

Abstract
Meiotic recombination enables reciprocal exchange of genetic information between parental chromosomes and is essential for fertility. MEIOB, a meiosis-specific ssDNA-binding protein, regulates early meiotic recombination. Here we report that the human infertility-associated missense mutation (N64I) in MEIOB causes protein degradation and reduced crossover formation in mouse testes. Although the MEIOB N64I substitution is associated with human infertility, the point mutant mice are fertile despite meiotic defects. Meiob mutagenesis identifies serine 67 as a critical residue for MEIOB. Biochemically, these two mutations (N64I and S67 deletion) cause self-aggregation of MEIOB and sharply reduced protein half-life. Molecular genetic analyses of both point mutants reveal an important role for MEIOB in crossover formation in late meiotic recombination. Furthermore, we find that the MEIOB protein levels directly correlate with the severity of meiotic defects. Our results demonstrate that MEIOB regulates meiotic recombination in a dosage-dependent manner.
INTRODUCTION
Meiotic recombination, a hallmark of meiosis, is essential for faithful segregation of homologous chromosomes during the first meiotic cell division and enhances genetic diversity in gametes (1,2). This process begins with the formation of a large number of programmed DNA double strand breaks (DSBs) in germ cells (∼300 per cell in mouse), which are generated by SPO11 (3,4). DSBs are resected to produce 3′ single-stranded DNA (ssDNA) overhangs (2). One DSB ssDNA end invades the homologous chromosomes to produce the displacement loop (D-loop). The capture of the second DSB end results in the formation of double Holliday junctions (dHJs). Resolution of dHJs produces crossovers (∼22 crossovers per cell in mouse). Failures in meiotic recombination entail blockade in meiosis and thus sterility.
Meiotic recombination is temporally executed by a large number of proteins including ssDNA-binding proteins (1,2). These proteins typically localize to DSBs as foci on meiotic chromosomes. RPA, consisting of RPA1, RPA2 and RPA3, is a ubiquitously expressed ssDNA-binding complex (5,6). RPA coats ssDNA to prevent formation of secondary structures and protect ssDNA from degradation. Loss of RPA1 causes a failure in meiotic recombination (7). RAD51 and DMC1, ssDNA-binding recombinases, bind to DSB ssDNA to drive strand invasion into homologues, resulting in the formation of D-loops (8–12). Hop2-Mnd1 proteins stimulate the RAD51/DMC1 recombinase activity (13). HSF2BP is a meiosis-specific localizer of BRCA2, which in turn recruits RAD51/DMC1 to DSBs (14,15). RPA interacts with RAD51 and DMC1, suggesting that RPA may recruit RAD51/DMC1 to meiotic DSB ssDNA (16). This notion is supported by the loss of RAD51/DMC1 foci in RPA1-deficient spermatocytes (7). MEIOB, an ssDNA-binding protein, is a meiosis-specific paralogue of RPA1 (17,18). MEIOB forms a heterodimer with SPATA22 and the MEIOB/SPATA22 heterodimer interacts with the RPA heterotrimer (17,19). The protein stability of MEIOB and SPATA22 depends on each other in both testis and transfected cells (17,19). When MEIOB is absent or reduced, SPATA22 is absent or reduced as well and vice versa. Although the MEIOB/SPATA22 complex and RPA colocalize as foci on meiotic chromosomes, their focal localizations are independent of each other (7,17). Unlike RPA, MEIOB/SPATA22 are not required for the recruitment of RAD51/DMC1 to DSB sites (17,18,20–23). Instead, MEIOB and SPATA22 appear to function in the second-end capture to stabilize recombination intermediates after strand invasion. Several proteins such as MSH4, MSH5 and TEX11 are involved in processing of recombination intermediates (24–26). In particular, the MSH4/MSH5 complex recognizes dHJ structures (24). The MLH1/MLH3 complex exhibits endonuclease activity and is required for formation of the majority of crossovers (27–30). Mus81 generates the remaining small subset of crossovers (31,32). Other proteins such as RNF212, HEI10 and CNTD1 also regulate crossover formation (33–36). Genetic studies have shown that lack of crossovers results in formation of univalent chromosomes, followed by chromosome mis-segregation and subsequent apoptosis of meiotic germ cells. In addition, HEI10, RNF212 and TEX11 function in crossover formation in a dosage-sensitive manner (33,37,38). This dosage sensitivity may have implications in modulation of genome-wide recombination rates.
Meiosis-specific proteins have been studied genetically in model organisms from yeast to mouse. However, our understanding of their role in human meiosis is limited (39). Mutations in several meiosis-specific genes have been found in humans with primary ovarian insufficiency or non-obstructive azoospermia (39). For example, a homozygous splice site mutation in human MSH4 causes primary ovarian insufficiency (40). Mutations in the X-linked TEX11 gene cause azoospermia in men (38,41). Several human studies reported homozygous mutations in the human MEIOB gene. A frame-shift mutation in MEIOB causes primary ovarian insufficiency (42). A different frame-shift MEIOB mutation is a recurrent cause of azoospermia in men (43). Interestingly, a homozygous missense mutation (N64I) in MEIOB was found in azoospermic men (44). In this study, we generated a mouse mutant to model the human MEIOB N64I mutation. Our biochemical studies reveal that the N64I mutation causes MEIOB self-dimerization and degradation. Unexpectedly, cell biological studies uncover a distinct role for MEIOB in crossover formation. Importantly, genetic studies of various combinations of Meiob mutant alleles demonstrate that MEIOB functions in meiotic recombination in a dosage-dependent manner.
MATERIALS AND METHODS
Generation of MeiobN64I Mice
The Meiob N64I point mutation was generated by the CRISPR/Cas9-mediated genome editing (Supplementary Figure S1B). Guide RNA (sgRNA) was cloned into the px330 vector, in vitro transcribed and purified as described in the protocol (45). The sgRNA flanks codon 64 (underlined): ACTTTGTGAATGTATCCTCC. The single-stranded oligodeoxyribonucleotide (ssODN) template contains the N64I mutation (AAT to ATC): 5′-GGAATATAAAAGCGTTGGTTTTCCTCAGATATTGGATCAGAAAGATACACTTTCAGTTTCACCATTCGTGACTCACCATGTCACTTTGTGATCGTATCCTCCTGGGGCAGTGAGGACTACATCCGCTCGCTGTCCGAGAGCTTCAGGGTTGCTGAGTGTGGTAAGTAGGCAGGATTCTTTTGTTTGTTTG-3′. A mixture of Cas9 mRNA (Trilink, 100 ng/μl), sgRNA (50 ng/μl) and ssODN DNA (100 ng/μl) was used for microinjection of 50 zygotes on the DBA x C57BL/6 hybrid genetic background. Out of 20 founders, the following genotypes were obtained: 8 MeiobN64I/FS (2 males, 6 females) (FS, frame-shift mutation), 2 MeiobN64I/N64I (1 male, 1 female), 2 MeiobN64I/ΔS67 (1 male, 1 female), 2 MeiobΔS67/ΔS67 (2 males) and 2 MeiobΔS67/FS (1 male, 1 female). Founder MeiobN64I/FS females were mated to wild type C57BL/6J males to obtain heterozygous MeiobN64I/+ mice. Germline transmission of the point mutation was validated by PCR and sanger sequencing (PCR primers 5′-GCATGCAGAACCAAAGCTAAATAA-3′ and 5′-GAGATAGGGTTTTACTATATAACA-3′; PCR product, 451 bp). The MeiobN64I allele was genotyped by PCR (216 bp) with primers 5′-GACTCACCATGTCACTTTGTGATC-3′ and 5′-GTGGCTGGATTGAAACTGGTTATG-3′. The wild type Meiob allele was genotyped by PCR (216 bp) with primers 5′-GACTCACCATGTCACTTTGTGAAT-3′ and 5′-GTGGCTGGATTGAAACTGGTTATG-3′.
