


Abstract
Family D DNA polymerase (PolD) is the essential replicative DNA polymerase for duplication of most archaeal genomes. PolD contains a unique two-barrel catalytic core absent from all other DNA polymerase families but found in RNA polymerases (RNAPs). While PolD has an ancestral RNA polymerase catalytic core, its active site has evolved the ability to discriminate against ribonucleotides. Until now, the mechanism evolved by PolD to prevent ribonucleotide incorporation was unknown. In all other DNA polymerase families, an active site steric gate residue prevents ribonucleotide incorporation. In this work, we identify two consensus active site acidic (a) and basic (b) motifs shared across the entire two-barrel nucleotide polymerase superfamily, and a nucleotide selectivity (s) motif specific to PolD versus RNAPs. A novel steric gate histidine residue (H931 in Thermococcus sp. 9°N PolD) in the PolD s-motif both prevents ribonucleotide incorporation and promotes efficient dNTP incorporation. Further, a PolD H931A steric gate mutant abolishes ribonucleotide discrimination and readily incorporates a variety of 2′ modified nucleotides. Taken together, we construct the first putative nucleotide bound PolD active site model and provide structural and functional evidence for the emergence of DNA replication through the evolution of an ancestral RNAP two-barrel catalytic core.
INTRODUCTION
Polymerases are essential enzymes for life responsible for preserving genetic information by replicating and repairing nucleic acid molecules as well as for the expression of genes through transcription (1,2). Nucleotide polymerization has independently evolved several times during evolution. Most nucleotide polymerases are divided into three major groups based on structures of their catalytic cores (3,4). The first group contains nucleotide polymerases that structurally resemble Escherichia coli Pol-I Klenow-fold (5). Their overall fold is characterized by thumb, finger and palm subdomains and is shared by most eukaryotic, archaeal, and viral replicative DNA polymerases (DNAPs), bacteriophage single-subunit RNA polymerase (RNAP), mitochondrial RNAP, and reverse-transcriptases (6,7). The second group, often referred to as Polβ-like polymerases, includes bacterial replicative DNAPs and several eukaryotic DNAPs involved in DNA repair (8). While this second group shares a similar three-dimensional arrangement of catalytic aspartates in the active site with the first group, these polymerases show completely different topology of the palm subdomain. The third group includes nucleotide polymerases with a common two-barrel catalytic core. The largest constituent of this group is multisubunit RNA polymerases (msRNAPs), required for transcription of cellular genomes of Bacteria, Archaea, and Eukarya as well as nucleo-cytoplasmic DNA viruses (9). The two-barrel catalytic core of this group is located at the interface between two double-psi β-barrel (DPBB) subdomains, which contribute distinct amino acid residues to the active site in an asymmetrical fashion (10). Recent sequencing and structural studies have added a surprising variety of nucleotide polymerases to the two-barrel superfamily and demonstrated the two-barrel active site architecture in msRNAPs is not restricted to DNA-dependent transcription (11–13). The structures of two distinct nucleic acid polymerases, Qde-1 and PolD, revealed this structural framework can also support RNA-dependent RNA synthesis and DNA-dependent DNA synthesis, respectively (14,15).
The substrate specificity of the two-barrel polymerase superfamily was substantially diversified with the structure elucidation of PolD, an archaeal replicative DNAP, which extends the two-barrel superfamily to DNA-dependent DNAPs involved in replication (14,16). PolD is a heterodimeric replicative DNAP composed of a large catalytic subunit (DP2) and a smaller subunit with 3′–5′ proofreading exonuclease activity (DP1) (17–19). PolD is essential for cell viability (20–22) and is present in all four major superphyla of Archaea, including methanogenic human symbionts and the emerging Asgard superphylum (23). Strikingly, while all other two-barrel nucleotide polymerases identified so far are RNA polymerases, PolD is a replicative DNA polymerase that shows strong ribonucleotide discrimination (24). In fact, previous nucleotide incorporation kinetic studies on Thermococcus sp. 9°N PolD revealed ribonucleotide discrimination is due to weak ribonucleotide binding in the PolD active site, leading to a ∼2,300-fold nucleotide selectivity of dNTPs over rNTPs. Further, the cellular concentration of rNTPs is ∼20-fold higher than dNTPs in the Thermococcales (25). However, the molecular mechanism of PolD nucleotide selectivity is currently unknown. Moreover, despite its fundamental biological role in Archaea, PolD is the least characterized DNAP on the structural level. Indeed, the structural basis for DNA binding, nucleotide recognition, dNTP vs rNTP discrimination is well documented for other DNAPs, with the availability of many nucleotide-bound crystal structures. Such structural information is missing for PolD, as a nucleotide bound PolD structure is yet to be determined.
In all other DNA polymerase families (A, B, C, X, Y and Reverse Transcriptases) a bulky active site amino acid, referred to as the steric gate, is responsible for blocking ribonucleotide incorporation (26). In addition to the steric gate, DNA polymerases may also contain additional residues that aid in ribonucleotide discrimination, like the polar Threonine residue found in E. coli Family Y DNA polymerase (27). In this work, we use structure-based sequence alignment to identify three motifs shared between the two-barrel catalytic cores of msRNAPs and PolD: an acidic motif (a-motif), basic motif (b-motif) and nucleotide-selectivity motif (s-motif). While a- and b- motifs residues are shared across PolDs and all two-barrel RNAPs, s-motif residues are specific to PolD. We further characterize each motif using site-directed mutagenesis, biochemical nucleotide incorporation end-point assays and single-nucleotide incorporation kinetics to reveal the essential role of a- and b-motif residues in PolD catalysis and unveiled a critical steric gate role for a histidine residue within the s-motif of PolD in preventing ribonucleotide incorporation during DNA synthesis. Taken together, this work provides structural and functional evidence for the emergence of DNA replication through the evolution of a RNAP two-barrel catalytic core.
MATERIALS AND METHODS
Structure-based alignment analysis
Structure-based alignment of the shared motifs within the two-barrel catalytic core of PolD, msRNAPs and single-chain RNA-dependent RNAPs were performed with PyMOL (The PyMOL Molecular Graphics System, Version 1.8.2.0 Schrödinger, LLC). The RNA and DNA polymerases were aligned with respect to their two-barrels catalytic core. Multiple protein sequence alignments were performed with Clustal omega (28) and visualized in JALVIEW (29). Figures were rendered using PyMOL and UCSF Chimera (30).
Cloning of Thermococcus sp. 9°N PolD and site-directed mutagenesis
Previous biochemical and kinetic work performed by Gardner et al (24,31,32) on Thermococcus sp. 9°N PolD revealed a highly soluble, well-expressed and easily purified PolD, and therefore, this PolD was used as a scaffold for mutations. Further, while structural studies have been performed on PolD from P. abyssi, Thermococcus sp. 9°N PolD and P. abyssi PolD are >90% homologous in their two-barrel catalytic core residues (Supplementary Figure S1). The genes for PolD DP1 and DP2 with a 6x Histag were cloned into a dual expression plasmid. DP1 was under control of the T7 promoter and DP2-his was under control of the ptac promoter. Site directed mutagenesis was performed using the Q5® Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA) with mutagenetic primers designed using the webtool NEBasechangerTM (http://nebasechanger.neb.com; Supplementary Table S1). Mutations were verified by DNA sequencing. All PolD variants used in this study contain a DP1 active site H554A mutation to create an exonuclease- variant.
Small-scale expression of PolD and PolD variants
Plasmids containing PolD and variant PolDs were transformed into T7 Express competent cells (New England Biolabs, Ipswich, MA, USA). A 25 ml culture was grown in LB supplemented with 0.1 mg/ml ampicilin at 37°C to an OD600 of 0.6. Protein expression was induced with 0.2 mM (final) Isopropyl β-d-1-thiogalactopyranoside (IPTG). Growth was continued for 3.5 h at 37°C and cells were harvested by centrifugation at 3,800 rpm for 30 min. Cell pellets were resuspended in 150 ml of Buffer A (20 mM Tris–HCl at pH 7.5, 300 mM NaCl, 0.1 mM EDTA, 20 mM Imidazole) and lysed using a constant cell disruptor (Constant Systems LTD, Northants, UK). Cell lysates were heated to 80°C for 20 minutes and clarified by centrifugation at 30 000 × g for 30 min. The supernatant was filtered through a WhatmanTM grade filter paper (GE Healthcare, Buckinghamshire, UK) and loaded onto a 20 ml HisPrepTM FF 16/10 column (GE Healthcare, Uppsala, Sweden) (pre-equilibrated with Buffer A) and eluted with Buffer B (20 mM Tris–HCl at pH 7.5, 300 mM NaCl, 0.1 mM EDTA, 500 mM Imidazole) with an imidazole gradient from 20 to 500 mM. Fractions were run on a 10–20% Tris–glycine gel (Invitrogen, Carlsbad, CA, USA) to confirm the presence PolD DP1 and DP2. Polymerase containing fractions were pooled and dialyzed in storage buffer (10 mM Tris–HCl, 100 mM KCl, 1 mM DTT, 0.1 mM EDTA and 50% glycerol, pH 7.4).
