

Abstract
Background
Chronic cerebral hypoperfusion causes damage to the brain’s white matter underpinning vascular cognitive impairment. Inflammation and oxidative stress have been proposed as key pathophysiological mechanisms of which the transcription factor Nrf2 is a master regulator. We hypothesised that white matter pathology, microgliosis, blood-brain barrier breakdown and behavioural deficits induced by chronic hypoperfusion would be exacerbated in mice deficient in the transcription factor Nrf2.
Methods
Mice deficient in Nrf2 (male heterozygote or homozygous for Nrf2 knockout) or wild-type littermates on a C57Bl6/J background underwent bilateral carotid artery stenosis (BCAS) to induce chronic cerebral hypoperfusion or sham surgery and survived for a further 6 weeks. White matter pathology was assessed with MAG immunohistochemistry as a marker of altered axon-glial integrity; alterations to astrocytes and microglia/macrophages were assessed with GFAP and Iba1 immunohistochemistry, and blood-brain barrier breakdown was assessed with IgG immunohistochemistry. Behavioural alterations were assessed using 8-arm radial arm maze, and alterations to Nrf2-related and inflammatory-related genes were assessed with qRT-PCR.
Results
Chronic cerebral hypoperfusion induced white matter pathology, elevated microglial/macrophage levels and blood-brain barrier breakdown in white matter tracts that were increased in Nrf2+/− mice and further exacerbated by the complete absence of Nrf2. Chronic hypoperfusion induced white matter astrogliosis and induced an impairment in behaviour assessed with radial arm maze; however, these measures were not affected by Nrf2 deficiency. Although Nrf2-related antioxidant gene expression was not altered by chronic cerebral hypoperfusion, there was evidence for elevated pro-inflammatory related gene expression following chronic hypoperfusion that was not affected by Nrf2 deficiency.
Conclusions
The results demonstrate that the absence of Nrf2 exacerbates white matter pathology and microgliosis following cerebral hypoperfusion but does not affect behavioural impairment.
Keywords: Chronic cerebral hypoperfusion, Oxidative stress, Inflammation, White matter, Nrf2, Microglia, Astrocytes, C4, C1q, Ccl3
Background
Sustained cerebral hypoperfusion is one of the pathophysiological mechanisms contributing to cognitive decline in ageing, vascular cognitive impairment (VCI) and Alzheimer’s disease (AD) by causing damage to the brain’s white matter [1–3]. White matter hyperintensities (WMH) are frequently detected in ageing, AD and VCI, and MRI and neuropathological investigation suggest they can be attributed to cerebral hypoperfusion [3, 4]. Furthermore, the burden of WMH has been correlated with a decline in global cognitive performance, executive function and processing speed [5, 6]. The severity of hypoperfusion predicts conversion from mild cognitive impairment to dementia [7, 8].
Animal models of chronic cerebral hypoperfusion have provided evidence for a causal link between cerebral hypoperfusion, white matter pathology and cognitive deficits (reviewed in [1]). We demonstrated that bilateral carotid artery stenosis (BCAS) in mice causes chronic cerebral hypoperfusion [9], white matter alterations detected using diffusion-tensor MRI [10] and impaired axon-glial integrity [11, 12]. Furthermore, we showed that the white matter disruption caused by BCAS induces a selective deficit in spatial working memory [11], reminiscent of the disrupted frontal cortical circuitry found in VCI, that progresses to encompass spatial reference memory deficits in the longer term response [13].
An elevated inflammatory response and oxidative stress are implicated in the pathogenesis of white matter damage following cerebral hypoperfusion. Elevated levels of microglia/macrophages parallel white matter damage after chronic cerebral hypoperfusion and correlate with impaired white matter function and cognitive deficits [9, 11, 14–16]. At a mechanistic level, activated microglia may damage white matter after chronic hypoperfusion by sustained elevation of pro-inflammatory molecules such as TNF-α, IL-1β, IL-6, MCP-1 and complement pathways [16–18]. Genetic and pharmacological inhibition of complement pathways can reduce activated microglia and attenuate white matter pathology and cognitive deficits [16] induced by cerebral hypoperfusion. Similarly, other broad-spectrum anti-inflammatory approaches can reduce white matter pathology and protect white matter function and cognitive deficits in models of cerebral hypoperfusion [14, 19]. Inflammation causes oxidative stress which is also proposed to contribute mechanistically to white matter damage and can in turn exacerbate inflammation [20, 21]. Markers of oxidative damage to proteins, lipid and nucleic acids and increased levels of free radicals have been reported in models of chronic hypoperfusion [22–25]. Oxidative stress induced by chronic hypoperfusion may be mediated by decreased levels of antioxidant enzymes such as SOD, catalase and glutathione, through mitochondrial dysfunction or indirectly via free radical production from increased levels of inflammatory cells [26–28].
The transcription factor nuclear factor erythroid 2-related factor (Nrf2) is a master regulator of endogenous cytoprotective antioxidant and anti-inflammatory signalling pathways [29]. Our work and that of others have demonstrated that cerebral hypoperfusion can activate Nrf2 signalling [30–32]. Consistent with a mechanistic role for inflammation and oxidative stress in causing white matter damage after chronic hypoperfusion, boosting Nrf2 signalling has protective effects in models of chronic cerebral hypoperfusion. Pharmacological approaches to activate Nrf2 signalling, including dimethyl fumarate and sulforaphane, protect white matter function and cognitive deficits induced by cerebral hypoperfusion [15, 33]. Furthermore, other protective approaches in models of cerebral hypoperfusion such as environment enrichment and olfactory ensheathing cell transplantation are reported to act by activating Nrf2 signalling [34, 35]. We reported a role for astrocytic Nrf2 in protecting white matter and behavioural deficits following chronic cerebral hypoperfusion in mice [36]; these protective effects were paralleled by attenuated astrogliosis and pro-inflammatory gene expression.
Conversely, lack of Nrf2 has been shown to exacerbate pathology and functional outcome in other models of neurodegenerative diseases that, in common with chronic hypoperfusion, feature oxidative stress and inflammation as disease mechanisms. These include models of Parkinson’s disease [37], familial amyotrophic lateral sclerosis [38] and AD [39–42]. In models of white matter disease (sciatic nerve injury and EAE), lack of Nrf2 impairs re-myelination and functional recovery [43, 44] indicating that Nrf2 is involved in maintaining white matter during pathological conditions and promoting white matter repair. Furthermore, Nrf2 has been shown to play a role in maintaining white matter structural integrity under normal physiological conditions, as Nrf2 knock out mice display age-related white matter disruption [45]. Collectively, these studies suggest that Nrf2 plays a role regulating white matter in health and disease. However, it is presently unclear if Nrf2 knockout mice are more vulnerable to the pathology and cognitive deficits following chronic cerebral hypoperfusion. Therefore, in the present study, we hypothesised that in mice deficient in Nrf2, white matter pathology and behavioural deficits would be exacerbated following chronic hypoperfusion.
Methods
Animals
All experiments were conducted in accordance with the Animal (Scientific Procedures) Act 1986 and local ethical approval at the University of Edinburgh and were performed under personal and project licences granted by the UK Home Office according to ARRIVE guidelines. Nrf2 knockout mice were originally developed by Chan et al. [46]. The Nrf2 gene was knocked out in embryonic stem cells (129X1SvJ) by homologous recombination. Adult male and female Nrf2 knockout mice (homozygous and heterozygous for Nrf2 knockout) were obtained from the Jackson Laboratory (strain B6.129X1-Nfe2l2tm1Ywk/J stock number 017009; backcrossed onto C57Bl/6 mice for at least 10 generations). Imported male and female homozygous Nrf2−/− mice were bred with each other whereas Nrf2+/− were bred with wild-type C57Bl/6Jax mice. First generation mice from both Nrf2−/+ and Nrf2−/− breeding pairs were used for experiments. In accordance with information available from Jackson (https://www.jax.org/strain/017009), Nrf2−/− were poor breeders, and limited numbers of male Nrf2−/− were produced; therefore, a mix of Nrf2+/− and Nrf2−/− mice was used for experiments. Mice were initially group housed on a 12-h light/dark cycle with ad libitum access to food and water and assigned experimental groups by genotype then randomly assigned surgery; wild-type sham (n = 7), wild-type hypoperfused (n = 10), Nrf2+/− sham (n = 7), Nrf2+/− hypoperfused (n = 7), Nrf2−/− sham (n = 3), Nrf2−/− hypoperfused (n = 3). All mice were male, mean age 6 months (25–44 g). Three animals tolerated surgery poorly and had to be culled (2 wild-type hypoperfused, 1 Nrf2+/− hypoperfused). Experimenters were blind to genotype and surgery status of the mice throughout data collection and analysis.