Generation of MeiobΔS67 mice
The targeting vector was constructed as illustrated (Supplementary Figure S2C). The codon 67 deletion was introduced by overlapping PCR (46). The homologous arms were amplified from a Meiob-containing BAC clone (RP23–397O23) by PCR with high-fidelity DNA polymerase. V6.5 ES cells on the 129 × C57BL/6 hybrid genetic background were electroporated with the linearized targeting vector and cultured in the presence of 350 μg/ml G418 (postive selection) and 2 μM ganciclovir (negative selection). Homoglously targeted ES clones were screened by long-distance PCR. Two targeted ES cell clones (1A12 and 1B11) were injected into B6C3F1 (Taconic) blastocysts. Male chimeras from both clones were mated with ubiquitous Actb-Cre females (FVB genetic background) to remove the floxed PGK-neo selection cassette and germline transmission were obtained from both clones (47). The codon 67 deletion was confirmed by sequencing. The wild type (397 bp) and MeiobΔS67 (495 bp) alleles were genotyped by PCR with the following primers: 5′-CTTTCAGTTTCACCATTCGTGACT-3′ and 5′-AAGGAAGTTACCTAGCAAGTCTCA-3′.
Histological, surface nuclear spread, and TUNEL analyses
For histological analysis, testes and ovaries were fixed in Bouin's solution overnight, embedded with paraffin, and sectioned. Sections were stained with hematoxylin and eosin. For TUNEL analysis, testes were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated in sucrose, embedded in OCT and sectioned. Frozen sections were analyzed with the TUNEL Enzyme and Label Kit (Roche Boehringer Mannheim). Nuclear spread analysis of spermatocytes was performed as previously described (48). The primary antibodies used for immunofluorescence were listed in Supplementary Table S1.
Imaging
Color histological images were captured on a Leica DM5500B microscope with a DFC450 digital color camera (Leica Microsystems). Immunolabeled chromosome spreads and testis TUNEL assay images were captured with an ORCA Flash4.0 digital monochrome camera (Hamamatsu Photonics) on a Leica DM5500B microscope (Leica Microsystems) and processed using the Photoshop software (Adobe).
Sperm count
Cauda epididymis was dissected and placed in a small tissue dish with 2 ml of 1× PBS buffer without calcium/magnesium. We minced epididymis with forceps and squeezed out sperm as much as possible. Samples were incubated at room temperature for 10 min to allow sperm to disperse and transferred to a 10-ml falcon tube. 2 ml PBS was used to wash the dish and then added to the same tube. 2 ml 4% paraformaldehyde (in PBS) was added to the tube to fix at room temperature for 10 min. 10 μl was loaded to the Hausser Bright-Line hematocytometer (Fisher Scientific). Sperm in the 25 small squares (a total volume of 0.1 μl) were counted and the number was multiplied by 6 × 104 to give rise to the total sperm count per male.
Expression constructs
The Meiob and Spata22 expression constructs were previously described (19). Mouse and human Meiob cDNAs were subcloned into the pcDNA6 vector (Invitrogen) harboring an N-terminal Flag tag and a C-terminal Myc tag. Mouse and human Spata22 cDNAs were subcloned into the pcDNA6 vector harboring an N-terminal Flag tag. All Meiob mutations were generated by PCR and subcloned into the pcDNA6 vector. Green fluorescent protein (GFP)-Meiob, N64I, ΔS67, Mlh1, Msh4 and Rnf212 constructs were generated by subcloning into the pEGFP-C1 vector (Clontech). Tex11-V5 construct was generated by subcloning into the pcDNA™3.1/V5-His vector (Invitrogen). All the constructs were verified by Sanger sequencing on an ABI 3730 DNA analyzer.
Cell culture and transfections
HEK293T cells were maintained in DMEM/high glucose (Mediatech) supplemented with 10% FBS (Sigma) and penicillin/streptomycin (Invitrogen). Plasmid DNA transfections in HEK293T cells were carried out using a standard calcium phosphate method. Briefly, transfections were performed when HEK293T cells are 50–90% confluent. To make 100 μl plasmid mixture, add 2 μg plasmid and 12.5 μl 2 M CaCl2. Mix the HEPES buffered saline solution (2× HEPES, pH 7.15, 100 μl) by vortexing at 300 RPM, add dropwise to the plasmid solution, and incubate at room temperature for 30 min. The entire 200 μl mixture was added directly into one well of a six-well plate in a dropwise manner. Cells were collected after 24–48 h.
Cycloheximide chase assays
HEK293T cells in one well of a six-well plate were transfected with 1 μg of flag-mMEIOB-myc, flag-mMEIOBN64I-myc, flag-mMEIOBΔS67-myc, flag-hMEIOB-myc, flag-hMEIOBN64I-myc or flag-mMEIOBIAA-myc, incubated at 37°C for 24 h, and treated with cycloheximide (20 μg/ml) for 0–24 h to inhibit de novo protein synthesis. Cells were collected in 2× SDS PAGE buffer for immunoblotting analysis with anti-flag antibody (Figure 7).
Figure 7.

Reduction in the half-life of MEIOB mutant proteins. (A) Western blot analysis and half-life of mouse MEIOB (wt), MEIOBN64I and MEIOBΔS67 in HEK293T cells treated with cycloheximide. The plot on the right shows the half-life. (B) Western blot analysis of human MEIOB (wt), human MEIOBN64I and mouse MEIOBIAA (as in Figure 6A) in HEK293T cells treated with cycloheximide. The plot on the right shows the half-life. ACTB serves as a loading control. hrs, hours after addition of cycloheximide. Experiments were done three times. Curve fitting was performed with one-phase exponential decay.
For protein degradation assays, HEK293T cells in one well of a six-well plate were transfected with 1 μg of flag-mMEIOB-myc, flag-mMEIOBN64I-myc or flag-mMEIOBΔS67-myc, incubated at 37°C for 24 h, and treated with cycloheximide (20 μg/ml) in the presence of 20 μM MG132 (M8699, Sigma) or 200 nM bafilomycin A1 (B1793, Sigma) for 0–24 h (Supplementary Figure S10).
Immunoprecipitation and immunoblotting assays
A total of 1 × 107 cells were harvested at 24–36 h post transfection and lysed in 1 ml RIPA buffer (140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM Tris–HCl, pH 8.0, and 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) supplemented with 1× protease inhibitor cocktail (Sigma). For immunoprecipitation experiments, the lysates were incubated on a rocking platform for 1 h at 4°C, followed by centrifugation at 16 100 g for 15 min at 4°C. The supernatant was transferred to a new tube, incubated with primary antibodies overnight, and then incubated with protein A or G dynabeads (Thermo Fisher Scientific) for 2–3 h. The immunoprecipitated complexes were washed with the RIPA buffer four times, boiled in 50 μl 2× SDS-PAGE loading buffer for 12 min, and subjected to PAGE electrophoresis and western blotting. For Western blot analysis, testes or cells were homogenized in 500 μl protein extraction buffer (62.5 mM Tris–HCl (pH 6.8), 3% SDS, 10% glycerol, 5% 2-mercaptoethanol). About 30 μg of protein extracts were resolved by SDS-PAGE, transferred onto nitrocellulose membranes using iBlot (Invitrogen), and immunoblotted with indicated antibodies (Supplementary Table S1).
Quantitative RT-PCR assays
Total RNA was extracted from postnatal day 20 testes with Trizol reagent (Thermo Fisher Scientific). 1 μg of total RNA was treated with DNaseI (Amp grade 1 U/μl, Invitrogen RNase free) and was reverse transcribed with PrimeScript™RT Master Mix (RR036A, TaKaRa, Japan). For quantitative RT-PCR, diluted cDNA was used for real-time PCR reactions containing SYBR Green Premix Ex Taq II (RR820A, TaKaRa). Gene expression was normalized to Actb (β-actin). The Meiob mRNA was assayed using primers 5′-CTGCATGGCAGCAACTGTAATCTC-3′ and 5′-GCATAAAGGATGCCATAGAAAGGA-3′ (exon 9–11). The Spata22 mRNA was assayed using primers 5′-GCCTGTGCCATTGTTCAATCAGAA-3′ and 5′-CGCCCAATCTGTTGGAAAAGAAGG-3′ (exon 3–4).
Statistics
All data were analyzed using GraphPad Prism (GraphPad Software Inc). All data are reported as mean ± SD. Statistical analysis was performed with Student's t-test.