PolD primer extension activity assay
A capillary electrophoresis-based assay was used to determine the polymerase incorporation activity of each PolD (33). A primer-template was prepared by annealing a 50mer 5′-FAM primer (5 μM) (5′-FAM-AGT GAA TTC GAG CTC GGT ACC CGG GGA TCC TCT AGA GTC GAC CTG CAG GT-3′) to a 67mer template (6.25 μM) (5′-AAG CAC GAA AGC AGG GTG CCT GCA GGT CGA CTC TAG AGG ATC CCC GGG TAC CGA GCT CGA ATT CAC T-3′) in 1× annealing buffer (20 mM Tris–HCl, 100 mM NaCl, pH 7.5) by heating to 95°C for 3 min followed by cooling to room temperature. 1 μl of each polymerase was added to 20 μl of 1× Thermopol buffer (20 mM Tris–HCl, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1% Triton X-100, pH 8.8 at 25 °C) in a 96-well plate and six  -fold serial dilutions in 1× Thermopol buffer were performed. An equal volume of 20 nM DNA (10 nM final), 2 μM dNTPs (1 μM final) was added to each PolD and the reactions were incubated at 65°C for 30 min followed by quenching with equal volume of 50 mM EDTA. Reaction products were separated by capillary electrophoresis using a 3730xl Genetic Analyzer (Applied Biosystems) and fluorescent peaks were analyzed using Peak Scanner software version 1.0 (Applied Biosystems). All assays were performed in triplicate to ensure experiment reproducibility.
-fold serial dilutions in 1× Thermopol buffer were performed. An equal volume of 20 nM DNA (10 nM final), 2 μM dNTPs (1 μM final) was added to each PolD and the reactions were incubated at 65°C for 30 min followed by quenching with equal volume of 50 mM EDTA. Reaction products were separated by capillary electrophoresis using a 3730xl Genetic Analyzer (Applied Biosystems) and fluorescent peaks were analyzed using Peak Scanner software version 1.0 (Applied Biosystems). All assays were performed in triplicate to ensure experiment reproducibility.
Single-nucleotide incorporation assay for steric gate identification
To determine the location of the steric gate in Thermococcus sp. 9°N PolD, a nucleotide incorporation assay was used to compare the ability of WT PolD and variant PolDs to incorporate rATP compared to dATP. 1 μl of the purified protein was added to 20 μl of 1× Thermopol buffer in a 96-well plate and six  fold serial dilutions were made in 1× Thermopol buffer. An equal volume of 20 nM DNA (10 nM final), 2 μM dATP or rATP (1 μM final) was added to each variant PolD and the reactions were incubated at 65°C for 15 min followed by quenching with equal volume of 50 mM EDTA. Reaction products were separated and analyzed the same method explained above. The percentage of ribonucleotide incorporation was divided by the percentage of deoxyribonucleotide incorporation to obtain the rA:dA ratio. All assays were performed in triplicate to ensure experiment reproducibility.
fold serial dilutions were made in 1× Thermopol buffer. An equal volume of 20 nM DNA (10 nM final), 2 μM dATP or rATP (1 μM final) was added to each variant PolD and the reactions were incubated at 65°C for 15 min followed by quenching with equal volume of 50 mM EDTA. Reaction products were separated and analyzed the same method explained above. The percentage of ribonucleotide incorporation was divided by the percentage of deoxyribonucleotide incorporation to obtain the rA:dA ratio. All assays were performed in triplicate to ensure experiment reproducibility.
Large-scale Expression and Purification of Thermococcus sp. 9°N PolD and PolD H931A
The plasmid containing PolD and PolD H931A were transformed into T7 Express competent cells (New England Biolabs, Ipswich, MA). Two liters of cells were grown at 37°C until an OD600 of 0.4 was reached, at which time the cells were induced with 0.4 mM (final concentration) of IPTG. Growth was continued for 3 h at 37°C and cells were harvested by centrifugation. The cell pellet was resuspended in Buffer A and lysed using a constant cell disruptor. The cell lysate was heated to 80°C for 20 min in a water bath and the lysate was clarified by centrifugation at 4,000 × g for 20 min. The clarified supernatant was loaded onto a HiPrep 16/10 DEAE column (GE Lifesciences, Pittsburg, PA, USA) and the flow-through was collected and loaded onto a 5 ml HisPrep FF column equilibrated with Buffer A. The polymerases were eluted off the column in fractions using Buffer B and an imidazole gradient from 50–250 mM. Fractions containing PolD were identified by SDS-PAGE analysis, followed by a primer extension activity assay to confirm the presence of active enzyme (described above). Active enzyme was pooled and dialyzed into 10 mM Tris–HCl, 100 mM KCl, 1 mM DTT, 0.1 mM EDTA and 50% glycerol, pH 7.4 and protein concentration was quantified using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Pre-steady-state single nucleotide incorporation assay
To obtain the rates of dATP and rATP incorporation by PolD H931A, pre-steady-state single nucleotide assays were performed. The primer-template substrates used in these assays were prepared as described above. A 150 μl PolD H931A and DNA aliquot was prepared by mixing 1× ThermoPol buffer, 60 nM primer/template DNA, and a 3-fold excess of PolD H931A (180 nM active enzyme concentration). Steady-state incorporation kinetics were preformed to determine the amount of active PolD H931A prior to pre-steady-state experiments (data not shown). A second 150 μl aliquot was prepared by mixing ThermoPol buffer (1× final concentration) and dATP or rATP (50–4000 μM). Using a RQF instrument, PolD H931A/DNA construct was rapidly mixed with dATP or rATP and quenched with 50 mM EDTA at 60°C. After mixing an equal volume of PolD H931A and DNA with the nucleotide solution by the RQF, the final reaction concentrations were 30 nM DNA, 90 nM active polymerase and 25–2000 μM dATP/rATP in 1× ThermoPol buffer. Two control reactions were performed. The first control reaction, which served as a negative control, dATP was replaced with 1× ThermoPol buffer and reacted in the RQF with PolD and DNA for 10 s. The second control reaction, which served as a quench control, dATP and EDTA were swapped in the RQF, and EDTA was mixed with PolDH931A and DNA for 0.05 seconds followed by addition of dATP. Reaction products were separated by capillary electrophoresis as described above. All time courses were performed in triplicate to ensure experiment reproducibility. The product concentration was graphed as a function of time and the data were fit to the single-exponential Equation (1) to obtain the observed rate constant of nucleotide incorporation (kobs) using the nonlinear regression program Kaleidagraph (Synergy Software).
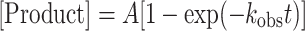 |
(1) |
To obtain the maximum rate of polymerization constant (kpol) and apparent equilibrium dissociation constant (Kd), the kobs were graphed as a function of dATP or rATP concentration and the data were fit to the hyperbolic Equation (2) using Kaleidagraph.
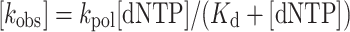 |
(2) |
The specific activities for dATP and rATP incorporation were calculated using Equation (3), and the nucleotide selectivity for rATP incorporation was calculated using Equation (4).
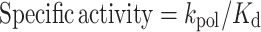 |
(3) |
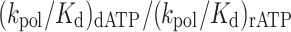 |
(4) |
Successive nucleotide incorporation assay
A nucleotide incorporation assay was performed to determine the ability of PolD and PolDH931A to incorporate successive ribonucleotides into a primer/template. The 50mer DNA primer/67mer DNA template was created as described above. In addition to the 50mer DNA primer, a construct with a 50 nucleotide RNA primer (same sequence) was also annealed to the 67mer DNA template as described above. A 20 μl reaction containing 20 nM nucleotide construct, 100 nM WT PolD or PolD H931A, and 1 μM rNTPs in 1× Thermopol buffer was placed at 65°C for 15 min. The reaction was quenched with equal volume of 50 mM EDTA and analyzed by capillary electrophoresis as described above. All assays were performed in triplicate to ensure experiment reproducibility.
Incorporation of 2′and 3′-modified-nucleotides
Modified nucleotides, 3′-dATP (Cordycepin), 2′-amino-ATP, 2′-azido-ATP, araATP and 2′-O-methyl-ATP were from Trilink Biotechnologies, TNP–ATP was from Invitrogen, and LNA–ATP was from Jena Biosciences. The 50mer DNA primer/ 67mer DNA template was created as described above. For each modified nucleotide incorporation reaction, a 100 μL reaction mix was prepared containing 1× ThermoPol Buffer, 10 nM primer-template, 25 nM wild-type WT PolD or PolD H931A and 1 μM modified nucleotide. Reactions were incubated for 0, 1 or 15 min at 65°C and 10 μl aliquots were removed and mixed with 50 mM EDTA to quench the reaction. Reaction products were analyzed by capillary electrophoresis as described above. The concentration of product (51 nt DNA with a FAM label) was graphed for each nucleotide. All assays were performed in triplicate to ensure experiment reproducibility.
