

Abstract
Mathematical models of biological reactions at the system-level lead to a set of ordinary differential equations with many unknown parameters that need to be inferred using relatively few experimental measurements. Having a reliable and robust algorithm for parameter inference and prediction of the hidden dynamics has been one of the core subjects in systems biology, and is the focus of this study. We have developed a new systems-biology-informed deep learning algorithm that incorporates the system of ordinary differential equations into the neural networks. Enforcing these equations effectively adds constraints to the optimization procedure that manifests itself as an imposed structure on the observational data. Using few scattered and noisy measurements, we are able to infer the dynamics of unobserved species, external forcing, and the unknown model parameters. We have successfully tested the algorithm for three different benchmark problems.
Author summary
The dynamics of systems biological processes are usually modeled using ordinary differential equations (ODEs), which introduce various unknown parameters that need to be estimated efficiently from noisy measurements of concentration for a few species only. In this work, we present a new “systems-informed neural network” to infer the dynamics of experimentally unobserved species as well as the unknown parameters in the system of equations. By incorporating the system of ODEs into the neural networks, we effectively add constraints to the optimization algorithm, which makes the method robust to noisy and sparse measurements.
Introduction
Systems biology aims at a system-level understanding of biological systems, which is a holistic approach to deciphering the complexity of biological systems [1]. To understand the biological systems, we must understand the structures of the systems (both their components and the structural relationships), and their dynamics [2]. The dynamics of a biological system is usually modeled by a system of ordinary differential equations (ODEs), which describes the time evolution of the concentrations of chemical and molecular species in the system. After we know the pathway structure of the chemical reactions, then the ODEs can be derived using some kinetic laws, e.g., the law of mass action or the Michaelis-Menten kinetics [3].
System-level biological models usually introduce some unknown parameters that are required to be estimated accurately and efficiently. Thus, one central challenge in computational modeling of these systems is the estimation of model parameters (e.g., rate constants or initial concentrations) and the prediction of model dynamics (e.g., time evolution of experimentally unobserved concentrations). Hence, a lot of attention has been given to the problem of parameter estimation in the systems biology community. In particular, extensive research has been conducted on the applications of different optimization techniques, such as linear and nonlinear least-squares fitting [4], genetic algorithms [5], evolutionary computation [6], and more [7]. Considerable interest has also been raised by Bayesian methods [8, 9], which could extract information from noisy data. The main advantage of Bayesian methods is the ability to infer the whole probability distributions of the unknown parameters, rather than just a point estimate. More recently, parameter estimation for computational biology models has been tackled by the algorithms in the framework of control theory. These algorithms were originally developed for the problem of estimating the time evolution of the unobserved components of the state of a dynamical system. In this context, extended Kalman filtering [10], unscented Kalman filtering [11], and ensemble Kalman methods [12] have been applied as well. In addition, different methods have also been developed to address the issue of hidden variables and dynamics [13, 14], but in their examples the number of observable variables is almost one half of the number of total variables, while as we will show in the results below our proposed method requires less observable variables (e.g., one out of eight in the cell apoptosis model) to correctly infer unknown parameters and predict all unobserved variables.
Due to technical limitations, however, biological reaction networks are often only partially observable. Usually, experimental data are insufficient considering the size of the model, which results in parameters that are non-identifiable [15] or only identifiable within confidence intervals (see more details in [16]). Furthermore, a large class of models in systems biology are sensitivity to the parameter values that are distributed over many orders of magnitude. Such sloppiness is also a factor that makes parameter estimation more difficult [17]. In the process of parameter inference, two issues accounting for system’s (non-)identifiability have to be considered: structural identifiability that is related to the model structure independent of the experimental data [18, 19]; and practical identifiability that is related to the amount and quality of measured data. The a priori structural identifiability can be used to address the question of unique estimation of the unknown parameters based on the postulated model. However, a parameter that is structurally identifiable may still be practically non-identifiable assuming that the model is exact, but the measurements are noisy or sparse [20].
In this work, we introduce a new deep learning [21] method—systems-informed neural networks, based on the method of physics-informed neural networks [22, 23], to infer the hidden dynamics of experimentally unobserved species as well as the unknown parameters in the system of equations. By incorporating the system of ODEs into the neural networks (through adding the residuals of the equations to the loss function), we effectively add constraints to the optimization algorithm, which makes the method robust to measurement noise and few scattered observations. In addition, since large system-level biological models are typically encountered, our algorithm is computationally scalable and feasible, and its output is interpretable even though it depends on a high-dimensional parameter space.
Materials and methods
Throughout this paper, we assume that the systems biological process can be modeled by a system of ordinary differential equations (ODEs) of the following form
| (1a) |
| (1b) |
| (1c) |
where the state vector x = (x1, x2, …, xS) represents the concentration of S species, and p = (p1, p2, …, pK) are K parameters of the model, which remain to be determined. Hence, the system of ODEs will be identified once p is known. y = (y1, y2, …, yM) are the M measurable signals (consistent with the ODE system), which we can measure experimentally and could possibly be contaminated with a white noise ϵ of Gaussian type with zero mean and standard deviation σ. The output function h is determined by the design of the experiments that are used for parameter inference. While h could, in general, be any function, it is assumed to be a linear function with M ≤ S in most models as follows:
| (2) |
i.e., y1, y2, …, yM are the noisy measurements of the species among all S species (1 ≤ s1 < s2 < ⋯ < sM ≤ S).
Systems-informed neural networks and parameter inference
Based on the method of physics-informed neural networks proposed in [22], we introduce a deep learning framework that is informed by the systems biology equations that describe the kinetic pathways (Eq (1a)). A neural network with parameters θ takes time t as the input and outputs a vector of the state variables as a surrogate of the ODE solution x(t) (Fig 1). In our network, in addition to the standard layers, e.g., the fully-connected layer, we add three extra layers to make the network training easier, described as follows.
Fig 1. Neural network architecture.