Bilateral carotid artery stenosis
Bilateral carotid artery stenosis (BCAS) surgery was carried out as previously described [11]. All surgical procedures were conducted using aseptic techniques while under isoflurane anaesthesia (5% induction, 1.5% maintenance). In brief, a small cervical midline incision was made, and the common carotid arteries were exposed. A 0.18-mm internal diameter microcoil (Sawane Spring Co, Japan) was placed around one common carotid artery; the animal was allowed to recover and was 30 min later re-anesthetised for the placement of the second microcoil (0.18 mm). Sham surgery was performed in the same manner excluding the placement of the microcoils.
Eight-arm radial arm maze to assess behavioural alterations
Behavioural testing commenced 4 weeks post-BCAS. Animals were singly housed and food restricted (maintained at 85% of initial body weight) 1 week prior to and throughout the radial arm maze test to promote motivation (12-h light/dark cycle, ad libitum access to water). Following the last trial, animals were again provided food ad libitum.
The radial arm maze comprises a central platform (20 cm in diameter) surrounded by 8 arms (47-cm long by 7-cm wide with 20-cm Plexiglas walls). Each arm has a 2-cm deep plastic well for placement of a sugar pellet, and all arms can be isolated from the central platform by Plexiglas doors (remotely controlled using the ANY-Maze software, Stoelting, UK). Large visual cues were placed on each of the four walls surrounding the maze, and a camera mounted on the ceiling was used for data acquisition (ANY-Maze software, Stoelting, UK).
Pre-training consisted of one 5-min trial of free exploration with sugar pellets scattered at random, and one where each animal was allowed to walk down each arm from the central platform to retrieve sugar pellets from the plastic cups. The training was carried out for 16 consecutive days (1 trial/day). Each arm was baited with a sugar pellet, and the animal was placed in the central platform at the start of the trial. The animal was confined to the central platform for 5 s between each arm choice, and the trial finished when the animal had retrieved all 8 pellets or when 25 min had elapsed. The number of revisiting errors (visits into unbaited arms) during each trial were recorded and analysed as a measure of behaviour.
Nrf2+/− and Nrf2−/− were grouped together for radial arm maze analysis. Animals that explored less than 75% of the maze during 2 of the first 4 trials were excluded from analysis due to lack of motivation resulting in skewed learning profile. Two animals were excluded according to this criterion (one wild type BCAS and one Nrf2 −/− sham). Therefore, final group sizes for behavioural analysis were wild-type sham (n = 7), wild-type BCAS (n = 7), Nrf2-deficient sham (Nrf2+/− n = 7, Nrf2−/− n = 2), Nrf2-deficient BCAS (Nrf2+/− n = 6, Nrf2−/− n = 3).
Immunohistochemistry
Six weeks after the onset of BCAS, mice were culled under deep anaesthesia by transcardiac perfusion, and hemibrains were snap frozen in liquid nitrogen for qPCR or post-fixed in 4% paraformaldehyde for 24 h, placed in 30% sucrose for 72 h, then frozen in isopentane at − 42 °C for 2 min. Coronal sections (30 μm) were cut using a cryostat and stored in a cryoprotective medium at − 20 °C. Sections were washed in PBS (3 × 15 mins), TB (2 × 15 mins) then mounted onto Superfrost slides (VWR International) and air dried then dehydrated through a series of alcohols (70%, 90%, 100%) then placed in xylene for 10 min. Sections were then placed in 100% ethanol, and endogenous peroxidases were quenched in 3% hydrogen peroxide in methanol. Antigen retrieval was undertaken for Iba1 and GFAP immunohistochemistry; sections were incubated in 10 mM citric acid buffer (pH 6.0) at 95 °C for 10 min. Sections were blocked with 10% normal serum and 0.5% bovine albumin serum before overnight incubation with primary antibody at 4 °C. Following block, immunoglobulin G (IgG) immunostained sections were incubated overnight with horse anti-mouse IgG antibody (Vector Labs BA-2000; 1:2500). Primary antibodies and concentrations were as follows: anti-myelin-associated glycoprotein (MAG; Abcam ab89780 1:15000); anti-glial fibrillary acidic protein (GFAP; Life technologies 13-0300 1:1000); anti-ionised calcium-binding adaptor molecule-1 (Iba-1; Menarini MP290-CR05 1:1000). The appropriate biotinylated secondary antibody (Vector Labs) was incubated for 1 h at room temperature and then further amplified by incubating with Vector ABC solution before visualisation of peroxidase activity using 3,3′-diaminobenzidine tetrahydrochloride (DAB, Vector Labs, UK). IgG immunostained sections were counterstained with haematoxylin to aid region of interest analysis. Sections were then dehydrated through a series of alcohols, placed in xylene then coverslips mounted. Images were acquired using a BX51 microscope (Olympus, UK) or a Zeiss Axio Scope.A1 (Zeiss, UK). Images were analysed for percentage area of MAG, Iba1, GFAP and IgG as described in the corpus callosum, internal capsule and optic tract which were manually delineated by the experimenter. Briefly, the ImageJ software (v1.46, NIH, Bethesda, MD, USA) was used to apply a global manual threshold followed by quantification of the positive signal detected above the selected threshold. IgG immunostained sections underwent colour deconvolution in Image J (H&E DAB) to remove blue counterstained nuclei, and the Renyi Entropy threshold was applied. Occasionally, animals were excluded from analysis as the anatomical area was missing from the slide (1 wild-type BCAS and 1 Nrf2+/− animal omitted from Iba1 analyses of internal capsule and optic tract; 1 wild-type BCAS animal omitted from GFAP internal capsule analysis).
RNA extraction, reverse transcription-PCR, and quantitative (q)-PCR
RNA was isolated from optic tract-enriched samples dissected from snap-frozen hemibrains. RNA was extracted using the QIAGEN RNeasy Lipid Tissue Mini Kit according to manufacturer’s instruction. Briefly, < 100 mg fresh frozen tissue was homogenised in 1 ml QIAzol® lysis reagent using the Qiagen automated tissue lyser system and metal beads. The homogenate was transferred to fresh RNase/DNase free tubes and incubated for 2 min at room temperature with 200 μl chloroform. The upper aqueous phase was collected following 15 min centrifugation at 4 °C (12,000×g), and RNA was subsequently purified in mini spin columns and washed with a series of buffers before it was eluted in RNase free water. RNase-free DNase I (Thermo Scientific/QIAGEN) was used to remove genomic DNA according to the manufacturer’s instruction. cDNA was synthesised from 1 to 3 μg RNA using the Roche Transcriptor First Strand cDNA Synthesis Kit, according to manufacturer’s instruction. Briefly, RNA was added to reverse transcriptase reaction mix and cycled through 10 min 25 °C, 30 min 55 °C and 5 min 85 °C. No RT control was run alongside, and cDNA was diluted to the equivalent of 3 ng initial RNA per 15 μl qPCR reaction. The CFX96 Real-Time PCR Machine (Bio Rad) was used with the DyNAmo ColorFlash SYBR Green qPCR kit according to manufacturer’s instructions (Thermo Scientific). cDNA template was mixed with SYBR green master mix, water and forward and reverse primer (200 nM each final concentration). Samples were run in duplicates alongside no template and no RT negative controls. Primers were validated to confirm efficiency prior to use, and sequences used are as follows: Gapdh-F 5′-GGGTGTAACCACGAGAAAT-3′ Gapdh-R 5′-CCTTCCACAATGCCAAAGTT-3′ Nrf2-F 5′-CAGCTCAAGGGCACAGTGC-3′ Nrf2-R 5′-GTGGCCCAAGTCTTGCTCC-3′ Slc7a11-F 5′-ATACTCCAGAACACGGGCAG-3′ Slc7a11-R 5′-AGTTCCACCCAGACTCGAAC-3′ Gclm-F 5′-GCACAGCGAGGAGCTTC-3′ Gclm-R 5′-GAGCATGCCATGTCAACTG-3′ Ccl3-F 5′-GCCAGGTGTCATTTTCCTGACT-3′ Ccl3-R 5′-TCAGGCATTCAGTTCCAGGTC-3′ C1q-F a 5′-CAAGGACTGAAGGGCGTGAA-3′ C1q-R 5′-CAAGCGTCATTGGGTTCTGC-3′ C4-F 5′-ACAACAAGGGAGACCCCCAG-3′ C4-R 5′-GCTCAGAGAGCCAGAGTCCTA-3′.
