

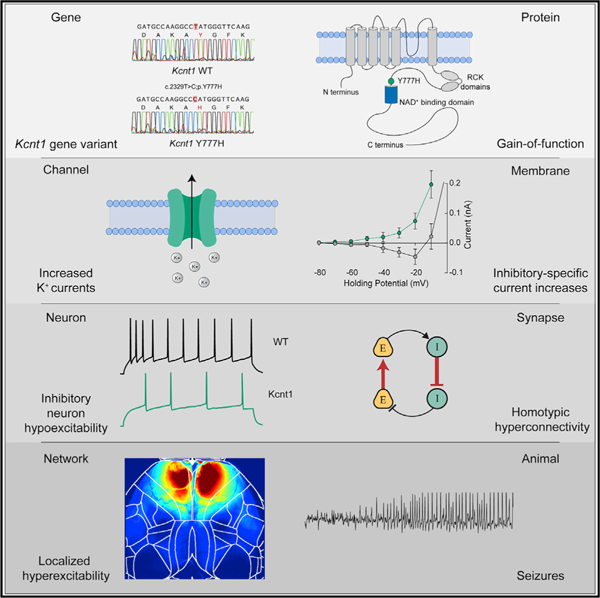
SUMMARY
Gain-of-function (GOF) variants in K+ channels cause severe childhood epilepsies, but there are no mechanisms to explain how increased K+ currents lead to network hyperexcitability. Here, we introduce a human Na+-activated K+ (KNa) channel variant (KCNT1-Y796H) into mice and, using a multiplatform approach, find motor cortex hyperexcitability and early-onset seizures, phenotypes strikingly similar to those of human patients. Although the variant increases KNa currents in cortical excitatory and inhibitory neurons, there is an increase in the KNa current across subthreshold voltages only in inhibitory neurons, particularly in those with non-fast-spiking properties, resulting in inhibitory-neuron-specific impairments in excitability and action potential (AP) generation. We further observe evidence of synaptic rewiring, including increases in homotypic synaptic connectivity, accompanied by network hyperexcitability and hypersynchronicity. These findings support inhibitory-neuron-specific mechanisms in mediating the epileptogenic effects of KCNT1 channel GOF, offering cell-type-specific currents and effects as promising targets for therapeutic intervention.
Graphical Abstract
In Brief
Shore et al. generate a mouse model of a GOF variant in the Na+-activated K+ channel gene KCNT1 that causes a childhood epilepsy disorder. In mice, KCNT1 GOF reduces the excitability of cortical GABAergic neurons and increases homotypic synaptic connectivity, resulting in disrupted E/I balance, network hyperexcitability, and seizures.
INTRODUCTION
K+ channels play essential roles in regulating the membrane excitability of neurons, and their dysfunction causes a wide range of neurological diseases, particularly epilepsy (Köhling and Wolfart, 2016; Villa and Combi, 2016; Oyrer et al., 2018). Traditionally, this mechanism has been conceptualized as loss-of-function (LOF) variants decreasing K+ currents and leading to neuronal hyperexcitability. Recent human gene discovery efforts, however, have identified novel, neurological-disease-associated K+ channel variants that increase peak K+ current magnitudes when expressed in heterologous cells, suggesting that they are gain-of-function (GOF) variants (Yang et al., 2013; Syrbe et al., 2015; Simons et al., 2015; Millichap et al., 2017; Du et al., 2005; Miceli et al., 2015; Lee et al., 2014). Although these variants have been biophysically characterized, how they function in neurons and networks to cause hyperexcitability is unknown (Niday and Tzingounis, 2018).
