

Abstract
The progressive increase in lifespan over the past century carries with it some adversity related to the accompanying burden of debilitating diseases prevalent in the older population. This review focuses on oxidative stress as a major mechanism limiting longevity in general, and healthful aging, in particular. Accordingly, the first goal of this review is to discuss the role of oxidative stress in limiting longevity, and compare healthful aging and its mechanisms in different longevity models. Secondly, we discuss common signaling pathways involved in protection against oxidative stress in aging and in the associated diseases of aging, e.g., neurological, cardiovascular and metabolic diseases, and cancer. Much of the literature has focused on murine models of longevity, which will be discussed first, followed by a comparison with human models of longevity and their relationship to oxidative stress protection. Finally, we discuss the extent to which the different longevity models exhibit the healthful aging features through physiological protective mechanisms related to exercise tolerance and increased β-adrenergic signaling and also protection against diabetes and other metabolic diseases, obesity, cancer, neurological diseases, aging-induced cardiomyopathy, cardiac stress and osteoporosis.
Keywords: Healthful Longevity, Oxidative Stress, Reactive Oxygen Species
I. Introduction
There has been a progressive increase in lifespan over the past century. At the turn of the 20th century the average lifespan was 48 years for men and 51 years for women. More than 100 years of progress has led to a life expectancy of 76 years for men and 81 years for women as of 2017 (Xu, Murphy et al. 2020). Prima facie, these data are encouraging, as almost everyone wishes to live longer. However, an extended lifespan carries with it the burden of concomitant debilitating diseases and accompanying morbidity, which limit ambulation and enjoyment of life. It is important to recognize that most of these diseases are mediated, in part, by increased oxidative stress (Beck 2000, Cervantes Gracia, Llanas-Cornejo et al. 2017, Guzik and Touyz 2017, Butterfield and Halliwell 2019, Goncalves and Romeiro 2019, Hosseinabadi and Khanjani 2019, Katerji, Filippova et al. 2019, Gyuraszova, Gurecka et al. 2020, Zhang, Li et al. 2020). Oxidative stress is thought to be a major mechanism limiting longevity in general (Finkel and Holbrook 2000, Yoon, Yun et al. 2002), and limiting healthful longevity, in particular, which has not been examined in depth. Accordingly, the first goal of this review is to discuss the role of oxidative stress in limiting healthful longevity. The second goal is to discuss the extent to which the different longevity models are protected against the associated disabling diseases of aging, e.g., diabetes and other metabolic diseases, obesity, cancer, neurological disorders, aging-induced cardiomyopathy, cardiac stress, osteoporosis, reduced exercise tolerance and increased β-adrenergic signaling.
II. Oxidative Stress in Aging
Oxidative stress impairs healthful aging (Szilard 1959), and accordingly, protection against oxidative stress is a common mechanism mediating the phenotype observed in animal models of longevity (Table I). Reactive oxygen species (ROS) are mainly a byproduct of oxygen metabolism and adenosine triphosphate (ATP) production. When ROS exceed antioxidant capacity, as occurs with radiation exposure, high fat diet, sugar, processed foods, cigarette smoking, other tobacco products, alcohol consumption, certain medications, pollution, exposure to pesticides or industrial chemicals, they induce oxidative stress that is directly linked to the development of many diseases that limit healthful aging (Valko, Leibfritz et al. 2007, Alfadda and Sallam 2012). A major mechanism by which oxidative stress causes tissue damage is through ROS induced apoptosis and necrosis by opening of the mitochondrial membrane permeability transition pore and releasing factors which limit cell survival, such as cytochrome c (Wei and Lee 2002). In order to maintain cellular equilibrium, mammalian cells scavenge ROS to nontoxic forms through a complex antioxidant defense system that includes superoxide dismutase (SOD), catalase, and glutathione peroxidase (Gpx) (Wei and Lee 2002, Valko, Leibfritz et al. 2007, Alfadda and Sallam 2012). Several cellular/biochemical signaling mechanisms mediate increase oxidative stress in aging; one mechanism is a decrease in mitochondrial efficiency, resulting increased ROS production, in order to maintain sufficient ATP production. The significance of the mitochondria in aging arises from their particular susceptibility to DNA damage. Most of an organism’s DNA is stored in the cell’s nucleus which, because of its membrane, is more protected from DNA damage by free radicals. The mitochondrial DNA (mtDNA) located within mitochondria, lacks protection provided by the nucleosomes and DNA repair mechanisms; which makes mtDNA highly susceptible to oxidant induced damage during aging (Mikhed, Daiber et al. 2015). ROS also play a vital role in several physiological functions as a second messenger and are critical in neutrophil function, impairing the host defense against micro-organisms and in the innate immune response. Neutrophils express and release cytokines, which in turn, amplify inflammatory reactions in several other cell types (Winterbourn, Kettle et al. 2016). Furthermore, similar to ROS, free-radicals also result in oxidative stress (Lobo, Patil et al. 2010), and the activity of free-radical scavenging enzymes diminishes with advancing age and contributes to the adverse effects of oxidative stress with aging (Inal, Kanbak et al. 2001).
Table I:
Oxidative Stress Protection in Longevity Models
| Longevity Model | Median Survival Age Wild Type (Months) | % Increase Lifespan Compared to Wild Type |
|---|---|---|
| Longevity Models with Known Protection Against Oxidative Stress | ||
| Calorie Restriction | 30 | Up to 50 |
| Snell Dwarf | 18 | 50 |
| Adenylyl Cyclase Type 5 Knockout (AC5 KO) | 25 | 32 |
| Growth Hormone Receptor Knockout (GHR KO) | 27 | 31 |
| p66 SHC-Transforming Protein 1 (p66 shc) Knockout | 26 | 30 |
| Angiotensin II Receptor Type 1 Alpha Knockout (Agtr1 α−/− KO) | 26 | 26 |
| Regulator of G Protein Signaling 14 Knockout (RGS14 KO) | 23 | 26 |
| Insulin-like Growth Factor Type 1 Receptor Hetero (IGF-1R+/− Hetero) | 21 | 24 |
| Klotho Overexpression | 32 | 22 |
| Mitochondrial Targeted Catalase (MCAT) Overexpression | 25 | 19 |
| Ribosomal Protein S6 Kinase β−1 (S6K1) Knockout | 28 | 19 |
| A Longevity Model Not Protected Against Oxidative Stress | ||
| Growth Hormone Receptor/Binding Protein Knockout (GHR/BP KO) | 23 | 47 |
| Longevity Models With Oxidative Stress Protection Controversial | ||
| Ames Dwarf | 21 | 57 |
| Mitochondrial 5-Demethoxyubiquinone Hydroxylase Hetero (Mclk1+/−) | 22 | 31 |
| Insulin Receptor Substrate 1 Knockout (IRS1 KO) | 24 | 18 |
| Longevity Models With Oxidative Stress Not Studied | ||
| Phosphoenolpyruvate Carboxykinase (PEPCK-C) Overexpression | Age unspecified, but the mice still reproduce after age 30 months | |
| Pregnancy-Associated Plasma Protein-A Knockout (PAPPA KO) | 21 | 26 |
There are genetically modified animal models designed to help elucidate the role of the antioxidant system. Manganese superoxide dismutase (MnSOD), which is also known as superoxide dismutase 2 (SOD2), is one of the main mitochondrial antioxidant proteins. The genetic ablation of SOD2 leads to early postnatal death in mice which exhibit a dilated cardiomyopathy, metabolic acidosis, steatosis of the liver and skeletal muscle, increased oxidative damage, and mitochondrial enzymatic abnormalities (Li, Huang et al. 1995, Melov, Coskun et al. 1999). Partial loss of SOD2 (SOD2 heterozygous mice) also induces increased oxidative stress (Kokoszka, Coskun et al. 2001). Treatment of SOD2 knockout (KO) mice with a synthetic SOD mimetic rescues their mitochondrial defects and prolongs their survival (Melov, Schneider et al. 1998). Other KO mice for other antioxidant proteins, e.g., Cu/Zn SOD or SOD1, also exhibit increased oxidative damage of DNA, lipid peroxidation, and an increase in mitochondrial aconitase, which is a biomarker of oxidative stress (Elchuri, Oberley et al. 2005). In contrast, mice with SOD1 overexpression tend to be more resistant to paraquat induced oxidative stress (Mele, Van Remmen et al. 2006). Similarly, investigations of other antioxidants, such as catalase, show that increasing the body’s defense against free radicals can extend life (Table I).
In summary, the mechanism of oxidative stress induced in aging is multifactorial, with mitochondrial damage playing a significant role. The final common pathway for premature cell death involves apoptosis, necrosis, and cell cycle arrest (Slater, Stefan et al. 1995, Sastre, Pallardo et al. 2003, Barzilai and Yamamoto 2004), all of which induce cell death (Choi, Kim et al. 2001, Yuan, Long et al. 2012) (Figures 1 and 2).
Figure 1:
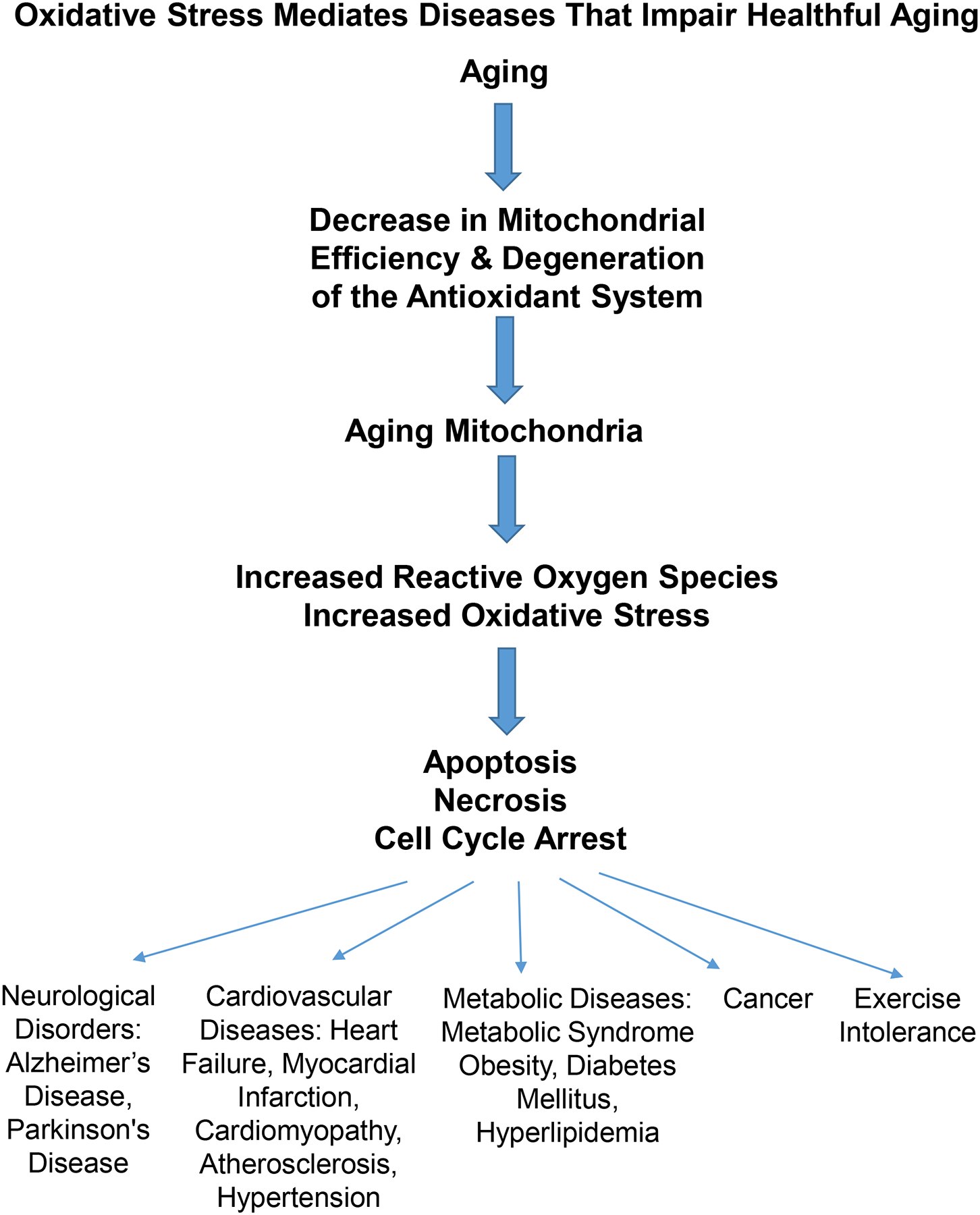
In aging, mitochondrial dysfunction and degeneration of the antioxidant system induce production of reactive oxygen species, which result in increased oxidative stress and cell dysfunction and death (e.g. apoptosis, necrosis, and cell cycle arrest). These deficiencies are involved in mediation of neurological disorders, cardiovascular diseases, metabolic disorders, exercise intolerance and cancer.
Figure 2:
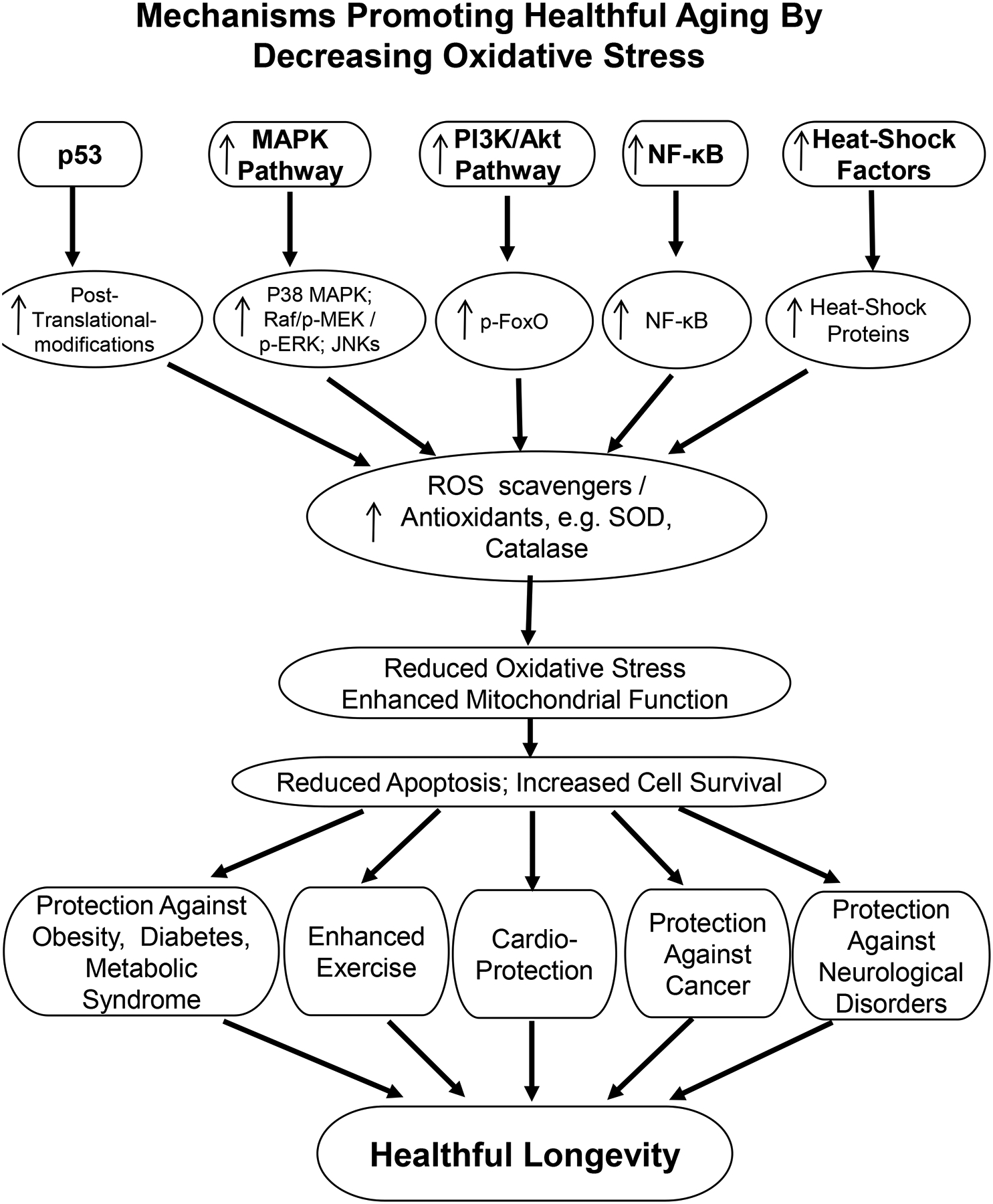
Activated MAPK, PI3K/Akt pathways, NF-κB, and heat-shock factors lead to upregulation of their downstream pathways, all acting to increase anti-oxidants, which then reduce oxidative stress and increase mitochondrial function, resulting in increased cell survival and healthful longevity. p53 has dual roles in oxidative stress. In this figure, under low oxidative stress condition, p53 activation provides an antioxidant defense to protect against oxidative stress.
III. Common Signaling Pathways Involved in Oxidative Stress Protection (Figure 2):
The major mechanisms mediating protection against oxidative stress are noted in Figure 2 and are discussed next. Many, but not all, of these mechanisms mediating oxidative stress protection are involved in each model of longevity with oxidative stress protection. These are the major mechanisms that lead to reduced oxidative stress, enhanced mitochondrial function and reduced apoptosis and cell death, ultimately culminating in protection against most diseases, which increase morbidity and mortality in the aging population.
Mitogen-Activated Protein Kinase (MAPK) Signaling Pathways:
Activator protein-1 (AP-1) activity is regulated by a variety of stimuli, which include oxidative stress and signals through the MAPK cascades leading to extracellular signal-related kinases (ERKs), c-jun NH2-terminal kinases (JNKs) and p38 MAPK. These pathways are known to be influenced not only by receptor ligand interactions, but also by different stressors placed on the cell. One type of stress that induces activation of MAPK pathways is the oxidative stress caused by ROS, which in turn activate the Proto-Oncogene, Serine/Threonine Kinase (Raf)/MEK/ERK, JNKs, or p38 MAPKs (Son, Cheong et al. 2011).
Phosphoinositide 3-Kinases (PI3K)/ Protein Kinase B (Akt) Pathway:
Oxidative stress activation of the PI3K/Akt pathway targets the nuclear factor kappa-light-chain-enhancer of the activated B cells (NF-κB) site in the SOD1 promoter, resulting in upregulation of SOD1 gene expression that lowers ROS levels and protects cells against oxidative stress (Noshita, Sugawara et al. 2003, Rojo, Salinas et al. 2004). The inhibition of Akt activation reduces production of superoxide and SOD1 levels (Song, Narasimhan et al. 2008). Aberrant PI3K/Akt signaling drives many of the molecular mechanisms to increase ROS levels by modulation of mitochondrial bioenergetics and activation of NADPH oxidases (Murphy 2009). NADPH oxidases activated by PI3K/Akt contribute to higher ROS levels in cancer cells (Chatterjee, Browning et al. 2012). The Forkhead box O (FoxO) family is an important regulator of the cellular stress response and is involved in the cellular antioxidant defense, and is downstream of the PI3K/Akt pathway (Brunet, Bonni et al. 1999, Martins, Lithgow et al. 2016). Phosphorylation of FoxOs induced by PI3K/Akt stimulates and regulates the transcription and expression of antioxidant enzymes in mitochondria and plasma, e.g., MnSOD, catalase, thioredoxin and stress-related gene products (Nemoto and Finkel 2002, Tran, Brunet et al. 2003, Olmos, Sanchez-Gomez et al. 2013). FoxOs also regulate the expression of ROS scavengers in response to ROS (Kops, Dansen et al. 2002, Nemoto and Finkel 2002). Reduced expression of FoxO transcription factors increase susceptibility to cell death induced by oxidative stress (Akasaki, Alvarez-Garcia et al. 2014). In addition, transcriptional and post-transcriptional control of FoxO gene expression is sensitive to ROS (Klotz, Sanchez-Ramos et al. 2015). Oxidative stress mediated by ROS is a key mediator of the acetylation and ubiquitination state of FoxOs (Brunet, Sweeney et al. 2004, Frescas, Valenti et al. 2005, Dansen, Smits et al. 2009).