The qPCR cycling programme was 10 min at 95 °C; 40 cycles of 30 s at 95 °C, 30 s at 65 °C (/30 s at 62.5 °C for C1q and Ccl2 experiment/40 s at 60 °C for Gclm experiment, 62.5 °C) with detection of fluorescence, 30 s at 72 °C; 1 cycle (for dissociation curve) of 1 min at 95 °C and 30 s at 55 °C with a ramp up to 30 s at 95 °C with continuous detection of fluorescence. The C4 experiment was run at 7 min at 95 °C initially and then 40 cycles of 10 s at 95 °C and 30 s at 65 °C (/30 s at 60 °C for Ccl3 experiment) with detection of fluorescence followed by the dissociation curve. Data was normalised to Gapdh expression as reference and expressed as fold change of wild-type sham expression.
Due to the very small volumes of optic tract-enriched RNA samples, more variation was present in these qPCR experiments. Criteria were therefore defined to exclude samples from qPCR analysis if housekeeper gene was detected > 3Ct away from the mean or if gene of interest expression was 1.5 interquartile ranges less than the first quartile or 1.5 interquartile ranges more than the third quartile for each group mean. The Ccl3 experiment therefore had one sample excluded (wild-type sham), and the Slc7a11 experiment had one sample excluded (Nrf2+/− BCAS). All other qPCR experiments included all samples.
Statistical analysis
Statistical analysis was performed using SPSS (v22, IBM Corp.) or Graphpad Prism (v5, GraphPad Software Inc, La Jolla, USA). Data are presented as mean ± SEM. Repeated measures ANOVA was used to analyse radial arm maze data. Statistical analysis was performed on the first and second half of the radial arm maze separately because the first half is primarily a learning phase. Two-way ANOVA was used to investigate the effect of BCAS surgery and genotype on immunohistochemistry and gene expression data. Bonferroni adjustment was used for post hoc analysis. Associations between microglial density and white matter pathology were analysed with Pearson’s correlation analysis. Significance was determined at p < 0.05.
Results
White matter pathology is more extensive in Nrf2-deficient mice compared to wild-type mice post-BCAS
We previously demonstrated alterations in the intensity of myelin-associated glycoprotein (MAG) immunostaining in response to chronic cerebral hypoperfusion that is indicative of altered axon-glial integrity [11] and associated with alterations in white matter integrity assessed with diffusion tensor imaging [10]. To determine if deficiency of Nrf2 exacerbated MAG pathology, the percentage area of MAG immunostaining was quantified in key white matter tracts: corpus callosum, internal capsule and optic tract (Fig. 1). There was significant white matter disruption in the corpus callosum following BCAS (F(1,28) = 6.04, p = 0.02) with an additional effect of genotype (F(2,28) = 4.47, p = 0.02), but no interaction or significant post hoc effects (Fig. 1a). There was no significant white matter disruption in the internal capsule as a result of BCAS surgery or genotype (F(1,28) = 2.99, p = 0.09, F(2,28) = 1.89, p = 0.17 respectively) (Fig. 1b). Notably in the optic tract, there was prominent white matter disruption post-BCAS (F(1,28) = 35.84, p < 0.0001) with a further effect of genotype (F(2,28) = 3.96, p = 0.03) and a significant interaction between the two variables (F(2,28) = 3.87, p = 0.03) (Fig. 1c). Post hoc analysis found significant differences between all BCAS groups and their respective sham controls (WT p = 0.04, Nrf2+/− p = 0.004, Nrf2−/− p < 0.001). Furthermore, there was significantly greater white matter disruption in the Nrf2−/− BCAS group compared to both Nrf2+/− (p = 0.02) and wild-type BCAS (p = 0.001) (Fig. 1c). Collectively, these data show that cerebral hypoperfusion induced by BCAS surgery causes white matter disruption in two major white matter tracts and that this disruption is exacerbated in the absence of Nrf2.
Fig. 1.

White matter disruption is more extensive in Nrf2-deficient mice following BCAS surgery. a White matter disruption was detected post-BCAS in the corpus callosum, with a significant effect of genotype (no post hoc differences). b There was no significant alteration in the levels of white matter disruption in the internal capsule when comparing experimental groups. c The optic tract displayed prominent white matter disruption post-BCAS with a further effect of genotype, with the Nrf2−/− group displaying the most severe disruption. Mean ± SEM. Group size presented in each bar. *p < 0.05, **p < 0.01, ***p < 0.001 (asterisk indicates post hoc differences between sham and BCAS), #p < 0.05, ##p < 0.01 (number sign indicates post hoc differences between BCAS groups). d Representative images of MAG immunostaining in the optic tract. Scale bar 50 μm
The density of microglia/macrophages is increased following BCAS to a greater extent in Nrf2−/− mice compared to wild-type and Nrf2+/− mice
We previously reported that deficits in white matter function and pathology following chronic cerebral hypoperfusion was associated with increased density of microglia/macrophages [14, 15] Similarly, Nrf2−/− mice have been reported to have an increased number of microglia at baseline [47] and an exacerbated microglial response in disease models such as Parkinson’s disease [47] and Alzheimer’s disease [39–41]. Therefore, we next investigated whether deficiency of Nrf2 exacerbated levels of microglia/macrophages in response to BCAS using Iba1 immunohistochemistry. Microglia/macrophage density was quantified by assessing percentage area of Iba1 immunostaining, which is an index of microglial activation [48], in corpus callosum, internal capsule and optic tract (Fig 2, Fig S1). There was a significant increase in density of microglia post-BCAS in the corpus callosum (F(1,28) = 7.63, p = 0.01) with a further effect of genotype (F(2,28) = 4.25, p = 0.02) but no interaction (Fig. 2a). Post hoc analysis identified significantly greater density in the Nrf2−/− BCAS group compared to sham controls (p = 0.004) and other BCAS groups (Nrf2+/− p = 0.005 and wild types p = 0.003 respectively) (Fig. 2a). The effect of BCAS surgery and genotype narrowly missed accepted levels of statistical significance in the internal capsule (F(1,26) = 3.66, p = 0.07, F(2,26) = 3.15, p = 0.06 respectively) (Fig. 2b). However, similar to the white matter disruption, there was a profound microglial response in the optic tract post-BCAS (Fig. 2c). There was a significant effect of both BCAS surgery and genotype (F(1,26) = 61.12, p < 0.0001, F(2,26) = 7.21, p = 0.003 respectively) and a significant interaction (F(2,26) = 5.8, p = 0.008). Post hoc analysis found significantly greater density of microglia in all BCAS groups compared to their sham controls (WT p = 0.005, Nrf2+/− p < 0.001, Nrf2−/− p < 0.001) and in the Nrf2−/− BCAS group compared to both Nrf2+/− (p = 0.002) and wild-type BCAS (p < 0.001) (Fig. 2c). These data show that cerebral hypoperfusion induced by BCAS surgery causes an increased density of microglia in two major white matter tracts which exhibit white matter pathology and that this increase is exacerbated in the absence of Nrf2.
Fig. 2.

The density of microglia/macrophages is increased after BCAS to a greater extent in Nrf2−/− mice. There was an effect of BCAS surgery and genotype on density of Iba1 in the corpus callosum, with the Nrf2−/− BCAS group displaying the highest density. b The effect of BCAS and genotype narrowly missed accepted levels of statistical significance in the internal capsule. c The optic tract displayed an increased density of Iba1 post-BCAS, with a further effect of genotype, with the Nrf2−/− BCAS group displaying the highest density. Mean ± SEM. Group size presented in each bar. **p < 0.01, ***p < 0.001 (asterisk indicates post hoc differences between sham and BCAS), #p < 0.05, ##p < 0.01, ###p < 0.001 (number sign indicates post hoc differences between BCAS groups). d Representative images of Iba1 staining in the optic tract. Scale bar 50 μm
We previously showed that was a correlation between impaired white matter function and microglial numbers after BCAS [14, 15], and others have suggested that aberrant activated microglia aggravate white matter injury during chronic hypoperfusion [16]. Therefore, we next investigated the association between microglial density and white matter disruption by correlating Iba1 and MAG immunostaining (Fig. 3). There was a significant positive correlation between microglial density and altered MAG disruption in the corpus callosum (r = 0.85, p < 0.0001; Fig. 3a), internal capsule (r = 0.78, p < 0.0001; Fig. 3b) and optic tract (r = 0.96, p < 0.0001; Fig. 3c). This indicates that there is an association between microglial activation and white matter pathology following BCAS surgery in WT and Nrf2-deficient mice.
Fig. 3.