Among K+ channels, variants in the gene encoding the Na+-activated K+ (KNa) channel KCNT1 (Slack, KNa 1.1) cause a range of epilepsies, including early infantile epileptic encephalopathies (EIEEs) (Ohba et al., 2015), malignant migrating partial seizures of infancy (MMPSI) (Barcia et al., 2012), and autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (Heron et al., 2012; Møller et al., 2015). These variants increase peak K+ current magnitude, potentially by increasing the cooperativity (Kim et al., 2014), enhancing the Na+ sensitivity, increasing the open probability, and/or shifting the voltage dependence of the mutant KCNT1 channel (Tang et al., 2016; McTague et al., 2018). The broad-spectrum K+ channel blocker quinidine can normalize the increased K+ currents caused by pathogenic KCNT1 variants (McTague et al., 2018; Milligan et al., 2014) and thus has been tried as a precision therapy to treat KCNT1-associated neurodevelopmental disease. Although some early cases were promising (Bearden et al., 2014), further reports showed limited therapeutic benefit of quinidine in KCNT1-related epilepsy (Mullen et al., 2018; Chong et al., 2016; Fitzgerald et al., 2019), emphasizing that an understanding of disease mechanisms beyond channel biophysics is necessary to advance clinical treatment.
In normal neuronal physiology, there is evidence that the KNa current mediated by KCNT1 can activate in three distinct activity regimes: during bursts of action potentials (APs) to reduce neuronal excitability and control interburst timing (Schwindt et al., 1989; Wallén et al., 2007; Kim and McCormick, 1998; Yang et al., 2007), after a single AP to increase afterhyperpolarization (AHP) and enhance bursting (Franceschetti et al., 2003; Liu and Stan Leung, 2004), or at subthreshold voltages to modify intrinsic neuronal excitability (Martinez-Espinosa et al., 2015; Reijntjes et al., 2019; Hage and Salkoff, 2012). The effect of a KCNT1 GOF variant on neuronal physiology and network activity would therefore depend on which of these regimes were affected. Following this, it has been hypothesized that KCNT1 GOF variants lead to network hyperexcitability by (1) accelerating AP repolarization selectively in excitatory neurons, thereby limiting sodium channel inactivation and causing a higher AP firing frequency; (2) reducing membrane excitability or inducing spike frequency adaptation selectively in inhibitory neurons, resulting in disinhibition; or (3) causing developmental alterations in synaptic connectivity, thus leading to a hyperexcitable network (Kim and Kaczmarek, 2014; Niday and Tzingounis, 2018; Milligan et al., 2014). To test these hypotheses, we generated a mouse model with a human-disease-causing GOF variant in the Kcnt1 gene and used a multiplatform approach to identify a time point, brain region, and neuron type that demonstrated strong functional alterations. We found that the Kcnt1 variant selectively increases KNa currents in inhibitory neurons at subthreshold voltages to reduce their intrinsic excitability and alters synaptic connectivity to promote a hyperexcitable and hypersynchronous network, thus providing a paradigm for how GOF variants in K+ channels cause neurodevelopmental disease.
RESULTS
The Kcnt1-Y777H Mouse Model Exhibits Epileptic and Behavioral Phenotypes Similar to Those of Human Patients
The genetic variant Y796H (c.2386T > C; p.[Tyr796His]) in KCNT1 has been identified as a heterozygous mutation causing both inherited and de novo cases of a severe, early-onset form of ADNFLE (Heron et al., 2012; Mikati et al., 2015). In heterologous systems, the Y796H variant enhances the Na+ sensitivity of KCNT1 and increases the peak KNa current from 3- to 11-fold over the WT channel (Tang et al., 2016; Milligan et al., 2014). To model this KCNT1 GOF variant-associated intractable childhood epilepsy in mice, we introduced a missense mutation (c.2329T > C) into the endogenous Kcnt1 allele on the C57BL/6NJ (B6NJ) strain background (Figure 1A). This mutation encodes a Y777H (p.[Tyr777His]) alteration corresponding to Y796H that lies adjacent to the NAD+-binding site of KCNT1 (Figure 1B). We generated cohorts of wild-type (WT), heterozygous (Kcnt1m/+), and homozygous (Kcnt1m/m) littermates. Genetic transmission of both alleles was normal, and no selective lethality was seen in either mutant genotype at any age. Western blot analysis of membrane and cytosolic protein fractions isolated from the cortices of WT, Kcnt1m/+, and Kcnt1m/m mice detected KCNT1 only in the membrane fraction from WT mice, and importantly, levels and localization of the Y777H variant were unaltered in Kcnt1m/+ and Kcnt1m/m mice (Figure 1C). Finally, dissected brains from 10- to 12-week-old Kcnt1m/+ and Kcnt1m/m mice appeared similar in morphology and size to their WT littermates (Figure 1D), suggesting that expression of the Kcnt1 GOF variant does not grossly alter brain development.
