, 9.4, 5.7 Hz, 1H), 6.80 (dtt, J = 8.1, 5.1, 3.0 Hz, 2H), 3.92 (bs, 2H), 3.59 (bs, 2H), 3.03 (m, 4H); 13C NMR (101 MHz, CDCl3, including 19F-coupling peaks) δ 166.7, 159.7, 159.6, 157.2, 157.1, 157.1, 156.95, 154.6, 154.5, 147.75, 135.8, 135.8, 135.75, 135.7, 135.3, 132.3, 131.8, 128.6, 124.6, 120.2, 120.1, 120.1, 120.0, 111.1, 111.03, 110.85, 110.8, 105.2, 104.92, 104.91, 104.7, 51.3, 50.6, 47.95, 42.55; HRMS calculated for C17H14ClF2N3O3 [M + H]+ 382.0765, found 382.0742.
(4-Chloro-3-nitrophenyl)(4-(2,4-dimethylphenyl)piperazin-1-yl)methanone (14)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (76 mg, 0.34 mmol), 1-(2,4-dimethylphenyl)piperazine (122.0 mg, 0.64 mmol), and Et3N (0.30 mL, 2.15 mmol) in THF (4 mL). Yield 120 mg (93%) as a light-yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 1.2 Hz, 1H), 7.63 (d, J = 1.3 Hz, 2H), 7.03 (d, J = 2.2 Hz, 1H), 6.99 (dd, J = 8.1, 2.1 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 3.92 (bs, 2H), 3.71–3.42 (m, 2H), 2.91 (m, 4H), 2.30 (s, 3H), 2.29 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.75, 148.1, 147.8, 135.6, 133.6, 132.6, 132.3, 132.0, 131.9, 128.45, 127.2, 124.7, 119.2, 52.25, 51.7, 48.5, 43.1, 20.7, 17.65; HRMS calculated for C19H20ClN3O3 [M + H]+ 374.1266, found 374.1241.
(4-Chloro-3-nitrophenyl)(4-(4-methoxyphenyl)piperazin-1-yl)methanone (15)
Prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (75.0 mg, 0.34 mmol), 1-(4-methoxyphenyl)piperazine (120.0 mg, 0.62 mmol), and Et3N (0.60 mL, 4.30 mmol) in THF (4 mL). Yield 65 mg (51%) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 1.8 Hz, 1H), 7.68–7.54 (m, 2H), 6.89 (d, J = 9.2 Hz, 2H), 6.84 (d, J = 9.0 Hz, 2H), 4.03–3.80 (m, 2H), 3.76 (s, 3H), 3.57 (bs, 2H), 3.07 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 166.65, 154.6, 147.75, 144.9, 135.4, 132.3, 131.8, 128.6, 124.6, 119.1, 114.55, 55.5, 51.3, 50.9, 47.9, 42.6; HRMS calculated for C18H18ClN3O4 [M + H]+ 376.1059, found 376.1045.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(4-chloro-2-nitrophenyl)methanone (16)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (61.0 mg, 0.29 mmol), 4-chloro-2-nitrobenzoic acid (93.0 mg, 0.46 mmol), DMAP (8.0 mg, 0.07 mmol), and EDC·HCl (143.0 mg, 0.75 mmol) in DMF (1 mL). Yield 62 mg (54%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 2.1 Hz, 1H), 7.72 (dd, J = 8.1, 2.1 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 7.05–6.91 (m, 2H), 3.97 (s, 2H), 3.38 (t, J = 5.0 Hz, 2H), 3.14–2.75 (m, 4H), 2.28 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.62, 151.7, 146.0, 135.9, 134.6, 132.1, 131.9, 131.1, 131.05, 129.3, 125.1, 123.85, 119.9, 51.3, 51.2, 47.3, 42.4, 17.4; HRMS calculated for C18H17Cl2N3O3 [M + H]+ 394.0722, found 394.0720.