Correlation between microglial activation and white matter pathology following BCAS surgery in WT and Nrf2 deficient mice. There was a significant positive correlation between levels of Iba1 immunostaining and MAG immunostaining in the a corpus callosum, b internal capsule, c optic tract
Reactive astrocytes are elevated in the optic tract following BCAS and are unaltered by deficiency of Nrf2
Aged Nrf2 knockout mice display elevated levels of reactive astrocytes in white matter compared with wild-type aged mice [45]. Similarly, elevated levels of reactive astrocytes are reported with Nrf2 deficiency in models of Parkinson’s disease [47] and Alzheimer’s disease [39]. Therefore, we investigated the effects of Nrf2 deficiency on levels of reactive astrocytes following chronic cerebral hypoperfusion by undertaking GFAP immunostaining of white matter tracts (Fig. 4, Fig S2). BCAS surgery did not affect the percentage area of GFAP immunostaining in the corpus callosum or the internal capsule (F(1,28) = 1.08, p = 0.31; F(1,27) = 0.30, p = 0.59 respectively) (Fig. 4a, b). However, there was an effect of genotype in the corpus callosum (F(2,28) = 3.96, p = 0.03), and post hoc analysis found that GFAP expression in Nrf2−/− animals was overall higher compared with wild types (p = 0.03). This genotype effect was not present in the internal capsule (F(2,27) = 1.52, p = 0.24). Consistent with the white matter disruption and the microglial response, there was a significant effect of BCAS surgery in the optic tract (F(1,28) = 43.61, p < 0.0001), although the effect of genotype narrowly missed accepted levels of statistical significance (F(2,28) = 3.26, p = 0.053) (Fig. 4c). Post hoc analysis found significant astrogliosis in all three BCAS groups compared to their sham controls (WT p = 0.005, Nrf2+/− p < 0.001, Nrf2−/− p < 0.001). These data show that Nrf2 deficiency but not hypoperfusion exacerbated levels of reactive astrocytes in the corpus callosum, whereas cerebral hypoperfusion induced by BCAS surgery causes astrogliosis in the optic tract that was not altered with deficiency of Nrf2.
Fig. 4.

Elevated levels of reactive astrocytes after BCAS that are not exacerbated by deficiency of Nrf2. a There was a significant effect of genotype on GFAP immunostaining (% area) in the corpus callosum but there was no effect of BCAS surgery. b Percent area of GFAP was unchanged in the internal capsule. c There was a significant effect of BCAS surgery on percent area of GFAP immunostaining in the optic tract, but the effect of genotype narrowly missed accepted levels of statistical significance. Mean ± SEM. **p < 0.01, ***p < 0.001 (asterisk indicates post hoc differences between sham and BCAS), #p < 0.05 (number sign indicates post hoc differences between wild type and Nrf2−/−). Group sizes presented in each bar. d Representative images of GFAP staining in the optic tract. Scale bar 50 μm
Blood-brain barrier breakdown is increased following BCAS to a greater extent in Nrf2−/− mice compared to wild-type and Nrf2+/− mice
We previously showed that BCAS induced blood-brain barrier breakdown (BBB), although this occurred in the chronic response to surgery (6 months) and was not evident at earlier timepoints (1 month) [13]. Because Nrf2 deficiency exacerbated white matter pathology and microglial/macrophage levels, we next investigated if this induced elevated levels of blood-brain barrier breakdown by labelling endogenous immunoglobulin G (IgG) within brain tissue, which would normally be excluded by an intact BBB [49] (Fig. 5). BCAS surgery did not induce blood-brain barrier breakdown in the corpus callosum (Fig. 5a) (F(1,28) = 0.24, p = 0.068), which contained negligible levels of IgG immunostaining that were not altered by Nrf2 deficiency (F(2,28) = 0.24, = 0.79). In contrast, in the internal capsule (Fig. 5b), there was a significant increase in IgG levels with BCAS surgery (F(1,28) = 8.99, p = 0.006), an effect of genotype (F(2,28) = 4.9, p = 0.015) and a significant interaction (F(2,28) = 4.8, p = 0.016). Post hoc analysis found a significantly greater density of IgG in the Nrf2−/− BCAS group compared to its sham control (p < 0.05) and in the Nrf2−/− BCAS group compared to both Nrf2+/− (p < 0.05) and wild-type BCAS (p < 0.01) (Fig. 5b). BCAS surgery also caused a significant elevation in IgG levels in the optic tract (Fig. 5c) (F(1,28) = 6.02, p = 0.02), although there was no effect of genotype (F(2,28) = 2.2, p = 0.13). Post hoc analysis found a significant increase in IgG levels in the Nrf2−/− BCAS group compared to its sham control (p < 0.05). Collectively, these data show that BCAS surgery causes blood-brain barrier breakdown in the internal capsule and optic tract, an effect that is exacerbated with deficiency of Nrf2.
Fig. 5.

Blood-brain barrier breakdown after BCAS is exacerbated by Nrf2 deficiency. a There were no significant alterations in IgG levels in the corpus callosum. b There was a significant effect of surgery and genotype on IgG levels in the internal capsule, with Nrf2−/− displaying highest density of IgG. (Double asterisk indicates post hoc significant difference between sham and BCAS Nrf2−/− mice, double number sign and number sign indicate a difference between Nrf2−/− BCAS and wild type or Nrf2+/− BCAS groups respectively) c There was a significant increase in IgG levels in the optic tract with BCAS surgery (asterisk indicates post hoc significant difference between sham and BCAS Nrf2−/− mice). Mean ± SEM. **p < 0.01, #p < 0.05, ##p < 0.01. d Representative images of IgG staining in the optic tract, brown stain depicts IgG, blue is haematoxylin counterstain. Scale bar 50 μm
Nrf2 and Nrf2-related antioxidant genes are unaltered post-BCAS
To confirm that Nrf2 levels were reduced in Nrf2-deficient mice, and to determine if BCAS surgery altered Nrf2 levels in the experimental groups, quantitative PCR analysis was used to assess gene expression levels (Fig. 6). Optic tract-enriched samples were used, as this was the area with most prominent pathology. As expected, Nrf2 levels were significantly reduced to about 50% in Nrf2+/− mice (p < 0.001) and nearly undetectable in Nrf2−/− compared to wild-type mice (p < 0.001) (genotype effect; F(2,28) = 29.6, p < 0.0001) (Fig. 6a). However, BCAS surgery did not alter the expression of Nrf2 in either WT or Nrf2-deficient mice (F(1,28) = 0.01, p = 0.91). To further investigate the effect of BCAS surgery on Nrf2-mediated signalling, two genes regulated by Nrf2-signalling that are involved in glutathione synthesis were investigated, Slc7a11 (xCT; encoding the glutamate/cystine antiporter) and Gclm, (glutamate-cysteine ligase enzyme subunit) that were previously reported to play a role in Nrf2-mediated neuroprotection against ischaemia and hypoxia [30, 31, 50]. However, there was no effect of BCAS surgery or genotype on Slc7a11 levels (F(1,27) = 0.23, p = 0.63, F(1,27) = 2.25, p = 0.13 respectively) or Gclm expression (F(1,27) = 0.39, p = 0.54, F(1,27) = 0.24 p = 0.79 respectively) (Fig. 6 b, c). Collectively, these results suggest that Nrf2 was reduced as expected in Nrf2-deficient mice, but Nrf2 and Nrf2-regulated genes (Slc7a11 and Gclm) were not altered in wild-type or Nrf2-deficient mice in response to BCAS.
Fig. 6.

Nrf2, Nrf-2-regulated antioxidant and pro-inflammatory genes in the optic tract after BCAS surgery. a There was a significant effect of genotype on Nrf2 expression but no effect of BCAS surgery. Nrf2 was reduced by ~ 50% in Nrf2+/− mice and almost undetectable in Nrf2−/− mice. b There was no effect of BCAS surgery or genotype on Slc7a11 or c Gclm expression; however, BCAS surgery increased the expression of d Ccl3 and e C1q. f The effect of BCAS surgery just missed accepted levels of statistical significance for C4 expression and there was no genotype effect on any gene. Gene expression was normalised to Gapdh and expressed as fold change compared to wild-type shams. Mean ± SEM. Group size presented in each bar. Dashed line indicates WT sham mean. *p < 0.05 (asterisk indicates post hoc differences between sham and BCAS groups), ##p < 0.01, ###p < 0.001 (number sign indicates differences between genotypes)
Therefore, to further investigate alternative mechanisms that may be responsible for the elevated white matter pathology in Nrf2-deficient mice in response to BCAS, we investigated levels of 3 pro-inflammatory genes, complement component C4 and C1q and chemokine ligand 3 (Ccl3) that we previously found to be elevated in response to hypoperfusion [9, 15]. The expression of Ccl3 and C1q was increased post-BCAS (F(1,28) = 7.96, p = 0.009, F(1,28) = 4.61, p = 0.04 respectively), whereas the effect of BCAS surgery on C4 expression narrowly missed accepted levels of statistical significance (F(1,28) = 3.9, p = 0.058) (Fig. 6d–f). However, there was no significant effect of genotype for these 3 genes (Ccl3-F(2,28) = 0.68, p = 0.52; C1q-F(2,28) = 0.28, p = 0.76; C4-F(2,28) = 0.48, p = 0.63), although post hoc analysis identified significantly increased expression of Ccl3 in the Nrf2−/− BCAS group compared to sham controls (p = 0.048). These data show that BCAS surgery caused modest elevations in pro-inflammatory gene expression that were not altered with Nrf2 deficiency with the exception of the chemokine ligand C3 that was further elevated in Nrf2−/− mice following BCAS.
