

Abstract

Dearomative functionalization of heteroaromatics, a readily available chemical feedstock, is one of the most straightforward approaches for the synthesis of three-dimensional, chiral heterocyclic systems, important synthetic building blocks for both synthetic chemistry and drug discovery. Despite significant efforts, direct nucleophilic additions to heteroaromatics have remained challenging because of the low reactivity of aromatic substrates associated with the loss of aromaticity, as well the regio- and stereoselectivities of the reaction. Here we present a catalytic system that leads to unprecedented, high-yielding dearomative C-4 functionalization of quinolines with organometallics with nearly absolute regio- and stereoselectivities and with a catalyst turnover number (TON) as high as 1000. The synergistic action of the chiral copper catalyst, Lewis acid, and Grignard reagents allows us to overcome the energetic barrier of the dearomatization process and leads to chiral products with selectivities reaching 99% in most cases. Molecular modeling provides important insights into the speciation and the origin of the regio- and enantioselectivity of the catalytic process. The results reveal that the role of the Lewis acid is not only to activate the substrate toward a potential nucleophilic addition but also to subtly control the regiochemistry by preventing the C-2 addition from happening.
Introduction
Optically active, nitrogen-containing heterocycles are important structural constructs found in a myriad of natural products, biologically active molecules, and pharmaceutically active ingredients.1,2 It has been shown that complex three-dimensional chiral molecules are more likely to show useful bioactive properties than achiral, flat aromatic compounds, because of improved interactions with the target proteins.3 The chiral hydroquinoline motif is a particularly relevant heterocyclic system, as it is present in many natural and unnatural compounds with interesting biological properties, and therefore, their synthesis is of great interest for synthetic chemistry, the agrochemical industry, and for drug discovery (Scheme 1a).1,2 As a result, both noncatalytic and catalytic strategies have been described for the synthesis of chiral hydroquinoline derivatives.2
Scheme 1. Importance of Chiral Tetrahydroquinoline Derivatives with Methods for Their Asymmetric Synthesis and Current Work: (a) Natural Products and Drugs Incorporating Chiral Tetrahydroquinolines; (b) State of the Art in Nucleophilic Additions to Quinoline and Quinolinium Salts; (c) Main Idea Behind the Current Work.

Nucleophilic dearomative addition to nitrogen-containing heteroaromatics is arguably one of the most straightforward approaches to synthesize chiral azaheterocycles.4 However, direct nucleophilic addition to heteroaromatic compounds is challenging: their reactivity is low because the addition is only possible at the cost of loss of aromaticity.5 The most common strategy to circumvent this reactivity issue consists of activating the heteroaromatic ring with a reactive electrophile to form a heteroarenium salt, followed by a nucleophilic attack. This approach has also been applied to quinolines.5,6 In nucleophilic dearomative additions to quinolines and quinolinium salts, apart from reactivity issues, enantioselectivity and regiocontrol are additional hurdles, since there are two electrophilic positions that can undergo nucleophilic attack, namely, the C-2 and C-4 position (Scheme 1b).6a−6g Interestingly, analysis of the literature reveals not only that all successful strategies for nucleophilic additions to quinoline derivatives require quinolinium salts but also that the overwhelming majority of both racemic and enantioselective methods feature a nucleophilic attack at the C-2 position.6g,6h,7 The only reported example of direct asymmetric addition to quinolines to date takes advantage of the high reactivity of organolithium reagents but provides the C-2-addition products with a modest enantioselectivity (58–79%) while requiring a large (20–100 mol %) loading of a chiral ligand (Scheme 1b).6h,7 Moreover, there are no precedents of catalytic asymmetric additions to the C-4 position of both quinolines or quinolinium salts. Only diastereoselective C-4 addition to quinolines has been reported, but these methodologies require chiral auxiliaries or preformed chiral nucleophiles and suffer from severe scope limitation.8
Realizing that the N–C-2–C-4 fragment of quinoline can be viewed as a masked α,β-unsaturated conjugated imine moiety, we envisioned a possible strategy for the enantioselective dearomative C-4 functionalization of quinolines (Scheme 1c). Rather than relying on premade quinolinium salts, a suitable Lewis acid (LA) could act as a transient activator of the quinoline ring toward nucleophilic addition.9 A catalyst can then be used to direct the nucleophile to the required C-4 position with a high level of enantiodiscrimination, as the C-3–C-4 double bond should be susceptible to copper-catalyzed conjugate addition of organometallics, a classic method for asymmetric conjugate additions to α,β-unsaturated carbonyl compounds.10 Thus, it should be possible to use copper(I)-based catalysts to direct the addition of organometallics toward the C-4 position. We anticipated that the unique reactivity provided by the combination of LA, copper(I) catalysis, and Grignard reagents previously demonstrated by our group11 would be able to overcome the energetic barrier of nucleophilic dearomative addition to quinolines.
Herein, we present the first catalytic system that allows highly selective catalytic enantioselective nucleophilic dearomatization of quinolines with organometallics (Scheme 1c). The synergistic combination of LA, chiral copper(I) catalyst, and Grignard reagents allows access to the C-4-addition products with nearly absolute regio- and stereocontrol. Remarkably, the amount of chiral copper catalyst can be reduced to 0.1 mol % in a preparative-scale reaction, representing the highest turnover number (TON) reported among any enantioselective reactions involving Grignard reagents. Comprehensive speciation and conformational analysis using molecular modeling provides insights into the role of the LA and the origin of the regio- and stereoselectivity.
Results and Discussion
Our investigation of the enantioselective dearomatization of quinolines began with the model reaction of quinoline 1a with EtMgBr in CH2Cl2 at −78 °C (Table 1) quenched with acetyl chloride upon reaction completion. The two anticipated regioisomeric products under these reaction conditions are products 2a and 3a resulting from nucleophilic attack of EtMgBr at either the C-2 or C-4 position, respectively, but neither was observed in the absence of any metal catalyst nor LA (Table 1, entry 1). To boost the reactivity of quinoline 1a we added the highly reactive LA Me3SiOTf, which resulted in a significant reaction rate acceleration and nearly full conversion of the substrate 1a. Analysis of the reaction crude revealed that the major product of the reaction derives from the nucleophilic attack at the C-2 position (Table 1, entry 2). Anticipating that Cu(I) salt may recognize the α,β-unsaturated conjugated imine motif in the quinoline molecule, we also carried out the addition of EtMgBr in the presence of Cu(I) salt and Cu(I) complex with chiral ligand L1, but no conversion to either product 2a or 3a was observed (Table 1, entry 3). Combining the chiral copper catalyst system L1/Cu(I) with Me3SiOTf resulted, similarly as when adding the LA alone, in an immense acceleration of the addition reaction, but now with improved selectivity toward the C-4-addition product, providing products 2a and 3a in a ratio of 79:18 (Table 1, entry 4).