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(2-chloro-3-nitrophenyl)methanone (17)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (53.0 mg, 0.25 mmol), 2-chloro-3-nitrobenzoic acid (102.0 mg, 0.50 mmol), DMAP (3.0 mg, 0.03 mmol), and EDC·HCl (114.0 mg, 0.59 mmol) in DMF (1.5 mL). Yield 45 mg (45%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 7.79 (dd, J = 7.6, 2.1 Hz, 1H), 7.51–7.40 (m, 2H), 7.03 (d, J = 8.1 Hz, 1H), 6.91 (dd, J = 8.1, 2.1 Hz, 1H), 6.87 (d, J = 2.1 Hz, 1H), 4.00–3.81 (m, 2H), 3.44–3.21 (m, 2H), 3.00–2.68 (m, 4H), 2.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.9, 151.6, 148.7, 138.8, 132.2, 131.9, 131.1, 131.0, 128.2, 125.7, 123.9, 123.3, 119.8, 51.8, 51.5, 47.2, 42.3, 17.4; HRMS calculated for C18H17Cl2N3O3 [M + Na]+ 416.0539, found 416.0513.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(3-chloro-5-nitrophenyl)methanone (18)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (57.0 mg, 0.27 mmol), 3-chloro-5-nitrobenzoic acid (103.0 mg, 0.51 mmol), DMAP (3.0 mg, 0.03 mmol), and EDC·HCl (115.0 mg, 0.60 mmol) in DMF (1.5 mL). Yield 92 mg (86%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 8.19 (t, J = 2.0 Hz, 1H), 8.10 (dd, J = 2.1, 1.4 Hz, 1H), 7.69 (t, J = 1.7 Hz, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.95–6.84 (m, 2H), 3.85 (s, 2H), 3.49 (s, 2H), 2.85 (d, J = 27.0 Hz, 4H), 2.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.4, 151.5, 148.6, 138.6, 136.1, 133.2, 132.2, 131.9, 131.0, 124.85, 124.0, 120.4, 119.9, 51.8, 51.5, 48.2, 42.9, 17.4; HRMS calculated for C18H17Cl2N3O3 [M + H]+ 394.0722, found 394.0671.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(3-chloro-4-nitrophenyl)methanone (19)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (58.0 mg, 0.27 mmol), 3-chloro-4-nitrobenzoic acid (103.0 mg, 0.51 mmol), DMAP (3.0 mg, 0.03 mmol), and EDC·HCl (115.0 mg, 0.60 mmol) in DMF (1.5 mL). Yield 96 mg (90%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 1.7 Hz, 1H), 7.46 (dd, J = 8.3, 1.7 Hz, 1H), 7.10 (d, J = 8.1 Hz, 1H), 6.98 (dd, J = 8.0, 2.1 Hz, 1H), 6.94 (s, 1H), 3.91 (s, 2H), 3.53 (s, 2H), 2.91 (d, J = 40.9 Hz, 4H), 2.26 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.7, 151.5, 148.2, 140.7, 132.2, 131.85, 131.0, 130.55, 127.7, 126.2, 125.95, 123.9, 119.8, 51.9, 51.45, 48.1, 42.7, 17.4; HRMS calculated for C18H17Cl2N3O3 [M + H]+ 394.0720, found 394.0701.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(4-fluoro-3-nitrophenyl)methanone (20)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (52.0 mg, 0.25 mmol), 4-fluoro-3-nitrobenzoic acid (75.0 mg, 0.40 mmol), DMAP (3.0 mg, 0.025 mmol), and EDC·HCl (100.0 mg, 0.52 mmol) in DMF (1.5 mL). Yield 77 mg (83%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 7.0, 2.2 Hz, 1H), 7.78 (ddd, J = 8.6, 4.2, 2.2 Hz, 1H), 7.40 (dd, J = 10.4, 8.5 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.05–6.91 (m, 2H), 3.77 (m, 6 Hz, 4H), 3.12–2.73 (m, 4H), 2.29 (s, 3H); 13C NMR (101 MHz, CDCl3, including 19F-coupling peaks) δ 166.9, 157.4, 154.7, 151.6, 137.2, 137.1, 134.6, 134.5, 132.6, 132.5, 132.2, 132.1, 131.9, 131.05, 125.4, 125.4, 123.9, 119.9, 119.8, 119.1, 118.9, 51.7, 51.6, 51.4, 48.3, 43.0, 17.4; HRMS calculated for C18H17ClFN3O3 [M + H]+ 378.1015, found 378.0985.