Behaviour assessed with radial arm maze is impaired post-BCAS but is not exacerbated by deficiency of Nrf2
We previously showed that chronic cerebral hypoperfusion caused an impairment in behavioural performance assessed with an 8-arm radial arm maze, due to the disruption of frontocortical circuity [11], and furthermore, boosting Nrf2 signalling specifically in astrocytes caused less pronounced behavioural impairments [36]. Since white matter pathology is exacerbated in Nrf2-deficient mice, the next aim was to determine if this was associated with more severe behavioural impairment. There was a significant effect of trial during both the first and second half of the test (F(1,84) = 7.03, p < 0.0001, F(3,84) = 3.62, p = 0.02 respectively) indicating that mice were able to learn the task successfully (Fig. 7). The effect of BCAS surgery was not significant during the first half of the test (F(1,28) = 0.10, p = 0.76) but was significant during the second half (F(1,28) = 4.21, p = 0.049) (Fig. 7). This indicates that while both sham and BCAS groups were learning the task initially, the BCAS groups’ learning plateaus, and they commit significantly more revisiting errors than sham groups during the second half of the test, indicating that BCAS caused significantly more revisiting errors. However, there was no effect of genotype on spatial working memory (first half, F(1,28) = 0.07, p = 0.79; second half, F(1,28) = 0.001, p = 0.98) (Fig. 7), which contests the hypothesis that reductions in Nrf2 exacerbates the behavioural impairment caused by BCAS-induced cerebral hypoperfusion.
Fig. 7.
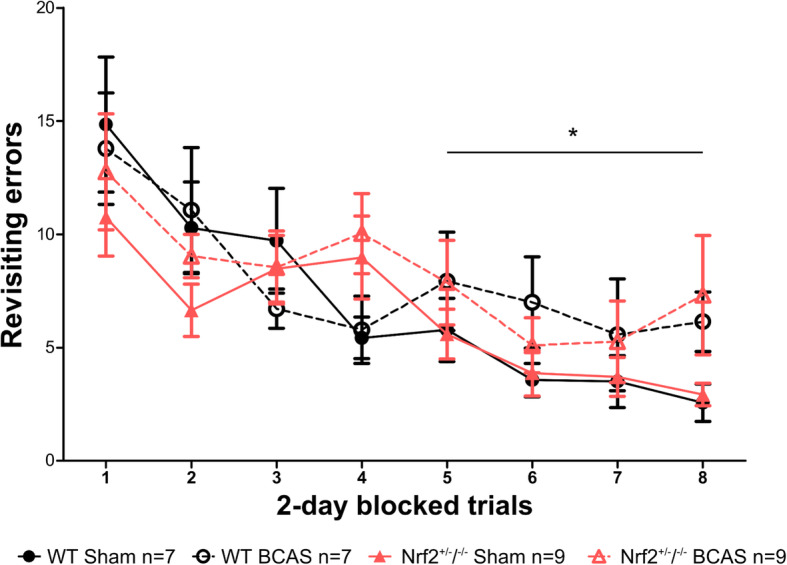
Behaviour assessed with radial arm maze is impaired after BCAS but is not exacerbated by deficiency in Nrf2. There was an effect of trial during both first (blocks 1–4) and second half (blocks 5–8) of the test indicating learning over time. During the second half of the test, revisiting errors were significantly higher post-BCAS demonstrating impaired spatial working memory. However, there was no effect of genotype on revisiting errors during either the first or second part of the test. Post hoc analyses identified no further significant differences. Mean ± SEM. *p < 0.05 (asterisk indicates main effect of BCAS surgery during second half of test). Group sizes are indicated in the graph
Discussion
In the current study, we demonstrated that deficiency of Nrf2 resulted in exacerbated white matter pathology in response to chronic hypoperfusion, an effect that was most marked with complete absence of Nrf2. Furthermore, this was associated with elevated levels of microgliosis and paralleled by elevated levels of blood-brain barrier breakdown. Although Nrf2-related antioxidant gene expression was not altered by chronic cerebral hypoperfusion, there was evidence for elevated pro-inflammatory-related gene expression following hypoperfusion that was not affected by Nrf2 deficiency. However, despite these molecular and pathological alterations post-BCAS in Nrf2 deficient mice, there was no worsening of the behavioural impairment of mice in the radial arm maze.
We found elevated levels of white matter disruption in the present study in the corpus callosum and optic tract 6 weeks after chronic hypoperfusion, evident by increased density of MAG-positive debris. MAG is expressed at the axon-glial interface and is sensitive to hypoxia-induced injury [51]. Human post-mortem studies identify loss of MAG, in particular its ratio to the more stable proteolipid protein (PLP), as a measure of white matter ischaemia [52]. Our previous work showed that chronic cerebral hypoperfusion in mice induces alterations to MAG as early as 3 days post-surgery, and at least initially in the absence of damage using other markers of myelin such as myelin basic protein and FluoroMyelin [9, 12], consistent with hypoxia-induced white matter injury in humans [51]. In contrast to our previous work and that of others [9, 11, 53], the optic tract was most severely damaged after BCAS whereas the corpus callosum and internal capsule were minimally damaged. However, this is consistent with the pattern of damage we recently reported in mice that overexpress Nrf2 specifically in astrocytes [36]. The background strain of the mice may account for these differences, as we report diffuse damage to multiple white matter tracts using mice on a pure C57Bl6/J background, whereas the severe optic tract damage was reported using mice originating from mixed backgrounds (129X1/SvJ or FVB/C57Bl6/J F1 mice). These strains may have a different circle of Willis patency, and therefore different spatial distribution and severity of hypoperfusion [54]. In addition, we cannot discount that the heightened vulnerability of the optic tract may account for the impaired behaviour in the radial arm maze in the hypoperfused mice.
The MAG debris induced by cerebral hypoperfusion, indicative of altered axon-glial integrity, was exacerbated in the absence of Nrf2 in the corpus callosum and optic tract. This is consistent with earlier studies investigating the role of Nrf2 in the maintenance of white matter integrity in health and disease [44, 55]. Hubbs et al. [45] found that the absence of Nrf2 in aged mice (> 10 months) is associated with vacuolar leukoencephalopathy causing white matter degeneration and myelin-specific oxidative injury. Furthermore, patients with de novo missense mutations in NFE2L2 that lead to accumulation of Nrf2 and increased expression of Nrf2-related genes present with leukoencephalopathy and diminished myelin content [56]. Therefore, it would appear that not only lack of Nrf2 but also chronic activation may cause pathology in white matter, highlighting the importance of the elaborate mechanisms that control Nrf2 activity.
White matter disruption after BCAS has previously been shown to be paralleled by cellular inflammation primarily characterised by a microglial response [9, 53, 57, 58], and here, we report correlations between white matter damage and microglial activation. White matter inflammation is proposed to contribute to age and disease-related white matter pathology through excessive pro-inflammatory signalling, phagocytosis and oxidative stress [59]. The data presented in the current study are consistent with previous work displaying increased density of microglia/macrophages in the corpus callosum, correlating with the severity of white matter functional deficit [15]. The results also demonstrate that hypoperfusion-induced increases in microglial/macrophage density are further increased in the absence of Nrf2, suggesting that signalling through Nrf2 normally acts to limit upregulation of microglia following cerebral hypoperfusion.
Microglia in white matter are important for myelin repair both through phagocytosis of myelin degradation products which are neurotoxic and prevent recovery if not cleared [52] and through release of growth factors [60]. A study of peripheral nerve crush injury found that Nrf2−/− mice had a reduced ability to clear myelin debris [43]. Similarly, Rojo et al. [47] identified reduced levels of phagocytosis in Nrf2−/− mice, suggesting a mechanism by which white matter disruption may be exacerbated in Nrf2−/− mice in the current study. Nrf2 has been suggested to act as a regulator of microglial dynamics [47, 61].
In the present study, we showed differential effects of hypoperfusion and Nrf2 deficiency on reactive astrocyte density in white matter, depending on the anatomical area analysed. Interestingly, there was an increase in the density of GFAP-positive astrocytes in both sham and hypoperfused Nrf2−/− mice in the corpus callosum. This finding is in agreement with Hubbs et al. [45] who found that aged Nrf2−/− mice display widespread astrogliosis in white matter including corpus callosum.