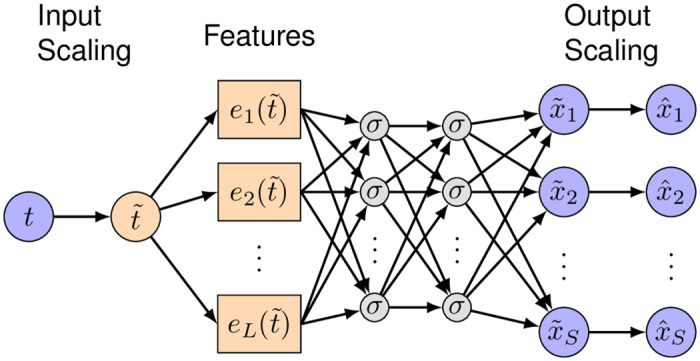
The network consists of an input-scaling layer, a feature layer, several fully-connected layers, and an output-scaling layer. The input-scaling layer and output-scaling layer are used to linearly scale the network input and outputs such that they are of order one. The feature layer is used to construct features explicitly as the input to the first fully-connected layer.
Input-scaling layer. Because the ODE system could have a large time domain, i.e., the input time t could vary by orders of magnitude, we first apply a linear scaling function to t, i.e., , such that . We can simply choose T as the maximum value of the time domain.
Feature layer. In many models, the ODE solution may have a certain pattern, e.g., periodicity in the yeast glycolysis model and fast decay in the cell apoptosis model, and thus it is beneficial to the network training by constructing a feature layer according to these patterns. Specifically, we employ the L functions e1(⋅), e2(⋅), …, eL(⋅) to construct the L features . The choice of the features is problem dependent, and we can select the features based on the available observations. For example, if the observed dynamics has certain periodicity (e.g., the yeast glycolysis model), then we can use sin(kt) as the features, where k is selected based on the period of the observed dynamics; if the observed dynamics decays fast (e.g., the cell apoptosis model), then we can use e−kt as a feature. However, if there is no clear pattern observed, the feature layer can also be removed.
Output-scaling layer. Because the outputs may have different magnitudes, similar to the input-scaling layer, we add another scaling layer to transform the output of the last fully-connected layer (of order one) to , i.e., , , . Here, k1, k2, …, kS are chosen as the magnitudes of the mean values of the ODE solution x1, x2, …, xS, respectively.
The next key step is to constrain the neural network to satisfy the scattered observations of y as well as the ODE system (Eq (1a)). This is realized by constructing the loss function by considering terms corresponding to the observations and the ODE system. Specifically, let us assume that we have the measurements of y1, y2, …, yM at the time , and we enforce the network to satisfy the ODE system at the time point . We note that the times and are not necessarily on a uniform grid, and they could be chosen at random. Then, the total loss is defined as a function of both θ and p:
| (3) |
where
| (4) |
| (5) |
| (6) |
is associated with the M sets of observations y given by Eq (1c), while enforces the structure imposed by the system of ODEs given in Eq (1a). We employ automatic differentiation (AD) to analytically compute the derivative in (see more details of AD in [23]). The third auxiliary loss term is introduced as an additional source of information for the system identification, and involves two time instants T0 and T1. It is essentially a component of the data loss; however, we prefer to separate this loss from the data loss, as in the auxiliary loss data are given for all state variables at these two time instants. We note that and are the discrepancy between the network and measurements, and thus they are supervised losses, while is based on the ODE system, and thus is unsupervised. In the last step, we infer the neural network parameters θ as well as the unknown parameters of the ODEs p simultaneously by minimizing the loss function via gradient-based optimizers, such as the Adam optimizer [24]:
| (7) |
We note that our proposed method is different from meta-modeling [25, 26], as we optimize θ and p simultaneously.
The M + 2S coefficients in Eq (4), in Eq (5), and in Eq (6) are used to balance the M + 2S loss terms. In this study, we manually select these weight coefficients such that the weighted losses are of the same order of magnitude during the network training. We note that this guideline makes the weight selection much easier, although there are many weights to be determined. These weights may also be automatically chosen, e.g., by the method proposed in [27]. In this study, the time instants for the observations are chosen randomly in the time domain, while the time instants used to enforce the ODEs are chosen in an equispaced grid. Additionally, in the auxiliary loss function, the first set of data is the initial conditions at time T0 for the state variables. The second set includes the values of the state variables at any arbitrary time instant T1 within the training time window (not too close to T0); in this work, we consider the midpoint time for the cell apoptosis model, and the final time instant for the yeast glycolysis model and ultradian endocrine model. If data is available at another time point, alternatively this point can be considered.
Analysis of system’s identifiability
A parameter pi is identifiable if the confidence interval of its estimate is finite. In systems identification problems, two different forms of identifiability namely, structural and practical are typically encountered. Structural non-identifiability arises from a redundant parameterization in the formal solution of y due to insufficient mapping h of internal states x to observables y in Eq (1) [15]. A priori structural identifiability has been studied extensively, e.g., using power series expansion [28] and differential algebraic methods [29], yet mostly limited to linear models as the problem is particularly difficult for nonlinear dynamical systems. Furthermore, practical non-identifiability cannot be detected with these methods, since experimental data are disregarded.
A parameter that is structurally identifiable may still be practically non-identifiable. Practical non-identifiability is intimately related to the amount and quality of measured data and manifests in a confidence interval that is infinite. Different methods have been proposed to estimate the confidence intervals of the parameters such as local approximation of the Fisher-Information-Matrix (FIM) [20] and bootstrapping approach [30]. Another approach is to quantify the sensitivity of the systems dynamics to variations in its parameters using a probabilistic framework [31]. For identifiability analysis, we primarily use the FIM method, which is detailed in S1 Text.
Implementation
The algorithm is implemented in Python using the open-source library DeepXDE [23]. The width and depth of the neural networks (listed in Table 1) depend on the size of the system of equations and the complexity of the dynamics. We use the function [32] as the activation function σ shown in Fig 1, and the feature layer is listed in Table 2.
Table 1. Hyperparameters for the problems in this study.