(4-Bromo-3-nitrophenyl)(4-(5-chloro-2-methylphenyl)piperazin-1-yl)methanone (21)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (59.0 mg, 0.28 mmol), 4-bromo-3-nitrobenzoic acid (105.0 mg, 0.43 mmol), DMAP (3.0 mg, 0.03 mmol), and EDC·HCl (100.0 mg, 0.52 mmol) in DMF (1.5 mL). Yield 115 mg (93%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 2.0 Hz, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.53 (dd, J = 8.2, 2.0 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.04–6.92 (m, 2H), 4.04–3.46 (m, 4H), 2.92 (bs, 4H), 2.28 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.9, 151.6, 149.7, 136.1, 135.5, 132.2, 131.9, 131.75, 131.05, 124.5, 123.9, 119.9, 116.0, 51.7, 48.2, 42.9, 17.4; HRMS calculated for C18H17BrClN3O3 [M + H]+ 438.0215, found 438.0190.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(4-iodo-3-nitrophenyl)methanone (22)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (55.0 mg, 0.26 mmol), 4-iodo-3-nitrobenzoic acid (143.0 mg, 0.49 mmol), DMAP (3.0 mg, 0.03 mmol), and EDC·HCl (100.0 mg, 0.52 mmol) in DMF (1.5 mL). Yield 115 mg (91%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.1 Hz, 1H), 7.85 (d, J = 1.9 Hz, 1H), 7.27 (dd, J = 8.1, 2.0 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.96–6.84 (m, 2H), 4.02–3.18 (m, 3H), 2.85 (m, 5H), 2.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 167.0, 152.9, 151.6, 142.4, 136.9, 132.2, 131.9, 131.8, 131.05, 124.2, 123.9, 119.9, 88.0, 51.7, 48.2, 42.8, 17.4; HRMS calculated for C18H17ClIN3O3 [M + H]+ 486.0084, found 486.0076.
(4-(5-Chloro-2-methylphenyl)piperazin-1-yl)(3-nitro-4-(trifluoromethoxy)phenyl)methan-one (23)
Prepared according to Procedure B using 1-(5-chloro-2-methylphenyl)piperazine (51.0 mg, 0.24 mmol), 3-nitro-4-(trifluoromethoxy)benzoic acid (51.0 mg, 0.20 mmol), DMAP (4.0 mg, 0.03 mmol), and EDC·HCl (47.0 mg, 0.24 mmol) in DMF (1 mL). Yield 54 mg (60%) as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 2.1 Hz, 1H), 7.69 (dd, J = 8.5, 2.1 Hz, 1H), 7.46 (dq, J = 8.6, 1.5 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.97–6.86 (m, 2H), 4.00–3.38 (m, 4H), 2.84 (m, 4H), 2.20 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.5, 151.5, 142.4, 142.0, 142.0, 135.3, 133.0, 132.2, 131.9, 131.0, 125.05, 124.0, 123.38, 123.37, 121.4, 119.9, 118.8, 51.55, 48.3, 47.25, 42.9, 17.42, 17.36; HRMS calculated for C19H17ClF3N3O4 [M + H]+ 444.0932, found 444.0920.
(4-Chloro-3-nitrophenyl)(4-(3-(prop-2-yn-1-yloxy)phenyl)piperazin-1-yl)methanone (24)
Intermediate (4-chloro-3-nitrophenyl)(4-(3-hydroxyphenyl)piperazin-1-yl)methanone was prepared according to Procedure A using 4-chloro-3-nitrobenzoyl chloride (1.05 g, 4.75 mmol), 1-(3-hydroxyphenyl)piperazine (1.30 g, 7.29 mmol), and Et3N (4 mL, 28.69 mmol) in THF (20 mL). The reaction mixture was diluted with EtOAc (50 mL) and washed with H2O (3 × 50 mL) and brine (50 mL). The collected organics were dried (Na2SO4) and concentrated under reduced pressure to give (4-chloro-3-nitrophenyl)(4-(3-hydroxyphenyl)piperazin-1-yl)methanone as a light-yellow solid. Without further purification, (4-chloro-3-nitrophenyl)(4-(3-hydroxyphenyl)piperazin-1-yl)methanone (180.0 mg, 0.49 mmol) was dissolved in DMF (5 mL). After addition of K2CO3 (500.0 mg, 3.62 mmol) and propargyl bromide (80% in PhMe, 0.25 mL, 2.24 mmol), the resulting mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and quenched with H2O (20 mL). The organic layer was washed with H2O (3 × 20 mL) and brine (20 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (gradient of 0–50% EtOAc in hexane) to yield 97 mg (49%) of 24 as a brown gummy solid. 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 1.8 Hz, 1H), 7.71–7.47 (m, 2H), 7.19 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 6.64–6.28 (m, 3H), 4.66 (d, J = 2.4 Hz, 2H), 3.73 (m, 4H), 3.19 (m, 4H), 2.52 (t, J = 2.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 166.7, 158.6, 152.0, 147.8, 135.3, 132.3, 131.85, 130.0, 128.6, 124.7, 110.2, 106.25, 104.2, 78.6, 75.6, 55.8, 49.6, 47.6, 42.35; HRMS calculated for C20H18ClN3O4 [M + H]+ 400.1059, found 400.1056.