Heat Shock Proteins:
Heat shock proteins are induced by a family of heat shock transcription factors and are known for their cytoprotective role in response to a variety of cellular insults through their chaperoning activities, which include protein folding, assembly, and the degradation of irreparable peptides (Mayer and Bukau 2005, Shiber and Ravid 2014). DNA fragmentation is induced in cells undergoing ROS-mediated genotoxicity, but this effect is rescued with the addition of the Hsp70 family suggesting that the cytoprotective effects of heat shock proteins protect against DNA breaks in response to ROS-induced insults (Jacquier-Sarlin, Fuller et al. 1994). Heat shock proteins work hand-in-hand with the antioxidant system to inhibit or neutralize the cellular effects of ROS (Trott, West et al. 2008, Wu, Biggar et al. 2015). Several studies investigated the role of heat shock proteins in reducing oxidative stress by increasing ROS scavenger genes (Wyttenbach, Sauvageot et al. 2002, Driedonks, Xu et al. 2015, Ghosh, Sarkar et al. 2018).
p53:
P53 is a tumor suppressor protein and regulates the expression of genes involved in cell cycle regulation, redox homeostasis, DNA replication and repair, autophagy, and apoptosis (Levine, Feng et al. 2006, Holley and St Clair 2009, Saleem, Adhihetty et al. 2009). In addition, p53 is also an important regulator of mitochondrial biogenesis (Yoshida, Izumi et al. 2003, Saleem, Adhihetty et al. 2009). This is observed in p53 KO mice, which have impaired mitochondrial function and elevated ROS (Saleem, Adhihetty et al. 2009). Furthermore, p53 has dual roles with respect to mediating oxidative stress. Under no oxidative stress or low oxidative stress conditions, p53 and its negative regulators are post-translationally modified, and lead to p53 activation, which provides an antioxidant defense (Liu and Xu 2011). For example, fasting from calorie restriction induced oxidative stress, activates p53 to enhance both antioxidant production and fatty acid oxidation mechanisms to reduce the overall oxidative stress (Beyfuss and Hood 2018). In contrast, under conditions of severe oxidative stress, p53 is activated to induce pro-oxidant genes and inhibit antioxidant genes to further increase ROS levels and sensitize cells to oxidative stress, which will then eliminate damaged cells via p53- dependent apoptosis and senescence. (Johnson, Yu et al. 1996, Donald, Sun et al. 2001, Drane, Bravard et al. 2001).
NF-κB:
NF-κB plays a protective role in response to oxidative stress by suppressing ROS accumulation (Lingappan 2018). One of the main signaling pathways that intersects with NF-κB related to ROS and cell death is the crosstalk that occurs between NF-κB and JNK (Morgan and Liu 2011). Decreases in NF-κB-mediated inhibition of JNK activation contributes to TNF-alpha-induced apoptosis (Tang, Tang et al. 2002). Thus, the anti-apoptotic function of NF-κB is also mediated by downregulation of the JNK pathway. NF-κB also has many anti-oxidant targets such as MnSOD, Glutathione S-transferase pi, Metallothionein-3, NAD(P)H dehydrogenase [quinone]1, heme oxygenase-1 and Gpx-1 (Kairisalo, Korhonen et al. 2007, Morgan and Liu 2011). The NF-κB pathway also has a pro-oxidant role by induction of genes such as NADPH oxidase 2 (NOX2) subunit gp91phox (Anrather, Racchumi et al. 2006).
IV. Oxidative Stress in Longevity Models
To further understand the importance of protection against oxidative stress in aging, the major longevity models and their relation to oxidative stress protection are enumerated in Table I followed by a description of the animal and human longevity models.
Longevity Animal Models (Tables I and II)
Table II-a:
Protective Features of Longevity Models Protected Against Oxidative Stress
| MODEL | IMPROVED EXERCISE | OBESITY PROTECTION | DIABETES PROTECTION | CV STRESS PROTECTION | CANCER PROTECTION | PROTECTION OBSERVED IN OLD AGE | DECREASED β-ADRENERGIC SIGNALING |
|---|---|---|---|---|---|---|---|
| Human Models of Longevity | + | + | + | + | + | + | + |
| Calorie Restriction | − | + | + | + | + | + | − |
| Snell Dwarf | ? | +/− | + | ? | + | + | − |
| Adenylyl Cyclase 5 KO | + | + | + | + | + | + | + |
| Growth Hormone Receptor (GHR) KO | ? | − | + | + | + | ? | ? |
| p66shc Protein KO | +/− | +/− | + | + | + | − | ? |
| Angiotensin II Receptor Type 1 alpha (Agtr1α) KO | ? | + | ? | +/− | + | ? | ? |
| Regulator of G Protein Signaling 14 KO | + | + | + | + | + | + | + |
| Insulin Like Growth Factor Type 1 Receptor +/− | ? | +/− | − | ? | +/− | ? | ? |
| Klotho Overexpression | ? | + | − | + | + | − | ? |
| Mitochondrial Targeted Catalase (MCAT) Overexpression | + | − | + | + | + | +/− | + |
| Ribosomal Protein S6 Kinase β-1 (S6K1) KO | + | + | + | ? | + | + | ? |
Most longevity models have evidence of protection against oxidative stress. Accordingly, this section and Table I are divided into those longevity models with known oxidative stress protection, those longevity models lacking such protection, and models where protection is controversial or has not been studied. Table I is organized by listing the longevity models from those with longest % increase in median survival, compared to their wild type (WT) littermates, to those with the least. This section and Table I include the median age of survival in the longevity models and the median age of survival in their WT littermates. Table II compares the protective features, observed in healthful aging, in these longevity models. The data for this review were collected by examining the literature for all longevity models and then determining whether they are protected from oxidative stress and those mechanisms and diseases that involve oxidative stress and the morbidity and mortality that limit healthful longevity.
Limitations of Longevity Models
Many longevity models discussed below only reflect longevity compared to their WT, and not compared with most mouse strains studied. When evaluating the differences in lifespan of mice from different genetic backgrounds, it is also necessary to understand the inherent differences in lifespan of the background inbred strains of mice that are used. Some of the most commonly used inbred strains include C57BL/6, BALB/c, and DBA/2J (Overbeek 2014). These mice exhibit differences in their median lifespan. For example, male and female BALB/cJ mice have a median lifespan of 19 and 18 months respectively, whereas male and female C57BL/6J male mice have a median lifespan of 22 and 21 months respectively, while both male and female DBA/2J male mice have a median lifespan of 22 months (Storer 1966).
Both molecular and environmental factors affect lifespan. For example, it has been shown that differences in lifespan among strains may be due to the plasma IGF1 level, which has been shown to be negatively correlated with median lifespan (Yuan, Tsaih et al. 2009). IGF1 is also known as an important regulator of longevity in transgenic mice, including Ames Dwarf mice, GHR KO mice, p66shc KO mice, and IGF-1R+/− mice (Brown-Borg, Borg et al. 1996, Migliaccio, Giorgio et al. 1999, Coschigano, Holland et al. 2003, Holzenberger, Dupont et al. 2003). Lifespan may also be affected by the environment, e.g. housing conditions. The median lifespan from some WT strains may vary among different studies, e.g., median lifespan of C57BL/6J mice was reported as 21–22 months in one study (Storer 1966), and 28–29 months in another study (Yuan, Tsaih et al. 2009). In our laboratory, we found the median lifespan of this strain to be around 24–25 months (Yan, Vatner et al. 2007). Another study showed that environmental enrichment, specifically auditory and acoustic stimulation, prolonged lifespan by roughly 17% in C57BL/6J mice (Yamashita, Kawai et al. 2018).
Thus, differences in longevity across transgenic models using different background inbred strains presents an additional layer of complexity in evaluating optimal models of longevity. Another limitation to mouse longevity studies is that in many of the longevity models there is a discrepancy between the average increase in lifespan compared to WT, depending on whether lifespan is evaluated by estimating median lifespan from the survival curve at 50% survival rate, vs. the longevity data noted in the publication (Schriner, Linford et al. 2005, Ortega-Molina, Efeyan et al. 2012). The reasons for these discrepancies are not clear. In addition, there are some mouse longevity models with increased average lifespan of males and females less than 16%. These models are not discussed and include: 1) models that are protected against oxidative stress: sirtuin 1 overexpressed mice, metallothionein overexpressed mice; 2) models that are not protected against oxidative stress: α-murine urokinase-type plasminogen activator overexpressed mice, glutathione peroxidase 4 hetero KO mice; 3) models that have controversial data on protection against oxidative stress: macrophage migration inhibitory factor KO mice, fat-specific insulin receptor KO mice; and 4) models with oxidative stress not studied: sirtuin 6 overexpressed mice, RIIβ KO mice, phosphatase and tensin overexpressed mice, and hypocretin neurons-specific uncoupling protein 2 overexpressed mice (Miskin and Masos 1997, Bluher, Kahn et al. 2003, Conti, Sanchez-Alavez et al. 2006, Yang, Doser et al. 2006, Ran, Liang et al. 2007, Enns, Morton et al. 2009, Harper, Wilkinson et al. 2010, Kanfi, Naiman et al. 2012, Ortega-Molina, Efeyan et al. 2012, Satoh, Brace et al. 2013, Steckler, Shabtay-Yanai et al. 2016).
(1). Longevity Models with Known Protection Against Oxidative Stress:
Calorie Restriction in Mice (Median Age of Survival of Calorie Restriction is Increased up to 50% Compared to Normal Diet Median Age of Survival of 30 Months):
Calorie restriction, extends lifespan up to 50% in rodents (McCay, Crowell et al. 1989, Fontana, Partridge et al. 2010) and generally ameliorates disease in mammals (Fontana, Partridge et al. 2010). The effect on lifespan with calorie restriction may vary by sex and genetic background in rodents (Liao, Rikke et al. 2010). The majority of studies showed calorie restriction leads to enhanced mitochondrial biogenesis and function as well as lower ROS production and greater antioxidant enzyme activity in wild-type mice (Sohal, Ku et al. 1994, Lopez-Lluch, Hunt et al. 2006, Swindell 2009, Qiu, Brown et al. 2010, Martin-Montalvo, Villalba et al. 2011, Chen, Hagopian et al. 2012, Donato, Walker et al. 2013, Walsh, Shi et al. 2014, Mitchell, Delville et al. 2015, Noyan, El-Mounayri et al. 2015) and in genetically altered mice (Harrison, Archer et al. 1984, Guo, Mitchell-Raymundo et al. 2002). However, differences in oxidative stress levels among different organs exists with calorie restriction (Sohal, Agarwal et al. 1994, Gong, Shang et al. 1997, Rebrin, Kamzalov et al. 2003, Rebrin, Forster et al. 2007, Walsh, Shi et al. 2014), and differences have been noted that can be attributed to the duration of calorie restriction (Wu, Sun et al. 2003). There are even some studies that reported calorie restriction either reduced lifespan in wild-type mice (Harrison and Archer 1987), or failed to further prolong lifespan in longevity mouse models (Bonkowski, Rocha et al. 2006, Bonkowski, Dominici et al. 2009). The most striking example is when calorie restriction was added to a mouse model of longevity, the AC5 KO mice. All the AC5 KO mice on a calorie restricted diet, that normally increases longevity, died within a month due to higher metabolism of the AC5 KO mice, which could not tolerate reduction in food intake, indicating that calorie restriction is deleterious in models of increased metabolism (Yan, Park et al. 2012).
Calorie Restriction Protects Against Cardiovascular Diseases:
Long term calorie restriction can alter the gene expression profile related to protection against myocardial remodeling, fibrosis and decreases in contractility (Swindell 2009, Yan, Gao et al. 2013). Short term calorie restriction results in smaller myocardial infarct size, improved cardiac function and survival in mice with myocardial infarction, as well as enrichment of anti-oxidative stress pathways (Noyan, El-Mounayri et al. 2015). Calorie restriction also reduces oxidative stress and atherosclerosis in genetically altered atherosclerosis mice (Guo, Mitchell-Raymundo et al. 2002, Yang, Zeng et al. 2020).
Calorie Restriction Protects Against Metabolic Diseases:
Mice undergoing calorie restriction exhibit lower bodyweight (Schmeisser, Priebe et al. 2013), a reduction in body fat (Yan, Gao et al. 2013, Mitchell, Tang et al. 2015), and an improvement in glucose homeostasis with lower oxidative stress (Mitchell, Delville et al. 2015). In one study, calorie restriction also lowered fasting blood glucose levels in diabetic mice (Wei, Zhao et al. 2019). Furthermore, a recent metabolomics study demonstrated brown adipose tissue activation in mice with calorie restriction (Green, Mitchell et al. 2020). Brown adipose tissue is a novel mechanism of longevity and protection against oxidative stress (Vatner, Zhang et al. 2018) and also protects against obesity and diabetes (Stanford, Middelbeek et al. 2013).
Calorie Restriction Protects Against Cancer:
The protective effects of calorie restriction on the incidence of cancer have been observed in numerous studies of spontaneous and chemically induced tumors in rodents (Weindruch, Walford et al. 1986, Hursting, Smith et al. 2010, Longo and Fontana 2010, De Lorenzo, Baljinnyam et al. 2011). Decreasing growth factor signaling as well as vascular perturbations and inflammation attenuates cancer risk and progression (Hursting, Dunlap et al. 2013). The model is further protected against cancer when calorie restriction is maintained throughout the entire lifespan compared to when initiated in adulthood (Hursting, Smith et al. 2010). Although the overall incidence of tumors is not reduced with calorie restriction later in life, tumorigenesis is delayed and survival is improved (Dhahbi, Kim et al. 2004). Calorie restriction prevents tumorigenesis by reducing metabolic rate and oxidative damage (Martin-Montalvo, Villalba et al. 2011). A review of rodent studies on cancer and calorie restriction revealed the protective role of calorie restriction against cancer in 40 out of 44 studies whereas only 4 studies reported negative or no difference results in mammary tumors (Tagliaferro, Ronan et al. 1996), prostate cancer (Birt, Pour et al. 1997, McCormick, Johnson et al. 2007), and intestinal tumors (Tsao, Dudley et al. 2002).
Non-human primate studies and human studies on calorie restriction are discussed in Sections VI and VII.
Snell Dwarf Mice (Median Age of Survival, of Snell Dwarf is Increased by 50% in Females, but Is Decreased in Males by 6% Compared to WT Median Age of Survival of 18 Months):
Snell dwarf mice have a mutation in the gene encoding pituitary-specific positive transcription factor 1 (Pit-1) and result in an altered hormone profile featuring primary deficiencies in growth hormone, thyroid-stimulating hormone, and prolactin and thus secondary deficiencies in insulin-like growth factor 1 and thyroxine (Bartke and Brown-Borg 2004). They exhibit longevity compared to their WT, with gender differences also apparent. In females, maximal lifespan is increased by 38% and median lifespan is increased by 50%; whereas in males, maximal lifespan is increased by 25% but median lifespan is 6% less compared to their WT mice (Flurkey, Papaconstantinou et al. 2002).
Snell dwarves’ protection from oxidative stress is two-fold. First, they have an altered response to oxidative stress with reduced MEK-ERK cascade activation and a lack of phosphorylation of c-Jun at Ser63 as well as heightened catalase response to hydrogen peroxide. Second, Snell dwarves have enhanced base excision repair activity (Madsen, Hsieh et al. 2004, Page, Salmon et al. 2009). Fibroblast cells from Snell dwarf mice also have reduced generation of ROS, improved antioxidant defenses, increased resistance to oxidative stress, and reduced oxidative damage (Murakami, Salmon et al. 2003, Bartke and Brown-Borg 2004, Salmon, Murakami et al. 2005, Maynard and Miller 2006). Snell Dwarf mice are protected against diabetes and cancer, with reduced bodyweight in the adults compared to WT mice, and with elevated β3-adrenergic signaling (Bielschowsky and Bielschowsky 1959, Flurkey, Papaconstantinou et al. 2002, Boylston, Gerstner et al. 2004, Alderman, Flurkey et al. 2009, Bartke and Westbrook 2012, Cannavo and Koch 2017, Simona Negreș, Cornel Chiriță et al. 2017). β3-adrenergic agonist drugs have been suggested for treatment of diabetes and heart failure, and in contrast to β1-adrenergic signaling, β3-adrenergic stimulation reduces myocardial contractility and myocardial oxygen consumption (Gauthier, Tavernier et al. 1996, Kitamura, Onishi et al. 2000, Varghese, Harrison et al. 2000, Cheng, Zhang et al. 2001, Masutani, Cheng et al. 2013, Belge, Hammond et al. 2014, Balligand 2016), salutary features for longevity. Exercise capacity and cardioprotection have not been examined in this model.
Adenylyl Cyclase Type 5 (AC5) KO Mice (Median Age of Survival of AC5 KO is Increased by 32% Compared to WT With Median Age of Survival of 25 Months):
AC5 is one of 10 AC isoforms, which are key to sympathetic regulation and cyclic AMP (cAMP) generation (Vatner, Pachon et al. 2015). AC5 KO mice extend lifespan by 32% compared to WT with no gender differences (Yan, Vatner et al. 2007). AC5 KO mice are protected against oxidative stress via the Sirtuin (SIRT) 1 /FoxO3a and the MEK/ERK pathways (Yan, Vatner et al. 2007, Lai, Yan et al. 2013) (Figure 2). AC5 KO mice, with their decreased β-adrenergic signaling, exhibit enhanced exercise capacity in both young and old animals, are protected against obesity and diabetes when challenged with high fat diet against osteoporosis, cancer, aging-induced cardiomyopathy, and heart failure induced by pressure overload or chronic catecholamine cardiomyopathy (Okumura, Takagi et al. 2003, Yan, Vatner et al. 2007, Lai, Yan et al. 2013, De Lorenzo, Chen et al. 2014, Ho, Zhao et al. 2015, Vatner, Yan et al. 2015, Bravo, Vatner et al. 2016). It is important to note that the protective features of decreased β-adrenergic signaling, primarily refer to β1 and β2-adrenergic signaling, which are the major β-adrenergic subtypes responsible for its adverse effects which include tachycardia, increased myocardial contractility and increased myocardial oxygen consumption, all of which act to intensify the adverse effects of heart failure and myocardial ischemia. AC5 KO mice also share organ specific mechanisms in the liver, heart, brain and skeletal muscle via genes and pathways common to the healthful aging model of calorie restriction (Yan, Park et al. 2012). This is particularly related to the metabolic phenotype, which suggests a unified theory for longevity and stress resistance. A pharmacological inhibitor of AC5 also exhibits protection against myocardial ischemia-reperfusion injury (Zhang, Levy et al. 2018).
Growth Hormone Receptor (GHR) KO Mice (Median Age of Survival, of GHR KO is: Increased by 31% Compared to WT Median Age of Survival of 27 Months):
GHR is known for regulation of growth and other biological functions including metabolism and control of physiological processes related to the cardiovascular, renal, and reproductive systems (Dehkhoda, Lee et al. 2018). GHR KO mice exhibit an increased lifespan in both male and female by 40% and 21% respectively, Ola-BALB/cJ background mice (Coschigano, Holland et al. 2003). However, when the GHR gene is disrupted at 6 weeks of age, GHR KO female mice, but not male mice, exhibit increased lifespan (Junnila, Duran-Ortiz et al. 2016). Calorie restriction (Bonkowski, Dominici et al. 2009) and intermittent fasting (Arum, Bonkowski et al. 2009) failed to extend lifespan further in GHR KO mice. Under hyperthermic conditions, female GHR KO mice exhibit a 14% increase in lifespan compared to when they are housed at room temperature, while male GHR KO mice do not exhibit increased lifespan in the hyperthermic environment. (Fang, McFadden et al. 2020). The further increase in lifespan in female GHR KO mice under hyperthermic conditions, may relate to improved insulin sensitivity when compared to male GHR KO mice (Fang, McFadden et al. 2020), though the mechanisms behind the gender differences in GHR KO mice need further study. The mechanism mediating longevity in GHR KO mice is complex, but involves upregulation of free-radical scavengers, increased insulin sensitivity, and activation of mitochondrial biogenesis through increased levels of AMPK, sirtuins, and PGC1α levels (Basu, Qian et al. 2018). In GHR KO mice, SOD2 and FoxO1 levels are also upregulated and p38 MAPK is activated (Al-Regaiey, Masternak et al. 2005). These mechanisms lead to protection against a variety of age-related diseases. GHR KO mice exhibit protection against diabetes, with increased insulin sensitivity and glucose tolerance, but do not show protection against obesity (List, Sackmann-Sala et al. 2011). GHR KO mice also show protection against a variety of neoplasms (Ikeno, Hubbard et al. 2009), hypertension and cardiomegaly (Egecioglu, Andersson et al. 2007). The effects of GHR KO on exercise performance and β-adrenergic signaling have yet to be examined.
p66 SHC-Transforming Protein 1 (p66shc) KO Mice (Male Median Age of Survival, of p66 shc KO is Increased by 30% Compared to WT Median Age of Survival of 26 Months):
p66 SHC is a protein isoform of the SHC1 gene located on chromosome 1 and has been shown to be involved in maintaining the redox balance (Nemoto and Finkel 2002, Giorgio, Berry et al. 2012) which makes it an important regulator of oxidative stress, apoptosis, and cellular proliferation (Galimov 2010). P66shc is an adapter protein, which induces the production of ROS through direct upregulation of NADPH oxidase and inhibition of antioxidant enzymes (Galimov 2010). This leads to ROS as secondary messengers resulting in apoptosis of cells. Deletion of the p66 SHC gene (p66shc KO) eliminates these features, decreases oxidative stress (Napoli, Martin-Padura et al. 2003, Menini, Amadio et al. 2006, Carpi, Menabo et al. 2009) with activation of NF-κB signaling (Menini, Amadio et al. 2006) and increases lifespan in male mice by up to 30% when compared to WT in normal housing environments (Migliaccio, Giorgio et al. 1999). Lifespan was not investigated in female mice in this model. Interestingly, when studied under natural conditions, i.e. non-laboratory conditions, low temperature, and reduced food availability, p66shc KO mice exhibited shorter survival (Giorgio, Berry et al. 2012). This is most likely due to the function of p66shc as important adaptor protein to increased environmental stress and its absence can be detrimental in situations of environmental stress, e.g., extreme temperatures and lack of food. Therefore, deletion of p66shc proves to be an effective model in extending lifespan under conditions of ambient temperature, lack of predators, and excessive food availability.