The fist and second number in the number of iterations correspond to the first and second training stage.
| Model | NN depth | NN width | #Iterations | |
|---|---|---|---|---|
| Yeast glycolysis | Noiseless | 4 | 128 | 1000, 9 × 104 |
| Noisy | 4 | 128 | 1000, 2 × 106 | |
| Cell apoptosis | Survival | 5 | 256 | 0, 1.5 × 106 |
| Death | 5 | 256 | 0, 1.5 × 106 | |
| Ultradian endocrine | Parameters only | 4 | 128 | 2000, 6 × 105 |
| Hidden nutrition | 4 | 128 | 2000, 1.5 × 106 |
Table 2. The feature layer used in the network for each problem.
| Model | Features |
|---|---|
| Yeast glycolysis | , , , , , , |
| Cell apoptosis | , |
| Ultradian endocrine | , , , , , |
For the training, we use an Adam optimizer [24] with default hyperparameters and a learning rate of 10−3, where the training is performed using the full batch of data. As the total loss consists of two supervised losses and one unsupervised loss, we perform the training using the following two-stage strategy:
-
Step 1
Considering that supervised training is usually easier than unsupervised training, we first train the network using the two supervised losses and for some iterations, such that the network can quickly match the observed data points.
-
Step 2
We further train the network using all the three losses.
We found empirically that this two-stage training strategy speeds up the network convergence. The number of iterations for each stage is listed in Table 1.
Results
Yeast glycolysis model
The model of oscillations in yeast glycolysis [33] has become a standard benchmark problem for systems biology inference [34, 35] as it represents complex nonlinear dynamics typical of biological systems. We use it here to study the performance of our deep learning algorithm used for parsimonious parameter inference with only two observables. The system of ODEs for this model as well as the target parameter values and the initial conditions are included in S2 Text. To represent experimental noise, we corrupt the observation data by a Gaussian noise with zero mean and the standard deviation of σϵ = cμ, where μ is the standard deviation of each observable over the observation time window and c = 0 − 0.1.
We start by inferring the dynamics using noiseless observations on two species S5 (the concentration of NADH) and S6 (the concentration of ATP) only. These two species are the minimum number of observables we can use to effectively infer all the parameters in the model. S1 Fig shows the noiseless synthetically generated data by solving the system of ODEs in S2 Text with the parameters listed in S1 Table. We sample data points within the time frame of 0 − 10 minutes at random and use them for training of the neural networks, where the neural network is informed by the governing ODEs of the yeast model as explained above. S2 Fig shows the inferred dynamics for all the species predicted by the systems-informed neural networks, and plotted against the exact dynamics that are generated by solving the system of ODEs. We observe excellent agreement between the inferred and exact dynamics within the training time window. The neural networks learn the input data given by scattered observations (shown by symbols in S2 Fig) and is able to infer the dynamics of other species due to the constraints imposed by the system of ODEs.
Next, we verify the robustness of the algorithm to noise. For that purpose, we introduce Gaussian additive noise with the noise level c = 10% to the observational data. The input training data are shown in Fig 2 for the same species (S5 and S6) as the observables, where similar to the previous test, we sample random scattered data points in time. Results for the inferred dynamics are shown in Fig 3. The agreement between the inferred and exact dynamics is excellent considering the relatively high level of noise in the training data. Our results show that the enforced equations in the loss function act as a constraint of the neural networks that can effectively prevent the overfitting of the network to the noisy data. One advantage of encoding the equations is their regularization effect without using any additional L1 or L2 regularization.
Fig 2. Glycolysis oscillator noisy observation data given to the algorithm for parameter inference.

500 measurements are corrupted by a zero-mean Gaussian noise and standard deviation of σ = 0.1μ. Only two observables S5 and S6 are considered and the data are randomly sampled in the time window of 0 − 10 minutes.
Fig 3. Glycolysis oscillator inferred dynamics from noisy measurements compared with the exact solution.

500 scattered observations are plotted using symbols for the two observables S5 and S6.
Our main objective in this work, however, is to infer the unknown model parameters p. This can be achieved simply by training the neural networks for its parameters θ as well as the model parameters using backpropagation. The results for the inferred model parameters along with their target values are given in Table 3 for both test cases (i.e., with and without noise in the observations). First thing to note is that the parameters can be identified within a confidence interval. Estimation of the confidence intervals a priori is the subject of structural identifiability analysis, which is not in the scope of this work. Second, practical identifiability analysis can be performed to identify the practically non-identifiable parameters based on the quality of the measurements and the level of the noise. We have performed local sensitivity analysis by constructing the Fisher Information Matrix (FIM) (S1 Text) and the correlation matrix R derived from the FIM.
Table 3. Parameter values for yeast glycolysis model and each corresponding inferred values.
The standard deviations are estimated using Eq. (S3) in S1 Text as practical non-identifiability analysis based on the FIM.
| Parameter | Target value | Inferred value (Noiseless observations) | Inferred value (Noisy observations) | Standard deviation |
|---|---|---|---|---|
| J0 | 2.5 | 2.50 | 2.49 | 0.18 |
| k1 | 100 | 99.9 | 86.1 | 62.0 |
| k2 | 6 | 6.01 | 4.55 | 21.3 |
| k3 | 16 | 15.9 | 14.0 | 21.9 |
| k4 | 100 | 100.1 | 97.1 | 103.6 |
| k5 | 1.28 | 1.28 | 1.24 | 0.25 |
| k6 | 12 | 12.0 | 12.7 | 5.1 |
| k | 1.8 | 1.79 | 1.55 | 4.34 |
| κ | 13 | 13.0 | 13.4 | 25.9 |
| q | 4 | 4.00 | 4.07 | 0.27 |
| K1 | 0.52 | 0.520 | 0.550 | 0.091 |
| ψ | 0.1 | 0.0994 | 0.0823 | 0.317 |
| N | 1 | 0.999 | 1.29 | 2.94 |
| A | 4 | 4.01 | 4.25 | 2.28 |
The inferred parameters from both noiseless and noisy observations are in good agreement with their target values. The most significant difference (close to 30% difference) can be seen for the parameter N (the total concentration of NAD+ and NADH). However, given that the glycolysis system (S2 Text) is identifiable (c.f. [33, 35] and S3 Fig), and the inferred dynamics shown in S2 Fig and Fig 3 show that the learned dynamics match very well with the exact dynamics, the inferred parameters are valid. We used Eq. (S3) in S1 Text to estimate the standard deviations of the model parameters. The σi estimates for the parameters are the lower bounds, and thus, may not be informative here. Further, these estimates are derived based on a local sensitivity analysis. A structural/practical identifiability analysis [15] or a bootstrapping approach to obtain the parameter confidence intervals is probably more relevant here. Using the FIM, we are able to construct the correlation matrix R for the parameters. Nearly perfect correlations (|Rij| ≈ 1) suggest that the FIM is singular and the correlated parameters may not be practically identifiable. For the glycolysis model, as shown in S3 Fig, no perfect correlations can be found in R (except for the anti-diagonal elements), which suggests that the model described by S2 Text is practically identifiable. In the example above, we considered 500 data measurements, but in systems biology we often lack the ability to observe system dynamics at a fine-time scale. To investigate the performance of our method to a set of sparse data points, we used only 200 data points, and still have a good inferred dynamics of the species S1, S2, S5 and S6 (S4 Fig).
