

Mallory-Denk-bodies (MDBs) are hepatic inflammation-associated protein aggregates. Similar protein aggregation in neurodegenerative diseases also triggers inflammation and NF-κB-activation by an as yet uncharacterized non-canonical mechanism. Herein, employing an MDB-inducing cell model, we uncovered a novel mechanism for NF-κB-activation via cytoplasmic IκBα-sequestration into insoluble aggregates. Additionally, through affinity pull-down, proximity labeling, and other proteomic approaches, we identified 10 proteins that interact with IκBα and form insoluble aggregates upon ZnPP- treatment. This novel mechanism would account for the protein aggregate-induced inflammation.
Keywords: Mallory-Denk-bodies, IκB, α, IκB, β, NF-κB, p62, erythropoietic protoporphyria, X-linked protoporphyria, liver inflammation, NMPP, PPIX, ZnPP, proteomics., affinity proteomics, hepatotoxicity, inflammation, inflammatory response, knockouts*, label-free quantification, liver disease, mass spectrometry, protein-protein interactions, immunoaffinity, IkBa, NF-kB, protein aggregation, ZnPPIX
Graphical Abstract

Highlights
Liver Mallory-Denk-Body inducers elicited an IκBα-loss and NF-κB-activation.
IκBα-loss was due to its sequestration into insoluble cytoplasmic aggregates.
Four proteomic approaches identified 10 IκBα-interacting/aggregating proteins.
Nup153/RanBP2-aggregation prevented IκBα nuclear entry for ending NF-κB-activation.
Abstract
Mallory-Denk-bodies (MDBs) are hepatic protein aggregates associated with inflammation both clinically and in MDB-inducing models. Similar protein aggregation in neurodegenerative diseases also triggers inflammation and NF-κB activation. However, the precise mechanism that links protein aggregation to NF-κB-activation and inflammatory response remains unclear. Herein we find that treating primary hepatocytes with MDB-inducing agents (N-methylprotoporphyrin (NMPP), protoporphyrin IX (PPIX), or Zinc-protoporphyrin IX (ZnPP)) elicited an IκBα-loss with consequent NF-κB activation. Four known mechanisms of IκBα-loss i.e. the canonical ubiquitin-dependent proteasomal degradation (UPD), autophagic-lysosomal degradation, calpain degradation and translational inhibition, were all probed and excluded. Immunofluorescence analyses of ZnPP-treated cells coupled with 8 M urea/CHAPS-extraction revealed that this IκBα-loss was due to its sequestration along with IκBβ into insoluble aggregates, thereby releasing NF-κB. Through affinity pulldown, proximity biotinylation by antibody recognition, and other proteomic analyses, we verified that NF-κB subunit p65, which stably interacts with IκBα under normal conditions, no longer binds to it upon ZnPP-treatment. Additionally, we identified 10 proteins that interact with IκBα under baseline conditions, aggregate upon ZnPP-treatment, and maintain the interaction with IκBα after ZnPP-treatment, either by cosequestering into insoluble aggregates or through a different mechanism. Of these 10 proteins, the nucleoporins Nup153 and Nup358/RanBP2 were identified through RNA-interference, as mediators of IκBα-nuclear import. The concurrent aggregation of IκBα, NUP153, and RanBP2 upon ZnPP-treatment, synergistically precluded the nuclear entry of IκBα and its consequent binding and termination of NF-κB activation. This novel mechanism may account for the protein aggregate-induced inflammation observed in liver diseases, thus identifying novel targets for therapeutic intervention. Because of inherent commonalities this MDB cell model is a bona fide protoporphyric model, making these findings equally relevant to the liver inflammation associated with clinical protoporphyria.
Protein aggregates and inclusion bodies are linked to various neurodegenerative, muscular and hepatic diseases [Alzheimer's, Parkinson's, Desmin-related myopathies, and Mallory-Denk-bodies (MDBs) (1)]. Although different aggregates vary with each tissue source and in predominant protein composition, they contain common components such as p62/Sequestosome-1, ubiquitin, chaperones and proteasome constituents, which are often misfolded, highly insoluble, and cross-linked (1, 2).
Hepatic MDBs consisting largely of phosphorylated cytokeratins and p62 are commonly found in patients with alcoholic steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH), primary biliary cirrhosis, nonalcoholic cirrhosis, hepatocellular carcinoma, morbid obesity, and copper-related disorders (3). MDBs can be reproduced in mice by long term feeding of griseofulvin (GF) or 3,5-dicarbethoxy-1,4-dihydrocollidine (DDC) (4, 5). Both compounds inactivate certain hepatic cytochromes P450, converting their prosthetic heme into N-methylprotoporphyrin(s) (NMPP) that inhibit hepatic ferrochelatase (Fech) resulting in hepatic heme depletion and protoporphyrin IX (PPIX) accumulation (6, 7) (Fig. 1A). Fechm1Pas mutant (fech/fech) mice with <5% of normal enzyme activity also develop spontaneous MDBs at 20-weeks of age, validating their use as an experimental model for MDB induction (8).
Fig. 1.

NMPP treatment results in concurrent PPIX-accumulation, NF-κB activation and IκBα-loss. A, A scheme for NMPP-mediated inhibition of ferrochelatase with consequent accumulation of the heme precursor PPIX. The PPIX content of lysates from mouse hepatocytes treated with two different commercial lots of NMPP for 24 h was quantified fluorometrically (Mean ± S.D., n = 3). B, IB analyses of p62 and IκBα with histone H3 as the loading control in lysates from mouse hepatocytes treated as indicated. NMPP (30 μM), hemin (10 μM added fresh every 4 h), and ALIs, autophagy/lysosomal inhibitors 3MA (5 mM) + NH4Cl (30 mM), and MG132 (20 μM) for 1 h before NMPP-addition. C, Mouse hepatocytes were treated as indicated. Representative EMSA of nuclear extracts (NER, 5 μg) with NF-κB-specific oligonucleotides is shown. Bar chart represents mean ± S.D. of NF-κB activation relative to controls, n = 3. D, Quantification of IκBα levels upon IB analyses of lysates from WT (WT) and HRI−/− mouse hepatocytes treated with NMPP (30 μM) for the indicated times (Mean ± S.D., n = 3). E, IB analyses of p62 and IκBα in lysates from mouse hepatocytes treated with PPIX (10 μM), ZnPP (10 μM) or NMPP (30 μM) with or without hemin (10 μM) for 8 h. NS, nonspecific band. F, EMSA of nuclear extracts (NER) from mouse hepatocytes treated as indicated for 24 h. G. IL-6 RT-PCR analyses of mRNA from mouse hepatocytes treated with ZnPP (10 μM) for the indicated times (Mean ± S.D., n = 3).
ASH and NASH patient liver histology reveals that MDBs are associated with inflammatory responses, with MDB-containing hepatocytes often surrounded by neutrophils (1, 9). All MDB-inducing mouse models also accumulate hepatic PPIX and exhibit liver inflammation and injury at an early stage when MDBs are not yet detectable (10, 11). Cumulative biochemical evidence indicates that in these mouse models as well as in DDC-, NMPP- or PPIX-treated cell models, certain proteins such as p62, cytokeratins CK8/18 and lamin begin to aggregate at a much earlier stage before MDBs are detected (12, 13). Similarly, many neurodegenerative diseases are also associated with inflammation at an early stage, and amyloid protein aggregation has been shown to initiate an inflammatory response (14). Moreover, induction of various protein aggregates, as in myofibrillar myopathy and Amyotrophic Lateral Sclerosis (ALS), triggers the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) via a noncanonical pathway independent of IκBα-phosphorylation/degradation (15). However, the precise molecular link between protein aggregation and NF-κB-activation and inflammatory response remains unclear.
Of the existing NF-κB/Rel transcription factors (16), p65/p50 is not only the most ubiquitous and biologically active NF-κB-heterodimer, but also the major hepatic species (17). p65/p50 is normally sequestered in the cytoplasm by NF-κB inhibitors (IκBs). Of these, in human hepatocytes, HepG2 cells and cultured mouse hepatocytes, IκBα and IκBβ are the major forms, each regulated by different signals (18–21). IκB-binding masks the NF-κB nuclear localization signal (NLS) and DNA-binding domain (16). Signal-induced IκB-unleashing of cytoplasmic NF-κB most commonly via phosphorylation and subsequent ubiquitin-dependent proteasomal degradation (UPD) of both IκBα and IκBβ (22–24), results in the nuclear translocation of DNA-binding competent NF-κB, with consequent transcriptional activation of its target genes including that of IκBα (25), but not of IκBβ (26). IκBα is then rapidly de novo synthesized, and following nuclear import, binds the DNA-bound NF-κB, masking its NLS and accelerating its nuclear export. This feedback process is required for the down-regulation and eventual termination of NF-κB-mediated transcriptional activation (27). Thus, while both hepatic IκBs are largely involved in NF-κB-cytoplasmic retention, only IκBα is involved in nuclear NF-κB-transcriptional suppression (19).
Our findings herein reveal a loss of IκBα and IκBβ with subsequent NF-κB activation upon treatment of primary hepatocytes with NMPP (an established MDB inducer), or ZnPPIX (ZnPP). We probed the role of four plausible mechanisms for NF-κB-activation via reduction of intracellular IκBα-levels in this NMPP-elicited IκBα-loss and excluded them all (supplemental Results). Instead, we found that the apparent loss of IκBα upon NMPP- or ZnPP-treatment is actually due to its phase sequestration into insoluble cellular aggregates which abolished its stable interaction with NF-κB-subunit p65. This, in addition to its ZnPP-elicited concurrent aggregation with the nucleoporins Nup153 and Nup358/RanBP2 precluded all IκBα nuclear import and subsequent termination of NF-κB activation.
To our knowledge, this is the first evidence that NF-κB is also activated through IκB-sequestration into insoluble aggregates. We believe such a novel mechanism accounts for the persistent hepatic NF-κB activation that may directly contribute to the severe inflammatory responses and liver injury observed not only in various MDB-featuring liver diseases and experimental MDB-models but also in acute erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLPP) (28–30).
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
C57BL/6 WT male mice (8-12-week old) purchased from the Jackson Laboratory (Bar Harbor, ME) were used for primary hepatocyte preparation. Hepatocytes were isolated by in situ collagenase perfusion and purified by Percoll-gradient centrifugation by the UCSF Liver Center Cell Biology Core, as described previously (31, 32). Fresh primary mouse hepatocytes were cultured on Type I collagen-coated 60 mm Permanox plates (Thermo Scientific, Grand Island, NY) in William's E Medium (Invitrogen, Grand Island, NY) supplemented with 2 mM l-glutamine, insulin-transferrin-selenium (Invitrogen), 0.1% bovine albumin Fraction V (Invitrogen), Penicillin-Streptomycin (Invitrogen) and 0.1 μM dexamethasone. Cells were allowed to attach for 4 to 6 h and then overlaid with Matrigel (Corning, Oneonta, NY). From the 2nd day after plating, the medium was replaced daily, and cells were further cultured for 4–5 days with daily light microscopic examination for any signs of cell death and/or cytotoxicity. On day 5, some cells were treated with 30 μM NMPP (Frontier Scientific, Logan, UT; dissolved in DMSO), 10 μM PPIX (Sigma-Aldrich, St. Louis, MO; dissolved in DMSO with sonication) or 10 μM zinc(II)-PPIX (ZnPP, Sigma-Aldrich, St. Louis, MO; dissolved in DMSO and complexed with BSA (BSA) at a molar ratio of 4:1 to keep it solubilized in the medium) for various times as indicated (Results). In some cases, hepatocytes were pretreated with inhibitors of various cellular processes as indicated (Results) for 1 h before treatment with NMPP, PPIX or ZnPP.
HepG2 and HeLa cells were cultured in minimal Eagle's medium (MEM) containing 10% v/v fetal bovine serum (FBS) and supplemented with nonessential amino acids and 1 mM sodium pyruvate. HEK293T and MEF cells were cultured with Dulbecco's Modified Eagle high glucose medium (DMEM) containing 10% v/v FBS. For transfection experiments, cells were seeded on 6-well plates, when cells were 60% confluent, each cell well was transfected with 3 μg plasmid DNA complexed with TurboFect transfection reagent (ThermoFisher, Grand Island, NY) for HEK293T cells and X-tremeGENE HP transfection reagent (Roche, Indianapolis, IN) for HepG2 cells according to the manufacturers' instructions. At 40-72 h after transfection, cells were either treated as indicated or directly harvested for assays.