Furthermore, p66shc KO mice also reduce the onset of age-related diseases including diabetic glomerulopathy (Menini, Amadio et al. 2006), cardiac stress (Carpi, Menabo et al. 2009), and vascular disease (Napoli, Martin-Padura et al. 2003). Additionally, p66shc deletion has been shown to be associated with decreases in proliferation of ovarian, breast, and prostate cancer (Veeramani, Igawa et al. 2005, Bhat, Baba et al. 2014, Muniyan, Chou et al. 2015). In terms of obesity, the protection of p66shc KO mice is not as clear. While these mice exhibit a decreased body mass (Berniakovich, Trinei et al. 2008, Ciciliot, Albiero et al. 2015), they do not exhibit the improved glucose tolerance and insulin sensitivity that would be expected (Ciciliot, Albiero et al. 2015). Similar results were found in humans, such that patients with low levels of p66shc were leaner, but were not protected from diabetes (Ciciliot, Albiero et al. 2015). Exercise capacity is also complex. On a normal diet no difference in exercise capacity was noted between p66shc KO mice and their WT, but on a high fat diet the p66shc KO mice maintain their exercise capacity, whereas WT on a high fat diet gain weight and exhibit reduced exercise capacity with greater ROS production (Granatiero, Gherardi et al. 2017), suggesting p66shc KO mice can tolerate the increased ROS associated with high-fat diets. β-adrenergic signaling has not been examined.
Angiotensin II Receptor Type 1a (Agtr1a) KO Mice (Male Median Age of Survival, of Agtr1a KO is Increased by 26% Compared to WT with Median Age of Survival of 26 Months):
Angiotensin II receptor Type 1 is a central component of the renin-angiotensin system and mediates the biological actions of angiotensin II (Guo, Sun et al. 2001). Lack of Agtr1a extends lifespan by 26% in male mice, reduces oxidative damage and prevents age-induced mitochondrial loss with upregulated expression of the pro-survival genes nicotinamide phosphoribosyl transferase and SIRT3 (Benigni, Corna et al. 2009). There are no data available on lifespan in female Agtr1a KO mice. This reduction in oxidative stress is mediated by a reduction in Ang-II-dependent upregulation of p47phox which functions to activate NAD(P)H oxidase, resulting in increased endothelial O2− production (Nickenig and Harrison 2002, Hirono, Yoshimoto et al. 2007).
Cardioprotection in Agtr1a KO is controversial (Zhu, Zhu et al. 2003). The salutary effects include permanent coronary artery occlusion (CAO) which caused 13.6% of WT mice deaths in the first week post-op in contrast with 5.9% of deaths in Agtr1a KO mice. More impressive is that no KO mice died in the following three weeks, during which period 9.1% of WT mice died (Harada, Sugaya et al. 1999). However, another study of male and female Agtr1a KO mice with 6-week CAO showed that there was no difference in survival between WT and Agtr1a KO mice, and myocardial infarct size among WT males, WT females, KO males and KO females (Bridgman, Aronovitz et al. 2005). Although both males and females were studied, the data were not separated by gender. In that study, the extent of hypertrophy, reflected by left ventricular weight to body weight (LV/BW) and myocyte cross sectional area following permanent CAO were also examined. LV/BW and myocyte cross-sectional area did not change in female Agtr1a KO after permanent CAO compared to the sham group, but all male and female WT mice and male Agtr1a KO mice developed approximately a 50% increase in LV/BW and myocyte cross-sectional area following CAO, compared to sham. The mechanism mediating these gender differences was not discussed (Bridgman, Aronovitz et al. 2005).
With high fat diet, both WT and Agtr1a KO mice gain body weight similarly, but Agtr1a KO mice exhibited lower body fat (Ma, Corsa et al. 2011). Obesity-related kidney injury was observed in the Agtr1a KO mice, which was ascribed to an increase in Agtr1b expression, suggesting a role for the broader Angiotensin II type 1 receptor (Ma, Corsa et al. 2011). Agtr1a KO mice also developed significant systolic and diastolic dysfunction with streptozotocin induced diabetes (Yong, Thomas et al. 2013). In terms of cancer, endothelium-specific Agtr1a KO mice have been shown to be protected from metastasis of melanoma to the lungs (Ishikane, Hosoda et al. 2018).
The studies on overexpression of Agtr1a in mice have shown deleterious effects such as increased hypertrophy, interstitial fibrosis (Paradis, Dali-Youcef et al. 2000), and arrhythmogenicity (Rivard, Paradis et al. 2008). Exercise performance and the role of β-adrenergic signaling have not been examined in this model.
Regulator of G Protein Signaling 14 (RGS14) KO Mice (Median Age of Survival, of RGS14 KO is Increased by 26% Compared to WT Median Age of Survival of 23 Months):
RGS14 is a complex RGS family member that contains a canonical RGS domain and a tandem (R1 and R2) Ras/Rap binding domain. The RGS14 KO mice have enhanced brown adipose tissue with improved metabolism (Vatner, Zhang et al. 2018). RGS14 KO extended the median lifespan by 17% in males and 29% in females. RGS14 KO mice are protected against oxidative stress, obesity, diabetes, cancer, exercise intolerance and cardiac stress with decreased β-adrenergic signaling (Zhang, Vatner et al. 2016, 134:A18750, Guers, Oydanich et al. 2017, 136:A18790, Oydanich, Zhang et al. 2018, Vatner, Zhang et al. 2018).
Insulin-like Growth Factor (IGF) Type 1 Receptor (1R) Heterozygote Mice (Median Age of Survival, of IGF-1R +/−is Increased by 24% Compared to WT Median Age of Survival of 21 Months):
IGF-1R is one of the two IGF cell surface receptors. IGF-1R regulates the effects of IGF-1, and its signaling involves auto-phosphorylation and subsequent tyrosine phosphorylation of the insulin receptor substrate and Shc (Shc represents Src homology/collagen proteins, which are encoded by the SHC1 gene) which is also important in the p66Shc KO model of longevity (Delafontaine, Song et al. 2004). Increased longevity is sex-specific, with females having 33% and males having 15.9% longer lifespans than controls with a 129/J background (Holzenberger, Dupont et al. 2003). In another lifespan study by the same group using mice with a C57BL/6J background, although females still live 11% longer than controls, males have a maximum lifespan reduced by 13% (Xu, Gontier et al. 2014). This discrepancy is explained by the different background strains in the longevity phenotypes, e.g., the IGF1 levels, which correlate inversely with longevity. The C57BL/6J WT have lower levels of IGF and live longer than 129/J WT (Yuan, Tsaih et al. 2009, Xu, Gontier et al. 2014).
The differences on lifespan between males and females may relate to the fact that female IGF-1R+/− mice are more resistant to oxidative stress with higher survival compared to their WT littermates when challenged with paraquat, whereas this improved resistance to oxidative stress was not found in male IGF-1R+/− mice (Holzenberger, Dupont et al. 2003, Bokov, Garg et al. 2011). The protection observed in the heterozygote mice is mediated by activation of the MAPK pathway along with a reduction in p66Shc and ERK1/2 phosphorylation, and alanine-leucine transaminase levels (Holzenberger, Dupont et al. 2003, Bokov, Garg et al. 2011). In vitro, myoblasts from IGF-1R+/− mice are resistant to oxidative stress induced by H2O2 and UV light (Thakur, Garg et al. 2013). Both aged male and female IGF-1R+/− mice exhibit insulin resistance (Holzenberger, Dupont et al. 2003), and male, but not female, IGF-1R+/− mice also display glucose intolerance (Holzenberger, Dupont et al. 2003, Bokov, Garg et al. 2011). With high fat diet, both male and female IGF-1R+/− mice develop insulin resistance and impaired glucose tolerance (Garg, Thakur et al. 2011). Both adult male and female IGF-1R+/− mice display a lower body weight than WT mice, however, when challenged with a high fat diet, female IGF-1R+/− mice have greater body weight than female WT but male IGF-1R+/− mice have lower body weight than male WT mice (Garg, Thakur et al. 2011).
This model also protects against cancer. One study showed that heterozygotes are protected from colitis and tumorigenesis, due to reduced oxidative damage and accordingly, prevention of TNF-α, IL-6, NF-κB and STAT3 cascade activation in colonic epithelial cells (Wang, Yang et al. 2019). IGF-1R antibodies are also used clinically in treating cancer (Beltran, Mitchell et al. 2009, Tolcher, Sarantopoulos et al. 2009, Tap, Demetri et al. 2012). However, the lower number of fatalities in IGF-1R+/− males and females did not attain statistical significance (Bokov, Garg et al. 2011).
Exercise capacity, cardioprotection, and the role of β-adrenergic signaling have not been examined in this model.
Klotho Overexpressed Mice (Median Age of Survival, of Klotho Overexpressed is: Increased by 22% Compared to WT Median Age of Survival of 32 Months):
The Klotho proteins, α-Klotho, β-Klotho, and γ-Klotho, are important transmembrane proteins involved in aging (Kuro-o, Matsumura et al. 1997). Klotho overexpression has been shown to increase lifespan in both male and female mice similarly (Kurosu, Yamamoto et al. 2005). There are two Klotho overexpressed mice lines that were studied, EFmKL46 or EFmKL48. In EFmKL46 mice, lifespan is extended in males by 20% and in females by 18.8%. In EFmKL48, mice lifespan is extended in males by 30.8% and in females by 19% (Kurosu, Yamamoto et al. 2005). Higher levels of Klotho correlate with higher levels of the p53 gene as well as antioxidant enzymes including catalase, SOD, peroxiredoxin, and glutathione peroxidase (Kimura, Shiizaki et al. 2018).
However, Klotho, seems to have differing results in protection against age-related diseases. Klotho overexpression protects against diet-induced obesity (38), but not diabetes (Samms, Cheng et al. 2016). In terms of cardioprotection, Klotho KO mice develop exaggerated cardiac hypertrophy in response to chronic isoproterenol administration, consistent with the protection observed in the Klotho overexpressed mice (Xie, Cha et al. 2012). Klotho is also upregulated in human hearts with cardiomyopathy compared to its level in normal human hearts, but it is not known if this is a cause of or a response to the cardiomyopathy (Poelzl, Ghadge et al. 2018). Another clinical study revealed higher Klotho levels associated with the absence of atrial fibrillation in hemodialysis patients (Nowak, Friedrich et al. 2014).
In terms of cancer, Klotho overexpression acts as a tumor suppressor, e.g. it decreases tumor growth in diffuse large B-cell lymphoma (Zhou, Fang et al. 2017) and suppresses growth and pulmonary metastasis of osteosarcoma (Li, Xiao et al. 2020). Furthermore, while there have been studies showing Klotho is increased after exercise training in Sprague Dawley rats compared to their control group (Ji, Luan et al. 2018) and in humans compared to their baseline level before exercise training (Tan, Chu et al. 2018), there haven’t been any studies showing that increases in Klotho directly lead to improvement in exercise performance. Therefore, its role in mediating exercise capacity requires further study. The role of β-adrenergic signaling has also not been examined in this model.
Mitochondrial Targeted Catalase (MCAT) Overexpressed Mice (Median Age of Survival, of MCAT Overexpressed is increased by 19% Compared to WT with Median Age of Survival of 25 Months):
Catalase (CAT) is a ubiquitously expressed antioxidant enzyme primarily located in peroxisomes, that catalyzes the decomposition of H2O2 to O2 and H2O (Halliwell and Gutteridge 1989). Catalase has a central role in regulating the cellular level of H2O2, and this catabolic action protects cells from oxidative stress (Gaetani, Ferraris et al. 1996, Nandi, Yan et al. 2019). Lifespan of MCAT overexpressed mice is increased similarly in males and females (Schriner, Linford et al. 2005).
In MCAT overexpressed mice, oxidative damage, H2O2 production and H2O2-induced aconitase inactivation are attenuated. Furthermore, the development of mitochondrial deletions is reduced in this murine model (Schriner, Linford et al. 2005, Dai, Santana et al. 2009). MCAT overexpressed mice are also protected from age-associated decline in AMP-activated protein kinase (AMPK) activity and mitochondrial density (Lee, Choi et al. 2010), resulting in reduced DNA oxidation in all the tissues compared to control mice (Perez, Bokov et al. 2009). CAT overexpressed-derived embryonic fibroblasts are resistant to H2O2 toxicity, demonstrating increased catalase-specific activity (Mele, Van Remmen et al. 2006). Aged MCAT overexpressed mice exhibit improved exercise performance with enhanced mitochondrial antioxidant activity and improved muscle function (Umanskaya, Santulli et al. 2014). MCAT overexpressed mice are also protected against lipid induced insulin resistance in muscle and demonstrate better muscle mitochondrial function (Lee, Choi et al. 2010). Body weights and body fat composition were similar in WT and MCAT overexpressed mice (Lee, Choi et al. 2010). MMTV-PyMT, (mouse mammary tumor virus-polyoma middle tumor-antigen) is a mouse breast cancer model. When MMTV-PyMT mice are crossed with MCAT overexpressed mice, they exhibit lower incidence of tumor invasiveness than PyMT breast cancer mice without MCAT overexpression (Goh, Enns et al. 2011).
Ang II-induced heart failure and the cardiovascular consequences of aging, e.g. increased LV mass, decreased diastolic function and myocardial performance, are attenuated in MCAT overexpressed mice with reduced oxidative stress and enhanced mitochondrial function (Dai, Santana et al. 2009, Dai, Johnson et al. 2011). MCAT overexpressed mice also exhibit decreased β-adrenergic signaling which protects against cardiac remolding (Uribe and Andersson 2019).
Ribosomal Protein S6 Kinase β−1 (S6K1) KO Mice (Median Age of Survival, of S6K1 KO is Increased by 19% in Females Compared to WT Median Age of Survival of 28 Months):
S6K1 is the principal kinase effector downstream of mammalian target of rapamycin complex 1 (mTORC1), which lowers mitochondrial ROS levels (Binsch, Jelenik et al. 2017). Longevity as measured by median lifespan was enhanced in female S6K1 KO mice by 19%, but no such difference was observed in males (Selman, Tullet et al. 2009). These gender differences could be due to estrogen regulation of S6K1 activation (Jaffer, Shynlova et al. 2009). Young male S6K1 KO mice, due to a marked increase in metabolic rate and lipolysis, exhibit lower body weight and have improved glucose tolerance and insulin sensitivity compared to WT in response to either normal diet (Um, Frigerio et al. 2004) or high fat diet (Um, Frigerio et al. 2004, Binsch, Jelenik et al. 2017). In addition, improved glucose tolerance and insulin sensitivity were also observed in 20 month old female S6K1 KO compared to WT mice in response to high fat diet, though this protective feature was not observed in young female S6K1 KO mice (Selman, Tullet et al. 2009).
S6K1 KO with high fat diet also exhibited greater running distance, longer running times, and reduced triglyceride contents in liver and skeletal muscle after 4 weeks of exercise training compared to WT mice (Binsch, Jelenik et al. 2017).
Two mechanisms mediate increased oxidative stress by S6K1: 1) mitochondrial superoxide production is directly increased, and 2) S6K1 activation mediates uncoupling of eNOS, decreasing antioxidant capacity (Rajapakse, Yepuri et al. 2011). In addition, S6K1 KO mice exhibit less mitochondrial ROS production (Binsch, Jelenik et al. 2017). In vitro, overexpression of S6K1 results in increased superoxide and decreased NO production (Rajapakse, Yepuri et al. 2011).
S6K1 levels are elevated in breast cancer, a result that is mediated by estrogen receptors, whereas S6K1 also regulates ligand independent estrogen receptor activity. Together the two form a feed-forward loop, promoting breast cancer cell proliferation (Maruani, Spiegel et al. 2012). In addition, S6K1 inhibitors induce autophagy and apoptosis in cervical cancer cells (Nam, Yi et al. 2019) and BT474 breast cancer cells (Park, Jin et al. 2015) in vitro. Cardioprotection, and the role of β-adrenergic signaling have not been examined in this model.
(2). A Longevity Model Not Protected Against Oxidative Stress:
Growth Hormone Receptor/Binding Protein (GHR/BP) KO Mice (Median Age of Survival, of GHR/BP is Increased by 47% Compared to WT Median Age of Survival of 23 Months):
GHR/BP KO mice have a longer lifespan in both males by 55% and females by 38% (Coschigano, Clemmons et al. 2000). Growth hormone binding protein (GHBP), is a truncated form of the GHR, which modulates growth hormone action (Baumann 2002). GHR regulates growth and other biological functions including metabolism and controlling physiological processes related to the cardiovascular, renal, and reproductive systems (Dehkhoda, Lee et al. 2018). GHR/BP KO mice with disrupted GHR and GHBP, exhibit severe postnatal growth impairment, proportionate dwarfism, decreased levels of IGF1 and increased GH levels (Zhou, Xu et al. 1997).
GHR/BP KO mice do not confer longevity by improved free-radical scavenging in the liver and kidney. In the kidney SOD1 is lower and Gpx is higher in GHR/BP KO mice. Lipid peroxidation is higher only in female GHR/BP KO mice. GHR/BP KO males are more susceptible to paraquat toxicity compared to females or normal males (Hauck, Aaron et al. 2002). GHR/BP KO are protected against cancer, with a lower percentage of tumor-bearing mice compared to WT and live longer than WT even when tumors are present. GHR/BP KO are also less susceptible to metastasis (Ikeno, Hubbard et al. 2009). GHR/BP KO mice have improved grip strength, balance, and motor coordination in middle and old age in both males and females (Arum, Rickman et al. 2014). Although GHR/BP KO mice exhibit smaller body size and lower body weight than WT, there are no studies on protection against obesity when challenged with high fat diet. Protection from diabetes, cardioprotection, and the role of β-adrenergic signaling have also not been examined in this model.
(3). Longevity Models With Oxidative Stress Protection Data Controversial:
Ames Dwarf Mice (Median Age of Survival, of Ames Dwarf is Increased by 57% Compared to WT Median Age of Survival of 21 Months):
Ames Dwarf mice are characterized as having deficiencies in hormones of the anterior pituitary, most notably growth hormone, prolactin, and thyroid-stimulating hormone (Bartke and Brown-Borg 2004), which is similar to the Snell Dwarf mice and GHR KO mice (Bartke and Brown-Borg 2004, Rohrbach, Teichert et al. 2008). These mice also have an extended lifespan by 64% in males and 49% in females (Brown-Borg, Borg et al. 1996). The mechanism for the shorter median lifespan of WT mice was not discussed. These mice show resistance to oxidative stress induced by paraquat and diquat, both of which are known producers of reactive oxygen species (Bokov, Lindsey et al. 2009). These mice also have elevated levels of catalase and SOD1 in the hypothalamus (Hauck and Bartke 2000). However, another study showed the opposite, with increased vascular oxidative stress with decreased expression of SOD and Gpx in the aortas of Ames dwarf mice (Csiszar, Labinskyy et al. 2008). Ames dwarf mice show protection against diabetic risk factors, cancer, and dobutamine-induced cardiac stress (Dominici, Hauck et al. 2002, Ikeno, Bronson et al. 2003, Bokov, Lindsey et al. 2009). While there are no studies on exercise performance, Ames dwarf mice do show increased antioxidant defense in skeletal muscle following acute and chronic exercise (Romanick, Rakoczy et al. 2004). The role of β-adrenergic signaling in this model has not been examined.
