

Abstract
Purpose:
The current paradigm in the development of high-grade serous ovarian carcinoma (HGSC) proposes that the majority of HGSCs arise from precursor serous tubal intraepithelial carcinoma (STIC) lesions of the fallopian tube. Here we survey genome-wide methylation in HGSC precursor lesions to identify genomic regions that exhibit high-specificity differential hypermethylation for potential use as biomarkers for detecting STIC and HGSC at stages when curative intervention likely remains feasible.
Experimental Design:
We first identified quality control criteria for performing reliable methylomic analysis of DNA-limited tubal precursor lesions with the Illumina Infinium MethylationEPIC array. We then used this platform to compare genome-wide methylation among 12 STICs with paired adjacent-normal epithelia, one p53 signature lesion and 2 samples of concurrent HGSC. The resulting methylomic data were analyzed by unsupervised hierarchical clustering and multidimensional analysis. Regions of high-confidence STIC-specific differential hypermethylation were identified using selective bioinformatic criteria and compared with published MethylationEPIC data from 23 HGSC tumors and 11 healthy fallopian tube mucosae.
Results:
Unsupervised analysis showed that STICs largely clustered with HGSCs, but were clearly distinct from adjacent normal fallopian tube epithelia. Forty-two genomic regions exhibited high-confidence STIC-specific differential hypermethylation, of which 17 (40.5%) directly overlapped with HGSC-specific differentially-methylated regions. Methylation at these shared loci were able to completely distinguish STIC and HGSC samples from normal and adjacent normal specimens.
Conclusions:
Our results suggest that most STICs are epigenetically similar to HGSCs and share regions of differential hypermethylation that warrant further evaluation for potential use as biomarkers for early detection of ovarian HGSC.
Keywords: ovarian cancer, DNA methylation, epigenetics, field cancerization, STIC
Introduction
Early detection of ovarian high-grade serous carcinoma (HGSC), the most common type of epithelial ovarian cancer, is crucial to reducing disease-associated morbidity and mortality (1,2). This is because while radical surgery and cytotoxic chemotherapy have extended survival, overall mortality rates from the disease have not improved (3). There is currently no effective screening strategy for HGSC in clinical practice, partly due to an inadequate understanding of the early events of ovarian carcinogenesis. Recently, it has become increasingly clear that most ovarian HGSCs originate from epithelial precursor lesions of the fallopian tubes rather than from the ovary itself. This new paradigm of ovarian HGSC genesis was proposed on the basis of the initial observation of dysplastic epithelium in the fallopian tubes among women carrying BRCA1 and BRCA2 germline mutations (4,5). Serous tubal intraepithelial carcinoma (STIC), a term currently used to denote the precursor lesion of HGSC, is characterized by a continuation of non-ciliated tubal epithelial cells showing marked nuclear atypia, mitotic figures, apoptotic bodies, and loss of cellular polarization with p53 staining abnormality and increased Ki-67 labeling index (6-10).
Since it was initial posited, the tubal paradigm has been supported by a wealth of epidemiologic, clinical, pathological and molecular studies. It has been reported that as much as 60% or more of HGSC cases are associated with STICs (11,12) and that STIC is more frequently detected in women at an increased risk of developing HGSC than those at an average risk (13). Transcriptomic analysis has demonstrated that HGSCs molecularly resemble fallopian tube epithelium as opposed to ovarian surface epithelium and peritoneal mesothelium (14). Molecular genetic analyses have further revealed that STICs harbor the same TP53 mutations as their concurrent HGSCs, thereby establishing a clonal relationship between the two (8). Moreover, phylogenetic studies of somatic mutations and DNA copy number profiles indicate a complex evolutionary history involving the development of STIC and concurrent HGSC, suggesting that STIC precedes HGSC in many, if not most, cases (12,15,16). Many STICs associated with HGSC have shorter telomeres and amplified centrosomes (17,18), further supporting their precursor roles in tumor progression. Incidental STICs without synchronous HGSC frequently present as multiples and also exhibit molecular alterations similar to HGSC (16).
Based on the number of somatic mutations acquired in STICs and clinicopathological observation, researchers have estimated that it takes approximately six to seven years for a STIC to progress into an invasive carcinoma (12,16). This suggests that there exists a critical window of time in which STICs might be detected before they progress to advanced and often incurable stages of the disease. While molecular genetic alterations such as somatic mutations have been extensively studied as surrogate markers for detecting HGSC and other cancers in body fluids such as Pap specimens and blood, some recent studies have called into question the specificity of frequently mutated genes, such as TP53, for diagnosing the disease. For example, ultra-high sensitivity approaches such as duplex sequencing have shown that TP53 mutations are readily detectable in the majority of apparently heathy women (19,20). More recently, it has also been shown that the majority of mutations detected in circulating cell-free DNA are actually derived from clonal hematopoiesis, not cancer (21). Thus, although TP53 mutations are present in essentially all STICs and HGSCs, they cannot be considered specific due to their occurrence in nominally-healthy and nonmalignant tissues, including normal-appearing fallopian tube epithelium (16,22,23). This portends potential limitations to achieving desirable diagnostic performance with mutation-only detection schemes and indicates that there remains a need for the identification of alternative biomarkers of HGSC, particularly at early stages of the disease.
Discovery of candidate methylation biomarkers by genome-wide methylomic analyses of STIC lesions has been largely hereto precluded by technical challenges. Nonetheless, we previously reported that ovarian HGSC-specific differentially-hypermethylated regions (DHMRs) could be readily detected in STIC lesions using locus-specific techniques (24), thereby providing prima facie evidence that epigenetic aberrations take place in early ovarian cancer carcinogenesis and are potentially useful as biomarkers for early-stage HGSC diagnostics. In this work, we seek to validate and significantly extend these observations by performing genome-wide assessment of methylation directly on select pathology-confirmed precursor lesions associated with the development of ovarian cancer. Bioinformatic analyses of these results should have several biological and clinical implications, most notably the identification of a set of non-mutation-based biomarkers that occur prevalently at precursor stages and persist throughout the progression HGSC that might be implemented to facilitate development of early diagnostic approaches for HGSC.
Material and Methods
Tissue Samples and Laser Capture Microdissection
This was a retrospective study with samples obtained according to protocols approved by the Institutional Review Boards (IRB) of Johns Hopkins Medical Institutions (Baltimore, MD). Formalin-fixed paraffin-embedded (FFPE) tissue blocks were selected from patients undergoing gynecological surgeries between 2010 and 2019. The tissues selected included lesions of STIC and p53 signature from women with or without HGSC. The diagnosis was confirmed independently by experienced gynecological pathologists according to previously-published criteria (10). Normal fallopian tube epithelium away from the lesion on the same tissue block was selected as the control. In cases with concurrent HGSC involving the fimbriated ends where the STICs were located, normal epithelium from the contralateral fallopian tube was selected as the control.
FFPE tissue blocks were cut onto PEN-Membrane Slides (Carl-Zeiss # 415909041000) for laser capture microdissection (LCM). Three additional unstained sections above and below the LCM membrane sections were reserved for immunohistological confirmation of lesion location by hematoxylin and eosin (H&E) staining, immunohistochemistry (IHC) for p53 (clone BP53-11, cat # 760-2542, Ventana Medical Systems, Tucson, AZ, USA), and IHC for Ki-67 (clone 30-7, cat # 790-4286, Ventana Medical Systems, Tucson, AZ, USA), respectively. Epithelial cells were micro-dissected using the adjacent slide pre-stained with p53 as a guide.
Post-LCM DNA extraction, quantification and bisulfite conversion
Genomic DNA were extracted using the QIAmp DNA FFPE tissue kit (Qiagen, 56404) according to the manufacturer’s instructions. Post-LCM DNA yields were then quantified by qPCR using forward primer sequence, 5’- AGG GTT TTT ATG GTT TTA GGT T −3’ and reverse primer sequence, 5’- ATC CCT TCC TTA CAC C −3’, targeting a shared 82 bp locus within the consensus sequences of the L1PA1-L1PA5 LINE-1 families (25). Assays were performed using 10X Master Mix to yield a final volume of 25 μl and final working concentrations of 16.6 mM (NH4)2SO4, 67 mM Tris pH 8.8, 2.7 mM MgCl2 10mM β-mercaptoethanol, 200 μM of each deoxynucleotide triphosphate (dNTP; ThermoFisher) and 0.04 U/μl of Platinum Taq polymerase (ThermoFisher). Cycling conditions were 95°C for 5 minutes, followed by 50 cycles of (95°C for 5 seconds, 50°C for 30 seconds and 72°C for 30 seconds). Standards for quantification were created by serial dilution of human male genomic DNA (Promega). PCR was performed in duplicate in 96-well plates using a CFX96 Touch Real-time PCR Detection System (Bio-Rad) and analyzed using the accompanying stock software, Bio-Rad CFX Manager (v3.1). DNA concentrations were determined by cycle of quantification (Cq) values with reference to a standard curve created via serial dilution of human male genomic DNA (Promega).
