

Stoichiometry of nucleotide binding to AAA+ ATPase PAN was established using native MS. Utilization of a Walker A mutant PANKA, which is impaired in nucleotide binding as an internal standard minimized the impact of residual solvation on mass measurement accuracy. PAN hexamer dissociation products and over-charged species – both maintaining nucleotide binding – were observed at highly variable levels. We hypothesize that these phenomena originated at the final stages of ESI droplet evolution, their extent dependent on minute changes in emitter performance.
Keywords: proteasome, AAA+ ATPase, nucleotide binding, stoichiometry, native mass spectrometry, cooperativity, mass spectrometry, Archaebacteria*, electron microscopy, macromolecular complex analysis, non-covalent interaction MS*, proteasome
Graphical Abstract

Highlights
All six binding sites in PANWT are occupied by ADP- or ATP-type nucleotides.
PANKA Walker A mutant substoichiometrically binds ATP- but not ADP-type nucleotides.
PAN hexamer dissociation of the solution origin characteristics was observed in MS.
We posit that the PAN hexamer dissociation proceeds within the ESI droplets.
Abstract
AAA+ ATPases constitute a large family of proteins that are involved in a plethora of cellular processes including DNA disassembly, protein degradation and protein complex disassembly. They typically form a hexametric ring-shaped structure with six subunits in a (pseudo) 6-fold symmetry. In a subset of AAA+ ATPases that facilitate protein unfolding and degradation, six subunits cooperate to translocate protein substrates through a central pore in the ring. The number and type of nucleotides in an AAA+ ATPase hexamer is inherently linked to the mechanism that underlies cooperation among subunits and couples ATP hydrolysis with substrate translocation. We conducted a native MS study of a monodispersed form of PAN, an archaeal proteasome AAA+ ATPase, to determine the number of nucleotides bound to each hexamer of the WT protein. We utilized ADP and its analogs (TNP-ADP and mant-ADP), and a nonhydrolyzable ATP analog (AMP-PNP) to study nucleotide site occupancy within the PAN hexamer in ADP- and ATP-binding states, respectively. Throughout all experiments we used a Walker A mutant (PANK217A) that is impaired in nucleotide binding as an internal standard to mitigate the effects of residual solvation on mass measurement accuracy and to serve as a reference protein to control for nonspecific nucleotide binding. This approach led to the unambiguous finding that a WT PAN hexamer carried – from expression host – six tightly bound ADP molecules that could be exchanged for ADP and ATP analogs. Although the Walker A mutant did not bind ADP analogs, it did bind AMP-PNP, albeit at multiple stoichiometries. We observed variable levels of hexamer dissociation and an appearance of multimeric species with the over-charged molecular ion distributions across repeated experiments. We posit that these phenomena originated during ESI process at the final stages of ESI droplet evolution.
AAA+ ATPases constitute a large superfamily of chaperone-like proteins that are involved in many cellular processes, including DNA disassembly, protein degradation and protein complex disassembly (1–4). Typically, they assemble into hexameric structures and combine with other proteins to form molecular machines of highly versatile functions, all powered by the energy released in the process of the AAA+ ATPase-assisted nucleotide triphosphate – typically ATP – hydrolysis. Sequence alignment (5) followed by an extensive mutational analysis of archaeal Thermoplasma acidophilum Lon protein (6) allowed for defining the core sequence “motifs” of the AAA+ proteins that are crucial for function: Walker A, Walker B, sensors 1 and 2, arginine finger and pore loop. Specifically, the Besche et al. (6) mutational analysis of Lon protein demonstrated that Walker A and B motifs and the sensor 1 and sensor 2' residues were essential for the ATPase activity, whereas the sensor 2 and the arginine finger were involved in activation of the protease domain. Numerous AAA+ protein mutants have been analyzed since, many listed in the Wendler et al. review (3). The fact that the structural blueprint for AAA+ ATPases has been maintained from archaea to eukaryotes suggests that this protein “class is ancient and has undergone considerable functional divergence prior to the emergence of the major divisions of life” (1), (see abstract).
The archaeal proteasome-activating nucleotidase (PAN) that shares extensive sequence homology with the six regulatory particles of triple-ATPase (Rpt) subunits of eukaryotic 26S proteasome – presumably, their evolutionary precursor – has been extensively studied as a prototypic AAA+ ATPase. PAN, which was first described by Zwickl et al. (7) in Methanocaldococcus jannaschii functions as a regulatory particle (RP) that interacts with archaeobacterial 20S core particle (CP) to facilitate protein degradation. However, in contrast to its eukaryotic counterpart, archaeal PAN-20S proteasome assembly functions in a ubiquitin-independent fashion. Early studies on PAN demonstrated that its oligomeric form was highly stable in the absence of ATP. An interaction between PAN and its CP appeared to be transient (8) and a formation of PAN-20S assembly required the presence of ATP or its nonhydrolyzable analogs (9). It was also demonstrated that although ATP hydrolysis was necessary for a degradation of globular proteins, it was not required for a translocation of the unfolded substrates through a central pore of the proteasome assembly (9). When expressed in E. coli, PAN was produced without the N-terminal methionine and was accompanied by its N-terminally truncated version, the latter resulting from the presence of an internal initiation site at M74 (8).
The early cryo-EM (9) and x-ray (10, 11) analyses of archaeal AAA+ proteins, the latter employing truncated forms of PAN, demonstrated that the six AAA-ATPase subunits assemble in two stacked rings: the AAA ring formed by the AAA domains is of pseudo 6-fold symmetry and it is positioned proximal to the CP, whereas the distal N-terminal domains form the N ring with a pseudo 3-fold symmetry with coiled-coil pairs protruding from it. The underlying key to the asymmetry within the N ring is the main chain configuration of Pro91 (in M. jannaschii PAN), which alternates between the cis- and trans-type peptide bond across subunits within a hexameric ring (10). The N ring facilitates substrate unfolding whereas the AAA ring is believed to actively translocate substrates into the CP in an ATP-dependent manner.
For a subset of AAA+ ATPases that facilitate protein unfolding and degradation, e.g. PAN, it is generally held that the process of substrate translocation for further processing is enabled by a cooperation among six AAA+ subunits that cycle through the nucleotide binding-dependent conformational states. Numerous studies aimed at establishing a stoichiometry of nucleotide binding to AAA+ ATPases were performed over the years and surprisingly, they delivered disparate results. As the ATP binding sites are positioned at the interface between the adjacent subunits, there are six potential ligand binding sites per hexamer. Importantly however, X-Ray studies, reviewed in Wendler et al. (3), and recent cryo-EM studies, e.g. Huang et al. (2016) (12) suggested a structure with two pairs of the protomer-nucleotide units acting separately from each other in the process of nucleotide binding (Fig. 1). For M jannaschii PAN the interacting amino acid residues were identified as K217 (Walker A), D270 (Walker B), T316 (Sensor I), and R328, R331 (Arginine finger) (3). The classical biochemical approach that employed radioactively labeled nucleotides found a maximum of four nucleotides per hexamer for ClpX, an RP of the bacterial AAA+ protease while pointing to cooperativity in ATP binding to distinct classes of ATP-binding sites (13). Smith et al. (14) reported similar results for PAN using P32-labeled ATP. Likewise, incubation of PAN with P32-labeled ADP pointed to the presence of four P32-labeled ADP ligands bound to the PAN hexamer. They further suggested that out of six potential sites, two remained unoccupied whereas the other four formed two pairs of high- and low-affinity ATP binding sites. The further study from the same laboratory utilized fluorescently labeled ATP analogs and confirmed their previous results (15). However, two independent studies on crystallization of E. coli HslU ATPase demonstrated the presence of 3, 4 or 6 nucleotides depending on the crystallization conditions, the type of nucleotide used and the presence or absence of its proteasomal partner CP, i.e. HslV (16, 17). After completion of our studies, several high-resolution cryo-EM studies on human p97 (18) and human proteasome (12, 19, 20) were published, each demonstrating the presence of six nucleotides bound per hexamer. The latest cryo-EM study on archaeal (A. fulgidus) proteasome pointed to the coexisting nucleotide states with a hexamer occupied by five nucleotides and revealed “the structural basis for a sequential around-the ring-ATPase cycle” (21), (see abstract).
Fig. 1.

Graphic rendering of the ADP binding by each of six subunits of PAN.
To address an apparent variability of nucleotide/protein stoichiometry information reported to date, we employed native MS to study nucleotide binding to the archaeal M. jannaschii proteasome AAA+ ATPase PAN homohexamer. Of note, native MS analysis of PAN from Methanosarcina mazei and M. jannaschii were published but the issue of protein-nucleotide occupancy was not addressed (22).
The term “native MS” was coined (23) to refer to MS assays performed under pseudo-physiological conditions that were likely to preserve macromolecular interactions upon their transfer from solution to the gas phase. Most of the work in the native MS field to date has employed electrospray ionization (ESI) in its nanoflow (nanoESI) format. Compared with other established techniques widely used for analysis of noncovalent assemblages, native MS offers unique advantages in terms of high throughput and the ability to handle mixtures, inclusive of differentiating among distinct folds of the same species. At the same time, native MS technology presents with challenges, of which an inherent heterogeneity of noncovalently bound molecular ion populations because of incomplete desolvation upon their transfer from the ESI droplet to the gas phase (24) needs to be addressed in studies on ligand binding stoichiometry. Nevertheless, as outlined in recent reviews (25–28), the long-term stability of native-like structures in the gas phase enabled numerous informative native MS studies aimed at characterizing solution-phase properties, e.g. stoichiometry, topology, dynamics and binding affinities of interactions of diverse types of macromolecular assemblages, including nucleotide-binding complexes (listed in supplemental Material).
In view of successful studies referred to above, we utilized ADP and its analogs (TNP-ADP and mant-ADP), and a nonhydrolyzable ATP analog (AMP-PNP) to study nucleotide site occupancy within the PAN hexamer in ADP- and ATP-binding states, respectively. The approach presented here can be applied to a broad range of other AAA+ ATPase machineries and nonAAA+ helicase hexamers as well, providing more accurate measurement of nucleotide stoichiometry than is achievable using traditional biochemical methods. In addition to reporting on stoichiometry of PAN-nucleotide binding, we will also discuss the possibility that the variable level of hexamer dissociation and possibly, an appearance of multimer species of over-charged molecular ion distributions that were observed in our experiments originated during ESI process at the final stages of ESI droplet evolution.
MATERIALS AND METHODS
PAN Proteins Production and Purification
Proteasome activating nucleotidase (PAN) gene from the thermophilic archaea M. jannaschii (PAN_METJA [Q58576]) was cloned into pET-15b expression vector in between NcoI and BamHI cutting sites, leaving out the His-tag and linker region on the vector. Protein expression was induced in E. coli BL21(DE3) by isopropyl β-d-1-thiogalactopyranoside (IPTG) when the cells were grown at 37 °C to a mid-log phase (OD 600 nm = 0.6). The 74th residue, methionine, was mutated to alanine to prevent alternative protein translation starting from this residue (7). Based on this variant, further mutations were made to the ATPase active site including: Walker A mutant (PANK217A), Walker B mutant (PANE271Q), an arginine finger mutant (PANR328A), and a double mutant (PANK217A/R328A).