(4-Methyl-3-nitrophenyl)(4-(3-(prop-2-yn-1-yloxy)phenyl)piperazin-1-yl)methanone (25)
Intermediate (4-(3-hydroxyphenyl)piperazin-1-yl)(4-methyl-3-nitrophenyl)methanone was prepared according to Procedure B using 4-methyl-3-nitrobenzoic acid (500.0 mg, 2.76 mmol), 1-(3-hydroxyphenyl)piperazine (740.0 mg, 4.14 mmol), DMAP (34.0 mg, 0.27 mmol), and EDC·HCl (800.0 mg, 4.14 mmol) in DMF (5 mL). The reaction mixture was diluted with EtOAc (40 mL) and washed with H2O (3 × 40 mL) and brine (40 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure to yield crude (4-(3-hydroxyphenyl)piperazin-1-yl)(4-methyl-3-nitrophenyl)methanone as a light-yellow solid. Without further purification, crude (4-(3-hydroxyphenyl)piperazin-1-yl)(4-methyl-3-nitrophenyl)methanone (71.0 mg, 0.50 mmol) was dissolved in DMF (1.5 mL). After addition of K2CO3 (207.0 mg, 1.50 mmol) and propargyl bromide (80% in PhMe, 150 □L, 1.20 mmol), the mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and quenched with H2O (20 mL). The organic layer was washed with H2O (3 × 20 mL) and brine (20 mL), dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by flash chromatography (gradient of 0–50% EtOAc in hexane) to yield 35 mg (71%) of 25 as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 1.7 Hz, 1H), 7.52 (dd, J = 7.8, 1.7 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.13 (t, J = 8.5 Hz, 1H), 6.55–6.41 (m, 3H), 4.60 (d, J = 2.4 Hz, 2H), 3.68 (m, 4H), 3.14 (bs, 4H), 2.57 (s, 3H), 2.45 (t, J = 2.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 167.8, 158.65, 152.1, 149.0, 135.4, 134.55, 133.25, 131.6, 130.0, 123.7, 110.2, 106.25, 104.2, 78.6, 75.5, 55.8, 49.5, 47.6, 42.3, 20.4; HRMS calculated for C21H21N3O4 [M + H]+ 380.1605, found 380.1604.
4-(4-(3,5-Dimethoxyphenyl)piperazine-1-carbonyl)-2-nitrobenzaldehyde (26)
Prepared according to Procedure B using 1-(3,5-dimethoxyphenyl)piperazine (130.0 mg, 0.58 mmol), 4-formyl-3-nitrobenzoic acid (125.0 mg), DMAP (6.0 mg, 0.05 mmol), and EDC·HCl (200.0 mg, 1.04 mmol) in DMF (1 mL). Yield 57 mg as a colorless thick oil. 1H NMR (400 MHz, CDCl3) δ 10.35 (s, 1H), 8.10 (d, J = 1.5 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.75 (dd, J = 7.8, 1.5 Hz, 1H), 6.00 (s, 3H), 4.01–3.76 (m, 2H), 3.70 (s, 6H), 3.49 (d, J = 19.3 Hz, 2H), 3.14 (d, J = 53.0 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 187.3, 166.5, 161.6, 152.55, 149.5, 141.0, 132.4, 131.9, 130.2, 123.5, 95.88, 92.6, 55.3, 49.8, 49.4, 47.5, 42.3; HRMS calculated for C20H21N3O6 [M + H]+ 400.1503, found 400.1501.
Cytotoxicity Assay
Mouse SCLC cell
lines were seeded
in duplicate in 96-well plates, 10,000 cells and 200 μL of DMEM
media (5% FBS) per well. After overnight incubation, compounds were
dispensed using a D300e Digital Dispenser (TECAN) in 10-point dose
response manner using 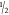 log dilutions. Cell
viability was assessed
after 72 h using CellTiter-Glo luminescent cell viability assay (Promega,
#G7571). The CellTiter-Glo reagent was diluted by adding PBS/Triton-X
(1%) (1:1 ratio). Each value was normalized to cells treated with
DMSO, and the IC50 values were calculated using GraphPad
Prism software.
log dilutions. Cell
viability was assessed
after 72 h using CellTiter-Glo luminescent cell viability assay (Promega,
#G7571). The CellTiter-Glo reagent was diluted by adding PBS/Triton-X
(1%) (1:1 ratio). Each value was normalized to cells treated with
DMSO, and the IC50 values were calculated using GraphPad
Prism software.