50 ng of DNA from each sample underwent bisulfite conversion using EZ DNA Methylation Kit (Zymo, D5001) according to the manufacturer’s protocol. In brief, 5 μl of M-dilution buffer was mixed with 45 μl of sample and incubated at 37°C for 15 mins; 100 μl of CT conversion reagent was then added to the mixture and incubated in the dark overnight. The sample was transferred into the spin column with M-binding buffer. Following the wash and spin down procedure, 200 μl of Desulphonation buffer was added. The sample was then eluted with 10 μl of elution buffer.
TP53 mutation analysis
The TP53 somatic mutation status in most of the micro-dissected samples was obtained either from the whole-exon sequencing or from TP53 amplicon sequencing data in our previous study (16). Some were obtained by Sanger sequencing targeting the TP53 DNA binding domain (from exon 4 to exon 9). Mutations were analyzed by the Mutation Explorer software.
DNA restoration and MethylationEPIC assay
Bisulfite-treated (BST) DNA was restored to amplifiable length using the Illumina Infinium FFPE DNA Restoration Kit (WG321-1004) according to the manufacturer’s instructions. Restored BST sample DNA was then analyzed using Illumina’s Infinium Human MethylationEPIC BeadChip Kit (WG-317-1002) by following manufacturer’s manual. Briefly, 8 μl of bisulfite-converted DNA was added to a 0.8 ml 96-well storage plate (Thermo-Fisher Scientific), denatured in 0.014N sodium hydroxide, neutralized and amplified with kit-provided reagents and buffer at 37°C for 20-24 hours. Samples were enzymatically fragmented by addition of 50 μl FMS fragmentation solution (Illumina) to each well. Plates were then vortexed at 1600 rpm for 1 minute and pulse centrifuged at 280g. The sealed plates were then incubated on a heat block at 37°C for one hour before precipitation of DNA by addition of 2-propanol to each sample-well. Re-suspended samples were denatured in a 96-well plate heat block at 95°C for 20 minutes. 26 μl of each sample was loaded onto an 8-sample chip and the chips were assembled into hybridization chamber as instructed in the manual. After incubation at 48°C for 16-20 hours, chips were briefly washed and then assembled and placed in a fluid flow-through station for primer-extension and staining procedures. Polymer-coated chips were image-processed in Illumina’s iScan scanner.
Data analysis
DNA methylation data containing the Illumina EpicArray iDat files were processed using the functional normalization (funNorm) algorithm, as implemented in the minfi package (26). The ilm10b2.hg19 package from Bioconductor provided current hg19 annotations for the probe sequences represented on the array, which were remapped to hg38 based on the UCSC liftOver mapping (4).The R studio software was used to analyze data obtained from the MethylationEPIC assay to identify differentially hypermethylated regions (DHMRs) between STIC and corresponding normal-appearing tubal epithelium. In order to identify DHMRs that are most specific to HGSC and STIC, we incorporated the MethylationEPIC array data from our previous study (24), which include 23 HGSC and 11 normal fallopian tube mucosae derived from patients having bilateral salpingo-oophorectomy (BSO) due to benign disease such as myoma or adenomyosis. Probes with detection p values greater than 0.05 were filtered out. Locus methylation was calculated as a β-value, low to high ranging from 0-1, respectively. Probes with a [mean β-value + 2 standard deviations] ≥ 0.2 in normal fallopian tube epithelium derived from women without malignancy were filtered out. To increase the confidence that the genomic region was methylated in HGSC and STIC, only genes with at least two probes hypermethylated in the promoter region were selected.
Data and code availability
All Illumina MethylationEPIC BeadArray data from this study , and our prior study (24), have been deposited to the NCBI GEO database as a SuperSeries (accession number GSE155761). An R Markdown file detailing all bioinformatic analyses is available for download at https://github.com/lesliecope/STICmanuscript.
Results
Case cohorts and study overview
The primary goal of this study was to survey the genome-wide landscape of methylation alterations that occur during the development of fallopian tube precursor lesions and to identify candidate differentially-hypermethylated region (DHMR) biomarkers potentially suitable for early-stage identification of HGSC. However, the diminutive nature and need for the tedious processing to unambiguously identify rare precursor lesions in formalin-fixed and paraffin-embedded (FFPE) fallopian tube tissues poses considerable challenges to identifying a suitable cohort of lesions in which genomic DNA of sufficient quantity and quality can be extracted to perform reliable whole genome methylation analysis. In light of these concerns, we first performed preliminary validation experiments to identify appropriate sample requirements that would ensure reliable analysis by the MethylationEPIC BeadArray platform. Toward this end, we performed MethylationEPIC profiling using three concentrations (500 ng, 50 ng and 10 ng per sample-volume) of serially-diluted DNA obtained from two separate laser-captured microdissected (LCM) FFPE HGSOC tumor specimens. We then examined the respective Illumina QC methylation profiling (Supplementary Fig. S1), sample clustering dendrograms (Supplementary Fig. S2) and CpG detection p-values and subsequently determined 50 ng of high-purity, laser-captured microdissected (LCM) DNA to be sufficient for consistent and reliable analysis using the MethylationEPIC platform, which is also in accord with previous studies (27).
From an extended library of 36 histopathologically-confirmed formalin-fixed and paraffin-embedded specimens containing tubal precursor lesions, we identified 13 tubal lesions whose post-LCM DNA yields were sufficient to meet the predetermined sample requirements (Supplementary Fig. S3). Established histopathological criteria were used to classify each lesion (10), of which representative cases can be seen in Supplementary Fig. S4. For each respective case, LCM was also performed, and DNA extracted, from normal-appearing, adjacent fallopian tube epithelia (normal FTE) control tissues, as well as from microdissected tumors when concomitant HGSC was present. In total, the initial sample cohort for the present study comprised 27 DNA samples, including: 12 STIC lesions (including 1 “dormant STIC”), 12 adjacent-normal FTE, 1 p53 signature and 2 HGSCs from a total of 12 women. Fig. 1 provides a pictorial overview of the overall sample cohort. Table 1 presents the relevant clinical information, mutational statuses (TP53 and BRCA1/2) and Ki-67 proliferation index for each of the lesions.
Figure 1. Study Overview.
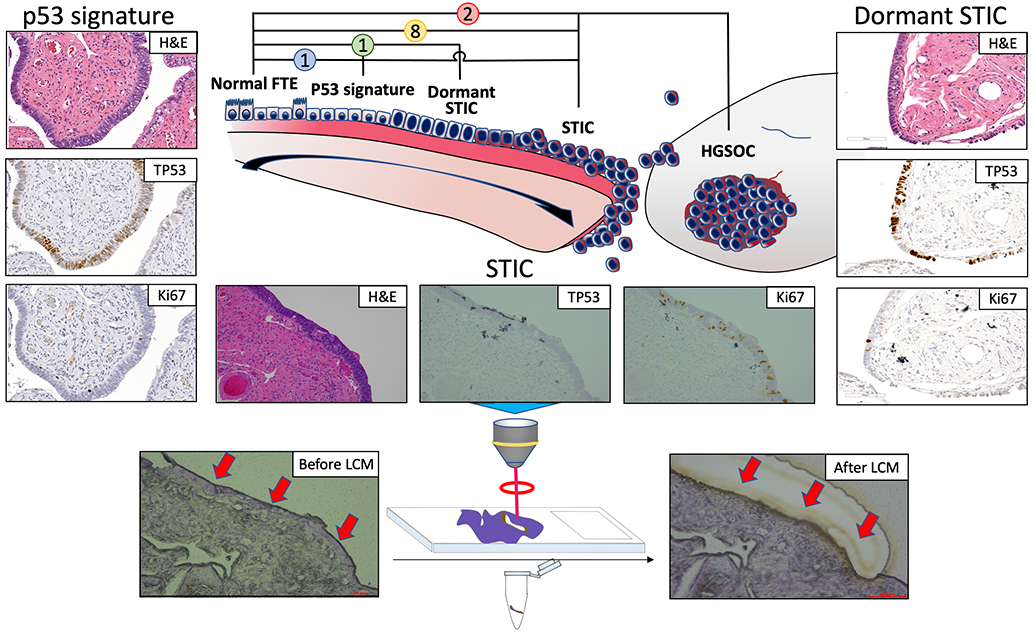
(A) MethylationEPIC analysis was performed on DNA obtained from 12 patients with histopathologically-classified precursor lesions of the distal fallopian tube, paired with corresponding normal-appearing adjacent epithelia and concomitant HGSC tumors (when present) from FFPE tissue. (B) All FFPE samples were dissected by laser capture microdissection to isolate the corresponding epithelia.