Table 1. Development of the Catalytic System for Dearomative C-4-Selective Functionalization of Quinolinea.

| entry | L | Cu(I) | LA | solvent | 1a:2a:3ab | ee (2a) [%]c | ee (3a) [%]c |
|---|---|---|---|---|---|---|---|
| 1d | CH2Cl2 | 100:0:0 | |||||
| 2 | Me3SiOTf | CH2Cl2 | 1:93:6 | ||||
| 3 | L1 | CuBr·SMe2 | CH2Cl2 | 100:0:0 | |||
| 4 | L1 | CuBr·SMe2 | Me3SiOTf | CH2Cl2 | 3:79:18 | 7 | 77 |
| 5 | L2 | CuBr·SMe2 | Me3SiOTf | CH2Cl2 | 0:87:13 | rac | rac |
| 6 | L3 | CuBr·SMe2 | Me3SiOTf | CH2Cl2 | 0:81:19 | rac | rac |
| 7 | L4 | CuBr·SMe2 | Me3SiOTf | CH2Cl2 | 0:88:12 | 7 | 45 |
| 8 | L1 | CuTc | Me3SiOTf | CH2Cl2 | 0:67:33 | 4 | 83 |
| 9 | L1 | CuTc | Et3SiOTf | CH2Cl2 | 36:19:45 | 6 | 32 |
| 10 | L1 | CuTc | t-BuMe2SiOTf | CH2Cl2 | 85:2:13 | 68 | |
| 11 | L1 | CuTc | i-Pr3SiOTf | CH2Cl2 | 87:9:4 | ||
| 12 | L1 | CuTc | t-BuPh2SiOTf | CH2Cl2 | 78:2:20 | 80 | |
| 13e | BF3·Et2O | CH2Cl2 | 80:14:6 | ||||
| 14e,f,g | L1 | CuTc | BF3·Et2O | CH2Cl2 | 1:0:99 | >99 | |
| 15e | L4 | CuTc | BF3·Et2O | CH2Cl2 | 26:5:69 | 88 | |
| 16e | L1 | CuTc | BF3·Et2O | THF | 12:81:7 | 42 | |
| 17e | L1 | CuTc | BF3·Et2O | 2-Me-THF | 1:0:99 | >99 | |
| 18e,h | L1 | CuTc | BF3·Et2O | t-BuOMe | 2:0:98 | >99 | |
| 19e,h | L1 | CuTc | BF3·Et2O | toluene | 1:0:99 | >99 | |
| 20e,h | L1 | CuTc | BF3·Et2O | Et2O | 2:0:98 | >99 |
Reaction conditions: 0.1 M 1a, Cu(I) (5 mol %), L (6 mol %), LA (2 equiv), EtMgBr (2 equiv) at −78 °C for 16 h, and then ClCOMe (5 equiv) at RT for 2 h.
The ratio was determined by 1H NMR of reaction crude.
Enantiomeric excess was determined by HPLC on a chiral stationary phase.
In this case the reaction was quenched directly without trapping with ClCOMe.
In this case THF (4 mL) was added after addition of ClCOMe: when using BF3·Et2O trapping was only successful in THF.
A 58% isolated yield of 3a and a complex mixture of byproducts was obtained.
Similar results were obtained when using CuBr·SMe2 instead of CuTc (see the Supporting Information).
In this case 3 equiv of LA and 3 equiv of EtMgBr were used.
Although still the minor product, the chiral C-4-addition product 3a was obtained with 77% enantiomeric excess (ee), while in contrast, the C-2-addition product 2a was obtained with only 7% ee. This result is crucial, since it indicates that the C-4 addition is mainly due to the catalytic reaction, while the C-2 addition is mainly produced by the noncatalyzed background reaction initiated by Me3SiOTf. This gave us confidence that further optimization of the catalyst and/or reaction conditions toward the catalytic C-4-addition pathway could lead to improved enantio- and regioselectivity. Bearing this in mind, we kicked off the optimization process by exploring the reactivity of several chiral ligands, L2–L4, in the presence of Me3SiOTf (Table 1, entries 5–7). These reactions afforded mainly the C-2-addition product 2a as well as decreased enantioselectivity, leading us to conclude that ligand L1 is an outstanding choice for achieving high enantioselectivity. Further screening of Cu(I) salts revealed that slightly higher C-4 regioselectivity and enantioselectivity are obtained when using copper(I) thiophene-2-carboxylate (CuTc) as the copper source instead of CuBr·SMe2 (Table 1, entry 8). Adopting ligand L1 in combination with CuTc as the optimal catalyst, we continued with screening of LAs. While Me3SiOTf activates quinoline toward both the C-2 and C-4 attacks, we hypothesized that increasing the steric bulk of the LA might reduce the background C-2 attack and direct EtMgBr toward C-4 addition. Testing several silyl-based LAs, all with TfO– as the counterion but with different degrees of steric hindrance (Table 1, entries 9–12), we observed that the catalytic system is very sensitive to this feature of the LA. When bulky LAs are used, the C-4 attack is the main reaction pathway, albeit at the cost of dramatically decreased reactivity and enantioselectivity. At this point we decided to switch from silicon-based LAs to the slightly less reactive boron-based LA BF3·Et2O, hoping that the diminished activation of the quinoline ring toward nucleophilic attack would allow the Cu(I) catalyst to outcompete the noncatalytic C-2 attack. Using BF3·Et2O as the LA in the reaction without a copper catalyst (Table 1, entry 13), only 20% of the quinoline was converted to the mixture of C-2- (major product) and C-4-addition products, with 80% of the substrate remaining. Much to our delight, when combining BF3·Et2O with the L1/Cu(I) complex, the reaction proceeded with absolute C-4 regioselectivity, providing the corresponding product 3a with 58% isolated yield and more than 99% ee (Table 1, entry 14).