RESULTS
The human MeiobN64I mutation destabilizes the MEIOB protein in testis
The human MEIOBN64I missense mutation was found in azoospermic men with meiotic arrest (44). The N64 residue is conserved in MEIOB orthologues across species including Drosophila, Xenopus, and mouse (Supplementary Figure S1A). To study the requirement of this residue, we generated MeiobN64I mutation in mice by CRISPR-mediated genome editing (Supplementary Figure S1B). In contrast with the meiotic arrest observed in infertile men, the MeiobN64I/N64I males had normal testis size, weight, and sperm count (Figure 1A–C), highlighting the species-specific requirement of this residue. We reduced the gene dosage by crossing this point mutant allele with the Meiob null allele, which was previously generated (17). Interestingly, the MeiobN64I/− males exhibited significant reduction in both testis weight and sperm count (Figure 1B and C), suggesting that MEIOB is a dosage-sensitive regulator of spermatogenesis. The abundance of MEIOB protein was reduced by >70% in the MeiobN64I/N64I testes and by 90% in MeiobN64I/− testes (Figure 1D). Accordingly, SPATA22, the obligate MEIOB-binding partner, was also dramatically reduced in abundance, whereas the abundance of RPA2, which interacts with MEIOB, remained relatively constant (Figure 1D) (17,19). These data suggest that the N64I substitution causes destabilization of the MEIOB protein and subsequent destabilization of SPATA22 in vivo.
Figure 1.

The Meiob N64I mutation causes stage XII-specific meiotic defects in males. (A) Testis size of 8-week-old MeiobN64I/+, MeiobN64I/N64I and MeiobN64I/− mice. (B, C) Reduced testis weight (B) and sperm count (C) in MeiobN64I/− males. n = 3, 8-week-old males. Data are presented as mean ± S.D., *P < 0.05 by Student's t-test. NS, non-significant. (D) The N64I substitution destabilizes the MEIOB protein in testes (postnatal day 20, P20). Band quantification was normalized to β-actin (ACTB). Western blotting was performed in triplicates. *P < 0.05. (E) Histological analysis of testes at P20 and P60. Stage XII tubules are shown. Apoptotic metaphase spermatocytes are marked by black arrowheads. Scale bar, 20 μm. (F) TUNEL analysis of testicular sections at P20. H3S10ph (Phosphorylated H3 at serine 10) is a metaphase marker. Scale bar, 20 μm. (G) Quantification of TUNEL-positive spermatocytes at P20. n, the number of TUNEL-positive stage XII tubules (TUNEL-negative stage XII tubules were excluded). Three mice per genotype were examined. *P < 0.05 by Student's t-test.
Meiob N64I mutation causes apoptosis in metaphase I spermatocytes in mice
Meiotic cell divisions occur at the stage XII of seminiferous tubules, which contain metaphase spermatocytes. Histological analysis revealed a sharply increased number of apparently apoptotic spermatocytes only in stage XII tubules in the MeiobN64I/N64I testes at postnatal day 20 (P20; juvenile) and P60 (adult), in contrast with the absence or rare appearance of apoptotic cells in the MeiobN64I/+ testes (Figure 1E). TUNEL analysis and quantification confirmed this observation (Figure 1F). In addition, the number of apoptotic spermatocytes was higher in MeiobN64I/− testes than in MeiobN64I/N64I testes (Figure 1E–G), showing that the level of apoptosis increases with reduced MEIOB protein abundance.
MEIOB regulates the number of crossovers
As previously reported, inactivation of Meiob leads to an early failure in meiotic recombination, prior to the formation of crossovers (17,18). Crossovers are formed in mid to late pachytene spermatocytes. Whether MEIOB plays a second role in meiotic recombination such as crossover formation cannot be studied, because the Meiob−/− mice lack mid to late pachytene spermatocytes, due to meiotic arrest (17,18). The increased apoptosis of metaphase I spermatocytes in MeiobN64I/N64I and MeiobN64I/− testes indicates a chromosome segregation defect. HEI10, an ubiquitin E3 ligase, is required for crossover formation and localizes as foci on meiotic chromosomes in pachytene spermatocytes (Figure 2A) (34,37). We found that the number of HEI10 foci was reduced in mid to late pachytene spermatocytes from MeiobN64I/N64I testes in comparison to MeiobN64I/+ testes (Figure 2A). Each chromosome from MeiobN64I/+ spermatocytes had at least one HEI10 focus, in contrast, some chromosomes from MeiobN64I/N64I testes lacked HEI10 foci (Figure 2A). Likewise, the number of MLH1 foci was reduced in MeiobN64I/N64I spermatocytes than MeiobN64I/+ spermatocytes (Figure 2B). In addition, MeiobN64I/− spermatocytes exhibited a further reduction in the number of HEI10 and MLH1 foci than MeiobN64I/N64I spermatocytes (Figure 2A and B).
Figure 2.
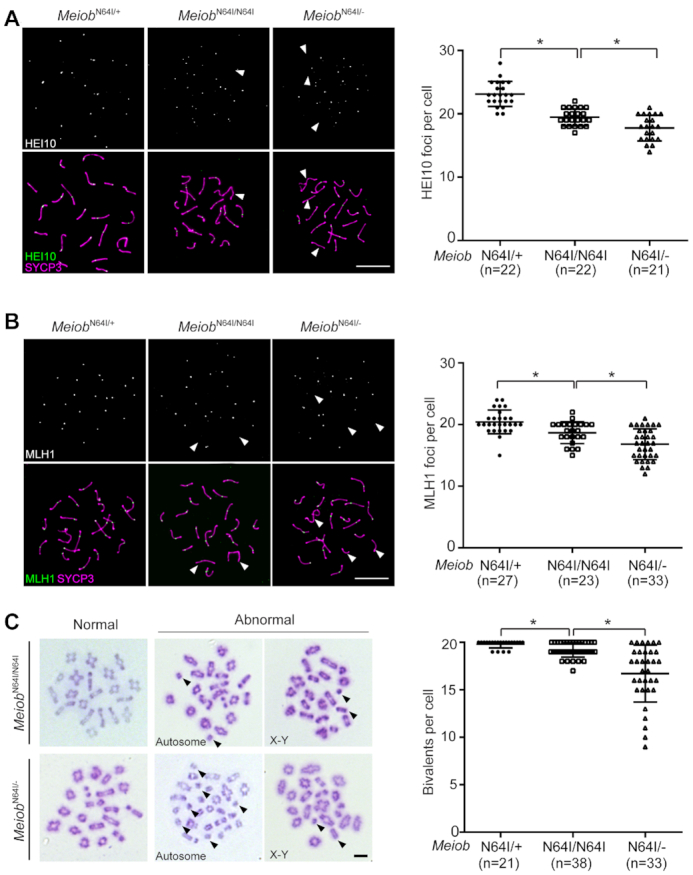
The Meiob N64I mutation reduces crossover formation. (A, B) Analysis of HEI10 foci (A), and MLH1 foci (B) in pachytene spermatocytes from wild type, MeiobN64I/N64I, and MeiobN64I/− mice at P20. n, number of mid to late pachytene spermatocytes counted. (C) Analysis of chiasmata in metaphase I spermatocytes from MeiobN64I/N64I and MeiobN64I/− testes at P20. Univalents are indicated by arrowheads. n, number of metaphase I spermatocytes counted. The data are from three to five mice per genotype. *P < 0.05 by Student's t-test. Scale bars, 10 μm.
We next examined metaphase I spermatocytes. While normal metaphase I spermatocytes with all 20 bivalents were observed in the mutant testes, the number of univalent chromosomes was significantly higher in MeiobN64I/N64I testes than MeiobN64I/+ testes (Figure 2C). Univalent chromosomes were even more prevalent in spermatocytes from MeiobN64I/− testes (Figure 2C). Both autosomes and sex chromosomes were affected by univalence. The presence of univalent chromosomes in MeiobN64I/N64I and MeiobN64I/− testes was consistent with the reduced number of MLH1 foci (the sites of crossovers) in the mutant testes. Taken together, the presence of univalent chromosomes leads to chromosome mis-segregation, activates the spindle assembly checkpoint, and thus causes increased apoptosis of metaphase I spermatocytes in MeiobN64I/N64I and MeiobN64I/− testes (Figure 1E–G). These results demonstrate that MEIOB plays a second and late role in meiotic recombination - crossover formation.