Cell apoptosis model
Although the glycolysis model is highly nonlinear and difficult to learn, we have shown that its parameters can be identified. To investigate the performance of our algorithm for non-identifiable systems, we study a cell apoptosis model, which is a core sub-network of the signal transduction cascade regulating the programmed cell death-against-survival phenotypic decision [36]. The equations defining the cell apoptosis model and the values of the rate constants for the model are taken from [36] and listed in S2 Table.
Although the model is derived using simple mass-action kinetics and its dynamics is easy to learn with our algorithm, most of the parameters are not identifiable due to both structural and practical non-identifiability. To infer the dynamics of this model, we only use 120 random samples of measurements collected for one observable (x4), where we assume that the measurements are corrupted by a zero-mean Gaussian noise and 5% standard deviation as shown in Fig 4. Furthermore, it is possible to use different initial conditions in order to produce different cell survival outcomes. The initial conditions for all the species are given in S3 Text, while we use x7(0) = 2.9 × 104 (molecules/cell) to model cell survival (Fig 4(top)) and x7(0) = 2.9 × 103 (molecules/cell) to model cell death (Fig 4(bottom)).
Fig 4. Cell apoptosis noisy observation data given to the algorithm for parameter inference.
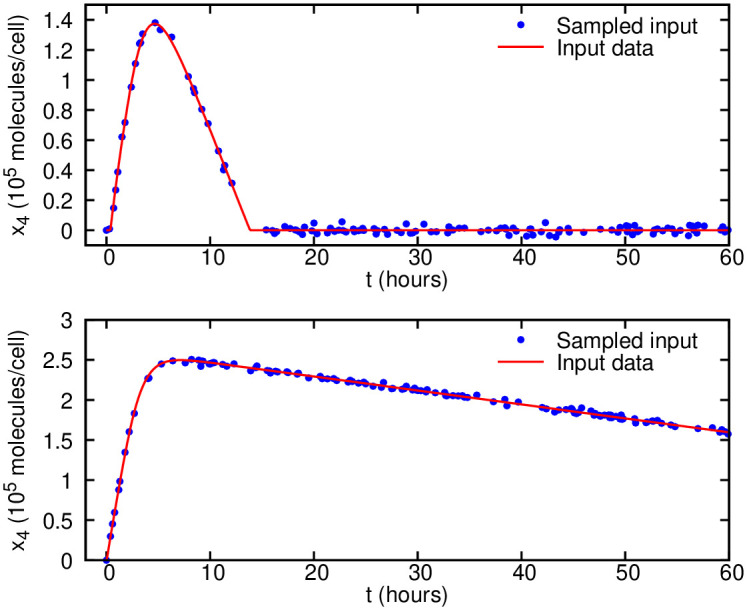
120 measurements are corrupted by a zero-mean Gaussian noise and standard deviation of σ = 0.05μ. Data for the observable x4 only are randomly sampled during the time window of 0 − 60 hours for two scenarios: (top) cell survival with the initial condition x7(0) = 2.9 × 104 (molecules/cell) and (bottom) cell death with x7(0) = 2.9 × 103 (molecules/cell).
Using the systems-informed neural networks and the noisy input data, we are able to infer most of the dynamics (including x3, x4, x6, x7 and x8) of the system as shown in S5 Fig and Fig 5. These results show a good agreement between the inferred and exact dynamics of the cell survival/apoptosis models using one observable only.
Fig 5. Cell apoptosis inferred dynamics from noisy observations compared with the exact solution.

Predictions are performed on equally-spaced time instants in the interval of 0 − 60 hours. The scattered observations are plotted using symbols only for the observable x4. The exact data and the scattered observations are computed by solving the system of ODEs given in S3 Text.
We report the inferred parameters for the cell apoptosis model in Table 4, where we have used noisy observations on x4 under two scenarios of cell death and survival for comparison. The results show that four parameters (k1, kd2, kd4 and kd6) can be identified by our proposed method with relatively high accuracy, as indicated by the check mark (✓) in the last column of Table 4. We observe that the standard deviations for most of the parameter estimates are orders of magnitude larger than their target values, and thus the standard deviations estimated using the FIM are not informative in the practical identifiability analysis. The only informative standard deviation is for kd6 (indicated by the symbol †), and kd6 is inferred with relatively high accuracy by our method.
Table 4. Parameter values for cell apoptosis model and their corresponding inferred values.
The standard deviations are estimated using Eq. (S3) in S1 Text as practical identifiability analysis using the Fisher Information Matrix. The symbols ✓, † and ⋆ in the last column denote that the corresponding variable is identifiable using our proposed method, FIM standard deviation, and null-eigenvector analysis.