Table 1.
Clinical information, p53 and germline mutation status, and proliferation index of lesions studied.
| Case Number |
Age | Race | Sample Name | Histopathology | p53 pattern | Ki-67(%) | TP53 mutation | Concurrent HGSOC | Other tumors | Germline mutations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 66 | W | N1 | NFT | No | BRCA2 mutation (+) c.26del (p.Pro9fs) | ||||
| S1 | STIC | Nonsense | 70 | P191fs | ||||||
| 2 | 50 | W | N2 | NFT | No | MMMT | N/A | |||
| S2 | STIC | Nonsense | 38 | H82fs | ||||||
| 3 | 79 | W | N3 | NFT | No | APST | N/A | |||
| S3 | STIC | Missense | 20 | L62P | ||||||
| 4 | 58 | W | N4 | NFT | No | Uterine endometrioid carcinoma | N/A | |||
| S4 | STIC | Nonsense | 65 | c.782+14del | ||||||
| * 5 | NA | NA | N5 | NFT | No | N/A | ||||
| S5 | STIC | N/A | N/A | N/A | ||||||
| 6 | 75 | W | N6 | NFT | Yes | N/A | ||||
| S6 | STIC | Missense | 31 | C145F | ||||||
| T6 | HGSC | C145F | ||||||||
| 7 | 71 | W | N7 | NFT | No | N/A | ||||
| S7 | STIC | Missense | 18 | R379H c.920-2 A>C c.920-0 GC>G | ||||||
| 8 | 61 | W | N8 | NFT | Yes | BRCA2 mutation (+) c.H89_1190ins | ||||
| S8 | STIC | Nonsense | 54 | D281N | ||||||
| T8 | HGSC | D281 N/N235N | ||||||||
| 9 | 74 | W | N9 | NFT | No | Uterine serous carcinoma | N/A | |||
| S9 | STIC | Missense | 22 | R175H | ||||||
| 11 | 56 | W | N11 | NFT | No | N/A | ||||
| P11 | p53 | Missense | 3 | A29D | ||||||
| S11 | STIC | Nonsense | 20 | Y88X | ||||||
| 12 | 49 | W | N12 | NFT | No | Breast cancer; hodgekin lymphoma | BRCA2 mutation (+) Mutation status N/A | |||
| S12 | STIC | Nonsense | 10 | C141* | ||||||
| 13 | 70 | W | N13 | NFT | No | N/A | ||||
| L13 | Dorm. STIC | Missense | 4 | N239S |
NFT: normal fallopian tube; STIC: serous tubal intraepithelial carcinoma; p53: p53 signature; Dorm. STIC: dormant STIC; MMMT: malignant Mixed Müllerian Tumors; APST: atypical proliferative serous tumor; N/A: not available
Patient information not available
Methylomic analysis of fallopian tube precursor lesions
We performed MethylationEPIC analyses on 50 ng of DNA extracted from each of the 27 FFPE tissue samples. QC of the resultant data indicated that all samples exhibited bimodal β-value distributions (Supplementary Fig. S5) as well as a similar or greater number of probes detected at statistical significant p-value (e.g., p < 0.05) than in the preliminary validation samples, thereby indicating the quality of the resultant methylation data to be sufficient for downstream analyses. A heatmap of the initial, unsupervised analysis of the 1000 most-variable CpG-sites between the 12 STIC – adjacent-normal-FTE pairs showed that all tumors and the majority of lesions were readily distinguishable from adjacent-normal FTE according to sample clustering (Fig. 2A). Unsupervised multi-dimensional analysis (MDA; Fig. 2B) and the sample dendrogram (Fig. 2C) of the array data also demonstrated congruent clustering patterns. Additionally, these data illustrate that the majority of normal FTE samples (and incidentally the p53 signature lesion) exhibit strong overlap in their respective methylomes, as compared to the STIC and HGSC samples, which are more heterogeneous in comparison. Taken as a whole, these results imply significant divergence between the genome-wide methylation patterns of precursor lesions, which is possibly reflective of the broader genomic instability known to arise during the progression of precursor lesions during ovarian cancer carcinogenesis (28). Of particular note is the direct subclustering seen in Fig. 2C between the two ovarian carcinomas (T6 and T8) and their associated STIC lesions (S6 and S8, respectively). This strongly suggests the existence of a clonal relationship between each cognate pair and provides additional anecdotal evidence supporting a fallopian tube precursor origin for the respective ovarian carcinomas.
Figure 2. Unsupervised analysis of genome-wide methylation of HGSC-precursor lesions of the fallopian tube.
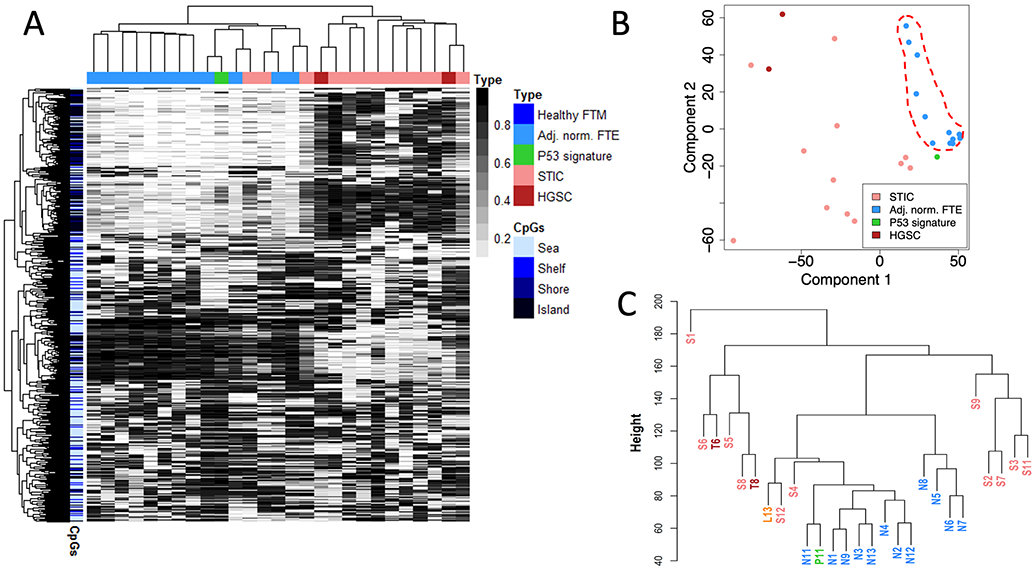
(A) Methylation heatmap showing the 1000 most variable CpG probes as determined by MethylationEPIC analysis. (B) Corresponding multidimensional analysis showing relative clustering of adjacent-normal epithelia (dashed red line). (C) Sample dendrogram showing that the majority of adjacent-normal epithelia and p53 signature lesion are readily distinguishable from STIC lesions and HGSC tumors. STIC-HGSC pairs are shown in bold red. FTM – Fallopian tube mucosa, FTE – Fallopian Tube Epithelium
We next employed a bioinformatic pipeline, described in our prior work (24), for identifying loci that exhibit high-confidence cancer-specific DHMRs that might be amenable for use as methylation biomarkers for identifying STIC lesions when compared to normal-appearing FTE. Using this algorithm, high specificity was prioritized by selecting only those CpG-probe-sites that exhibited minimal methylation [(mean β + 2 S.D.) < 0.2] in all tested control tissues (i.e., adjacent-normal FTE). Second, a so-called “high-confidence” criterion was employed to identify only those high-specificity loci containing a high-density of CpG-sites (CpG islands, shores and shelves within 1500-bp of the transcription start site) exhibiting contiguous STIC-specific hypermethylation within a maximum inter-probe distance of 400 bp. The combination of both high specificity and high confidence criteria are aimed at identifying only those loci that are most likely to exhibit dense, disease-specific hypermethylation that are thereby well-suited to commonly-employed locus-specific methylation analysis techniques such methylation-specific PCR or targeted bisulfite sequencing. Analysis of STIC and adjacent-normal methylome data with this pipeline identified a total of 42 loci that exhibited high-confidence, STIC-specific hypermethylation (Supplementary Table S1). A heatmap demonstrated that methylation at these loci were able to not only fully discriminate all STIC from HGSC specimens, but incidentally classified even the p53 signature sample as distinct from both healthy and malignant tissues (Fig. 3).