Cross-Linking Experiments
Murine SCLC cells, 518T2, were plated in 12-well plates (1 million cells per well) in 1 mL of DMEM media (5% FBS) per well. The following day, probe 24 or probe 25 dissolved in DMSO was dispensed at increasing concentrations (50 nM to 50 μM). After 1.5 h incubation at 37 °C in 5% CO2, cells were washed gently with PBS and lysed in 1% SDS Buffer A (50 mM HEPES, pH 7.4, 10 mM KCl, 2 mM MgCl2), freshly supplemented with 1:10,000 benzonase (Sigma-Aldrich). For competition experiments, analogs dissolved in DMSO were dispensed at increasing concentrations (100 nM to 50 μM) first and murine SCLC cells, 518T2, were plated in 12-well plates (1 million cells per well) in 1 mL DMEM media (5% FBS) per well. The following day, either probe 24 or probe 25 was dispensed in DMSO at increasing concentrations (50 nM to 50 μM). After 1.5 h incubation at 37 °C in 5% CO2, cells were washed gently with PBS and lysed in 1% SDS Buffer A (50 mM HEPES, pH 7.4, 10 mM KCl, 2 mM MgCl2), freshly supplemented with 1:10,000 benzonase (Sigma-Aldrich). Click chemistry was performed as previously described.29
Protein Purification
For purification experiments, cells were plated in 15 cm dishes at a large scale to obtain 100 mg of protein per condition and allowed to adhere overnight. The following day, cells were subjected to photocross-linking experiments described above. Instead of 25 μM Alexa Fluor 532 azide, 100 μM diazo biotin azide (Click Chemistry Tools, #1041) was used for the click reaction. Proteins were precipitated with four volumes of acetone cooled to −80 °C, and the insoluble protein pellets were centrifuged at 6000g and then resolubilized in PBS (4% SDS) overnight. The insoluble material was pelleted by centrifugation at 20,000g, and the soluble supernatant was filtered using 0.45 μm and 0.22 μm filters. Streptavidin agarose beads (Solu-link) were added and incubated with the resolubilized protein for 1 h at 25 °C. Beads were washed three times with 4% SDS in PBS, and the protein was eluted at 95 °C in Laemmli sample buffer. Proteins were resolved by SDS-PAGE, and the gel was stained with silver stain (Pierce, #24612). For proteomics analysis, samples were run until proteins enter into the SDS-PAGE gel, and each sample was cut and sent for shotgun analysis using LC–MS/MS.
Proteomics Analysis
Gel band samples were digested overnight with trypsin (Pierce) following reduction and alkylation with DTT and iodoacetamide (Sigma-Aldrich). The samples then underwent solid-phase extraction cleanup with an Oasis HLB plate (Waters), and the resulting samples were injected onto an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 μm i.d., 75 cm-long EasySpray column (Thermo) and eluted with a gradient from 0 to 28% buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer was operated in positive ion mode with a source voltage of 1.5 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap, and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.4 SP1 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, respectively, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate cutoff was 1% for all peptides.
Proteomic data for samples 24 and 25 are accessible at MassIVE accession number: MSV000085897 (ftp://massive.ucsd.edu/MSV000085897/).
Expression and Purification of β3 Human αβ-Tubulin
Human αβ-tubulin (non-tagged TUBA1B gene and TUBB3 gene with a cleavable His-tag at the C-terminus) was expressed in insect cells as described previously.23 Briefly, Tni cells (Expression Systems) and ESF-921 insect cell medium (Expression Systems) were used for expression. Cells were harvested approximately ∼42 h post-infection, re-suspended in 3 volumes of lysis buffer (25 mM Hepes, pH 7.4, 30 mM imidazole, 1 mM MgSO4, 50 μM GTP), and lysed using a glass dounce. Lysate was clarified by centrifugation, and recombinant tubulin was purified by Ni-affinity (5 mL Ni-NTA column, TaKaRA) and anion exchange (4 mL Source-Q column, GE Amersham) chromatography. The His-tag was removed by TEV protease (2 h on ice using a TEV at 0.2 mg/mL final concentration) prior to anion exchange chromatography. Peak fractions were pooled, concentrated to 15–20 μM, buffer-exchanged to BRB80 with 50 μM GTP, flash-frozen on liquid nitrogen in 100 μL aliquots, and stored at −80 °C.