Although levels of reactive astrocytes in the optic tract were elevated in response to chronic hypoperfusion, their levels were not altered by Nrf2 deficiency. In contrast, chronic hypoperfusion did not alter levels of reactive astrocytes in the corpus callosum or internal capsule, despite elevated levels of white matter damage and microglia/macrophage in the corpus callosum that were further increased in Nrf2 knockout mice. Similarly, in a cuprizone model of demyelination, loss of myelin was exacerbated in Nrf2-deficient mice, and although this was associated with increased microglia/macrophage levels, there was a 50% reduction in levels of reactive astrocytes when compared with wild-type cuprizone-treated mice. Importantly, alterations to levels of reactive astrocytes, as assessed with GFAP immunostaining, does not inform on the antioxidant and pro/anti-inflammatory capacity of those astrocytes, which may differ between genotypes. Transcriptomic analysis of isolated astrocytes display a different profile based on the activating insult, suggested to indicate the functional phenotype of that population of reactive astrocytes [62]. Additionally, astrocytes have also been shown to display both regional and age-dependent heterogeneity [63, 64]. Therefore, RNA sequencing of isolated astrocytes from different white matter regions would provide more comprehensive information about phenotype alterations to astrocytes with chronic hypoperfusion and Nrf2 deficiency.
As expected, the present study showed that there was a 50% reduction of Nrf2 in Nrf2+/− mice, and Nrf2 expression was nearly undetectable in Nrf2−/− mice compared to wild-type controls. However, cerebral hypoperfusion did not alter the expression of Nrf2 in Nrf2+/− or wild type mice. The glutathione system has been shown to be a major pathway mediating neuroprotection against oxidative stress by Nrf2 [50]. However, we did not report any alterations in Slc7a11 and Gclm, genes required for the synthesis of glutathione, with either cerebral hypoperfusion or Nrf2 deficiency. It is possible that Nrf2 and Nrf2-related gene expression may have increased acutely following the reduction of CBF. This is observed following middle cerebral artery occlusion where Nrf2 peaks between 8 [65] and 24 h [66] but is reduced again at 72 h. A similar phenomenon is observed following traumatic brain injury [67] and indicates that regulatory mechanisms of the Nrf2 system may prevent it from being chronically activated, particularly given the white matter lesions and neurological consequences of chronic Nrf2 elevation reported in patients with mutations in the gene encoding Nrf2 [56].
Because we did not report alterations in Nrf2 or Nrf2-related gene expression following cerebral hypoperfusion in the present study, an alternative explanation is that pro-inflammatory gene expression may be altered, consistent with the exacerbated levels of microgliosis that we report with both hypoperfusion and Nrf2 deficiency. Pro-inflammatory gene signalling is exacerbated in Nrf2−/− mice in other disease models [42, 44, 47]. Here, we found elevated levels of chemokine Ccl3 and complement component C1q following cerebral hypoperfusion, consistent with our previous work [12, 15, 36]. Post hoc analysis showed that Ccl3 levels were significantly increased in Nrf2−/− mice in comparison with shams. CCL3 regulates the recruitment of microglia as well as peripheral monocytes and macrophages [68]. We also reported that an Nrf2-activating drug, dimethyl fumarate, dampened levels of CCL3 (MIP-1α), an effect paralleled by decreased levels of microglia/macrophage and improved white matter function [15]. Hypoperfusion induced a significant increase in C1q, C4 expression just missed accepted levels of significance, although there was no genotype effect. Activation of the complement pathway has been suggested to potentiate chronic inflammation and neurodegeneration [69]. Interestingly, the complement cascade has also been shown to be activated by myelin-associated products [70], suggesting that white matter disruption in itself may potentiate inflammatory changes observed following cerebral hypoperfusion.
Cerebral hypoperfusion has previously been shown to cause an impairment in spatial working memory [11, 53] attributed to disruption of frontal-subcortical circuitry. Consistent with previous reports, the current study demonstrated that cerebral hypoperfusion impairs behaviour assessed with the radial arm maze both in wild-type and Nrf2-deficient mice. However, contrary to the study hypothesis, Nrf2 deficiency did not exacerbate revisiting errors despite the exacerbation of white matter damage. This contrasts with the protective effects that we demonstrated using the radial arm maze in mice with astrocyte-specific overexpression of Nrf2 that underwent cerebral hypoperfusion [36]. One limitation of the current study is the limited number of Nrf2−/− mice, due to breeding problems. Therefore, it is possible that the study is underpowered to detect exacerbated white matter pathology in more subtly damaged white matter tracts that may have impacted on the behavioural performance. There is a very limited number of studies investigating cognition in models of CNS disease with Nrf2-deficient mice, with only one study [40] reporting an exacerbated spatial learning and memory deficits and working and associative memory deficits in an Nrf2-deficient APP/PS1 mutant model of Alzheimer’s disease. However, different disease mechanisms, anatomical differences in pathology and the older age of the mice may account for the increased vulnerability of these mice to cognitive deficits.
It is also possible that in the present study the exacerbation of white matter damage was of an insufficient level to worsen the behavioural alterations detected using the radial arm maze at the 6-week time point. Longer duration of hypoperfusion would be expected to intensify the pathological differences between wild-type and Nrf2-deficient animals, and a later time point in aged animals may have identified functional effects of Nrf2 deficiency. Indeed, with ageing, the cerebral blood vessels of Nrf2-deficient mice have elevated levels of senescence markers, ageing-induced vascular inflammation and blood-brain barrier leakage [71, 72] and white matter leukoencephalopathy [45].
Conclusions
To conclude, we have demonstrated that chronic cerebral hypoperfusion induces white matter pathology, elevated levels of microglia/macrophages and blood-brain barrier breakdown that were exacerbated with deficiency of Nrf2, thus confirming a role for Nrf2 in protecting white matter from pathology and in microglial dynamics. Reactive astrocytes and pro-inflammatory gene expression were increased with cerebral hypoperfusion but were not altered by Nrf2 deficiency. Despite elevated levels of white matter pathology, there was no exacerbation of the impaired behavioural performance in the radial arm maze induced by cerebral hypoperfusion in Nrf2-deficient mice.
Supplementary Information
Acknowledgements
We thank Mr James Febery for the assistance with the imaging of sections.
Abbreviations
- AD
Alzheimer’s disease
- BBB
Blood-brain barrier
- BCAS
Bilateral common carotid artery stenosis
- C1q
Complement component 1q
- C4
Complement component 4
- Ccl3
Chemokine ligand 3
- EAE
Experimental autoimmune encephalomyelitis
- GFAP
Glial fibrillary acidic protein
- Iba1
Ionised calcium-binding adaptor molecule-1
- IgG
Immunoglobulin G
- IL-1β
Interleukin 1-beta
- IL-6
Interleukin 6
- MAG
Myelin-associated glycoprotein
- MCP-1
Monocyte chemoattractant protein-1 (also called CCL2 = chemokine ligand 2).
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- ROI
Region of interest
- SOD
Superoxide dismutase
- TNF-α
Tumour necrosis factor alpha
- VCI
Vascular cognitive impairment
Authors’ contributions
E.S. and M.M. performed behavioural testing. E.S. performed immunohistochemical, biochemical and qPCR experiments and analysis of data, prepared figures and wrote the manuscript. G.E.H. provided feedback on the data and manuscript. K.H. planned, designed and supervised the studies and data analysis, performed the surgeries and wrote the manuscript. J.H.F. provided input to the qPCR studies, performed immunohistochemical experiments, analysis, prepared figures, contributed to planning, design and supervision of the study and wrote the manuscript. The authors read and approved the final manuscript.
Funding
We gratefully acknowledge the grant support from the Alzheimer Society (152 (PG-157); 290 (AS-PG-15b-018); 228 (AS-DTC-2014-017)), Alzheimer Research UK (ART-PG2010-3; ARUK-PG2013-22; ARUK-PG2016B-6) and The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, part of the cross council Lifelong Health and Wellbeing Initiative (G0700704/84698), Biogen and The Wellcome Trust. E.S. was supported by an Alzheimer’s Society Doctoral Training PhD studentship with additional support from RS Macdonald Trust. J.H.F was funded by an Alzheimer’s Society Fellowship (Grant 297) and is currently supported by an ARUK Senior Fellowship (ARUK-SRF-2013-4 and ARUK-SRF2018B-005). Funding bodies did not contribute to design of study or collection, analysis, interpretation of data or writing of manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
All experiments were conducted in accordance with the Animal (Scientific Procedures) Act 1986 and local ethical approval at the University of Edinburgh and were performed under personal and project licences granted by the UK Home Office.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Emma Sigfridsson, Email: emma.sigfridsson@ed.ac.u.
Martina Marangoni, Email: maramarti@hotmail.it.