PANWT and mutants were purified from cell lysate of induced E. coli first by heat precipitation in 80˚C water bath. The supernatant of the heated material was then loaded on a HiTrap Q 5 ml column (GE Healthcare) for anion exchange chromatography and was eluted in a gradient of 150 mm to 1.0 M NaCl. The ion-exchange column fractions containing PAN were pooled and loaded onto a HiLoad Superose 6 column (GE Healthcare, 323 ml column volume) for gel filtration in a buffer containing 50 mm Tris pH 8.5, 150 mm NaCl, 1 mm EDTA at a flow rate of 2 ml/min. Alternatively, gel filtration was conducted on an analytical gel filtration column Superose 6 10/300 GL (GE Healthcare, 23 ml column volume). A comparative gel filtration analysis of PANWT protein in Tris versus 0.5 M ammonium acetate buffer used Superdex 200 Increase 10/300 column (GE Healthcare, bed volume ∼24 ml). Flowrate used for both analytical columns was 0.25 ml/min. Elute was monitored by UV at 260 nm and 280 nm. The purity of purified protein was examined by SDS-PAGE, electron microscopy (EM), and MS. All PAN proteins were examined by the UV absorbance scan within 240–330 nm. Protein concentrations were determined by using a Bradford protein assay kit (Bio-Rad Life Science, Hercules CA), a Pierce BCA protein assay kit (ThermoFisher Scientific, Waltham, MA), and a Modified Lowry Protein Assay kit (ThermoFisher Scientific). Proteins were brought to ∼10 μm hexamer concentration in 50 mm Tris pH 8.5, 150 mm NaCl, 1 mm EDTA, 50% glycerol, aliquoted to small portions and stored at −80˚C.
Preparation of ADP and ATP Analogs
Fluorescently labeled ADP analogs: 2′-(or-3′)-O-(N-methylanthraniloyl)adenosine 5′-diphosphate (mant-ADP) (Cat. No. M12416) and 2′-(or-3′)-O-(trinitrophenyl)adenosine 5′-diphosphate (TNP-ADP) (Cat. No. T7601), both as disodium salts in 50 mm Tris pH 7.5 solution at concentrations of 5 mm and 7 mm respectively, were purchased from Thermo Fisher Scientific. Adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) (Cat. No. A2647) in its lithium hydrate form of ∼95% purity as well as the unlabeled ADP and ATP were purchased from Sigma-Aldrich (St. Louis, MO). Each of the three labeled nucleotides was dissolved in distilled water to form a 100 mm stock solution, aliquoted in 20 µL portions, flash frozen in liquid nitrogen and stored at −80˚C. The frozen aliquots were thawed immediately before use and any reagent remainders were discarded. The molecular weights of these nucleotides excluding metal ions and water are: (ADP) 427.2 Da, (ATP) 507.2 Da, (AMP-PNP) 506.2 Da, (mant-ADP) 560.4 Da, (TNP-ADP) 637.3 Da.
Nucleotide Exchange and Sample Preparation for Native MS
Aliquots of purified PANWT at 20 μm stored at -80˚C were thawed, mixed with a nucleotide of interest in a buffer containing 50 mm Tris pH 8.0, 150 mm NaCl and 10 mm MgCl2. The PAN hexamer concentration in the mixture was kept above 5 μm to ensure reliable signal in nanoESI-MS. The nucleotide concentration in the mixtures ranged from 0.5 mm to 10 mm, i.e. at 100 to 2000-fold excess over the PAN hexamer concentration: typically, the nucleotide analog concentration was set to afford a ∼80-fold molar excess over the number of nucleotide binding sites. The mixtures were left at 4˚C overnight (for ADP or its analogs) or for 15 min (for AMP-PNP) before MS experiments. After incubation, buffer was exchanged to 0.5 M ammonium acetate and free nucleotide was concomitantly removed from PANWT by using the Micro Bio-Spin P-6 column (Bio-Rad), which according to the Manufacturer's instructions was expected to retain 98% of unincorporated nucleotides at 20 µL load volume. The procedures followed manufacturer instructions. Prior to nanoESI MS analysis, internal standard PANKA mutant was buffer exchanged to 0.5 M ammonium acetate using the same type of a spin column and PANWT and PANKA were mixed at a 1:1 ratio.
ESI MS Analysis of PAN Proteins under Denaturing Conditions
QTOF mass spectrometer (SYNAPT G2 HDMS, Waters, Milford, MA) equipped with a nanoAcquity ultra performance liquid chromatography (UPLC) system (Waters) was employed. Samples (2–4 µL) of PANWT and PANKA, each at a monomer concentration of ∼5 μm were injected into nanoAcquity UPLC and separated on a Symmetry C18 trap column (180 μm × 20 mm, Waters) at a flow rate of 5 µL/min using a 10 min linear gradient of 10–70% Phase B (0.1% formic acid in acetonitrile) in Phase A (0.1% formic acid in water). Column elute was delivered to the nanospray source of mass spectrometer using a precut TaperTip Emitter (360 μm O.D. × 20 mm I.D., 2.5” length, Waters) at a potential of 3–3.5 kV. The instrument was mass calibrated using sodium iodide for m/z range 50–2000. The mass spectrometer was operated in “high resolution” mode and spectra were acquired within 500–2000 m/z range. MassLynx software (Version SCN781, Waters) was used for data analysis. Specifically, mass spectra were smoothed, background-subtracted and centroided for manual assignment of components. Alternatively, raw data were submitted to MaxEnt-driven deconvolution of molecular ion envelopes to identify sample components and measure their molecular masses.
nanoESI MS Analysis of Native PAN Proteins
Samples were introduced into the QTOF mass spectrometer (SYNAPT G2 HDMS, Waters) via homemade borosilicate glass nanoESI emitters that were prepared as described in (29). Typically, a 3 µL PAN sample (hexamer concentration 5–10 μm) was loaded into the emitter and nanospray was initiated by applying 0.6–1.0 kV potential via a platinum wire (0.127 mm diameter, Sigma-Aldrich) that was inserted into a spray capillary and remained in contact with the solution. Typical instrumental settings were as follows: source temperature = 80–100˚C, desolvation temperature = 150˚C, sampling cone = 100–200 V, trap gas flow = 4 ml/min, backing pressure = 7 mbar, detector = 2700–3000 V, m/z range = 500–14,000, scan time 1 s, interscan time 0.024 s. The instrument was mass calibrated using cesium iodide for m/z range of 500–5000. When required, data acquired in few separate MS experiments that were performed under the same conditions were combined – using MassLynx “Combine All Files” tool – to increase signal-to-noise ratio. Native MS data analysis utilized various modules of Massign software (30). Specifically, data were smoothed and linearized (settings: smooth curve 20, linearization setting one datapoint per Da), background-subtracted (step function 100, smooth curve 200), protein components were manually assigned and then simulated using a Levenberg-Marquardt algorithm; Lorentzian peak shape trailing edge settings were used. Detailed parameters generated for the measured mixture components using Massign simulations are listed in supplemental Tables S1C, S2C, S3C for analyses of mixtures of PANKA with PANWT before and after nucleotide exchange with mantADP, and TNP-ADP. Supplemental Tables S4B and S5 show Massign results for the AMP-PNP exchange experiments of PANWT with either PANKA or double mutant PANK217A/R328A, respectively.
CRYO-EM Analysis
For cryo-EM experiments, purified proteins of PANWT and PANK217A were each diluted to a final concentration ∼1 μm PAN hexamer. 2.5 µL sample was applied to a glow discharge Quantifoil R1.2/1.3 holey carbon grid. The grids were blotted and plunge-frozen in liquid ethane that was kept slightly above -180˚C by using a Vitrobot IV (FEI Co., Hillsborough OR). The proteins in vitreous ice were inspected on a Tecnai TF20 transmission electron microscope (FEI Co.) equipped with a field emission gun operating at 200 kV. Images were collected using a TemF816 8K x 8K CMOS camera (TVIPS GmbH, Gauting, Germany) at a nominal magnification of 62 kX (pixel size 1.204 Å). The defocus values of the micrographs were determined by using the software CTFFIND. For each protein, ∼4000 particles in the defocus range of 2.5–3.5 μm were classified into 64 class averages using the K-mean method.
Experimental Design and Statistical Rationale
All studies utilized the PAN sequence modified by M74A substitution, to prevent alternative protein translation starting from the M74 residue (7). For simplicity, we refer to this M74A variant of PAN as WT PAN (PANWT) whereas mutants are annotated with their secondary mutations, e.g. PANK217A (PANKA) carrying KA mutation at position 217 in addition to the M74A mutation that was shared by all the constructs.
Limitations in resolution caused by an inevitable presence of residual solvation (i.e. an attachment of residual solvent/salt molecules to protein molecular ions) in the process of native ESI MS (24) poses a challenge in studies on stoichiometry of ligand binding. Differentiating between mass increase due to ligand binding versus that caused by residual solvation might be difficult when ligand-related and residual solvation-related mass increments are of the same order of magnitude. The extent of desolvation is sensitive to even small variations in electrospray conditions and hence might fluctuate among experiments. Notably however—within the same experiment—the residual solvation effects are expected to be uniform for all sample components, and their extent correlates with protein size (31). A residual solvation mass increment is defined as a ratio of the difference between the experimental (MW exp) and theoretical (MW theor) protein mass to the theoretical protein mass: [(MW exp – MW theor)/MW theor]. As all proteins in the mixture will be affected by residual solvation to the same extent, their respective residual solvation mass increments are expected to be the same. Hence, establishing residual solvation mass increment for one protein—with a caveat that the residual solvation can reliably be expected to be solely responsible for the observed increase in the control protein experimental mass—will allow to calculate the residual solvation related mass increases for all other proteins in the mixture. This way an increase in an experimental mass that exceeds the level established for the control protein will point to the presence of other factors responsible for the observed mass difference, e.g. the presence of ligands or errors in amino acid sequence used to calculate protein MW theor. Therefore, we included Walker A mutant PANKA as an internal standard in all analyses of PANWT samples. As PANKA was expected to be devoid of bound nucleotides, all mass increase observed for this protein was due to residual solvation. Hence, after accounting for mass difference between the two proteins because of the K217A mutation, we were able to separate the measured [(MW exp – MW theor)] difference for PANWT into a portion related to the residual solvation and a portion related to the presence of bound ligands – see details in step 6 of the experimental scenario below. At the same time, PANKA served as a control for nonspecific ligand-protein interactions.
The following experimental scenario was used:
Express PANWT and PAN mutants in E. coli and purify the proteins by a combination of anion exchange and size exclusion chromatography.
Incubate PANWT proteins with ligands of interest in Tris-buffered saline (TBS).
Exchange buffer to 0.5 M ammonium acetate while removing excess of nucleotides using spin column. Perform the same buffer exchange process for PANKA.