Figure 3. STIC-specific methylation.
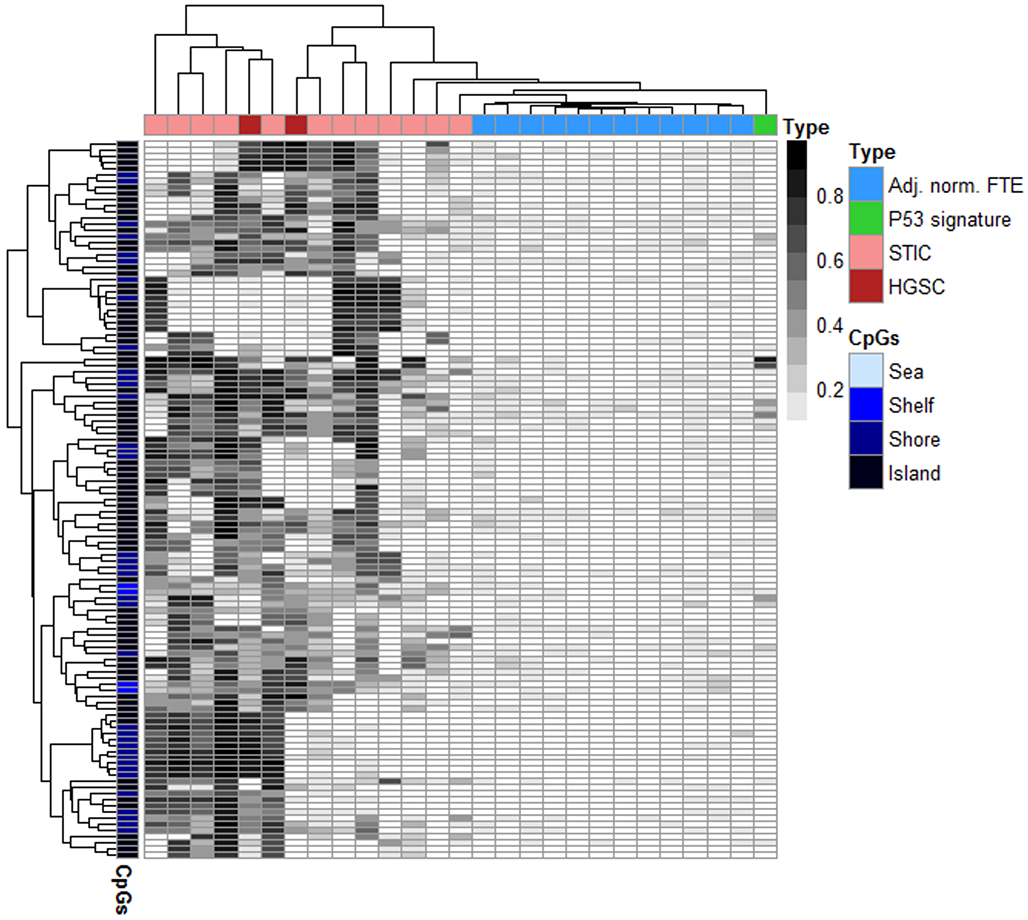
Methylation heatmap of the 42 differentially-hypermethylated regions (DHMRs) exhibiting high-confidence, STIC-specific hypermethylation in comparison with adjacent normal epithelia. FTM – Fallopian tube mucosa, FTE – Fallopian Tube Epithelium
Correlation between the STIC and HGSC methylome
We next proceeded to further compare the methylomic patterns between STIC lesions and HGSC. Toward this end, we combined the MethylationEPIC dataset from the current study with MethylationEPIC datasets from HGSCs (n = 23) and normal gynecological mucosal tissues (n = 11) derived from our previous study (24). A corresponding methylation heatmap of the unsupervised analysis of the top 1000 most variable CpG sites of all samples in the conjoined datasets is shown in Fig. 4. There are a number of notable observations regarding the sample clustering from this analysis. First, our data demonstrate that the methylomes of HGSC and STIC lesions were clearly and reproducibly distinct from healthy and normal-appearing gynecological tissues. Notably, the methylomes of most STICs also formed a subcluster distinct from both the adjacent-normal FTE and HGSC samples, possibly reflective of their intermediate state of malignancy. It was also notable that the p53 signature specimen clustered well within the adjacent normal FTE specimens, indicating a largely nonmalignant epigenotype.
Figure 4. Unsupervised analysis of HGSC compared with precursor lesions and healthy tissues of the fallopian tube.
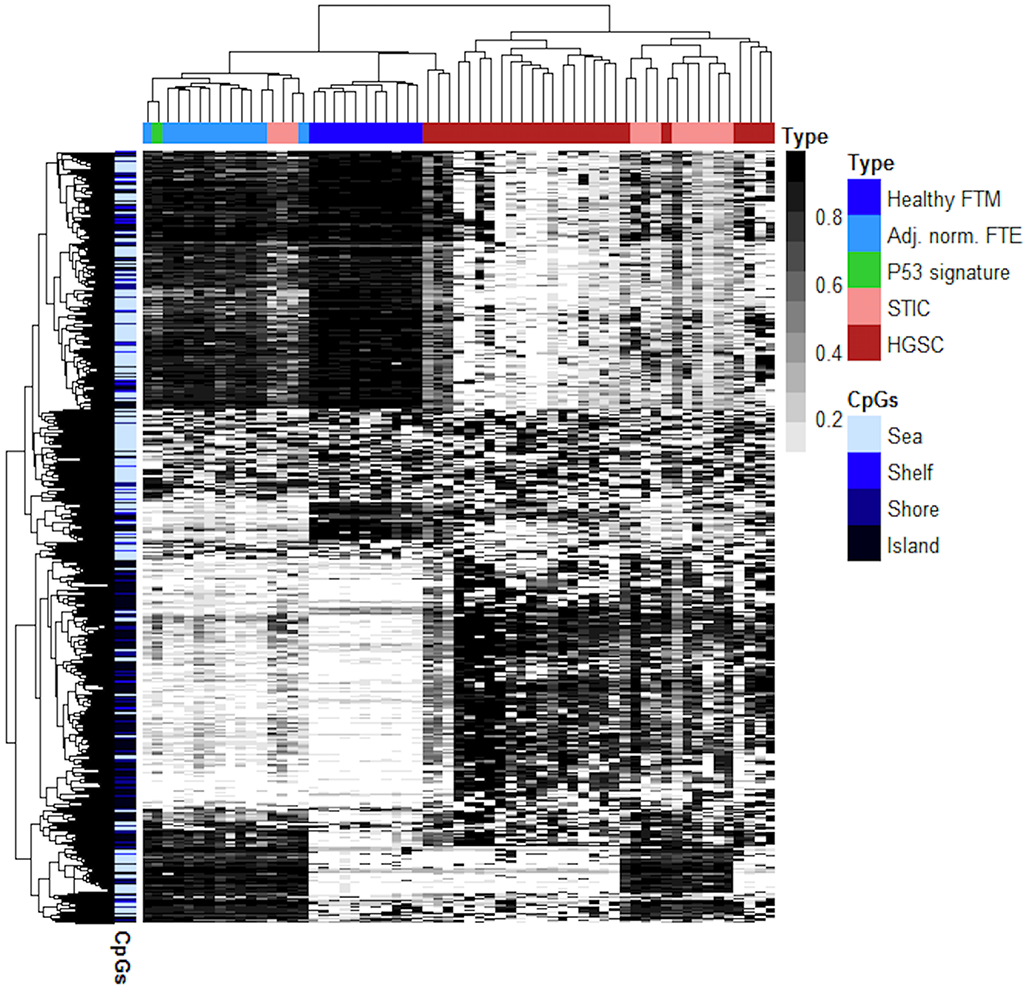
Methylation heatmap showing the 1000 most variable CpG probes as determined by MethylationEPIC analysis of the present study with conjoined data derived from previously-reported methylomic of analysis of HGSC. FTM – Fallopian tube mucosa, FTE – Fallopian Tube Epithelium
Shared Cancer and STIC-specific hypermethylation
We next sought to determine whether there might be a convergence between HGSC-specific DHMRs with those regions exhibiting hypermethylation in the putative precursor, STIC lesions. To achieve this, we compared the 42 high-confidence STIC-specific DHMRs identified in the present study with the 91 high-confidence HGSC-specific DHMRs independently identified in the previous study (24). Overall, we observed a striking overlap between the two candidate biomarker sets, with a total of 17 DHMRs in common (Fig. 5A and Table 2), accounting for a total of 40.5% of all high-confidence STIC-specific differentially-hypermethylated regions.