When BF3·Et2O was used together with L4/Cu(I), excellent regioselectivity was obtained as well, with 88% ee, but much lower conversion (Table 1, entry 15). Settling on BF3·Et2O and L1/Cu(I) as the best LA and catalyst system we turned to the screening of various solvents (Table 1, entries 16–20). Remarkably, except for THF, all the solvents tested, including 2-methyltetrahydrofuran (2-Me-THF), t-BuOMe, toluene, and Et2O, afforded absolute regioselectivity toward the desired addition product 3a with almost full conversion and over 99% ee. However, the NMR yields are lower than with CH2Cl2 (see the Supporting Information), which is therefore the solvent of choice.
Having established the optimal reaction conditions for this dearomatization protocol, we addressed the problems associated with the isolated yields of the addition product 3a. Because of the instability of the addition product, only 58% isolated yield of 3a with a complex mixture of byproducts was obtained, despite achieving nearly full conversion of the substrate (Table 1, entry 14). Continued attempts to optimize the reaction to eliminate the byproducts did not lead to an improved isolated yield of 3a. Thus, we decided to change the isolation strategy and instead of trapping the addition product with acetyl chloride we attempted to reduce the addition intermediate with BH3·THF to form the tetrahydroquinoline 4a (Scheme 2).
Scheme 2. Scope of Grignard Reagents and Substrates.
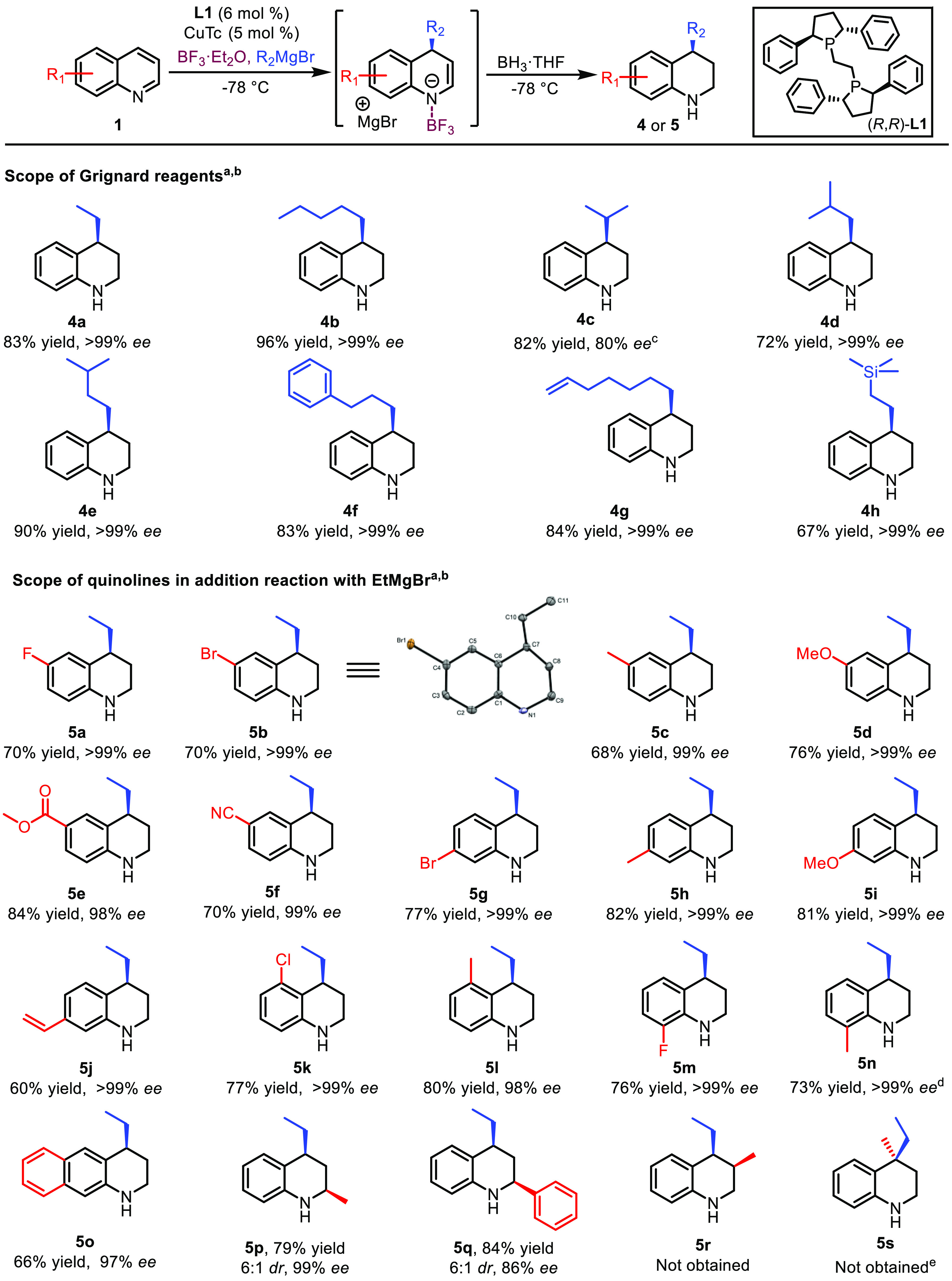
Only C-4-addition products are formed. The isolated yields of C-4-addition products are shown. The absolute configuration of 5b was determined by single-crystal X-ray crystallography, and the relative configuration of 5q was determined by NOE experiments. The absolute configurations of other compounds were assigned by analogy (for details, see the Supporting Information).
Reaction conditions: 0.1 M 1 in CH2Cl2, CuTc (5 mol %), L1 (6 mol %), BF3·Et2O (1.2–3 equiv), RMgBr (2–3 equiv) at −78 °C for 2–16 h (depending on the substrate and the Grignard), and then BH3·THF (5 equiv) at −78 °C for 16 h.
In this case 10 mol % of L1 and 12 mol % of CuTc were used.
In this case the regioselectivity C-2/C-4 is 1/5.
In this case 15% conversion toward the C-2-addition product was observed.
This strategy worked well at −78 °C, avoiding the formation of byproducts and providing an 83% isolated yield, thus establishing this final optimized strategy: BF3·Et2O is used as LA in the presence of 6 mol % of chiral ligand L1 and 5 mol % of CuTc, with CH2Cl2 as solvent at −78 °C, and after addition of Grignard reagents, BH3·THF is used to reduce the dearomatized intermediate.