RESULTS
Structure-based alignment reveals three critical active site motifs in PolD and msRNAPs
The extended two-barrel polymerase superfamily encompasses DNA-dependent RNAPs responsible for DNA transcription, the eukaryotic RNA-dependent RNAPs (quelling defective phenotype (QDE-1)) involved in gene silencing, and the PolD DNA-dependent replicative DNAPs (12,34). Comparison of the recent near-atomic resolution cryo-EM structure of the P. abyssi PolD-PCNA complex to the many structures of two-barrel polymerases shows that all distantly related two-barrel nucleotide polymerases share a common catalytic core located at the interface between two double-psi β-barrel (DPBB) subdomains (Figure 1A) (16,35). Superimposing the active site of PolD onto the active site of the two-barrel RNAPs reveals that the relative positioning of their DPBB subdomains is highly conserved. Remarkably, the active sites of both PolD and RNAPs orient the DNA template entry and the nascent duplex exit in a same relative axis with respect to the two-barrel catalytic core (16). A structure-based alignment of the two-barrel polymerases in the structurally homologous catalytic core reveals that their common architecture is strengthened by the conservation of hydrophobic, polar and charged residues (Figure 1B). No structure of PolD in complex with an incoming nucleotide has been reported; however, superimposing the recent near-atomic resolution structure of the P. abyssi PolD-PCNA-DNA ternary complex (35) with the crystal structure of Thermus thermophilus RNAP (36) in complex with an incoming nucleotide sheds light on the molecular basis for their respective substrate specificities. Interestingly, each DPBB contributes several invariant amino acids located in the immediate vicinity of their catalytic Mg2+ cations and nucleotide binding sites. These residues can be divided into three distinct motifs, arbitrarily named the acidic motif (a-motif), the basic motif (b-motif) and the nucleotide-selectivity motif (s-motif) (Figure 1C).
Figure 1.

Structure-based comparison of two-barrel RNA and DNA polymerases. (A) Shared structural elements between two-barrel RNA and DNA polymerases. The structures of Pyrococcus abyssi PolD and Thermus thermophilus (41) msRNAP (PDBid: 2o5j)] are aligned with respect to their two-barrel catalytic core. Top panels show a schematic representation of the T. thermophilus msRNAP (left) and the P. abyssi PolD (right). Bottom panels show a cartoon representation of their respective two-barrel catalytic cores. The side chains of their consensus catalytic residues are colored in yellow. The RNAP- and DNAP-specific selectivity motifs are colored in red and blue, respectively. (B) Structure-based alignment of the shared motifs within the two-barrel catalytic core of PolD, msRNAPs and single-chain RNA-dependent RNAPs. Grey boxes highlight hydrophobic residues, aromatic, or polar residues. Black boxes highlight the consensus residues within the acidic and basic motifs. Residues within the RNAP- and DNAP-specific selectivity motifs are highlighted by Red and Blue boxes, respectively. The following structures were used for the structure-based alignment: Saccharomyces cerevisiae RNAPI(43)(PDBid:5m5x), Saccharomyces cerevisiae RNAPII(44)(PDBid:2e2i), Saccharomyces cerevisiae RNAPIII(45)(PDBid:6f41), Thermococcus kodakarensis RNAP(46)(PDBid:4qiw), E. coli RNAP(47)(PDBid:6asx), Thermus thermophylus RNAP(41)(PDBid:2o5i), Neurospora crassa QDE-1(15)(PDBid:2j7o), and P. abyssi PolD(35) (PDBid:6hmf). (C) Superimposition of the basic motif (left), selectivity motif (middle) and acidic motif (right) in the structures of T. thermophilus msRNAP (pink) and the P. abyssi PolD (blue). Side chain, nucleotide and RNA are shown as sticks. C-alphas and magnesium ions are shown as spheres. Amino acid numbering is for Thermococcus sp. 9°N PolD for manuscript clarity.
All two-barrel RNAPs contain the consensus a-motif DxDGD, whose three carboxylate groups (β′-D739, β′-D741, and β′-D743 in T. thermophilus RNAP) bind the catalytic Mg2+ ions (37). The consensus PolD a-motif, NxDGD, has only two canonical aspartic residues (DP2-D964 and DP2-D966 in Thermococcus sp. 9°N PolD) and a conserved asparagine (DP2-N962 in Thermococcus sp. 9°N PolD), with the notable exception of species in the archaeal phylum Thaumarchaeota, where the canonical three aspartic residues are conserved. Importantly, the orientation of the aspartic acid residues is conserved in both P. abyssi PolD and T. thermophilus RNAP active sites (Figure 1C). Additionally, the N-terminus of the PolD a-motif carries two conserved arginine residues (DP2-R960 and DP2-R961 in Thermococcus sp. 9°N PolD), specific to only PolD, that may provide additional contacts with the phosphate moiety of the incoming nucleotide (Figure 1B).
The consensus b-motif carries two conserved positively charged lysine residues (β-K838 and β-K846 in T. thermophilus RNAP and DP2-K392 and DP2-K398 in Thermococcus sp. 9°N PolD). In both DNA-bound structures, the positively charged side chains of these residues occupy similar positions in the polymerase active sites (Figure 1C). In msRNAPs, these two conserved lysine residues bind the growing RNA transcript via contacts between the lysine side chains and the RNA phosphodiester backbone (38–40). In addition to the two conserved lysine residues, the b-motif contains a highly conserved arginine residue (β-R842 in T. thermophilus RNAP and DP2-R395 in Thermococcus sp. 9°N PolD) which is oriented away from the bound DNA in both msRNAP and PolD structures(data not shown).
Structural comparison of two-barrel polymerases also reveals unique structural determinants (s-motif) that may account for the distinct substrate specificities of PolD for dNTPs and RNAPs for rNTPs. X-ray crystallography studies on msRNAP indicated that incoming nucleotides are loaded through a pore leading to the active site center. The incoming nucleotide first binds to an entry site and then rotates to reach the final nucleotide addition site (41). This rotation brings the ribose ring close to the side chain of a conserved arginine residue (β′-R704 in T. thermophilus RNAP). This arginine is part of the RxPxLH motif and is strictly conserved among all two-barrel msRNA polymerases, but is not found in PolD. This arginine forms hydrogen bonds with the incoming ribonucleotide and belongs to a catalytic triad of residues that are proposed to stabilize incorporation of rNTPs and exclude dNTPs in the msRNAP active site (41,42). PolD instead contains a different conserved GLAPHT motif at the same position, with PHT invariable across all PolDs (Figure 1B). Importantly, PHT residues are located on a structurally similar loop as the arginine residue of the RxPxLH motif found in msRNAPs (Figure 1C). Further, superimposition with RNAP suggests that the side chain of the histidine residue in the PHT s-motif (H931 in Thermococcus sp. 9°N PolD) points toward the ribose ring of the incoming nucleotide. We therefore speculate that the histidine residue of the PolD s-motif accounts for the distinct substrate specificity of the PolD two-barrel active site.
Functional characterization of acidic and basic motifs of Thermococcus sp. 9°N PolD
Structural alignment revealed conservation of the a- and b-motifs within the two-barrel catalytic core of all DNAPs (PolDs) and RNAPs (multi-subunit and single-chain RNAPs)(Figure 1B). Both motifs contain a set of highly conserved amino acids that are potentially critical for PolD polymerase catalysis. To assess the contributions of each motif to PolD polymerization, we utilized a well-expressing, highly soluble PolD from Thermococcus sp. 9°N to mutate critical residues and perform end-point primer extension assays (Figure 2A, Supplementary Table S1). Further, Thermococcus sp. 9°N and P. abyssi PolD DP2 share significant homology (>90%) of two-barrel catalytic core residues and previous studies report similar biochemical properties between the two PolDs (Supplementary Figure S1) (25,31,32,48,49). To understand the role of the acidic motif in PolD catalysis, we mutated each conserved residue, 959-KRRNCDGDD-966, to alanine and assessed the ability of each mutant to extend a 50nt/67nt DNA primer/template with dNTPs. We observed the most drastic effects on PolD catalysis with the mutation of the two highly conserved aspartate residues, D964 and D966, of the NxDGD motif. While wild type PolD synthesized full length (17 nt) DNA primer extension products, mutants D964A or D966A Thermococcus sp. 9°N PolD (Figure 2B) or D957A or D959A Pyrococcus horikoshii PolD (50) are completely abolished of DNA polymerization activity. Similarly, in msRNAPs these two structurally equivalent aspartates (D741 and D743 in the β-subunit of T. thermophilus msRNAP) are known to coordinate catalytic Mg2+ required for ribonucleotide addition (Figure 2C) and mutation of these aspartates leads to a catalytically inactive msRNAP (37). We therefore speculate the two PolD aspartates within the a-motif are similarly required for Mg2+ binding within the PolD active site (Figure 2C).
Figure 2.

Polymerase primer extension activity of Family D DNA polymerase mutants. (A) Schematic of the primer extension assay used to assess the catalytic activity of PolD mutants. A 5′ FAM labeled 50 nt primer annealed to a 67 nt template was incubated with each PolD mutant and dNTPs at 65°C for 30 min. Samples were analyzed by capillary electrophoresis (B) Table displaying the corresponding motif, primer extension activity, and representative primer extension CE trace for each PolD mutant. Extensions activity −, +, ++, and ++ + indicate no detectable DNA polymerase activity, 1–20%, 20–60% and 61–100% of wild type enzyme activity after 30 min incubation, respectively (C) Organization and putative organization of critical acidic, basic and nucleotide selectivity amino acids in the two-barrel catalytic core of RNAPs and PolD, respectively.