Figure 5. HGSC/STIC-specific hypermethylation.
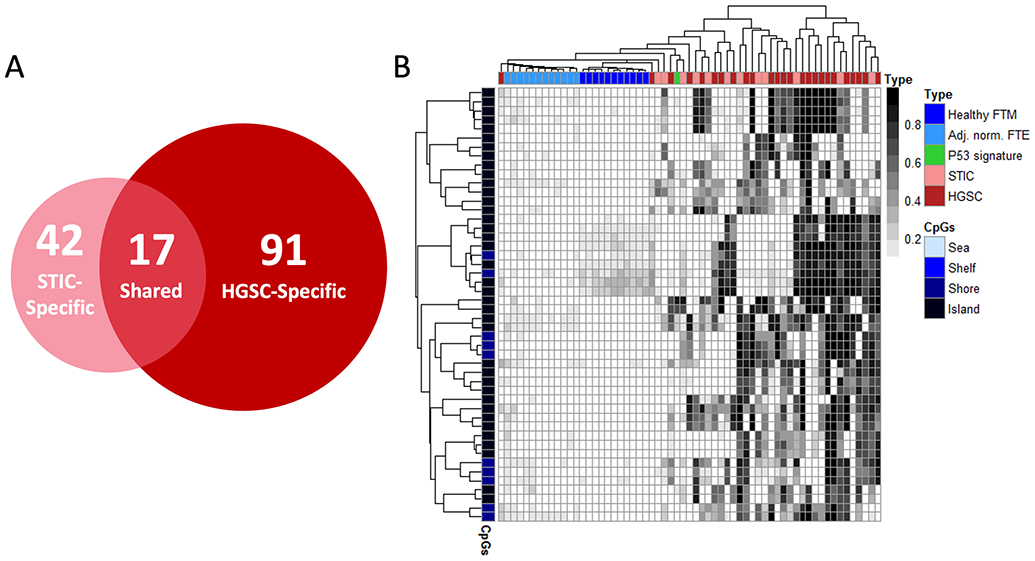
(A) Venn diagram of STIC- and HGSC-specific high-confidence DHMRs identified by MethylationEPIC analysis in the present and previous, HGSC, study, respectively. Seventeen DHMRs were found to exhibit both STIC- and HGSC-specific hypermethylation. (B) Methylation heatmap of the 17 shared, high-confidence HGSC-/STIC-specific DHMRs using merged datasets and samples from both studies. FTM – Fallopian tube mucosa, FTE – Fallopian Tube Epithelium
Table 2.
High-confidence DHMRs exhibiting HGSC/STIC-specific hypermethylation.
| Genomic Location* | Gene | STIC vs. Adj. norm. FTE AUC |
HGSC VS. Healthy FTM AUC |
HGSC & STIC vs. Healthy FTM & Adj. norm FTE AUC |
|---|---|---|---|---|
| chr5:141208649-141208659 | PCDHB12 | 0.958 | 0.673 | 0.723 |
| chr5:141407937-141407940 | PCDHGB6,PCDHGA1,PCDHGA2,PCDHGA3,PCDHGA4, | 0.875 | 0.793 | 0.813 |
| chr5:141417605-141417623 | PCDHGB7,PCDHGA1,PCDHGA10,PCDHGA2,PCDHGA3 | 0.833 | 0.88 | 0.871 |
| chr19:50667559-50667804 | SHANK1 | 0.826 | 0.745 | 0.761 |
| chr3:187737583-187737946 | BCL6 | 0.826 | 0.807 | 0.81 |
| chr17:60421510-60421683 | C17orf64 | 0.819 | 0.924 | 0.879 |
| chr11:2271280-2271282 | ASCL2 | 0.812 | 0.789 | 0.753 |
| chr11:131910988-131911000 | NTM | 0.806 | 0.855 | 0.804 |
| chr17:37745219-37745526 | HNF1B | 0.778 | 0.633 | 0.737 |
| chr2:153871394-153871412 | GALNT13 | 0.771 | 0.782 | 0.746 |
| chr6:26183280-26183344 | HIST1H2BE | 0.771 | 0.782 | 0.765 |
| chr12:38905914-38906042 | CPNE8 | 0.722 | 0.836 | 0.784 |
| chr12:24903146-24903193 | BCAT1 | 0.694 | 0.676 | 0.699 |
| chr6:26043976-26044177 | HIST1H2BB,HIST1H3C | 0.632 | 0.815 | 0.76 |
| chr17:37743962-37744227 | HNF1B | 0.625 | 0.531 | 0.555 |
| chr2:42792857-42792873 | HAAO | 0.549 | 0.829 | 0.717 |
| chr6:31815145-31815220 | HSPA1A,HSPA1L | 0.507 | 0.716 | 0.651 |
| Mean: | 0.753 | 0.769 | 0.755 | |
| Standard Deviation: | 0.118 | 0.098 | 0.077 | |
| Wilcoxan p-value: | 0.679 | |||
Coordinates refer to the human reference genome hg38 release (Genome Reference Consortium GRCh38, Dec 2013).
A methylation heatmap demonstrated that the 17 shared DHMRs readily distinguished malignant (STIC and HGSC) from all normal-appearing gynecological tissues without pathological changes (Fig. 5B). It is, however, particularly notable that HGSC could not be distinguished from STIC based on the presence and/or extent of methylation in these regions. While the inability to distinguish precursor lesions from high grade ovarian carcinomas provides additional anecdotal support for the fallopian tube origin of HGSC, it also implies that, collectively speaking, methylation at these loci likely accrues primarily during the progression of precursor lesions prior to metastasizing to the ovary.
A plot of the average β-value of the 3 candidate biomarker sets (HGSC-, STIC- and HGSC/STIC-specific DHMRs), as shown in Fig. 6A and Supplementary Tables S1-S3, also showed that although malignant tissues exhibited considerably higher methylation than normal-appearing fallopian tube tissues, HGSC specimens were only marginally more methylated in the majority (76%) of STIC-specific and HGSC/STIC-specific DHMRs (paired t-test, p = 0.13 and 0.33, respectively). On the other hand, we observed moderately higher mean methylation at 88% of HGSC-specific DHMRs in HGSC than in STIC specimens (paired t-test, p = 2E-10), indicating that hypermethylation at many of these loci either arises late or continues to accrue following dissemination to the ovary. In comparison, when all CpG-probes in islands, shores and shelves located near promotor regions were considered, no correlation between hypermethylation and disease status (STIC versus HGSC) was observed. A similar trend was observed when the when the ROC performance the DHMR sets was considered (Fig. 6B and Supplementary Table S1-S3). These data revealed that while STIC-specific DHMRs performed equally well at discriminating STIC and HGSC from corresponding healthy tissues (mean AUC 0.816 and 0.815, respectively), they actually slightly outperformed HGSC-specific DHMRs at discriminating HGSC tissues (mean HGSC-specific DHMR AUC 0.799) and greatly outperformed HGSC-specific DHMRs in identifying STIC (mean HGSC-specific DHMR AUC 0.702). These results further support the notion that STIC-specific hypermethylation likely accrues before, and is largely retained following, dissemination to the ovary, while some HGSC-specific differential hypermethylation arises during subsequent progression of ovarian carcinoma. Lastly, it is worth noting that although STIC-specific DHMRs appear to perform better in terms of distinguishing both HGSC and STIC from healthy tissues than HGSC/STIC-specific DHMRs (mean HGSC/STIC-specific AUC 0.769 and 0.753, respectively), these loci exhibit a considerably lower mean and variance in β-value in healthy fallopian tube mucosae. This implies that the HGSC/STIC-specific DHMRs would likely outperform STIC-specific DHMRs in terms of clinical specificity for the detection of STIC or HGSC in samples such as blood or Pap specimens that would nominally contain DNA derived from heterogeneous populations of cells within the mucosae of the fallopian tube, endometrium and cervix, which were included as controls in our previous study (24).
Figure 6. Hypermethylation with respect to disease stage.