Serine 67 is critical for MEIOB function in meiosis
In the process of generating the N64I allele using the CRISPR/Cas9 approach, the serine 67 deletion allele (MeiobΔS67) was also produced. From the CRISPR injection, we obtained MeiobN64I/ΔS67 and MeiobΔS67/ΔS67 founders, both of which were sterile (Supplementary Figure S2). The adult MeiobΔS67/ΔS67 testes showed complete meiotic arrest at the pachytene-like stage (Supplementary Figure S2A) and the adult MeiobΔS67/ΔS67 ovaries lacked oocytes (Supplementary Figure S2B). The adult MeiobN64I/ΔS67 testes exhibited metaphase I arrest (Supplementary Figure S2A), a phenotype different from the Meiob null or MeiobΔS67/ΔS67 phenotypes. Therefore, the serine 67 residue may provide important insights into the function of MEIOB. However, the MeiobΔS67 allele could not be transmitted through the germline from the founders due to sterility and MeiobΔS67/+ founder mice (heterozygous) were never produced from the CRISPR injections. To validate the finding on the critical role of serine 67, we generated MeiobΔS67 allele through homologous recombination in embryonic stem (ES) cells (Supplementary Figure S2C and S2D).
While MeiobΔS67/+ males and females were fertile, both MeiobΔS67/ΔS67 males and females were sterile (Figure 3). Histological analysis showed meiotic arrest at the pachytene-like stage in the testis and lack of spermatozoa in the epididymis from MeiobΔS67/ΔS67 males (Figure 3A). The adult MeiobΔS67/ΔS67 ovary exhibited a complete loss of oocytes (Figure 3B). We further analyzed the spermatocytes from the adult mutant testes by assessing chromosomal synapsis with immunostaining of synaptonemal complex (SC) proteins SYCP1 and SYCP3 (Figure 3C–H). The percentage of zygotene-like spermatocytes was significantly higher in the MeiobΔS67/ΔS67 testes than the MeiobΔS67/+ testes. Pachytene spermatocytes with normal synapsis were abundant in MeiobΔS67/+ testes (Figure 3G and H) but absent in MeiobΔS67/ΔS67 testes. Instead, pachytene-like spermatocytes with well-formed SC lateral elements but lack of synapsis were observed in MeiobΔS67/ΔS67 testes (Figure 3F and H). γH2AX-positive XY body was present in wild type pachytene spermatocytes but absent in MeiobΔS67/ΔS67 pachytene-like spermatocytes (Figure 3I) (49). These results demonstrate a failure in chromosome synapsis in the MeiobΔS67/ΔS67 mice.
Figure 3.
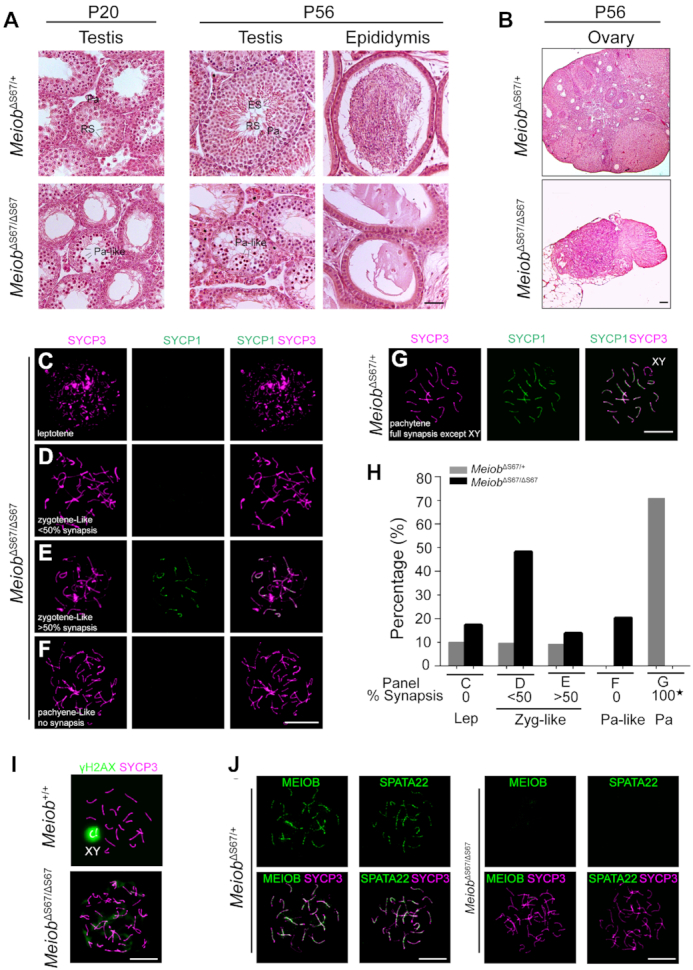
The S67 residue of MEIOB is essential for meiosis and fertility in mice of both sexes. (A) Histological analysis of testis and epididymis from P20 and 8-week-old mice. Zyg-like, zygotene-like spermatocytes; Pa, pachytene spermatocytes; RS, round spermatids; ES, elongated spermatids. Scale bar, 50 μm. (B) Histological analysis of ovaries from 8-week-old mice. Scale bar, 100 μm. (C–H) Spread nuclei of spermatocytes from 8-week-old males were immunostained with anti-SYCP1 and anti-SYCP3 antibodies. 247 MeiobΔS67/+ and 201 MeiobΔS67/ΔS67 spermatocytes were counted and categorized into different groups (three mice per genotype): leptotene (C), zygotene-like with <50% synapsis (D), zygotene-like with >50% synapsis (E), pachytene-like with no synapsis (F) and MeiobΔS67/+ pachytene spermatocytes (G). (H) Percentage of spermatocytes at each stage of meiotic prophase I with representative images shown in panels C-G. The 100% of synapsis (asterisk) in MeiobΔS67/+ pachytene spermatocytes refer to the full synapsis of all autosomes excluding XY chromosomes. (I) γH2AX localization in spermatocytes from 8-week-old males. Note the γH2AX-positive XY body in the wild type pachytene spermatocyte and cloud-like γH2AX signals in pachytene-like MeiobΔS67/ΔS67 spermatocyte. (J) MEIOB and SPATA22 foci are present in MeiobΔS67/+ late zygotene spermatocytes but absent in MeiobΔS67/ΔS67 pachytene-like spermatocytes (I). Scale bars (C–J), 10 μm.
MEIOB and SPATA22 interact with each other and form foci on SC in meiotic germ cells (Figure 3J) (17). In addition, the stability of these two proteins depends on each other in both cultured cells and testis (19,50). Interestingly, MEIOB foci were not detected in MeiobΔS67/ΔS67 spermatocytes (Figure 3J). As expected, the SPATA22 foci were also absent in MeiobΔS67/ΔS67 spermatocytes (Figure 3J). These MeiobΔS67/ΔS67 phenotypes were similar to the Meiob−/− phenotypes (17), showing that serine 67 is required for the MEIOB function.
Impaired spermatogenesis in MeiobN64I/ΔS67 males
To test the dosage effect of these two Meiob mutant alleles, we generated MeiobN64I/ΔS67 males through breeding of MeiobΔS67/+ and MeiobN64I/N64I mice in anticipation that the MEIOB protein levels in MeiobN64I/ΔS67 testis may lie between those in MeiobN64I/N64I and MeiobΔS67/ΔS67 testes. The MeiobN64I/ΔS67 testis was smaller than MeiobN64I/+ testis but larger than MeiobΔS67/ΔS67 testis (Figure 4A). Consistently, the testis weight of adult MeiobN64I/ΔS67 males was significantly lower than that of MeiobN64I/+ males but higher than that of MeiobΔS67/ΔS67 males (Figure 4B). The sperm count in MeiobN64I/ΔS67 males was much lower than MeiobN64I/+ males, whereas MeiobΔS67/ΔS67 males lacked sperm (Figure 4C). We performed mating tests for both males and females (Supplementary Table S2). While all four MeiobN64I/+ males were fertile, two of five MeiobN64I/ΔS67 males did not sire any offspring but the remaining three MeiobN64I/ΔS67 males produced fewer litters but with normal litter size (Supplementary Table S2A). The MeiobN64I/ΔS67 females produced normal litter size but Meiob/ΔS67/ΔS67 females were sterile (Supplementary Table S2B).