Figure 1. Homozygous Kcnt1-Y777H Mice Have Frequent, Sleep-Associated, Spontaneous Seizures.
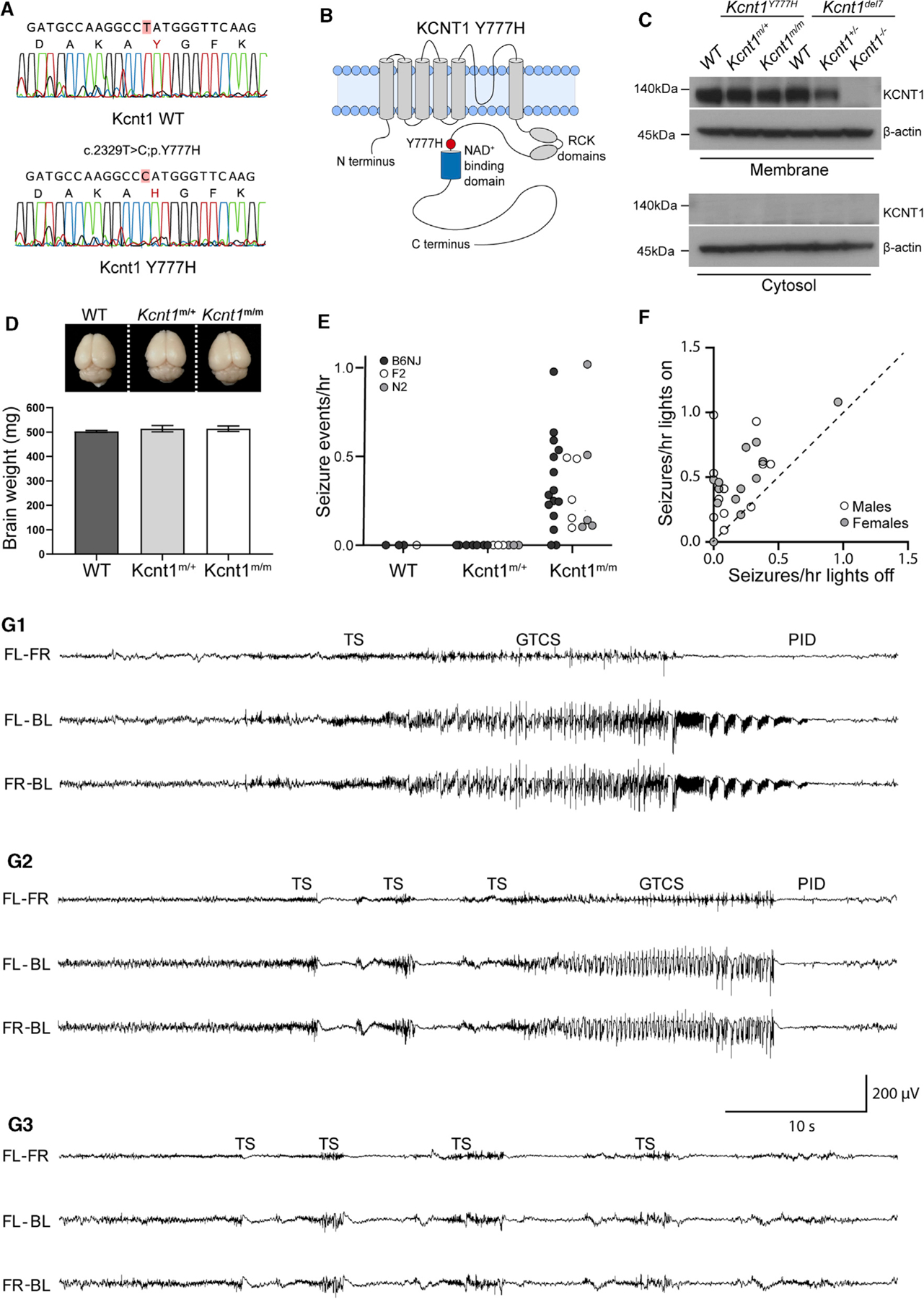
(A) Sanger sequencing chromatograms show the WT (top) and Y777H (bottom) alleles obtained from direct sequencing of PCR products amplified from genomic DNA from a WT and a Kcnt1m/m mouse.