Knockout Mice and Cell Lines
HRI-knockout mice: Were generated as described (33). p62 knockout mice: Originally generated as detailed (34) and provided by Dr. T. Yanagawa at the University of Tsukuba, through Prof. Do-Hyung Kim, University of Minnesota. p62 MEF cells: Were generated from p62 KO mice by Prof. Masaaki Komatsu's laboratory (35), Niigata University, Japan, and provided by Prof. Haining Zhu, University of Kentucky.
ATG5 MEF cells: Were generated by Prof. Noboru Mizushima's laboratory (36), University of Tokyo, and provided by Prof. Randy Schekman's laboratory (UC Berkeley).
CAPN4 MEF cells: Were generated as described (37).
PentaKO (p62/NBR1/OPTN/NDP52/TAX1BP1) HeLa cells: Were generated and provided by Dr. Richard J. Youle's laboratory NIH (38).
Hepatic PPIX Content
PPIX content was determined using the intrinsic PPIX fluorescence as described (39). Briefly, 50 μl of cell lysates were first extracted with 400 μl of an ethyl acetate and acetic acid mixture (3:1, EtOAc-HAc) and then re-extracted with another 400 μl of EtOAc-HAc. The extracts were pooled and re-extracted with 400 μl of 3 M HCl. After centrifugation, the aqueous phase was recovered for fluorescent determination in a SpectrumMax M5 plate reader at an excitation of 405 nm. The intensity at an emission of 610 nm was quantified relative to a standard curve prepared with known concentrations of pure PPIX.
Plasmids
pCMV-3HA-IκBα and pCMV-3HA-IκBα-S32A/S36A were gifts from Dr. Warner Greene (plasmids #21985, #24143; Addgene, Cambridge, MA). C1-Emerald was a gift from Dr. Michael Davidson (Addgene plasmid #54734). pcDNA6 and pcDNA3 vectors were from Invitrogen (Grand Island, NY). pcDNA6-HA-rHRI, C1-Emerald-IκBα, pcDNA6-p62-myc and pcDNA3-NBR1 were constructed by us. The primers, templates, vectors and restriction enzymes (RE) used are summarized below (Table I).
Table I.
| Plasmid | Template | PCR Primers (Restriction Sites Underlined) | Vector | RE |
|---|---|---|---|---|
| C1-Emerald- IκBα | pCMV-3HA- IκBα | GCTCAAGCTTCGttccaggcggccgagcg | C1-Emerald | HindIII |
| ggaggccagcgtctgacgttatgaCCCGGGATCC | Smal | |||
| pcDNA6-p62-Myc | cDNA clone (IMAGE:4298142) | AAGCTTatggcgtcgctcaccg | pcDNA6.1Myc/His6 | HindIII |
| gcatcccccgccgttgCTCGAG | XhoI | |||
| pcDNA3-NBR1 | cDNA clone (IMAGE: 2989212) | TAGACTCGAGGCCACCatggaaccacaggttactc | pcDNA3.1 | XhoI |
| ctggtacagccaacgctattgaCTCGAGTCTA | KpnI |
Electrophoretic Mobility Shift Assay (EMSA)
Nuclear fractions of cells were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher, Grand Island, NY). EMSA were performed using LightShift Chemiluminescent EMSA Kit (ThermoFisher). Briefly, 2–5 μg of nuclear extracts were incubated with 1 μl of biotin-labeled NF-κB DNA probes (Panomics, Cleveland, OH) in binding buffer [10 mM Tris, pH 7.5, 50 mM KCl, 1 mM DTT, 5 mM MgCl2, 0.2 mM EDTA, 5% glycerol, 1 μg poly dI-dC]. The final volume of the mixture was adjusted to 20 μl and incubated at room temperature for 30 min, and then mixed with 5 μl loading buffer for loading onto 5% TBE Polyacrylamide Gel (Bio-Rad, Hercules, CA). The gel was run at 120 V until the dye reached the gel bottom, and then transferred to Hybond-N+ positively charged nylon membrane (GE Life Sciences, Marlborough, MA). The NF-κB complex shifted probes were detected by blotting with HRP-coupled streptavidin. For super-shift EMSA, the binding mixture (in the absence of the biotin-labeled probes) was first incubated with p65 antibody for 20 min on ice, and then the biotin-labeled probes were added and further incubated at room temperature for another 30 min before gel loading.
RNA Isolation and Semi-Quantitative RT-PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacturers' instructions. Total RNA (2 μg) was used to perform reverse transcription using SuperScript VILO Master Mix (Invitrogen, Grand Island, NY) in a 20 μl-reaction. Reverse transcribed first strand cDNA (1 μl) was used in PCRs. GAPDH was used as the internal control. PCR primers were as follows: IL6-RT forward: ACAACCACGGCCTTCCCTACTT; IL6-RT reverse: CACGATTTCCCAGAGAACATGT; GAPDH-RT forward: ACCACAGTCCATGCCATCAC; GAPDH-RT reverse: CACCACCCTGTTGCTGTAGCC.
Western Immunoblotting (IB) Analyses
For Western IB analyses, whole-cell extracts were prepared with Cell Lysis buffer (Cell Signaling Technology, Danvers, MA) containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin and supplemented with 10% glycerol and protease/phosphatase inhibitor mixture (Pierce, Grand Island, NY). Cell lysates were sonicated for 10 s and then cleared by centrifugation at 4 °C in a tabletop centrifuge at 14,000 × g for 10 min. Protein concentrations were determined by BCA assay and equal amounts of proteins were separated on 4–15% Tris-Glycine eXtended (TGX) polyacrylamide gels. Proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA) for IB analyses.
The following primary antibodies were employed: c-Myc (9E10, Santa Cruz Technology, Dallas, TX), HA (C29F4, Cell Signaling Technology), IκBα (N terminus, E130, Abcam, Cambridge, MA), IκBα (C terminus, 44D4, Cell Signaling Technology), p62 (2C11, Abnova, Taipei City, Taiwan), phospho-eIF2α (Ser52) (polyclonal, Invitrogen), eIF2α (Invitrogen), IκBα (Mouse mAb for IP, 112B2, Cell Signaling Technology), NF-κB p65 (D14E12, Cell Signaling Technology), LC3B (Novus, Littleton, CO), β-Actin (Sigma, St. Louis, MO), Histone H3 (Abcam), Nup153 (Bethyl, Montgomery, TX), Nup153 (QE5, BioLegend, San Diego, CA). The following secondary antibodies were also used: Rabbit anti-mouse IgG H&L (HRP) (Abcam) and Goat anti-rabbit IgG H&L (HRP) (Abcam). The immunoblots were developed using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Grand Island, NY).
Co-Immunoprecipitation (Co-IP) Analyses
Whole-cell extracts were prepared as described above. Cell lysates (1 mg) were then incubated with indicated antibodies (2 µg) or control IgGs at 4 °C overnight. Antibody-antigen complexes were then captured by protein G Dynabeads (Invitrogen, Grand Island, NY) at room temperature for 1 h, and then eluted by heating at 95 °C for 10 min in 2 × SDS-loading buffer. Eluates were subjected to IB analyses as described above.
Sequential Solvent Extraction of Cell Lysates
Cells were harvested in cell lysis buffer (≈ 300 µl) as described above and the cell lysates were cleared by centrifugation at 14,000 × g. The pellet was then solubilized in RIPA buffer (Cell Signaling Technology, Danvers, MA) supplemented with 0.1% SDS, 10% glycerol and protease/phosphatase inhibitor mixture with sonication followed by centrifugation at 14,000 × g for 10 min. The resulting pellet was then solubilized in half the volume (≈150 µl) of cell lysis buffer with sonication in urea/CHAPS buffer containing 8 M urea, 2 M thiourea, 4% CHAPS, 20 mM Tris-base, and 30 mM DTT and supplemented with protease/phosphatase inhibitor mixture. High salt buffer (HSB)-extraction (Fig. 3C) was carried out as described previously (40). Briefly, cells were first harvested in cell lysis buffer containing 1% Triton as described above, the resulting pellet was then suspended in a cell lysis buffer supplemented with 1.5 M KCl (HSB) with sonication. Upon sedimentation, the resulting pellet was solubilized in Laemmli buffer containing 4% SDS or in urea/CHAPS buffer as described above.
Fig. 3.

ZnPP-elicited IκBα-loss is due to its sequestration into insoluble cellular aggregates. A, HEK293T cells were transfected with pCMV4-3HA-IκBα for 40 h, and then treated with ZnPP (10 μM) for 4 h. Cells were then subjected to sequential extraction. Extracts (10 μg) were used for IB analyses. B, The same extracts from (A) were immunoblotted using three different antibodies each targeting a different region of the same HA-IκBα protein: Anti-HA-tag, rabbit mAb targeting IκBα N terminus (N), and rabbit mAb targeting IκBα-C terminus (C). C, HEK293T cells were transfected and treated as in A. and then subjected to sequential extraction with the following buffers: Cell Signaling lysis buffer (Triton), Cell Signaling lysis buffer supplemented with 1.5 M KCl (HSB). The pellet obtained after HSB-extract was either solubilized with heating in a Laemmli buffer with 4% SDS (SDS) or in urea buffer. Extracts (10 μg) were used for IB analyses.
Confocal Immunofluorescence Microscopy (CIFM)
Cells were grown on collagen-coated glass coverslips and treated as indicated. Cells were fixed with 4% formaldehyde for 20 min at room temperature followed by methanol at −20 °C for 1 min. Cells were then rinsed with PBS and blocked for 1 h with 10% normal goat serum in PBS/0.1% Tween at room temperature, and then stained with indicated primary antibodies at 4 °C overnight. Cells were then washed in PBS/0.1% Tween three times and then stained with secondary antibodies for 1 h at room temperature. Cells were further washed three times in PBS/0.1% Tween and then mounted using ProLong Diamond Antifade Mountant with DAPI nuclear stain (Molecular Probes, Grand Island, NY). The following primary antibodies were used: IκBα (rabbit monoclonal, E130, Abcam), IκBα (mouse monoclonal, 44D4, Cell Signaling Technology), p62 (2C11, mouse monoclonal, Abnova, Taipei City, Taiwan), p65 (D14E12, rabbit monoclonal, Cell Signaling Technology). The following secondary antibodies were applied: Goat anti-rabbit IgG Alexa Fluor 488 (Invitrogen), anti-mouse IgG Alexa Fluor 647 (Cell Signaling Technology). These fluor dyes were selected to circumvent any specific interference from intrinsic ZnPP-fluorescence. Images were taken with a Nikon Yokogawa CSU-22 Spinning Disc Confocal Microscope using a Plan Apo VC 100 ×/1.4 oil lens or on a Nikon high-speed wide-field Andor Borealis CSU-W1 spinning disc confocal microscope using a Plan Apo VC 60 ×/1.4 oil lens. Images were processed using ImageJ software. For quantification, at least 600 cells at each condition were evaluated. P-values were calculated using the two-sided unpaired Student's t test.
Immunoaffinity Purification (IAP)
We employed high-affinity alpaca Nanobody cross-linked beads (GFP-Trap, ChromoTek, Germany; #gta-20) for IAP, which not only enabled a high-level enrichment of target proteins but also eliminated IgG contaminants that confound downstream LC–MS/MS analyses. N-terminally mEmerald (GFP)-tagged IκBα (GFP-IκBα) was transiently transfected into HEK293T cells, with mEmerald-transfected cells as background control (C1-GFP). Two wells of HEK293T cells grown on 6-well plates were pooled and lysed in 1 ml lysis buffer supplemented with 10% glycerol, protease/phosphatase inhibitor mixture and 20 mM N-ethylmaleimide (NEM). Centrifugation-cleared cell lysates were incubated with 50 μl GFP-trap agarose beads at 4 °C overnight. Subsequently, GFP-trap beads were collected by centrifugation at 3000 × g for 30 s and then washed 5 times with cell lysis buffer. Co-immunoprecipitated proteins were eluted by incubating beads with 2 × Laemmli buffer at 70 °C for 15 min. Eluates were then subjected to SDS-PAGE and stained with Coomassie Blue to visualize the bands for subsequent in-gel digestion. Three biological replicates were performed.