Giles E. Hardingham, Email: Giles.Hardingham@ed.ac.uk
Karen Horsburgh, Email: Karen.Horsburgh@ed.ac.uk.
Jill H. Fowler, Email: Jill.Fowler@ed.ac.uk
Supplementary Information
Supplementary information accompanies this paper at 10.1186/s12974-020-02038-2.
References
- 1.Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: a key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci (Lond) 2017;131:2451–2468. doi: 10.1042/CS20160727. [DOI] [PubMed] [Google Scholar]
- 2.Hase Y, Horsburgh K, Ihara M, Kalaria RN. White matter degeneration in vascular and other ageing-related dementias. J Neurochem. 2018;144:617–633. doi: 10.1111/jnc.14271. [DOI] [PubMed] [Google Scholar]
- 3.Makedonov I, Black SE, MacIntosh BJ. Cerebral small vessel disease in aging and Alzheimer’s disease: a comparative study using MRI and SPECT. Eur J Neurol. 2013;20:243–250. doi: 10.1111/j.1468-1331.2012.03785.x. [DOI] [PubMed] [Google Scholar]
- 4.Fernando MS, O'Brien JT, Perry RH, English P, Forster G, McMeekin W, Slade JY, Golkhar A, Matthews FE, Barber R, et al. Comparison of the pathology of cerebral white matter with post-mortem magnetic resonance imaging (MRI) in the elderly brain. Neuropathol Appl Neurobiol. 2004;30:385–395. doi: 10.1111/j.1365-2990.2004.00550.x. [DOI] [PubMed] [Google Scholar]
- 5.Bolandzadeh N, Davis JC, Tam R, Handy TC, Liu-Ambrose T. The association between cognitive function and white matter lesion location in older adults: a systematic review. BMC Neurol. 2012;12:126. doi: 10.1186/1471-2377-12-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11:157–165. doi: 10.1038/nrneurol.2015.10. [DOI] [PubMed] [Google Scholar]
- 7.Chao LL, Buckley ST, Kornak J, Schuff N, Madison C, Yaffe K, Miller BL, Kramer JH, Weiner MW. ASL perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis Assoc Disord. 2010;24:19–27. doi: 10.1097/WAD.0b013e3181b4f736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y, Matsuda H, Nemoto K, Imabayashi E, Yamada M, et al. The prediction of rapid conversion to Alzheimer’s disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage. 2005;28:1014–1021. doi: 10.1016/j.neuroimage.2005.06.066. [DOI] [PubMed] [Google Scholar]
- 9.McQueen J, Reimer MM, Holland PR, Manso Y, McLaughlin M, Fowler JH, Horsburgh K. Restoration of oligodendrocyte pools in a mouse model of chronic cerebral hypoperfusion. PLoS One. 2014;9:e87227. doi: 10.1371/journal.pone.0087227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holland PR, Bastin ME, Jansen MA, Merrifield GD, Coltman RB, Scott F, Nowers H, Khallout K, Marshall I, Wardlaw JM, et al. MRI is a sensitive marker of subtle white matter pathology in hypoperfused mice. Neurobiol Aging. 2011;32:2325. doi: 10.1016/j.neurobiolaging.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Coltman R, Spain A, Tsenkina Y, Fowler JH, Smith J, Scullion G, Allerhand M, Scott F, Kalaria RN, Ihara M, et al. Selective white matter pathology induces a specific impairment in spatial working memory. Neurobiol Aging. 2011;32:2324. doi: 10.1016/j.neurobiolaging.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Reimer MM, McQueen J, Searcy L, Scullion G, Zonta B, Desmazieres A, Holland PR, Smith J, Gliddon C, Wood ER, et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci. 2011;31:18185–18194. doi: 10.1523/JNEUROSCI.4936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holland PR, Searcy JL, Salvadores N, Scullion G, Chen G, Lawson G, Scott F, Bastin ME, Ihara M, Kalaria R, et al. Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. J Cereb Blood Flow Metab. 2015;35:1005–1014. doi: 10.1038/jcbfm.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manso Y, Holland PR, Kitamura A, Szymkowiak S, Duncombe J, Hennessy E, Searcy JL, Marangoni M, Randall AD, Brown JT, et al. Minocycline reduces microgliosis and improves subcortical white matter function in a model of cerebral vascular disease. Glia. 2018;66:34–46. doi: 10.1002/glia.23190. [DOI] [PubMed] [Google Scholar]
- 15.Fowler JH, McQueen J, Holland PR, Manso Y, Marangoni M, Scott F, Chisholm E, Scannevin RH, Hardingham GE, Horsburgh K. Dimethyl fumarate improves white matter function following severe hypoperfusion: involvement of microglia/macrophages and inflammatory mediators. J Cereb Blood Flow Metab. 2018;38:1354–1370. doi: 10.1177/0271678X17713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang LY, Pan J, Mamtilahun M, Zhu Y, Wang L, Venkatesh A, Shi R, Tu X, Jin K, Wang Y, et al. Microglia exacerbate white matter injury via complement C3/C3aR pathway after hypoperfusion. Theranostics. 2020;10:74–90. doi: 10.7150/thno.35841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou X, Liang X, Chen JF, Zheng J. Ecto-5’-nucleotidase (CD73) is involved in chronic cerebral hypoperfusion-induced white matter lesions and cognitive impairment by regulating glial cell activation and pro-inflammatory cytokines. Neuroscience. 2015;297:118–126. doi: 10.1016/j.neuroscience.2015.03.033. [DOI] [PubMed] [Google Scholar]
- 18.Su SH, Wu YF. Lin Q, Wang DP, Hai J: URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J Neuroinflammation. 2019;16:260. doi: 10.1186/s12974-019-1668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hei Y, Zhang X, Chen R, Zhou Y, Gao D, Liu W. High-mobility group box 1 neutralization prevents chronic cerebral hypoperfusion-induced optic tract injuries in the white matter associated with down-regulation of inflammatory responses. Cell Mol Neurobiol. 2019;39:1051–1060. doi: 10.1007/s10571-019-00702-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HA, Miller AA, Drummond GR, Thrift AG, Arumugam TV, Phan TG, Srikanth VK, Sobey CG. Vascular cognitive impairment and Alzheimer’s disease: role of cerebral hypoperfusion and oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:953–959. doi: 10.1007/s00210-012-0790-7. [DOI] [PubMed] [Google Scholar]
- 21.Daulatzai MA. Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J Neurosci Res. 2017;95:943–972. doi: 10.1002/jnr.23777. [DOI] [PubMed] [Google Scholar]
- 22.Kasparová S, Brezová V, Valko M, Horecký J, Mlynárik V, Liptaj T, Vancová O, Ulicná O, Dobrota D. Study of the oxidative stress in a rat model of chronic brain hypoperfusion. Neurochem Int. 2005;46:601–611. doi: 10.1016/j.neuint.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Washida K, Ihara M, Nishio K, Fujita Y, Maki T, Yamada M, Takahashi J, Wu X, Kihara T, Ito H, et al. Nonhypotensive dose of telmisartan attenuates cognitive impairment partially due to peroxisome proliferator-activated receptor-gamma activation in mice with chronic cerebral hypoperfusion. Stroke. 2010;41:1798–1806. doi: 10.1161/STROKEAHA.110.583948. [DOI] [PubMed] [Google Scholar]
- 24.Dong YF, Kataoka K, Toyama K, Sueta D, Koibuchi N, Yamamoto E, Yata K, Tomimoto H, Ogawa H, Kim-Mitsuyama S. Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension. 2011;58:635–642. doi: 10.1161/HYPERTENSIONAHA.111.173534. [DOI] [PubMed] [Google Scholar]
- 25.Miyamoto N, Maki T, Pham LD, Hayakawa K, Seo JH, Mandeville ET, Mandeville JB, Kim KW, Lo EH, Arai K. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke. 2013;44:3516–3521. doi: 10.1161/STROKEAHA.113.002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji HJ, Hu JF, Wang YH, Chen XY, Zhou R, Chen NH. Osthole improves chronic cerebral hypoperfusion induced cognitive deficits and neuronal damage in hippocampus. Eur J Pharmacol. 2010;636:96–101. doi: 10.1016/j.ejphar.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 27.Sayan-Ozacmak H, Ozacmak VH, Barut F, Jakubowska-Dogru E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem Int. 2012;61:287–290. doi: 10.1016/j.neuint.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 28.Du J, Ma M, Zhao Q, Fang L, Chang J, Wang Y, Fei R, Song X. Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience. 2013;231:345–352. doi: 10.1016/j.neuroscience.2012.11.062. [DOI] [PubMed] [Google Scholar]
- 29.Dinkova-Kostova AT, Kostov RV, Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. Febs J. 2018;285:3576–3590. doi: 10.1111/febs.14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, Chowdhry S, Patani R, Chandran S, Horsburgh K, et al: Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc Natl Acad Sci U S A 2011, 108:E1-E2; author reply E3-4. [DOI] [PMC free article] [PubMed]
- 31.Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE: Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxid Med Cell Longev 2011, 2011:689524. [DOI] [PMC free article] [PubMed]