(B) Diagram of the KCNT1 protein shows location of the Y777H residue.
(C) Western blots show KCNT1 expression in Kcnt1Y777H (WT, Kcnt1m/+, and Kcnt1m/m) P0 cortices following membrane and cytosol fractioning. The corresponding lysates from the Kcnt1 mouse knockout Kcnt1del7 (WT, Kcnt1+/−, and Kcnt1−/−) were used as a negative control.
(D) Representative image shows similar gross morphology of brains dissected from 10- to 12-week-old WT, Kcnt1m/+, and Kcnt1m/m littermate mice. Bar graph shows that brain weight is unaltered by the Y777H variant (WT; n = 3, Kcnt1m/+; n = 4, and Kcnt1m/m; n = 3). Data are represented as mean ± SEM.
(E) Dot plot shows the combined spontaneous generalized tonic-clonic seizure (GTCS) and tonic seizure (TS) frequency in WT, Kcnt1m/+, and Kcnt1m/m mice in three genetic backgrounds indicated by shaded dots.
(F) Scatterplot shows that seizures are more likely to occur when lights are on. Each dot represents one Kcnt1m/m mouse and is shaded to indicate sex.
(G1–G3) Representative EEG traces show seizure episodes from B6NJ Kcnt1m/m mice, including GTCS, TS, epileptiform discharges (EDs), and post-ictal depression (PID) episodes as noted in each panel. The traces shown are differential recordings between front left (FL), front right (FR), and back left (BL) EEG electrodes.
Patients with KCNT1-associated ADNFLE present with frequent, often nocturnal seizures with a variety of phenotypes, including hypermotor, focal with tonic posturing, and secondary generalized tonic-clonic (Gertler et al., 2018; Mikati et al., 2015; McTague et al., 2018; Fitzgerald et al., 2019). To test for the presence of spontaneous seizures, we monitored adult mice (≥7 weeks of age) of each genotype using video electroencephalography (video-EEG) recordings. Epileptiform activity was not observed in WT (n = 3) or Kcnt1m/+ (n = 8) mice. However, 87% (13/15) of Kcnt1m/m mice had at least one spontaneous seizure in an average of 38 h of continuous recording (Figure 1E, black dots). To test the impact of the Kcnt1 GOF variant on seizure activity in a broader genetic context, we performed video-EEG on WT, Kcnt1m/+, and Kcnt1m/m mice on a mixed background using an FVB-based partner strain (see STAR Methods). Of the F2 and N2 hybrid Kcnt1m/m mice, 100% (10/10) had at least one spontaneous seizure in an average of 47 h of continuous recording, whereas seizures were not observed in WT (n = 1) or Kcnt1m/+ (n = 6) mice (Figure 1E, gray and white dots). Thus, although heterozygous expression of the Y796H variant is sufficient to cause frequent seizures in patients, Kcnt1-Y777H heterozygous mice did not exhibit any detectable epileptiform activity. Therefore, we focused the remainder of our study on Kcnt1-Y777H homozygous mice to understand the contribution of K+ channel GOF to seizure disorders.