Figure 4.

Meiob N64I/ΔS67 males exhibit age-dependent meiotic defects in juvenile spermatogenesis. (A) Testis size of adult (8-week-old) males. (B, C) Testis weight (B) and sperm count (C) of adult (8-week-old) males. n, number of males. Data are shown as mean ± S.D., *P < 0.05 by Student's t-test. (D) Histological analysis of testis and epididymis at P20, P24, P28 and P35. Stage IV and stage XII tubules are labeled. Apoptotic spermatocytes are marked by arrowheads. Scale bar, 50 μm. (E, F) TUNEL analysis of MeiobN64I/+ and MeiobN64I/ΔS67 testes at P24 (E) and quantification of TUNEL-positive spermatocytes (F). n, the number of TUNEL-positive stage XII tubules (TUNEL-negative stage XII tubules were excluded). Three mice per genotype were examined. *P < 0.05 by Student's t-test. Scale bar, 20 μm. (G) Western blot analysis of MEIOB, SPATA22, and RPA2 in P20 testes. The levels of SYCP3 and γH2AX are constant across samples. Relative band intensity (normalized to ACTB) is shown.
In spermatogenesis, the pachytene checkpoint functions at stage IV tubules and the spindle checkpoint operates at stage XII tubules. Histological analysis revealed increased apoptosis of spermatocytes in stage XII but not stage IV seminiferous tubules from MeiobN64I/ΔS67 males at P24, 28 and 35 (Figure 4D). The increased apoptosis at stage XII was confirmed by TUNEL (Figure 4E and F). However, MeiobN64I/ΔS67 males (generated through ES cells) still produced sperm and thus their meiotic defects were similar to but less severe than those observed in MeiobN64I/ΔS67 founder males (generated through CRISPR injection), which showed complete metaphase I arrest in stage XII (Supplementary Figure S2A). It was possible that CRISPR founders harbored unknown off-target mutations or had a different (DBA × C57BL/6) hybrid genetic background. Interestingly, increased apoptosis of spermatocytes was also observed in stage IV tubules from MeiobN64I/ΔS67 males at a younger age (P20), which did not have stage XII tubules possibly due to a delay in spermatogenesis (Figure 4D). In contrast, spermatogenesis progressed to stage XII and there was increased apoptosis in stage XII but not stage IV tubules in MeiobN64I/− testes at P20 (Figure 1E), suggesting that the MeiobN64I/ΔS67 testes exhibited slightly more severe meiotic defects than MeiobN64I/− testes.
To investigate the cause of increased apoptosis in stage IV tubules in P20 MeiobN64I/ΔS67 testis, we analyzed chromosome synapsis. P20 MeiobN64I/+ testis contained spermatocytes from leptotene to metaphase I, however, P20 MeiobN64I/ΔS67 testis lacked diplotene and metaphase I spermatocytes, suggesting a delay in spermatogenesis (Supplementary Figure S3A). In P20 MeiobN64I/ΔS67 testis, two types of abnormal spermatocytes were observed: zygotene-like (no synapsis) and pachytene-like (condensed SC with <50% synapsis) (Supplementary Figure S3A). The pachytene-like spermatocytes were γH2AX-positive throughout the chromatin and lacked the typical XY body (Supplementary Figure S3B). Consistent with the histological result (Figure 4D), massive apoptosis was observed in stage IV tubules from P20 MeiobN64I/ΔS67 testis, presumably to eliminate the pachytene-like spermatocytes (Supplementary Figure S3C). However, zygotene-like and pachytene-like spermatocytes were absent in P24 MeiobN64I/ΔS67 testis (Supplementary Figure S3D). One possible explanation is that meiosis in the first wave of spermatogenesis at P20 is more sensitive to perturbation than the subsequent waves (P24 and beyond).
To probe the nature of gene dosage effect, Western blot analysis was performed on testes of various combinations of Meiob alleles (Figure 4G). The MEIOB protein was nearly absent but detectable in MeiobΔS67/ΔS67 testes, providing an explanation for the complete meiotic arrest phenotype. As expected, the MEIOB protein abundance was reduced by nearly 50% in MeiobΔS67/+ testes compared to wild type. MeiobN64I/ΔS67 testes exhibited significantly lower MEIOB protein abundance than MeiobN64I/N64I testes and slightly lower than MeiobN64I/− testes. The SPATA22 abundance correlated with the MEIOB abundance (Figure 4G). Consistent with their low protein abundance, MEIOB and SPATA22 foci on meiotic chromosomes were much weaker in MeiobN64I/ΔS67 spermatocytes (Supplementary Figure S4A). In contrast, RPA2, another MEIOB-interacting protein, did not change in protein abundance in these Meiob mutant testes (Figure 4G). The Meiob and Spata22 transcript levels were measured by real-time RT-PCR (Supplementary Figure S4B). The slight decrease in the transcript abundance could not account for the near absence of MEIOB and SPATA22 proteins in MeiobΔS67/ΔS67 testes. Therefore, these results demonstrate that the S67 deletion severely affects the MEIOB protein stability in vivo.
Reduced crossover formation in MeiobN64I/ΔS67 testes
We examined meiotic recombination in P24 MeiobN64I/ΔS67 spermatocytes. The number of RPA2 foci was comparable between MeiobN64I/ΔS67 and MeiobN64I/+ spermatocytes (Supplementary Figure S5A). The number of γH2AX flares was also comparable between MeiobN64I/ΔS67 and MeiobN64I/+ spermatocytes (Supplementary Figure S5B). The increased apoptosis of metaphase spermatocytes in stage XII tubules from MeiobN64I/ΔS67 testes suggested a defect in chromosome segregation. Therefore, we assessed crossover formation in spermatocytes from MeiobN64I/ΔS67 testes. TEX11, a component of intermediate meiotic recombination nodules, forms foci on meiotic chromosomes and regulates crossover formation (26). The number of TEX11 foci was reduced in both early- and mid-pachytene spermatocytes from MeiobN64I/ΔS67 testes (Figure 5A). The number of HEI10 and MLH1 foci was also significantly reduced in mid-late pachytene spermatocytes from MeiobN64I/ΔS67 testes (Figure 5B and C). Metaphase spread analysis revealed a significant increase in the number of univalent chromosomes in MeiobN64I/ΔS67 testes and univalence affected both autosomes and sex chromosomes (Figure 5D).
Figure 5.

MEIOB dosage-dependent regulation of crossover formation. (A–C) Analysis of TEX11, HEI10, and MLH1 foci in pachytene spermatocytes at P24. n, number of pachytene spermatocytes counted. HEI10 and MLH1 foci were counted in mid-late pachytene spermatocytes. (D) Spread analysis of metaphase I spermatocytes at P24. Univalents are indicated by arrowheads. n, number of metaphase I spermatocytes counted. Data are from three to five mice per genotype. *P < 0.05 by Student's t-test. Scale bars, 10 μm.
At the diplotene stage, homologous chromosomes are connected by chiasmata (the sites of crossover) and remain as bivalents (Supplementary Figure S6). However, univalent chromosomes were present in diplotene spermatocytes from MeiobN64I/N64I, MeiobN64I/−, and MeiobN64I/ΔS67 testes at an increasing frequency (Supplementary Figure S6). These data demonstrate that both MeiobN64I and MeiobΔS67 mutations reduce the formation of crossovers in spermatocytes.
Females are usually more tolerant of meiotic errors than males (51). We examined meiosis in oocytes from MeiobN64I/+ and MeiobN64I/ΔS67 ovaries at E18.5, when pachytene oocytes are most abundant (Supplementary Figure S7). Meiotic progression was slightly delayed in MeiobN64I/ΔS67 ovaries, indicated by the lower percentage of pachynema and the concurrently increased percentage of leptonema and zygonema (Supplementary Figure S7A). However, the numbers of RPA2 foci and MLH1 foci, and γH2AX disappearance were comparable between MeiobN64I/+ and MeiobN64I/△S67 ovaries at E18.5, showing normal crossover formation in females (Supplementary Figure S7B–D).