Biotinylation by Antibody-Recognition (BAR)
Traditional approaches for in vivo protein-protein interactions such as co-IP are based on affinity capture of stable protein complexes that will be disrupted under harsh denaturing conditions. By contrast, biotinylation proximity labeling circumvents this limitation by introducing an enzyme to the target protein that can generate distance-constrained reactive biotin molecules to covalently link neighboring proteins, providing a permanent tag that survives purification under harsh conditions for downstream identification (41). We chose BAR/APEX over BioID (the two popular methods for in vivo biotinylation) because the BAR approach (42) enables quicker capture of interacting proteins (as the peroxidase-mediated biotinylation takes only a few minutes) than the more protracted BioID methodology (requiring 18–24 h tagging time), which could overlook short-term/transient interactors. BAR analyses were performed according to Bar et al. (42). Briefly, two wells of HEK293T cells grown on collagen-coated 6-well plates were first treated as indicated and then fixed with 4% formaldehyde for 10 min at room temperature and permeabilized for 7 min with 0.5% Triton-X in PBS. After rinsing with PBS, cells were incubated with 0.5% H2O2 for 10 min. After further rinsing with PBS, cells were blocked for 1 h with 10% normal goat serum in PBS/0.1% Tween, and then stained with IκBα-antibody (mouse monoclonal, L35A5) at a 1:200 v:v dilution in 1% normal goat serum in PBS/0.1% Tween at 4 °C overnight. Negative control staining with no antibody or staining with mouse IgG1 isotype controls (adjusted to the same final concentration as that of IκBα-antibody) was also performed. Cells were then washed in PBS/0.1% Tween for over 1 h with at least 5 buffer changes and then stained with secondary poly-HRP-conjugated goat anti-mouse IgGs (Biotin XX Tyramide SuperBoost™ Kit; Invitrogen, Grand Island, NY, B40911) at a 1:1000 v:v dilution in 1% normal goat serum in PBS/0.1% Tween for 1 h. Cells were further washed for over 2 h in PBS/0.1% Tween with at least 5 buffer changes. After that, cells were labeled with biotin using Biotin XX Tyramide SuperBoost™ Kit following product instructions. Briefly, cells were pre-incubated with 300 μl of reaction buffer containing biotin-XX-tyramide (Biotin-Phenol) for 15 min; H2O2 was then added to a final concentration of 0.5 mm and incubated for another 5 min. The labeling reaction was then stopped by quickly exchanging the reaction solution with 1 ml of 500 mM sodium ascorbate. Cells were washed 3 times with PBS and then lysed in RIPA buffer supplemented with 2% SDS and boiled for 1 h to reverse formaldehyde-cross-linking. The cell lysates were cleared by centrifugation at 14,000 × g for 10 min, the supernatants were used as whole cell lysates (WCL). The resulting pellet was further solubilized in urea/CHAPS buffer as described above. Aliquots (10 µg) of WCL (∼ 0.4%) and pellet fraction (∼7.5%) were used for streptavidin-HRP blot (Invitrogen, #S911), to characterize the specificity and efficiency of BAR. The remaining WCL was diluted 1:2 (v:v) in RIPA buffer for streptavidin (SA) pulldown. SA pull-downs were performed as detailed (43). Briefly, SA-magnetic beads (50 μl) were added to the lysates and the mixture incubated at 4 °C overnight on a rotator. Beads were then washed twice with RIPA buffer, once with 1 M KCl, once with 2 M urea in 20 mM Tris base, and lastly, twice with RIPA buffer. Biotinylated proteins were then eluted by incubating beads in 40 μl of 2 × Laemmli buffer supplemented with 2 mM biotin at 95 °C for 5 min. Eluates were then subjected to SDS-PAGE and immunoblotting (IB) analyses with p65 antibody. For mass spectrometric analyses after SA pulldown, 2 additional washes were performed using 100 mM ammonium bicarbonate (ABC) before proceeding with on-bead digestion.
Mass Spectrometric (MS) Analyses
For in-gel digestion, each gel lane was sliced into 8–10 sections and processed separately using a standard in-gel digestion procedure (44, 45). Briefly, each gel piece was reduced and alkylated, and then digested overnight with 300 ng of Trypsin/Lys-C Mix (Promega, Madison, WI). For on-bead digestion after SA pulldown, beads were first resuspended in 6 M urea in 100 mM ammonium bicarbonate (ABC), and reduced by adding 10 mM DTT (final) and incubation at 37 °C for 30 min. The samples were then alkylated by adding 15 mM iodoacetamide (final) with incubation at room temperature in the dark for 30 min. Trypsin/Lys-C Mix (100 ng) was then added and the mixture incubated for 4 h at 37 °C. The mixture was then diluted 1:6 (v:v) with 100 mM ABC to reduce the urea concentration to 1 M and then further incubated at 37 °C overnight. The resulting peptide mixture was desalted using C18-Zip tips (EMD Millipore, Billerica, MA), speed-vacuumed to dryness and suspended in 0.1% formic acid and analyzed in data-dependent acquisition mode on either an LTQ-Orbitrap Velos (for the IAP) or a Q-Exactive Plus (for the aggregation and the BAR experiments) mass spectrometer (Thermo Fisher, San Jose, CA) coupled to a nano-Acquity UPLC (Waters, Milford, MA) with an EASY-Spray column (75 μm × 15 cm column packed with 3 μm, 100 Å PepMap C18 resin; Thermo Scientific). Separation of peptides derived from gel band analyses used a 60-min linear gradient from 3–28% B at 300 nl/min (Solvent A: water + 0.1% formic acid; Solvent B: acetonitrile + 0.1% formic acid). Separations from on-bead digestions used a 103-min gradient from 2–23% B followed by a second 9-min gradient from 23–40% B at 400 nl/min. Precursor ion spectra were recorded in the Orbitrap analyzer (QExactive: resolving power = 70,000, range = 350–1500 m/z, AGC target = 3e6, max injection time = 100 ms; Velos: resolving power = 30,000, range = 300–1800 m/z, AGC target = 2e6, max injection time = 250 ms; both instruments used the ambient polydimethylcyclosiloxane ion at m/z 445.120025 as a lock mass). The n most intense ion signals per precursor scan (QExactive: n = 10; Velos: n = 6) were isolated with 4 m/z bandwidth, dissociated by HCD (QExactive: NCE = 25; Velos: NCE = 30) and measured in the Orbitrap analyzer (QExactive: resolving power = 17,500, AGC target = 5e4, max injection time = 120 ms; Velos: resolving power = 7500, AGC target = 9e4, max injection time = 500 ms). Singly charged and unassigned precursor ions were excluded from analysis and additional selection criteria were applied (QExactive: intensity threshold = 17,000 counts, dynamic exclusion = 15 s, “peptide match preferred” and “exclude isotopes” were enabled; Velos: intensity threshold = 3000 counts, dynamic exclusion = 30 s, exclusion width from −0.5 m/z to +1.5 m/z). Peak lists were extracted using PAVA, an in-house software developed by UCSF Mass Spectrometry Facility. Peak lists from 8–10 MS fractions corresponding to gel bands from a given experimental condition were pooled to search against the SwissProt human database (SwissProt.2015.12.1; 20194 entries searched) using ProteinProspector (version 5.19.1; http://prospector.ucsf.edu/prospector/mshome.htm) (46). A fully randomized database (additional 20194 protein sequences) was used to estimate false discovery rates (FDR) (47). ProteinProspector search parameters were as follows: Tolerance for precursor and product ions were 20 ppm and 25 ppm, respectively; a maximum of 1 missed cleavage of trypsin was allowed; carbamidomethylation of cysteine was set as a fixed modification; variable modifications were set for: N-terminal Met loss and/or acetylation, Met oxidation, peptide N-terminal Gln to pyroGly conversion; the number of variable modifications was 2. Reporting thresholds were as follows: Minimum score of protein: 22.0; minimum score of peptide: 15.0; maximum E value of protein: 0.01; maximum E value of peptide: 0.05. These score thresholds resulted in a <1% FDR at the peptide level.
Experimental Design and Statistical Rationale
The goal of the proteomic experiments was to identify proteins that interact with IκBα under basal conditions as well as continue to interact with IκBα and/or aggregate upon ZnPP-treatment. We first used the SAINTexpress software package (48) to independently assess the specificity with which a protein was identified in one of four experimental workflows: (i) AP-MS purifications of GFP-tagged IκBα targets versus GFP controls to assess interactions under baseline soluble conditions, 3 biological replicates were performed (n = 3) (supplemental Tables S1–S2); (ii) Proximity labeling BAR experiments to document proximity to IκBα without or upon ZnPP-treatment. Three biological replicates were performed (n = 3), each biological replicate contained 2 negative controls, (IgG-staining, or IκBα-staining in cells with IκBα siRNA-knockdown/-ZnPP, n = 6), (supplemental Tables S3–S6). BAR analyses following ZnPP-treatment were employed in the main analyses (Figs. 5C, 5D) and BAR analyses without ZnPP-treatment were employed in the supplemental analyses (supplemental Fig. S6E). (iii) MS analyses of protein aggregates induced by 2 h ZnPP-treatment in HEK293 cells, with +ZnPP as the target condition and -ZnPP as the control, 3 biological replicates (n = 3) were performed (supplemental Tables S7–S8); and (iv) MS analyses of protein aggregates in HepG2 cells [as in #3; 3 biological replicates (n = 3)], were also performed (supplemental Tables S9–S10). The Protein Prospector search results were reformatted and analyzed by SAINTexpress on the CRAPome website (www.crapome.org) (49). The SAINT output from the 4 separate analyses was first filtered to a BFDR (Bayesian False Discovery Rate) score of 0.05 and then merged into a single data frame. Bait versus Prey hierarchical clustering was performed using the ProHitsVis Web Tool (http://prohits-viz.lunenfeld.ca) (50) using AvgSpec (average spectral counts) as the abundance measure and BFDR as the score measure. The clustering was repeated using the full list of all proteins matching the BFDR selection criteria (supplemental Table S11, supplemental Fig. S8) as well as employing only the proteins identified under all 4 experimental conditions (10 genes) (Fig. 5D). Venn diagrams illustrating the overlap between the experimental conditions were created using a VENN DIAGRAM on-line tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Fig. 5.

IAP-MS of IκBα-interactome, BAR-analyses of IκBα-interactome after ZnPP-induction, and overlap analyses with ZnPP-induced aggregate proteome. A, Flow-chart of the strategy used to identify a high-confidence IκBα-interactome. Cells expressing GFP (mEmerald) alone were used as the background control. B, Flow-chart of the strategy used to identify potential IκBα interactors upon ZnPP-induction using biotinylation via antibody recognition (BAR) analyses under denaturing conditions. C, Venn diagram of proteins specifically found employing 4 different experimental approaches. Well-known IκBα stable interactors are listed in the box. D, Clustering analyses of the 10 proteins found in all 4 approaches using average spectral counts as the abundance measure and Bayesian false discovery rate (BFDR) as the score measure. Proteins previously found in aggregates from other disease models and/or human pathological samples are indicated in red. Proteins previously found in protein aggregates of pathological samples from human MS and/or Lewy bodies of Parkinson's disease (76, 78). Proteins previously found in aggregates or mislocalized in ALS/FTLD patient samples (77).
Network Analyses of Aggregated Proteins
225 commonly found proteins in ZnPP-induced aggregates from both cell lines were searched against a STRING database version 11.0 (51) for protein-protein interactions. We employed the highest confidence score (≥ 0.9) to obtain a protein-interaction network. The network was exported to Cytoscape software (52) for pathway annotation and visualization.
siRNA-Knockdown Analyses
HEK293T cells were transfected using DharmaFECT 4 Transfection Reagent, with 10 nM siRNA specific for NUP153 or RanBP2. Treatments were performed 48 h after transfection. HEK293T cells or HepG2 cells were transfected using DharmaFECT 4 Transfection Reagent with 50 nM siGENOME Human NFKBIA (IκBα) siRNA-Smartpool or control nontargeting siRNA. Treatments were carried out at 48 h after transfection. Primary mouse hepatocytes were transfected using DharmaFECT 4 Transfection Reagent with 50-100 nM siGENOME Mouse Nfkbia siRNA-SMARTpool or control siRNA nontargeting siRNA, 16 h before overlay with Matrigel. Cells were harvested 72–96 h after transfection.