Mix same amounts of PANWT and PANKA in ammonium acetate buffer and acquire mass spectra of proteins by employing native nanoESI MS.
Use Massign software to assign spectra components, i.e., to identify charge state ion series and determine their molecular masses.
Assuming that PANWT and PANKA are affected by incomplete desolvation to the same extent (see above), use the following formula to calculate the number “n” of ADP molecules nascently bound to PANWT: n = (Δ-FKA)/MWADP, where MW stands for molecular mass of species annotated by a subscript, Δ= (exp MWPANWT-exp MWPANKA), and FKA serves as a normalizing factor to account for the mutant protein mass decrease of 6 × 57, as compared to WT protein, because of the presence of the K->A mutation in each of six monomers.
The number (“x”) of nucleotide analogs that were exchanged for the nascently bound ADP was calculated as follows: x = (ΔN-6xMWADP)/MWanalog-MWADP), where ΔN is a normalized experimental mass difference between the WT and KA protein, as calculated above.
In total, multiple PANWT and PANK217A preparations were used to measure the number of ADP bound to PANWT. The nucleotide exchange experiments using TNP-ADP, mantADP and AMP-PNP were performed on four, five and three separate occasions, respectively. Each time, multiple sets of nanoESI MS data were acquired and data sets with the highest resolution and S/N ratio were selected for analysis and are presented here.
RESULTS
Integrity of PAN Protein Preparations
PAN protein preparations to be used for native MS studies were first examined by SDS-PAGE (not shown), reversed phase (RP) liquid chromatography (LC) nanoESI MS, UV absorbance profiling and cryo-EM. In RPLC nanoESI MS, both PANWT and PANK217A were detected as monomers and generated broad charge state distributions characteristic for ESI spectra of denatured proteins (Fig. 2A). The experimental average masses of 48,499 ± 0.5 Da and 48,441 ± 0.4 Da matched—with very high degree of accuracy and precision—the theoretical values of 48,499 Da and 48,442 Da calculated for PANWT and PANKA amino acid sequences lacking initiator methionine, respectively. Thus, the nanoESI MS analysis demonstrated that PAN proteins were faithfully expressed, were not truncated (except for N-terminal M removal), carried no post-translational modifications, and samples contained no ESI-detectable contaminating proteins, except for a single preparation used for the TNP-ADP exchange experiment, which contained an unknown impurity of Mr ∼69 k presenting with molecular ion envelope within 4k m/z range, annotated in a supplemental Table S3A.
Fig. 2.

Evaluation of PAN protein preparations. A, ESI MS analysis of PAN proteins under denaturing conditions. RP LC nanoESI raw mass spectra of denatured WT and K217A (KA) PAN proteins are shown in the left and right panels, respectively. Molecular ion envelopes spanning 24-74 charge states are seen (positions of charge states 30+, 40+, 50+, 60+, 70+ are annotated). The insets show data deconvoluted to a zero-charge state using MaxEnt software. B, Electron micrographs of frozen hydrated PAN proteins. PANWT and PANKA are shown in the left and right panels, respectively. The insets show four representative class averages enlarged twice for clarity. The first is the top view with a (pseudo) 6-fold symmetry, followed by oblique PAN hexamer. Scale bar, 50 nm.
The cryo-EM micrographs of frozen hydrated PAN proteins in TBS buffer showed a monodispersed population, which—as indicated by class averages—was composed of fully assembled hexamers, consistent with their (pseudo) 6-fold symmetry (10) (Fig. 2B).
The UV spectra of PAN proteins carrying WT P-loop motif (PANWT and arginine finger mutant PANR328A) showed higher absorbance at 260 nm than at 280 nm, pointing to the presence of nucleotides in those two preparations. In contrast, the UV profile of the PAN Walker A mutant (PANKA) was distinctly different showing maximum absorbance at 280 nm, consistent with the absence of nucleotides (supplemental Fig. S1A).
As native MS experiments were performed in 0.5 M ammonium acetate buffer and not in TBS buffer where the presence of PAN hexamers was demonstrated by cryoEM, we compared size exclusion chromatography elution profiles of PANWT in both TBS and AA buffers (supplemental Fig. S1B). The PAN elution profiles were essentially the same thus pointing to the comparable stability of PAN hexamer in both buffers.
Characteristics of Native Mass Spectra of PAN Hexamers
NanoESI MS spectra of native PANWT and PANK217A demonstrated narrow molecular ion distributions (Fig. 2A) that are typical for ESI spectra of folded protein structures. The data were analyzed with Massign software to detect and to model the component species of the spectra by using a process of simulation based on a Gaussian distribution of relative intensities of the charge state series (Fig. 3B).
Fig. 3.
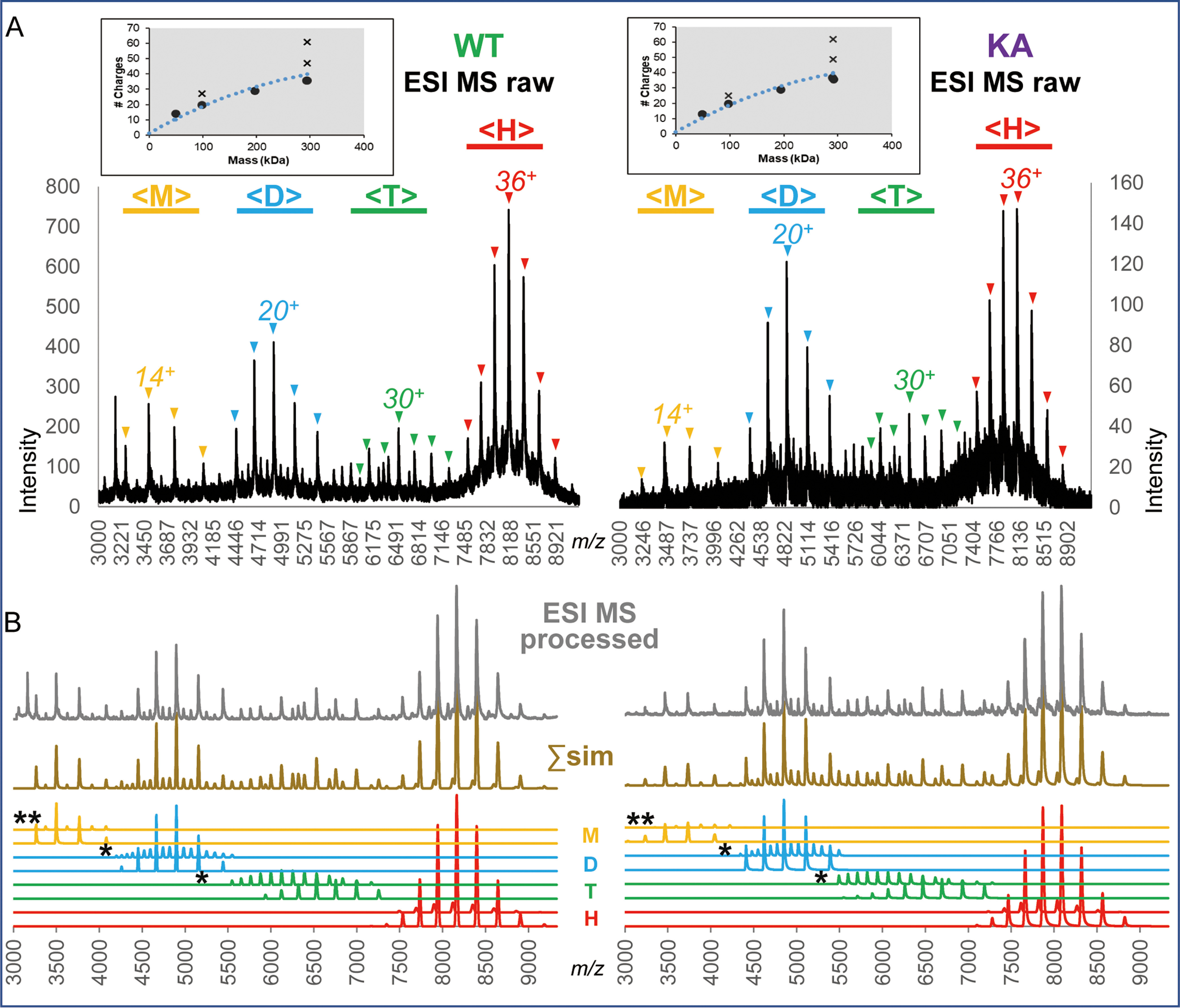
Native nanoESI mass spectra of separate analyses of PANWT (left) and PANKA (right). A, Raw mass spectra of proteins electrosprayed from 0.5 M ammonium acetate solution. The m/z ranges within the spectra where molecular ion envelopes consistent with monomers, dimers, tetramers and hexamers are observed are marked with yellow, blue, green and red horizontal bars and annotated with <M>, <D>, <T> and <H>, respectively. The charge state series corresponding to native-like monomers, dimers, tetramers, and hexamers are marked with “▾” and the most abundant charge state for each of the above species is annotated. The insets show the plots of relationships between the most abundant charge state and molecular mass of protein species analyzed under native ESI conditions (generated by Massign software): the blue dotted line shows a typical relationship between protein mass and the number of charges for the apex of native molecular ion envelope, experimental charge states are shown with black dots. Charge states observed for over-charged species, i.e. dimers within <M> range and hexamers within <D> and <T> ranges are annotated with “x” symbols. Of note, no species carrying a reduced number of charges for its mass were detected, pointing to the solution – rather than gas phase – origin of all the observed ions. B, The results of the Massign simulation of components detected in the native nanoESI MS analyses of PANWT and PANKA shown in panel A. The simulated spectra for species identified within the <M> (yellow), <D> (blue), <T> (green) and <H> (red) mass ranges are shown in the bottom part of the figure. The spectra marked with the “*” and “**” symbols show simulated spectra of over-charged hexamers and dimers, respectively. The spectra in the middle annotated Σsim and drawn in light brown represent a sum of the simulated spectra of components presented below. The top spectra are the experimental data that were processed, i.e. linearized, smoothed and background-subtracted using Massign software (see Methods).
The native MS spectra of PANWT and PANKA were typically dominated by the charge state series within a 7200–9300 m/z range (annotated with <H> in the Fig. 3A), which were consistent with being derived from hexameric (H) structures carrying 33-39 charges (dominant charge 36), as shown in red in the simulated spectra in the bottom of Fig. 3B. In addition to hexameric PAN species, nanoESI spectra contained varying levels of “satellite” narrow charge state distributions within m/z ranges of 3000–4200, 4200–5500, and 5500–7000, annotated with <M>, <D> and <T> (Fig. 3A), whose molecular masses were consistent with the products of PAN dissociation, i.e. the monomeric (M), dimeric (D), and tetrameric (T) species, respectively. The simulated spectra of these species are shown in Fig. 3B and marked in yellow, blue, and green for the <M>, <D>, and <T> m/z ranges, respectively. Relative levels of hexamer (H) versus satellite native-like structures tended to vary, sometimes significantly, between experiments and in some cases, within the course of a single data acquisition. Fig. 4 and supplemental Fig. S2A, S2B show some cases of significant changes in relative intensities of intact hexamer and products of its dissociation occurring during a single acquisition, generally accompanied by an abrupt alteration in TIC. We found no correlation between the extent of PAN dissociation and global instrumental parameters, e.g. cone and capillary voltages.