In addition to the two conserved aspartates in the acidic motif, msRNAPs contain a third conserved aspartate required for ribonucleotide addition (739DFDGD743 in T. thermophilus msRNAP) (37). Structural alignment of PolD with msRNAPs revealed that PolD contains a conserved asparagine (962NCDGD966) instead of this aspartate (Figure 1B). The PolD N962A mutant retained partial catalytic activity, but DNA polymerization primer extension products were limited to 8 nt in length, confirming the requirement of N962 for efficient PolD catalysis (Figure 2B). We further mutated N962 to aspartate (N962D) to mimic the catalytic aspartate triad observed in msRNAPs and observed full length extension products (data not shown). Indeed, species of Thaumarchaeota PolD contain an aspartate residue instead of an asparagine at this position, like msRNAPs, suggesting PolD functions with either an asparagine or aspartate (Figure 1B). Interestingly, about half of archaeal species that encode PolD, including Thermococcus sp. 9°N, contain an intein in the native DP2 precursor inserted in-frame between acidic motif amino acids N962 and C963. Previous work suggests the asparagine and cysteine residues play a critical role in excising the intein to form the mature PolD DP2 subunit (50–52).
Mutation of residues R960 and R961, structurally predicted to interact with the incoming nucleotide, had reduced primer extension activity with products limited to ∼10 nt (Figure 2B). This observation confirms these two residues are ideally positioned to stabilize the incoming nucleotide triphosphate in the active site (Figures 1C and 2C). In addition to R960 and R961, the conserved amino acids K959 and C963 are in close proximity to the incoming nucleotide and catalytic Mg2+. Mutation of these residues to alanine (K959A and C963A) had minimal effect on DNA primer extension activity suggesting that while these amino acids flank critical residues required for PolD catalysis, they do not play a direct catalytic role (data not shown). Together, these data demonstrate that msRNAP and PolD share structurally and functionally homologous amino acids required for Mg2+ coordination and stabilization of the incoming nucleotide triphosphate in the a-motif.
The basic motif contains three highly conserved residues: two lysine residues and an arginine residue (Figure 1B). One lysine residues (K398 in Thermococcus sp 9°N PolD and β-K846 in T. thermophilus RNAP) and the arginine residue (R395 in Thermococcus sp. 9°N PolD and β-R842 in T. thermophilus RNAP) are strictly conserved throughout DNA- and RNA-dependent RNAPs and PolD. A second lysine residue (K392 in Thermococcus sp. 9°N PolD and β-K838 in T. thermophilus RNAP) is limited to DNA-dependent msRNAPs and PolD (Figure 1B). To understand the role of the three conserved basic motif residues in PolD catalysis, we mutated each to alanine and performed an end-point primer/template extension assay (Figure 2A). Mutation of the K398A in Thermococcus sp. 9°N PolD severely reduced polymerase extension activity with <15% of primers extended by 1 nt (Figure 2B). In both T. thermophilus RNAP and P. abyssi PolD DNA-bound structures, the positively charged side chain of this lysine residue contacts the phosphate moiety of the 3′-OH end of the nascent RNA or DNA, suggesting this residue helps stabilize the growing nucleotide polymer (Figures 1B and 2C) (16,36). Furthermore, previous studies of the basic motif in the β-subunit of E. coli RNAP showed that mutating the equivalent invariant β-K1065R (β-K846 in T. thermophilus RNAP) abolished transcriptional activity (40). Mutation of the second conserved lysine, K392, to alanine in PolD did not significantly alter polymerase primer extension, as we observed full length DNA extension products (Figure 2B). Similarly, mutation of the homologous lysine, β-K1051, to arginine in the β-subunit of E. coli RNA polymerase had minimal impact on transcriptional activity (40). A third conserved b-motif residue (R395) points away from the active site in both PolD and msRNAPs and retained full extension activity when mutated to alanine (R395A) (Figure 2B). These data suggest that in both two-barrel RNAPs and DNAPs, a single lysine residue that binds to the 3′ end of the growing nucleotide chain is critical for polymerization activity while the second conserved lysine and highly conserved arginine of the b-motif in PolD play minor roles in DNA polymerization.
Ribonucleotide discrimination by the nucleotide selectivity motif in PolD
Despite structural and functional conservation of the a- and b-motifs and shared amino acids for Mg2+ binding and primer stabilization, a major difference between msRNAPs and PolD remains: msRNAPs incorporate rNTPs while PolD incorporates dNTPs. In all DNA polymerase families, including Family D, a mechanism is present within the polymerase active site to discriminate against ribonucleotide incorporation. In families A, B, C, X, Y and RTs, an active site amino acid, referred to as the steric gate, creates a steric clash with the 2′-OH of the ribose ring of an incoming ribonucleotide and blocks ribonucleotide incorporation (26). Comparison of two-barrel polymerases revealed a selectivity s-motif that is unique to DNAPs versus RNAPs and may account for their distinct substrate specificities for dNTPs or rNTPs, respectively (Figure 1B). Examination of the PolD active site shows the s-motif forms a loop that contains the strictly conserved PHT residues and is oriented toward the incoming nucleotide with the histidine residue (H931 in Thermococcus sp. 9°N) pointing towards the ribose ring of the incoming nucleotide (Figure 1C and 2C). We therefore speculated this amino acid, or amino acids in this region of the s-motif, may account for the distinct substrate specificities of PolD versus msRNAPs. We first assessed the contributions of the PHT residues of the s-motif in PolD polymerase catalysis by mutating each residue to alanine to test the impact on DNA polymerization using end-point primer/template extension assays (Figure 2A). All three mutants, P930A, H931A and T932A, were capable of synthesizing full length (17 nt) primer extension products after 30 min incubation, suggesting these residues are not required for PolD catalysis (Figure 2B).
Similar to other DNA polymerases, wild-type PolD likely blocks ribonucleotide incorporation through a steric clash between an active site steric gate amino acid and the 2′-OH of the ribose ring. Mutation of the steric gate amino acid to a smaller side chain (like alanine) would allow ribonucleotide incorporation and has been demonstrated for other DNA polymerase families (53–55). We therefore employed the same strategy using PHT mutants (P903A, H931A, and T932A) and compared their ability to incorporate dATP and rATP (Figure 3 A). These experiments were performed at a nucleotide concentration (1 μM) ∼100-fold below the Kd for PolD rNTP binding to more readily observe nucleotide discrimination and control for rNTP incorporation at high nucleotide concentration (24). WT PolD readily incorporates dATP into a primer/template but minimally (<25%) incorporates rATP after incubation for 15 min (Figure 3B). Similar to WT PolD, mutants P930A and T932A blocked rATP incorporation, where <25% of primers were extended by rATP (Figure 3B). Notably, the H931A mutant dramatically increased rATP incorporation and with similar efficiency to dATP incorporation suggesting H931 plays a role in discriminating between rNTP and dNTP incorporation and is likely the PolD steric gate amino acid (Figure 3B). Moreover, PolD H931A incorporates all other rNTPs (rCTP, rGTP and rUTP) with similar efficiency as the counterpart dNTPs (dCTP, dGTP and dTTP) (Supplementary Table S2). We further mutated H931 to a variety of other amino acids, including serine, valine, aspartate, arginine, and tyrosine to observe the effect of varying amino acid side chains on ribonucleotide incorporation (Figure 3B). Amino acids with small side chains, such as serine and valine, allowed for increased ribonucleotide incorporation compared to WT, while bulky or charged residues either prevented ribonucleotide incorporation (aspartate) or completely abolished PolD polymerase activity all together (arginine and tyrosine) (Figure 3B). These results suggest a critical role for H931 in the PolD s-motif for efficient selection and incorporation of the incoming nucleotide. In addition to the PHT motif mutants, we performed alanine substitutions for residues surrounding the putative nucleotide binding pocket and assessed their ability to incorporation rATP versus dATP (Supplementary Figure S2). Importantly, all of these active site mutants still blocked rATP incorporation at low (1 μM) nucleotide concentration.
Figure 3.