Bioinformatic Analyses
GRAVY scores estimating average hydrophobicity were calculated using Kyte and Doolittle hydropathic analyses (http://web.expasy.org/protparam/) (53). Positive GRAVY scores suggested greater hydrophobicity. Proteins with long intrinsically disordered regions were predicted using the SLIDER method (http://biomine.cs.vcu.edu/servers/SLIDER/) (54). This computational method is based on the physicochemical properties of amino acids, sequence complexity, and amino acid composition. A SLIDER score of >0.538 indicates a high likelihood that the protein contains a long (>30 residues) disordered segment. HEK293 proteome was defined as described (55). Briefly, we used the HEK293 cell data from The Human Protein Atlas's RNA-seq data (The Human Protein Atlas version 19 and Ensemble version 92.38, https://www.proteinatlas.org/download/rna_celline.tsv.zip). Genes with a pTPM (protein-coding transcripts per million) value ≥ 1 were considered part of the HEK293 proteome. This list contained 10905 genes, matched to 10783 reviewed UniProtKB/Swiss-Prot proteins. 10783 protein sequences were downloaded to calculate the GRAVY and SLIDER scores.
RESULTS
NMPP-Elicited PPIX Accumulation with Concurrent IκBα-Loss and NF-κB Activation in Cultured Mouse Hepatocytes
NMPP-treatment of cultured mouse hepatocytes as expected from its ferrochelatase inhibition, resulted in cellular PPIX accumulation (Fig. 1A). In parallel, a gradual IκBα-loss was also observed in hepatic lysates upon NMPP-treatment (Fig. 1B). This IκBα-loss was reversed upon inclusion of hemin, but not that of the proteasomal inhibitor MG-132, or the dual autophagic-lysosomal degradation (ALD) inhibitors 3-methyladenine (3MA) + NH4Cl. These findings thus excluded both ubiquitin-dependent proteasomal degradation (UPD) and ALD involvement in this IκBα-loss. Further supportive evidence was provided by studies in HepG2 cells transfected with a S32A/S36A-IκBα mutant that is normally resistant to phosphorylation and consequent UPD (supplemental Fig. S1A) as NMPP caused a similar time-dependent loss of both WT IκBα and its mutant. Parallel EMSA of nuclear extracts from these NMPP-treated hepatocytes revealed a time-dependent hepatic NF-κB activation, indicating that this hepatic IκBα-loss was indeed physiologically relevant (Figs. 1C and supplemental Fig. S1B). This NF-κB activation was also attenuated by hemin (Fig. 1C), consistent with the hemin-mediated inhibition of IκBα-loss (Fig. 1B). These findings suggested that this culture model was a valid model to interrogate the association of PPIX accumulation with NF-κB-activation and inflammation.
Additional mechanisms for the loss of intracellular IκBα-levels and consequent NF-κB-transcriptional activation also exist and were all probed. Thus, we excluded two such established mechanisms: (i) Translational suppression of IκBα upon activation of the heme-regulated inhibitor (HRI) eIF2α kinase through NMPP-elicited heme-deficiency, by documenting that HRI-genetic ablation failed to affect such NMPP-elicited IκBα-loss in mouse hepatocytes [(56); Figs. 1D and supplemental Fig. S2A]; and (ii) enhanced IκBα autophagy by documenting that such an IκBα-loss in MEF cells was unaffected by ATG5-knockout (KO) [(57); supplemental Fig. S2B]. Additionally, we also excluded two plausible mechanisms: (iii) Enhanced calpain-mediated proteolysis of IκBα by documenting that such a loss was unaffected in MEF cells upon genetic calpain-KO [(58); supplemental Fig.S2C]; and (iv) PPIX-photoactivation and consequent ROS-mediated oxidative stress (28) in NMPP-elicited IκBα-loss through the use of specific ROS scavengers, antioxidants and/or inhibitors (Supplemental Results; supplemental Fig. S3).
PPIX and ZnPP Are Even More Potent Inducers of IκBα-Loss, NF-κB-Activation and p62 Oligomerization/Aggregation than NMPP
Because NMPP-elicited ferrochelatase inhibition results in PPIX-accumulation, we examined whether this accumulation (and not heme deficiency per se) was mainly responsible for the observed IκBα-loss and NF-κB activation. We observed a greater IκBα-loss after either PPIX- or ZnPP-treatment relative to that seen with NMPP alone (Fig. 1E). While the NMPP-elicited IκBα-loss was to a great extent prevented by hemin inclusion, the IκBα-loss after PPIX or ZnPP was not1 (Fig. 1E). EMSA analyses revealed a NF-κB activation that was proportional to the observed IκBα-loss, ranking in order as follows: ZnPP>PPIX>NMPP (Fig. 1F). Consistent with this NF-κB activation, RT-PCR analyses of mRNA from ZnPP-treated hepatocytes indicated marked up-regulation of the inflammatory cytokine IL-6 at 24 h (Fig. 1G). Furthermore, marked hepatic up-regulation of not only IL-6 mRNA, but also that of IL-1β, TNFα and osteopontin (SPP-1) was observed upon in vivo ZnPP-treatment of mice (supplemental Fig. S1C).
Because the autophagic receptor p62 is a major component of MDB and various other pathological aggregates (1), we monitored hepatic p62 response upon NMPP, PPIX and ZnPP ± hemin treatments (Figs. 1B, 1E). In parallel with IκBα-loss, p62 formed aggregates and possibly cross-linked species, with a concurrent decline in the monomeric species (∼ 62 kDa; Ctrl), evident at later (8 h) rather than earlier (2–4 h) time points. This apparent time-lag is most likely due to an initial counteractive compensatory p62 transcriptional induction in response to PPIX-elicited oxidative stress and Nrf2 activation (8). Notably, given its superior potency and low potential for phototoxicity, we employed ZnPP in most of our subsequent studies.
ZnPP-Elicited IκBα-Loss is Independent of p62 or Other Autophagic Receptors/Adapters
The capacity of p62 to stabilize IκBα upon co-expression in HEK293T cells (Fig. 2A) and the close temporal relationship between IκBα-loss and p62 aggregation (Fig. 1B) suggested these proteins were intimately associated. We thus examined their interdependence in ZnPP-treated p62 WT and p62 null mouse (p62KO) hepatocytes or MEF cells (Figs. 2B and supplemental Fig. S4A), and found that this IκBα-loss was independent of p62 aggregation. The additional possibility that in p62-lacking cells, IκBα-loss was due to its degradation via UPD, ALD or calpain pathways was excluded with specific inhibitors of these pathways (supplemental Fig. S4B). We also examined the plausible compensation by NBR1 (Neighbor of Braca 1 gene), a redundant autophagic receptor that can effectively substitute for p62 in its absence (59) (supplemental Figs. S4C, S4D). Furthermore, we also conclusively excluded other plausible redundant functional mimics of p62 in this IκBα-loss (Fig. 2C) by examining ZnPP-elicited IκBα-loss in HeLa cells with a combined KO of five autophagic receptors/adapters (p62, NBR1, TAX1BP1, NDP52, and OPTN) (38).
Fig. 2.

ZnPP-elicited IκBα-loss is p62-independent. A, HEK293T cells were transfected with pCMV4-3HA-IκBα, or co-transfected with either pcDNA6-p62-Myc or pcDNA6-Myc empty vector for 48 h. Cell lysates were used for IB analyses with actin as the loading control. B, Primary hepatocytes from WT (p62WT) and p62 knockout (p62KO) mice were treated with ZnPP (10 μM) for the indicated times. Cell lysates were used for IB analyses. C, Wild-type Hela cells (WT) and CRISPR-engineered Hela cells with five autophagic adapters genetically deleted (5KO) were treated with ZnPP (10 μM) for the indicated times. Lysates were used for IB analyses. D, CIFM analyses. p62WT and p62KO MEF cells were treated with ZnPP (10 μM) or vehicle control for 2 h, and then fixed and stained with anti-IκBα (green), anti-p62 (magenta) and DAPI (Blue). Insets depict enlarged regions of IκBα and p62 colocalization.
Cytoplasmic IκBα-Loss is Due to Its Physical Sequestration into Insoluble Cellular Aggregates
Confocal microscopic immunofluorescence (CMIF) analyses of ZnPP-treated WT and p62-/--MEF cells provided an informative clue (Fig. 2D). In WT-cells, both proteins were largely localized to the cytoplasm, with some colocalization (Fig. 2D, insets). Surprisingly, following ZnPP-treatment, despite the immunochemical IκBα-loss (Fig. 2B), IκBα-associated immunofluorescence signal persisted comparably in both WT and p62-/--MEF cells (Fig. 2D). This finding provided the first clue that upon ZnPP-treatment, IκBα was not irretrievably lost, but just undetectable in detergent-solubilized cell lysates routinely employed for immunoblotting (IB) analyses. It was thus likely that such an IκBα physical inaccessibility was due to its intracellular ZnPP-triggered protein aggregation. To examine this possibility, HEK293T cells co-transfected with both HA-IκBα and p62-Myc plasmids, were treated with ZnPP and then sequentially extracted using detergents of increasing strengths (Experimental Procedures). IB analyses showed that upon ZnPP-treatment, in parallel with p62-aggregation, HA-IκBα became undetectable in soluble Triton and RIPA fractions (Fig. 3A), however, the majority of HA-IκBα as well as monomeric p62 and p62-aggregates could be recovered from the RIPA-insoluble pellet by heat extraction with 8 M urea/CHAPS buffer. Furthermore, similar results were found upon IB analyses of these urea extracts irrespective of whether antibodies to the HA-tag, N- or C terminus of IκBα were employed (Fig. 3B). IB analyses of high salt buffer (HSB)-extracts from ZnPP-treated cell lysates also revealed detectable monomeric and aggregated IκBα and p62 species, albeit to a much lesser extent than corresponding urea extracts (Fig. 3C). Together, these findings indicated that upon ZnPP-treatment, p62 and IκBα along with other cellular proteins aggregate, and these aggregates are sequestered from the cytoplasm into an insoluble cellular fraction, essentially unavailable to function physiologically.
Proteomic Identification/Analyses of IκBα-Interactors and ZnPP-Aggregated Proteins
PPIX-treatment is causally associated with the highly selective aggregation of many cellular proteins (60). Thus, IκBα could either be the prime target of ZnPP-elicited physical sequestration or a “silent partner dragged along for the ride” by one or more of its cellular interactors. To identify any plausible mediators of ZnPP-elicited IκBα-sequestration into insoluble cellular aggregates, we employed a proteomic approach to comprehensively identify the proteins that both normally interact with IκBα, and either concurrently aggregate or form protein co-aggregates with IκBα upon ZnPP-treatment. That is, these aggregation-prone proteins upon IκBα-interaction could drive its sequestration and co-aggregation.
Previous studies in fech/fech mouse and protoporphyric zebra fish models have identified several (≈ 30) aggregated proteins in the high molecular mass (HMM)-regions upon SDS-PAGE of Nonidet P-40-soluble and/or SDS-soluble liver fractions (8, 13, 60, 61). Our discovery that ZnPP-elicited protein aggregates could be re-solubilized by heating in urea buffer, prompted comprehensive proteomic analyses of these urea-solubilized protein aggregates. For this purpose, HEK293T and HepG2 cells were first treated with ZnPP for 2 h (Fig. 4A). The entire urea-solubilized fraction as well as the HMM-bands from SDS-PAGE of Triton and RIPA soluble extracts were subjected to LC–MS/MS proteomic analyses (Figs. 4B, supplemental Fig. S5A). Overlap analyses revealed that 225 proteins commonly aggregated in both cell lines upon ZnPP-treatment (Fig. 4B, supplemental Tables S7–S10). Eight of them overlapped with aggregated proteins in fech/fech mouse model and 2 of them overlapped with the aggregated proteins in protoporphyric zebra fish models (supplemental Fig. S5B). Our analyses of the physicochemical properties of these 225 aggregated proteins revealed that they contain a strikingly higher fraction of >30-residue-long intrinsically disordered regions and exhibited lower hydrophobicity compared with the whole HEK293T cell proteome (Fig. 4C). To gain functional insight into these 225 ZnPP-aggregated proteins and their possible contribution to cell toxicity, we undertook a comprehensive protein network analyses using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) coupled with biological process and pathway overrepresentation analyses (Fig. 4D). ZnPP-elicited physical aggregation clustered these proteins under 4 critical biological processes. The proteins occupying a network hub/node position within their individual clusters, thus could disrupt multiple vital cellular interactions, leading to global hepatic functional collapse.