With the optimized conditions in hand, we started to explore the scope of this transformation, first concentrating on the addition of various types of Grignard reagents (Scheme 2). We were delighted to find that, when quinoline 1a is used as the substrate, excellent results are obtained with both linear and sterically demanding α-, β-, and γ-branched Grignard reagents (4a–4e). Grignard reagents containing a terminal phenyl, a terminal double bond, or a silyl functional group are also well-tolerated, providing the corresponding products 4f, 4g, and 4h with high yields and over 99% ee. It is remarkable that, except for the C-4 product obtained with i-PrMgBr (80% enantiomeric excess), the addition of all the tested Grignard reagents to 1a led exclusively to the C-4-addition products with over 99% ee, proving that our catalytic system has excellent control over the reactivity as well as the regio- and enantioselectivity. Subsequently, we explored the scope of the substrates, focusing on investigating the effect of different substitutions on the quinoline upon addition with EtMgBr.
Evaluating the effect of substituents at the C-6 position, we found that addition to substrates bearing either an electron-withdrawing group (fluoro and bromo) or an electron-donating group (methyl and methoxy) leads to the corresponding C-4-addition products 5a–5d with excellent results. The ester and cyano functional groups at the C-6 position are also tolerated, generating products 5e and 5f with excellent yields and ee, indicating that this catalytic system can efficiently control the regioselectivity to avoid C-2 addition as well as chemoselectivity to avoid addition to the functional groups. C-4-addition products 5g–5j, derived from the addition of EtMgBr to substrates bearing bromo-, methyl-, methoxy-, and vinyl-substituents at the C-7 position, respectively, were obtained with high yields and enantiopurities over 99%.
Our catalytic system can even tolerate the presence of substituents at the sterically demanding 5- and 8-positions, providing the corresponding addition products 5k–5n with high yields and excellent ee. Also, an example of a 6,7-disubstituted quinoline, bearing a conjugated phenyl ring, was successfully subjected to our catalytic system, leading to the formation of 1,4-addition product 5o with excellent regio- and enantioselectivity. Significantly, quinolines bearing substituents at the C-2 position are also excellent substrates for this reaction, allowing access to tetrahydroquinolines 5p and 5q with multiple stereocenters with 6:1 diastereoselectivity and high enantiomeric excess. Contrary to these findings, no conversion was observed with C-3- and C-4-substituted quinolines. This kind of sensitivity to the substituents aligns with our observations in copper-catalyzed additions to α,β-unsaturated amides and carboxylic acids, where it was shown that additional substitutions in the α- or β-positions with respect to the electron-withdrawing group diminishes the reactivity of the substrates drastically and prevents copper catalysis from operating.11
It is also important to note that the reaction rate decreases with increased steric hindrance of either the substrate or the Grignard reagent, as well as with enhanced electron density of the substrate. For these reasons, increased amounts of Grignard reagent and BF3·Et2O (3 equiv each) are required to reach full conversion toward products 4b, 4d–4h, 5d, 5i, 5l, 5n, 5o, and 5q. When using an aryl Grignard reagent, namely, PhMgBr, no conversion to the addition product was observed, even in the presence of a large excess of the corresponding Grignard reagent and BF3·Et2O. Conversely, for substrate 5e, the amount of LA had to be reduced from 2 to 1.2 equiv to avoid possible binding to the ester group and thus to ensure that the addition occurs selectively at the quinoline ring. For substrate 5j, the amount of LA was also reduced to 1.2 equiv in order to reduce the formation of byproducts.
To showcase the efficacy of our catalytic protocol, we determined the reaction time needed to achieve full conversion of the substrate and studied the effect of the catalyst loading on the reaction outcome. These studies were performed on the model reaction of addition of EtMgBr to quinoline 1a in the presence of the catalyst L1/Cu(I) and BF3·Et2O, followed by trapping with acetyl chloride (Table 2).
Table 2. Effect of the Catalyst Loading on the Reaction Outcomea.

| entry | L1/Cu(I) [mol %] | 1a:2a:3ab | ee (3a) [%]c |
|---|---|---|---|
| 1 | 5 | 1:0:99 | >99 |
| 2d | 5 | 6:0:94 | >99 |
| 3 | 1 | 5:0:95 | >99 |
| 4 | 0.1 | 5:0:95 | 99 |
| 5 | 0.01 | 58:19:23 | 40 |
Reaction conditions: 0.1 M 1a in CH2Cl2, BF3·Et2O (2 equiv), EtMgBr (2 equiv) at −78 °C for 16 h, and then THF (4 mL), ClCOMe (5 equiv) at RT for 2 h.
The ratio was determined by 1H NMR of reaction crude.
Enantiomeric excess was determined by HPLC on a chiral stationary phase.
The reaction time of addition step is 10 min.
First, we tested the reaction with 5 mol % of catalyst loading for a reaction time of 10 min and found that product 3a was obtained with 94% conversion and over 99% ee (Table 2, entry 2). We were also pleasantly surprised by the fact that reducing the catalyst loading from 5 to 1 mol %, and even further to 0.1 mol %, did not affect the reaction outcome, still promoting the conversion of the substrate to the C-4-addition product with nearly full conversion and 99% enantiopurity (Table 2, entries 3 and 4). These results are remarkable when compared to other catalytic enantioselective methods using Grignard reagents: a TON of 1000 represents the highest TON among all the catalytic asymmetric reactions performed with Grignard reagents reported today. We also tested the scaling up of our catalytic system by performing the addition reaction of EtMgBr to 1.3 g of quinoline 1a (a 20-fold scale-up) with 0.1 mol % of L1/Cu(I), followed by reduction with BH3·THF. The corresponding product 4a, which is a key intermediate for the synthesis of drug candidate (S)-LG121071, a selective androgen receptor modulator (SARM),12 was obtained with 71% yield and 96% ee (Scheme 3).
Scheme 3. Twenty-Fold Scale-Up Synthesis of Product 4a, a Key Intermediate for Preparation of Drug Candidate (S)-LG121071.

The mechanism of this transformation might follow a pathway analogous to the one proposed for the copper-catalyzed addition of organocuprates to α,β-unsaturated carbonyl compounds.13 However, since in our system the α,β-unsaturated imine moiety is part of an aromatic ring, a LA and a copper complex (L1/Cu) are needed to activate the substrate and overcome the high energy barrier induced by the dearomatization during the addition reaction. The number of reagents involved, and the fact that this allows complete enantio- and regiocontrol, implies a complex mechanistic picture in which subtle details control the reaction outcome. To gain insight into the reaction mechanism and specifically the role of LA and the L1/Cu(I) catalyst system on the reactivity and selectivity (regio- and stereoselectivities) we have carried out thorough molecular modeling studies (Figures 1 and 2). For the modeling we adopted the quinoline (1a)/BF3/L1–CuBr/EtMgBr reaction system.