Mitochondrial 5-Demethoxyubiquinone Hydroxylase (MCLK1) Heterozygote Mice (Median Age of Survival, of MCLK1+/− is Increased by 31% Compared to WT Median Age of Survival of 22 Months):
MCLK1 heterozygous mice exhibit enhanced longevity in both male and female mice (Liu, Jiang et al. 2005). On average, both sexes live 31% longer than WT. The cause of the short median lifespan in WT mice was not discussed. As previously mentioned, this may be due to the different background of WT mice and housing conditions. Inactivation of MCLK1 in embryonic stem cells has been shown to incur protection from oxidative stress and damage to DNA (Liu, Jiang et al. 2005). However, in the MCLK1 +/− mouse model, a different, more complex result was discovered. These mice have increased production of ROS and decreased antioxidant production resulting in increased mitochondrial oxidative stress and mitochondrial dysfunction, but have decreases in cytosolic oxidative damage and low non-mitochondrial oxidative damage with reduced systemic oxidative stress markers associated with aging (Lapointe and Hekimi 2008). A mechanism proposed for these differences involves a reduction in the rate of cytoplasmic ROS producing processes due to the decreased levels of ATP and total NAD (Lapointe and Hekimi 2008). This model shares some congruence with the theory of mitochondrial hormesis, suggesting that early increases in oxidative stressors can lead to adaptive responses that promote longevity. This mechanism also promotes protection against cancer in this model, via an increase in tumor latency (Wang, Wang et al. 2012). Further studies on protection against diabetes, obesity, cardiac stress, exercise intolerance, and the role of β-adrenergic signaling have yet to be conducted.
Insulin Receptor Substrate 1 (IRS1) KO Mice: (Median Age of Survival, of IRS1 KO is Increased by 18% Compared to WT Median Age of Survival of 24 Months):
IRS1 protein is one of the primary mediators of insulin-dependent mitogenesis and regulation of glucose metabolism in most cell types involving PI3K/Akt and ERK/MAP kinase pathways (White 2002, Mardilovich, Pankratz et al. 2009). It has been shown to increase longevity in IRS1 KO male by 15% and female mice by 21% (Selman, Partridge et al. 2011). The role of IRS1 in regulating oxidative stress is controversial. Overexpression of IRS1 has been shown to reduce oxidative stress and prevent autophagy (Chan, Kikkawa et al. 2012). However, IRS1 KO mice maintain normal catalase levels with aging leading to better oxidative stress defense, while WT mice exhibit decreases in catalase levels (Selman, Lingard et al. 2008). IRS1 KO mice are protected against obesity with less body weight and fat mass during aging (Selman, Lingard et al. 2008), but not against diabetes with impaired glucose tolerance, insulin sensitivity (Araki, Lipes et al. 1994, Selman, Lingard et al. 2008) and cardiac stress (Furumoto, Fujii et al. 2005). The findings on cancer protection are controversial. IRS1 KO (Ma, Gibson et al. 2006) and IRS1 overexpressed (Dearth, Cui et al. 2006) mice both exhibit increases in mammary tumor metastasis making the role of IRS1 in cancer difficult to interpret. No studies have examined either the KO or overexpressed models in terms of β-adrenergic signaling.
(4). Longevity Models With Oxidative Stress Not Studied:
Cytosolic Form of Phosphoenolpyruvate Carboxykinase (PEPCK-C) Overexpressed Mice (Median Age of Survival, unspecified):
PEPCK-C is present in the liver, kidney cortex and brown and white adipose tissue; and is involved in gluconeogenesis and/or glyceroneogenesis (Croniger, Olswang et al. 2002). Both male and female PEPCK-C overexpressed mice are still able to reproduce after the age of 21 months (Hakimi, Yang et al. 2007). PEPCK-C overexpressed mice have enhanced exercise capacity and are able to maintain respiratory exchange ratios below unity over 40 minutes of strenuous exercise. Conversely, WT mice exceed RER unity after less than 10 minutes. Performance was improved even in old animals compared to young WT. Lactate levels in PEPCK-C overexpressed mice were reduced during exercise compared to baseline whereas WT had a 4-fold increase in lactate in response to exercise (Hakimi, Yang et al. 2007). PEPCK-C overexpressed mice have lower body weight despite a 60% increase in food intake, lower percentage of adipose tissue even when aged, but greater blood glucose levels compared to WT mice (Hakimi, Yang et al. 2007). Many of these attributes result in longevity, but the median age of survival of PEPCK-C overexpressed mice was not reported, Cancer, cardioprotection, β-adrenergic signaling and oxidative stress have not been examined in this model.
Pregnancy-Associated Plasma Protein-A (PAPPA) KO Mice (Median Age of Survival, of PAPPA KO is increased by 26% Compared to WT Median Age of Survival of 21 Months):
PAPPA is the largest of the pregnancy associated proteins, produced by both the embryo and the placenta during pregnancy. This protein has several different functions, including preventing recognition of the fetus by the maternal immune system, matrix mineralization and angiogenesis (Fialova and Malbohan 2002). PAPPA-KO increases longevity to a similar extent in females and in males (Conover, Bale et al. 2010). The median lifespan of the WT was only 21 months. The cause of the short median lifespan of WT mice was not discussed. PAPPA KO mice are protected against cancer with a reduction in neoplasms observed at necropsy compared to wild-type (Conover and Bale 2007). Reducing PAPPA protein expression decreases tumor growth in-vivo whereas overexpression and secretion PAPPA protein in the xenograft cell-line resulted in augmented tumor growth via upregulation of the IGF pathway. Accordingly, the over-expression of PAPPA alone did not result in augmented growth of the tumor cell line. Only when PAPPA protein secretion was also upregulated did the proliferative phenotype become apparent (Pan, Hanada et al. 2012). Spontaneous pathology of the heart discovered at necropsy was absent in aged KO mice, whereas 50% of WT mice had evidence of disease (Conover and Bale 2007). Female KO mice were more tolerant of glucose challenge and had increased insulin sensitivity with high fat and high sucrose diet (Hill, Arum et al. 2015). PAPPA KO mice exhibit partial protection from obesity with a reduction in visceral fat depot weight and an increase in circulating adiponectin but no difference in percent-body-fat compared to WT (Conover and Bale 2007, Heitzeneder, Sotillo et al. 2019). There are no studies on exercise capacity, cardioprotection, β-adrenergic signaling and oxidative stress.
V. Caenorhabditis elegans (C. elegans) Models of Longevity Associated with Oxidative Stress Protection
In C. elegans, as with other models, the accumulation of oxidative damage is deleterious to both the lifespan and health span of the animal. Several mutant models of protection from oxidative stress in C. elegans are long-lived compared to their wild type counterparts (Friedman and Johnson 1988, Friedman and Johnson 1988, Kenyon, Chang et al. 1993, Li, Gao et al. 2007, Mehta, Steinkraus et al. 2009). Mutations in AGE-1, daf2, RLE-1, VHL-1, and HIF-1 all yielded longer lives and are resistant to oxidative stress (Friedman and Johnson 1988, Friedman and Johnson 1988, Kenyon, Chang et al. 1993, Li, Gao et al. 2007, Mehta, Steinkraus et al. 2009). In addition, several other C. elegans models would have been expected to have deleterious effects from their oxidative stress response, based on data from mouse models, but did not demonstrate significant improvement in longevity from these C. elegans models (Oh, Mukhopadhyay et al. 2005, Lehtinen, Yuan et al. 2006, Leiser, Begun et al. 2011). For example, JNK1 activation by oxidative stress in mice results in a pro-inflammatory phenotype in the kidney (Wu, Mei et al. 2009) and lack of JNK1 in mouse embryonic fibroblasts results in increased sensitivity to oxidative stress (Ventura, Cogswell et al. 2004). However, overexpression of JNK1 in C. elegans yields a significant extension in life-span (Oh, Mukhopadhyay et al. 2005). Similarly, MST-1 overexpression in the hearts of mice resulted in a fibrotic phenotype, which suggests that MST-1 can mediate oxidative stress induced damage to the heart (Yamamoto, Yang et al. 2003). In contrast, overexpression of the CST-1, the MST-1 orthologue in C. elegans, resulted in longer lifespan (Lehtinen, Yuan et al. 2006). Finally HIF-1a protects against oxidative stress in mice (Li, Zhou et al. 2019), but the HIF-1 KO in C. elegans resulted in enhanced lifespan (Leiser, Begun et al. 2011). In each of these models, protection was conferred via indirect activation of daf-16, which both alone and as part of a complex of proteins, is protective against oxidative stress (Oh, Mukhopadhyay et al. 2005, Lehtinen, Yuan et al. 2006, Leiser, Begun et al. 2011, Lin, Sen et al. 2018). Changes in environmental conditions, particularly temperature, often affected the lifespan in these models, sometimes eliminating it entirely (Van Voorhies and Ward 1999, Xiao, Zhang et al. 2013). Additional studies on C. elegans, where oxidative stress is increased, are discussed in the mitochondrial hormesis section, which follows the Section VII.
VI. Non-Human Primate Models of Longevity Associated with Oxidative Stress Protection
Among the non-human primate studies on calorie restriction and lifespan, only those studies carried out at the Wisconsin National Primate Research Center in Rhesus monkeys showed an increase in lifespan, as well as age-related diseases (Colman, Anderson et al. 2009, Colman, Beasley et al. 2014). In contrast, the National Institute on Aging study on lifespan, also in Rhesus monkeys, did not find an extended lifespan (Mattison, Roth et al. 2012). However, both studies reported beneficial effects of caloric restriction on age-related diseases (Colman, Anderson et al. 2009, Mattison, Roth et al. 2012, Colman, Beasley et al. 2014), with reduced adiposity (Colman, Roecker et al. 1998, Edwards, Rudel et al. 1998), increased insulin sensitivity (Gresl, Colman et al. 2003), and reduced oxidative damage (Zainal, Oberley et al. 2000). The differences of lifespan in the two studies in response to calorie restriction have been attributed to study design, husbandry and diet composition (Mattison, Colman et al. 2017). However, we also think that lifespan studies in primates in captivity may be complicated by many factors not observed when the primates are in their natural environment.
VII. Human Models of Longevity Associated with Oxidative Stress Protection
While there are immense challenges to mounting a randomized controlled study on the effects of calorie restriction on human lifespan, existing studies do suggest that calorie restriction enhances human longevity (Trepanowski, Canale et al. 2011, Ravussin, Redman et al. 2015, Redman, Smith et al. 2018). This is due, in large part, to the reduction in caloric intake, which protects against obesity and subsequently a decrease in obesity-associated morbidity and mortality. Calorie restriction also protects against other diseases and pathologic processes of aging that increase morbidity and mortality, e.g., cardiovascular diseases (Meyer, Kovacs et al. 2006, Hammer, Snel et al. 2008, Riordan, Weiss et al. 2008), diabetes and metabolic diseases (Larson-Meyer, Heilbronn et al. 2006, Weiss, Racette et al. 2006, Ryan, Ortmeyer et al. 2012), inflammation (Imayama, Ulrich et al. 2012) and cancer (Wei, Brandhorst et al. 2017, Lope, Martín et al. 2019, de Groot, Lugtenberg et al. 2020).
Calorie restriction reduces ROS production in healthy people (Redman, Smith et al. 2018). Even in those that are already obese, calorie restriction is linked to a reduction in oxidative stress markers (Buchowski, Hongu et al. 2012). Calorie restriction also slows metabolic rate (Heilbronn, de Jonge et al. 2006, Civitarese, Carling et al. 2007), and lowers oxidative damage (Heilbronn, de Jonge et al. 2006, Buchowski, Hongu et al. 2012, Redman, Smith et al. 2018) in both healthy and overweight individuals.
However, in one of the human studies, resting metabolic rate significantly decreased in the calorie restriction group after 12 months compared to the control group, but no difference was observed at 24 months between the two groups (Ravussin, Redman et al. 2015). Increased mitochondrial biogenesis in healthy humans under calorie restriction is one mechanism to explain the decreased oxidative stress and decreased DNA damage (Civitarese, Carling et al. 2007).
Exercise training also prolongs lifespan, but enhanced exercise performance is not a feature of calorie restriction (Martin, Das et al. 2011, Racette, Rochon et al. 2017). When exercise intolerance occurs in the older population, its severity is augmented by chronic diseases, including heart failure (Del Buono, Arena et al. 2019), diabetes (Poitras, Hudson et al. 2018), lung diseases (Vogiatzis and Zakynthinos 2012) and cancer (Jones, Eves et al. 2009), which all share increased oxidative stress as a mechanism in their pathophysiology. For example, exercise intolerance in patients with heart failure is linked to increased mitochondrial ROS (Shirakawa, Yokota et al. 2019), increased plasma lipid peroxidation (Keith, Geranmayegan et al. 1998, Sawyer 2011), and increased plasma malondialdehyde (Nishiyama, Ikeda et al. 1998) and decreased SOD activity (Nishiyama, Ikeda et al. 1998). However, the relation between exercise and oxidative stress is complex. Acute exercise can actually increase oxidative stress (Fisher-Wellman and Bloomer 2009), whereas long-term exercise training reduces oxidative stress (Liu, Yeo et al. 2000, Elosua, Molina et al. 2003, Vollaard, Shearman et al. 2005) by upregulating antioxidant enzymes such as SOD, Gpx, and glutathione reductase. As noted, exercise also stimulates cellular ROS production in muscle, liver, and other organs; this is due to mitochondrial sources of ROS such as NADPH oxidase or xanthine oxidase and ultimately leads to more efficient mitochondrial ATP production (Coggan, Spina et al. 1990, Lanza, Short et al. 2008, Gomez-Cabrera, Salvador-Pascual et al. 2015, Bouzid, Filaire et al. 2018). The effects of antioxidant supplementation on mitochondrial biogenesis in skeletal muscle are controversial, suggesting that antioxidant supplementation either has little effect or is deleterious to the beneficial adaptations to exercise training in skeletal muscle, highlighting the important role of ROS as second messengers (Gomez-Cabrera, Salvador-Pascual et al. 2015). This role for ROS is supported by the observation that the geographic distribution in the prevalence of glucose-6-phosphate dehydrogenase (G6PD) deficiency correlates with the severity of malaria (Ruwende, Khoo et al. 1995). G6PD deficient red blood cells are unable to provide the optimum red blood cell redox status that is required by malaria parasites for their survival (Vega-Rodriguez, Franke-Fayard et al. 2009). Lower survival for malaria parasites can explain the lower risk for severe malaria in Africans with G6PD deficiency.
Mitochondrial myopathies serve as a valuable clinical model for the study of oxidative stress. These diseases generally result in increased oxidative stress and higher levels of ROS (Esposito, Melov et al. 1999). Coenzyme Q10 (CoQ10) and its synthetic analogue, idebenone, are examples of antioxidants that have been used successfully to treat patients with mitochondrial myopathies. There is modest evidence to support the rationale of these therapies that scavenging reactive oxygen species can lead to clinical improvement. In a randomized, placebo-controlled, double-blind trial of 85 patients with Leber hereditary optic neuropathy (LHON) (Klopstock, Yu-Wai-Man et al. 2011), idebenone showed a trend for improved vision. Even though the trial demonstrated no benefit from idebenone by intention-to-treat analysis for the primary outcome of visual acuity, these data led to the approval of idebenone in 2015 by the European Medicines Agency for the treatment of visual impairment in adolescent and adult patients with LHON (Erin O’Ferrall 2020). CoQ10 has also been used to treat mitochondrial disorders that lack CoQ10. It should be mentioned that besides its function as an antioxidant, CoQ10 also serves as an integral part of the mitochondrial respiratory function via its role as an electron acceptor. It is not clear which of its two roles, as an antioxidant or mitochondrial electron acceptor, produces a benefit in CoQ10 supplementation for patients with mitochondrial disorders or CoQ10 deficiency. Although not all clinical trials of CoQ10 have been positive (Muller 1990), the consensus is that the treatment is effective with few side effects for patients with mitochondrial disorders including infants and children (Chen, Huang et al. 1997). Some of the controversy related to the role of mitochondria in longevity could be due to a biological phenomenon known as mitochondrial hormesis (Ristow and Zarse 2010), which will be discussed next.
Mitochondrial Hormesis
The controversial relationship between antioxidant supplementation and longevity increases the complexity of understanding the role of ROS in mediating longevity and has led to different theories, such as mitochondrial hormesis. Mitochondrial hormesis refers to conditions whereby increased levels of oxidative stress can lead to healthful aging (Ristow and Zarse 2010). Whereas it is recognized that high levels of oxidative stress are detrimental and lead to increased morbidity and mortality in aging, mitochondrial hormesis, advances the interesting argument that small increases in levels of ROS may not always be detrimental, but rather may function to modulate several signaling pathways, including activation of sirtuins and AMPK, and inhibition of mTOR signaling (Ristow and Schmeisser 2014). All of these pathways have been shown to be involved in extending longevity through increased stress resistance, resulting in a long-term reduction of oxidative stress.
Exercise is a classic example of mitochondrial hormesis (Ristow, Zarse et al. 2009, Hood, Zhang et al. 2018, Musci, Hamilton et al. 2019) which acutely increases ROS production but its chronic influence is to induce healthful aging (Fisher-Wellman and Bloomer 2009, Ristow, Zarse et al. 2009). Such observations of opposing effects under acute and chronic conditions are not an unusually rare phenomenon clinically. For instance, smoking cessation acutely leads to a more reactive airway and increased secretions and is therefore not recommended for patients undergoing surgery (Bluman, Mosca et al. 1998), whereas chronically the beneficial effects of smoking cessation are enormous. Another example is alcohol exposure which acutely decreases anesthetic requirements, but chronic alcohol consumption, increases anesthetic requirements (Lobo and Lopez 2020) and also has opposing effects on inflammation, with acute exposure reducing inflammation whereas chronic exposure increases inflammation (Mandrekar, Bala et al. 2009). The beneficial effects of regular exercise are partly based on the exercise induced ROS production, which then induce expression of antioxidants, DNA repair and protein degrading enzymes, and result in decreases of oxidative stress (Liu, Yeo et al. 2000, Elosua, Molina et al. 2003, Vollaard, Shearman et al. 2005) and oxidative stress related diseases (Radak, Chung et al. 2005). For example, xanthine oxidase increases ROS, and inhibition of xanthine oxidase impairs gene transcription in response to acute exercise, but does not cause a major effect on long-term exercise training, which induces mitochondrial biogenesis and antioxidant defenses (Wadley, Nicolas et al. 2013). Studies directly linking mitochondrial hormesis and exercise training are still pending (Merry and Ristow 2016).
Calorie restriction is also considered a model of mitochondrial hormesis (Schmeisser, Priebe et al. 2013, Ristow and Schmeisser 2014, Hood, Zhang et al. 2018) by improved mitochondrial function mediated partially through increased mitochondrial turnover. This is based on studies in C. elegans (Schmeisser, Priebe et al. 2013) or yeast (Mesquita, Weinberger et al. 2010, Zuin, Carmona et al. 2010) demonstrating extended lifespan conferred by causing mitochondria to emit more reactive oxygen species which, though a defensive response which senses the higher levels, and induces mechanisms to lower oxidative stress (Ristow and Schmeisser 2014). Another example is glucose restriction, which promotes increases in ROS, which induce catalase activity, resulting in increased oxidative stress resistance and in extended lifespan in C. elegans (Schulz, Zarse et al. 2007). Finally, extended lifespan with impaired insulin/IGF-1 signaling also involves the mitochondrial hormesis mechanism. Examples include the Ames Dwarf mice, Snell Dwarf mice, GHR KO mice, p66shc KO mice, and IGF-1R+/− mice (Brown-Borg, Borg et al. 1996, Quarrie and Riabowol 2004). Studies in the long-lived C. elegans mutant daf-2 model. i.e., the worm orthologue of the insulin/IGF-1 receptor, demonstrate that increased ROS activates the ROS defense enzymes, culminating in reduced ROS levels and extended lifespan (Brys, Castelein et al. 2010, Zarse, Schmeisser et al. 2012). It is important to note that the mechanism of mitochondrial hormesis has not been found in humans or large mammalian models of calorie restriction and extended longevity.