Fig. 4.

STRING network analyses of ZnPP-induced cellular aggregate proteome. A, Flow-chart of the strategy employed to identify ZnPP-induced aggregate proteome of both HepG2 and HEK293T cells. B, Venn diagram of proteins enriched in ZnPP-induced aggregates from HEK293T and HepG2 cells indicated 225 proteins as common hits. C, Box plots of the proteins with long intrinsic disordered regions (regions with >30 consecutive disordered residues) and relative protein hydrophobicity. Long intrinsic disordered regions were predicted by the SLIDER score. The higher the score the more likely is a protein to contain a long-disordered segment. Scores above 0.538 indicate that a given protein has a long-disordered segment. Hydrophobicity was measured by the GRAVY score (grand average of hydropathy), The hydropathy values range from −2 to +2 for most proteins, with the positively rated proteins being more hydrophobic. D, The complete interaction network of 225 common ZnPP-induced protein aggregates was obtained via STRING analyses with the highest confidence score (≥ 0.9). Network statistics are summarized in the inset. Functional enrichments are annotated with color-lined boxes with the false discovery rates (FDR) included.
To obtain a high-confidence IκBα-interactome under basal conditions, we performed rigorous immunoaffinity purification coupled with MS (IAP-MS) analyses, with extensive biological repetitions and strict filtering (Figs. 5A, supplemental Fig. S5C, supplemental Tables S1–S2). This approach identified 259 high-confidence IκBα-interactors, including well-established IκBα-interactors (Fig. 5C) [i.e. NF-κB subunits (RelA, NFKB1, NFKB2), and IKK-complex proteins (CHUK, IKBKAP)], thus validating our IAP-MS approach. However, most of the proteins identified were novel IκBα-interactors.
To identify specific proteins interacting with IκBα upon ZnPP-treatment, we employed a biotinylation proximity labeling approach that enabled the identification of IκBα-interactors even under denaturing conditions required to solubilize aggregated IκBα. After due consideration of various proximity labeling approaches, we selected HRP-biotinylation by antibody recognition (BAR) analyses (42) for identification of IκBα-interacting proteins in 2 h ZnPP-treated and untreated cells (Figs. 5B, supplemental Fig. S6, supplemental Tables S3–S6). After ZnPP-treatment, cells were first fixed and then immunostained with IκBα-antibody, followed by HRP-conjugated secondary antibody. Biotinylation was then initiated by adding biotin-phenol and H2O2 to immunostained cells. After which, cells were collected and boiled with 2% SDS for 1 h to reverse the cell fixation and release proteins for the streptavidin-bead enrichment of biotinylated proteins. Under these harsh denaturing conditions, both soluble and insoluble proteins were extracted, enabling us to assess IκBα-interacting proteins before and after ZnPP-treatment. Interestingly, immunoblotting of streptavidin-bead enriched proximity-labeled cell lysates revealed that the IκBα stable interactor NF-κB subunit p65 (RelA) was specifically labeled in untreated cells, but not in ZnPP-treated cells (supplemental Fig. S6B), suggesting that NF-κB p65 no longer binds to IκBα once the latter aggregates upon ZnPP-treatment. Furthermore, p65 (RelA) and other above mentioned stable, well-known IκBα-interactors were not detected among the IκBα-interactors identified upon ZnPP-treatment (Figs. 5C and S6E). Notably, they were also absent from the ZnPP-induced aggregate proteome of either cell type (Fig. 5C), indicating that novel transient interactors may instead participate in the ZnPP-elicited IκBα-aggregation.
We then performed hierarchical clustering analyses of the proteins derived from IκBα-IAP-MS, 2 h ZnPP-induced aggregate MS in two cell lines, and IκBα-BAR MS following 2 h ZnPP-treatment. We identified 10 proteins in common under all of these four experimental conditions (Figs. 5C, 5D). Interestingly, 2 of these proteins (Nup153, Nup358/SUMO E3-ligase RANBP2) are nuclear pore complex (NPC) nucleoporins, suggesting that IκBα upon direct interaction may either co-aggregate or concurrently aggregate with these nucleoporins during its nuclear-cytoplasmic transport.
Nucleoporin Nup153 is an IκBα-Interactant and ZnPP-Target
We therefore first verified the presence of Nup153 through IB analyses of Triton- and urea-solubilized cellular aggregates upon a 2 h-ZnPP-treatment of HEK293 and HepG2 cells. Indeed, whereas native Nup153 exhibited its intrinsic 153 kDa-mobility in untreated cells, upon ZnPP-treatment it was found as HMM-protein aggregates in Triton- and urea extracts, along with endogenous IκBα (Fig. 6A). However, to determine the specific role of either Nup153- or RanBP2, if any, in ZnPP-elicited IκBα-sequestration, we examined this IκBα-sequestration upon Nup153- or RanBP2-siRNA-knockdown and found that it was unaffected (Figs. 6B, 6C). This suggested that although they concurrently aggregate, Nup153 and RanBP2 are not inducers of IκBα-sequestration.
Fig. 6.
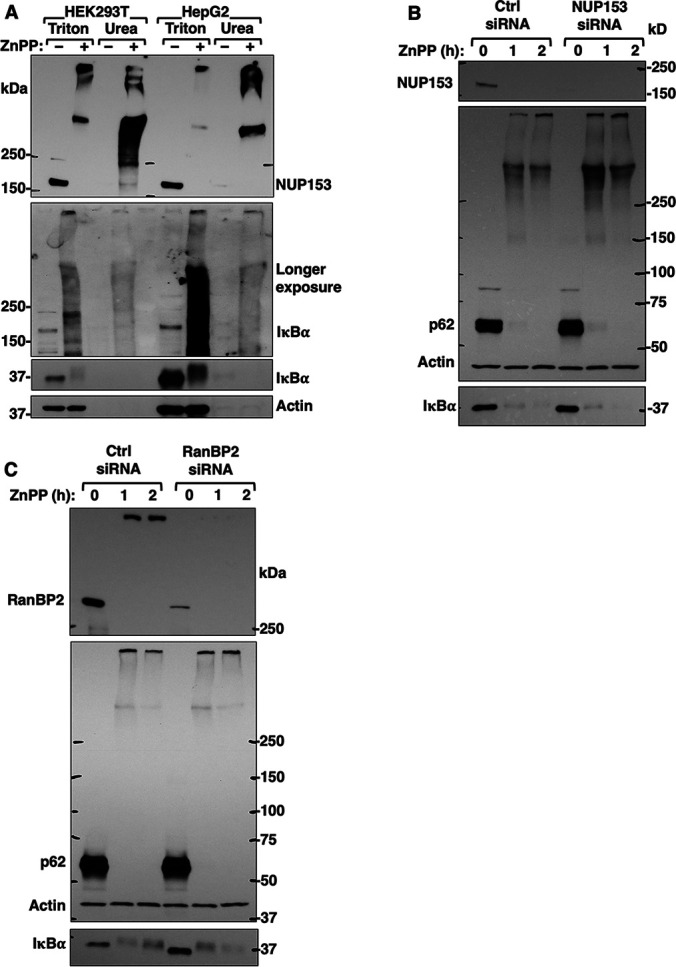
Nup153 is a major target of ZnPP-elicited aggregation but not an inducer of IκBα aggregation. A, HEK293T cells and HepG2 cells were treated with ZnPP (10 μM) for 2 h. Cells were sequentially extracted with Triton and urea buffers. Extracts (10 µg) were used for Nup153 and IκBα IB analyses, with actin as the loading control. B, HEK293T cells were transfected with control siRNA (Ctrl) or NUP153 siRNA for 48 h, and then treated with ZnPP (10 μM) for the indicated times. Cell lysates were used for IB analyses of Nup153, IκBα and co-IB analyses of p62 and actin (loading control). C, HEK293T cells were transfected with control siRNA (Ctrl) or RanBP2 siRNA for 48 h, and then treated with ZnPP (10 μM) for the indicated times. Cell lysates were used for IB analyses of RanBP2, IκBα and co-IB analyses of p62 and actin (loading control).
To determine the physiological significance of IκBα-NUP153 interaction and their concurrent aggregation, we monitored their interaction through co-immunoprecipitation (Co-IP) using HEK293T cells. This revealed that a significant fraction of endogenous Nup153 interacted with IκBα both in the cytoplasmic and nuclear extracts of GFP-IκBα-transfected cells, but not in mock- or C1-GFP-transfected cells (Fig. 7A). A similar interaction was also evident among the endogenous counterparts under basal conditions. However, this Nup153-IκBα-interaction was greatly enhanced upon TNFα-treatment at times (1 and 1.5 h) when rapid translocation of de novo synthesized IκBα across the nuclear pore would be expected to trigger its post-induction repression of NF-κB-activation (Fig. 7B). Additional insight into the critical importance of such IκBα-interaction during its nuclear import was provided by our Nup153 and RanBP2 siRNA-knockdown analyses that revealed the marked attenuation of this IκBα nuclear import upon TNFα-activation (Figs. 7C, 7D). By contrast, no differences were found in the newly synthesized cytoplasmic IκBα levels upon Nup153 or RanBP2 siRNA-knockdown. This cytoplasmic restoration of IκBα upon TNFα-activation suggested that the nuclear import of NF-κB as well as the nuclear export of IκBα mRNA were unaffected. Thus, siRNA-knockdown of either Nup153 or RanBP2, albeit impairing IκBα nuclear import, apparently does not affect nuclear pore integrity. Furthermore, ZnPP-treatment of TNFα-pretreated cells led to the accelerated IκBα-sequestration, whose timing revealed that ZnPP may preferentially target de novo synthesized IκBα as it increasingly interacts with cytoplasmic Nup153 during its nuclear import (Figs. 7B and 8A). Consistent with this, our CMIF analyses of HepG2 cells revealed that under basal conditions, both NF-κB (p65-Rel A subunit) and IκBα are localized in the cytoplasm (Fig. 8B). But upon TNFα-treatment, IκBα-UPD results in NF-κB nuclear translocation within 0.5 h. Subsequently, upon transcriptional activation of the NF-κB-responsive IκBα-gene, newly synthesized IκBα enters the nucleus resulting in NF-κB dissociation and nuclear export, all within 1 h of TNFα-treatment. By contrast, upon ZnPP-treatment, despite this normal IκBα-restoration at 1 h of TNFα-treatment, it is apparently functionally defective as a nuclear NF-κB-repressor, as NF-κB persisted in the nucleus (Fig. 8B). This stalled IκBα appears prominently both in the cytoplasm as well as clustered around the outer nuclear envelope rim (Fig. 8B)
Fig. 7.

Nup153 is an IκBα-interacting protein important for its nuclear import. A, Co-immunoprecipitation (Co-IP) of overexpressed IκBα with endogenous Nup153. HEK293T cells were transfected with C1-GFP control vector, or C1-GFP-IκBα vector, or a mock control for 40 h. Co-IP of cytosolic fractions (CER) and nuclear fractions (NER) with GFP-trap followed by Nup153 and IκBα IB analyses, with actin as the loading control, and GAPDH and HistoneH3 (H3) as fractionation markers. B, Co-IP of endogenous IκBα with endogenous Nup153. HepG2 cells were treated with TNFα (20 ng/ml) for the indicated times, followed by co-IP of whole cell lysates with IκBα-antibody or control IgG, and IB of Nup153 and IκBα with actin as the loading control. HEK293T cells were transfected with control siRNA (Ctrl) or NUP153 siRNA (C) or RanBP2 siRNA (D) for 48 h, and then treated with TNFα (20 ng/ml) for the indicated times. Cytosol and nuclear extracts were used for IB analyses. Relative IκBα-content (Mean ± S.D.) from three biological replicates is plotted over time.
Fig. 8.