Figure 1.
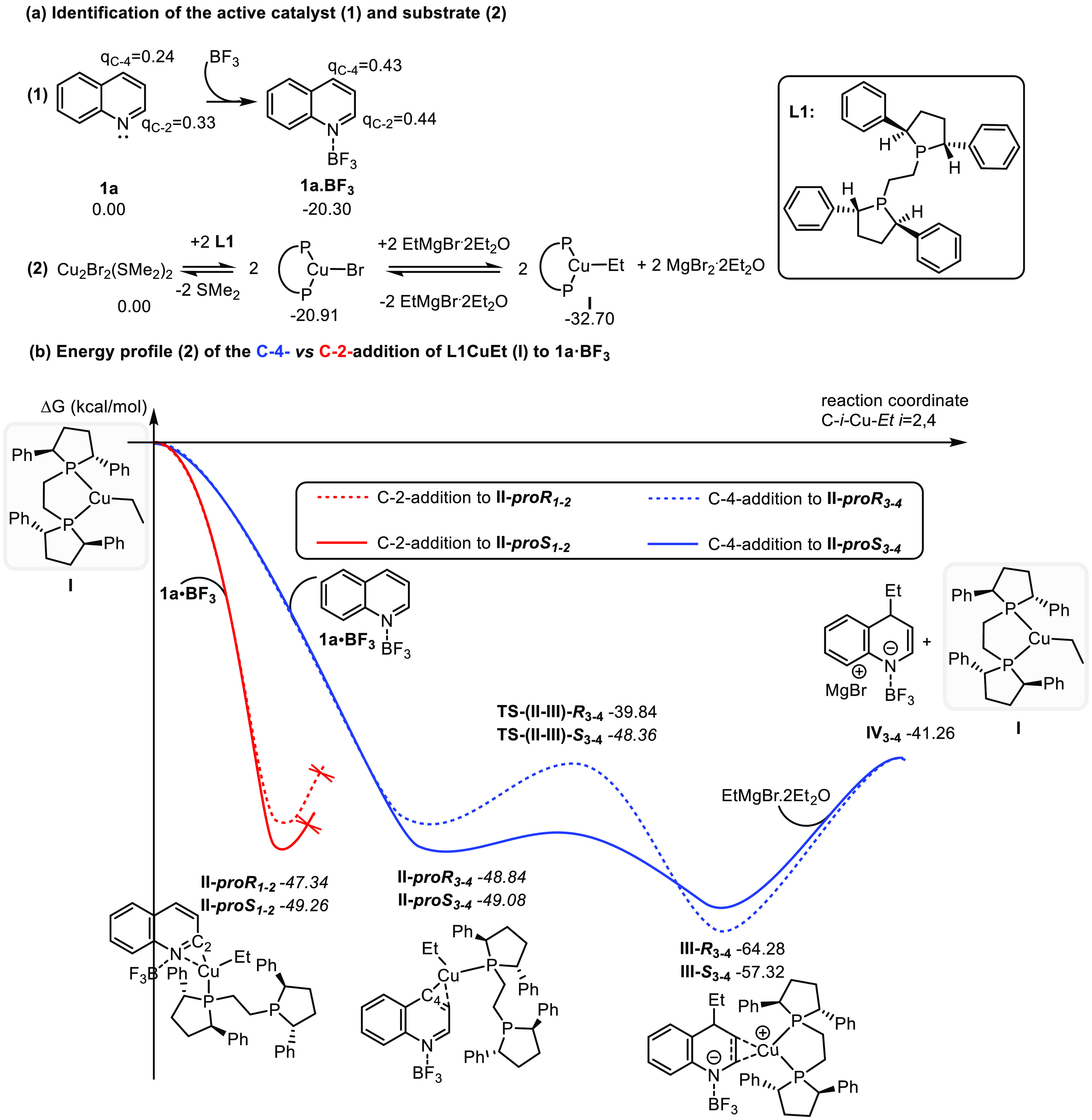
DFT mechanistic studies for the copper-catalyzed asymmetric C-4 addition of EtMgBr to quinoline. (a) Identification of the active forms of the quinoline (1) and the catalyst (2) in the reaction media. The ligand L1 is represented by an arc at the proposed equilibriums for simplicity. (b) Energy profile for the copper-catalyzed asymmetric C-2 and C-4 addition of EtMgBr to 1a–BF3. Calculations were performed at the PCM14 (CH2Cl2)/M0615/def2svpp16 computational level using the Gaussian 09 program.17 The thermochemistry was obtained at 1 atm and 195 K. Red lines mark the C-2-addition path, while blue lines indicate the C-4-addition path. The depicted charges correspond to APT charges with hydrogens summed into heavy atoms. The reported relative energies are computed wrt 1a/BF3/2xEtMgBr–2Et2O/I and expressed in kcal/mol.
Figure 2.