Microtubule Dynamics Assays
Microtubule dynamics were measured by time-lapse differential interference contrast microscopy, as described previously.30−32 Temperature was maintained at 30 °C using an enclosure fit to the microscope body, and the microscope was controlled using a MicroManager.33 Briefly, assays were performed in flow chambers using GMPCPP seeds made from brain tubulin (5% biotinylated; PurSolutions) attached to coverslips using neutravidin (Invitrogen). Microtubule dynamics were measured in the following buffer (144 mM PIPES, pH 6.9, 3.6 mM MgCl2, 50 mM KCl, 1 mM GTP, 0.1 mg/mL BSA, 0.07% methylcellulose, and 4% DMSO final concentration). DMSO was the solvent where compounds were dissolved in. We used a higher than typical concentration of PIPES because microtubule dynamics were not robustly observable at lower concentrations for the recombinant human tubulin. Images were acquired every 50 ms (using a Photometrics Prime95B camera) and averaged in batches of 30 to improve signal to noise. Recordings typically lasted for 30 min. Microtubule growing rates were obtained from kymographs prepared using ImageJ.33
Acknowledgments
This work was funded through a UT Southwestern Dean’s Circle of Friends Pilot Synergy Grant. J.K.D.B. acknowledges support from the Welch Foundation (Grant I-1422) and holds the Julie and Louis Beecherl, Jr., Chair in Medical Science. D.G.M. acknowledges support from the Welch Foundation (Grant I-2040), the Disease Oriented Scholars Program at UT Southwestern, a Clinical Investigator Award from the Damon Runyon Cancer Research Foundation (102-19), the Cancer Prevention and Research Institute of Texas (RR140084 and RP190141), and the National Cancer Institute of the NIH (U54CA231649). J.M.P. is supported by a fellowship from the Chilton Family Foundation. HRMS data were obtained from the Shimadzu Center for Advanced Analytical Chemistry (SCAAC) at UT Arlington. Work in the laboratory of L.M.R. was supported by the NIH (R01-GM098543) and the Welch Foundation (Grant I-1908). Andrew Lemoff and colleagues at the UT Southwestern Medical Center Proteomics Core Facility performed mass spectrometry analysis on benzamide-bound proteins. We thank John Minna and Adi Gazdar for sharing human SCLC cell lines. We thank Deepak Nijhawan for critically reading the manuscript and for numerous helpful discussions. We thank Kuanqing Liu for critically reading the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01482.
List of proteins by mass spectrometry analysis in tubulin-probe purification with analog 24 compared to compound 25 (XLSX)
Somatic mutations identified in resistant clones to compound 27 compared to parental 319N1-M2KO cells (XLSX)
Supplementary tables (benzamide analogs and proteins enriched), supplementary figures (dose–response curves, SDS-PAGE full gel images, and kymographs), methods (Selection of Resistant Clones; Tubb5T238A Point Mutation; Whole Exome Sequence Analysis; and Identification of Recurrently Mutated Genes in Sulfonamide 27-Resistant Clones), and copies of 1H and 13C spectra, HPLC chromatograms and HRMS data (PDF)
Molecular formula strings (CSV)
Author Contributions
∇ J.M.P. and R.R. contributed equally to this work
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- McFadden D. G.; Papagiannakopoulos T.; Taylor-Weiner A.; Stewart C.; Carter S. L.; Cibulskis K.; Bhutkar A.; McKenna A.; Dooley A.; Vernon A.; Sougnez C.; Malstrom S.; Heimann M.; Park J.; Chen F.; Farago A. F.; Dayton T.; Shefler E.; Gabriel S.; Getz G.; Jacks T. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbo J.; Meuwissen R.; van Montfort E.; van Tellingen O.; Berns A. Genotype-phenotype relationships in a mouse model for human small-cell lung cancer. Cold Spring Harbor Symp. Quant. Biol. 2005, 70, 225–232. 10.1101/sqb.2005.70.026. [DOI] [PubMed] [Google Scholar]