| Parameter | Target Value | Cell Survival | Cell Death | Identifiable | ||
|---|---|---|---|---|---|---|
| Inferred Value | Standard Deviation | Inferred Value | Standard Deviation | |||
| k1 | 2.67 × 10−9 | 0.59 × 10−9 | 6.2 × 10−6 | 0.34 × 10−9 | 1.9 × 10−5 | ✓⋆ |
| kd1 | 1 × 10−2 | 7.06 × 10−10 | 35.5 | 3.37 × 10−7 | 96.7 | |
| kd2 | 8 × 10−3 | 1.72 × 10−3 | 4.9 | 2.38 × 10−3 | 23.8 | ✓ |
| k3 | 6.8 × 10−8 | 0.15 × 10−8 | 1.0 × 10−4 | 0.16 × 10−8 | 2.1 × 10−4 | |
| kd3 | 5 × 10−2 | 4.19 × 10−10 | 62.8 | 4.23 × 10−10 | 78.0 | |
| kd4 | 1 × 10−3 | 0.92 × 10−3 | 0.20 | 1.28 × 10−3 | 1.2 | ✓⋆ |
| k5 | 7 × 10−5 | 1.49 × 10−7 | 0.019 | 7.25 × 10−8 | 0.37 | |
| kd5 | 1.67 × 10−5 | 6.92 × 10−11 | 0.034 | 8.51 × 10−11 | 13.7 | |
| kd6 | 1.67 × 10−4 | 1.81 × 10−4 | 0.35 × 10−4 | 1.57 × 10−4 | 1.57 × 10−4 | ✓†⋆ |
To have a better picture of the practical identifiability analysis, we have plotted the correlation matrix R in S6 Fig. We observe perfect correlations |Rij| ≈ 1 between some parameters. Specifically, parameters k1 − kd1, and k3 − kd3 have correlations above 0.99 for cell survival model, which suggests that these parameters may not be identified. This is generally in agreement with the parameter inference results in Table 4 with some exceptions. Our parameter inference algorithm suggests that k1 is identifiable, whereas kd1 is not for the cell survival model. Thus, in order to increase the power of the practical identifiability analysis and to complement the correlation matrix, we have computed the FIM null eigenvectors and for each eigenvector we identified the most dominant coefficients, which are plotted in Fig 6. We observe that there are six null eigenvectors associated with the zero eigenvalues of the FIM for both the cell survival and cell death models. The most dominant coefficient in each null eigenvector is associated with a parameter that can be considered as practically non-identifiable. The identifiable parameters include k1, kd4 and kd6 (indicated by the symbol ⋆ in Table 4), which agree well with the results of our algorithm. On the contrary, our algorithm successfully infers one more parameter kd2 than the above analysis. This could be due to the fact that checking practical identifiability using the FIM can be problematic, especially for partially observed nonlinear systems [37]. We have similar results for the cell death model.
Fig 6. Fisher information matrix null eigenvectors of the cell apoptosis model.

The most dominant component in each null eigenvector associated with a specific parameter suggests that the parameter may not be practically identifiable: (left) cell survival and (right) cell death.
Ultradian endocrine model
The final test case for assessing the performance of the proposed algorithm is to infer parameters of the ultradian model for the glucose-insulin interaction. We use a relatively simple ultradian model [38] with 6 state variables and 30 parameters. This model is developed in a non-pathophysiologic context and represents relatively simple physiologic mechanics. In this model, the main variables are the plasma insulin concentration Ip, the interstitial insulin concentration Ii, the glucose concentration G, and a three stage filter (h1, h2, h3) that reflects the response of the plasma insulin to glucose levels [38]. The resulting system of ODEs, the nominal values for the parameters of the ultradian model along with the initial conditions for the 6 state variables are given in S4 Text.
The nutritional driver IG(t) is the systematic forcing of the model that represents the external sources of glucose from nutritional intake. Although the nutritional intake (modeled by the N discrete nutrition events) is required to be defined and properly recorded by the patients, it is not always accurately recorded or may contain missing values. Therefore, it would be useful to employ systems-informed neural networks to not only infer the model parameters given the nutrition events, but also to assume that the intake is unknown (hidden forcing) and infer the nutritional driver in Eq. S7f (S4 Text) as the same time.
Model parameter inference given the nutrition events
We consider an exponential decay functional form for the nutritional intake , where the decay constant k is the only unknown parameter and three nutrition events are given by (tj, mj) = [(300, 60) (650, 40) (1100, 50)] (min, g) pairs. The only observable is the glucose level measurements G shown in Fig 7 (generated here synthetically by solving the system of ODEs), which are sampled randomly to train the neural networks for the time window of 0 − 1800 minutes. Because we only use the observation of G and have more than 20 parameters to infer, we limit the search range for these parameters. The range of seven parameters is adopted from [38], and the range for other parameters is set as (0.2x, 1.8x), where x is the nominal value of that parameter (S3 Table).
Fig 7. Ultradian glucose-insulin model observation data given to the algorithm for parameter inference.
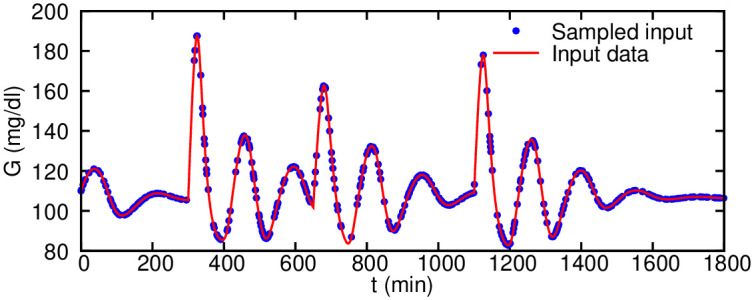
360 noiseless measurements on glucose level (G) only are randomly sampled in the time window of 0 − 1800 minutes (∼ one day).
For the first test case, we set the parameters Vp, Vi and Vg to their nominal values and infer the rest of the parameters. The inferred values are given in Table 5 (column Test 1), where we observe good agreement between the target and inferred values. For the second test, we also infer the values of Vp, Vi and Vg (Table 5). Although the inferred parameters are slightly worse than the Test 1, when using the inferred parameters, we are able to solve the equations for unseen time instants with high accuracy. We perform forecasting for the second test case after training the algorithm using the glucose data in the time interval of t = 0 − 1800 min and inferring the model parameters. Next, we consider that there is a nutrition event at time tj = 2000 min with carbohydrate intake of mj = 100 g. As shown in Fig 8, we are able to forecast with high accuracy the glucose-insulin dynamics, more specifically, the glucose levels following the nutrition intake.