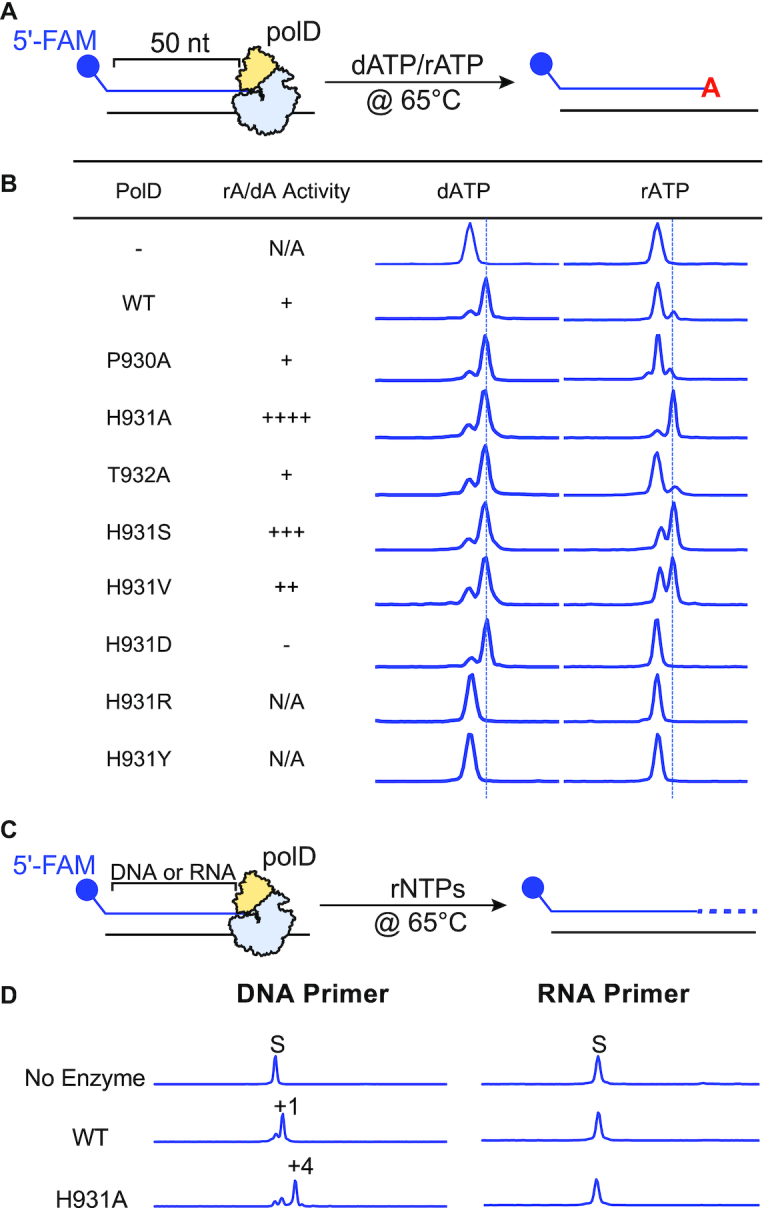
Ribonucleotide incorporation screening assay to identify Family D DNA polymerase steric gate. (A) Schematic of the assay used to assess single ribonucleotide incorporation activity of s-Motif PolD mutants. A 5′ FAM labeled 50 nt primer annealed to a 67 nt template was incubated with each S-motif PolD mutant and either dATP or rATP at 65°C for 15 min. Samples were analyzed by capillary electrophoresis. (B) Table displaying the rA/dA incorporation activity and representative dATP and rATP CE incorporation traces for each S-motif PolD mutant. The rA/dA activity −, +, ++, ++ + and ++ + + indicates no detectable DNA polymerase activity, 1-25%, 25–50%, and 50–75%, and 75-100% ratio of rA incorporation compared to dA incorporation, respectively. (C) Schematic of the assay used to assess multiple nucleotide incorporation activity of PolD and H931A PolD. A 5′ FAM labeled 50 nt primer annealed to a 67 nt template was incubated with PolD or H931A PolD and either dNTPs or NTPs at 65°C for 30 min. (D) Representative CE traces for DNA or RNA primer extension with rNTPs by WT and H931A PolD.
With the observed increase in single ribonucleotide incorporation activity of PolD H931A, we further explored the ability to incorporate multiple rNTPs and determine if this single mutation (H931A) turned PolD into an RNA polymerase. We monitored WT PolD and PolD H931A extension of a DNA or RNA primer with rNTPs (Figure 3C). While WT PolD extends a DNA primer by just one ribonucleotide, the H931A variant could efficiently incorporate four successive ribonucleotides (as confirmed by oligonucleotide size controls; data not shown) (Figure 3D) and extension did not proceed beyond 4 ribonucleotides after prolonged incubation (data not shown). Like Thermococcus sp. 9°N PolD, previous experiments show P. abyssi PolD also minimally extends a DNA primer with rNTPs (25). Neither WT nor H931A PolD could extend an RNA primer by a single ribonucleotide (Figure 3D) and only a small percentage of RNA primers were extended by a single ribonucleotide for both WT and H931A PolD after prolonged incubation (data not shown). Taken together, these results suggest other structural checkpoints are in place to prevent RNA synthesis by PolD besides the steric gate amino acid. Indeed, in Family B DNA polymerases, additional checkpoints, besides the steric gate, limit incorporation of successive rNTPs (56). To enable long ribonucleotide synthesis, additional mutations were engineered into a Family B DNA polymerase steric gate mutant (Y409G) from Thermococcus gorgonarius that permitted synthesis of A-form RNA:DNA molecules and increased ribonucleotide incorporation up to a 1.7 kb (57).
Kinetic Characterization of PolD and PolD H931A ribonucleotide incorporation
Steady-state end point assays revealed that PolD H931A incorporates dATP and rATP with similar efficiency. To further characterize rNTP discrimination by PolD H931A, we performed single-nucleotide pre-steady-state ([Enzyme]>>>[DNA]) incorporation kinetics. Pre-steady-state kinetics provide a quantitative assessment of enzyme catalysis that is otherwise masked by steady-state endpoint assays. Previous pre-steady-state single-nucleotide incorporation kinetics of Thermococcus sp. 9°N PolD revealed that ribonucleotide discrimination arises due to reduced ribonucleotide binding within the PolD active site as demonstrated by an over 100-fold increase in the Kd for rATP incorporation compared to dATP (360 μM versus 2.5 μM, respectively), and a 10-fold decrease in kpol (0.16 s–1 versus 2.6 s–1) (24). Therefore, the nucleotide kinetic selectivity [(kpol/Kd)dATP/(kpol/Kd)rATP] of dATP over rATP for Thermococcus sp. 9°N PolD is ∼2,300. These results are consistent with the presence of a WT PolD steric gate amino acid blocking ribonucleotide incorporation.
We performed pre-steady-state single-nucleotide incorporation assays to obtain the Kd and kpol for dATP and rATP incorporation by PolD H931A. PolD H931A was preincubated with fluorescently labeled 50 nt primer/67 nt template and rapidly mixed with varying concentrations of dATP or rATP from 0.05 to 30 s and quenched using a rapid quench flow (RQF) instrument (Figure 4 A). With increasing incubation time, we observed the accumulation of 51 nt product (Figure 4B). The amount of 51 nt product was graphed as a function of time for each nucleotide concentration to obtain the kobs (Figure 4C, D). Finally, the kobs was graphed as a function of nucleotide concentration to obtain the kpol and Kd of PolD H931A dATP and rATP incorporation (Figure 4E, F). Unlike WT PolD which discriminated against rATP by ∼2,300-fold, H931A showed similar dATP and rATP incorporation rates (kpol = 1.3 ± 0.05 s–1 and 1.4 ± 0.08 s–1) and binding constants (Kd = 56 ± 10 and 67 ± 12 μM), respectively, and has a nucleotide selectivity of dATP over rATP of 1 (Figure 4G). These kinetic data demonstrate that like steric gate mutants from other DNA polymerase families, PolD H931A has lost the ability to discriminate between dATP and rATP during binding and incorporation (53–55).
Figure 4.

Single-nucleotide incorporation kinetics of PolD H931A. (A) Schematic of the single-nucleotide primer extension assay used to assay PolD H931A incorporation kinetics. A 5′ FAM labeled 50 nt primer annealed to a 67 nt template was preincubated with a 3-fold excess of active H931A PolD, followed by rapid mixing at 60°C with various concentrations of dATP or rATP from 0.05–30 s using a RQF instrument. (B) Representative CE traces showing the accumulation of 51-nt product with increasing incubation time. The 51-nt product was graphed as a function of time for 25 ( ), 50 (
), 50 ( ), 250 (X), 500 (
), 250 (X), 500 ( ), 1000 (
), 1000 ( ) and 2000 (
) and 2000 ( ) nM (C) dATP and (E) rATP and fit to Equation (1) to obtain kobs. The dependence of reaction rate kobs on nucleotide concentration was fit to Equation (2) to obtain kpol and Kd for (D) dATP:T and (F) rATP:T. (G) Table displaying the kinetic parameters kpol, Kd, incorporation efficiency (kpol/Kd) for dATP and rATP incorporation, and the overall nucleotide selectivity for WT (24) and H931A PolD.
) nM (C) dATP and (E) rATP and fit to Equation (1) to obtain kobs. The dependence of reaction rate kobs on nucleotide concentration was fit to Equation (2) to obtain kpol and Kd for (D) dATP:T and (F) rATP:T. (G) Table displaying the kinetic parameters kpol, Kd, incorporation efficiency (kpol/Kd) for dATP and rATP incorporation, and the overall nucleotide selectivity for WT (24) and H931A PolD.
Kinetic data also show that in addition to blocking rNTP incorporation, H931 may also be important for correct dNTP binding in the active site. WT PolD binds dATP with a ∼20-fold lower Kd (2.5 μM) compared to H931A (Kd = 56 ± 10 μM). Furthermore, the dATP incorporation rate was 2-fold faster for WT PolD (kpol = 2.6 s-1) compared to H931A (kpol = 1.3 ± 0.05 s–1). These results suggest that H931 potentially plays a dual-role in not only preventing ribonucleotide incorporation, but also stabilizing dNTP incorporation through efficient binding. Similar effects on dNTP kinetic efficiencies were also observed in steric gate mutants in Family A, B and Y DNA polymerases (53–55).
Incorporation of 2′-modified nucleotides by PolD and PolD H931A
Nucleotide incorporation kinetics of PolD H931A demonstrate mutation of H931 to alanine removes the steric gate and allows efficient ribonucleotide incorporation. Further, the increase in Kd and decrease in kpol for dATP incorporation by H931A PolD compared to WT suggest an additional role for H931 in stabilizing the incoming dNTP. To further understand the putative dual-role of H931 in PolD nucleotide selectivity, we tested the incorporation of a wide variety of modified nucleotides with 2′ or 3′ modifications by WT and H931A PolD onto a 50nt primer/67nt template as probes of the active site (Figure 5A).
Figure 5.