Fig. 4.

Examples of fluctuations in the extent of PAN hexamer dissociation observed during a single nanoESI MS acquisition. The relative abundance of the partially dissociated versus native hexamer levels of the PAN proteins varied significantly while maintaining the same instrumental parameters. On each panel, the top shows the Massign-processed spectra that were derived by summing up the scans marked at the total ion current (TIC) traces shown in the insets. Data showing low and high level of hexamer dissociation are shown in green and red, respectively. PANKA is shown here, more examples of both PANKA and PANWT dissociation are shown in supplemental Fig. S2A, S2B.
Charge states at the apices of molecular ion envelopes of the H as well as M, D, and T species correlated with their respective molecular masses in a manner characteristic for ESI spectra of folded globular proteins (31) (annotated with black dots in Fig. 3A insets) thus suggesting that the M, D and T species represented the native-like products of the solution- rather than gas phase-driven dissociation of PAN hexamer (see Discussion). Importantly, partially dissociated PAN species did maintain ligand binding. To this end, ligand loss that could be triggered by an increase in buffer concentration, was clearly detectable under the MS conditions used in the study. When using 1 M ammonium acetate buffer, the ADP concentration-dependent dissociation of PANWT-bound ADP was well resolved at a monomer and dimer levels (Fig. 5A, 5B) and resulting heterogeneity was clearly discerned at a tetramer level (data not shown). Under the same MS conditions, no nucleotide-protein dissociation was ever observed when using lower concentration of the same buffer (0.5 M), which ultimately was employed for all further studies. As PANKA species carried a mutated Walker A binding site making it defective in ADP binding, no parallel appearance of a PANKA-related peak at lower mass was observed in 1 M ammonium acetate buffer thus substantiating the reliability of using PANKA as a ADP-free control in these studies.
Fig. 5.

ESI MS analysis of PAN in 1 M ammonium acetate buffer. A, PANWT dimers in the presence of various levels of added ADP. Although the main species is consistent with PANWT dimer carrying 2 ADP molecules, the presence of a minor charge state envelope that is consistent with PANWT dimer carrying 1 ADP is seen in the presence of low concentration of added ADP (bottom and middle spectra, marked with red arrows). The minor component disappears in the presence of 5 μm ADP (top spectrum). B, Dimers detected for the mixture of PANWT and PANKA proteins in the absence of added ADP. The Massign-processed nano ESI MS data and a sum of the Massign-modeled major components of the spectra are shown on top and bottom parts of panel B. Signals corresponding to PANKA dimer carrying no ADP, PANWT dimer carrying 2 ADP and the PANWT carrying single ADP, the latter series annotated with red arrows are clearly resolved. Low intensity signals seen in the top part of panel B correspond to the overcharged hexamers and they were not modeled with Massign in this experiment.
The observed dissociation pattern of PAN hexamer (dimers and tetramers but not trimers or pentamers) is consistent with the known PAN architecture: PAN hexamer is built as a trimer of identical dimers (AB, CD, and EF), each dimer being assembled from two monomers of the dissimilar Pro91 configuration (cis for monomers A, C, E versus trans for monomers B, D, F) (10).
We note that species carrying higher number of charges than expected for the folded globular protein mass (annotated with “x” in Fig. 3A insets) were also detected at variable levels throughout the study. Specifically, over-charged hexamers spanning <D> and <T> m/z regions and over-charged dimers appearing within the <M> region, (annotated with “*” and “**” in Fig. 3B, respectively) were seen. As the over-charged species were not used to establish nucleotide-binding stoichiometry, their further description is provided in the supplemental material.
In summary, PAN spectra show the presence of native hexamers (H), which—in most experiments—constituted the major species accompanied by various levels of partially dissociated native-like as well as over-charged multimers. At the same time, our data point to the stability of protein-nucleotide interactions within all the observed species under MS conditions employed in the study. Therefore, we posit that notwithstanding the presence of the partially dissociated species in the spectra, not only did our analyses provide valid information on the stoichiometry of PAN-nucleotide binding but it benefited from the ability to analyze representative dimer species at the m/z region that afforded higher mass resolution than would otherwise be feasible to achieve for the higher m/z native hexamer region. To this end, whenever warranted by their good quality, we used the dimer- in addition to the hexamer-derived data to calculate PANWT nucleotide occupancy.
PANWT Hexamer Binds Six ADP Ligands
The PANWT and PANKA proteins were expressed and purified on multiple occasions and their mixtures analyzed to generate five high quality native nanoESI MS datasets that are presented here. An example raw MS spectrum shown in Fig. 6 demonstrates that data generated for a PAN protein mixture has the same general characteristics as previously observed in MS analyses of individual protein components, i.e. an appearance of partially dissociated species (ranges <M>, <D> and <T>) in addition to folded hexamers (range <H>). Insets show the Massign software-processed spectra (grey lines) and mock spectra derived by summing up contributions of the simulated deconvoluted species for native-like monomers in m/z region <M>, native-like dimers and over-charged hexamers in m/z region <D>, and native hexamers in m/z region <H> (shown in yellow, blue and red, respectively).
Fig. 6.
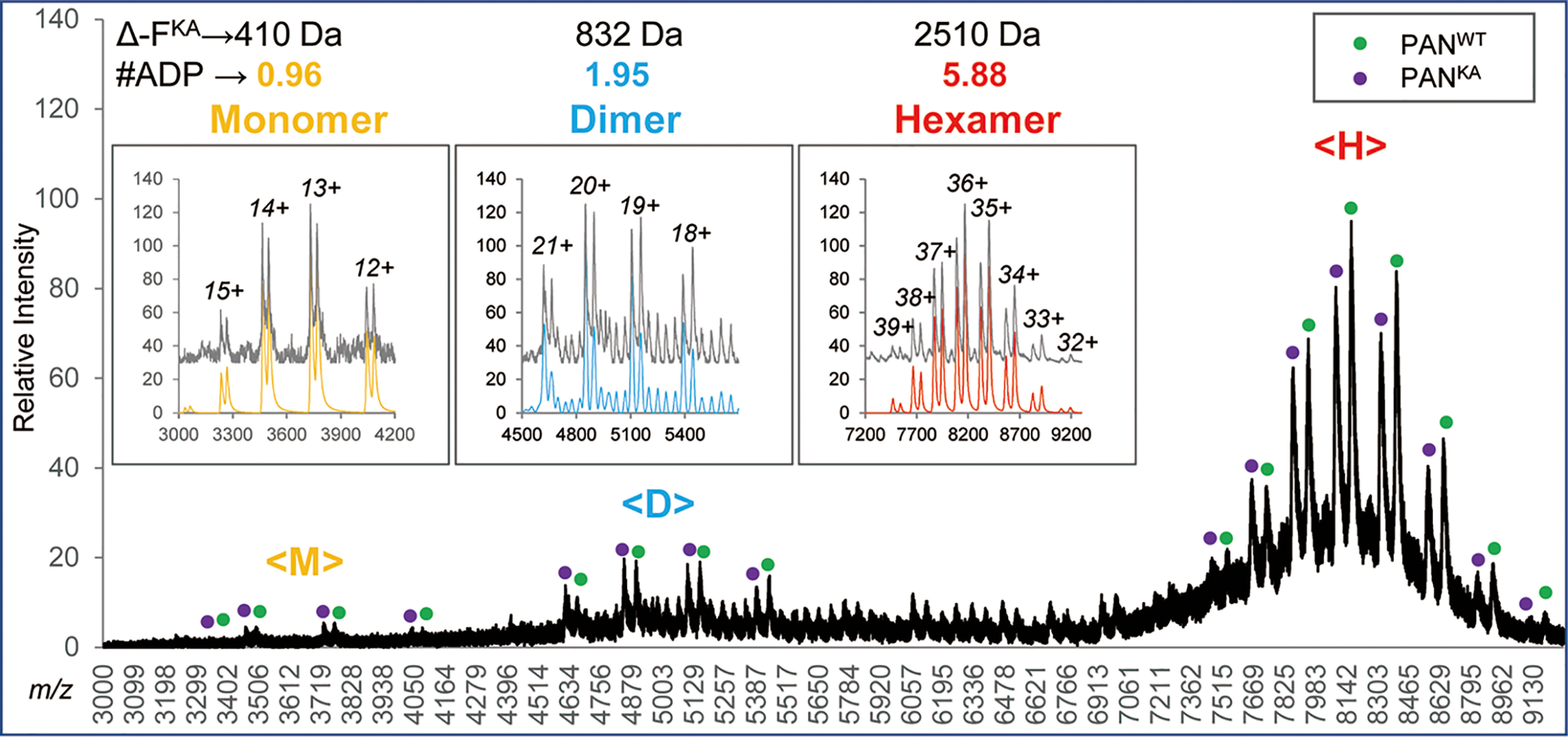
Native nanoESI MS analysis of a mixture of WT and K217A PAN proteins. Raw MS data are shown; molecular ions generated from WT and K217A proteins are annotated with green and purple dots, respectively. Insets show Massign-processed data for species detected within m/z ranges <M>, <D> and <H> of the spectrum: data that were linearized, smoothed and background-subtracted are shown in grey, the simulated spectra representing sums of protein components inferred through a process of Massign data deconvolution are shown in yellow, blue and red for the <M>, <D> and <H> m/z ranges, and show the presence of native-like monomers, dimers and hexamers, respectively. The over-charged hexamers within the <D> range were also detected and modeled.
We have analyzed the hexamer and dimer data by considering all potential forms of PANWT hexamer fully and partially loaded with either nucleotide or their mixtures (result summary and detailed calculations are shown in supplemental Tables S1A and S1D, respectively). All calculations were based on a premise that control protein PANKA carried no nucleotides, as supported by the experiments performed in 1 M ammonium acetate buffer (Fig. 5), and that the residual solvation level measured for PANKA could serve as a reliable approximation of the residual solvation of the WT protein after accounting for the presence of the KA mutation. Hence, we interpreted the experimental mass difference between the WT and KA proteins as representing bound nucleotides, as described in Materials and Methods. Formulas shown in the steps 6 and 7 listed in the Experimental Design and Statistical Rationale section of Material and Methods were used to calculate the number of ADP and/or ATP for PANWT carrying a single type or two types of nucleotides, respectively.
We considered the consistency between the residual solvation mass increments of KA and WT proteins as a measure of the reliability of the PANWT composition assignment. To this end, the ratios between the KA and WT protein residual solvation mass increments—normalized to the same protein molecular mass—were expected to be close to 1.0 and to be uniform across all five experiments. Out of a total of eighteen potential compositions of PANWT carrying 4, 5 and 6 nucleotides, four nucleotide compositions listed below in Table I stood out, as they met the above criteria the best, with CV of PANKA/PANWT residual solvation mass increment ratios across all five experiments below 10 and 20% for hexamer and dimer results, respectively.