Table 5. Parameter values for the ultradian glucose-insulin model and their corresponding inferred values.
| Parameter | Nominal value | Inferred value (Test 1) | Inferred value (Test 2) |
|---|---|---|---|
| Vp | 3 | – | 2.97 |
| Vi | 11 | – | 9.25 |
| Vg | 10 | – | 11.3 |
| E | 0.2 | 0.209 | 0.216 |
| tp | 6 | 6.58 | 6.30 |
| ti | 100 | 96.6 | 136 |
| td | 12 | 11.8 | 11.6 |
| k | 0.0083 | 0.00837 | 0.00833 |
| Rm | 209 | 232 | 198 |
| a1 | 6.6 | 6.56 | 6.48 |
| C1 | 300 | 321 | 277 |
| C2 | 144 | 52.6 | 44.9 |
| C4 | 80 | 67.1 | 73.9 |
| C5 | 26 | 25.2 | 25.6 |
| Ub | 72 | 68.3 | 73.6 |
| U0/C3 | 0.04 | 0.0464 | 0.0463 |
| Um/C3 | 0.9 | 0.790 | 0.975 |
| Rg | 180 | 182 | 182 |
| α | 7.5 | 7.89 | 7.94 |
| β | 1.772 | 1.91 | 1.85 |
Fig 8. Ultradian glucose-insulin inferred dynamics and forecasting compared with the exact solution given nutrition events.

600 scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. Note that the parameter k in the intake function IG is considered to be unknown, while the timing and carbohydrate content of each nutrition event are given. Given the inferred parameters, we can accurately forecast the glucose levels following the event at time t = 2000 min.
Model parameter inference with hidden nutrition events
As detailed in the following, one of the significant advantages of the systems-informed neural network is its ability to infer the hidden systematic forcing in the model. For example, in the glucose-insulin model, the nutritional driver IG is the forcing that we aim to infer as well. Here, we use the glucose measurements to train the model for the time interval t = 0 − 1800 min shown in Fig 7, while we assume that the time instants and quantities of three nutritional events are additionally unknown.
We found that it is difficult to infer all the parameters as well as the the timing and carbohydrate content of each nutrition event. However, given Vp, Vi, Vg and the timing of each nutrition event, the algorithm is capable of inferring the other model parameters as well as the carbohydrate content (S4 Table column Test 1). Having the nutrition events as well as all other unknown parameters estimated, we are able to forecast the glucose levels for t = 1800 − 3000 min assuming there has been a nutritional intake of (tj, mj) = (2000, 100). The predictions for the glucose G and the nutritional driver IG are shown in S7 Fig, which show excellent agreement in the forecasting of glucose levels. For the second test, we also infer the values of Vp, Vi and Vg, and the result is slightly worse (S4 Table column Test 2 and S8 Fig).
If both the timing and carbohydrate content of each nutrition event are unknown, the algorithm is also capable to infer them by assuming that certain model parameters are known. We found that the selection of the known parameters is important. As shown in S4 Table, we consider different combinations of parameters to be known in Test 3 and Test 4; Test 3 leads to good prediction accuracy (S9 Fig) while Test 4 does not.
Discussion
We presented a new and simple to implement “systems-biology-informed” deep learning algorithm that can reliably and accurately infer the hidden dynamics described by a mathematical model in the form of a system of ODEs. The system of ODEs is encoded into a plain “uninformed” deep neural networks and is enforced through minimizing the loss function that includes the residuals of the ODEs. Enforcing the equations in the loss function adds additional constraints in the learning process, which leads to several advantages of the proposed algorithm: first, we are able to infer the unknown parameters of the system of ODEs once the neural network is trained; second, we can use a minimalistic amount of data on a few observables to infer the dynamics and the unknown parameters; third, the enforcement of the equations adds a regularization effect that makes the algorithm robust to noise (we have not used any other regularization technique); and lastly, the measurements can be scattered, noisy and just a few.
The problem of structural and practical non-identifiability (such as the one encountered in the cell apoptosis model) is a long-standing problem in the field of systems identification, and has been under extensive research, e.g., [39]. Structural non-identifiabilities originate from incomplete observation of the internal model states. Because the structural non-identifiability is independent of the accuracy of experimental data measurements, we cannot resolve it by a refinement of existing measurements, and one possible way to resolve this issue is increasing the number of observed species. Our focus in this study is mostly on practical identifiability, which can guide us to redesign the experiment, improve the model, or collect more experimental measurements. In this study, we used FIM and local sensitivity analysis for the identifiability analysis, but we note that FIM has many limitations and can be problematic, especially for partially observed nonlinear systems [37], and hence other advanced alternatives [15, 40] should be used in future works. However, our goal in this work was not to do systematic identifiability analysis, but rather to use identifiability analysis to explain some of our findings.
Conclusion
We have used three benchmark problems to assess the performance of the algorithm including a highly nonlinear glycolysis model, a non-identifiable cell apoptosis model, and an ultradian glucose-insulin model for glucose forecasting based on the nutritional intake. Given the system of ODEs and initial conditions of the state variables, the algorithm is capable of accurately inferring the whole dynamics with one or two observables, where the unknown parameters are also inferred during the training process. An important and very useful outcome of the algorithm is its ability to infer the systematic forcing or driver in the model such as the nutritional intake in the glucose-insulin model. In this work, we considered the synthetic data of three small problems to test the performance and limitation of the proposed method. We will apply our method to larger problems and real data (e.g., the dataset introduced in [41]) in future work.
Supporting information
(PDF)
(PDF)
(PDF)
(PDF)
500 noiseless measurements of two observables S5 and S6 are randomly sampled in the time window of 0 − 10 minutes.
(PDF)
Predictions are performed on equally-spaced time instants in the interval of 0 − 10 minutes. The scattered observations are plotted using symbols for the two observables S5 and S6. The exact data and the scattered observations are computed by solving the system of ODEs given in S2 Text.
(PDF)
The correlation matrix is computed using the local sensitivity analysis and the FIM for the parameters involved in the glycolysis oscillator model assuming 10% noise in the observation data. We observe no perfect correlations suggesting that FIM is not singular and the parameters in the glycolysis model are practically identifiable.
(PDF)
200 scattered observations are plotted using symbols for the two observables S5 and S6.
(PDF)
Predictions are performed on equally-spaced time instants in the interval of 0 − 60 hours. The scattered observations are plotted using symbols only for the observable x4. The exact data and the scattered observations are computed by solving the system of ODEs given in S3 Text.