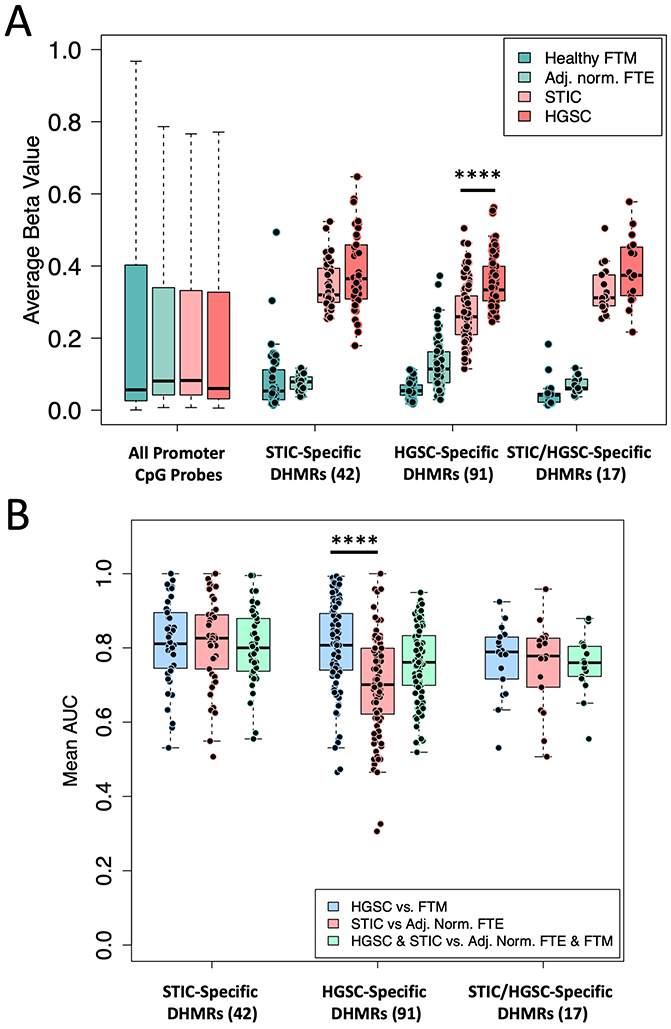
(A) Mean β-value of high-confidence DHMR sets compared with all promotor regions with respect to pathologically-classified disease stage. All promoter CpG probes include CpG probes located in CpG island, shelves and shores within 1500 bp of the transcription site. (B) Area under the ROC curve (AUC) of DHMRs in each set with respect to disease stage. ****p << 0.0001 (Wilcoxan Test)
Discussion
There continues to be a critical unmet need for biomarkers and diagnostic methods capable of detecting asymptomatic ovarian HGSC, particularly at early, precursor and/or incipient stages of the disease when surgical intervention is most effective. While considerable effort has been made toward this goal, particularly in the implementation of sequencing technologies with noninvasively-collected samples such as blood, uterine lavage or Pap specimens (20,29-31), the increasing analytical sensitivity of these technologies has also begun to reveal that cancer-free individuals and healthy tissues commonly harbor these same genetic abnormalities (19,20). This issue is further confounded by the fact that TP53 is the only significantly mutated gene (SMG) present in most HGSC tumors (32), indicating that targeted sequencing of additional frequently-mutated genes should provide only limited improvement in diagnostic performance. These issues ultimately imply that alternative biomarkers and/or multivariate approaches will likely be necessary to fully leverage advances in molecular diagnostic techniques while maintaining acceptable clinical specificity.
Biomarkers based on cancer-specific alterations in DNA methylation are a particularly attractive option for the noninvasive detection of HGSC. First, in comparison to non-synonymous mutations, which typically occur in no more than two SMGs per HGSC tumor (32), cancer-specific aberrant DNA methylation is far more abundant, typically occurring in thousands of CpG dinucleotides within hundreds of CpG islands in the typical tumor genome (33). This redundancy is particularly useful for noninvasive diagnostic applications, as panels of methylation biomarkers can be utilized to dramatically improve clinical sensitivity in challenging samples, such as liquid biopsies, that normally contain only limited quantities of tumor DNA (34,35). A second key advantage of DNA methylation is that it is highly cell-type specific (36). A number of key studies have likewise demonstrated that not only are DNA methylation biomarkers useful for early detection, but also for identifying corresponding tissues of origin and even subtypes of many human cancers (35,37-40), including etiologically-diverse epithelial ovarian carcinomas (41).
This study directly builds upon our previous efforts to identify a discrete set of genomic regions exhibiting ovarian HGSC-specific differential hypermethylation through genome-wide methylomic analysis. Using locus-specific methylation analyses, our prior study also provided initial evidence that hypermethylation at the HGSC-specific loci was likely present in STIC lesions, the putative precursors of ovarian HGSC, thereby indicating that that epigenetic aberrations are likely an early event in, and possibly contribute to, ovarian cancer carcinogenesis. In the present study, we sought to validate and significantly extend these observations by directly evaluating genome-wide assessment of methylation in pathology-confirmed precursor lesions associated with the development of ovarian cancer. Overall, we found that HGSCs and its precursor lesions are readily distinguishable from normal-appearing fallopian tube tissues based solely on the methylation status of a limited set of genomic loci.
In unsupervised analyses, all but 2 STIC samples clustered together with the 25 HGSC samples, which is reflective of the common origin between the two malignancies. Perhaps more surprisingly, these analyses also demonstrated that the methylomes of STIC samples could also be distinguished from HGSCs, primarily due to hypermethylation of certain loci otherwise associated with normal FTE, as well as notably less prevalent and extensive degrees of hyper- and hypomethylation in HGSC-specific DHMRs. This subclustering implies that while, for the most part, the methylomes of STIC lesions largely resemble that of HGSC tumors, there appears to be notable hypomethylation within certain genomic regions that occurs in HGSC, but not in STIC, samples. However, an open question remains as to whether the observed HGSC-specific hypomethylation is a result of cellular heterogeneity within the tumor samples, epigenetic reprogramming that occurs within malignant non-ciliated fallopian tube epithelial cells following dissemination to the ovary or, possibly, an FFPE-related artifact (samples from the prior study were fresh-frozen rather than FFPE). Nonetheless, taken at face value, our results imply that assessment of methylation might be potentially leveraged to differentiate early, premalignant disease from HGSC.
In general, HGSCs appeared to exhibit only marginally more hypermethylation at STIC-specific DHMRs than precursor STIC lesions, implying that the majority of hypermethylation at these loci likely occurs before dissemination to the ovary. This is also supported by the observation that STIC-specific DHMRs perform equally well at discriminating STIC and HGSC from healthy tissues. We also observed that microdissected adjacent-normal fallopian tube epithelium exhibited a modest, but detectable degree of HGSC-specific methylation. This observation is in general accord with the results of our previous study (24) that indicated the potential existence of STIC-associated methylation field effect or broader, genetically-driven, epigenomic reprogramming (42) that may occur even prior to the development of early-stage, p53 signatures. Likewise, because the high-confidence STIC-specific DHMRs identified in the present study were selected using adjacent-normal FTE as a negative control, it is probable that at least some DHMRs positively associated with the development of precursor lesions and subsequent HGSC have been excluded as a result of hypermethylation due to a methylation field effect.
p53 signature is a tubal lesion of unknown clinical significance that, while likely related to STIC lesions, is often detected in fallopian tubes in the absence of disease (16). p53 signature is defined by an abnormal p53 immunostaining pattern compatible with a missense TP53 mutation in a minute stretch of otherwise normal-appearing non-ciliated tubal epithelium. p53 signatures are thought to represent an evolutionary “dead end” and do not have any known clinical significance but a few of them may precede STIC as the earliest precursor lesions. Because of their diminutive nature, the DNA obtained from p53 signatures is rarely sufficient for global methylation studies. Nevertheless, in this study, we, for the first time, were able to profile the methylome of this incipient lesion. The fact that p53 signature is epigenetically closer to normal-appearing fallopian tube epithelium than to STIC and HGSC suggests that TP53 mutations may not significantly affect the methylation profiles in epithelial cells and reassure that p53 signatures unlike STIC have yet to undergo malignant transformation. Although more p53 signatures will need to be analyzed in follow-up studies, this initial study indicates that methylation analyses might ultimately be leveraged to both detect and distinguish precursor lesions with respect to their malignant potential.