Mitochondrial hormesis is also involved in hypothermia, in which low temperatures can induce transient increases in ROS production. Hypothermia can stimulate longevity but has only been shown to induce an extended lifespan in lower animals, mainly C. elegans and Drosophila models (Baxter, Allard et al. 2014, Angstman, Frank et al. 2018). There are some studies suggesting that hypothermia is associated with increased ROS production in piglets (Langley, Chai et al. 2000), and lower levels of antioxidants in rats (Gamez, Alva et al. 2008) with decreases in oxygen availability (Alva, Palomeque et al. 2013) higher levels of malondialdehyde, a marker of oxidative stress, and lower levels of GSH, but no differences in the antioxidants, SOD and Gpx (Dede, Deger et al. 2002). Another rat study concluded hypothermia causes oxidative stress with higher lipid peroxidation, lower activities of catalase, Gpx, and GSH, but higher SOD (Gumuslu, Sarikcioglu et al. 2002). However, there are also some studies that reported a reduction in oxidative stress as a result of hypothermia in vitro (Hasegawa, Ogihara et al. 2009), ex vivo (Mihara, Dohi et al. 2004) and in vivo from rats with brain injury, (Maier, Sun et al. 2002, Kuo, Lo et al. 2011), ischemia-reperfusion injury of skeletal muscle (Ozkan, Ekinci et al. 2015), and cardiac arrest in pigs (Ahn, Lee et al. 2020), (Gong, Li et al. 2012), (Ostadal, Mlcek et al. 2013).
Hypothermia is also an important protective mechanism in humans, e.g., hypothermia is used to protect patients in anesthesia and in critical care (Corry 2012). In addition, in a human study with cardiac arrest, therapeutic hypothermia reduced both reactive oxygen metabolites and antioxidant activity, whereas, the opposite is observed with rewarming, where both were increased (Dohi, Miyamoto et al. 2013).
This is consistent with well documented clinical observations of complete recovery of some patients with accidental hypothermia and cardiac arrest despite prolonged resuscitation (Giesbrecht 2000). These neuroprotective effects of hypothermia are now well recognized and therapeutic hypothermia is a common clinical tool in management of certain post-cardiac arrest patients (Rittenberger 2020). Another human study reported malondialdehyde is reduced and antioxidants levels are increased except for paraoxonase-1, in hypothermia patients compared to normothermia patients (Hackenhaar, Medeiros et al. 2017). It has also been shown that hypothermia can induce transient increases in ROS production in a variety of human cell lines (Brinkkoetter, Song et al. 2008, Hendriks, Bruggenwirth et al. 2019) and therefore can be speculated to activate similar adaptive responses leading to increases in longevity.
Blue Zones (Regions with Longer Lifespan in Their Inhabitants)
The goal of extending lifespan in humans has already been successful in certain parts of the world. Some countries like, Monaco, Japan and Switzerland have higher life expectancies than others (The-World-Factbook 2020). However, while it is tempting to attribute the long-life phenotype in these populations to their national origin or ethnicity, this is likely to be misleading because of the heterogeneity of life-style factors, even within individuals of a single country or ethnicity, which also profoundly affect longevity. Blue zones are relatively small regions where people routinely become centenarians. Rather than being limited to a nation or ethnicity, they are geographically widespread, and include locations in the United States, Costa Rica, the Mediterranean, and East-Asia (Buettner and Skemp 2016, Huang and Mark Jacquez 2017); for example, Loma Linda, USA; the Nicoya peninsula in Costa Rica; Sardinia, Italy; Ikaria, Greece; Okinawa, Japan (Buettner and Skemp 2016, Huang and Mark Jacquez 2017). In these areas the number of centenarians, i.e., those reaching the age of 100 is 10 times greater than the average in the United States. These regions are characterized by cultural preferences which discourage over-eating and excessive alcohol consumption, while encouraging active lifestyles. While the characteristics shared by these regions extend beyond diet and exercise, both a healthy diet and exercise training have been shown to reduce oxidative stress and protect against the deleterious effects of aging.
Common Specific Characteristics in Blue Zones
Ambulation: The inhabitants live in environments with motivation for frequent ambulation.
Healthy social relationships and psychological well-being: 1) many belong to some faith-based community, 2) seniors are valued members in their families and community, giving them a rich sense of purpose.
Diet: 1) 80% rule: stop eating when stomachs are 80% full; 2) Plant based diet: beans are the cornerstone of most centenarian diets. Meat is eaten on average only 5 times per month. 3) Drink alcohol moderately but regularly, 1–2 glasses per day, with friends and/or with food.
A cultural environment that reinforces healthy lifestyle and behavior.
VIII. Human Models of Reduced Longevity Associated with Increased Oxidative Stress
Perhaps the most convincing evidence for the role of oxidative stress in aging is progeria, a unique medical condition characterized by premature aging (Sinha, Ghosh et al. 2014). Teenagers with this condition often suffer from atherosclerosis, cardiomyopathy, coronary artery disease and premature death. The etiology involves the LMNA gene on chromosome 1q. Chromosome 1 is the largest human chromosome, and chromosome 1q is the q-arm on chromosome 1, which contains about 150 genes, one of these genes is LMNA, which encodes prelamin A. Prelamin A is ultimately converted to lamin A, a structural protein component of the nuclear lamina that stabilizes the nuclear membrane (Elzeneini and Wickström 2017). Pathogenic variants of LMNA cause degenerative disorders collectively known as laminopathies. The prototypical laminopathy is the classic Hutchinson-Gilford progeria syndrome (HGPS), which is commonly referred to simply as progeria. Accumulation of oxidized proteins causing DNA damage has been described as a causative mechanism in this disease (Berlett and Stadtman 1997, Lattanzi, Marmiroli et al. 2012).
Sources of oxidative stress include obesity, high fat diet or diets rich in sugar and processed foods, exposure to radiation, cigarette smoking, other tobacco products, alcohol consumption, certain medications, pollution, exposure to pesticides or industrial chemical and even oxygen itself. Supplemental oxygen can be deleterious, and in fact can contribute to increased mortality (Girardis, Busani et al. 2016, Chu, Kim et al. 2018). Both human and animal studies show that high concentrations of inspired oxygen can induce pulmonary complications, spanning from mild airway insult such as tracheobronchitis to severe parenchymal injury with diffuse alveolar damage that is histologically indistinguishable from acute respiratory distress syndrome (Hedley-Whyte 1970, Deneke and Fanburg 1980, Davis, Rennard et al. 1983). Excessive supplemental oxygen is also known to cause retinopathy of prematurity, an important cause of severe visual impairment in childhood. The etiology of retinopathy of prematurity involves low serum IGF-I values (Hellstrom, Perruzzi et al. 2001) which is associated with both suppression of vascular growth and proliferative retinopathy of prematurity (Hellstrom, Engstrom et al. 2003). Treating preterm infants with recombinant human (rh) IGF-I can prevent retinopathy of prematurity (Hellström, Smith et al. 2013). Interestingly, the IGF-1R+/− Hetero mouse is a longevity model with protection against oxidative stress (Table I).
Metabolic syndrome is a serious clinical condition, comprised of a collection of cardiometabolic risk factors that includes obesity, insulin resistance, hypertension and dyslipidemia, mediated, in part, by increased oxidative stress (Roberts and Sindhu 2009, Mahjoub and Masrour-Roudsari 2012, Vona, Gambardella et al. 2019). Patients with metabolic syndrome have increased oxidative damage with increased lipid peroxidation malondialdehyde levels, protein carbonyls, and xanthine oxidase activity and decreased antioxidant protection, and decreased levels of antioxidant, e.g. superoxide dismutase activity, vitamin A, vitamin C, and carotenoids (Ford, Mokdad et al. 2003, Palmieri, Grattagliano et al. 2006, Armutcu, Ataymen et al. 2008). In addition, higher levels of oxidized low-density lipoproteins, carbonylation of cellular proteins, and NADPH oxidase activity, which induce oxidative stress, are associated with metabolic syndrome (Holvoet, Lee et al. 2008, Vona, Gambardella et al. 2019).
Cigarette smoking is another cause of increased ROS and decreased antioxidant status in tissues, and similarly, fine particulate matter in polluted air is also associated with increased ROS and oxidative stress (Lodovici and Bigagli 2011, Jansen EHJM, Beekhof P et al. 2014). According to the CDC, the life expectancy for smokers is at least 10 years shorter than for nonsmokers (Jha, Ramasundarahettige et al. 2013, NCCDPHP 2014).
Almost all of the diseases that limit longevity and healthful aging are associated with increased oxidative stress, e.g., myocardial infarction (Zweier and Talukder 2006), heart failure (Tsutsui, Kinugawa et al. 2011), exercise intolerance (Witman, McDaniel et al. 2012), hypertension (Rodrigo, Gonzalez et al. 2011), cancer (Reuter, Gupta et al. 2010), neurological diseases, e.g., Alzheimer’s Disease and Parkinson’s Disease (Uttara, Singh et al. 2009, Patel 2016) and metabolic syndrome (Vona, Gambardella et al. 2019), diabetes (Giacco and Brownlee 2010), obesity (Manna and Jain 2015) and hyperlipidemia (Yang, Shi et al. 2008). There are two important points; 1) all of these diseases limit healthful aging beyond limiting only lifespan, and 2) many of these diseases of aging are linked to others, augmenting the negative impact of oxidative stress on healthful aging.
Increased β-adrenergic signaling is a major mechanism increasing oxidative stress and mediating reduced longevity, as it results in increased myocardial oxygen consumption, which in the presence of atherosclerosis or hypertension leads to myocardial ischemic disease and heart failure, major causes of disability and death in the older population. Increased β-adrenergic signaling via chronic administration of β-adrenergic receptor agonists increase morbidity and mortality in humans and other mammals (Ho, Yan et al. 2010).
Conversely, low resting heart rate, a key feature of reduced β-adrenergic signaling is associated with healthful longevity (Jensen 2019). There are physiological methods to reduce increased β-adrenergic signaling, e.g., exercise and weight loss. There are also many therapeutic modalities to reduce increased β-adrenergic signaling. Most commonly β-adrenergic blocking drugs are administered to patients to accomplish this, e.g., metoprolol or bisoprolol. β-adrenergic receptor blockade therapy has become increasingly important in the aging population, since it decreases mortality after acute myocardial infarction and chronic stable angina post-myocardial infarction (Gibbons, Chatterjee et al. 1999), and protects against hypertension (Wiysonge and Opie 2013) and reduces both the incidence and severity of Alzheimer’s Disease (Bristow 2000, Rosenberg, Mielke et al. 2008). β-adrenergic receptor blockade therapy also reduces mortality in type 2 diabetics with concomitant hypertension (UK-Prospective-Diabetes-Study-Group 1998), heart failure (Bell, Lukas et al. 2006), or myocardial infarction (Gottlieb, McCarter et al. 1998). On the other hand, these drugs have major adverse side effects including decreased cardiac output, increased airway resistance which can worsen asthma, exacerbation of peripheral vascular disease, and sexual dysfunction. These drugs also alter glucose metabolism and lead to hypoglycemia and are associated with weight gain. Accordingly, new pharmaceutical modalities are required to alleviate enhanced β-adrenergic signaling and protect against oxidative stress.
One approach to develop new pharmacological agents to reduce β-adrenergic signaling, can be derived from prior sections in this review involving longevity models, particularly the adenylyl cyclase type 5 KO mice, which have decreased β-adrenergic signaling, resulting in increased maximal lifespan (Yan, Vatner et al. 2007). This is further supported in humans, in which individuals with the genetic haplotype A-C, a known haplotype of decreased β2AR signaling, were more likely to become centenarians (Zhao, Yang et al. 2012).
Summary
Protection against oxidative stress is a major mechanism mediating increased longevity. Other mechanisms mediating increased longevity are summarized in Table II and include exercise, protection against obesity, increased β-adrenergic signaling, and against concomitant diseases of aging, e.g., diabetes, cancer, cardiovascular diseases, and neurological diseases including Alzheimer’s Disease. This review describes the animal models of increased longevity, relating these findings to human longevity. The review of these animal models identifies gaps in knowledge to facilitate further research and drug development. New pharmacologic agents could be designed based on any of the healthful longevity models noted in Table II.
Table II-b:
Protective Features of A Longevity Model Not Protected Against Oxidative Stress
| MODEL | IMPROVED EXERCISE | OBESITY PROTECTION | DIABETES PROTECTION | CV STRESS PROTECTION | CANCER PROTECTION | PROTECTION OBSERVED IN OLD AGE | DECREASED β-ADRENERGIC SIGNALING |
|---|---|---|---|---|---|---|---|
| Growth Hormone Receptor and Binding Protein KO | + | + | ? | ? | + | + | ? |
Table II-c:
Protective Features of Longevity Models With Oxidative Stress Protection Controversial
| MODEL | IMPROVED EXERCISE | OBESITY PROTECTION | DIABETES PROTECTION | CV STRESS PROTECTION | CANCER PROTECTION | PROTECTION OBSERVED IN OLD AGE | DECREASED β-ADRENERGIC SIGNALING |
|---|---|---|---|---|---|---|---|
| Ames Dwarf | ? | − | + | + | + | +/− | ? |
| Mitochondrial 5-Demethoxyubiquinone Hydroxylase Hetero (Mclk1+/−) | ? | ? | ? | ? | + | ? | ? |
| Insulin Receptor Substrate 1 KO | ? | + | − | − | +/− | +/− | + |
Table II-d:
Protective Features of Longevity Models With Oxidative Stress Not Studied
| MODEL | IMPROVED EXERCISE | OBESITY PROTECTION | DIABETES PROTECTION | CV STRESS PROTECTION | CANCER PROTECTION | PROTECTION OBSERVED IN OLD AGE | DECREASED β-ADRENERGIC SIGNALING |
|---|---|---|---|---|---|---|---|
| Phosphoenolpyruvate Carboxykinase (PEPCK-C) Overexpression | + | + | − | ? | ? | + | ? |
| Pregnancy Associated Protein Plasma A KO | ? | +/− | + | ? | + | ? | ? |
Table II:+: positive results, −: negative results; +/−: controversial results; ?: not studied.
Highlights:
The role of protection against oxidative stress mediating longevity and more importantly, healthful aging.
The role of protection against those debilitating diseases that are prevalent in the elderly and negatively impact healthful aging, e.g., diabetes and other metabolic diseases, obesity, cancer, neurological diseases, aging-induced cardiomyopathy, cardiac stress, osteoporosis and reduced exercise capacity and increased β-adrenergic signaling.
The common cellular signaling pathways involved in protection against oxidative stress in aging and in the associated diseases of aging
SOURCES OF FUNDING
This study was supported by National Institutes of Health grants R01HL106511, R01HL130848, R01HL137368, and R01HL137405.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
None
References
- Ahn JH, et al. (2020). “Therapeutic Hypothermia Improves Hind Limb Motor Outcome and Attenuates Oxidative Stress and Neuronal Damage in the Lumbar Spinal Cord Following Cardiac Arrest.” Antioxidants (Basel) 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasaki Y, et al. (2014). “FoxO transcription factors support oxidative stress resistance in human chondrocytes.” Arthritis Rheumatol 66(12): 3349–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Regaiey KA, et al. (2005). “Long-lived growth hormone receptor knockout mice: interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction.” Endocrinology 146(2): 851–860. [DOI] [PubMed] [Google Scholar]
- Alderman JM, et al. (2009). “Neuroendocrine inhibition of glucose production and resistance to cancer in dwarf mice.” Exp Gerontol 44(1–2): 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadda AA and Sallam RM (2012). “Reactive oxygen species in health and disease.” J Biomed Biotechnol 2012: 936486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alva N, et al. (2013). “Oxidative stress and antioxidant activity in hypothermia and rewarming: can RONS modulate the beneficial effects of therapeutic hypothermia?” Oxid Med Cell Longev 2013: 957054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angstman NB, et al. (2018). “Hypothermia ameliorates blast-related lifespan reduction of C. elegans.” Sci Rep 8(1): 10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, et al. (2006). “NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox.” Journal of Biological Chemistry 281(9): 5657–5667. [DOI] [PubMed] [Google Scholar]
- Araki E, et al. (1994). “Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene.” Nature 372(6502): 186–190. [DOI] [PubMed] [Google Scholar]
- Armutcu F, et al. (2008). “Oxidative stress markers, C-reactive protein and heat shock protein 70 levels in subjects with metabolic syndrome.” Clin Chem Lab Med 46(6): 785–790. [DOI] [PubMed] [Google Scholar]
- Arum O, et al. (2009). “The growth hormone receptor gene-disrupted mouse fails to respond to an intermittent fasting diet.” Aging Cell 8(6): 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arum O, et al. (2014). “The slow-aging growth hormone receptor/binding protein gene-disrupted (GHR-KO) mouse is protected from aging-resultant neuromusculoskeletal frailty.” Age (Dordr) 36(1): 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balligand JL (2016). “Cardiac salvage by tweaking with beta-3-adrenergic receptors.” Cardiovasc Res 111(2): 128–133. [DOI] [PubMed] [Google Scholar]
- Bartke A and Brown-Borg H (2004). “Life extension in the dwarf mouse.” Curr Top Dev Biol 63: 189–225. [DOI] [PubMed] [Google Scholar]
- Bartke A and Westbrook R (2012). “Metabolic characteristics of long-lived mice.” Front Genet 3: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai A and Yamamoto K (2004). “DNA damage responses to oxidative stress.” DNA Repair (Amst) 3(8–9): 1109–1115. [DOI] [PubMed] [Google Scholar]
- Basu R, et al. (2018). “MECHANISMS IN ENDOCRINOLOGY: Lessons from growth hormone receptor gene-disrupted mice: are there benefits of endocrine defects?” Eur J Endocrinol 178(5): R155–R181. [DOI] [PubMed] [Google Scholar]
- Baumann G (2002). “Growth hormone binding protein. The soluble growth hormone receptor.” Minerva Endocrinol 27(4): 265–276. [PubMed] [Google Scholar]
- Baxter SL, et al. (2014). “Cold temperature improves mobility and survival in Drosophila models of autosomal-dominant hereditary spastic paraplegia (AD-HSP).” Dis Model Mech 7(8): 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck MA (2000). “Nutritionally induced oxidative stress: effect on viral disease.” Am J Clin Nutr 71(6 Suppl): 1676S–1681S. [DOI] [PubMed] [Google Scholar]
- Belge C, et al. (2014). “Enhanced expression of beta3-adrenoceptors in cardiac myocytes attenuates neurohormone-induced hypertrophic remodeling through nitric oxide synthase.” Circulation 129(4): 451–462. [DOI] [PubMed] [Google Scholar]
- Bell DS, et al. (2006). “The effect of carvedilol on mortality risk in heart failure patients with diabetes: results of a meta-analysis.” Curr Med Res Opin 22(2): 287–296. [DOI] [PubMed] [Google Scholar]
- Beltran PJ, et al. (2009). “AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells.” Mol Cancer Ther 8(5): 1095–1105. [DOI] [PubMed] [Google Scholar]
- Benigni A, et al. (2009). “Disruption of the Ang II type 1 receptor promotes longevity in mice.” J Clin Invest 119(3): 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlett BS and Stadtman ER (1997). “Protein oxidation in aging, disease, and oxidative stress.” Journal of Biological Chemistry 272(33): 20313–20316. [DOI] [PubMed] [Google Scholar]
- Berniakovich I, et al. (2008). “p66Shc-generated oxidative signal promotes fat accumulation.” J Biol Chem 283(49): 34283–34293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyfuss K and Hood DA (2018). “A systematic review of p53 regulation of oxidative stress in skeletal muscle.” Redox Rep 23(1): 100–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat HF, et al. (2014). “Role of SNTA1 in Rac1 activation, modulation of ROS generation, and migratory potential of human breast cancer cells.” Br J Cancer 110(3): 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielschowsky F and Bielschowsky M (1959). “Carcinogenesis in the pituitary dwarf mouse. The response to methylcholanthrene injected subcutaneously.” Br J Cancer 13: 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binsch C, et al. (2017). “Absence of the kinase S6k1 mimics the effect of chronic endurance exercise on glucose tolerance and muscle oxidative stress.” Mol Metab 6(11): 1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birt DF, et al. (1997). “Dietary energy restriction does not inhibit pancreatic carcinogenesis by N-nitrosobis-2-(oxopropyl)amine in the Syrian hamster.” Carcinogenesis 18(11): 2107–2111. [DOI] [PubMed] [Google Scholar]
- Bluher M, et al. (2003). “Extended longevity in mice lacking the insulin receptor in adipose tissue.” Science 299(5606): 572–574. [DOI] [PubMed] [Google Scholar]
- Bluman LG, et al. (1998). “Preoperative smoking habits and postoperative pulmonary complications.” Chest 113(4): 883–889. [DOI] [PubMed] [Google Scholar]
- Bokov AF, et al. (2011). “Does reduced IGF-1R signaling in Igf1r+/− mice alter aging?” PLoS One 6(11): e26891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokov AF, et al. (2009). “Long-lived ames dwarf mice are resistant to chemical stressors.” J Gerontol A Biol Sci Med Sci 64(8): 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski MS, et al. (2009). “Disruption of growth hormone receptor prevents calorie restriction from improving insulin action and longevity.” PLoS One 4(2): e4567. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bonkowski MS, et al. (2006). “Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction.” Proc Natl Acad Sci U S A 103(20): 7901–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzid MA, et al. (2018). “Lifelong Voluntary Exercise Modulates Age-Related Changes in Oxidative Stress.” Int J Sports Med 39(1): 21–28. [DOI] [PubMed] [Google Scholar]
- Boylston WH, et al. (2004). “Altered cholesterologenic and lipogenic transcriptional profile in livers of aging Snell dwarf (Pit1dw/dwJ) mice.” Aging Cell 3(5): 283–296. [DOI] [PubMed] [Google Scholar]
- Bravo CA, et al. (2016). “A Food and Drug Administration-Approved Antiviral Agent that Inhibits Adenylyl Cyclase Type 5 Protects the Ischemic Heart Even When Administered after Reperfusion.” J Pharmacol Exp Ther 357(2): 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgman P, et al. (2005). “Gender-specific patterns of left ventricular and myocyte remodeling following myocardial infarction in mice deficient in the angiotensin II type 1a receptor.” Am J Physiol Heart Circ Physiol 289(2): H586–592. [DOI] [PubMed] [Google Scholar]
- Brinkkoetter PT, et al. (2008). “Hypothermic injury: the mitochondrial calcium, ATP and ROS love-hate triangle out of balance.” Cell Physiol Biochem 22(1–4): 195–204. [DOI] [PubMed] [Google Scholar]