To determine whether the seizures observed in Kcnt1m/m mice recapitulate the signature features of those in KCNT1-associated ADNFLE patients, we analyzed the relationship between seizure activity and sleep in Kcnt1m/m mice. Strikingly, regardless of mouse strain or sex, the incidence of seizure episodes was ~2-fold higher when the vivarium lights were on, corresponding to the sleep period of mice (Figure 1F). Moreover, we compared the incidence of seizures in wake, NREM sleep, and REM sleep in seven B6NJ Kcnt1m/m mice and found that 90% (131/145) occurred during NREM sleep (Table S1). Next, we analyzed the video-EEG of all Kcnt1m/m mice to assess their seizure phenotypes. Two distinct seizure types were observed: generalized tonic-clonic seizures (GTCSs), typically lasting 30–60 s, and tonic seizures (TSs), typically lasting 1–2 s. TSs often preceded GTCSs (Figures 1G1 and 1G2), but TSs also occurred without further progression, sometimes as single events but often in clusters of two or more episodes within a few seconds of each other (Figure 1G3). Together, these data highlight meaningful parallels between pathology observed in the homozygous mouse model and the human disease caused by the KCNT1 GOF variant, including seizure frequency, brain state-association, and phenotype.
Because KCNT1 GOF variants are also associated with intellectual disability and psychiatric conditions, including anxiety and attention-deficit hyperactive disorder, we next performed a series of tests to assess the effects of the Y777H variant on mouse behavior. In an open field test, Kcnt1m/m mice exhibited a robust hyperactivity phenotype, traveling a further distance compared to WT littermates; however, they were not different from WT on the elevated plus maze (Figures S1A and S1B), indicating hyperactivity without changes in anxiety-like behaviors. Kcnt1m/m mice also exhibited decreased freezing behaviors in the contextual and cued fear conditioning tests, without changes in post-shock freezing behavior or acoustic startle responses (Figures S1C and S1D), suggesting learning deficits unconfounded by sensorimotor deficits. As a global indicator of mouse well-being, we examined nest building, which is often reduced in neurodegenerative and psychiatric murine disease models (Jirkof, 2014). Both Kcnt1m/m females and males were poor nest builders compared with WT, as assessed using a score of nest quality and the weight of unshredded nesting material (Figure S1E). These data suggest that, in addition to recapitulating the seizure phenotypes, Kcnt1m/m mice exhibit several behavioral phenotypes similar to those found in ADNFLE patients.
Widefield Ca2+ Imaging Reveals Network Hyperactivity in the Secondary Motor Cortex of Kcnt1m/m Mice
To determine which cortical regions were most strongly impacted by the Kcnt1 variant, we next crossed Kcnt1-Y777H mice to the Snap25-GCaMP6s line to generate Kcnt1m/m; Snap25G6s/+ and Kcnt1+/+; Snap25G6s/+ (hereafter referred to as Kcnt1m/m-G6s and WT-G6s) littermates. We then imaged spontaneous neural activity across the dorsal cortices of awake, head-fixed, behaving Kcnt1m/m-G6s and WT-G6s adult mice using a custom tandem-lens epifluorescence macroscope (Figure 2A). Both Kcnt1m/m-G6s and WT-G6s mice exhibited dynamic, spatially complex patterns of spontaneous cortical activity. To quantify this activity, we performed event detection and plotted the width versus prominence profile of all detected events (Figure 2B). In all Kcnt1m/m-G6s mice (n = 6), there were narrow, large-amplitude calcium events, similar in appearance to previous reports of interictal epileptiform discharges (IEDs) captured using the same technique (Steinmetz et al., 2017, Rossi et al., 2017). Importantly, these events were rarely observed in WT-G6s littermates (n = 5). Across Kcnt1m/m-G6s mice, these events were consistently localized to the anterior medial cortex (Figure 2C; Video S1), consistent with previous reports of IEDs localized to the anterior medial cortex in human patients carrying the Y796H variant (Mikati et al., 2015). Alignment of the activity maps from Kcnt1m/m-G6s mice to the Allen Mouse Common Coordinate Framework (Allen Mouse Brain Connectivity Atlas, Wang, 2020) showed that these events localized primarily to the secondary motor cortex region (M2; Figures 2C and S2), suggesting a regional specificity to the interictal hyperexcitability in Kcnt1m/m mice.