Table I. Evaluation of ratios between the residual solvation mass increments for PANKA and PANWT of various potential nucleotide compositions across five experiments.
| #ADP | #ATP | Code | Hexamer |
Dimer |
||
|---|---|---|---|---|---|---|
| Avg. | CV | Avg. | CV | |||
| 0 | 5 | 0|5 | 1.09 | 5.3% | 1.10 | 15.3% |
| 1 | 4 | 1|4 | 1.02 | 5.5% | 0.98 | 14.7% |
| 2 | 3 | 2|3 | 0.97 | 7.7% | 0.89 | 16.3% |
| 6 | 0 | 6|0 | 1.12 | 6.0% | 1.15 | 16.2% |
Except for the composition (6|0) calling for the presence of 6 ADP (and 0 ATP), all other compositions carried only five nucleotides, thus calling for one of six potential binding sites to be empty. For those forms, we should be able to observe the presence of apo PANWT monomer at a level of ∼16.7% in addition to the ADP/ATP carrying species. Likewise, the presence of a singly occupied dimer at ∼33.3% level was expected in addition to a doubly occupied form(s) carrying various nucleotide combinations. We evaluated the spectra of WT protein on its own to avoid a potential partial overlap between the KA and WT-related signals (mass difference 57 Da per monomer). As it is shown in Fig. 7A, no signal of the apo PANWT form is seen in the monomer spectrum. There is also no evidence of the presence of two forms of PANWT carrying ADP and ATP, albeit the presence of a low level of the ATP form at the tailing part of the ADP-carrying peaks cannot be unequivocally dismissed. The dimer spectrum suggests that dimer carrying 2 ADP is the major species. The resolution at the dimer level does not allow for dismissing a possibility that the WT dimer carrying 2 ADP is accompanied by ADP/ATP and/or 2 ATP low abundance forms. However, the absence of the singly occupied dimers—either with ADP or with ATP—can conclusively be discerned (Fig. 7B). Taken together, these results strongly suggest that the presence of the PANWT hexamer carrying only 5 nucleotides is highly unlikely and hence we conclude that the PANWT is fully loaded with ADP.
Fig. 7.

Evaluation of the native monomer (panel A) and native dimer (panel B) regions of PANWT spectrum. PANWT spectra are shown in green, for comparison, spectra of PANWT/PANKA mixtures are shown in grey on the bottom of each panel. Should PANWT carry any of the top three nucleotide combinations listed in Table I, i.e. compositions with a total of 5 nucleotides, the presence of an apo form and a dimer carrying either single ADP (1) or ATP (2) is expected to be discernable in the spectrum—none were detected.
The results of the experiment shown in Fig. 6 showed the presence of 0.96, 1.95, and 5.88 ADP ligands per PANWT native-like monomer, native-like dimer, and native hexamer, respectively. Combination of the dimer- and hexamer-based results across all five data sets generated for three distinct PANWT and PANKA preparations pointed to the presence of 0.96 ± 0.04 ADP molecules per PANWT monomer (supplemental Table S1B). The ratio between the residual solvation for PANKA versus PANWT was 1.12 ± 0.07 and 1.15 ± 0.19 for the hexamer and dimer forms, respectively. The results of analysis of PANWT that was partially exchanged to TNP-ADP, described below (Fig. 8) provides further support for the nascent ADP presence in PANWT. We point that analyses of PANWT and PANKA mixtures reflected the presence of the host-derived ADP attached to the protein; addition of ADP to the buffer prior to MS analysis did not change the result (data not shown).
Fig. 8.

Native nanoESI MS analysis of a mixture of WT and KA PAN proteins following the process of ligand exchange with TNP-ADP reagent. The left and right panels show the data of the selected charge states for the monomer (13+ and 14+) and dimer (20+ and 21+) species, respectively. The Massign-processed experimental data are shown in grey on top. At the bottom, the Massign-simulated spectra of each of the software-deconvoluted components of the mixture are shown in violet for KA-derived molecular ions and various shades of green for WT-derived ions. Molecular ions superscripts indicate the type and number of bound ligands.
To verify the ability of the WTPAN to bind six ADP ligands, we performed nucleotide exchange experiments using fluorescently labeled ADP analogs: mant-ADP (Mr 560.4) and TNP-ADP (Mr 637.3). The nucleotide-exchanged samples were mixed with internal standard PANKA and analyzed by nanoESI MS. The number of exchanged nucleotides was calculated as shown in the step 7 listed in the Experimental Design and Statistical Rationale section of Material and Methods. The nucleotide exchange results varied across experiments to deliver 57.3 ± 20.6% (n = 5) and 57.5 ± 6.5% (n = 4) conversation rates for mantADP and TNP-ADP, respectively (supplemental Tables S2 and S3). An incomplete ligand exchange was apparent in view of the significantly higher residual solvation observed for the KA mutant versus that calculated for the WT protein carrying full complement of ADP analogs of higher mass than ADP (supplemental Tables S2 and S3). In one of the TNP-ADP exchange experiments, high signal intensity and good resolution within the M and D regions of the spectrum allowed for detecting discrete forms of incompletely exchanged PANWT monomers and dimers (Fig. 8A, 8B). Fig. 8A demonstrates the presence of two PANWT-related monomer species (WTADP and WTTNP) spaced by 211 Da, with their mass difference closely reflecting molecular mass difference of 210.1 between ADP and its TNP-ADP analog. In accord with the PANWT ligand content detected on a monomer level, three discrete PANWT- related dimer species of similar abundances were observed, including a hybrid dimer carrying one ADP and one TNP-ADP molecule (WTADP-WTTNP, Fig. 8B).
In conclusion, the above experiments demonstrated that WT PAN protein carried 6 ligands and that it was ADP rather than ATP that was bound to a nascent protein. It is noteworthy that across all experiments described above there was no indication of PANKA protein carrying any ligands, neither from the expression host nor captured from a solution carrying excess of free ADP or its analogs (data not shown). Likewise, no species consistent with PANWT protein carrying more than one ADP (or its analog) per monomer were seen. Notably, no PANWT-PANK217A hybrid multimers were ever observed across all experiments performed in the study.
Both PANWT and Walker A Mutant Bind AMP-PNP
We have used AMP-PNP, a nonhydrolyzable ATP analog, to establish stoichiometry of ATP binding to PAN. PANWT was mixed with AMP-PNP for 15 min on ice and the reaction mixture was filtered through a fast spin column pre-equilibrated with 0.5 M ammonium acetate; the filtered PANWT was then mixed with PANKA prior to MS. Three top spectra in Fig. 9 show the results of native MS analysis of the PANKA and PANWT mixture, PANKA and PANWT separately each carrying AMP-PNP. For comparison, the spectrum of the PANKA and PANWT mixture carrying the expression host-derived ADP is shown at the bottom. The appearance of a ladder of closely spaced species in the spectra for PANKA-AMP-PNP containing samples suggests that in contrast to ADP and its analogs, which interacted exclusively with the WT protein, AMP-PNP did bind to the PANKA mutant as well. There is no indication of PANWT carrying varying numbers of AMP-PNP at the achieved resolution (the third spectrum from the top). Although because of limitations of resolution we cannot dismiss a possibility that during analysis of the PANKA and PANWT mixture, Walker A mutant was stripping PANWT of some of its ligands, we propose that the more likely source of a nucleotide was a leftover of AMP-PNP in a flow-through after spin-column buffer exchange of the PANWT- AMP-PNP reaction mixture (∼2% free nucleotide according to the manufacturer's specifications).
Fig. 9.

Comparison of PANKA spectra in the presence and absence of AMP-PNP. Massign software processed experimental data for the three most abundant charge states of native hexamer molecular ion envelopes are shown. Top three spectra show data for PANWT+PANKA (WT/KA), PANKA (KA) and PANWT (WT), each carrying AMP-PNP. For comparison, the bottom spectrum shows WT/KA mixture carrying ADP. A ladder of KA protein carrying various numbers of AMP-PNP is seen at the top two spectra.
Fig. 10A–C summarizes the results of Massign deconvolution analysis of the [PANWT + PANKA + AMP-PNP] ESI spectrum. Fig. 10A shows the presence of PANKA (KA) carrying peaks P1-P4 in addition to PANWT fully loaded with ligands (peak P5). The composition of PANKA and PANWT-derived species is annotated as KA or WT followed by (N1|N2) where N1 and N2 stand for the number of bound ADP and AMP-PNP nucleotides, respectively (Fig. 10C). Although peak P5 representing PANWT protein appears as a single species, its heterogeneity because of the presence of low level of unresolved contaminants, including potential PANKA protein carrying 6 AMP-PNP, i.e. KA (6|0) cannot be ruled out. Of note, relative abundance distribution of PANKA-associated peaks (P1-P4) suggests that small amount of KA (0|6) might be present (Fig. 10B). Likewise, the presence of unresolved partially converted PANWT carrying a mixture of ADP and AMP-PNP ligands within P5 cannot be ruled out at achievable resolution. Thus, we used residual solvation mass increments calculated for P1-P4 components of the composition shown in Fig. 10C as a reference to approximate a potential composition of PANWT represented by peak P5 and hence, an efficiency of the ADP to AMP-PNP ligand exchange.
Fig. 10.

Detailed analysis of the PANWT/PANKA exchange with AMP-PNP. A, The experimental data (top) and simulated spectra of the Massign-deconvoluted components in the WT/KA AMP-PNP exchanged sample (bottom). The identified components P1-P5 are annotated for the 36+ charge state molecular ion on the experimental spectrum. B, Massign deconvolution-derived relative abundance of species modeled in peaks P1-P5 for Exp. 2. C, Analysis of the residual solvation data generated for the Massign-simulated species P1-P5. The composition of PANKA- and PANWT-derived species is annotated as KA or WT followed by (N1|N2) where N1 and N2 stand for the number of bound ADP and AMP-PNP nucleotides, respectively. Residual solvation values were normalized to molecular masses of each species to generate residual solvation mass increments. Species P1-P4 contain KA protein carrying various numbers of bound ligands (listed at the top of panel C). Data for P1-P4 residual solvation increment values are shown with circles of various shades of grey for experiments 1 and 2 (Exp.1 and Exp.2). The average values for the KA-derived residual solvation mass increments were 0.07%± 0.03% and 0.09 ± 0.02% for the Exp.1 and Exp. 2, respectively and are shown as yellow circles. The residual solvation mass increment values calculated using P5 experimental molecular mass and assuming various possible components of this peak are shown as green and purple squares for WT- and KA-related species, respectively.
As molecular mass of ADP is lower than that of AMP-PNP by 79 Da, a contribution of a partially exchanged PANWT to P5 would have resulted in lowering its experimental mass thus leading to an apparent increase in the measured residual solvation mass increment that was calculated for the fully exchanged PANWT; likewise, because of the mass decrement of 6 × 57 Da per each PANKA hexamer as compared with the WT protein, an admixture of unresolved KA (0|6) within P5 would have had the same effect of decreasing experimental mass of P5 and thus increasing solvation-related mass increment calculated for the PANWT carrying 6 AMP-PNP ligands.