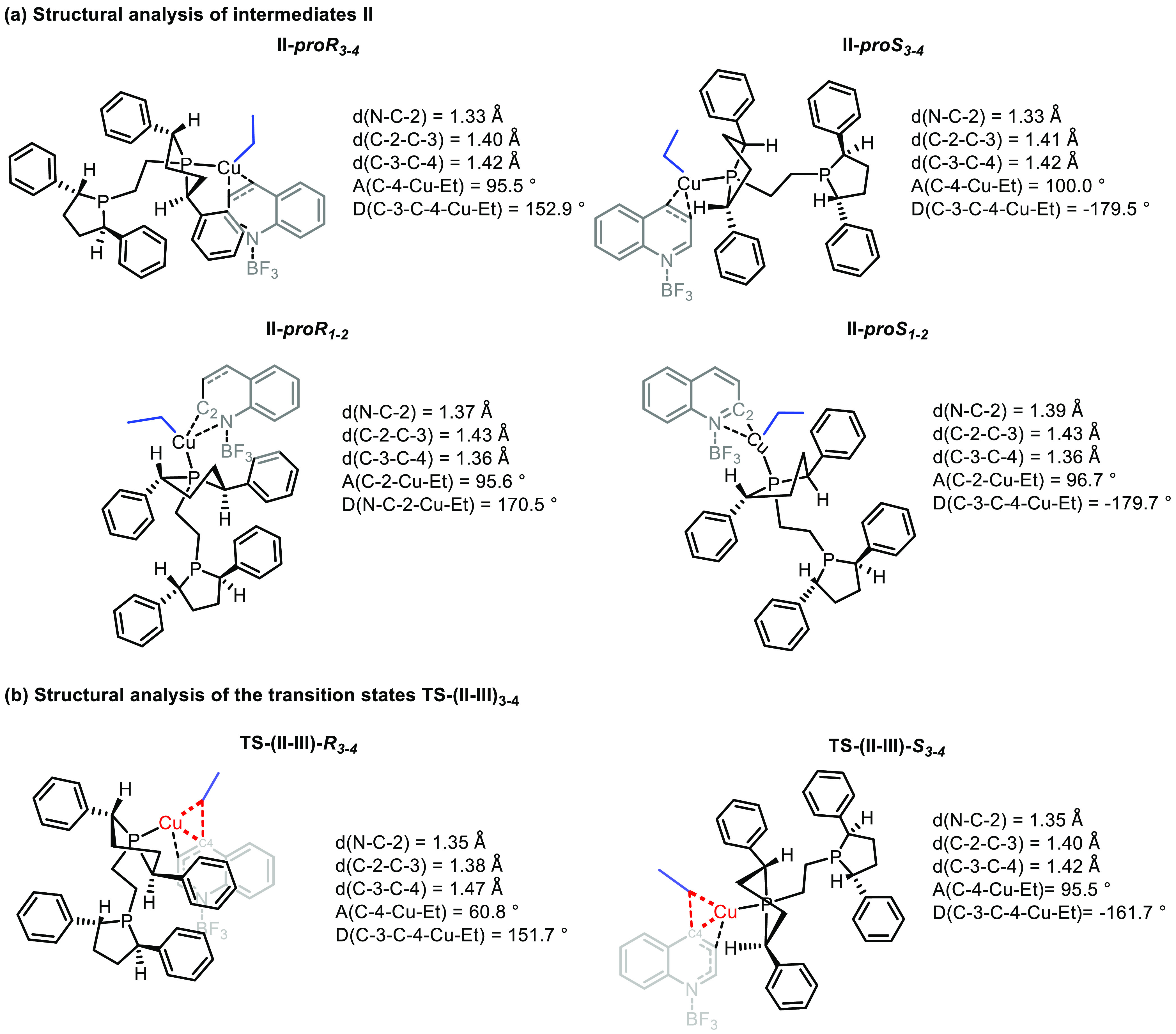
Structural analysis and 3D representation of relevant intermediates. (a) Intermediates II. (b) Transition states TS-(II–III)3–4. The copper center and the bonds participating in the reaction coordinate (decreasing of C-4–Cu–Et angle) are marked in red. For visualization purposes we have depicted the ligand in black, the substrate in gray (the substrate is always oriented in the plane of the paper), and the ethyl group being transferred in blue. The letters “d”, “A”, and “D” are used to indicate bond distances in angstroms (Å) and angles and dihedral angles in degrees (deg), respectively.
Upon addition of BF3 to quinoline 1a a new complex 1a–BF3 is formed in a highly exergonic process (ΔG = −20.3 kcal/mol). This complexation induces a change of charges at carbons C-2 and C-4 from 0.33 and 0.24 au in 1a to 0.44 and 0.43 au in 1a–BF3, confirming significant activation of quinoline toward nucleophilic additions but also explaining why the addition of Grignard reagent to 1a–BF3 in the absence of copper complex catalyst is not regioselective: the charges on both carbons are rather similar (see Figure 1a and Table 1, entry 13).
A systematic density functional theory (DFT) analysis on the relative stability of the copper species that might be present in the reaction media revealed that, once the copper source and L1 can interact, they will form the complex L1–CuBr (ΔG = −20.91 kcal/mol). At the same time, the large excess of Grignard reagent wrt L1–CuBr leads us to propose that direct transmetalation of L1–CuBr will afford the organcopper species I, which is thermodynamically preferred (−32.70 kcal/mol) and also the starting point for the catalytic cycle (see Figure 1a and the Supporting Information). Having identified the active forms of the substrate (1a–BF3) and the catalyst (I), we explored their interaction: species I can approach the LA-activated substrate 1a–BF3 to form either of species II depending on the regioselectivity (C-2 vs C-4) of the reaction. Once species II has been formed, it evolves further via the transfer of the Et group from the copper center to either the C-2 or C-4 position of quinoline to form the respective species III. This step is followed by the addition of the second molecule of EtMgBr to the copper center of species III, thus promoting the release of either C-4- or C-2-addition product intermediate IV and the recovery of the organocopper species I. Our modeling studies described below provide full rationalization for the experimentally observed C-4 selectivity and the stereochemistry of the process.
In the first step of the catalytic cycle species I can, a priori, bind to either of the two faces of 1a–BF3 and either to the N–C-2 or the C-3–C-4 region, thus leading to two sets of possible diastereomeric complexes (II-proR1–2, II-proS1–2, II-proR3–4, II-proS3–4), all of which are thermodynamically favorable and very close in energy (from −47.34 to −49.26 kcal/mol); see Figure 1b.
A detailed structural analysis of these four complexes reveals that the dangling uncoordinated branch in the ligand in species II3–4 is oriented toward the outside of the molecule, preventing destabilizing steric interactions with the substrate while also inhibiting a differentiation between both diastereomers. Alternatively, in species II1–2 the ligand is located on top of the molecule (toward the LA) but too far from the quinoline for an energetic differentiation (Figure 2). Interestingly, we found that, in order for the complexes II to be formed, one of the hemilabile phosphorus atoms of the ligand has to abandon the first-coordination sphere of the copper center so that a new coordination vacancy is created and the quinoline molecule can be accommodated. Once the complex II is formed, the evolution of the resulting diastereomeric pairs via the transfer of the Et group from the copper center to either the C-2 or C-4 position of quinoline to form species III differs in terms of energy.
Turning first to the C-4 pathway with species II3–4: the evolution of II-proR3–4 involves an energy barrier of 9.0 kcal/mol, while the analogous evolution of II-proS3–4 is only 0.7 kcal/mol. Thus, the formation of species III constitutes the enantiodetermining step. The difference in energy between the two diastereoisomers can be rationalized by analyzing the transition state structures TS-(II–III)-R3–4 and TS-(II–III)-S3–4. The delivery of the ethyl group in TS-(II–III)3–4 requires a decrease of the C-4–Cu–Et angle due to the approach of the ligand moiety to the quinoline part. In the case of TS-(II–III)-R3–4, a phenyl group in the ligand moiety is pointing to the quinoline; thus, the decrease of the C-4–Cu–Et angle results in a slight energy penalization due to the clash between the phenyl group and the quinoline in the transition state. In the TS-(II–III)-S3–4 the phenyl group is pointing away from the quinoline, and thus the resulting lower steric hindrance makes this transition state energetically much more favorable. On the other hand, our attempts to localize the transition states for the C-2-addition case (from II1–2 to III1–2) resulted surprisingly in the release of organocopper species I from II1–2.