Reduced crossover formation in spermatocytes was also reported in other meiotic mutants such as Rpa1, Tex11 and Rnf212 (7,26,33). MEIOB colocalizes and interacts with RPA1 (17,19). MEIOB colocalizes with MSH4 in a subset of foci (17). Immunofluorescent analysis showed that the majority of MEIOB foci (83%) overlapped with TEX11 foci in pachytene spermatocytes (Supplementary Figure S8A). We performed co-transfection/co-immunoprecipitation experiments in HEK293T cells. MEIOB was not associated with any of the proteins tested (TEX11, MSH4, RNF212 and MLH1) (Supplementary Figure S8). Although it is possible that there are unknown protein interactions involving MEIOB, the MEIOB-RPA1 interaction might be the main contributor to crossover regulation.
N64I and ΔS67 mutations destabilize the MEIOB protein
The MEIOB protein abundance was dramatically reduced in MeiobN64I/N64I testes and further reduced in MeiobN64I/ΔS67 testes (Figure 4G), suggesting that the mutant MEIOB proteins are unstable. To further test the effect of these mutations on protein stability, we constructed expression plasmids with various mutations for expression in HEK293T cells (Figure 6A). In transfected cells, the MEIOBN64I protein was less abundant than the wild type, and the MEIOBΔS67 protein abundance was even lower (Figure 6B). Therefore, these two mutations reduced the MEIOB protein stability both in testes and in transfected somatic cells. It is puzzling that the N64I mutation causes meiotic arrest in the infertile man but not in mice (44). The difference in mouse and human phenotypes could be attributed to the context of the mutation, because the mouse and human MEIOB proteins exhibit only 85% identity and noticeably have two different residues adjacent to N64 (Supplementary Figure S1A). To mimic the human protein sequence, we made the partially humanized IAA mouse mutant (Figure 6A). Indeed, the mouse MEIOBIAA protein was much less stable than the MEIOBN64I protein, and was even less stable than the MEIOBΔS67 protein (Figure 6B). In the presence of SPATA22, the abundance of these MEIOB mutant proteins was still significantly reduced (Figure 6B). Human MEIOB increased human SPATA22 abundance when co-expressed (Figure 6C). The human MEIOBN64I protein was less stable than wild type in the absence or presence of hSPATA22 (Figure 6D). In conclusion, the severity of these mutations on MEIOB protein stability provides an explanation for the meiotic arrest phenotypes in MeiobΔS67/ΔS67 male mice and MEIOBN64I/N64I man.
Figure 6.

Meiob mutations destabilize the MEIOB protein and cause self-association. Full-length mouse and human MEIOB proteins were expressed in HEK293T cells in all transfection experiments. The number below each band is the fold change of protein abundance compared to the wild type. (A) Wild type and mutant MEIOB proteins. m, mouse; h, human. (B) Decreased stability of mutant mouse MEIOB proteins in the presence and absence of mSPATA22. The experiments were performed three times. *P < 0.05 by Student's t-test. (C) Human MEIOB stabilizes hSPATA22. (D) The N64I substitution destabilizes hMEIOB. hMEIOB bands were quantified. The experiments were performed three times. *P < 0.05 by Student's t-test. (E, F) Wild type MEIOB does not self-associate (E) but associate in the presence of SPATA22 (F). (G–K) MEIOBN64I interacts with itself (G), MEIOBΔS67 interacts with itself (H), MEIOBN64I interacts with MEIOBΔS67 (I), MEIOBN64I interacts with wild type MEIOB (J), MEIOBΔS67 interacts with wild type MEIOB (K).
N64I and ΔS67 mutations cause MEIOB self-association
To investigate the destabilizing effects of N64I and ΔS67 mutations, we tested protein aggregation. The wild type MEIOB protein did not dimerize in the absence of SPATA22 (Figure 6E) but were associated with each other in the presence of SPATA22 (Figure 6F). However, in the absence of SPATA22, MEIOBN64I protein alone formed a complex (Figure 6G). MEIOBΔS67 alone also formed a complex (Figure 6H). In addition, MEIOBN64I and MEIOBΔS67 interacted with each other (Figure 6I) and each mutant protein formed a complex with the wild type MEIOB (Figure 6J and K). We performed reciprocal immunoprecipitations using GFP antibody and obtained the same results (Supplementary Figure S9).
We measured the half-life of wild type mouse MEIOB, mouse MEIOBN64I, and mouse MEIOBΔS67 proteins in transfected HEK293T cells (Figure 7A). The wild type MEIOB protein was very stable with a half-life of 19.9 h (Figure 7A). However, the MEIOBN64I protein had a significantly shortened half-life (9.4 h). The S67 deletion further reduced the MEIOB half-life to 7.5 h, showing that S67 deletion is more deleterious to the MEIOB protein than the N64I substitution. We also measured the half-life of wild type human MEIOB, human MEIOBN64I, and mouse MEIOBIAA proteins in transfected HEK293T cells (Figure 7B). The half-life of human wild-type MEIOB (19.1 h) was comparable to that of mouse MEIOB. The human MEIOBN64I had a shortened half-life (9.1 h), which was comparable to the half-life of mouse MEIOBN64I. In addition, the mouse MEIOBIAA protein had the shortest half-life (3.7 h). Taken together, these data demonstrate that N64I and ΔS67 mutations cause abnormal self-aggregation of MEIOB, which could be targeted for degradation.
We sought to investigate the mechanism of accelerated degradation of mutant MEIOB proteins in HEK293T cells by inhibiting the proteasome or the autophagy protein degradation pathways. Treatment with MG132, a proteasome inhibitor, caused the accumulation of c-Jun, an endogenous proteasome target (52), but did not extend the half-life of wild type MEIOB, MEIOBN64I and MEIOBΔS67 proteins (Supplementary Figure S10A). Likewise, treatment with bafilomycin A1, an autophagy inhibitor, increased abundance of LC3A/B-II, an autophagosome marker (53), but lacked any effect on the turnover of wild type MEIOB, MEIOBN64I, and MEIOBΔS67 proteins (Supplementary Figure S10B). These results indicate that instability of MEIOBN64I and MEIOBΔS67 proteins is dependent on proteases, which are likely not associated with proteasome or lysosome activities. While many examples of such proteases (including calpain and caspases) are known, the exact nature of enzymes facilitating rapid degradation of MEIOB mutants is yet to be determined.
DISCUSSION
Our results demonstrate that MEIOB is a dosage-sensitive regulator of meiotic recombination (Figure 8). As described in this study and previous studies (17,18), Meiob−/− and MeiobΔS67/ΔS67 spermatocytes lack or nearly completely lose the MEIOB protein, show a complete failure in DSB repair in early meiotic recombination, and as a result, undergo apoptosis at stage IV due to the activation of the pachytene checkpoint (Figure 8A). In contrast, the MeiobN64I/N64I spermatocytes retaining 30% of the protein progress through the pachytene stage but exhibit reduced crossovers and thus undergo apoptosis at stage XII due to activation of the spindle assembly checkpoint. The reduction in crossovers is more pronounced in MeiobN64I/− and MeiobN64I/ΔS67 spermatocytes, which have ∼10% of the protein. It is striking that even 10% of the MEIOB protein is sufficient to complete early meiotic recombination and most crossovers in late meiotic recombination. We postulate that, at a low MEIOB/SPATA22 abundance, second ends are captured but a subset of captured second ends are not stable enough to form double Holliday junctions, resulting in a lower number of crossovers (Figure 8B). Notably, meiosis-specific proteins RNF212, HEI10 and TEX11 are also dosage-sensitive regulators of crossovers (33,37,38). Therefore, crossover formation appears to be particularly sensitive to protein abundance.
Figure 8.

Dosage-dependent and stage-specific regulation of meiotic recombination by MEIOB. (A) The gradient of the MEIOB protein level in testis is depicted (WT, 100%; Meiob−/−, 0%). Two stages of meiotic defects are diagrammed: pachytene defect at stage IV and metaphases I defect at stage XII. (B) Working model on the role of MEIOB/SPATA22 in crossover formation. MEOIB/SPATA22 (dimer) and RPA (trimer) interact with each other but bind to ssDNA independently. The interaction between these two complexes facilitates second end capture and enhances dHJ formation. Reduced MEIOB/SPATA22 abundance decreases the stability of captured second ends and thus fewer crossovers. Lack of MEIOB/SPATA22 blocks the formation of dHJs.