We have calculated average value of residual solvation mass increment in the Exp. 1 and Exp. 2 using data for peaks P1-P4 containing PANKA loaded with 2 to 5 AMP-PNP. We then used experimental mass of peak P5 to calculate residual solvation mass increments for several potential components of this peak. The following potential species were evaluated: PANWT carrying six nucleotides fully WT (0|6), and partially (WT (1|5); WT (2|4) exchanged for AMP-PNP, KA (0|6) and for comparison, the PANWT prior to exchange, i.e. WT (6|0). We have also considered WT (0|5), i.e. PANWT carrying only 5 AMP-PNP nucleotides. The results are presented in Fig. 10C.
As expected, all partially resolved PANKA-AMP-PNP species (peaks P1-P4) demonstrated similar residual solvation mass increments (annotated with circles of various shades of gray), with average values of 0.07 ± 0.03% and 0.09 ± 0.02% (annotated with yellow circles) for experiments 1 and 2, respectively. The potential components of P5 containing PANWT and PANKA are represented with green and purple squares, respectively. The level of residual solvation mass increments calculated for WT (0|6), WT (1|5), and WT (2|4) was within two standard deviations from the experimental average observed for PANKA species within peaks P1-P4 (supplemental Table S4A). This finding combined with a high likelihood of the presence of low levels KA (6|0) species within P5 pointed to high efficiency of ADP to AMP-PNP ligand conversion for the WT protein. We thus concluded that WT PAN has a capacity to carry a total of 6 nucleotides, most likely 5-6 AMP-PNP ligands, in accord with its previously shown capacity to bind 6 ADP ligands. Of note, residual solvation mass increment calculated for a form of PANWT carrying a total of 5 ATP analogs, i.e. WT (0|5), as suggested by the recent cryo-EM study (21) was way outside two standard deviations from the experimental average in both experiments.
Examination of the spectra at the monomer and dimer levels confirmed that PANKA carried AMP-PNP, as the PANKA dimer with 1 AMP-PNP (peak 2) was clearly discerned in addition to apo PANKA monomer and dimer (peak 1) (see supplemental Fig. S3). Limitations of resolution did not allow for separating PANKA monomer carrying 1 AMP-PNP as well as PANKA dimer carrying 2 AMP-PNP from other possible components of the mixture. For the same reason, we were not able to unequivocally confirm or dismiss the potential presence of PANWT carrying ADP as the masses of PANWT-ADP and PANKA-AMP-PNP monomers and dimers were too close to afford their resolution in our experiments (mass difference 22 Da per monomer). The monomer and dimer spectra suggested the possible presence of PANKA carrying small level of nonspecifically bound AMP-PNP (2 and 3 bound for monomer and dimer, respectively), albeit no further experiments were performed to confirm the composition of the detected MS peaks.
To confirm a phenomenon of ATP-analog binding to PAN protein carrying Walker A mutation, we generated a double mutant PANK217A/R328A. The double mutant interacted with nucleotides in a similar manner to that observed for PANKA, i.e. it did not bind ADP and it did bind AMP-PNP (supplemental Fig. S4 bottom and top spectrum, respectively). However, the degree of AMP-PNP ligand binding to the double mutant was much lower, with a maximum of three ligands per hexamer, versus five to six ligands seen in Fig. 10 for PANK217A. We note that although distinct components of a double mutant-ATP analog ladder were not resolved, the presence of multiple species was apparent upon comparing peak widths of the mutant versus WT protein, as shown for two most abundant charge states (supplemental Fig. S4A). A ratio between peak width (full width at half maximum, FWHM) calculated for the six most abundant charge states (33+ - 38+) of a double mutant protein to its WT counterpart was 1.0 ± 0.14 and 1.5 ± 0.15 in the absence and presence of AMP-PNP (t test = 1.49E-04), respectively, (supplemental Fig. S4B).
DISCUSSION
We divide the result discussion to two sections: the first one focused on the structural aspects of PAN and the second one addressing the issues regarding the methodological aspects of the study.
ATPase Structure
The characteristics of nucleotide binding to PAN revealed by the mass spectra provide information that is critical to unveiling the proteasome ATPase hexamer cooperation mechanisms. Proteasome is a large protein assembly that removes misfolded proteins and short-lived signaling proteins (2, 4). The eukaryotic 26S proteasome consists of a 20S proteasome core particle (CP) and a 19S regulatory particle (RP). Six distinct Rpt subunits form the base of RP that interfaces with CP. PAN shares high homology with Rpt subunits and therefore it serves as a simpler model for the Rpt hexamer. Determining the precise number and type of nucleotides bound to proteasomal ATPases at a given state is important to understanding the details of the cooperation mechanism.
An earlier biochemical study of PAN and 26S proteasome suggested a pairwise model, in which a total of 4 nucleotides were bound to a hexameric proteasomal ATPase at any giving time, with two opposite subunits carrying ATP, another two opposite subunits carrying ADP and two remaining subunits in an apo state (14). Our results revealed that the purified WT PAN (PANWT) hexamer carried six tightly bound ADP molecules and that nascent ADP ligands could be replaced with ADP and ATP analogs. Of note, never did we observe an apo-form of PANWT. Specifically, our study points to the full occupancy of all six nucleotide-binding sites within the PANWT hexamer in both, the ADP- and ATP-binding states. We point out however, that we analyzed PAN in the absence of its functional partner, i.e. proteolytic core particle 20S and hence our results might not represent the PAN nucleotide loading in its active state in vivo. Binding of ADP and its analogs is not likely to be affected by the exclusion of CP because the PAN-20S proteasome does not form in the absence of ATP (9). However, it is conceivable that PAN-20S interaction might alter ATP binding stoichiometry. Our results point to binding of a total of 6 nucleotides, either 6 AMP-PNP or 5 AMP-PNP and 1 ADP (or potentially 4 AMP-PNP and 2 ADP). According to our data, the presence of 5 AMP-PNP with one nucleotide-free site is unlikely (Fig. 7C). Taken together, our results support a sequential model of the AAA+ ATPase hexamer, where nucleotide can bind to all subunits simultaneously and the cooperation seems closely related to the synergy between neighboring subunits in binding ATP.
We also show that although the Walker A mutant PANK217A hexamer carried no nascent ligands, it could be loaded with multiple copies of AMP-PNP. This unexpected AMP-PNP binding to Walker A mutant implies that the arginine finger might play a key role in ATP binding, which differs from the notion that arginine finger is not involved in ATP binding in a recent study of PAN (15). Indeed, introduction of an additional R328A mutation on one of the arginine finger residues reduced such binding, as shown by native MS of the protein mixture of PANWT and PANK217A/R328A treated with AMP-PNP (supplemental Fig. S4).
Our data are consistent with the recently published cryo-EM studies on AAA+ ATPases, which demonstrated the presence of either 5 or 6 bound nucleotides. Specifically, the single molecule cryo-EM study of A. fulgidus PAN-proteasome complex in the presence of ATPγS (nonhydrolyzable ATP analog) at 5.9 Å resolution suggested the presence of a total of 5 nucleotides, with 4 ATPγS occupying “closed” pockets, ADP in the “intermediate” pocket and leaving “open” pocket nucleotide-free (21). A. fulgidus PAN shares 57.54% identity with M. jannaschii PAN that we have studied. However, three independent cryo-EM studies of human 26S proteasome at high resolution (12, 19, 20) revealed that all six Rpt subunits in the AAA+ ATPase hexamer carried nucleotides: whereas Huang et al. (12) and Chen et al. (20) refrained from specifying the types of bound nucleotides, Schweitzer et al. (19) reported that Rpt6 subunit carried ADP and the remaining subunits were tentatively assigned as carrying ATP. Cryo-EM structure of P97 at 2.4 Å revealed that both D1 and D2 rings carried 6 ADP, and in the presence of ATPγS the exchange of ADP->ATPγS within either D1 only or within both D domains led to a generation of coexisting distinct conformations that were resolved at 3.2-3.3 Å resolution (18). Interestingly, the recent native ESI MS study demonstrated the presence of up to 11 nucleotides in p97 (32).
Our results are consistent with the hexameric structure of PAN, as demonstrated by cryo-EM data (9, 21). Early reports suggested a dodecameric structure of PAN, in view of its elution volume in gel filtration chromatography that was consistent with a mass of a dodecamer (>600 kDa) rather than that of a hexamer (∼300 kDa) (7). A notion of the possible weak association of PAN hexamers to form dodecameric structures was supported by an observation of PAN dodecamers in some of the cryo-EM studies on two species of archaeal PAN reported by Medalia et al (22). However, only hexameric structures were seen for the same proteins in the native MS analysis performed by the same authors. We note that on occasion we detected very weak signals that were consistent with PAN dodecamer structure (data not shown). However, we attribute their formation to an artifact of nonspecific protein-protein interactions in ESI droplets rather than to a representation of a native dodecamer arrangement. The emitters of the diameter of ∼1 μm that we used in this study were demonstrated to generate relatively long-lived ESI droplets thus encouraging nonspecific interactions at final stages of droplet evolution when final protein concentration gets very high (33).
The data pointing to the PAN (14, 15) and ClpX (13) binding a maximum of 4 nucleotides are baffling in view of the results derived from cryo-EM and our native MS studies. The somewhat trivial explanation might be that biochemical methods that were used relied on two separate calibration routines: one for a protein and another one for a nucleotide, each relying on their own set of standards, thus compounding the likelihood of errors. It is well established that the typically used dye-based protein determination assays are highly dependent on sample composition, protein sequence and the relative abundance of specific amino acid residues within the sequence (34). To explore this possibility, we measured protein concentration in two PAN preparations by the three most often used protein assays (supplemental Fig. S5). Using the same vial of BSA solution as a standard, the concentration of the same PANWT sample was determined to be 4.7 mg/ml by BCA assay, 8.2 mg/ml by Bradford assay, and 5.5 mg/ml by Lowry assay. Hence, depending on the assay used, an underestimation or overestimation of the nucleotide/protein stoichiometry is feasible. On the other hand, the recent cross-linking experiments performed using PANM87C mutant provided a surprising outcome of an asymmetric configuration of three identical coil-coil (CC) domains of PAN, with each CC adopting a different conformation (35). Although these results support a notion of a pairwise model of PAN carrying a total of 4 nucleotides (14), they are contradicted by the X-Ray analyses that were performed on truncated forms of PAN from A. fulgidus (11) and from M. jannaschii (AA 74-150) (10), which pointed to an in-register arrangement of 3 CC domains. Clearly, despite the progress in proteasome-related studies, some aspects of AAA+ ATPase structure and function remain unresolved.