Intrigued by these results we studied the evolution of the energy of II-proR1–2 with the variation of the reaction coordinate (decrease of the C-2–Cu–Et angle) and found that this movement entails the separation of the copper from the quinoline and results in a weak complex between I and 1a–BF3. In this complex the copper is already located in the C-3–C-4 region, thus predisposing the system to evolve via the C-4 addition. The main difference between the evolution of II3–4 via C-4 addition and II1–2 via C-2 addition resides in the ability of the environment to stabilize the transient structures during the reorganization of copper(I) species while the ethyl group moves from the copper center to the C-2 or C-4 position. In the case of the C-4 addition the decrease of the C-4–Cu–Et angle is accompanied by an incipient stabilizing interaction of the copper with the C-2–C-3 bond, whereas this interaction is not possible when the C-2–Cu–Et angle is decreased. On the contrary, the LA prevents the copper center from interacting with the nitrogen for possible stabilization, thus resulting in the release of the organocopper species I (for details see the Supporting Information).
Thus, our molecular modeling has shown that the regio- and the enantioselectivity in this reaction are determined in the second step of the proposed catalytic cycle and that the complex II is predisposed to evolve via C-4 addition. The results also reveal that the role of the LA is not only to activate the substrate 1a toward a potential nucleophilic addition but also to subtly control the regiochemistry by preventing the C-2 addition from happening. These findings are in line with the tolerance of the reaction to quinoline substituents at the C-2 position and the sensitivity to a substituent in the C-3 and C-4 positions.
Conclusions
We have presented the first copper-catalyzed enantioselective strategy for the dearomatization of nitrogen-containing heteroaromatics in the presence of Lewis acid. We have shown that the synergistic action of Lewis acid, chiral copper catalyst, and Grignard reagents provides direct access to the chiral dearomatized products with excellent regioselectivity and enantioselectivity and with a catalyst TON as high as 1000. Furthermore, we were able to unravel the roles of both the Lewis acid and the copper complex and prove that the former is responsible not only for the activation of the substrate but also for the control of the regioselectivity and the latter for controlling the enantioselectivity and further activating the substrate. Our results together with modeling studies support mechanistic connection between copper-catalyzed conjugate additions of organometallics to nonaromatic carbonyl-based Michael acceptors on the one hand and to aromatic quinolines on the other.
Acknowledgments
Financial support from the European Research Council (S.R.H. Grant No. 773264, LACOPAROM), the China Scholarship Council (CSC, to X.Y. and L.G.), and the Ministry of Education, Culture and Science (Gravity program 024.001.035 to S.R.H.) is acknowledged. M.C.R. thanks the Centro de Supercomputacion de Galicia (CESGA) for the free allocation of computational resources and the Xunta de Galicia (Galicia, Spain) for financial support through the ED481B-Axudas de apoio á etapa de formación posdoutoral (modalidade A) fellowship. This work was partially carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09974.
Author Contributions
† X.Y. and L.G. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Pozharskii A. F.; Soldatenkov A.; Katritzky A. R.. Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, 2nd ed.; Wiley: Chichester, U.K., 2011. [Google Scholar]; b Blacker J. A.; Williams M. T.. Pharmaceutical Process Development: Current Chemical and Engineering Challenges; Royal Society of Chemistry: Cambridge, U.K., 2011. [Google Scholar]
- a Mitchenson A.; Nadin A. Saturated nitrogen heterocycles. J. Chem. Soc., Perkin Trans. 2000, 1, 2862–2892. 10.1039/a908537h. [DOI] [Google Scholar]; b Muthukrishnan I.; Sridharan V.; Menéndez J. C. Progress in the Chemistry of Tetrahydroquinolines. Chem. Rev. 2019, 119, 5057–5191. 10.1021/acs.chemrev.8b00567. [DOI] [PubMed] [Google Scholar]; c Sridharan V.; Suryavanshi P. A.; Menendez J. C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]; d Katritzky A. R.; Rachwal S.; Rachwal B. Recent progress in the synthesis of 1,2,3,4,-tetrahydroquinolines. Tetrahedron 1996, 52, 15031–15070. 10.1016/S0040-4020(96)00911-8. [DOI] [Google Scholar]
- a Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; b Clemons P. A.; Bodycombe N. E.; Carrinski H. A.; Wilson J. A.; Shamji A. F.; Wagner B. K.; Koehler A. N.; Schreiber S. L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18787–18792. 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a You S.-L.Asymmetric Dearomatization Reactions; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]; b López Ortiz F. L.; Iglesias M. J.; Fernández I.; Andújar Sánchez C. M. A.; Ruiz Gómez G. R. Nucleophilic Dearomatizing (DNAr) Reactions of Aromatic C,H-Systems. Chem. Rev. 2007, 107, 1580–1691. 10.1021/cr030207l. [DOI] [PubMed] [Google Scholar]; c Zhuo C.; Zhang W.; You S.-L. Catalytic asymmetric dearomatization reactions. Angew. Chem., Int. Ed. 2012, 51, 12662–12686. 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]
- Jones G.The Chemistry of Heterocyclic Compounds; Wiley: Bristol, U.K., 1982. [Google Scholar]