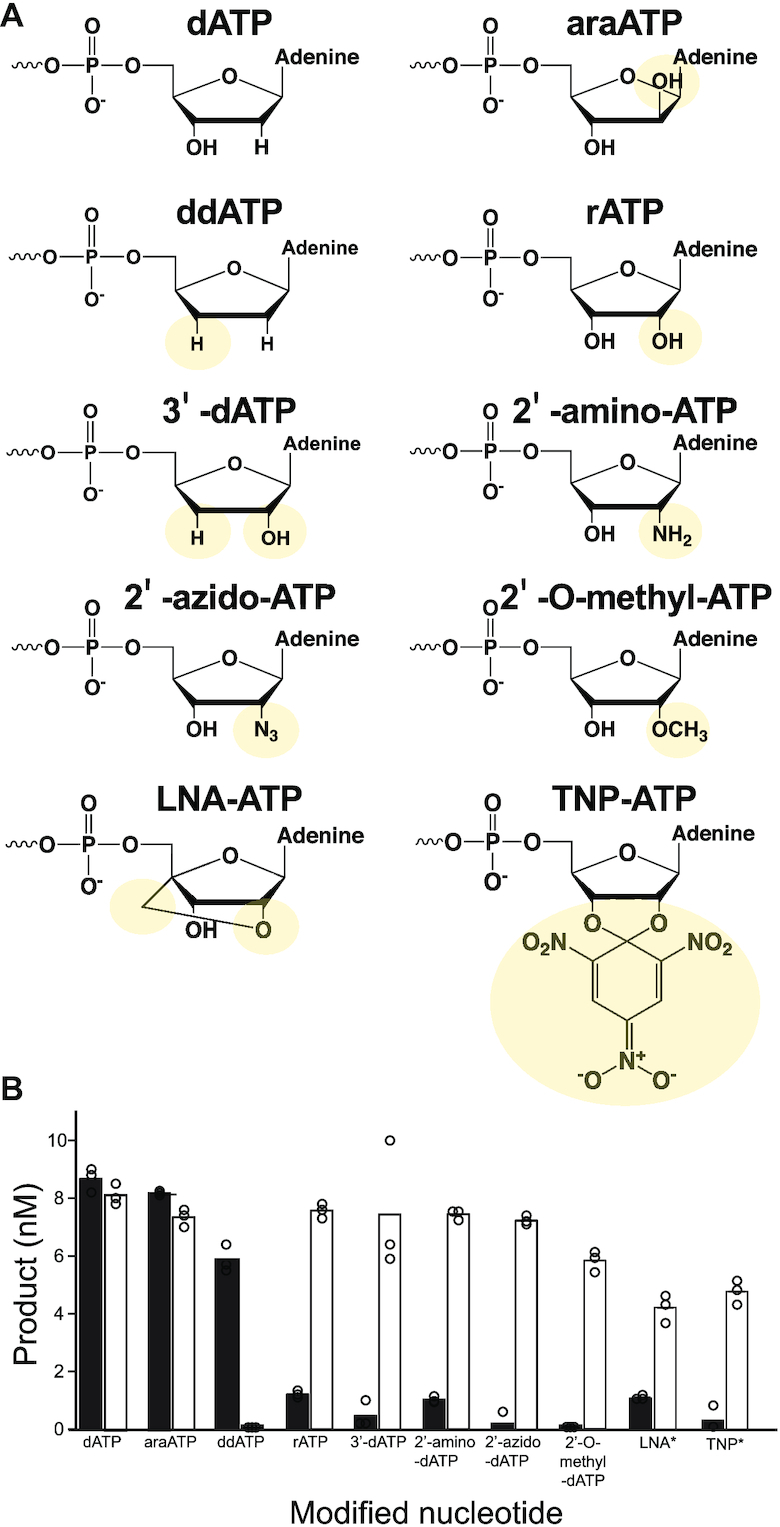
Incorporation of 2′- and 3′-modified nucleotides by WT and H931A PolD. (A) Structure of canonical dATP, 2′-, and 3′-modified dATPs where yellow circle denotes presence of modification. (B) Histogram displaying the amount of 51 nt product produced after 1 or 15 min (denoted with *) incubation of 50nt primer/67 nt template with dATP, 2′- or 3′-modified ATPs and WT PolD (black bars) or PolDH931A (white bars).
In general, for WT PolD, the size of the 2′-group determined the level of nucleotide incorporation, where larger 2′-groups (2′-OH, 2′-NH2, 2′-N3 and 2′-OCH3) were poorly incorporated, presumably due to a steric clash between the 2′-group and H931, while small 2′-groups (2′-H and 2′-ara-OH) were efficiently incorporated (Figure 5B). Supporting its role as a steric gate amino acid, the mutant PolD H931A efficiently incorporated nucleotides regardless of the 2′-group size (Figure 5B). Even large 2′-modifications such as 2′-N3 and 2′-OCH3 were efficiently incorporated demonstrating mutation H931A reduces active site steric clashes with 2′ modifications. PolD H931A was also able to incorporate the bulky 2′-2,4,6-trinitrophenol(TNP)-ATP and locked nucleic acid(LNA)-ATP after extended incubation time (15 min), while WT PolD was unable to efficiently incorporate either TNP or LNA-ATP. Importantly, both TNA-ATP and LNA-nucleotides have important biotechnological applications, including to use of LNA nucleotides to stabilize RNA aptamers that are used as therapeutics (58). We therefore speculate PolD H931A also has potential biotechnological applications for the synthesis of nucleic acids containing bulky nucleotides. It is important to note, neither WT PolD nor the H931A mutant were able to incorporate biotin-14-ATP, in which a large biotin moiety extends off the adenosine nucleobase (data not shown). Such results confirm the H931A mutation opens the PolD active site only by the ribose ring of the incoming nucleotide.
In WT PolD, we predict that a hydrogen bond between H931 imidazole N3 and the 3′-OH of the incoming nucleotide could stabilize and position the nucleotide in the proper geometry for efficient incorporation. The kinetics, described above, demonstrated PolD H931A incorporates dATP less efficiently compared to WT PolD likely due to loss of the hydrogen bond between H931 with the 3′-OH. We used 3′-modified ddATP, which lacks both 2′ and 3′-OH, to explore the role H931 plays in stabilizing the incoming nucleotide during incorporation (Figure 5A). Incorporation of ddATP by WT PolD was less efficient than dATP, presumably due to loss of a hydrogen bond between H931 and the 3′-OH of the incoming nucleotide (Figure 5B). We observed further decrease of ddATP incorporation when the active site hydrogen bonding network was further disrupted by loss of H931 to alanine (Figure 5B). Further structural analysis of PolD with bound nucleotide will shed light on the active site hydrogen bonding network within the PolD active site.
DISCUSSION
With the recent structural elucidation of Family D DNA polymerase, DNAPs are now divided into three groups based on the structure of their catalytic core. Further, replicative DNAPs from Bacteria, Archaea and Eukarya are not homologous and the origin of DNA replication remains a mystery (59). The recently discovered evolutionary connection between PolD and the universally conserved RNAP bridges together in cellular life DNA transcription and DNA replication within the same protein superfamily. The first RNAP transcriptase and the first replicative DNAP probably evolved from a two-DPBB common ancestor that functioned as an RNA-dependent RNAP (Figure 6). The ability of this catalytic core to acquire novel nucleotide polymerization activities, such as a steric gate, may have allowed DNA replication and transcription to jointly evolve from a common catalytic core, rather than being invented separately. Here, through both structural and functional characterization, we have constructed the first putative nucleotide-bound PolD active site model and reveal it bears a striking similarity with well-established msRNAP nucleotide-bound active site, with one critical difference, the presence of a steric gate histidine residue in PolD (Figure 2C). This putative active site supports the theory of a jointly evolving DPBB catalytic core through conserved catalytic motifs and the discovery and characterization of a novel motif which determines the nucleotide incorporation activity of these polymerases. The capacity of PolD to use RNA-primed DNA may be a property inherited from its RNA polymerase ancestor (48). PolD has also recruited two ancestral nucleic acids binding domains, a KH-domain (16) and an oligo-nucleotide binding domain (named OB) (Figure 6). Interestingly, the OB domain of PolD shows the highest similarity to the OB domain of class-II aminoacyl tRNA synthetases (14), thereby further linking PolD to the RNA world. Further acquisition of additional domains by PolD, including the calcineurin-like phosphoesterase DP1 exonuclease domain, has allowed PolD to evolve into the essential replicative polymerase found across the majority of archaeal phylum.
Figure 6.

Proposed scenario for the origin and early evolution of DNA replication and transcription from a common two-barrel nucleotide polymerase ancestor.