- Bristow MR (2000). “beta-adrenergic receptor blockade in chronic heart failure.” Circulation 101(5): 558–569. [DOI] [PubMed] [Google Scholar]
- Brown-Borg HM, et al. (1996). “Dwarf mice and the ageing process.” Nature 384(6604): 33. [DOI] [PubMed] [Google Scholar]
- Brunet A, et al. (1999). “Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor.” Cell 96(6): 857–868. [DOI] [PubMed] [Google Scholar]
- Brunet A, et al. (2004). “Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase.” Science 303(5666): 2011–2015. [DOI] [PubMed] [Google Scholar]
- Brys K, et al. (2010). “Disruption of insulin signalling preserves bioenergetic competence of mitochondria in ageing Caenorhabditis elegans.” BMC Biol 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchowski MS, et al. (2012). “Effect of modest caloric restriction on oxidative stress in women, a randomized trial.” PLoS One 7(10): e47079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner D and Skemp S (2016). “Blue Zones: Lessons From the World’s Longest Lived.” Am J Lifestyle Med 10(5): 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA and Halliwell B (2019). “Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease.” Nat Rev Neurosci 20(3): 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo A and Koch WJ (2017). “Targeting beta3-Adrenergic Receptors in the Heart: Selective Agonism and beta-Blockade.” J Cardiovasc Pharmacol 69(2): 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi A, et al. (2009). “The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury.” Biochim Biophys Acta 1787(7): 774–780. [DOI] [PubMed] [Google Scholar]
- Cervantes Gracia K, et al. (2017). “CVD and Oxidative Stress.” J Clin Med 6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, et al. (2012). “Insulin receptor substrate-1 prevents autophagy-dependent cell death caused by oxidative stress in mouse NIH/3T3 cells.” J Biomed Sci 19: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, et al. (2012). “Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS.” Am J Physiol Heart Circ Physiol 302(1): H105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RS, et al. (1997). “Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Short-term double-blind, crossover study.” Eur Neurol 37(4): 212–218. [DOI] [PubMed] [Google Scholar]
- Chen Y, et al. (2012). “The influence of dietary lipid composition on skeletal muscle mitochondria from mice following 1 month of calorie restriction.” J Gerontol A Biol Sci Med Sci 67(11): 1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HJ, et al. (2001). “Upregulation of functional beta(3)-adrenergic receptor in the failing canine myocardium.” Circ Res 89(7): 599–606. [DOI] [PubMed] [Google Scholar]
- Choi JA, et al. (2001). “Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin.” Int J Oncol 19(4): 837–844. [DOI] [PubMed] [Google Scholar]
- Chu DK, et al. (2018). “Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis.” Lancet 391(10131): 1693–1705. [DOI] [PubMed] [Google Scholar]
- Ciciliot S, et al. (2015). “p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans.” Diabetologia 58(10): 2352–2360. [DOI] [PubMed] [Google Scholar]
- Civitarese AE, et al. (2007). “Calorie restriction increases muscle mitochondrial biogenesis in healthy humans.” PLoS Med 4(3): e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggan AR, et al. (1990). “Histochemical and enzymatic characteristics of skeletal muscle in master athletes.” J Appl Physiol (1985) 68(5): 1896–1901. [DOI] [PubMed] [Google Scholar]
- Colman RJ, et al. (2009). “Caloric restriction delays disease onset and mortality in rhesus monkeys.” Science 325(5937): 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, et al. (2014). “Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys.” Nat Commun 5: 3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, et al. (1998). “The effect of dietary restriction on body composition in adult male and female rhesus macaques.” Aging (Milano) 10(2): 83–92. [DOI] [PubMed] [Google Scholar]
- Conover CA and Bale LK (2007). “Loss of pregnancy-associated plasma protein A extends lifespan in mice.” Aging Cell 6(5): 727–729. [DOI] [PubMed] [Google Scholar]
- Conover CA, et al. (2010). “Longevity and age-related pathology of mice deficient in pregnancy-associated plasma protein-A.” J Gerontol A Biol Sci Med Sci 65(6): 590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti B, et al. (2006). “Transgenic mice with a reduced core body temperature have an increased life span.” Science 314(5800): 825–828. [DOI] [PubMed] [Google Scholar]
- Corry JJ (2012). “Use of hypothermia in the intensive care unit.” World J Crit Care Med 1(4): 106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschigano KT, et al. (2000). “Assessment of growth parameters and life span of GHR/BP gene-disrupted mice.” Endocrinology 141(7): 2608–2613. [DOI] [PubMed] [Google Scholar]
- Coschigano KT, et al. (2003). “Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span.” Endocrinology 144(9): 3799–3810. [DOI] [PubMed] [Google Scholar]
- Croniger CM, et al. (2002). “Phosphoenolpyruvate carboxykinase revisited - Insights into its metabolic role.” Biochemistry and Molecular Biology Education 30(1): 14–20. [Google Scholar]
- Csiszar A, et al. (2008). “Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice.” Am J Physiol Heart Circ Physiol 295(5): H1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, et al. (2011). “Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure.” Circ Res 108(7): 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, et al. (2009). “Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging.” Circulation 119(21): 2789–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansen TB, et al. (2009). “Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity.” Nat Chem Biol 5(9): 664–672. [DOI] [PubMed] [Google Scholar]
- Davis WB, et al. (1983). “Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia.” N Engl J Med 309(15): 878–883. [DOI] [PubMed] [Google Scholar]
- de Groot S, et al. (2020). “Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial.” Nat Commun 11(1): 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorenzo MS, et al. (2011). “Caloric restriction reduces growth of mammary tumors and metastases.” Carcinogenesis 32(9): 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorenzo MS, et al. (2014). “‘Reduced malignancy as a mechanism for longevity in mice with adenylyl cyclase type 5 disruption’.” Aging Cell 13(1): 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearth RK, et al. (2006). “Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2.” Mol Cell Biol 26(24): 9302–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dede S, et al. (2002). “Effect of short-term hypothermia on lipid peroxidation and antioxidant enzyme activity in rats.” J Vet Med A Physiol Pathol Clin Med 49(6): 286–288. [DOI] [PubMed] [Google Scholar]
- Dehkhoda F, et al. (2018). “The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects.” Front Endocrinol (Lausanne) 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Buono MG, et al. (2019). “Exercise Intolerance in Patients With Heart Failure: JACC State-of-the-Art Review.” J Am Coll Cardiol 73(17): 2209–2225. [DOI] [PubMed] [Google Scholar]
- Delafontaine P, et al. (2004). “Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels.” Arterioscler Thromb Vasc Biol 24(3): 435–444. [DOI] [PubMed] [Google Scholar]
- Deneke SM and Fanburg BL (1980). “Normobaric oxygen toxicity of the lung.” N Engl J Med 303(2): 76–86. [DOI] [PubMed] [Google Scholar]
- Dhahbi JM, et al. (2004). “Temporal linkage between the phenotypic and genomic responses to caloric restriction.” Proc Natl Acad Sci U S A 101(15): 5524–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi K, et al. (2013). “Status of systemic oxidative stress during therapeutic hypothermia in patients with post-cardiac arrest syndrome.” Oxid Med Cell Longev 2013: 562429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici FP, et al. (2002). “Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice.” J Endocrinol 173(1): 81–94. [DOI] [PubMed] [Google Scholar]
- Donald SP, et al. (2001). “Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species.” Cancer Res 61(5): 1810–1815. [PubMed] [Google Scholar]
- Donato AJ, et al. (2013). “Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice.” Aging Cell 12(5): 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drane P, et al. (2001). “Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis.” Oncogene 20(4): 430–439. [DOI] [PubMed] [Google Scholar]
- Driedonks N, et al. (2015). “Multi-Level Interactions Between Heat Shock Factors, Heat Shock Proteins, and the Redox System Regulate Acclimation to Heat.” Front Plant Sci 6: 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards IJ, et al. (1998). “Caloric restriction in rhesus monkeys reduces low density lipoprotein interaction with arterial proteoglycans.” J Gerontol A Biol Sci Med Sci 53(6): B443–448. [DOI] [PubMed] [Google Scholar]
- Egecioglu E, et al. (2007). “Growth hormone receptor deficiency in mice results in reduced systolic blood pressure and plasma renin, increased aortic eNOS expression, and altered cardiovascular structure and function.” Am J Physiol Endocrinol Metab 292(5): E1418–1425. [DOI] [PubMed] [Google Scholar]
- Elchuri S, et al. (2005). “CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life.” Oncogene 24(3): 367–380. [DOI] [PubMed] [Google Scholar]
- Elosua R, et al. (2003). “Response of oxidative stress biomarkers to a 16-week aerobic physical activity program, and to acute physical activity, in healthy young men and women.” Atherosclerosis 167(2): 327–334. [DOI] [PubMed] [Google Scholar]
- Elzeneini E and Wickström SA (2017). “Lipodystrophic laminopathy: Lamin A mutation relaxes chromatin architecture to impair adipogenesis.” J Cell Biol 216(9): 2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns LC, et al. (2009). “Disruption of protein kinase A in mice enhances healthy aging.” PLoS One 4(6): e5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Ferrall Erin (2020). “Mitochondrial myopathies: Treatment.” UpToDate https://www.uptodate.com/contents/mitochondrial-myopathies-treatment.
- Esposito LA, et al. (1999). “Mitochondrial disease in mouse results in increased oxidative stress.” Proc Natl Acad Sci U S A 96(9): 4820–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, et al. (2020). “Lifespan of long-lived growth hormone receptor knockout mice was not normalized by housing at 30 degrees C since weaning.” Aging Cell 19(5): e13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fialova L and Malbohan IM (2002). “Pregnancy-associated plasma protein A (PAPP-A): theoretical and clinical aspects.” Bratisl Lek Listy 103(6): 194–205. [PubMed] [Google Scholar]
- Finkel T and Holbrook NJ (2000). “Oxidants, oxidative stress and the biology of ageing.” Nature 408(6809): 239–247. [DOI] [PubMed] [Google Scholar]
- Fisher-Wellman K and Bloomer RJ (2009). “Acute exercise and oxidative stress: a 30 year history.” Dyn Med 8: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, et al. (2002). “The Snell dwarf mutation Pit1(dw) can increase life span in mice.” Mech Ageing Dev 123(2–3): 121–130. [DOI] [PubMed] [Google Scholar]
- Fontana L, et al. (2010). “Extending healthy life span--from yeast to humans.” Science 328(5976): 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ES, et al. (2003). “The metabolic syndrome and antioxidant concentrations: findings from the Third National Health and Nutrition Examination Survey.” Diabetes 52(9): 2346–2352. [DOI] [PubMed] [Google Scholar]
- Frescas D, et al. (2005). “Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes.” Journal of Biological Chemistry 280(21): 20589–20595. [DOI] [PubMed] [Google Scholar]
- Friedman DB and Johnson TE (1988). “A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility.” Genetics 118(1): 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB and Johnson TE (1988). “Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene.” J Gerontol 43(4): B102–109. [DOI] [PubMed] [Google Scholar]
- Furumoto T, et al. (2005). “Loss of insulin receptor substrate-1 signaling induces the cardiovascular and proteo(fibrino)lytic system derangements typical of insulin resistance.” Coron Artery Dis 16(2): 117–123. [DOI] [PubMed] [Google Scholar]
- Gaetani GF, et al. (1996). “Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes.” Blood 87(4): 1595–1599. [PubMed] [Google Scholar]
- Galimov ER (2010). “The Role of p66shc in Oxidative Stress and Apoptosis.” Acta Naturae 2(4): 44–51. [PMC free article] [PubMed] [Google Scholar]
- Gamez A, et al. (2008). “Beneficial effects of fructose 1,6-biphosphate on hypothermia-induced reactive oxygen species injury in rats.” Eur J Pharmacol 590(1–3): 115–119. [DOI] [PubMed] [Google Scholar]
- Garg N, et al. (2011). “High fat diet induced insulin resistance and glucose intolerance are gender-specific in IGF-1R heterozygous mice.” Biochem Biophys Res Commun 413(3): 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier C, et al. (1996). “Functional beta3-adrenoceptor in the human heart.” J Clin Invest 98(2): 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, et al. (2018). Role of Heat Shock Proteins in Oxidative Stress and Stress Tolerance Heat Shock Proteins and Stress. Asea AAA and Kaur P. Cham, Springer International Publishing: 109–126. [Google Scholar]
- Giacco F and Brownlee M (2010). “Oxidative stress and diabetic complications.” Circ Res 107(9): 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, et al. (1999). “ACC/AHA/ACP-ASIM guidelines for the management of patients with chronic stable angina: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients With Chronic Stable Angina).” J Am Coll Cardiol 33(7): 2092–2197. [DOI] [PubMed] [Google Scholar]
- Giesbrecht GG (2000). “Cold stress, near drowning and accidental hypothermia: a review.” Aviat Space Environ Med 71(7): 733–752. [PubMed] [Google Scholar]
- Giorgio M, et al. (2012). “The p66Shc knocked out mice are short lived under natural condition.” Aging Cell 11(1): 162–168. [DOI] [PubMed] [Google Scholar]
- Girardis M, et al. (2016). “Effect of Conservative vs Conventional Oxygen Therapy on Mortality Among Patients in an Intensive Care Unit: The Oxygen-ICU Randomized Clinical Trial.” Jama 316(15): 1583–1589. [DOI] [PubMed] [Google Scholar]
- Goh J, et al. (2011). “Mitochondrial targeted catalase suppresses invasive breast cancer in mice.” BMC Cancer 11: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, et al. (2015). “Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training?” Free Radic Biol Med 86: 37–46. [DOI] [PubMed] [Google Scholar]
- Goncalves PB and Romeiro NC (2019). “Multi-target natural products as alternatives against oxidative stress in Chronic Obstructive Pulmonary Disease (COPD).” Eur J Med Chem 163: 911–931. [DOI] [PubMed] [Google Scholar]
- Gong P, et al. (2012). “Mild hypothermia attenuates mitochondrial oxidative stress by protecting respiratory enzymes and upregulating MnSOD in a pig model of cardiac arrest.” PLoS One 7(4): e35313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, et al. (1997). “Antioxidant enzyme activities in lens, liver and kidney of calorie restricted Emory mice.” Mech Ageing Dev 99(3): 181–192. [DOI] [PubMed] [Google Scholar]
- Gottlieb SS, et al. (1998). “Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction.” N Engl J Med 339(8): 489–497. [DOI] [PubMed] [Google Scholar]
- Granatiero V, et al. (2017). “Role of p66shc in skeletal muscle function.” Sci Rep 7(1): 6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CL, et al. (2020). “The Effects of Graded Levels of Calorie Restriction: XIV. Global Metabolomics Screen Reveals Brown Adipose Tissue Changes in Amino Acids, Catecholamines, and Antioxidants After Short-Term Restriction in C57BL/6 Mice.” J Gerontol A Biol Sci Med Sci 75(2): 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresl TA, et al. (2003). “Insulin sensitivity and glucose effectiveness from three minimal models: effects of energy restriction and body fat in adult male rhesus monkeys.” Am J Physiol Regul Integr Comp Physiol 285(6): R1340–1354. [DOI] [PubMed] [Google Scholar]
- Guers J, et al. (2017, 136:A18790). “Brown Adipose Tissue Enhances Exercise Performance.” Circulation (Suppl). [Google Scholar]
- Gumuslu S, et al. (2002). “Influences of different stress models on the antioxidant status and lipid peroxidation in rat erythrocytes.” Free Radic Res 36(12): 1277–1282. [DOI] [PubMed] [Google Scholar]
- Guo DF, et al. (2001). “The angiotensin II type 1 receptor and receptor-associated proteins.” Cell Res 11(3): 165–180. [DOI] [PubMed] [Google Scholar]
- Guo Z, et al. (2002). “Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E-deficient mice.” Mech Ageing Dev 123(8): 1121–1131. [DOI] [PubMed] [Google Scholar]
- Guzik TJ and Touyz RM (2017). “Oxidative Stress, Inflammation, and Vascular Aging in Hypertension.” Hypertension 70(4): 660–667. [DOI] [PubMed] [Google Scholar]
- Gyuraszova M, et al. (2020). “Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers.” Oxid Med Cell Longev 2020: 5478708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenhaar FS, et al. (2017). “Therapeutic Hypothermia Reduces Oxidative Damage and Alters Antioxidant Defenses after Cardiac Arrest.” Oxid Med Cell Longev 2017: 8704352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi P, et al. (2007). “Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse.” Journal of Biological Chemistry 282(45): 32844–32855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B and Gutteridge J (1989). “Free radicals in biology and medicine.” Oxford: Oxford University Press. [Google Scholar]
- Hammer S, et al. (2008). “Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function.” J Am Coll Cardiol 52(12): 1006–1012. [DOI] [PubMed] [Google Scholar]
- Harada K, et al. (1999). “Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction.” Circulation 100(20): 2093–2099. [DOI] [PubMed] [Google Scholar]
- Harper JM, et al. (2010). “Macrophage migration inhibitory factor-knockout mice are long lived and respond to caloric restriction.” FASEB J 24(7): 2436–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE and Archer JR (1987). “Genetic differences in effects of food restriction on aging in mice.” J Nutr 117(2): 376–382. [DOI] [PubMed] [Google Scholar]
- Harrison DE, et al. (1984). “Effects of food restriction on aging: separation of food intake and adiposity.” Proc Natl Acad Sci U S A 81(6): 1835–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, et al. (2009). “Hypothermic inhibition of apoptotic pathways for combined neurotoxicity of iron and ascorbic acid in differentiated PC12 cells: reduction of oxidative stress and maintenance of the glutathione redox state.” Brain Res 1283: 1–13. [DOI] [PubMed] [Google Scholar]
- Hauck SJ, et al. (2002). “Antioxidant enzymes, free-radical damage, and response to paraquat in liver and kidney of long-living growth hormone receptor/binding protein gene-disrupted mice.” Horm Metab Res 34(9): 481–486. [DOI] [PubMed] [Google Scholar]
- Hauck SJ and Bartke A (2000). “Effects of growth hormone on hypothalamic catalase and Cu/Zn superoxide dismutase.” Free Radic Biol Med 28(6): 970–978. [DOI] [PubMed] [Google Scholar]
- Hedley-Whyte J (1970). “Causes of pulmonary oxygen toxicity.” N Engl J Med 283(27): 1518–1519. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, et al. (2006). “Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial.” JAMA 295(13): 1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzeneder S, et al. (2019). “Pregnancy-Associated Plasma Protein-A (PAPP-A) in Ewing Sarcoma: Role in Tumor Growth and Immune Evasion.” J Natl Cancer Inst 111(9): 970–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom A, et al. (2003). “Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth.” Pediatrics 112(5): 1016–1020. [DOI] [PubMed] [Google Scholar]
- Hellstrom A, et al. (2001). “Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical retinopathy of prematurity.” Proc Natl Acad Sci U S A 98(10): 5804–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström A, et al. (2013). “Retinopathy of prematurity.” Lancet 382(9902): 1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks KDW, et al. (2019). “Renal temperature reduction progressively favors mitochondrial ROS production over respiration in hypothermic kidney preservation.” J Transl Med 17(1): 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CM, et al. (2015). “Female PAPP-A knockout mice are resistant to metabolic dysfunction induced by high-fat/high-sucrose feeding at middle age.” Age (Dordr) 37(3): 9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono Y, et al. (2007). “Angiotensin II receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: the possible role of local renin-angiotensin system.” Endocrinology 148(4): 1688–1696. [DOI] [PubMed] [Google Scholar]
- Ho D, et al. (2010). “Modulation of beta-adrenergic receptor signaling in heart failure and longevity: targeting adenylyl cyclase type 5.” Heart Fail Rev 15(5): 495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho D, et al. (2015). “Adenylyl Cyclase Type 5 Deficiency Protects Against Diet-Induced Obesity and Insulin Resistance.” Diabetes 64(7): 2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley AK and St Clair DK (2009). “Watching the watcher: regulation of p53 by mitochondria.” Future Oncol 5(1): 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holvoet P, et al. (2008). “Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome.” JAMA 299(19): 2287–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, et al. (2003). “IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice.” Nature 421(6919): 182–187. [DOI] [PubMed] [Google Scholar]
- Hood WR, et al. (2018). “Life History Trade-offs within the Context of Mitochondrial Hormesis.” Integr Comp Biol 58(3): 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinabadi MB and Khanjani N (2019). “The Effect of Extremely Low-Frequency Electromagnetic Fields on the Prevalence of Musculoskeletal Disorders and the Role of Oxidative Stress.” Bioelectromagnetics 40(5): 354–360. [DOI] [PubMed] [Google Scholar]
- Huang Y and Mark Jacquez G (2017). “Identification of a Blue Zone in a Typical Chinese Longevity Region.” Int J Environ Res Public Health 14(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursting SD, et al. (2013). “Calorie restriction and cancer prevention: a mechanistic perspective.” Cancer Metab 1(1): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursting SD, et al. (2010). “Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research.” Carcinogenesis 31(1): 83–89. [DOI] [PubMed] [Google Scholar]
- Ikeno Y, et al. (2003). “Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: correlation to extended longevity.” J Gerontol A Biol Sci Med Sci 58(4): 291–296. [DOI] [PubMed] [Google Scholar]
- Ikeno Y, et al. (2009). “Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice.” J Gerontol A Biol Sci Med Sci 64(5): 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayama I, et al. (2012). “Effects of a caloric restriction weight loss diet and exercise on inflammatory biomarkers in overweight/obese postmenopausal women: a randomized controlled trial.” Cancer Res 72(9): 2314–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inal ME, et al. (2001). “Antioxidant enzyme activities and malondialdehyde levels related to aging.” Clin Chim Acta 305(1–2): 75–80. [DOI] [PubMed] [Google Scholar]