Newly synthesized IκBα showed accelerated ZnPP-induced aggregation resulting in stalled nuclear import. A, Mouse hepatocytes were either untreated or pretreated with TNFα (20 ng/ml) for 1 h and then treated with ZnPP (10 μM) or left untreated for the indicated times. Cell lysates were used for IB analyses of Nup153 and IκBα with actin as the loading control. IκBα content relative to 0 h control was quantified (Mean ± S.D., n = 3, ** P < 0.01, * P < 0.05). B, CIFM analyses. HepG2 cells were treated with vehicle control (Ctrl), or TNFα (20 ng/ml) for 0.5 h or 1 h. Some TNFα-treated cells were also treated with ZnPP (10 μM) 20 min after TNFα-addition and incubated for another 40 min (TNFα + ZnPP, 1h). Cells were fixed and stained with anti-p65/anti-rabbit-Alexa-488 IgGs (green) and anti-IκBα/anti-mouse Alexa-647-conjugated IgGs (magenta) and DAPI. Images were obtained under 100 × lens. The same slides were then observed under wide-field microscope using 60 × lens to quantify the percentage of cells with predominant nuclear p65 accumulation. Cells (>600) were assessed at each experimental condition (Mean ± S.D., n = 3).
IκBα siRNA-Knockdown Analyses Reveal That ZnPP-Triggered NF-κB Activation May Additionally Involve IκBβ-Sequestration
Because of the functional redundancy of various IκBs in NF-κB cytoplasmic retention, IκBα-deficiency through genetic ablation, siRNA knockdown, or cycloheximide-inhibition of IκBα-synthesis fails to increase constitutive NF-κB transcriptional activation in many nonhepatic cell-types (19, 62, 63). Thus, upon our IκBα siRNA-knockdown, a similar slight, barely detectable constitutive NF-κB activation was observed in HepG2 cells (supplemental Fig. S7, left panel), whereas in primary mouse hepatocytes this activation was quite substantial (supplemental Fig. S7, right panel). However, the latter was not quite as marked as that observed upon ZnPP-treatment, possibly due to the appreciable compensatory IκBβ-up-regulation upon IκBα siRNA-knockdown (Fig. 9A). Because in hepatocytes both IκBα and IκBβ are the predominantly expressed IκBs (18, 21), and given the relatively more pronounced, sustained and consistently reproducible ZnPP-elicited NF-κB activation, we determined whether ZnPP also similarly sequestered IκBβ. Indeed, upon IκBα siRNA of HepG2 cells, the compensatorily increased IκBβ was similarly targeted to ZnPP-elicited aggregation, and thus, unable to appreciably mitigate the ZnPP-induced NF-κB activation post IκBα-knockdown (Fig. 9A). Furthermore, re-analyses of the RIPA-solubilized ZnPP-treated lysates by IB revealed that IκBβ also disappeared from its usual monomeric position, but was found in HMM protein aggregates (Fig. 9B). Such an apparent “IκBβ-loss” was also resistant to various UPD, calpain and ALD inhibitors (Fig. 9B), consistent with a sequestration process similar to that of IκBα. The ZnPP-targeting of both hepatic IκBs thus accounts for its potent NF-κB activation.
Fig. 9.

Effects of IκBα-siRNA knockdown on IκBβ-levels and constitutive and ZnPP-induced NFκB-activation. A, HepG2 cells were transfected with control siRNA (Ctrl) or IκBα siRNA. Cell lysates were used for IB analyses. Nuclear extracts were used for EMSA analyses. The marked hepatic IκBβ-up-regulation coupled with the relatively less pronounced NFκB-activation observed upon IκBα-siRNA knockdown versus ZnPP-treatment led to the examination of whether IκBβ was similarly sequestered by ZnPP as IκBα. B, Hepatocytes were similarly treated with proteolytic inhibitors as in supplemental Fig. S4B. Cell lysates were used for IB analyses.
DISCUSSION
Why is IκBα Vulnerable to ZnPP-Elicited Physical Sequestration from the Cytoplasm?
Recent evidence increasingly supports a sequestration and co-aggregation model of pathogenesis (64). In a “snowballing” effect, aggregation-prone disease proteins “hijack” their interacting partners with vital functions, sequestering them into cytotoxic insoluble inclusions (64). The protein scaffold p62 is one such IκBα-interacting and aggregating protein. However, scrutiny of ZnPP-treated p62-/- hepatocytes and MEF cells revealed that it is not essential for ZnPP-elicited IκBα-sequestration. This is consistent with the report that in p62-/- mouse liver, p62 is not required for MDB-formation, just for their maturation and stability (65). Similarly, individual knock-out of 2 out of the 10 newly identified IκBα-interacting and aggregating proteins also did not affect ZnPP-elicited IκBα-loss. This suggests that the aggregation process is unlikely to be mediated by any single protein, instead, it is more likely to be mediated through the interactions of several aggregation-prone proteins under pro-aggregation conditions. Aggregation-prone proteins share some common physicochemical properties: Preexistent proteins are relatively large in size, enriched in domains with high intrinsic disorder or unstructured regions, and exhibit low average hydrophobicity (66); newly synthesized proteins on the other hand, are more vulnerable due to prolonged exposure of their relatively hydrophobic domains either during or soon after synthesis during their subsequent folding, assembly, or transport (66).
None of the 10 proteins that interacted with IκBα and concurrently aggregated upon ZnPP-treatment are well-established stable IκBα-interactors. Although most of the known stable IκBα-interactors were repeatedly found in our co-IP analyses, they were never detected in the ZnPP-aggregate proteome. This suggests that these 10 proteins may be transient IκBα-interactors that are difficult to capture without highly sensitive and selective approaches. Our analyses of the physicochemical properties of these 10 proteins as well as IκB-proteins indicated that they ranged widely in size, with all of them exhibiting relatively low hydrophobicity, and all of them predicted to contain a long (>30 residues) disordered segment (Table II). These dual features of low hydrophobicity coupled with high intrinsic disorder could synergistically contribute to their ZnPP-elicited aggregation upon transient interaction, and may explain why ZnPP initially targets preexistent IκBα-species exhibiting both features (Table II). Similar vulnerability to ZnPP-elicited sequestration of the structurally related IκBβ thus accounts for the remarkably profound NF-κB-activation. Whether this vulnerability stems from their common structural ankyrin-repeat domain feature remains to be determined. Free IκBα-species is apparently unstable and requires NF-κB binding for folding to a stable conformation (67). Upon TNFα-stimulation, newly synthesized IκBα would be unstable and greatly susceptible to ZnPP-elicited aggregation without nuclear import to enable it to bind NF-κB and properly fold. Its ZnPP-elicited aggregation thus would result in the sustained ZnPP-triggered NF-κB-activation.
Table II. Structural properties of the 10 IκBα-interacting and ZnPP-inducible-aggregating proteins.
| Gene | Accession | Gravy* | Slider** | Protein Name |
|---|---|---|---|---|
| NUP153 | P49790 | −0.469 | 0.869 | Nuclear pore complex protein Nup153 |
| RANBP2 | P49792 | −0.596 | 0.840 | E3 SUMO-protein ligase RanBP2 |
| SMC4 | Q9NTJ3 | −0.691 | 0.8800 | Structural maintenance of chromosomes protein 4 |
| SMC2 | O95347 | −0.671 | 0.8320 | Structural maintenance of chromosomes protein 2 |
| SMC1A | Q14683 | −0.969 | 0.8980 | Structural maintenance of chromosomes protein 1A |
| POLR2A | P24928 | −0.534 | 0.7974 | DNA-directed RNA polymerase II subunit RPB1 |
| POLR2B | P30876 | −0.371 | 0.6869 | DNA-directed RNA polymerase II subunit RPB2 |
| SMARCA4 | P51532 | −0.839 | 0.9517 | Transcription activator BRG1 |
| TBC1D4 | O60343 | −0.553 | 0.8824 | TBC1 domain family member 4 |
| SLC3A2 | P08195 | −0.147 | 0.6931 | 4F2 cell-surface antigen heavy chain |
| NFKBIA | P25963 | −0.452 | 0.8195 | NF-kappa-B inhibitor alpha |
| NFKBIB | Q15653 | −0.388 | 0.7360 | NF-kappa-B inhibitor beta |
| NFKBIE | O00221 | −0.4336 | 0.8077 | NF-kappa-B inhibitor epsilon |
| SQSTM1 | Q13501 | −0.632 | 0.6246 | Sequestosome-1/p62 |
The GRAVY number of a protein is a measure of its hydrophobicity or hydrophilicity. The hydropathy values range from −2 to +2 for most proteins, with the positively rated proteins being more hydrophobic. Less hydrophobic proteins are labeled red.
Proteins are listed with SLIDER score, the higher the score the more likely a protein has a long (>= 30 AAs) disordered segment. Scores above 0.538 indicates that a given protein has a long disorder segment (labeled red).
Although NF-κB and IκBα form a stable complex, they are not static cytoplasmic residents but shuttle continuously between the nucleus and cytoplasm (68). Interestingly, p65 and IκBα translocate into the nucleus via different pathways. p65 enters the nucleus via the classical NLS-and importin (α3/α4)-dependent machinery (69), whereas IκBα and several other ankyrin repeat domain-containing proteins (ARPs) enter the nucleus via a NLS- and importin-independent machinery (70). This machinery depends on a direct interaction between the ARPs, RanGDP as well as mobile nucleoporins such as Nup153 and RanBP2 for entry across the NPC into the nucleus. Indeed, plausibly relevant to this IκBα-nucleocytoplasmic shuttling process, our proteomic analyses of the 10 common IκBα-interacting and concurrently ZnPP-aggregating proteins underscored significant enrichment of mobile NPC nucleoporins. Of these, hierarchical clustering analyses singled out Nup153 as both a robust direct IκBα-interactant and ZnPP-aggregation target (Fig. 5D). By contrast, importins α3 and α4 relevant to p65 nuclear import were not found in the ZnPP-aggregate proteome, which may explain why p65 nuclear import was unaffected and thus conducive to NF-κB activation.
Although we identified 10 IκBα-interacting proteins that concurrently aggregate with IκBα upon ZnPP-treatment, we are unable to definitively conclude that they co-aggregate. A limitation of this study is that the technical requirement for boiling in 2% SDS/1 h in the BAR approach led to the solubilization of many insoluble ZnPP-aggregated proteins that are otherwise isolated in the 14,000 × g-pellet. Thus, we could only examine IκBα-interacting proteins upon ZnPP-treatment in whole cell lysates. To conclusively identify IκBα-co-aggregating proteins, it would be necessary to specifically scrutinize IκBα-interacting proteins within the ZnPP-induced insoluble aggregate pellet, a task precluded by many currently available approaches that we have tried.
Potential Role of Nup153 in IκBα-Mediated NF-κB-Repression
The nucleoporin Nup153 primarily exists N-terminally anchored to the nuclear pore basket while its disordered and flexible FG-rich C terminus extends into the cytoplasm (71). Another Nup153 pool apparently exists that shuttles between the cytoplasmic and NPC faces (72), and interacts directly with cargos (i.e. Stat1, Smad2, and PU.1) for their nuclear import via a transporter-independent machinery (71). Nup153, in contrast to other FG-rich nucleoporins i.e. Nup98, Nup62, Nup214, also strongly binds ARs of iASPP and ASPP2, two representative substrates of the NLS-independent ARP-RanGDP nuclear import pathway (70). Structural/biochemical analyses indicate that Nup153 binds RanGDP with a higher affinity than RanGTP (73). This collective evidence suggests a critical role of Nup153 in facilitating ARP-RanGDP nuclear import. Our findings that IκBα co-immunoprecipitated not only with Nup153 in the nuclear extracts (NER), but also more appreciably with that in the cytoplasmic extracts (CER), even though the basal cytoplasmic Nup153-content is much lower than its nuclear content (compare inputs, Fig. 7A), are consistent with a minor, albeit highly dynamic, cytoplasmic Nup153 pool. This pool directly interacts with IκBα and may be responsible for the continuous IκBα-nucleocytoplasmic traffic at steady state, as indeed verified by our Nup153-knockdown analyses (Fig. 7C). The concurrent ZnPP-elicited aggregation of Nup153 and IκBα suggests that in addition to such IκBα-aggregation, the nuclear import of any residual soluble IκBα would be disrupted, leading to NF-κB nuclear persistence and prolonged response.