Generation of over-Charged Multimers and Partial Hexamer Dissociation in the Native nanoESI MS Spectra of PAN Hexamer
Many published native ESI mass spectra of protein-protein interactions show the presence of partially dissociated structure(s) of complexes in addition to their intact forms. In the great majority of these studies, the processes of protein complex dissociation either in the bulk solution prior to ESI MS or protein complex molecular ion fragmentation in the gas phase are invoked as the origin of the dissociated structures. Here we argue, that in case of PAN hexamers in our study, neither bulk solution nor gas phase fragmentation are responsible for the observed phenomena. Rather, we hypothesize that the conditions affecting very late stages of droplet decomposition or an “intermediate regime” (36) of the ESI process lead to a partial dissociation and potentially, to a generation of over-charged structures within ESI droplets prior to molecular ion transfer to the gas phase.
If the PAN dissociation were to happen in the gas phase, it would most likely have proceeded in the ESI source and would have been driven by the collisionally induced dissociation (CID). Yet, the types of the observed dissociation products are inconsistent with the typical mechanism of the protein complex disassembly under CID conditions, which involves an asymmetric separation of charge and mass (37, 38). Thus, the products of typical CID would have been an unfolded highly charged monomer and a charge-stripped pentamer rather than the observed folded tetramers, dimers, and monomers. Although atypical symmetric CID driven fragmentation was observed, we note that a generation of monomers and pentamers under CID conditions was demonstrated in the previous native MS study on PAN (22). In addition, the fact that we observed no correlation between the extent of dissociation and cone voltage (data not shown), highly supports a notion that in-source CID was not responsible for the PAN complex disassembly.
As argued above, the characteristics of charge state distributions of the partially dissociated “satellite” species points to their globular structures and solution origin (Fig. 3A insets). However, the question remains whether the dissociation happened in the bulk solution prior to ESI experiment or it was somehow triggered during the ESI process. The arguments against the bulk solution as the origin of PAN hexamer dissociation are as follows (1) the cryo-EM analyses (Fig. 2B) that were performed in TBS buffer demonstrated hexamers as predominant if not exclusive component of PAN protein preparations, and the same gel-filtration profiles of PAN in 0.5 M ammonium acetate and in TBS buffer were observed (supplemental Fig. S1B), jointly argue against any significant dissociation of PAN hexamers in bulk solution that would correspond to the relatively high level of “satellite” sub-oligomers observed in some of native MS; (2) no hybrid multimers resulting from the likely process of re-association of the partially dissociated WT and mutant PAN were ever detected while analyzing equimolar protein mixtures, even when potential constituents of such hybrids, i.e. monomers and dimers were observed in the spectra at substantial levels—of note, generation of hybrid hexamers between WT and mutant AAA+ ATPase species was reported (39); (3) levels of dissociated products might vary for the repeated analyses of the same sample while employing different emitters; (4) significant fluctuation of relative intensities of dissociated versus intact products were observed on a number of occasions during the single acquisition, with either a decrease or an increase of the levels of dissociation products over the acquisition time (Fig. 4 and supplemental Fig. S3). Taken together, the data suggest that ESI-droplets are the origin of the observed dissociation products: whereas some level of dissociation was always present across all the acquisitions, additional not yet identified factor(s) must have affected the generation, stability and/or manner of disintegration of ESI droplets, which in turn swayed the extent of the dissociation process.
Acidification of a droplet solution under conditions of positive ion ESI is a well-established phenomenon and it can only be partially abated by using volatile buffers of high concentration (40). Hence the stability of the complex (e.g. PAN hexamer) that is assessed in the bulk solution does not necessarily translate to the conditions within ESI droplets. Whether the ESI-related droplet acidification contributes to the PAN hexamer dissociation or not, the significant variability in the extent of this process that we have observed across multiple experiments suggests that additional factors might also be at play, specifically the forces that affect evolution of a droplet during ESI (41). As compared with the bulk solution, an environment of a nanodroplet “in the throes of what is now known as a Rayleigh Instability, and sometimes referred to as a Coulomb explosion” (42) (see Fig. 4 caption on page 3885) has distinct properties, e.g. high concentration of ions relative to that of the bulk solution; strong electric field because of the presence of charged macromolecules or complexes; a large surface to volume ratio (41). These unique nanodroplet characteristics impart significant changes onto chemical processes that occur in these small volume “reactors” (43) as opposed to the bulk solution. For example, chemical reactions, including unimolecular processes (44) occur much faster in nanodroplets; cases of acceleration up to the level of 10E6 were reported (45, 46). By the same token, the rate of protein complex dissociation might also be greatly enhanced in the course of ESI-driven droplet evolution from its initial bulk solvent-like stage upon release from a capillary emitter to its final offspring phase that gives rise to the gas phase molecular ions. Consequently, it is likely that even small changes in the droplet evolution process that might affect the rate of the offspring droplet generation, their size, final pH or morphology might be accompanied by the large differences in the reaction rates within these droplets, thus in turn leading to distinct differences in substrate-to-product ratios. The absence of hybrid multimers that potentially could have been generated via a re-assembly of the released subunits suggests that the dissociation might occur at the late stages of the droplet transformation process when a nanodroplet carries a single or a few analytes.
Although we have no clear-cut experimental evidence to support the above reasoning, we point to several studies that in our view give the presented hypothesis some credence. The sensitivity of noncovalent interactions stabilized by electrostatic forces to the ESI conditions was demonstrated by Gabelica et al. (47) in analysis of protein-DNA complexes. The authors found that in addition to the cone and capillary voltages and capillary-cone distance, the way in which the capillary was cut was also a crucial parameter in controlling the extent of complex dissociation, thus suggesting that minute changes to the capillary orifice, which potentially might occur during the spray might greatly affect ESI performance. Likewise, distinct differences in ESI spectra of noncovalent assemblages were seen in response to changes in a spraying mode (cone-jet versus pulsating mode), likely associated with the enhanced droplet acidification produced in the former mode (48), again suggesting that spray mode changes triggered by fluctuations in emitter performance are likely to result in variability of ESI mass spectra. The presence of products consistent with in-droplet fragmentation of model organic molecules (49), and products of protein complex dissociation occurring “in-solution or during the electrospray process at the front of the instrument” (50) (see page 2805) were reported. The recent MD simulation study outlined a mechanism for the in-droplet dissociation of a transient protein complex in response to pH changes (51). According to the “opinion piece” recently published by Chingin et al. (52) (see Concluding remarks), “the chemistry of proteins confined in a microenvironment proceeds in a strikingly different fashion from that in the bulk solution. Recent experimental evidence indicates that, as far as reaction rate is concerned, 1 ms in a microdroplet can be equal to seconds and even minutes or hours in solution”, thus disputing the commonly accepted notion “that the shift in the protein complex equilibrium in charged microdroplets is not likely to occur.”
In summary, we posit that the observed in-droplet dissociation of PAN hexamers has likely been driven by a number of factors affecting droplet transformation in ESI, i.e. controlling an “intermediate regime” (36) of the process, including but not necessarily limited to the level of droplet solution acidification. In view of the lack of a clear correlation between the extent of dissociation and global instrumental parameters, e.g. cone and capillary voltages, occasional deviations in emitter performance (because of their manufacturing and/or to aging effects in the course of the spray) likely represent the major trigger of the observed variability in the extent of PAN dissociation in the native nanoESI mass spectra.
CONCLUSIONS
Our results demonstrate that native MS can be utilized to determine the stoichiometry of nucleotide binding to an AAA+ ATPase hexamer. Its high precision and capability to maintain macromolecules in a native-like state allowed us to simultaneously measure the masses of protein and bound nucleotides whose molecular weights differ by ∼ three orders of magnitude. Use of a Walker A mutant as an internal standard minimized the impact of residual solvation on mass measurement accuracy, as both PANKA and PANWT were subjected to identical ESI conditions thus allowing us to decouple the effects of a ballast of extraneous adducts, expected to be roughly the same for both proteins, from those of the PANWT-specific binders. We propose that a generation of partially dissociated species observed in this study was driven by the chemistry of ESI droplets at the final stages of their evolution, rather than by the hexamer-dimer-tetramer equilibria in the bulk solution. Although the triggers of the dissociation are presently unknown, we note that this process did not adversely affect the integrity of the results and it actually aided our investigation by supporting information gathered on a hexamer level with the data generated for monomers, dimers and tetramers.
DATA AVAILABILITY
All raw MS files and associated Massign-analyzed result files as well spectra lists were uploaded to the UCSD Center for Computational Mass Spectrometry, MassIVE with the dataset identifier MSV000085155. The data set can be downloaded on line using the link ftp://massive.ucsd.edu/MSV000085155/. Refer to the supplemental Table “Guide to Results-Massive Data Deposition” to link specific results presented in figures and tables to their associated data.
Supplementary Material
Acknowledgments
We are indebted to Dr. Nina Morgner for her help in all matters related to the use of Massign software, which she had developed. We acknowledge Dr. Susan Fisher and colleagues at the UCSF Sandler-Moore Mass Spectrometry Core Facility for their support for these studies. Synapt G2 HDMS mass spectrometers (Waters) was funded by the National Institutes of Health High End Shared Instrumentation Program (Grant 1S10RR029446-01, Witkowska PI). We thank Roger Cook and Nariman Naber for help on electronic paramagnetic resonance spectrometry study of PAN.
Footnotes
This article contains supplemental data
Funding and additional information—This study has been supported in part by a grant from National Institutes of Health (R01GM082893) and a grant from UCSF Program for Breakthrough Biomedical Research (Opportunity Award in Basic Science) to YC and by grants from NIGMS (R01GM51923-13). YC is an Investigator of Howard Hughes Medical institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- PAN
- proteasome activating nucleotidase
- MS
- mass spectrometry
- ESI
- electrospray ionization
- TNP-ADP
- 2'-(or-3')-O-(trinitrophenyl)adenosine 5'-diphosphate
- mant-ADP
- 2'-(or-3')-O-(N-methylanthraniloyl)adenosine 5'-diphosphate
- AMP-PNP
- adenosine 5'-(β,γ-imido)triphosphate
- ATPγS
- adenosine 5'-O-(3-thio)triphosphate
- Rpt
- regulatory particle of triple-ATPase.