The MEIOBN64I mutation caused male infertility in humans but the biochemical basis of the deleterious effect of this missense mutation was unknown (44). Our previous study identifies D383 in MEIOB as a critical residue for interaction with SPATA22. The D383A mutation abolishes the MEIOB-SPATA22 interaction and causes sterility in mice (19,50). Asparagine 64 and serine 67 reside in one of the three OBDs (oligonucleotide-binding domain) but outside of the SPATA22-binding domain (19,44). We find that the N64I missense mutation and deletion of S67 result in self-aggregation of MEIOB and sharply reduced protein half-life. We also generated MeiobR235A mice through CRISPR/Cas9-mediated genome editing. Arginine 235 (R235) in MEIOB resides in one of its OBDs and is critical for its ssDNA-binding activity in vitro (17). However, the MeiobR235A/R235A mice had normal fertility, spermatogenesis, and sperm count (data not shown), showing that R235 is dispensable. Therefore, S67 deletion is not likely to affect meiosis through its effect on ssDNA-binding. Given the dramatic reduction in MEIOBΔS67 protein abundance, the reduced protein stability is most likely to be the main cause of the meiotic arrest phenotype in MeiobΔS67/ΔS67 testis. A large number of missense SNPs in humans have been identified from the thousand genome project and other genome sequencing projects. Each human genome contains 10 000–12 000 protein sequence-altering variants (54). However, the functional consequences of these variants are usually difficult to predict. A study of ∼2000 missense human variants show that ∼10% of them disrupt protein interactions (55). Our study of Meiob mutations suggest that some of human missense variants may affect protein stability.
The Meiob N64I substitution exerts different effects on fertility in human and mouse. Men with the homozygous MEIOBN64I mutation exhibit meiotic arrest and infertility (44), whereas MEIOBN64I/N64I mice display mild meiotic defects but are fertile. The human and mouse MEIOB proteins share 85% sequence identity. One possible explanation is that the N64I substitution may destabilize the human protein more than the mouse protein. However, this is not the case, because the human and mouse MEIOBN64I proteins have comparable half-lives in cultured cells (Figure 7). It is possible that the MEIOB threshold required for fertility in humans is much higher than in mouse. Intriguingly, both a homozygous mutation in PTX4 (400 kb from MEIOB) and MEIOBN64I/N64I co-segregated with infertility in the family (44). Therefore, the meiotic arrest phenotype in azoospermic men could be due to the compound effect of the MEIOBN64I mutation and the PTX4 mutation (44).
Intriguingly, both MEIOB and SPATA22 protein levels were reduced, but their transcript levels were increased in MeiobN64I/N64I testes (Figure 4G and Supplementary Figure S4B). Codon usage can affect transcript abundance through its effect on transcription (56). It is formally possible that the N64I codon change (AAT→ATC) causes increased transcription of Meiob through local chromatin modification. SPATA22 and MEIOB are binding partners with the same expression profiles. Functionally related mammalian genes could be co-regulated transcriptionally through topographically associated domains (TADs) even if located on different chromosomes (57). It is possible that the Spata22 transcription might be increased in MeiobN64I/N64I testes through high-order chromatin interactions. These possibilities warrant further investigation.
Supplementary Material
ACKNOWLEDGEMENTS
We thank H. Lin, R. Liu, Y. Guan and F. Yang for comments on the manuscript.
Contributor Information
Rui Guo, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA; State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, China.
Yang Xu, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA.
N Adrian Leu, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA.
Lei Zhang, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA.
Serge Y Fuchs, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA.
Lan Ye, State Key Laboratory of Reproductive Medicine, Nanjing Medical University, Nanjing, China.
P Jeremy Wang, Department of Biomedical Sciences, University of Pennsylvania School of Veterinary Medicine, Philadelphia, PA 19104, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH/National Institute of General Medical Sciences [R35GM118052 to P.J.W.]; National Natural Science Foundation of China [31871503 and 32070843 to L.Y.]; Innovative and Entrepreneurial Program of Jiangsu Province (to L.Y.); Project C088 from International Cooperation and Exchanges of Nanjing Medical University (to R.G.); Lalor foundation postdoctoral fellowship (to Y.X.); NIH/National Cancer Institute [R01CA902900 to S.Y.F.]. Funding for open access charge: NIH/National Institute of General Medical Sciences and National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Handel M.A., Schimenti J.C.. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 2010; 11:124–136. [DOI] [PubMed] [Google Scholar]
- 2. Hunter N. Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect. Biol. 2015; 7:a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Romanienko P.J., Camerini-Otero R.D.. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell. 2000; 6:975–987. [DOI] [PubMed] [Google Scholar]
- 4. Baudat F., Manova K., Yuen J.P., Jasin M., Keeney S.. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell. 2000; 6:989–998. [DOI] [PubMed] [Google Scholar]
- 5. Wold M.S. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 6. Ribeiro J., Abby E., Livera G., Martini E.. RPA homologs and ssDNA processing during meiotic recombination. Chromosoma. 2016; 125:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi B., Xue J., Yin H., Guo R., Luo M., Ye L., Shi Q., Huang X., Liu M., Sha J. et al.. Dual functions for the ssDNA-binding protein RPA in meiotic recombination. PLoS Genet. 2019; 15:e1007952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shinohara A., Ogawa H., Ogawa T.. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992; 69:457–470. [DOI] [PubMed] [Google Scholar]
- 9. Bishop D.K., Park D., Xu L., Kleckner N.. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell. 1992; 69:439–456. [DOI] [PubMed] [Google Scholar]
- 10. Yoshida K., Kondoh G., Matsuda Y., Habu T., Nishimune Y., Morita T.. The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol. Cell. 1998; 1:707–718. [DOI] [PubMed] [Google Scholar]
- 11. Pittman D.L., Cobb J., Schimenti K.J., Wilson L.A., Cooper D.M., Brignull E., Handel M.A., Schimenti J.C.. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell. 1998; 1:697–705. [DOI] [PubMed] [Google Scholar]
- 12. Dai J., Voloshin O., Potapova S., Camerini-Otero R.D.. Meiotic knockdown and complementation reveals essential role of RAD51 in mouse spermatogenesis. Cell. Rep. 2017; 18:1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petukhova G.V., Pezza R.J., Vanevski F., Ploquin M., Masson J.Y., Camerini-Otero R.D.. The Hop2 and Mnd1 proteins act in concert with Rad51 and Dmc1 in meiotic recombination. Nat. Struct. Mol. Biol. 2005; 12:449–453. [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., Fujiwara Y., Yamamoto S., Shibuya H.. A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination. Nat. Commun. 2019; 10:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brandsma I., Sato K., van Rossum-Fikkert S.E., van Vliet N., Sleddens E., Reuter M., Odijk H., van den Tempel N., Dekkers D.H.W. et al.. HSF2BP interacts with a conserved domain of BRCA2 and is required for mouse spermatogenesis. Cell. Rep. 2019; 27:3790–3798. [DOI] [PubMed] [Google Scholar]