- a Takamura M.; Funabashi K.; Kanai M.; Shibasaki M. Asymmetric Reissert-type reaction promoted by bifunctional catalyst. J. Am. Chem. Soc. 2000, 122, 6327–6328. 10.1021/ja0010352. [DOI] [Google Scholar]; b Takamura M.; Funabashi K.; Kanai M.; Shibasaki M. Catalytic enantioselective Reissert-Type reaction: development and application to the synthesis of a potent NMDA receptor antagonist (−)-L-689,560 using a solid-supported catalyst. J. Am. Chem. Soc. 2001, 123, 6801–6808. 10.1021/ja010654n. [DOI] [PubMed] [Google Scholar]; c Pappoppula M.; Cardoso F. S. P.; Garrett B. O.; Aponick A. Enantioselective copper-catalyzed quinoline alkynylation. Angew. Chem. 2015, 127, 15417–15421. 10.1002/ange.201507848. [DOI] [PubMed] [Google Scholar]; d Wang Y.; Liu Y.; Zhang D.; Wei H.; Shi M.; Wang F. Enantioselective rhodium-catalyzed dearomative arylation or alkenylation of quinolinium salts. Angew. Chem., Int. Ed. 2016, 55, 3776–3780. 10.1002/anie.201511663. [DOI] [PubMed] [Google Scholar]; e Yamaoka Y.; Miyabe H.; Takemoto Y. Catalytic enantioselective Petasis-type reaction of quinolines catalyzed by a newly designed thiourea catalyst. J. Am. Chem. Soc. 2007, 129, 6686–6687. 10.1021/ja071470x. [DOI] [PubMed] [Google Scholar]; f Zurro M.; Asmus S.; Beckendorf S.; Mück-Lichtenfeld C.; Mancheño O. G. Chiral helical oligotriazoles: new class of anion-binding catalysts for the asymmetric dearomatization of electron-deficient N-heteroarenes. J. Am. Chem. Soc. 2014, 136, 13999–14002. 10.1021/ja507940k. [DOI] [PubMed] [Google Scholar]; g Fischer T.; Duong Q.-N.; García Mancheño O. G. Triazole-based anion-binding catalysis for the enantioselective dearomatization of N-heteroarenes with phosphorus nucleophiles. Chem. - Eur. J. 2017, 23, 5983–5987. 10.1002/chem.201605660. [DOI] [PubMed] [Google Scholar]; h Cointeaux L.; Alexakis A. Enantioselective addition of organolithium reagents to quinoline catalyzed by 1,2-diamines. Tetrahedron: Asymmetry 2005, 16, 925–929. 10.1016/j.tetasy.2005.01.007. [DOI] [Google Scholar]
- Amiot F.; Cointeaux L.; Jan Silve E. J.; Alexakis A. Enantioselective nucleophilic addition of organometallic reagents to quinoline: regio-, stereo- and enantioselectivity. Tetrahedron 2004, 60, 8221–8231. 10.1016/j.tet.2004.06.088. [DOI] [Google Scholar]
- a Meyers A. I.; Wettlaufer D. G. Complete intramolecular transfer of a central chiral element to an axial chiral element. Oxidation of (S)-4-naphthyldihydroquinolines to (S)-4-naphthylquinolines. J. Am. Chem. Soc. 1984, 106, 1135–1136. 10.1021/ja00316a063. [DOI] [Google Scholar]; b Mohiti M.; Rampalakos C.; Feeney K.; Leonori D.; Aggarwal V. K. Asymmetric addition of chiral boron-ate complexes to cyclic iminium ions. Chem. Sci. 2014, 5, 602–607. 10.1039/C3SC52409D. [DOI] [Google Scholar]
- Mani N. S.; Chen P.; Jones T. K. Addition of Grignard reagents to quinolinium salts: evidence for a unique redox reaction between a 1,4-and a 1,2-dihydroquinoline. J. Org. Chem. 1999, 64, 6911–6914. 10.1021/jo990586b. [DOI] [PubMed] [Google Scholar]
- a Perlmutter P.Conjugate Addition Reactions in Organic Synthesis; Tetrahedron Organic Chemistry, Vol. 9; Pergamon: Oxford, U.K., 1992. [Google Scholar]; b Harutyunyan S. R.; den Hartog T.; Geurts K.; Minnaard A. J.; Feringa B. L. Catalytic asymmetric conjugate addition and allylic alkylation with Grignard reagents. Chem. Rev. 2008, 108, 2824–2852. 10.1021/cr068424k. [DOI] [PubMed] [Google Scholar]; c Alexakis A.; Krause N.; Woodward S.. Copper-Catalyzed Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]; d Howell G. P. Asymmetric and diastereoselective conjugate addition reactions: C–C bond formation at large scale. Org. Process Res. Dev. 2012, 16, 1258–1272. 10.1021/op200381w. [DOI] [Google Scholar]
- a Jumde R. P.; Lanza F.; Veenstra M. J.; Harutyunyan S. R. Catalytic asymmetric addition of Grignard reagents to alkenyl-substituted aromatic N-heterocycles. Science 2016, 352, 433–437. 10.1126/science.aaf1983. [DOI] [PubMed] [Google Scholar]; b Rodríguez-Fernández M.; Yan X.; Collados J. F.; White P. B.; Harutyunyan S. R. Lewis acid enabled copper-catalyzed asymmetric synthesis of chiral β-substituted amides. J. Am. Chem. Soc. 2017, 139, 14224–14231. 10.1021/jacs.7b07344. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rong J.; Oost R.; Desmarchelier A.; Minnaard A. J.; Harutyunyan S. R. Catalytic asymmetric alkylation of acylsilanes. Angew. Chem., Int. Ed. 2015, 54, 3038–3042. 10.1002/anie.201409815. [DOI] [PubMed] [Google Scholar]; d Yan X.; Harutyunyan S. R. Catalytic enantioselective addition of organometallics to unprotected carboxylic acids. Nat. Commun. 2019, 10, 3402. 10.1038/s41467-019-11345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann L. G.; Mani N. S.; Davis R. L.; Wang X.-N.; Marschke K. B.; Jones T. K. Discovery of a potent, orally active, nonsteroidal androgen receptor agonist: 4-ethyl-1,2,3,4-tetrahydro-6-(trifluoromethyl)-8-pyridono[5,6-g]-quinoline (LG121071). J. Med. Chem. 1999, 42, 210–212. 10.1021/jm9806648. [DOI] [PubMed] [Google Scholar]
- a Yoshikai N.; Nakamura E. Mechanisms of nucleophilic organocopper (I) reactions. Chem. Rev. 2012, 112, 2339–2372. 10.1021/cr200241f. [DOI] [PubMed] [Google Scholar]; b Yamanaka M.; Inagaki A.; Nakamura E. Theoretical studies on structures and reactivities of organocuprate (I) and organocopper (III) species. J. Comput. Chem. 2003, 24, 1401–1409. 10.1002/jcc.10132. [DOI] [PubMed] [Google Scholar]; c Harutyunyan S. R.; Lopez F.; Browne W. R.; Correa A.; Peña D.; Badorrey R.; Meetsma A.; Minnaard A. J.; Feringa B. L. On the mechanism of the copper-catalyzed enantioselective 1,4-addition of Grignard reagents to α,β-unsaturated carbonyl compounds. J. Am. Chem. Soc. 2006, 128, 9103–9118. 10.1021/ja0585634. [DOI] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision A.02; Gaussian, Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.