REFERENCES
- 1. Neuwald A. F., Aravind L., Spouge J. L., and Koonin E. V. (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 2. Tanaka K. (2009) The proteasome: Overview of structure and functions. Proc. Jpn. Acad, Ser. B, Phys. Biol. Sci. 85, 12–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wendler P., Ciniawsky S., Kock M., and Kube S. (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta - Mol. Cell Res. 1823, 2–14 [DOI] [PubMed] [Google Scholar]
- 4. Finley D., Chen X., and Walters K. J. (2016) Gates, channels, and switches: elements of the proteasome machine. Trends Biochem. Sci. 41, 77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walker J. E., Saraste M., Runswick M. J., and Gay N. J. (1982) Distantly related sequences in the a- and f-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Besche H., Tamura N., Tamura T., and Zwickl P. (2004) Mutational analysis of conserved AAA+ residues in the archaeal Lon protease from Thermoplasma acidophilum. FEBS Lett. 574, 161–166 [DOI] [PubMed] [Google Scholar]
- 7. Zwickl P., Ng D., Woo K. M., Klenk H. P., and Goldberg A. L. (1999) An archaebacterial ATPase, homologous to ATPases in the eukaryotic 26 S proteasome, activates protein breakdown by 20 S proteasomes. J. Biol. Chem. 274, 26008–26014 [DOI] [PubMed] [Google Scholar]
- 8. Wilson H. L., Ou M. S., Aldrich H. C., and Maupin-Furlow J. (2000) Biochemical and physical properties of the Methanococcus jannaschii 20S proteasome and PAN, a homolog of the ATPase (Rpt) subunits of the eucaryal 26S proteasome. J. Bacteriol. 182, 1680–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith D. M., Kafri G., Cheng Y., Ng D., Walz T., and Goldberg A. L. (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell. 20, 687–698 [DOI] [PubMed] [Google Scholar]
- 10. Zhang F., Zhang F., Hu M., Hu M., Tian G., Tian G., Zhang P., Zhang P., Finley D., Finley D., Jeffrey P. D., Jeffrey P. D., Shi Y., and Shi Y. (2009) Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol. Cell. 34, 473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Djuranovic S., Hartmann M. D., Habeck M., Ursinus A., Zwickl P., Martin J., Lupas A. N., and Zeth K. (2009) Structure and Activity of the N-Terminal Substrate Recognition Domains in Proteasomal ATPases. Mol. Cell. 34, 580–590 [DOI] [PubMed] [Google Scholar]
- 12. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 18, 1–10 [DOI] [PubMed] [Google Scholar]
- 13. Hersch G. L., Burton R. E., Bolon D. N., Baker T. A., and Sauer R. T. (2005) Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: Allosteric control of a protein machine. Cell 121, 1017–1027 [DOI] [PubMed] [Google Scholar]
- 14. Smith D. M., Fraga H., Reis C., Kafri G., and Goldberg A. L. (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim Y.-C., Snoberger A., Schupp J., and Smith D. M. (2015) ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat. Commun. 6, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bochtler M., Hartmann C., Song H. K., Bourenkov G. P., Bartunik H. D., and Huber R. (2000) The structures of HslU and the ATP-dependent protease HslU-HslV. Nature 403, 800–805 [DOI] [PubMed] [Google Scholar]
- 17. Wang J., Song J. J., Franklin M. C., Kamtekar S., Im Y. J., Rho S. H., Seong I. S., Lee C. S., Chung C. H., and Eom S. H. (2001) Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9, 177–184 [DOI] [PubMed] [Google Scholar]
- 18. Banerjee S., Bartesaghi A., Merk A., Rao P., Bulfer S. L., Yan Y., Green N., Mroczkowski B., Neitz R. J., Wipf P., Falconieri V., Deshaies R. J., Milne J. L. S., Huryn D., Arkin M., and Subramaniam S. (2016) 3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schweitzer A., Aufderheide A., Rudack T., Beck F., Pfeifer G., Plitzko J. M., Sakata E., Schulten K., Förster F., and Baumeister W. (2016) Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. U S A. 113, 7816–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen S., Wu J., Lu Y., Ma Y. B., Lee B. H., Yu Z., Ouyang Q., Finley D. J., Kirschner M. W., and Mao Y. (2016) Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U S A. 113, 12991–12996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Majumder P., Rudack T., Beck F., Danev R., Pfeifer G., Nagy I., and Baumeister W. (2019) Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc. Natl. Acad. Sci. U S A. 116, 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Medalia N., Sharon M., Martinez-Arias R., Mihalache O., Robinson C. V., Medalia O., and Zwickl P. (2006) Functional and structural characterization of the Methanosarcina mazei proteasome and PAN complexes. J. Struct. Biol. 156, 84–92 [DOI] [PubMed] [Google Scholar]
- 23. Van Den Heuvel R. H. H., and Heck A. J. R. (2004) Native protein mass spectrometry: From intact oligomers to functional machineries. Curr. Opin. Chem. Biol. 8, 519–526 [DOI] [PubMed] [Google Scholar]
- 24. Lössl P., Snijder J., and Heck A. J. R. (2014) Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 906–917 [DOI] [PubMed] [Google Scholar]
- 25. Mehmood S., Allison T. M., and Robinson C. V. (2015) Mass Spectrometry of Protein Complexes: From Origins to Applications. Annu. Rev. Phys. Chem. 66, 453–474 [DOI] [PubMed] [Google Scholar]
- 26. Kitova E. N., El-Hawiet A., Schnier P. D., and Klassen J. S. (2012) Reliable determinations of protein-ligand interactions by direct ESI-MS measurements. Are we there yet?. J. Am. Soc. Mass Spectrom. 23, 431–441 [DOI] [PubMed] [Google Scholar]
- 27. Chait B. T., Cadene M., Olinares P. D., Rout M. P., and Shi Y. (2016) Revealing higher order protein structure using mass spectrometry. J. Am. Soc. Mass Spectrom. 27, 952–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Snijder J., and Heck A. J. R. (2014) Analytical approaches for size and mass analysis of large protein assemblies. Annu Rev Anal. Chem. (Palo Alto Calif). 7, 43–64 [DOI] [PubMed] [Google Scholar]
- 29. Susa A. C., Mortensen D. N., and Williams E. R. (2014) Effects of cations on protein and peptide charging in electrospray ionization from aqueous solutions. J. Am. Soc. Mass Spectrom. 25, 918–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morgner N., and Robinson C. V. (2012) Massign: An assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal. Chem. 84, 2939–2948 [DOI] [PubMed] [Google Scholar]
- 31. de La Mora F., J. (2000) Electrospray ionization of large multiply charged species proceeds via Dole's charged residue mechanism. Anal. Chim. Acta 406, 93–104 [Google Scholar]
- 32. Schuller J. M., Beck F., Lössl P., Heck A. J. R., and Förster F. (2016) Nucleotide-dependent conformational changes of the AAA+ ATPase p97 revisited. FEBS Lett. 590, 595–604 [DOI] [PubMed] [Google Scholar]
- 33. Xia Z., and Williams E. R. (2018) Effect of droplet lifetime on where ions are formed in electrospray ionization. Analyst 144, 237–248 [DOI] [PubMed] [Google Scholar]
- 34. Noble J. E., Knight A. E., Reason A. J., Di Matola A., and Bailey M. J. A. (2007) A comparison of protein quantitation assays for biopharmaceutical applications. Mol. Biotechnol. 37, 99–111 [DOI] [PubMed] [Google Scholar]
- 35. Snoberger A., Brettrager E. J., and Smith D. M. (2018) Conformational switching in the coiled-coil domains of a proteasomal ATPase regulates substrate processing. Nat. Commun. 9, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogorzalek Loo R. R., Lakshmanan R., and Loo J. A. (2014) What protein charging (and supercharging) reveal about the mechanism of electrospray ionization. J. Am. Soc. Mass Spectrom. 25, 1675–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jurchen J. C., Garcia D. E., and Williams E. R. (2003) Gas-phase dissociation pathways of multiply charged peptide clusters. J. Am. Soc. Mass Spectrom. 14, 1373–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loo R. R., and Loo J. A. (2016) Salt bridge rearrangement (SaBRe) explains the dissociation behavior of noncovalent complexes. J. Am. Soc. Mass Spectrom. 27, 975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yakushiji Y., Nishikori S., Yamanaka K., and Ogura T. (2006) Mutational analysis of the functional motifs in the ATPase domain of Caenorhabditis elegans fidgetin homologue FIGL-1: Firm evidence for an intersubunit catalysis mechanism of ATP hydrolysis by AAA ATPases. J. Struct. Biol. 156, 93–100 [DOI] [PubMed] [Google Scholar]
- 40. Konermann L. (2017) Addressing a common misconception: ammonium acetate as neutral pH “buffer” for native electrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 28, 1827–1835 [DOI] [PubMed] [Google Scholar]
- 41. Sharawy M., and Consta S. (2015) How do non-covalent complexes dissociate in droplets? A case study of the desolvation of dsDNA from a charged aqueous nanodrop. Phys. Chem. Chem. Phys. 17, 25550–25562 [DOI] [PubMed] [Google Scholar]
- 42. Fenn J. B. (2003) Electrospray wings for molecular elephants (Nobel lecture). Angew. Chem. Int. Ed. Engl. 42, 3871–3894 [DOI] [PubMed] [Google Scholar]
- 43. Li Y., Yan X., and Cooks R. G. (2016) The role of the interface in thin film and droplet accelerated reactions studied by competitive substituent effects. Angew. Chem. Int. Ed. Engl. 55, 3433–3437 [DOI] [PubMed] [Google Scholar]
- 44. Mortensen D. N., and Williams E. R. (2016) Ultrafast (1 Μs) mixing and fast protein folding in nanodrops monitored by mass spectrometry. J. Am. Chem. Soc. 138, 3453–3460 [DOI] [PubMed] [Google Scholar]
- 45. Bain R. M., Pulliam C. J., and Cooks R. G. (2015) Accelerated Hantzsch electrospray synthesis with temporal control of reaction intermediates. Chem. Sci. 6, 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee J. K., Banerjee S., Nam H. G., and Zare R. N. (2015) Acceleration of reaction in charged microdroplets. Q Rev. Biophys. 48, 437–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gabelica V., Vreuls C., Filée P., Duval V., Joris B., and De Pauw E. (2002) Advantages and drawbacks of nanospray for studying noncovalent protein-DNA complexes by mass spectrometry. Rapid Commun. Mass Spectrom. 16, 1723–1728 [DOI] [PubMed] [Google Scholar]
- 48. Nemes P., Goyal S., and Vertes A. (2008) Conformational and noncovalent complexation changes in proteins during electrospray ionization. Anal. Chem. 80, 387–395 [DOI] [PubMed] [Google Scholar]
- 49. Banerjee S., Prakash H., and Mazumdar S. (2011) Evidence of molecular fragmentation inside the charged droplets produced by electrospray process. J. Am. Soc. Mass Spectrom. 22, 1707–1717 [DOI] [PubMed] [Google Scholar]
- 50. Olinares P. D. B., Dunn A. D., Padovan J. C., Fernandez-Martinez J., Rout M. P., and Chait B. T. (2016) A robust workflow for native mass spectrometric analysis of affinity-isolated endogenous protein assemblies. Anal. Chem. 88, 2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oh M. I., and Consta S. (2017) Stability of a transient protein complex in a charged aqueous droplet with variable pH. J. Phys. Chem. Lett. 8, 80–85 [DOI] [PubMed] [Google Scholar]
- 52. Chingin K., Barylyuk K., and Chen H. (2016) On the preservation of non-covalent protein complexes during electrospray ionization. Philos. Trans. R. Soc. London Ser. A 374, 20150377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw MS files and associated Massign-analyzed result files as well spectra lists were uploaded to the UCSD Center for Computational Mass Spectrometry, MassIVE with the dataset identifier MSV000085155. The data set can be downloaded on line using the link ftp://massive.ucsd.edu/MSV000085155/. Refer to the supplemental Table “Guide to Results-Massive Data Deposition” to link specific results presented in figures and tables to their associated data.