It is worth highlighting that in addition to the 17 HGSC-specific genomic loci identified in our previous study, several other identified genes associated with the STIC-specific hypermethylation have also previously been implicated by others in HGSC and associated carcinogenesis. Most notably, silencing of the genes HOXA11 (43,44), miR-124 (45), RUNX3 (46,47), SHANK1 (48), MN1 (49), SOCS2 (50) and OPCML (51) have all been documented to contribute to the development of HGSC. Interestingly, the methylation heatmaps of STIC-specific hypermethylation seem to indicate that in most lesions, methylation occurs in an “all or nothing” manner at various individual loci, reflecting considerable heterogeneity between the lesions, which are themselves, epigenetically clonal in nature.
The 91, 42 and 17 DHMRs identified here as highly specific to HGSC, STIC and both specimen types, respectively, represent a relatively conservative set of candidate hypermethylation biomarkers for further validation studies. It is ultimately anticipated that the performance of mutation-based diagnostics approaches such as the PapSEEK tests will likely benefit from incorporation of complementary epigenetic or protein biomarkers that can be concurrently evaluated in a single, noninvasively-collected specimen. A simple, unified test would be particularly attractive for early HGSC diagnostic or screening purposes, especially for women at increased risk of developing ovarian cancer due to germline mutations in BRCA1/2 and other DNA repair genes as well as a family history of breast and ovarian cancer.
There are a number of limitations to the present study that warrant discussion. First, the relative scarcity and minute nature of fallopian tube precursor lesions make it particularly difficult to obtain a large cohort of specimens with sufficient DNA for genome-wide methylation analysis. As such, the inclusion criteria for our study reflect a best effort to achieve an ideal compromise between sufficient data quality and analysis of a suitable number of specimens to provide an adequate representation of the overall methylomic landscape of precursor lesions. Another limitation inherent to the study is that precursor lesions must undergo FFPE processing for identification and accurate classification. This ultimately implies that the quality of downstream molecular analyses, methylomic or otherwise, are necessarily subject to potential artifacts associated with FFPE sample processing. Batch effects arising from such artifacts can be at least partially addressed through incorporation of appropriate FFPE controls, such as the adjacent normal FTE specimens used here. Similarly, the potential for false DMR discovery arising from spurious probe data can be further minimized by implementation of rigid discovery criteria, such as the “high-confidence” DHMR, which are characterized by minimal methylation in control specimens with statistically-significant hypermethylation in contiguous CpG probes in STIC specimens. Regardless, batch effects may still exist when comparing data from DNA obtained from FFPE specimens to those from fresh frozen samples, such as the HGSC and healthy fallopian tube mucosae samples of our prior study. However, this concern is somewhat mitigated by previous studies demonstrating the equivalence of EPIC datasets from DNA obtained from fresh-frozen samples and 50 ng of restored FFPE DNA (27).
The present study represents an important first initial survey of the methylomic landscape of fallopian tube lesions associated with high-grade serous ovarian carcinoma. Future studies are needed to validate our findings in an independent cohort of STIC and p53 signature lesions and to further determine the underlying biological mechanisms from which STIC and/or HGSC-specific methylation arises. Validation of DHMRs specific to STIC but not in fallopian tube epithelium will provide new insight into the very early pathogenesis in the development of ovarian HGSC. Furthermore, exploration into possible epigenetic “field” effects in normal-appearing tubal epithelium in reference to anatomical location might help to elucidate epigenetic contributions that might contribute to or accelerate lesion formation and/or progression.
Supplementary Material
Translational Relevance.
There is accumulating evidence that the majority of high-grade serous ovarian carcinomas (HGSCs) likely originate as precursor, serous tubal intraepithelial carcinoma (STIC) lesions of the fallopian tube. A better understanding of these lesions is likewise expected to be critically important to further elucidation of HGSC carcinogenesis and for the development of new methods for early detection. However, technical challenges associated with the rarity and diminutive nature of STIC lesions have made comprehensive and statistically-significant molecular analyses, such as genome-wide DNA methylation, difficult to perform. In the present study, we validate and employ a protocol to successfully perform methylomic analyses of 13 precursor fallopian tube lesions, 12 adjacent normal epithelia and 2 concurrent HGSC tumors and compare the resulting datasets to independent methylomic analyses of 23 HGSC tumors and 11 normal fallopian tube epithelia. Our results demonstrate that STIC lesions exhibit a high degree of epigenetic similarity to HGSC and enable identification of a discrete set of differentially-hypermethylated regions (DHMRs) that exhibit potential promise as sensitive and specific methylation biomarkers for identifying HGSC at early stages of carcinogenesis.
Acknowledgments
This work was supported by The Honorable Tina Brozman Foundation, The Department of Defense CDMRP (grant number W81XWH-11-2-0230), The National Institutes of Health/National Cancer Institute (grant numbers EDRN UO1CA200469, RO1CA215483), The Teal award, The Gray Foundation, Colleen’s Dream Foundation, The Johns Hopkins Discovery Team Award and The Richard W. TeLinde Endowment, Johns Hopkins University.
Financial Support: This work was supported by the Honorable Tina Brozman Foundation, National Institutes of Health/National Cancer Institute (grant numbers P50CA228991, UO1CA200469, RO1CA215483), Teal Award, Gray Foundation, Department of Defense CDMRP (grant number W81XWH-11-2-0230), Johns Hopkins Discovery Award and the Richard W. TeLinde Endowment, Johns Hopkins University.
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Peres LC, Cushing-Haugen KL, Köbel M, Harris HR, Berchuck A, Rossing MA, et al. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. JNCI: Journal of the National Cancer Institute 2018;111(1):60–8 doi 10.1093/jnci/djy071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cress RD, Chen YS, Morris CR, Petersen M, Leiserowitz GS. Characteristics of Long-Term Survivors of Epithelial Ovarian Cancer. Obstet Gynecol 2015;126(3):491–7 doi 10.1097/aog.0000000000000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurman RJ, Shih Ie M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol 2016;186(4):733–47 doi 10.1016/j.ajpath.2015.11.011S0002-9440(16)00008-0 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piek JMJ, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJJ, Menko FH, et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. The Journal of Pathology 2001;195(4):451–6 doi 10.1002/path.1000. [DOI] [PubMed] [Google Scholar]
- 5.Piek JMJ, Verheijen RHM, Kenemans P, Massuger LF, Bulten H, van Diest PJ. BRCA1/2-related ovarian cancers are of tubal origin: a hypothesis. Gynecologic Oncology 2003;90(2):491 doi 10.1016/S0090-8258(03)00365-2. [DOI] [PubMed] [Google Scholar]
- 6.CRUM CP, MCKEON FD, XIAN W. The Oviduct and Ovarian Cancer: Causality, Clinical Implications, and “Targeted Prevention”. Clinical Obstetrics and Gynecology 2012;55(1):24–35 doi 10.1097/GRF.0b013e31824b1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am J Surg Pathol 2007;31(2):161–9 doi 10.1097/01.pas.0000213335.40358.47. [DOI] [PubMed] [Google Scholar]
- 8.Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, et al. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high‐grade serous carcinoma—evidence supporting the clonal relationship of the two lesions. The Journal of Pathology 2012;226(3):421–6 doi doi: 10.1002/path.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vang R, Shih I-M, Kurman RJ. Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology 2013;62(1):44–58 doi 10.1111/his.12046. [DOI] [PubMed] [Google Scholar]
- 10.Vang R, Visvanathan K, Gross A, Maambo E, Gupta M, Kuhn E, et al. Validation of an Algorithm for the Diagnosis of Serous Tubal Intraepithelial Carcinoma. International Journal of Gynecological Pathology 2012;31(3):243–53 doi 10.1097/PGP.0b013e31823b8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Przybycin CG, Kurman RJ, Ronnett BM, Shih I-M, Vang R. Are All Pelvic (Nonuterine) Serous Carcinomas of Tubal Origin? The American Journal of Surgical Pathology 2010;34(10):1407–16 doi 10.1097/PAS.0b013e3181ef7b16. [DOI] [PubMed] [Google Scholar]