- Ishikane S, et al. (2018). “Angiotensin II promotes pulmonary metastasis of melanoma through the activation of adhesion molecules in vascular endothelial cells.” Biochem Pharmacol 154: 136–147. [DOI] [PubMed] [Google Scholar]
- Jacquier-Sarlin MR, et al. (1994). “Protective effects of hsp70 in inflammation.” Experientia 50(11–12): 1031–1038. [DOI] [PubMed] [Google Scholar]
- Jaffer S, et al. (2009). “Mammalian target of rapamycin is activated in association with myometrial proliferation during pregnancy.” Endocrinology 150(10): 4672–4680. [DOI] [PubMed] [Google Scholar]
- Jansen EHJM, et al. (2014). “The Effect of Smoking on Biomarkers of (Anti)oxidant Status.” J Mol Biomark Diagn 5(6): 207. [Google Scholar]
- Jensen MT (2019). “Resting heart rate and relation to disease and longevity: past, present and future.” Scand J Clin Lab Invest 79(1–2): 108–116. [DOI] [PubMed] [Google Scholar]
- Jha P, et al. (2013). “21st-Century Hazards of Smoking and Benefits of Cessation in the United States.” New England Journal of Medicine 368(4): 341–350. [DOI] [PubMed] [Google Scholar]
- Ji N, et al. (2018). “Aerobic exercise-stimulated Klotho upregulation extends life span by attenuating the excess production of reactive oxygen species in the brain and kidney.” Exp Ther Med 16(4): 3511–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TM, et al. (1996). “Reactive oxygen species are downstream mediators of p53-dependent apoptosis.” Proc Natl Acad Sci U S A 93(21): 11848–11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LW, et al. (2009). “Exercise intolerance in cancer and the role of exercise therapy to reverse dysfunction.” Lancet Oncol 10(6): 598–605. [DOI] [PubMed] [Google Scholar]
- Junnila RK, et al. (2016). “Disruption of the GH Receptor Gene in Adult Mice Increases Maximal Lifespan in Females.” Endocrinology 157(12): 4502–4513. [DOI] [PubMed] [Google Scholar]
- Kairisalo M, et al. (2007). “X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation.” Biochem Biophys Res Commun 364(1): 138–144. [DOI] [PubMed] [Google Scholar]
- Kanfi Y, et al. (2012). “The sirtuin SIRT6 regulates lifespan in male mice.” Nature 483(7388): 218–221. [DOI] [PubMed] [Google Scholar]
- Katerji M, et al. (2019). “Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field.” Oxid Med Cell Longev 2019: 1279250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith M, et al. (1998). “Increased oxidative stress in patients with congestive heart failure.” J Am Coll Cardiol 31(6): 1352–1356. [DOI] [PubMed] [Google Scholar]
- Kenyon C, et al. (1993). “A C. elegans mutant that lives twice as long as wild type.” Nature 366(6454): 461–464. [DOI] [PubMed] [Google Scholar]
- Kimura T, et al. (2018). “The impact of preserved Klotho gene expression on antioxidative stress activity in healthy kidney.” Am J Physiol Renal Physiol 315(2): F345–F352. [DOI] [PubMed] [Google Scholar]
- Kitamura T, et al. (2000). “The negative inotropic effect of beta3-adrenoceptor stimulation in the beating guinea pig heart.” J Cardiovasc Pharmacol 35(5): 786–790. [DOI] [PubMed] [Google Scholar]
- Klopstock T, et al. (2011). “A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy.” Brain 134(Pt 9): 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz LO, et al. (2015). “Redox regulation of FoxO transcription factors.” Redox Biol 6: 51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, et al. (2001). “Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis.” Proc Natl Acad Sci U S A 98(5): 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, et al. (2002). “Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress.” Nature 419(6904): 316–321. [DOI] [PubMed] [Google Scholar]
- Kuo JR, et al. (2011). “Attenuation of brain nitrostative and oxidative damage by brain cooling during experimental traumatic brain injury.” J Biomed Biotechnol 2011: 145214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, et al. (1997). “Mutation of the mouse klotho gene leads to a syndrome resembling ageing.” Nature 390(6655): 45–51. [DOI] [PubMed] [Google Scholar]
- Kurosu H, et al. (2005). “Suppression of aging in mice by the hormone Klotho.” Science 309(5742): 1829–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, et al. (2013). “Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway.” Circulation 127(16): 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley SM, et al. (2000). “The free radical spin trap alpha-phenyl-tert-butyl nitrone attenuates the cerebral response to deep hypothermic ischemia.” J Thorac Cardiovasc Surg 119(2): 305–313. [DOI] [PubMed] [Google Scholar]
- Lanza IR, et al. (2008). “Endurance exercise as a countermeasure for aging.” Diabetes 57(11): 2933–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J and Hekimi S (2008). “Early mitochondrial dysfunction in long-lived Mclk1+/− mice.” Journal of Biological Chemistry 283(38): 26217–26227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson-Meyer DE, et al. (2006). “Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects.” Diabetes Care 29(6): 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzi G, et al. (2012). “Nuclear damages and oxidative stress: new perspectives for laminopathies.” Eur J Histochem 56(4): e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, et al. (2010). “Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance.” Cell Metab 12(6): 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, et al. (2006). “A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span.” Cell 125(5): 987–1001. [DOI] [PubMed] [Google Scholar]
- Leiser SF, et al. (2011). “HIF-1 modulates longevity and healthspan in a temperature-dependent manner.” Aging Cell 10(2): 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, et al. (2006). “Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways.” Genes Dev 20(3): 267–275. [DOI] [PubMed] [Google Scholar]
- Li HS, et al. (2019). “HIF-1alpha protects against oxidative stress by directly targeting mitochondria.” Redox Biol 25: 101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, et al. (2007). “RLE-1, an E3 ubiquitin ligase, regulates C. elegans aging by catalyzing DAF-16 polyubiquitination.” Dev Cell 12(2): 235–246. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. (1995). “Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase.” Nat Genet 11(4): 376–381. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. (2020). “Overexpression of klotho suppresses growth and pulmonary metastasis of osteosarcoma in vivo.” Genet Mol Biol 43(2): e20190229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CY, et al. (2010). “Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening.” Aging Cell 9(1): 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XX, et al. (2018). “DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity.” Nat Commun 9(1): 4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingappan K (2018). “NF-kappaB in Oxidative Stress.” Curr Opin Toxicol 7: 81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List EO, et al. (2011). “Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse.” Endocr Rev 32(3): 356–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D and Xu Y (2011). “p53, oxidative stress, and aging.” Antioxid Redox Signal 15(6): 1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. (2000). “Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants.” J Appl Physiol (1985) 89(1): 21–28. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. (2005). “Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice.” Genes Dev 19(20): 2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo SA and Lopez J (2020). Minimum Alveolar Concentration. StatPearls; Treasure Island (FL). [PubMed] [Google Scholar]
- Lobo V, et al. (2010). “Free radicals, antioxidants and functional foods: Impact on human health.” Pharmacogn Rev 4(8): 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodovici M and Bigagli E (2011). “Oxidative stress and air pollution exposure.” J Toxicol 2011: 487074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD and Fontana L (2010). “Calorie restriction and cancer prevention: metabolic and molecular mechanisms.” Trends Pharmacol Sci 31(2): 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lope V, et al. (2019). “Overeating, caloric restriction and breast cancer risk by pathologic subtype: the EPIGEICAM study.” Scientific Reports 9(1): 3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lluch G, et al. (2006). “Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency.” Proc Natl Acad Sci U S A 103(6): 1768–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LJ, et al. (2011). “Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity.” Am J Physiol Renal Physiol 300(5): F1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, et al. (2006). “Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis.” Mol Cell Biol 26(24): 9338–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen MA, et al. (2004). “Altered oxidative stress response of the long-lived Snell dwarf mouse.” Biochem Biophys Res Commun 318(4): 998–1005. [DOI] [PubMed] [Google Scholar]
- Mahjoub S and Masrour-Roudsari J (2012). “Role of oxidative stress in pathogenesis of metabolic syndrome.” Caspian J Intern Med 3(1): 386–396. [PMC free article] [PubMed] [Google Scholar]
- Maier CM, et al. (2002). “Effects of mild hypothermia on superoxide anion production, superoxide dismutase expression, and activity following transient focal cerebral ischemia.” Neurobiol Dis 11(1): 28–42. [DOI] [PubMed] [Google Scholar]
- Mandrekar P, et al. (2009). “The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes.” J Immunol 183(2): 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna P and Jain SK (2015). “Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies.” Metab Syndr Relat Disord 13(10): 423–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardilovich K, et al. (2009). “Expression and function of the insulin receptor substrate proteins in cancer.” Cell Commun Signal 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, et al. (2011). “NRF2, cancer and calorie restriction.” Oncogene 30(5): 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CK, et al. (2011). “Effect of calorie restriction on the free-living physical activity levels of nonobese humans: results of three randomized trials.” J Appl Physiol (1985) 110(4): 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins R, et al. (2016). “Long live FOXO: unraveling the role of FOXO proteins in aging and longevity.” Aging Cell 15(2): 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruani DM, et al. (2012). “Estrogenic regulation of S6K1 expression creates a positive regulatory loop in control of breast cancer cell proliferation.” Oncogene 31(49): 5073–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani S, et al. (2013). “beta3-Adrenergic receptor antagonist improves exercise performance in pacing-induced heart failure.” Am J Physiol Heart Circ Physiol 305(6): H923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison JA, et al. (2017). “Caloric restriction improves health and survival of rhesus monkeys.” Nat Commun 8: 14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison JA, et al. (2012). “Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study.” Nature 489(7415): 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP and Bukau B (2005). “Hsp70 chaperones: cellular functions and molecular mechanism.” Cell Mol Life Sci 62(6): 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard SP and Miller RA (2006). “Fibroblasts from long-lived Snell dwarf mice are resistant to oxygen-induced in vitro growth arrest.” Aging Cell 5(1): 89–96. [DOI] [PubMed] [Google Scholar]
- McCay CM, et al. (1989). “The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935.” Nutrition 5(3): 155–171; discussion 172. [PubMed] [Google Scholar]
- McCormick DL, et al. (2007). “Null effect of dietary restriction on prostate carcinogenesis in the Wistar-Unilever rat.” Nutr Cancer 57(2): 194–200. [DOI] [PubMed] [Google Scholar]
- Mehta R, et al. (2009). “Proteasomal regulation of the hypoxic response modulates aging in C. elegans.” Science 324(5931): 1196–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele J, et al. (2006). “Characterization of transgenic mice that overexpress both copper zinc superoxide dismutase and catalase.” Antioxid Redox Signal 8(3–4): 628–638. [DOI] [PubMed] [Google Scholar]
- Melov S, et al. (1999). “Mitochondrial disease in superoxide dismutase 2 mutant mice.” Proc Natl Acad Sci U S A 96(3): 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, et al. (1998). “A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase.” Nat Genet 18(2): 159–163. [DOI] [PubMed] [Google Scholar]
- Menini S, et al. (2006). “Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress.” Diabetes 55(6): 1642–1650. [DOI] [PubMed] [Google Scholar]
- Merry TL and Ristow M (2016). “Mitohormesis in exercise training.” Free Radic Biol Med 98: 123–130. [DOI] [PubMed] [Google Scholar]
- Mesquita A, et al. (2010). “Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity.” Proc Natl Acad Sci U S A 107(34): 15123–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TE, et al. (2006). “Long-term caloric restriction ameliorates the decline in diastolic function in humans.” J Am Coll Cardiol 47(2): 398–402. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, et al. (1999). “The p66shc adaptor protein controls oxidative stress response and life span in mammals.” Nature 402(6759): 309–313. [DOI] [PubMed] [Google Scholar]
- Mihara Y, et al. (2004). Mild Brain Hypothermia Suppresses Oxygen Free Radicals in Patients with Neuroemergency: An Ex Vivo Electron Spin Resonance Study Hypothermia for Acute Brain Damage, Tokyo, Springer Japan. [Google Scholar]
- Mikhed Y, et al. (2015). “Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction.” Int J Mol Sci 16(7): 15918–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskin R and Masos T (1997). “Transgenic mice overexpressing urokinase-type plasminogen activator in the brain exhibit reduced food consumption, body weight and size, and increased longevity.” J Gerontol A Biol Sci Med Sci 52(2): B118–124. [DOI] [PubMed] [Google Scholar]
- Mitchell SE, et al. (2015). “The effects of graded levels of calorie restriction: II. Impact of short term calorie and protein restriction on circulating hormone levels, glucose homeostasis and oxidative stress in male C57BL/6 mice.” Oncotarget 6(27): 23213–23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SE, et al. (2015). “The effects of graded levels of calorie restriction: I. impact of short term calorie and protein restriction on body composition in the C57BL/6 mouse.” Oncotarget 6(18): 15902–15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ and Liu ZG (2011). “Crosstalk of reactive oxygen species and NF-kappaB signaling.” Cell Res 21(1): 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Reimers CD, Berninger T (1990). “Coenzyme Q10 in ophthalmoplegia plus—a double blind, cross over therapeutic trial.” J Neurol Sci. [Google Scholar]
- Muniyan S, et al. (2015). “p66Shc longevity protein regulates the proliferation of human ovarian cancer cells.” Mol Carcinog 54(8): 618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, et al. (2003). “Multiplex stress resistance in cells from long-lived dwarf mice.” FASEB J 17(11): 1565–1566. [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009). “How mitochondria produce reactive oxygen species.” Biochem J 417(1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musci RV, et al. (2019). “Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension.” Sports (Basel) 7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KH, et al. (2019). “Identification of a novel S6K1 inhibitor, rosmarinic acid methyl ester, for treating cisplatin-resistant cervical cancer.” BMC Cancer 19(1): 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi A, et al. (2019). “Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases.” Oxid Med Cell Longev 2019: 9613090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli C, et al. (2003). “Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet.” Proc Natl Acad Sci U S A 100(4): 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCCDPHP (2014). The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General. Atlanta (GA). [Google Scholar]
- Nemoto S and Finkel T (2002). “Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway.” Science 295(5564): 2450–2452. [DOI] [PubMed] [Google Scholar]
- Nickenig G and Harrison DG (2002). “The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis.” Circulation 105(3): 393–396. [DOI] [PubMed] [Google Scholar]
- Nishiyama Y, et al. (1998). “Oxidative stress is related to exercise intolerance in patients with heart failure.” Am Heart J 135(1): 115–120. [DOI] [PubMed] [Google Scholar]
- Noshita N, et al. (2003). “Copper-zinc superoxide dismutase affects Akt activation after transient focal cerebral ischemia in mice.” Stroke 34(6): 1513–1518. [DOI] [PubMed] [Google Scholar]
- Nowak A, et al. (2014). “Prognostic value and link to atrial fibrillation of soluble Klotho and FGF23 in hemodialysis patients.” PLoS One 9(7): e100688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyan H, et al. (2015). “Cardioprotective Signature of Short-Term Caloric Restriction.” PLoS One 10(6): e0130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyan H, et al. (2015). “Cardioprotective Signature of Short-Term Caloric Restriction.” PLoS One 10(6): e0130658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, et al. (2005). “JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16.” Proc Natl Acad Sci U S A 102(12): 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura S, et al. (2003). “Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload.” Proc Natl Acad Sci U S A 100(17): 9986–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos Y, et al. (2013). “SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex.” Antioxid Redox Signal 19(13): 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, et al. (2012). “Pten positively regulates brown adipose function, energy expenditure, and longevity.” Cell Metab 15(3): 382–394. [DOI] [PubMed] [Google Scholar]
- Ostadal P, et al. (2013). “Mild therapeutic hypothermia is superior to controlled normothermia for the maintenance of blood pressure and cerebral oxygenation, prevention of organ damage and suppression of oxidative stress after cardiac arrest in a porcine model.” J Transl Med 11: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek PA (2014). 3 - Factors Affecting Transgenic Animal Production Transgenic Animal Technology (Third Edition). Pinkert CA. London, Elsevier: 71–107. [Google Scholar]
- Oydanich M, et al. (2018). “Two Mechanisms Mediating Enhanced Glucose Tolerance with Regulator of GS Protein 14 Disruption; Increased Exercise Capacity and Increased Brown Adipose Tissue.” Diabetes (Suppl) 67. [Google Scholar]
- Ozkan H, et al. (2015). “Evaluation and comparison of the effect of hypothermia and ozone on ischemia-reperfusion injury of skeletal muscle in rats.” J Surg Res 196(2): 313–319. [DOI] [PubMed] [Google Scholar]
- Page MM, et al. (2009). “Mechanisms of stress resistance in Snell dwarf mouse fibroblasts: enhanced antioxidant and DNA base excision repair capacity, but no differences in mitochondrial metabolism.” Free Radic Biol Med 46(8): 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri VO, et al. (2006). “Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome.” J Nutr 136(12): 3022–3026. [DOI] [PubMed] [Google Scholar]
- Pan H, et al. (2012). “Protein secretion is required for pregnancy-associated plasma protein-A to promote lung cancer growth in vivo.” PLoS One 7(11): e48799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis P, et al. (2000). “Overexpression of angiotensin II type I receptor in cardiomyocytes induces cardiac hypertrophy and remodeling.” Proc Natl Acad Sci U S A 97(2): 931–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JA, et al. (2015). “S6K1 inhibition enhances the apoptotic cell death of breast cancer cells in response to Bcl-2/Bcl-xL inhibition by the downregulation of survivin.” Oncol Lett 10(2): 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M (2016). “Targeting Oxidative Stress in Central Nervous System Disorders.” Trends Pharmacol Sci 37(9): 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez VI, et al. (2009). “Is the oxidative stress theory of aging dead?” Biochim Biophys Acta 1790(10): 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelzl G, et al. (2018). “Klotho is upregulated in human cardiomyopathy independently of circulating Klotho levels.” Sci Rep 8(1): 8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitras VJ, et al. (2018). “Exercise intolerance in Type 2 diabetes: is there a cardiovascular contribution?” J Appl Physiol (1985) 124(5): 1117–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, et al. (2010). “Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation.” Cell Metab 12(6): 662–667. [DOI] [PubMed] [Google Scholar]
- Quarrie JK and Riabowol KT (2004). “Murine models of life span extension.” Sci Aging Knowledge Environ 2004(31): re5. [DOI] [PubMed] [Google Scholar]
- Racette SB, et al. (2017). “Effects of Two Years of Calorie Restriction on Aerobic Capacity and Muscle Strength.” Med Sci Sports Exerc 49(11): 2240–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak Z, et al. (2005). “Exercise and hormesis: oxidative stress-related adaptation for successful aging.” Biogerontology 6(1): 71–75. [DOI] [PubMed] [Google Scholar]
- Rajapakse AG, et al. (2011). “Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol.” PLoS One 6(4): e19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Q, et al. (2007). “Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis.” J Gerontol A Biol Sci Med Sci 62(9): 932–942. [DOI] [PubMed] [Google Scholar]
- Ravussin E, et al. (2015). “A 2-Year Randomized Controlled Trial of Human Caloric Restriction: Feasibility and Effects on Predictors of Health Span and Longevity.” J Gerontol A Biol Sci Med Sci 70(9): 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, et al. (2007). “Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice.” Brain Res 1127(1): 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, et al. (2003). “Effects of age and caloric restriction on glutathione redox state in mice.” Free Radic Biol Med 35(6): 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman LM, et al. (2018). “Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging.” Cell Metab 27(4): 805–815 e804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, et al. (2010). “Oxidative stress, inflammation, and cancer: how are they linked?” Free Radic Biol Med 49(11): 1603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan MM, et al. (2008). “The effects of caloric restriction- and exercise-induced weight loss on left ventricular diastolic function.” Am J Physiol Heart Circ Physiol 294(3): H1174–1182. [DOI] [PubMed] [Google Scholar]
- Ristow M and Schmeisser K (2014). “Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS).” Dose Response 12(2): 288–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M and Zarse K (2010). “How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis).” Exp Gerontol 45(6): 410–418. [DOI] [PubMed] [Google Scholar]
- Ristow M, et al. (2009). “Antioxidants prevent health-promoting effects of physical exercise in humans.” Proc Natl Acad Sci U S A 106(21): 8665–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittenberger J, Callaway CW., (2020). “Post-cardiac arrest management in adults.” UpToDate https://www.uptodate.com/contents/post-cardiac-arrest-management-in-adults/print.