(PDF)
The correlation matrix is computed using FIM for the practical identifiablity analysis of parameters involved in the cell apoptosis model assuming 5% noise in the observation data for two scenarios: (left) cell survival and (right) cell death. We observe perfect correlations of ≈ 1.0 between the parameters suggesting that the FIM is singular and some parameters in the cell apoptosis model are practically non-identifiable.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The parameter k in the intake function IG as well as carbohydrate content (mj) of each nutrition event are treated as unknown, while Vp, Vi, Vg and the timing (tj) of each nutrition event are given.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The parameter k in the intake function IG as well as carbohydrate content (mj) of each nutrition event are treated as unknown, while the timing (tj) of each nutrition event are given.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The timing (tj) and carbohydrate content (mj) of each nutrition event are treated as unknown, while the parameter k in the intake function IG is given.
(PDF)
(PDF)
(PDF)
The search range for the first 7 parameters is adopted from [38], and the range for the other parameters is (0.2x, 1.8x), where x is the nominal value of that parameter.
(PDF)
(PDF)
Data Availability
All relevant data are within the manuscript, its Supporting information files and available from https://github.com/alirezayazdani1/SBINNs.
Funding Statement
This work was supported by the National Institutes of Health U01HL142518 (GEK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wilkinson DJ. Stochastic modelling for systems biology. CRC press; 2018. [Google Scholar]
- 2. Kitano H. Systems biology: a brief overview. Science. 2002;295(5560):1662–1664. 10.1126/science.1069492 [DOI] [PubMed] [Google Scholar]
- 3. Cornish-Bowden A, Cornish-Bowden A. Fundamentals of enzyme kinetics. vol. 510 Wiley-Blackwell; Weinheim, Germany; 2012. [Google Scholar]
- 4. Mendes P, Kell D. Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics (Oxford, England). 1998;14(10):869–883. 10.1093/bioinformatics/14.10.869 [DOI] [PubMed] [Google Scholar]
- 5. Srinivas M, Patnaik LM. Genetic algorithms: A survey. Computer. 1994;27(6):17–26. 10.1109/2.294849 [DOI] [Google Scholar]
- 6. Moles CG, Mendes P, Banga JR. Parameter estimation in biochemical pathways: a comparison of global optimization methods. Genome research. 2003;13(11):2467–2474. 10.1101/gr.1262503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Villaverde AF, Fröhlich F, Weindl D, Hasenauer J, Banga JR. Benchmarking optimization methods for parameter estimation in large kinetic models. Bioinformatics. 2019;35(5):830–838. 10.1093/bioinformatics/bty736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkinson DJ. Bayesian methods in bioinformatics and computational systems biology. Briefings in bioinformatics. 2007;8(2):109–116. 10.1093/bib/bbm007 [DOI] [PubMed] [Google Scholar]
- 9. Calvetti D, Somersalo E. An introduction to Bayesian scientific computing: ten lectures on subjective computing. vol. 2 Springer Science & Business Media; 2007. [Google Scholar]
- 10. Lillacci G, Khammash M. Parameter estimation and model selection in computational biology. PLoS computational biology. 2010;6(3):e1000696 10.1371/journal.pcbi.1000696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quach M, Brunel N, d’Alché Buc F. Estimating parameters and hidden variables in non-linear state-space models based on ODEs for biological networks inference. Bioinformatics. 2007;23(23):3209–3216. 10.1093/bioinformatics/btm510 [DOI] [PubMed] [Google Scholar]
- 12. Albers DJ, Blancquart PA, Levine ME, Seylabi EE, Stuart A. Ensemble Kalman methods with constraints. Inverse Problems. 2019;35(9):095007 10.1088/1361-6420/ab1c09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engelhardt B, Frőhlich H, Kschischo M. Learning (from) the errors of a systems biology model. Scientific reports. 2016;6(1):1–9. 10.1038/srep20772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engelhardt B, Kschischo M, Fröhlich H. A Bayesian approach to estimating hidden variables as well as missing and wrong molecular interactions in ordinary differential equation-based mathematical models. Journal of The Royal Society Interface. 2017;14(131):20170332 10.1098/rsif.2017.0332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmüller U, et al. Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics. 2009;25(15):1923–1929. 10.1093/bioinformatics/btp358 [DOI] [PubMed] [Google Scholar]
- 16. Villaverde AF, Tsiantis N, Banga JR. Full observability and estimation of unknown inputs, states and parameters of nonlinear biological models. Journal of the Royal Society Interface. 2019;16(156):20190043 10.1098/rsif.2019.0043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP. Universally sloppy parameter sensitivities in systems biology models. PLoS computational biology. 2007;3(10):e189 10.1371/journal.pcbi.0030189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chis OT, Banga JR, Balsa-Canto E. Structural identifiability of systems biology models: a critical comparison of methods. PloS one. 2011;6(11):e27755 10.1371/journal.pone.0027755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Majda AJ. Challenges in climate science and contemporary applied mathematics. Communications on Pure and Applied Mathematics. 2012;65(7):920–948. 10.1002/cpa.21401 [DOI] [Google Scholar]
- 20. Rodriguez-Fernandez M, Mendes P, Banga JR. A hybrid approach for efficient and robust parameter estimation in biochemical pathways. Biosystems. 2006;83(2-3):248–265. 10.1016/j.biosystems.2005.06.016 [DOI] [PubMed] [Google Scholar]
- 21.Goodfellow I BY, A C. Deep Learning; 2017.
- 22. Raissi M, Perdikaris P, Karniadakis GE. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. Journal of Computational Physics. 2019;378:686–707. 10.1016/j.jcp.2018.10.045 [DOI] [Google Scholar]
- 23.Lu L, Meng X, Mao Z, Karniadakis GE. DeepXDE: A deep learning library for solving differential equations. arXiv preprint arXiv:190704502. 2019.
- 24.Kingma DP, Ba J. Adam: A method for stochastic optimization. arXiv preprint arXiv:14126980. 2014.
- 25.Caughlin D. Parameter identification methods for metamodeling simulations. In: Proceedings of the 28th conference on Winter simulation; 1996. p. 756–763.