The acquisition of a steric gate amino acid by PolD was an essential evolutionary step to minimize incorporation of ribonucleotides during DNA synthesis and to maintain the archaeal genome (60,61). Previous studies have shown that 1 rNTP per 2,000 nt in T. kodakarensis and 1 rNTP per 700 nt in Thermococcus barophilus are incorporated during replication (25,62,63). Importantly, the vast majority of rNTP incorporated during replication are then repaired by the ribonucleotide excision repair (RER) pathway (62). Ribonucleotides embedded in the genome have severe consequences and can lead to strand breaks, chromosomal rearrangements, replication fork stalling and activation of DNA damage response (64). A steric gate is especially critical due to the large cellular excess of rNTPs compared to dNTPs in the nucleotide pool (25,65). Further, many archaeal species that encode PolD, including Thermococcus sp. 9°N, thrive at high temperatures which facilitates strand breaks at labile bases like ribonucleotides; therefore, while all other DNA polymerase families contain steric gate residues to prevent ribonucleotide incorporation, the presence of a steric gate in the replicative PolD is especially critical for archaeal genome maintenance. Other reported steric gate amino acids are tyrosine, phenylalanine and glutamate (26). Structural studies on Family C DNA polymerases propose a histidine residue is the steric gate due to its structural proximity to the 2′ position of the bound nucleotide, however, mutational studies have not confirmed this hypothesis (66,67). In this work, extensive site-directed mutagenesis of residues bordering the PolD active site reveal a single histidine residue is crucial for nucleotide selectivity. The mutation of H931 to a variety of different amino acids reveal a clear correlation between amino acid side chain size and ribonucleotide incorporation efficiency (Figure 3B), and kinetics reveal abolished ribonucleotide discrimination in a PolD H931A mutant (Figure 4). Taken together, these results provide substantial evidence for H931 as the PolD steric gate. We further speculate H931 not only blocks rNTPs due to a clash between C2 of the histidine imidazole ring with the 2′-OH of an incoming ribonucleotide ribose ring, but uses this histidine residue to position dNTPs in the correct geometry for efficient incorporation by hydrogen bonding between N3 of the imidazole ring and the 3′-OH of an incoming dNTP deoxyribose ring. However, a structure of nucleotide bound PolD is required to clarify the steric gate mechanism of H931 at the molecular level. It is intriguing that H931A PolD has the ability to accommodate and incorporate bulky 2′ modified nucleotides, such as LNA-ATP and TNP-ATP (Figure 5). A structure of PolD bound to LNA-ATP and TNP-ATP would provide an understanding of how the H931A mutation alters the PolD active site for bulky nucleotide accommodation. Additionally, like PolD H931A, Family B DNA polymerase variants containing a small number of active site mutations, including the steric gate, can be engineered to incorporate bulky modified nucleotides used to synthesize synthetic molecules (68).
Despite similarities between the active sites of PolD and msRNAPs, and efficient single rNTP incorporation by PolD H931A, the loss of the PolD steric gate does not allow PolD to function as an RNA polymerase for RNA synthesis. In fact, incorporation of successive ribonucleotides is limited to short products suggesting that other (yet unknown) secondary PolD structural checkpoints sense A-form vs. B-form DNA and limit synthesis of long RNA transcripts. These checkpoints likely evolved in combination with the steric gate to confirm the DNAP activity of PolD. Additional structural and mutational studies will be required to identify and understand PolD amino acids responsible for limiting RNA synthesis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Danielle Fuchs, Laurie Mazzola and Kristen Augulewicz for technical assistance; Clément Madru for help with preparing the figures; William Jack, Zvi Kelman, Alexandra Gehring, Katharina Bilotti and John Pryor for helpful manuscript revision and discussion; and Don Comb for fostering a supportive research environment.
Contributor Information
Kelly M Zatopek, New England Biolabs, 240 County Road Ipswich, MA 01938, USA.
Ece Alpaslan, New England Biolabs, 240 County Road Ipswich, MA 01938, USA.
Thomas C Evans, Jr., New England Biolabs, 240 County Road Ipswich, MA 01938, USA
Ludovic Sauguet, Institut Pasteur, Unité de Dynamique Structurale des Macromolécules, 75015 Paris, France.
Andrew F Gardner, New England Biolabs, 240 County Road Ipswich, MA 01938, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale de la Recherche [ANR-17-CE11-0005-01]; Institut Pasteur; New England Biolabs. Funding for open access charge: New England Biolabs.
Conflict of interest statement. K.M.Z., E.A., T.C.E and A.F.G. are employed and funded by New England Biolabs, Inc., a manufacturer and vendor of molecular biology reagents, including DNA replication and repair enzymes. This affiliation does not affect the authors' impartiality, objectivity of data generation or its interpretation, adherence to journal standards and policies or availability of data.
REFERENCES
- 1. Kornberg A., Baker T.A.. DNA Replication. 2005; University Science Books. [Google Scholar]
- 2. Bebenek K., Kunkel T.. Function of DNA polymerases. Adv. Protein Chem. 2004; 69:137–165. [DOI] [PubMed] [Google Scholar]
- 3. Wu S., Beard W., Pederson L., Wilson S.. Structural comparison of DNA polymerase architecture suggest a nucleotide gateway to the polymerase active site. Chem. Rev. 2014; 114:2759–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raia P., Delarue M., Sauguet L.. An updated structural classification of replicative DNA polymerases. Biochem. Soc. T. 2019; 47:239–249. [DOI] [PubMed] [Google Scholar]
- 5. Ollis D., Brick P., Hamlin R., Xuong N., Steitz T.. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985; 313:762–766. [DOI] [PubMed] [Google Scholar]
- 6. Delarue M., Poch O., Todo N., Moras D., Argos P.. An attempt to unify the structure of polymerases. Protein Eng. 1990; 3:461–467. [DOI] [PubMed] [Google Scholar]
- 7. Patel P., Loeb L. A.. Getting a grip on how DNA polymerases function. Nat. Struct. Biol. 2001; 8:656–659. [DOI] [PubMed] [Google Scholar]
- 8. Bailey S., Wing R., Steitz T.. The structure of T. aquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006; 126:893–904. [DOI] [PubMed] [Google Scholar]
- 9. Werner F., Grohmann D.. Evolution of multisubunit RNA polymerases in the three domains of life. Nat. Rev. Microbiol. 2011; 9:85–98. [DOI] [PubMed] [Google Scholar]
- 10. Cramer P. Multisubunit RNA polymerases. Curr. Opin. Struct. Biol. 2002; 12:89–97. [DOI] [PubMed] [Google Scholar]
- 11. Ruprich-Robert G., Thuriaux P.. Non-canonical DNA transcription enzymes and the conservation of two-barrel RNA polymerases. Nucleic Acids Res. 2010; 38:4559–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fouqueau T., Blombach F., Werner F.. Evolutionary origins of two-barrel RNA polymerases and site-specific transcription initiation. Annu. Rev. Microbiol. 2017; 71:331–348. [DOI] [PubMed] [Google Scholar]
- 13. Forrest D. Unusual relatives of the multisubunit RNA polymerase. Biochem. Soc. T. 2018; 47:219–228. [DOI] [PubMed] [Google Scholar]
- 14. Sauguet L., Raia P., Henneke G., Delarue M.. Shared active site architecture between archaeal PolD and multi-subunit RNA polymerases revealed by X-ray crystallography. Nat. Commun. 2016; 7:12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salgado P., Koivunen M., Makeyev E., Bamford D., Stuart D., Grimes J.. The structure of an RNAi polymerase links RNA silencing and transcription. PLoS Biol. 2006; 4:e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raia P., Carroni M., Henry E., Pehau-Arnaudet G., Brule S., Beguin P., Henneke G., Lindahl E., Delarue M., Sauguet L.. Structure of the DP1-DP2 PolD complex bound with DNA and its implications for the evolutionary history of DNA and RNA polymerases. PLos Biol. 2019; 17:e3000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imamura M., Uemori T., Kato I., Ishino Y.. A non-alpha-like DNA polymerase from the hyperthermophilic archaeon Pyrococcus furiosus. Biological and Pharmaceutical Bulletin. 2019; 18:1647–1652. [DOI] [PubMed] [Google Scholar]
- 18. Ishino Y., Komori K., Cann I., Koga Y.. A novel DNA polymerase family found in Archaea. J. Bacteriol. 1998; 180:2232–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takashima N., Ishino S., Takafuji M., Yamagami T., Matsuo R., Mayanagi K., Ishino Y.. Elucidating functions of DP1 and DP2 subunits from the Thermococcus kodakarensis family D DNA polymerase. Extremophiles. 2019; 23:161–172. [DOI] [PubMed] [Google Scholar]
- 20. Berquist B., DasSarma P., DasSarma S.. Essential and non-essential DNA replication genes in the model halophilic Archaeon, Halobacterium sp. NRC-1. Extremophiles. 2007; 23:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cubonova L., Richardson T., Burkhart B., Kelman Z., Bernard C., Reeve J., Santangelo T.. Archaeal DNA polymerase D but not DNA polymerase B is required for genome replication in Thermococcus kodakarensis. J. Bacteriol. 2013; 195:2322–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Birien T., Thiel A., Henneke G., Flament D., Moalic Y., Jebbar M.. Development of an effective 6-methylpurine counterselection marker for genetic manipulation in Thermococcus barophilus. Genes. 2018; 9:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Castelle C., Banfield J.. Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell. 2018; 172:1181–1197. [DOI] [PubMed] [Google Scholar]