- 16. Golub E.I., Gupta R.C., Haaf T., Wold M.S., Radding C.M.. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res. 1998; 26:5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luo M., Yang F., Leu N.A., Landaiche J., Handel M.A., Benavente R., La Salle S., Wang P.J.. MEIOB exhibits single-stranded DNA-binding and exonuclease activities and is essential for meiotic recombination. Nat. Commun. 2013; 4:2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Souquet B., Abby E., Herve R., Finsterbusch F., Tourpin S., Le Bouffant R., Duquenne C., Messiaen S., Martini E., Bernardino-Sgherri J. et al.. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013; 9:e1003784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu Y., Greenberg R.A., Schonbrunn E., Wang P.J.. Meiosis-specific proteins MEIOB and SPATA22 cooperatively associate with the ssDNA-binding RPA complex and DNA double-strand breaks. Biol. Reprod. 2017; 96:1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. La Salle S., Palmer K., O’Brien M., Schimenti J.C., Eppig J., Handel M.A.. Spata22, a novel vertebrate-specific gene, is required for meiotic progress in mouse germ cells. Biol. Reprod. 2012; 86:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishishita S., Matsuda Y., Kitada K.. Genetic evidence suggests that Spata22 is required for the maintenance of Rad51 foci in mammalian meiosis. Sci. Rep. 2014; 4:6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hays E., Majchrzak N., Daniel V., Ferguson Z., Brown S., Hathorne K., La Salle S.. Spermatogenesis associated 22 is required for DNA repair and synapsis of homologous chromosomes in mouse germ cells. Andrology. 2017; 5:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrillo C., Barroca V., Ribeiro J., Lailler N., Livera G., Keeney S., Martini E., Jain D.. Shani mutation in mouse affects splicing of Spata22 and leads to impaired meiotic recombination. Chromosoma. 2020; 129:161–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Snowden T., Acharya S., Butz C., Berardini M., Fishel R.. hMSH4-hMSH5 recognizes holliday junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol. Cell. 2004; 15:437–451. [DOI] [PubMed] [Google Scholar]
- 25. Kolas N.K., Svetlanov A., Lenzi M.L., Macaluso F.P., Lipkin S.M., Liskay R.M., Greally J., Edelmann W., Cohen P.E.. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J. Cell Biol. 2005; 171:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang F., Gell K., van der Heijden G.W., Eckardt S., Leu N.A., Page D.C., Benavente R., Her C., Hoog C., McLaughlin K.J. et al.. Meiotic failure in male mice lacking an X-linked factor. Genes Dev. 2008; 22:682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rogacheva M.V., Manhart C.M., Chen C., Guarne A., Surtees J., Alani E.. Mlh1-Mlh3, a meiotic crossover and DNA mismatch repair factor, is a Msh2-Msh3-stimulated endonuclease. J. Biol. Chem. 2014; 289:5664–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edelmann W., Cohen P.E., Kane M., Lau K., Morrow B., Bennett S., Umar A., Kunkel T., Cattoretti G., Chaganti R. et al.. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996; 85:1125–1134. [DOI] [PubMed] [Google Scholar]
- 29. Baker S.M., Plug A.W., Prolla T.A., Bronner C.E., Harris A.C., Yao X., Christie D.M., Monell C., Arnheim N. et al.. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996; 13:336–342. [DOI] [PubMed] [Google Scholar]
- 30. Lipkin S.M., Moens P.B., Wang V., Lenzi M., Shanmugarajah D., Gilgeous A., Thomas J., Cheng J., Touchman J.W., Green E.D. et al.. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat. Genet. 2002; 31:385–390. [DOI] [PubMed] [Google Scholar]
- 31. Holloway J.K., Booth J., Edelmann W., McGowan C.H., Cohen P.E.. MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet. 2008; 4:e1000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de los Santos T., Hunter N., Lee C., Larkin B., Loidl J., Hollingsworth N.M.. The Mus81/Mms4 endonuclease acts independently of double-holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics. 2003; 164:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reynolds A., Qiao H., Yang Y., Chen J.K., Jackson N., Biswas K., Holloway J.K., Baudat F., de Massy B., Wang J. et al.. RNF212 is a dosage-sensitive regulator of crossing-over during mammalian meiosis. Nat. Genet. 2013; 45:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ward J.O., Reinholdt L.G., Motley W.W., Niswander L.M., Deacon D.C., Griffin L.B., Langlais K.K., Backus V.L., Schimenti K.J., O’Brien M.J. et al.. Mutation in mouse hei10, an e3 ubiquitin ligase, disrupts meiotic crossing over. PLoS Genet. 2007; 3:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rao H.B., Qiao H., Bhatt S.K., Bailey L.R., Tran H.D., Bourne S.L., Qiu W., Deshpande A., Sharma A.N., Beebout C.J. et al.. A SUMO-ubiquitin relay recruits proteasomes to chromosome axes to regulate meiotic recombination. Science. 2017; 355:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holloway J.K., Sun X., Yokoo R., Villeneuve A.M., Cohen P.E.. Mammalian CNTD1 is critical for meiotic crossover maturation and deselection of excess precrossover sites. J. Cell Biol. 2014; 205:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qiao H., Prasada Rao H.B., Yang Y., Fong J.H., Cloutier J.M., Deacon D.C., Nagel K.E., Swartz R.K., Strong E., Holloway J.K. et al.. Antagonistic roles of ubiquitin ligase HEI10 and SUMO ligase RNF212 regulate meiotic recombination. Nat. Genet. 2014; 46:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang F., Silber S., Leu N.A., Oates R.D., Marszalek J.D., Skaletsky H., Brown L.G., Rozen S., Page D.C., Wang P.J.. TEX11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol. Med. 2015; 7:1198–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang F., Wang P.J.. Cheng C.Y. Genetics of mammalian meiosis. Spermatogenesis - Biology and Clinical Implications. 2018; CRC Press; 113–124. [Google Scholar]
- 40. Carlosama C., El Zaiat M., Patino L.C., Mateus H.E., Veitia R.A., Laissue P.. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum. Mol. Genet. 2017; 26:3161–3166. [DOI] [PubMed] [Google Scholar]
- 41. Yatsenko A.N., Georgiadis A.P., Ropke A., Berman A.J., Jaffe T., Olszewska M., Westernstroer B., Sanfilippo J., Kurpisz M., Rajkovic A. et al.. X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N. Engl. J. Med. 2015; 372:2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caburet S., Todeschini A.L., Petrillo C., Martini E., Farran N.D., Legois B., Livera G., Younis J.S., Shalev S., Veitia R.A.. A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. Ebiomedicine. 2019; 42:524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gershoni M., Hauser R., Barda S., Lehavi O., Arama E., Pietrokovski S., Kleiman S.E.. A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum. Reprod. 2019; 34:666–671. [DOI] [PubMed] [Google Scholar]
- 44. Gershoni M., Hauser R., Yogev L., Lehavi O., Azem F., Yavetz H., Pietrokovski S., Kleiman S.E.. A familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genes. Genet. Med. 2017; 19:998–1006. [DOI] [PubMed] [Google Scholar]
- 45. Yang H., Wang H., Jaenisch R.. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014; 9:1956–1968. [DOI] [PubMed] [Google Scholar]
- 46. Wang P.J., Page D.C.. Functional substitution for TAFII250 by a retroposed homolog that is expressed in human spermatogenesis. Hum. Mol. Genet. 2002; 11:2341–2346. [DOI] [PubMed] [Google Scholar]
- 47. Lewandoski M., Meyers E.N., Martin G.R.. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 1997; 62:159–168. [PubMed] [Google Scholar]
- 48. Peters A.H., Plug A.W., van Vugt M.J., de Boer P.. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997; 5:66–68. [DOI] [PubMed] [Google Scholar]
- 49. Turner J.M. Meiotic silencing in mammals. Annu. Rev. Genet. 2015; 49:395–412. [DOI] [PubMed] [Google Scholar]
- 50. Xu Y., Liu R., Leu N.A., Zhang L., Ibragmova I., Schultz D.C., Wang P.J.. A cell-based high-content screen identifies isocotoin as a small molecule inhibitor of the meiosis-specific MEIOB-SPATA22 complex. Biol. Reprod. 2020; 103:333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hunt P.A., Hassold T.J.. Sex matters in meiosis. Science. 2002; 296:2181–2183. [DOI] [PubMed] [Google Scholar]
- 52. Nakayama K., Furusu A., Xu Q., Konta T., Kitamura M.. Unexpected transcriptional induction of monocyte chemoattractant protein 1 by proteasome inhibition: Involvement of the c-jun N-terminal kinase-activator protein 1 pathway. J. Immunol. 2001; 167:1145–1150. [DOI] [PubMed] [Google Scholar]
- 53. Redmann M., Benavides G.A., Berryhill T.F., Wani W.Y., Ouyang X., Johnson M.S., Ravi S., Barnes S., Darley-Usmar V.M., Zhang J.. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017; 11:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. 1000 Genomes Project Consortium Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fragoza R., Das J., Wierbowski S.D., Liang J., Tran T.N., Liang S., Beltran J.F., Rivera-Erick C.A., Ye K., Wang T.Y. et al.. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019; 10:4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou Z., Dang Y., Zhou M., Li L., Yu C.H., Fu J., Chen S., Liu Y.. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E6117–E6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.