- Gazdar A. F.; Savage T. K.; Johnson J. E.; Berns A.; Sage J.; Linnoila R. I.; MacPherson D.; McFadden D. G.; Farago A.; Jacks T.; Travis W. D.; Brambilla E. The comparative pathology of genetically engineered mouse models for neuroendocrine carcinomas of the lung. J Thorac Oncol 2015, 10, 553–564. 10.1097/JTO.0000000000000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R.; Linn S. C.; Linnoila R. I.; Zevenhoven J.; Mooi W. J.; Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. 10.1016/S1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Nile A. H.; Tripathi A.; Yuan P.; Mousley C. J.; Suresh S.; Wallace I. M.; Shah S. D.; Pohlhaus D. T.; Temple B.; Nislow C.; Giaever G.; Tropsha A.; Davis R. W.; St Onge R. P.; Bankaitis V. A. PITPs as targets for selectively interfering with phosphoinositide signaling in cells. Nat. Chem. Biol. 2014, 10, 76–84. 10.1038/nchembio.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The trifluoromethoxy group has been described as a leaving group in SNAr, see:; Marrec O.; Billard T.; Vors J.-P.; Pazenok S.; Langlois B. R. A New and direct trifluoromethoxylation of aliphatic substrates with 2,4-dinitro(trifluoromethoxy)benzene. Adv. Synth. Catal. 2010, 352, 2831–2837. 10.1002/adsc.201000488. [DOI] [Google Scholar]
- Theodoropoulos P. C.; Wang W.; Budhipramono A.; Thompson B. M.; Madhusudhan N.; Mitsche M. A.; McDonald J. G.; De Brabander J. K.; Nijhawan D. A medicinal chemistry-driven approach identified the sterol isomerase EBP as the molecular target of TASIN colorectal cancer toxins. J. Am. Chem. Soc. 2020, 142, 6128–6138. 10.1021/jacs.9b13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A. E.; Cravatt B. F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11, 535–546. 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Wright A. T.; Cravatt B. F. Chemical proteomic probes for profiling cytochrome p450 activities and drug interactions in vivo. Chem. Biol. 2007, 14, 1043–1051. 10.1016/j.chembiol.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.; Elwood F.; McNally J.; Weiszmann J.; Lindstrom M.; Amaral K.; Nakamura M.; Miao S.; Cao P.; Learned R. M.; Chen J. L.; Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem 2002, 277, 19649–19657. 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- Harris G.; Schaefer K. L. The microtubule-targeting agent T0070907 induces proteasomal degradation of tubulin. Biochem. Biophys. Res. Commun. 2009, 388, 345–349. 10.1016/j.bbrc.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Schaefer K. L.; Takahashi H.; Morales V. M.; Harris G.; Barton S.; Osawa E.; Nakajima A.; Saubermann L. J. PPARgamma inhibitors reduce tubulin protein levels by a PPARgamma, PPARdelta and proteasome-independent mechanism, resulting in cell cycle arrest, apoptosis and reduced metastasis of colorectal carcinoma cells. Int. J. Cancer 2007, 120, 702–713. 10.1002/ijc.22361. [DOI] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Yan W.; Li W.; Qiu Q.; Ye H.; Chen L. Covalent modification of Cys-239 in β-tubulin by small molecules as a strategy to promote tubulin heterodimer degradation. J Biol Chem 2019, 294, 8161–8170. 10.1074/jbc.RA118.006325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankmoelle W. P.; Medina J. C.; Shan B.; Narbut M. R.; Beckmann H. Glutathione S-transferase metabolism of the antineoplastic pentafluorophenylsulfonamide in tissue culture and mice. Drug Metab. Dispos. 2000, 28, 951–958. [PubMed] [Google Scholar]
- Shan B.; Medina J. C.; Santha E.; Frankmoelle W. P.; Chou T.-C.; Learned R. M.; Narbut M. R.; Stott D.; Wu P.; Jaen J. C.; Rosen T.; Timmermans P. B. M. W. M.; Beckmann H. Selective, covalent modification of β-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5686–5691. 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R.; Covell D. G.; Pei X.-F.; Ewell J. B.; Nguyen N. Y.; Brossi A.; Hamel E. Mapping the binding site of colchicinoids on β-tubulin. 2-Chloroacetyl-2-demethylthiocolchicine covalently reacts predominantly with cysteine 239 and secondarily with cysteine 354. J Biol Chem 2000, 275, 40443–40452. 10.1074/jbc.M005299200. [DOI] [PubMed] [Google Scholar]