- 32.Yang Y, Zhang J, Liu H, Zhang L. Change of Nrf2 expression in rat hippocampus in a model of chronic cerebral hypoperfusion. Int J Neurosci. 2014;124:577–584. doi: 10.3109/00207454.2013.863196. [DOI] [PubMed] [Google Scholar]
- 33.Mao L, Yang T, Li X, Lei X, Sun Y, Zhao Y, Zhang W, Gao Y, Sun B, Zhang F. Protective effects of sulforaphane in experimental vascular cognitive impairment: contribution of the Nrf2 pathway. J Cereb Blood Flow Metab. 2019;39:352–366. doi: 10.1177/0271678X18764083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, Zhang J, Xiong L, Deng M, Wang J, Xin J, Liu H. Cognitive improvement induced by environment enrichment in chronic cerebral hypoperfusion rats: a result of upregulated endogenous neuroprotection? J Mol Neurosci. 2015;56:278–289. doi: 10.1007/s12031-015-0529-2. [DOI] [PubMed] [Google Scholar]
- 35.Yu A, Mao L, Zhao F, Sun B. Olfactory ensheathing cells transplantation attenuates chronic cerebral hypoperfusion induced cognitive dysfunction and brain damages by activating Nrf2/HO-1 signaling pathway. Am J Transl Res. 2018;10:3111–3121. [PMC free article] [PubMed] [Google Scholar]
- 36.Sigfridsson E, Marangoni M, Johnson JA, Hardingham GE, Fowler JH, Horsburgh K. Astrocyte-specific overexpression of Nrf2 protects against optic tract damage and behavioural alterations in a mouse model of cerebral hypoperfusion. Sci Rep. 2018;8:12552. doi: 10.1038/s41598-018-30675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vargas MR, Burton NC, Kutzke J, Gan L, Johnson DA, Schäfer M, Werner S, Johnson JA. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS One. 2013;8:e56625. doi: 10.1371/journal.pone.0056625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer’s disease-like pathology in the APP/PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36:664–679. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Branca C, Ferreira E, Nguyen TV, Doyle K, Caccamo A, Oddo S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2017;26:4823–4835. doi: 10.1093/hmg/ddx361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojo AI, Pajares M, García-Yagüe AJ, Buendia I, Van Leuven F, Yamamoto M, López MG, Cuadrado A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018;18:173–180. doi: 10.1016/j.redox.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rojo AI, Pajares M, Rada P, Nuñez A, Nevado-Holgado AJ, Killik R, Van Leuven F, Ribe E, Lovestone S, Yamamoto M, Cuadrado A. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–451. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Johnson D, Johnson JA. Deletion of Nrf2 impairs functional recovery, reduces clearance of myelin debris and decreases axonal remyelination after peripheral nerve injury. Neurobiol Dis. 2013;54:329–338. doi: 10.1016/j.nbd.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol Sci. 2010;114:237–246. doi: 10.1093/toxsci/kfp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hubbs AF, Benkovic SA, Miller DB, O’Callaghan JP, Battelli L, Schwegler-Berry D, Ma Q. Vacuolar leukoencephalopathy with widespread astrogliosis in mice lacking transcription factor Nrf2. Am J Pathol. 2007;170:2068–2076. doi: 10.2353/ajpath.2007.060898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojo AI, Innamorato NG, Martín-Moreno AM, De Ceballos ML, Yamamoto M, Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010;58:588–598. doi: 10.1002/glia.20947. [DOI] [PubMed] [Google Scholar]
- 48.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.STR.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 49.Leonardi A, Gandolfo C, Caponnetto C, Arata L, Vecchia R. The integrity of the blood-brain barrier in Alzheimer’s type and multi-infarct dementia evaluated by the study of albumin and IgG in serum and cerebrospinal fluid. J Neurol Sci. 1985;67:253–261. doi: 10.1016/0022-510X(85)90121-2. [DOI] [PubMed] [Google Scholar]
- 50.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barker R, Wellington D, Esiri MM, Love S. Assessing white matter ischemic damage in dementia patients by measurement of myelin proteins. J Cereb Blood Flow Metab. 2013;33:1050–1057. doi: 10.1038/jcbfm.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- 53.Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598–2603. doi: 10.1161/01.STR.0000143725.19053.60. [DOI] [PubMed] [Google Scholar]
- 54.León-Moreno LC, Castañeda-Arellano R, Rivas-Carrillo JD, Dueñas-Jiménez SH. Challenges and improvements of developing an ischemia mouse model through bilateral common carotid artery occlusion. J Stroke Cerebrovasc Dis. 2020;29:104773. doi: 10.1016/j.jstrokecerebrovasdis.2020.104773. [DOI] [PubMed] [Google Scholar]
- 55.Nellessen A, Nyamoya S, Zendedel A, Slowik A, Wruck C, Beyer C, Fragoulis A, Clarner T. Nrf2 deficiency increases oligodendrocyte loss, demyelination, neuroinflammation and axonal damage in an MS animal model. Metab Brain Dis. 2020;35:353–362. doi: 10.1007/s11011-019-00488-z. [DOI] [PubMed] [Google Scholar]
- 56.Huppke P, Weissbach S, Church JA, Schnur R, Krusen M, Dreha-Kulaczewski S, Kühn-Velten WN, Wolf A, Huppke B, Millan F, et al. Activating de novo mutations in NFE2L2 encoding NRF2 cause a multisystem disorder. Nat Commun. 2017;8:818. doi: 10.1038/s41467-017-00932-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miki K, Ishibashi S, Sun L, Xu H, Ohashi W, Kuroiwa T, Mizusawa H. Intensity of chronic cerebral hypoperfusion determines white/gray matter injury and cognitive/motor dysfunction in mice. J Neurosci Res. 2009;87:1270–1281. doi: 10.1002/jnr.21925. [DOI] [PubMed] [Google Scholar]
- 58.Saggu R, Schumacher T, Gerich F, Rakers C, Tai K, Delekate A, Petzold GC. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. 2016;4:76. doi: 10.1186/s40478-016-0350-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, Mantingh-Otter IJ, Van Dam D, De Deyn PP, den Dunnen W, et al. Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front Mol Neurosci. 2017;10:206. doi: 10.3389/fnmol.2017.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lloyd AF, Davies CL, Miron VE. Microglia: origins, homeostasis, and roles in myelin repair. Curr Opin Neurobiol. 2017;47:113–120. doi: 10.1016/j.conb.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 61.Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D. Cuadrado A: α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet. 2012;21:3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 62.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morel L, Chiang MSR, Higashimori H, Shoneye T, Iyer LK, Yelick J, Tai A, Yang Y. Molecular and functional properties of regional astrocytes in the adult brain. J Neurosci. 2017;37:8706–8717. doi: 10.1523/JNEUROSCI.3956-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boisvert MM, Erikson GA, Shokhirev MN, Allen NJ. The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep. 2018;22:269–285. doi: 10.1016/j.celrep.2017.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka N, Ikeda Y, Ohta Y, Deguchi K, Tian F, Shang J, Matsuura T, Abe K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 66.Srivastava S, Alfieri A, Siow RC, Mann GE, Fraser PA. Temporal and spatial distribution of Nrf2 in rat brain following stroke: quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J Physiol. 2013;591:3525–3538. doi: 10.1113/jphysiol.2013.257964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller DM, Wang JA, Buchanan AK, Hall ED. Temporal and spatial dynamics of nrf2-antioxidant response elements mediated gene targets in cortex and hippocampus after controlled cortical impact traumatic brain injury in mice. J Neurotrauma. 2014;31:1194–1201. doi: 10.1089/neu.2013.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol. 2004;36:1882–1886. doi: 10.1016/j.biocel.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 69.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 70.Johns TG, Bernard CC. Binding of complement component Clq to myelin oligodendrocyte glycoprotein: a novel mechanism for regulating CNS inflammation. Mol Immunol. 1997;34:33–38. doi: 10.1016/S0161-5890(97)00005-9. [DOI] [PubMed] [Google Scholar]
- 71.Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, Bari F, Ungvari Z, Csiszar A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40:513–521. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tarantini S, Valcarcel-Ares MN, Yabluchanskiy A, Tucsek Z, Hertelendy P, Kiss T, Gautam T, Zhang XA, Sonntag WE, de Cabo R, et al. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J Gerontol A Biol Sci Med Sci. 2018;73:853–863. doi: 10.1093/gerona/glx177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.