Another noteworthy proteomic finding is our quite prominent detection of the SUMO E3-ligase RanBP2 in this 10-protein cohort. RanBP2, an outer NPC component, fans its FG-rich filaments into the cytoplasm (74). The proteomic detection of RanBP2, but no other cytoplasmic NPC nucleoporin i.e. Nup214 in this common 10-protein cohort is functionally intriguing because besides Nup153, RanBP2 is also involved in receptor-independent nuclear import through direct interactions with its specific cytoplasmic cargos (74). This is intriguing given that IκBα-SUMOylation is reportedly involved in its nuclear entry and subsequent post-induction NF-κB-repression (75). Because RanBP2 also strongly binds RanGDP, conceivably a RanBP2-RanGDP-Nup153-IκBα-SUMO complex is involved in IκBα-nuclear import, a possibility consistent with our RanBP2-knockdown analyses (Fig. 7D). The inviability of the actively dividing HEK293T cells precluded our simultaneous knockdown of both nucleoporins to mimic their concurrent ZnPP-elicited sequestration.
We find it quite instructive that 2 of the 10 IκBα-interacting aggregation proteins are well-established components of pathogenic neurodegenerative disease protein aggregates (MS and ALS) or inclusions (Parkinson's Lewy bodies)[Fig. 5D; (66, 76, 77). Nup153 is found to co-aggregate with cargo proteins in oligodendrocyte precursor cells in MS lesions (78), and is also detected as anterior horn cell cytoplasmic inclusions of an ADAR2-deficient ALS mouse model (79). These findings not only underscore the common intrinsic physicochemical and physiological properties of these aggregation-prone proteins, but also suggest that their interaction and aggregation with IκBα under disease stresses could account for the associated tissue inflammation and injury commonly encountered in both liver and neurodegenerative diseases.
Plausible Clinical Relevance to EPP and XLPP
Such ZnPP-elicited persistent NF-κB transcriptional activation may also account for the inflammation and injury stemming from chronic PPIX exposure not only in Fechm1Pas, DDC- and GF-protoporphyric rodent livers, but also in EPP and XLPP patient livers (10, 11, 28–30, 80), wherein avid Fe-overutilization may drive ZnPP-generation. ZnPP-generation is also a prominent feature of iron-deficiency anemias and lead poisoning (Lamola and Yamane, 1974). Although our studies were largely confined to primary hepatocytes, in intact protoporphyric human and mouse livers these parenchymal cells are juxtaposed to nonparenchymal Kupffer cells and hepatic stellate cells that are quite capable of both canonical and noncanonical NF-κB-signaling. Thus, upon chronic PPIX-exposure, their NF-κB-activation could induce proinflammatory cytokines, chemokines, growth factors and other mitogens (17). This additional paracrine cytokine stimulus could further potentiate the PPIX-elicited NF-κB-signaling activation within the hepatocyte, further aggravating the extent of EPP and XLPP liver injury. Indeed, exogenous TNFα greatly accelerated ZnPP-elicited IκBα-sequestration in hepatocytes (Fig. 8A).
The precise mechanism of PPIX-induced protein aggregation remains to be elucidated. Porphyrins trigger protein cross-linking through a secondary reaction between the photooxidative products of histidine, tyrosine and/or tryptophan and free NH2-groups of amino acids (81). Given the light-sheltered in vivo environment of the liver, such a photooxidative protein-cross-linking seems unlikely in PPIX-mediated hepatic MDB formation, although PPIX can trigger protein aggregation and cell toxicity in the dark, albeit at a slower rate (60). Additionally, porphyrins also induce structural changes in certain proteins through oxidation of select residues, such as methionine. Such structural changes could expose some hydrophobic regions thereby inducing protein aggregation and leading to their functional impairment (82, 83). In this context, the PPIX-targeted aggregate proteins identified herein, we propose, could provide valuable insight into the future elucidation of the precise mechanism of PPIX-induced protein aggregation.
DATA AVAILABILITY
The MS proteomics data have been deposited onto the MassIVE repository with the dataset identifier MSV000086042.
Supplementary Material
Acknowledgments
We gratefully acknowledge Mr. Chris Her, UCSF Liver Cell & Tissue Biology Core Facility (supported by NIDDK Grant P30DK26743) for hepatocyte isolation. We are most grateful to Dr. D. M. Bissell (UCSF) for valuable discussions and his critical review of our manuscript. We also gratefully acknowledge Prof. P. Ortiz de Montellano (UCSF) for valuable discussions of porphyrin chemistry, and Dr. G. Knudsen (UCSF) for her helpful comments on our IAP/MS methodology and critical review of our manuscript and proteomic data. We also sincerely thank Profs. T. Yanagawa, M. Komatsu, H. Zhu, N. Mizushima, R. Scheckman, and R. J. Youle for valuable cell-lines. Y. Liu is most grateful to Dr. D. Larsen, UCSF Nikon Imaging Center for her training in confocal immunofluorescence microscopy, and Drs. M. Laurance, UCSF HDF Comprehensive Cancer Center and A. Pico, The Gladstone Institute Bioinformatics Facility, for training on pathway analyses and visualization. Published as a BioRxiv preprint (March 21st, 2019): doi: https://doi.org/10.1101/585497.
Footnotes
This article contains supplemental data.
Funding and additional information—These studies were supported by NIH Grants GM44037 (M.A.C.), DK26506 (M.A.C.), GM25515 (P.O.M.), GM097057 (D.H.K.), DK087984 (J.J.C.), and CIHR grant #81189 (P.A.G.). We also acknowledge the UCSF Biomedical Mass Spectrometry and Proteomics Resource Center (Prof. A. L. Burlingame, Director) supported by the Adelson Medical Research Foundation.
Conflict of interest—The authors have no conflict of interest to declare.
The inhibition of NMPP-elicited IkBa-loss by exogenous hemin could also be due to inhibition of ALAS1 and consequent reduction of endogenously generated PPIX.
Abbreviations—The abbreviations used are:
- MBDs
- Mallory-Denk-bodies
- ASH
- alcoholic steatohepatitis
- NASH
- nonalcoholic steatohepatitis
- GF
- griseofulvin
- NLS
- nuclear localization signal
- UPD
- ubiquitin-dependent proteasomal degradation.
REFERENCES
- 1. Strnad P., Zatloukal K., Stumptner C., Kulaksiz H., and Denk H. (2008) Mallory-Denk-bodies: lessons from keratin-containing hepatic inclusion bodies. Biochim. Biophys. Acta 1782, 764–774 [DOI] [PubMed] [Google Scholar]
- 2. Zatloukal K., Stumptner C., Fuchsbichler A., Heid H., Schnoelzer M., Kenner L., Kleinert R., Prinz M., Aguzzi A., and Denk H. (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 160, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jensen K., and Gluud C. (1994) The Mallory body: morphological, clinical and experimental studies (Part 1 of a literature survey). Hepatology 20, 1061–1077 [DOI] [PubMed] [Google Scholar]
- 4. Denk H., Gschnait F., and Wolff K. (1975) Hepatocellar hyalin (Mallory bodies) in long term griseofulvin-treated mice: a new experimental model for the study of hyalin formation. Lab. Invest. 32, 773–776 [PubMed] [Google Scholar]
- 5. Yokoo H., Harwood T. R., Racker D., and Arak S. (1982) Experimental production of Mallory bodies in mice by diet containing 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Gastroenterology 83, 109–113 [PubMed] [Google Scholar]
- 6. Holley A. E., Frater Y., Gibbs A. H., De Matteis F., Lamb J. H., Farmer P. B., and Naylor S. (1991) Isolation of two N-monosubstituted protoporphyrins, bearing either the whole drug or a methyl group on the pyrrole nitrogen atom, from liver of mice given griseofulvin. Biochem. J. 274, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ortiz de Montellano P. R., Beilan H. S., and Kunze K. L. (1981) N-Alkylprotoporphyrin IX formation in 3,5-dicarbethoxy-1,4-dihydrocollidine-treated rats. Transfer of the alkyl group from the substrate to the porphyrin. J. Biol. Chem. 256, 6708–6713 [PubMed] [Google Scholar]
- 8. Singla A., Moons D. S., Snider N. T., Wagenmaker E. R., Jayasundera V. B., and Omary M. B. (2012) Oxidative stress, Nrf2 and keratin up-regulation associate with Mallory-Denk body formation in mouse erythropoietic protoporphyria. Hepatology 56, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goodman Z. D. (2011) Hepatic Histopathology. In: Schiff E. R., Maddrey W. C., and Sorrell M. F., eds. Schiff's Diseases of the Liver, pp. 280–282, Wiley [Google Scholar]
- 10. Davies R., Schuurman A., Barker C. R., Clothier B., Chernova T., Higginson F. M., Judah D. J., Dinsdale D., Edwards R. E., Greaves P., Gant T. W., and Smith A. G. (2005) Hepatic gene expression in protoporphyic Fech mice is associated with cholestatic injury but not a marked depletion of the heme regulatory pool. Am J Pathol 166, 1041–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gant T. W., Baus P. R., Clothier B., Riley J., Davies R., Judah D. J., Edwards R. E., George E., Greaves P., and Smith A. G. (2003) Gene expression profiles associated with inflammation, fibrosis, and cholestasis in mouse liver after griseofulvin. EHP Toxicogenomics 111, 37–43 [PubMed] [Google Scholar]
- 12. Stumptner C., Fuchsbichler A., Heid H., Zatloukal K., and Denk H. (2002) Mallory body–a disease-associated type of sequestosome. Hepatology 35, 1053–1062 [DOI] [PubMed] [Google Scholar]
- 13. Singla A., Griggs N. W., Kwan R., Snider N. T., Maitra D., Ernst S. A., Herrmann H., and Omary M. B. (2013) Lamin aggregation is an early sensor of porphyria-induced liver injury. J. Cell Sci. 126, 3105–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Currais A., Fischer W., Maher P., and Schubert D. (2017) Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J. 31, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nivon M., Fort L., Muller P., Richet E., Simon S., Guey B., Fournier M., Arrigo A. P., Hetz C., Atkin J. D., and Kretz-Remy C. (2016) NFkappaB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell 27, 1712–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liou H. C., and Baltimore D. (1993) Regulation of the NF-kappa B/rel transcription factor and I kappa B inhibitor system. Curr. Opin. Cell Biol. 5, 477–487 [DOI] [PubMed] [Google Scholar]
- 17. He G., and Karin M. (2011) NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 21, 159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Azimifar S. B., Nagaraj N., Cox J., and Mann M. (2014) Cell-type-resolved quantitative proteomics of murine liver. Cell Metab. 20, 1076–1087 [DOI] [PubMed] [Google Scholar]
- 19. Han Y., and Brasier A. R. (1997) Mechanism for biphasic rel A. NF-kappaB1 nuclear translocation in tumor necrosis factor alpha-stimulated hepatocytes. J. Biol. Chem. 272, 9825–9832 [DOI] [PubMed] [Google Scholar]
- 20. Rao P., Hayden M. S., Long M., Scott M. L., West A. P., Zhang D., Oeckinghaus A., Lynch C., Hoffmann A., Baltimore D., and Ghosh S. (2010) IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature 466, 1115–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wiśniewski J. R., Vildhede A., Norén A., and Artursson P. (2016) In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J. Proteomics. 136, 234–247 [DOI] [PubMed] [Google Scholar]
- 22. Beg A. A., Finco T. S., Nantermet P. V., and Baldwin A. S. Jr (1993) Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of I kappa B alpha: a mechanism for NF-kappa B activation. Mol. Cell Biol. 13, 3301–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Traenckner E. B., Wilk S., and Baeuerle P. A. (1994) A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 13, 5433–5441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weil R., Laurent-Winter C., and Israël A. (1997) Regulation of IkappaBbeta degradation. Similarities to and differences from IkappaBalpha. J. Biol. Chem. 272, 9942–9949 [DOI] [PubMed] [Google Scholar]
- 25. Arenzana-Seisdedos F., Thompson J., Rodriguez M. S., Bachelerie F., Thomas D., and Hay R. T. (1995) Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol. Cell Biol. 15, 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson J. E., Phillips R. J., Erdjument-Bromage H., Tempst P., and Ghosh S. (1995) I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell 80, 573–582 [DOI] [PubMed] [Google Scholar]