- Rivard K, et al. (2008). “Cardiac-specific overexpression of the human type 1 angiotensin II receptor causes delayed repolarization.” Cardiovasc Res 78(1): 53–62. [DOI] [PubMed] [Google Scholar]
- Roberts CK and Sindhu KK (2009). “Oxidative stress and metabolic syndrome.” Life Sci 84(21–22): 705–712. [DOI] [PubMed] [Google Scholar]
- Rodrigo R, et al. (2011). “The role of oxidative stress in the pathophysiology of hypertension.” Hypertens Res 34(4): 431–440. [DOI] [PubMed] [Google Scholar]
- Rohrbach S, et al. (2008). “Caloric restriction counteracts age-dependent changes in prolyl-4-hydroxylase domain (PHD) 3 expression.” Biogerontology 9(3): 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo AI, et al. (2004). “Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB.” J Neurosci 24(33): 7324–7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanick MA, et al. (2004). “Long-lived Ames dwarf mouse exhibits increased antioxidant defense in skeletal muscle.” Mech Ageing Dev 125(4): 269–281. [DOI] [PubMed] [Google Scholar]
- Rosenberg PB, et al. (2008). “Effects of cardiovascular medications on rate of functional decline in Alzheimer disease.” Am J Geriatr Psychiatry 16(11): 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwende C, et al. (1995). “Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria.” Nature 376(6537): 246–249. [DOI] [PubMed] [Google Scholar]
- Ryan AS, et al. (2012). “Exercise with calorie restriction improves insulin sensitivity and glycogen synthase activity in obese postmenopausal women with impaired glucose tolerance.” Am J Physiol Endocrinol Metab 302(1): E145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A, et al. (2009). “Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle.” Physiol Genomics 37(1): 58–66. [DOI] [PubMed] [Google Scholar]
- Salmon AB, et al. (2005). “Fibroblast cell lines from young adult mice of long-lived mutant strains are resistant to multiple forms of stress.” Am J Physiol Endocrinol Metab 289(1): E23–29. [DOI] [PubMed] [Google Scholar]
- Samms RJ, et al. (2016). “Overexpression of beta-Klotho in Adipose Tissue Sensitizes Male Mice to Endogenous FGF21 and Provides Protection From Diet-Induced Obesity.” Endocrinology 157(4): 1467–1480. [DOI] [PubMed] [Google Scholar]
- Sastre J, et al. (2003). “The role of mitochondrial oxidative stress in aging.” Free Radic Biol Med 35(1): 1–8. [DOI] [PubMed] [Google Scholar]
- Satoh A, et al. (2013). “Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH.” Cell Metab 18(3): 416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer DB (2011). “Oxidative stress in heart failure: what are we missing?” Am J Med Sci 342(2): 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, et al. (2013). “Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension.” Mol Metab 2(2): 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriner SE, et al. (2005). “Extension of murine life span by overexpression of catalase targeted to mitochondria.” Science 308(5730): 1909–1911. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, et al. (2007). “Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress.” Cell Metab 6(4): 280–293. [DOI] [PubMed] [Google Scholar]
- Selman C, et al. (2008). “Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice.” FASEB J 22(3): 807–818. [DOI] [PubMed] [Google Scholar]
- Selman C, et al. (2011). “Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1.” PLoS One 6(1): e16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, et al. (2009). “Ribosomal protein S6 kinase 1 signaling regulates mammalian life span.” Science 326(5949): 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A and Ravid T (2014). “Chaperoning proteins for destruction: diverse roles of Hsp70 chaperones and their co-chaperones in targeting misfolded proteins to the proteasome.” Biomolecules 4(3): 704–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa R, et al. (2019). “Mitochondrial reactive oxygen species generation in blood cells is associated with disease severity and exercise intolerance in heart failure patients.” Scientific Reports 9(1): 14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negreș Simona, et al. (2017). “New Potential Beta-3 Adrenergic Agonists with Beta-Phenylethylamine Structure, Synthesized for the Treatment of Dyslipidemia and Obesity” Adiposity - Epidemiology and Treatment Modalities Chapter 13. [Google Scholar]
- Sinha JK, et al. (2014). “Progeria: a rare genetic premature ageing disorder.” Indian J Med Res 139(5): 667–674. [PMC free article] [PubMed] [Google Scholar]
- Slater AF, et al. (1995). “Signalling mechanisms and oxidative stress in apoptosis.” Toxicol Lett 82–83: 149–153. [DOI] [PubMed] [Google Scholar]
- Sohal RS, et al. (1994). “Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice.” Mech Ageing Dev 76(2–3): 215–224. [DOI] [PubMed] [Google Scholar]
- Sohal RS, et al. (1994). “Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse.” Mech Ageing Dev 74(1–2): 121–133. [DOI] [PubMed] [Google Scholar]
- Son Y, et al. (2011). “Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways?” J Signal Transduct 2011: 792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YS, et al. (2008). “The role of Akt signaling in oxidative stress mediates NF-kappaB activation in mild transient focal cerebral ischemia.” J Cereb Blood Flow Metab 28(12): 1917–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, et al. (2013). “Brown adipose tissue regulates glucose homeostasis and insulin sensitivity.” J Clin Invest 123(1): 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckler R, et al. (2016). “Long-Lived alphaMUPA Mice Show Reduced Sexual Dimorphism in Lifespan, and in Energy and Circadian Homeostasis-Related Parameters.” J Gerontol A Biol Sci Med Sci 71(4): 451–460. [DOI] [PubMed] [Google Scholar]
- Storer JB (1966). “Longevity and gross pathology at death in 22 inbred mouse strains.” J Gerontol 21(3): 404–409. [DOI] [PubMed] [Google Scholar]
- Swindell WR (2009). “Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse.” BMC Genomics 10: 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard L (1959). “ON THE NATURE OF THE AGING PROCESS.” Proc Natl Acad Sci U S A 45(1): 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro AR, et al. (1996). “Cyclic food restriction alters substrate utilization and abolishes protection from mammary carcinogenesis female rats.” J Nutr 126(5): 1398–1405. [DOI] [PubMed] [Google Scholar]
- Tan SJ, et al. (2018). “High-intensity physical exercise increases serum alpha-klotho levels in healthy volunteers.” J Circ Biomark 7: 1849454418794582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, et al. (2002). “The absence of NF-kappaB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor alpha-induced apoptosis.” Mol Cell Biol 22(24): 8571–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap WD, et al. (2012). “Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors.” J Clin Oncol 30(15): 1849–1856. [DOI] [PubMed] [Google Scholar]
- Thakur S, et al. (2013). “Deficiency of insulin-like growth factor-1 receptor confers resistance to oxidative stress in C2C12 myoblasts.” PLoS One 8(5): e63838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The-World-Factbook (2020). “The World Factbook 2020 — Central Intelligence Agency: https://www.cia.gov/library/publications/the-world-factbook/rankorder/2102rank.html.” from https://www.cia.gov/library/publications/the-world-factbook/rankorder/2102rank.html.
- Tolcher AW, et al. (2009). “Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1.” J Clin Oncol 27(34): 5800–5807. [DOI] [PubMed] [Google Scholar]
- Tran H, et al. (2003). “The many forks in FOXO’s road.” Sci STKE 2003(172): RE5. [DOI] [PubMed] [Google Scholar]
- Trepanowski JF, et al. (2011). “Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: a summary of available findings.” Nutrition Journal 10(1): 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott A, et al. (2008). “Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule.” Mol Biol Cell 19(3): 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao JL, et al. (2002). “Diet, cancer and aging in DNA mismatch repair deficient mice.” Carcinogenesis 23(11): 1807–1810. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, et al. (2011). “Oxidative stress and heart failure.” Am J Physiol Heart Circ Physiol 301(6): H2181–2190. [DOI] [PubMed] [Google Scholar]
- UK-Prospective-Diabetes-Study-Group (1998). “Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group.” BMJ 317(7160): 703–713. [PMC free article] [PubMed] [Google Scholar]
- Um SH, et al. (2004). “Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity.” Nature 431(7005): 200–205. [DOI] [PubMed] [Google Scholar]
- Umanskaya A, et al. (2014). “Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging.” Proc Natl Acad Sci U S A 111(42): 15250–15255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe E and Andersson D (2019). “P6279Mitochondria targeted catalase overexpression protects against beta-adrenergic receptor mediated cardiac remodeling in mice.” European Heart Journal 40(Supplement_1). [Google Scholar]
- Uttara B, et al. (2009). “Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options.” Curr Neuropharmacol 7(1): 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, et al. (2007). “Free radicals and antioxidants in normal physiological functions and human disease.” Int J Biochem Cell Biol 39(1): 44–84. [DOI] [PubMed] [Google Scholar]
- Van Voorhies WA and Ward S (1999). “Genetic and environmental conditions that increase longevity in Caenorhabditis elegans decrease metabolic rate.” Proc Natl Acad Sci U S A 96(20): 11399–11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese P, et al. (2000). “beta(3)-adrenoceptor deficiency blocks nitric oxide-dependent inhibition of myocardial contractility.” J Clin Invest 106(5): 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatner DE, et al. (2015). “Type 5 adenylyl cyclase disruption leads to enhanced exercise performance.” Aging Cell 14(6): 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatner DE, et al. (2018). “Enhanced longevity and metabolism by brown adipose tissue with disruption of the regulator of G protein signaling 14.” Aging Cell: e12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatner SF, et al. (2015). “Inhibition of adenylyl cyclase type 5 increases longevity and healthful aging through oxidative stress protection.” Oxid Med Cell Longev 2015: 250310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramani S, et al. (2005). “Expression of p66(Shc) protein correlates with proliferation of human prostate cancer cells.” Oncogene 24(48): 7203–7212. [DOI] [PubMed] [Google Scholar]
- Vega-Rodriguez J, et al. (2009). “The glutathione biosynthetic pathway of Plasmodium is essential for mosquito transmission.” PLoS Pathog 5(2): e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, et al. (2004). “JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species.” Genes Dev 18(23): 2905–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzis I and Zakynthinos S (2012). “Factors limiting exercise tolerance in chronic lung diseases.” Compr Physiol 2(3): 1779–1817. [DOI] [PubMed] [Google Scholar]
- Vollaard NB, et al. (2005). “Exercise-induced oxidative stress:myths, realities and physiological relevance.” Sports Med 35(12): 1045–1062. [DOI] [PubMed] [Google Scholar]
- Vona R, et al. (2019). “Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases.” Oxid Med Cell Longev 2019: 8267234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vona R, et al. (2019). “Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases.” Oxid Med Cell Longev 2019: 8267234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadley GD, et al. (2013). “Xanthine oxidase inhibition attenuates skeletal muscle signaling following acute exercise but does not impair mitochondrial adaptations to endurance training.” Am J Physiol Endocrinol Metab 304(8): E853–862. [DOI] [PubMed] [Google Scholar]
- Walsh ME, et al. (2014). “The effects of dietary restriction on oxidative stress in rodents.” Free Radic Biol Med 66: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, et al. (2012). “An enhanced immune response of Mclk1(+)/(−) mutant mice is associated with partial protection from fibrosis, cancer and the development of biomarkers of aging.” PLoS One 7(11): e49606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SQ, et al. (2019). “Heterozygous knockout insulin-like growth factor-1 receptor (IGF-1R) regulates mitochondrial functions and prevents colitis and colorectal cancer.” Free Radic Biol Med 134: 87–98. [DOI] [PubMed] [Google Scholar]
- Wei M, et al. (2017). “Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease.” Sci Transl Med 9(377). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, et al. (2019). “Comparison of glycemic improvement between intermittent calorie restriction and continuous calorie restriction in diabetic mice.” Nutr Metab (Lond) 16: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YH and Lee HC (2002). “Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging.” Exp Biol Med (Maywood) 227(9): 671–682. [DOI] [PubMed] [Google Scholar]
- Weindruch R, et al. (1986). “The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake.” J Nutr 116(4): 641–654. [DOI] [PubMed] [Google Scholar]
- Weiss EP, et al. (2006). “Improvements in glucose tolerance and insulin action induced by increasing energy expenditure or decreasing energy intake: a randomized controlled trial.” Am J Clin Nutr 84(5): 1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF (2002). “IRS proteins and the common path to diabetes.” Am J Physiol Endocrinol Metab 283(3): E413–422. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, et al. (2016). “Reactive Oxygen Species and Neutrophil Function.” Annu Rev Biochem 85: 765–792. [DOI] [PubMed] [Google Scholar]
- Witman MA, et al. (2012). “A differing role of oxidative stress in the regulation of central and peripheral hemodynamics during exercise in heart failure.” Am J Physiol Heart Circ Physiol 303(10): H1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiysonge CS and Opie LH (2013). “beta-Blockers as initial therapy for hypertension.” JAMA 310(17): 1851–1852. [DOI] [PubMed] [Google Scholar]
- Wu A, et al. (2003). “Modulations by dietary restriction on antioxidant enzymes and lipid peroxidation in developing mice.” J Appl Physiol (1985) 94(3): 947–952. [DOI] [PubMed] [Google Scholar]
- Wu CW, et al. (2015). “Induction of Antioxidant and Heat Shock Protein Responses During Torpor in the Gray Mouse Lemur, Microcebus murinus.” Genomics Proteomics Bioinformatics 13(2): 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, et al. (2009). “Oxidative stress-induced JNK activation contributes to proinflammatory phenotype of aging diabetic mesangial cells.” Am J Physiol Renal Physiol 297(6): F1622–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, et al. (2002). “Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin.” Hum Mol Genet 11(9): 1137–1151. [DOI] [PubMed] [Google Scholar]
- Xiao R, et al. (2013). “A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel.” Cell 152(4): 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, et al. (2012). “Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart.” Nat Commun 3: 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, et al. (2014). “Longevity effect of IGF-1R(+/−) mutation depends on genetic background-specific receptor activation.” Aging Cell 13(1): 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, et al. (2020). “Mortality in the United States, 2018.” NCHS Data Brief 355. [PubMed] [Google Scholar]
- Yamamoto S, et al. (2003). “Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy.” J Clin Invest 111(10): 1463–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y, et al. (2018). “Induction of prolonged natural lifespans in mice exposed to acoustic environmental enrichment.” Sci Rep 8(1): 7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, et al. (2013). “Calorie restriction can reverse, as well as prevent, aging cardiomyopathy.” Age (Dordr) 35(6): 2177–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, et al. (2012). “Common mechanisms for calorie restriction and adenylyl cyclase type 5 knockout models of longevity.” Aging Cell 11(6): 1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, et al. (2007). “Type 5 adenylyl cyclase disruption increases longevity and protects against stress.” Cell 130(2): 247–258. [DOI] [PubMed] [Google Scholar]
- Yang J, et al. (2020). “Food with calorie restriction reduces the development of atherosclerosis in apoE-deficient mice.” Biochem Biophys Res Commun 524(2): 439–445. [DOI] [PubMed] [Google Scholar]
- Yang RL, et al. (2008). “Increasing Oxidative Stress with Progressive Hyperlipidemia in Human: Relation between Malondialdehyde and Atherogenic Index.” J Clin Biochem Nutr 43(3): 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, et al. (2006). “Metallothionein prolongs survival and antagonizes senescence-associated cardiomyocyte diastolic dysfunction: role of oxidative stress.” FASEB J 20(7): 1024–1026. [DOI] [PubMed] [Google Scholar]
- Yong QC, et al. (2013). “Angiotensin type 1a receptor-deficient mice develop diabetes-induced cardiac dysfunction, which is prevented by renin-angiotensin system inhibitors.” Cardiovasc Diabetol 12: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SO, et al. (2002). “Dose effect of oxidative stress on signal transduction in aging.” Mech Ageing Dev 123(12): 1597–1604. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, et al. (2003). “P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA.” Cancer Res 63(13): 3729–3734. [PubMed] [Google Scholar]
- Yuan R, et al. (2009). “Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels.” Aging Cell 8(3): 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, et al. (2012). “Quercetin-induced apoptosis of HL-60 cells by reducing PI3K/Akt.” Mol Biol Rep 39(7): 7785–7793. [DOI] [PubMed] [Google Scholar]
- Zainal TA, et al. (2000). “Caloric restriction of rhesus monkeys lowers oxidative damage in skeletal muscle.” FASEB J 14(12): 1825–1836. [DOI] [PubMed] [Google Scholar]
- Zarse K, et al. (2012). “Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal.” Cell Metab 15(4): 451–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. (2018). “A novel adenylyl cyclase type 5 inhibitor that reduces myocardial infarct size even when administered after coronary artery reperfusion.” J Mol Cell Cardiol 121: 13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. (2016, 134:A18750). “Increased Brown Adipose Tissue as a Novel Mechanism Mediating Cardioprotection.” Circulation (Suppl). [Google Scholar]
- Zhang P, et al. (2020). “Oxidative stress and diabetes: antioxidative strategies.” Front Med. [DOI] [PubMed] [Google Scholar]
- Zhao L, et al. (2012). “Common genetic variants of the beta2-adrenergic receptor affect its translational efficiency and are associated with human longevity.” Aging Cell 11(6): 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, et al. (2017). “Klotho, an anti-aging gene, acts as a tumor suppressor and inhibitor of IGF-1R signaling in diffuse large B cell lymphoma.” J Hematol Oncol 10(1): 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, et al. (1997). “A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse).” Proc Natl Acad Sci U S A 94(24): 13215–13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YC, et al. (2003). “Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and cardiac remodelling.” Clin Exp Pharmacol Physiol 30(12): 911–918. [DOI] [PubMed] [Google Scholar]
- Zuin A, et al. (2010). “Lifespan extension by calorie restriction relies on the Sty1 MAP kinase stress pathway.” EMBO J 29(5): 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier JL and Talukder MA (2006). “The role of oxidants and free radicals in reperfusion injury.” Cardiovasc Res 70(2): 181–190. [DOI] [PubMed] [Google Scholar]