- 26. Song Xm, Kong Fz, Zhan Cs, Han Jw, Zhang Xh. Parameter identification and global sensitivity analysis of Xin’anjiang model using meta-modeling approach. Water Science and Engineering. 2013;6(1):1–17. [Google Scholar]
- 27.Wang S, Teng Y, Perdikaris P. Understanding and mitigating gradient pathologies in physics-informed neural networks. arXiv preprint arXiv:200104536. 2020.
- 28. Pohjanpalo H. System identifiability based on the power series expansion of the solution. Mathematical biosciences. 1978;41(1-2):21–33. 10.1016/0025-5564(78)90063-9 [DOI] [Google Scholar]
- 29. Ljung L, Glad T. On global identifiability for arbitrary model parametrizations. Automatica. 1994;30(2):265–276. 10.1016/0005-1098(94)90029-9 [DOI] [Google Scholar]
- 30. Balsa-Canto E, Alonso AA, Banga JR. Computational procedures for optimal experimental design in biological systems. IET systems biology. 2008;2(4):163–172. 10.1049/iet-syb:20070069 [DOI] [PubMed] [Google Scholar]
- 31. Foo J, Sindi S, Karniadakis GE. Multi-element probabilistic collocation for sensitivity analysis in cellular signalling networks. IET systems biology. 2009;3(4):239–254. 10.1049/iet-syb.2008.0126 [DOI] [PubMed] [Google Scholar]
- 32.Ramachandran P, Zoph B, Le QV. Searching for activation functions. arXiv preprint arXiv:171005941. 2017.
- 33. Ruoff P, Christensen MK, Wolf J, Heinrich R. Temperature dependency and temperature compensation in a model of yeast glycolytic oscillations. Biophysical chemistry. 2003;106(2):179–192. 10.1016/S0301-4622(03)00191-1 [DOI] [PubMed] [Google Scholar]
- 34. Schmidt M, Lipson H. Distilling free-form natural laws from experimental data. science. 2009;324(5923):81–85. 10.1126/science.1165893 [DOI] [PubMed] [Google Scholar]
- 35. Daniels BC, Nemenman I. Efficient inference of parsimonious phenomenological models of cellular dynamics using S-systems and alternating regression. PloS one. 2015;10(3):e0119821 10.1371/journal.pone.0119821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aldridge BB, Haller G, Sorger PK, Lauffenburger DA. Direct Lyapunov exponent analysis enables parametric study of transient signalling governing cell behaviour. IEE Proceedings-Systems Biology. 2006;153(6):425–432. 10.1049/ip-syb:20050065 [DOI] [PubMed] [Google Scholar]
- 37. Joshi M, Seidel-Morgenstern A, Kremling A. Exploiting the bootstrap method for quantifying parameter confidence intervals in dynamical systems. Metabolic engineering. 2006;8(5):447–455. 10.1016/j.ymben.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 38. Sturis J, Polonsky KS, Mosekilde E, Van Cauter E. Computer model for mechanisms underlying ultradian oscillations of insulin and glucose. American Journal of Physiology-Endocrinology And Metabolism. 1991;260(5):E801–E809. 10.1152/ajpendo.1991.260.5.E801 [DOI] [PubMed] [Google Scholar]
- 39. Hong H, Ovchinnikov A, Pogudin G, Yap C. SIAN: software for structural identifiability analysis of ODE models. Bioinformatics. 2019;35(16):2873–2874. 10.1093/bioinformatics/bty1069 [DOI] [PubMed] [Google Scholar]
- 40. Gábor A, Villaverde AF, Banga JR. Parameter identifiability analysis and visualization in large-scale kinetic models of biosystems. BMC systems biology. 2017;11(1):1–16. 10.1186/s12918-017-0428-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albers D, Levine M, Sirlanci M, Stuart A. A Simple Modeling Framework For Prediction In The Human Glucose-Insulin System. arXiv preprint arXiv:191014193. 2019. [DOI] [PMC free article] [PubMed]
- 42. Albers DJ, Levine M, Gluckman B, Ginsberg H, Hripcsak G, Mamykina L. Personalized glucose forecasting for type 2 diabetes using data assimilation. PLoS computational biology. 2017;13(4):e1005232 10.1371/journal.pcbi.1005232 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
(PDF)
(PDF)
500 noiseless measurements of two observables S5 and S6 are randomly sampled in the time window of 0 − 10 minutes.
(PDF)
Predictions are performed on equally-spaced time instants in the interval of 0 − 10 minutes. The scattered observations are plotted using symbols for the two observables S5 and S6. The exact data and the scattered observations are computed by solving the system of ODEs given in S2 Text.
(PDF)
The correlation matrix is computed using the local sensitivity analysis and the FIM for the parameters involved in the glycolysis oscillator model assuming 10% noise in the observation data. We observe no perfect correlations suggesting that FIM is not singular and the parameters in the glycolysis model are practically identifiable.
(PDF)
200 scattered observations are plotted using symbols for the two observables S5 and S6.
(PDF)
Predictions are performed on equally-spaced time instants in the interval of 0 − 60 hours. The scattered observations are plotted using symbols only for the observable x4. The exact data and the scattered observations are computed by solving the system of ODEs given in S3 Text.
(PDF)
The correlation matrix is computed using FIM for the practical identifiablity analysis of parameters involved in the cell apoptosis model assuming 5% noise in the observation data for two scenarios: (left) cell survival and (right) cell death. We observe perfect correlations of ≈ 1.0 between the parameters suggesting that the FIM is singular and some parameters in the cell apoptosis model are practically non-identifiable.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The parameter k in the intake function IG as well as carbohydrate content (mj) of each nutrition event are treated as unknown, while Vp, Vi, Vg and the timing (tj) of each nutrition event are given.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The parameter k in the intake function IG as well as carbohydrate content (mj) of each nutrition event are treated as unknown, while the timing (tj) of each nutrition event are given.
(PDF)
Scattered observations of glucose level are randomly sampled from 0 − 1800 min and used for training. The timing (tj) and carbohydrate content (mj) of each nutrition event are treated as unknown, while the parameter k in the intake function IG is given.
(PDF)
(PDF)
(PDF)
The search range for the first 7 parameters is adopted from [38], and the range for the other parameters is (0.2x, 1.8x), where x is the nominal value of that parameter.
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the manuscript, its Supporting information files and available from https://github.com/alirezayazdani1/SBINNs.