- 27. Arenzana-Seisdedos F., Turpin P., Rodriguez M., Thomas D., Hay R. T., Virelizier J. L., and Dargemont C. (1997) Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 110, 3, 369–378 [DOI] [PubMed] [Google Scholar]
- 28. Cox T. M. (2003) Protoporphyria. In: Karl M. Kadish K. M. S., Roger Guilard, ed. The Porphyrin Handbook: Medical aspects of porphyrins, Elsevier Science, U. S. A. [Google Scholar]
- 29. Thapar M., and Bonkovsky H. L. (2008) The diagnosis and management of erythropoietic protoporphyria. Gastroenterol. Hepatol. (N Y) 4, 561–566 [PMC free article] [PubMed] [Google Scholar]
- 30. Balwani M., Doheny D., Bishop D. F., Nazarenko I., Yasuda M., Dailey H. A., Anderson K. E., Bissell D. M., Bloomer J., Bonkovsky H. L., Phillips J. D., Liu L., Desnick R. J., and Porphyrias Consortium Of The National Institutes Of Health Rare Diseases Clinical Research, N, Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network, (2013) Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol. Med. 19, 26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han X. M., Lee G., Hefner C., Maher J. J., and Correia M. A. (2005) Heme-reversible impairment of CYP2B1/2 induction in heme-depleted rat hepatocytes in primary culture: translational control by a hepatic alpha-subunit of the eukaryotic initiation factor kinase? J. Pharmacol. Exp. Ther. 314, 128–138 [DOI] [PubMed] [Google Scholar]
- 32. Liao M., Pabarcus M. K., Wang Y., Hefner C., Maltby D. A., Medzihradszky K. F., Salas-Castillo S. P., Yan J., Maher J. J., and Correia M. A. (2007) Impaired dexamethasone-mediated induction of tryptophan 2,3-dioxygenase in heme-deficient rat hepatocytes: translational control by a hepatic eIF2alpha kinase, the heme-regulated inhibitor. J. Pharmacol. Exp. Ther. 323, 979–989 [DOI] [PubMed] [Google Scholar]
- 33. Han A. P., Yu C., Lu L., Fujiwara Y., Browne C., Chin G., Fleming M., Leboulch P., Orkin S. H., and Chen J. J. (2001) Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 20, 6909–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okada K., Yanagawa T., Warabi E., Yamastu K., Uwayama J., Takeda K., Utsunomiya H., Yoshida H., Shoda J., and Ishii T. (2009) The alpha-glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol. Res. 39, 490–500 [DOI] [PubMed] [Google Scholar]
- 35. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., and Tanaka K. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 36. Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., and Yoshimori T. (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan Y., Dourdin N., Wu C., De Veyra T., Elce J. S., and Greer P. A. (2006) Conditional disruption of ubiquitous calpains in the mouse. Genesis 44, 297–303 [DOI] [PubMed] [Google Scholar]
- 38. Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., Sideris D. P., Fogel A. I., and Youle R. J. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chisolm J. Jr., and Brown D. H. (1975) Micro-scale photofluorometric determination of “free erythrocyte pophyrin” (protoporphyrin IX). Clin. Chem. 21, 1669–1682 [PubMed] [Google Scholar]
- 40. Ku N. O., Toivola D. M., Zhou Q., Tao G. Z., Zhong B., and Omary M. B. (2004) Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 78, 489–517 [DOI] [PubMed] [Google Scholar]
- 41. Che Y., and Khavari P. A. (2017) Research Techniques Made Simple: Emerging Methods to Elucidate Protein Interactions through Spatial Proximity. J. Invest. Dermatol. 137, e197–e203 [DOI] [PubMed] [Google Scholar]
- 42. Bar D. Z., Atkatsh K., Tavarez U., Erdos M. R., Gruenbaum Y., and Collins F. S. (2018) Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 15, 127–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hung V., Udeshi N. D., Lam S. S., Loh K. H., Cox K. J., Pedram K., Carr S. A., and Ting A. Y. (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 11, 456–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosenfeld J., Capdevielle J., Guillemot J. C., and Ferrara P. (1992) In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 203, 173–179 [DOI] [PubMed] [Google Scholar]
- 45. Hellman U., Wernstedt C., Gonez J., and Heldin C. H. (1995) Improvement of an “In-Gel” digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 224, 451–455 [DOI] [PubMed] [Google Scholar]
- 46. Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., and Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell. Proteomics 7, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 48. Teo G., Liu G., Zhang J., Nesvizhskii A. I., Gingras A. C., and Choi H. (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics. 100, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., and Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Knight J. D. R., Choi H., Gupta G. D., Pelletier L., Raught B., Nesvizhskii A. I., and Gingras A. C. (2017) ProHits-viz: a suite of web tools for visualizing interaction proteomics data. Nat. Methods 14, 645–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kyte J., and Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 54. Peng Z., Mizianty M. J., and Kurgan L. (2014) Genome-scale prediction of proteins with long intrinsically disordered regions. Proteins 82, 145–158 [DOI] [PubMed] [Google Scholar]
- 55. Youn J. Y., Dunham W. H., Hong S. J., Knight J. D. R., Bashkurov M., Chen G. I., Bagci H., Rathod B., MacLeod G., Eng S. W. M., Angers S., Morris Q., Fabian M., Cote J. F., and Gingras A. C. (2018) High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell. 69, 517–532.e11 [DOI] [PubMed] [Google Scholar]
- 56. Chen J. J. (2007) Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood 109, 2693–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cuervo A. M., Hu W., Lim B., and Dice J. F. (1998) IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 9, 1995–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen F., Lu Y., Kuhn D. C., Maki M., Shi X., Sun S. C., and Demers L. M. (1997) Calpain contributes to silica-induced I kappa B-alpha degradation and nuclear factor-kappa B activation. Arch. Biochem. Biophys. 342, 383–388 [DOI] [PubMed] [Google Scholar]
- 59. Kirkin V., Lamark T., Sou Y. S., Bjorkoy G., Nunn J. L., Bruun J. A., Shvets E., McEwan D. G., Clausen T. H., Wild P., Bilusic I., Theurillat J. P., Overvatn A., Ishii T., Elazar Z., Komatsu M., Dikic I., and Johansen T. (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 33, 505–516 [DOI] [PubMed] [Google Scholar]
- 60. Maitra D., Elenbaas J. S., Whitesall S. E., Basrur V., D'Alecy L. G., and Omary M. B. (2015) Ambient Light Promotes Selective Subcellular Proteotoxicity after Endogenous and Exogenous Porphyrinogenic Stress. J. Biol. Chem. 290, 23711–23724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Elenbaas J. S., Maitra D., Liu Y., Lentz S. I., Nelson B., Hoenerhoff M. J., Shavit J. A., and Omary M. B. (2016) A precursor-inducible zebrafish model of acute protoporphyria with hepatic protein aggregation and multiorganelle stress. FASEB J. 30, 1798–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Klement J. F., Rice N. R., Car B. D., Abbondanzo S. J., Powers G. D., Bhatt P. H., Chen C. H., Rosen C. A., and Stewart C. L. (1996) IkappaBalpha deficiency results in a sustained NF-kappaB response and severe widespread dermatitis in mice. Mol. Cell Biol. 16, 2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tergaonkar V., Correa R. G., Ikawa M., and Verma I. M. (2005) Distinct roles of IkappaB proteins in regulating constitutive NF-kappaB activity. Nat. Cell Biol. 7, 921–923 [DOI] [PubMed] [Google Scholar]
- 64. Yang H., and Hu H. Y. (2016) Sequestration of cellular interacting partners by protein aggregates: implication in a loss-of-function pathology. FEBS J. 283, 3705–3717 [DOI] [PubMed] [Google Scholar]
- 65. Lahiri P., Schmidt V., Smole C., Kufferath I., Denk H., Strnad P., Rulicke T., Frohlich L. F., and Zatloukal K. (2016) p62/Sequestosome-1 Is Indispensable for Maturation and Stabilization of Mallory-Denk Bodies. PLoS ONE. 11, e0161083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., and Vabulas R. M. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 [DOI] [PubMed] [Google Scholar]
- 67. Ferreiro D. U., and Komives E. A. (2010) Molecular mechanisms of system control of NF-kappaB signaling by IkappaBalpha. Biochemistry 49, 1560–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ghosh S., and Karin M. (2002) Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl, S81–S96 [DOI] [PubMed] [Google Scholar]
- 69. Fagerlund R., Kinnunen L., Kohler M., Julkunen I., and Melen K. (2005) NF-{kappa}B is transported into the nucleus by importin {alpha}3 and importin {alpha}4. J. Biol. Chem. 280, 15942–15951 [DOI] [PubMed] [Google Scholar]
- 70. Lu M., Zak J., Chen S., Sanchez-Pulido L., Severson D. T., Endicott J., Ponting C. P., Schofield C. J., and Lu X. (2014) A code for RanGDP binding in ankyrin repeats defines a nuclear import pathway. Cell 157, 1130–1145 [DOI] [PubMed] [Google Scholar]
- 71. Ball J. R., and Ullman K. S. (2005) Versatility at the nuclear pore complex: lessons learned from the nucleoporin Nup153. Chromosoma 114, 319–330 [DOI] [PubMed] [Google Scholar]
- 72. Nakielny S., Shaikh S., Burke B., and Dreyfuss G. (1999) Nup153 is an M9-containing mobile nucleoporin with a novel Ran-binding domain. EMBO J. 18, 1982–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Partridge J. R., and Schwartz T. U. (2009) Crystallographic and biochemical analysis of the Ran-binding zinc finger domain. J. Mol. Biol. 391, 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Walde S., Thakar K., Hutten S., Spillner C., Nath A., Rothbauer U., Wiemann S., and Kehlenbach R. H. (2012) The nucleoporin Nup358/RanBP2 promotes nuclear import in a cargo- and transport receptor-specific manner. Traffic 13, 218–233 [DOI] [PubMed] [Google Scholar]
- 75. Desterro J. M., Rodriguez M. S., and Hay R. T. (1998) SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell. 2, 233–239 [DOI] [PubMed] [Google Scholar]
- 76. Ofengeim D., Ito Y., Najafov A., Zhang Y., Shan B., DeWitt J. P., Ye J., Zhang X., Chang A., Vakifahmetoglu-Norberg H., Geng J., Py B., Zhou W., Amin P., Berlink Lima J., Qi C., Yu Q., Trapp B., and Yuan J. (2015) Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Boeynaems S., Bogaert E., Van Damme P., and Van Den Bosch L. (2016) Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 132, 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakahara J., Kanekura K., Nawa M., Aiso S., and Suzuki N. (2009) Abnormal expression of TIP30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J. Clin. Invest. 119, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yamashita T., Aizawa H., Teramoto S., Akamatsu M., and Kwak S. (2017) Calpain-dependent disruption of nucleo-cytoplasmic transport in ALS motor neurons. Sci. Rep 7, 39994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tutois S., Montagutelli X., Da Silva V., Jouault H., Rouyer-Fessard P., Leroy-Viard K., Guénet J. L., Nordmann Y., Beuzard Y., and Deybach J. C. (1991) Erythropoietic protoporphyria in the house mouse. A recessive inherited ferrochelatase deficiency with anemia, photosensitivity, and liver disease. J. Clin. Invest. 88, 1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dubbelman T. M., de Goeij A. F., and van Steveninck J. (1978) Photodynamic effects of protoporphyrin on human erythrocytes. Nature of the cross-linking of membrane proteins. Biochim. Biophys. Acta 511, 141–151 [DOI] [PubMed] [Google Scholar]
- 82. Afonso S. G., Enriquez de Salamanca R., and Batlle A. M. (1999) The photodynamic and non-photodynamic actions of porphyrins. Braz. J. Med. Biol. Res. 32, 255–266 [DOI] [PubMed] [Google Scholar]
- 83. Maitra D., Carter E. L., Richardson R., Rittie L., Basrur V., Zhang H., Nesvizhskii A. I., Osawa Y., Wolf M. W., Ragsdale S. W., Lehnert N., Herrmann H., and Omary M. B. (2019) Oxygen and conformation dependent protein oxidation and aggregation by porphyrins in hepatocytes and light-exposed cells. Cell. Mol. Gastroenterol. Hepatol. 8, 659–682.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited onto the MassIVE repository with the dataset identifier MSV000086042.