- Steinmetz M. O.; Prota A. E. Microtubule-targeting agents: strategies to hijack the cytoskeleton. Trends Cell Biol 2018, 28, 776–792. 10.1016/j.tcb.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Brouhard G. J.; Rice L. M. Microtubule dynamics: an interplay of biochemistry and mechanics. Nat Rev Mol Cell Biol 2018, 19, 451–463. 10.1038/s41580-018-0009-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010, 9, 790–803. 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlini C.; Raspaglio G.; Mozzetti S.; Cicchillitti L.; Filippetti F.; Gallo D.; Fattorusso C.; Campiani G.; Scambia G. The seco-taxane IDN5390 is able to target class III β-tubulin and to overcome paclitaxel resistance. Cancer Res. 2005, 65, 2397–2405. 10.1158/0008-5472.CAN-04-3065. [DOI] [PubMed] [Google Scholar]
- Mozzetti S.; Ferlini C.; Concolino P.; Filippetti F.; Raspaglio G.; Prislei S.; Gallo D.; Martinelli E.; Ranelletti F. O.; Ferrandina G.; Scambia G. Class III β-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res 2005, 11, 298–305. [PubMed] [Google Scholar]
- Arnst K. E.; Banerjee S.; Chen H.; Deng S.; Hwang D. J.; Li W.; Miller D. D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. 10.1002/med.21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X.; Kim T.; Geyer E. A.; Rice L. M. Insights into allosteric control of microtubule dynamics from a buried β-tubulin mutation that causes faster growth and slower shrinkage. Protein Sci. 2020, 29, 1429–1439. 10.1002/pro.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M.; Chen Y.; Gilbert L. A.; Horlbeck M. A.; Krenning L.; Menchon G.; Rai A.; Cho M. Y.; Stern J. J.; Prota A. E.; Kampmann M.; Akhmanova A.; Steinmetz M. O.; Tanenbaum M. E.; Weissman J. S. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol. Cell 2017, 68, 210–223.e6. e6 10.1016/j.molcel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yan W.; Li Y.; Niu L.; Ye H.; Chen L. The natural compound withaferin A covalently binds to Cys239 of β-tubulin to promote tubulin degradation. Mol. Pharmacol. 2019, 96, 711–719. 10.1124/mol.119.117812. [DOI] [PubMed] [Google Scholar]
- Povedano J. M.; Liou J.; Wei D.; Srivatsav A.; Kim J.; Xie Y.; McFadden D. G. Engineering forward genetics into cultured cancer cells for chemical target identification. Cell Chem. Biol. 2019, 1315–1321.e3. 10.1016/j.chembiol.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machin N. A.; Lee J. M.; Barnes G. Microtubule stability in budding yeast: characterization and dosage suppression of a benomyl-dependent tubulin mutant. Mol. Biol. Cell 1995, 6, 1241–1259. 10.1091/mbc.6.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. H.; Neff N. F.; Botstein D. Isolation and characterization of mutations in the β-tubulin gene of Saccharomyces cerevisiae. Genetics 1985, 111, 715–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhusudhan N.; Hu B.; Mishra P.; Calva-Moreno J. F.; Patel K.; Boriack R.; Ready J. M.; Nijhawan D. Target discovery of selective non-small-cell lung cancer toxins reveals inhibitors of mitochondrial complex I. ACS Chem. Biol. 2020, 15, 158–170. 10.1021/acschembio.9b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer E. A.; Burns A.; Lalonde B. A.; Ye X.; Piedra F. A.; Huffaker T. C.; Rice L. M. A mutation uncouples the tubulin conformational and GTPase cycles, revealing allosteric control of microtubule dynamics. Elife 2015, 4, e10113 10.7554/eLife.10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.; Rice L. M. Long-range, through-lattice coupling improves predictions of microtubule catastrophe. Mol. Biol. Cell 2019, 30, 1451–1462. 10.1091/mbc.E18-10-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S.; Kim T.; Chen Z.; Munyoki S.; Tso S. C.; Brautigam C. A.; Rice L. M. An isolated CLASP TOG domain suppresses microtubule catastrophe and promotes rescue. Mol. Biol. Cell 2018, 29, 1359–1375. 10.1091/mbc.E17-12-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A.; Amodaj N.; Hoover K.; Vale R.; Stuurman N. Computer control of microscopes using μManager. Curr Protoc Mol Biol 2010, 92, 14.20.1–14.20.17. 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.