- 24. Schermerhorn K., Gardner A.. Pre-steady-state kinetic analysis of a family D DNA polymerase from Thermococcus sp. 9°N reveals mechanisms for archaeal genomic replication and maintenance. J. Biol. Chem. 2015; 290:21800–21810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lemor M., Kong Z., Henry E., Brizard R., Laurent S., Bosse A., Henneke G.. Differential activities of DNA polymerases in processing ribonucleotides during DNA synthesis in archaea. J. Mol. Biol. 2018; 430:4908–4924. [DOI] [PubMed] [Google Scholar]
- 26. Brown J., Suo Z.. Unlocking the sugar ‘Steric Gate’ of DNA polymerases. Biochemistry. 2011; 50:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson M., Kottur J., Deepak T.. A polar filter in DNA polymerases prevents ribonucleotide incorporation. Nucleic Acids Res. 2019; 47:10693–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Madeira F., Park Y., Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A., Potter S., Finn R., Lopez R.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waterhouse A., Procter J., Martin D., Clamp M., Barton G.. TJalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peterson E., Goddard T., Huang C., Couch G., Greenblatt D., Meng E., Ferrin T.. UCSF chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 2009; 25:1605–1614. [DOI] [PubMed] [Google Scholar]
- 31. Greenough L., Menin J., Desai N., Kelman Z., Gardner A.. Characterization of family D DNA polymerase from Thermococcus sp. 9°N. Extremophiles. 2014; 18:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenough L., Kelman Z., Gardner A.. The roles of family B and D DNA polymerases in Thermococcus species 9°N Okazaki fragment maturation. J. Biol. Chem. 2015; 290:12514–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greenough L., Schermerhorn K., Mazzola L., Bybee J., Rivizzigno D., Cantin E., Slatko B., Gardner A.. Adapting capillary gel electrophoresis as a sensitive, high-throughput method to accelerate characterization of nucleic acid metabolic enzymes. Nucleic Acids Res. 2016; 44:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sauguet L. The extended ‘Two-barrel’ polymerases superfamily: structure, function and evolution. J. Mol. Biol. 2019; 431:4167–4183. [DOI] [PubMed] [Google Scholar]
- 35. Madru C., Henneke G., Raia P., Hugonneau-Beaufet I., Pehau-Arnaudet G., England P., Lindahl E., Delarue E., Carroni M., Sauguet L.. Structural basis for the increased processivity of D-family DNA polymerases in complex with PCNA. Nat. Commun. 2020; 11:1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vassylyev D., Sekine S.-I., Laptenko O., Lee J., Vassylyeva M., Borukhov S., Yokoyama S.. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature. 2002; 417:712–719. [DOI] [PubMed] [Google Scholar]
- 37. Zaychikov E., Martin E., Denissova L., Kozolv M., Markovtsov V., Kashelv M., Nikiforov V., Goldfarb A., Mustaev A.. Mapping of catalytic residues in the RNA polymerase active center. Science. 1996; 273:107–109. [DOI] [PubMed] [Google Scholar]
- 38. Treich I., Carles C., Sentenac A., Riva M.. Determination of lysine residues affinity-labeled in the active site of yeast RNA polymerase II(B) by mutagenesis. Nucleic Acids Res. 1992; 20:4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cramer P., Bushnell D., Kornberg R.. Structural basis of transcription: RNA polymerase at 2.8 Å resolution. Science. 2001; 292:1863–1876. [DOI] [PubMed] [Google Scholar]
- 40. Kashlev M., Lee J., Zalenskaya K., Nikiforov V., Goldfard A.. Blocking of the initiation-to-elongation transition by a transdominant RNA polymerase mutation. Science. 1990; 248:1006–1009. [DOI] [PubMed] [Google Scholar]
- 41. Vassylyev D., Vassylyeva M., Zhang J., Palangat M., Artsimovitch I., Landick R.. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007; 448:163–169. [DOI] [PubMed] [Google Scholar]
- 42. Mäkinen J., Shin Y., Vieras E., Virta P., Metsä-Ketelä M., Murakami K., Belogurov G.. The mechanism of the nucleo-sugar selection by multi-subunit RNA polymerases. 2020; bioRxiv doi:01 July 2020, preprint: not peer reviewed 10.1101/2020.06.30.179606. [DOI] [PMC free article] [PubMed]
- 43. Cramer P., Bushnell D., Kornberg R.. Molecular Structures of transcribing RNA polymerase I. Mol. Cell. 2016; 64:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang D., Bushnell D., Westover K., Kaplan C., Kornberg R.. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006; 127:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vorlander K., Khatter H., Wetzel R., Hagen W., Muller C.. Molecular mechanism of promoter opening by RNA polymerase III. Nature. 2018; 553:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jun S., Hirata A., Kanai T., Santangelo T., Imanaka T., Murakami K.. The X-ray crystal structure of the euryarchaeal RNA polymerase in an open-clamp configuration. Nat. Commun. 2014; 5:5132–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang J., Mishanina T., Bellecourt M., Mooney R., Darst S., Landick R.. RNA polymerase accommodates a pause RNA hairpin by global conformational rearrangements that prolong pausing. Mol. Cell. 2018; 69:802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Henneke G., Flament D., Hubscher U., Querellou J., Raffin J.. The hyperthermophilic euryarchaeota Pyrococcus abyssi likely requires the two DNA polymerases D and B for DNA replication. J. Mol. Biol. 2005; 350:53–64. [DOI] [PubMed] [Google Scholar]
- 49. Gueguen Y., Rolland J.-L., Lecompte O., Azam P., Le Romancer G., Flament D., Raffin J.-P., Dietrich J.. Characterization of two DNA polymerases from the hyperthermophilic euryarchaeon Pyrococcus abyssi. Eur. J. Biochem. 2001; 268:5961–5969. [DOI] [PubMed] [Google Scholar]
- 50. Shen Y., Musti K., Hiramoto M., Kikuchi H., Kawarabayashi Y., Matsui I.. Invariant Asp-1122 and Asp-1124 are essential residues for polymerization catalysis of family D DNA polymerase from Pyrococcus horikoshii. J. Biol. Chem. 2001; 276:27376–27383. [DOI] [PubMed] [Google Scholar]
- 51. Mills K., Johnson M., Perler F.. Protein splicing: how inteins escape from precursor proteins. J. Biol. Chem. 2014; 289:14498–14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Minteer C., Siegart N., Colelli K., Liu X., Linhardt R., Wang C., Gomez A., Reitter J., Mills K.. Intein-promoted cyclization of aspartic acid flanking the intein leads to atypical N-terminal cleavage. Biochemistry. 2017; 56:1042–1050. [DOI] [PubMed] [Google Scholar]
- 53. Astatka M., Ng K., Grindley N., Joyce C.. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Avad. Sci. U.S.A. 1998; 95:3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang G., Franklin M., Li J., Lin T., Konigsberg W.. A conserved Tyr residue is required for sugar selectivity in a Pol alpha DNA polymerase. Biochemistry. 2002; 41:10256–10261. [DOI] [PubMed] [Google Scholar]
- 55. Delucia A., Grindley N., Joyce C.. An error-prone family Y DNA polymerase (DinB homolog from Sulfolobus solfataricus) uses a ‘steric gate’ residue for discrimination against ribonucleotides. Nucleic Acids Res. 2003; 31:4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gardner A., Jack W.. Determinants of nucleotide sugar recognition in an archaeon DNA polymerase. Nucleic Acids Res. 1999; 27:2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cozens C., Pinheiro V., Vaisman A., Woodgate R., Holliger P.. A short adaptive path from DNA to RNA polymerases. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8067–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schmidt K., Borkowski S., Kurrek J., Stephens A., Bald R., Hecht M., Friebe M., Dinkelborg L., Erdman V.. Application of locked nucleic acids to improve aptamer in vivo stability and targeting function. Nucleic Acids Res. 2004; 32:5757–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koonin E., Krupovic M., Ishino S., Ishino Y.. The replication machinery of LUCA: common origin of DNA replication and transcription. BMC Biol. 2020; 9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Williams J., Kunkel T.. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair. 2014; 19:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Williams J., Lujan S., Kunkel T.. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell. Biol. 2016; 17:350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Heider M., Burkhart B., Santangelo T., Gardner A.. Defining the RNaseH2 enzyme-initiated ribonucleotide excision repair pathway in Archaea. J. Biol. Chem. 2017; 26:8835–8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zatopek K., Potapov V., Maduzia L., Alpaslan E., Chen L., Evans T., Ong J., Ettwiler L., Gardner A.. RADAR-seq: a RAre DAmage and repair sequencing method for detecting DNA damage on a genome-wide scale. DNA Repair. 2019; 80:36–44. [DOI] [PubMed] [Google Scholar]
- 64. Reijns M., Rabe B., Rigby R., Mill P., Astell K., Lettice L., Boyle S., Leitch A., Keighren M., Kilanowski F. et al.. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012; 149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Traut T. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994; 140:1–22. [DOI] [PubMed] [Google Scholar]
- 66. Evans R.J., Davies D.R., Bullard J.M., Christensen J., Green L.S., Guiles J.W., Patac J.D., Ribblea W.K., Janjica N., Jarvis T.C.. Structure of PolC reveals unique DNA binding and fidelity determinants. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:20695–20700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Parasuram R., Coulther T., Hollander J., Keston-Smith E., Ondrechen M., Beuning P.. Prediction of active site and distal residues in E. coli DNA polymerase III alpha polymerase activity. Biochemisty. 2018; 57:1063–1072. [DOI] [PubMed] [Google Scholar]
- 68. Gardner A., Jackson K., Boyle M., Buss J., Potapov V., Gehring A., Zatopek K., Correa I., Ong J., Jack W.. Therminator DNA polymerase: modified nucleotides and unnatural substrates. Front. Mol. Biosci. 2019; 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.