- 12.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nature Communications 2017;8(1):1093 doi 10.1038/s41467-017-00962-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Visvanathan K, Shaw P, May BJ, Bahadirli-Talbot A, Kaushiva A, Risch H, et al. Fallopian tube lesions in women at high risk for ovarian cancer: A multicenter study. Cancer Prevention Research 2018:canprevres.0009.2018 doi 10.1158/1940-6207.Capr-18-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nature Communications 2017;8(1):990 doi 10.1038/s41467-017-01217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert MA, Pan S, Hernandez KM, Loth RM, Andrade J, Volchenboum SL, et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discovery 2016. doi 10.1158/2159-8290.Cd-16-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu R-C, Wang P, Lin S-F, Zhang M, Song Q, Chu T, et al. Genomic landscape and evolutionary trajectories of ovarian cancer precursor lesions. The Journal of Pathology 2019;248(1):41–50 doi 10.1002/path.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asaka S, Davis C, Lin S-F, Wang T-L, Heaphy CM, Shih I-M. Analysis of Telomere Lengths in p53 Signatures and Incidental Serous Tubal Intraepithelial Carcinomas Without Concurrent Ovarian Cancer. The American Journal of Surgical Pathology 2019;43(8):1083–91 doi 10.1097/pas.0000000000001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhn E, Meeker A, Wang T-L, Sehdev AS, Kurman RJ, Shih I-M. Shortened Telomeres in Serous Tubal Intraepithelial Carcinoma: An Early Event in Ovarian High-grade Serous Carcinogenesis. The American Journal of Surgical Pathology 2010;34(6):829–36 doi 10.1097/PAS.0b013e3181dcede7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krimmel JD, Schmitt MW, Harrell MI, Agnew KJ, Kennedy SR, Emond MJ, et al. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proceedings of the National Academy of Sciences 2016;113(21):6005–10 doi 10.1073/pnas.1601311113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salk JJ, Loubet-Senear K, Maritschnegg E, Valentine CC, Williams LN, Higgins JE, et al. Ultra-Sensitive TP53 Sequencing for Cancer Detection Reveals Progressive Clonal Selection in Normal Tissue over a Century of Human Lifespan. Cell Reports 2019;28(1):132–44.e3 doi 10.1016/j.celrep.2019.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nature Medicine 2019;25(12):1928–37 doi 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019;364(6444):eaaw0726 doi 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visvanathan K, Shaw P, May BJ, Bahadirli-Talbott A, Kaushiva A, Risch H, et al. Fallopian Tube Lesions in Women at High Risk for Ovarian Cancer: A Multicenter Study. Cancer Prevention Research 2018;11(11):697–706 doi 10.1158/1940-6207.Capr-18-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pisanic TR, Cope LM, Lin S-F, Yen T-T, Athamanolap P, Asaka R, et al. Methylomic Analysis of Ovarian Cancers Identifies Tumor-Specific Alterations Readily Detectable in Early Precursor Lesions. Clinical Cancer Research 2018;24(24):6536–47 doi 10.1158/1078-0432.Ccr-18-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan H, Smit A, Boissinot S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Research 2006;16(1):78–87 doi 10.1101/gr.4001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fortin J-P, Triche JTJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017;33(4):558–60 doi 10.1093/bioinformatics/btw691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohara K, Arai E, Takahashi Y, Fukamachi Y, Ito N, Maeshima AM, et al. Feasibility of methylome analysis using small amounts of genomic DNA from formalin-fixed paraffin-embedded tissue. Pathology International 2018;68(11):633–5 doi 10.1111/pin.12716. [DOI] [PubMed] [Google Scholar]
- 28.Chene G, Ouellet V, Rahimi K, Barres V, Caceres K, Meunier L, et al. DNA Damage Signaling and Apoptosis in Preinvasive Tubal Lesions of Ovarian Carcinoma. International Journal of Gynecologic Cancer 2015;25(5):761–9 doi 10.1097/igc.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 29.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359(6378):926–30 doi 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinde I, Bettegowda C, Wang Y, Wu J, Agrawal N, Shih I-M, et al. Evaluation of DNA from the Papanicolaou Test to Detect Ovarian and Endometrial Cancers. Science Translational Medicine 2013;5(167):167ra4–ra4 doi 10.1126/scitranslmed.3004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Li L, Douville C, Cohen JD, Yen T-T, Kinde I, et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Science Translational Medicine 2018;10(433):eaap8793 doi 10.1126/scitranslmed.aap8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502(7471):333–9 doi 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumour-type–specific patterns. Nature Genetics 2000;24(2):132–8 doi 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 34.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563(7732):579–83 doi 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 35.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Liu MC, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology 2020. doi 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518(7539):317–30 doi 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo S, Diep D, Plongthongkum N, Fung H-L, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nature Genetics 2017;49(4):635–42 doi 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. The Lancet Oncology 2016;17(10):1386–95 doi 10.1016/S1470-2045(16)30297-2. [DOI] [PubMed] [Google Scholar]
- 39.Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jönsson G, Olsson H, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Research 2010;12(3):R36 doi 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D, et al. Methylation Subtypes and Large-Scale Epigenetic Alterations in Gastric Cancer. Science Translational Medicine 2012;4(156):156ra40–ra40 doi 10.1126/scitranslmed.3004504. [DOI] [PubMed] [Google Scholar]
- 41.Ishak CA, Lheureux S, De Carvalho DD. DNA Methylation as a Robust Classifier of Epithelial Ovarian Cancer. Clinical Cancer Research 2019;25(19):5729–31 doi 10.1158/1078-0432.Ccr-19-1797. [DOI] [PubMed] [Google Scholar]
- 42.Bartlett TE, Chindera K, McDermott J, Breeze CE, Cooke WR, Jones A, et al. Epigenetic reprogramming of fallopian tube fimbriae in BRCA mutation carriers defines early ovarian cancer evolution. Nature Communications 2016;7:11620 doi 10.1038/ncomms11620 https://www.nature.com/articles/ncomms11620#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiegl H, Windbichler G, Mueller-Holzner E, Goebel G, Lechner M, Jacobs IJ, et al. HOXA11 DNA methylation—A novel prognostic biomarker in ovarian cancer. International Journal of Cancer 2008;123(3):725–9 doi 10.1002/ijc.23563. [DOI] [PubMed] [Google Scholar]
- 44.Richards EJ, Permuth-Wey J, Li Y, Chen YA, Coppola D, Reid BM, et al. A functional variant in HOXA11-AS , a novel long non-coding RNA, inhibits the oncogenic phenotype of epithelial ovarian cancer. Oncotarget 2015;6(33). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Wang Q, Zhao Q, Di W. MiR-124 inhibits the migration and invasion of ovarian cancer cells by targeting SphK1. Journal of Ovarian Research 2013;6(1):84 doi 10.1186/1757-2215-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Häfner N, Steinbach D, Jansen L, Diebolder H, Dürst M, Runnebaum IB. RUNX3 and CAMK2N1 hypermethylation as prognostic marker for epithelial ovarian cancer. International Journal of Cancer 2016;138(1):217–28 doi 10.1002/ijc.29690. [DOI] [PubMed] [Google Scholar]
- 47.Bae S-C, Choi J-K. Tumor suppressor activity of RUNX3. Oncogene 2004;23(24):4336–40 doi 10.1038/sj.onc.1207286. [DOI] [PubMed] [Google Scholar]
- 48.Lilja J, Zacharchenko T, Georgiadou M, Jacquemet G, De Franceschi N, Peuhu E, et al. SHANK proteins limit integrin activation by directly interacting with Rap1 and R-Ras. Nat Cell Biol 2017;19(4):292–305 doi 10.1038/ncb3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kruzelock RP, Cuevas BD, Wiener JR, Xu F-J, Yu Y, Cabeza-Arvelaiz Y, et al. Functional evidence for an ovarian cancer tumor suppressor gene on chromosome 22 by microcell-mediated chromosome transfer. Oncogene 2000;19(54):6277–85 doi 10.1038/sj.onc.1204013. [DOI] [PubMed] [Google Scholar]
- 50.Sutherland KD, Lindeman GJ, Choong DYH, Wittlin S, Brentzell L, Phillips W, et al. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene 2004;23(46):7726–33 doi 10.1038/sj.onc.1207787. [DOI] [PubMed] [Google Scholar]
- 51.McKie AB, Vaughan S, Zanini E, Okon IS, Louis L, de Sousa C, et al. The OPCML Tumor Suppressor Functions as a Cell Surface Repressor–Adaptor, Negatively Regulating Receptor Tyrosine Kinases in Epithelial Ovarian Cancer. Cancer Discovery 2012;2(2):156–71 doi 10.1158/2159-8290.Cd-11-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All Illumina MethylationEPIC BeadArray data from this study , and our prior study (24), have been deposited to the NCBI GEO database as a SuperSeries (accession number GSE155761). An R Markdown file detailing all bioinformatic analyses is available for download at https://github.com/lesliecope/STICmanuscript.