Figure 2. Interictal Epileptiform Discharges (IEDs) in Kcnt1m/m Mice Localize to the Secondary Motor Cortex.
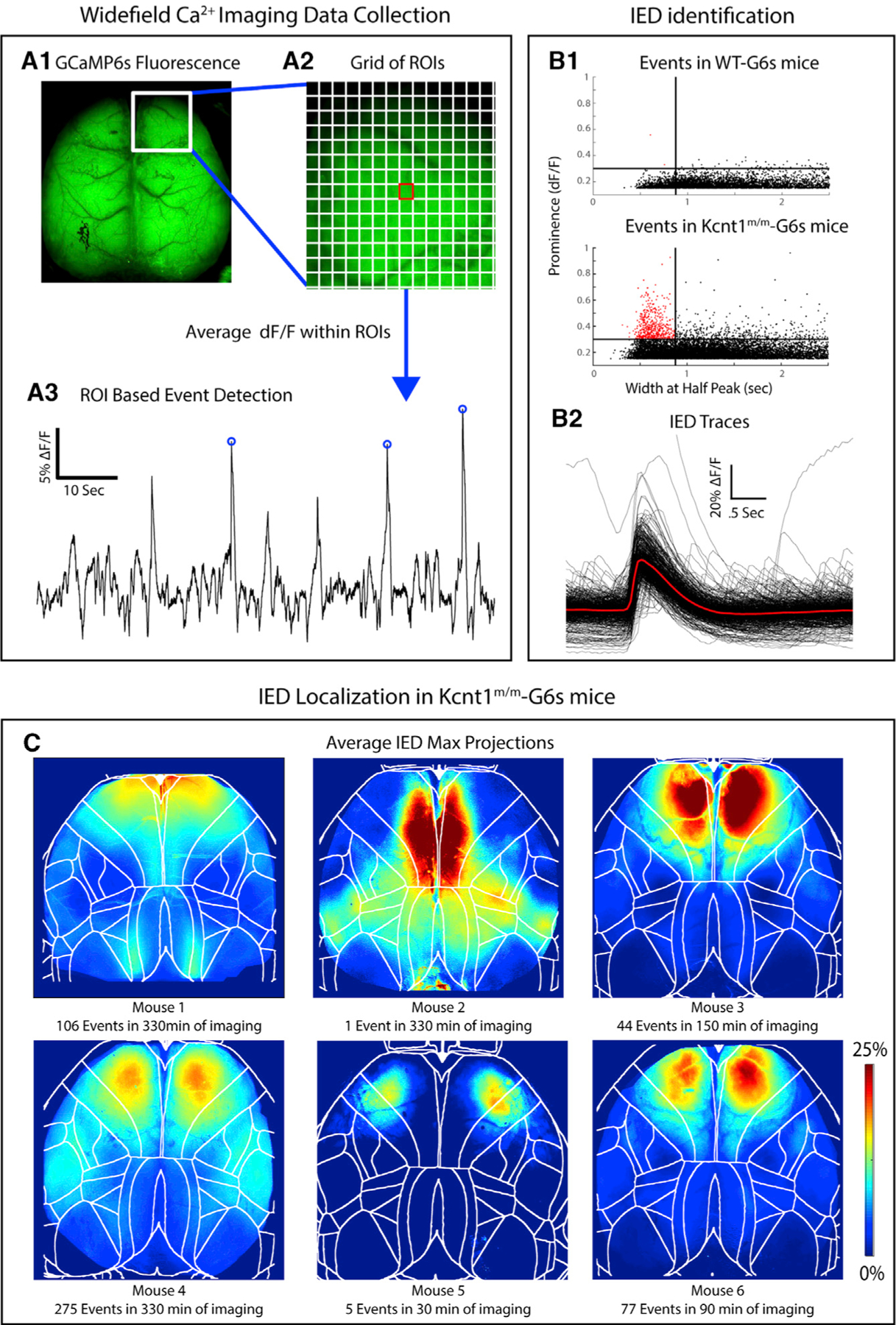
(A1) GCaMP6s fluorescence in the brain of a Kcnt1m/m-G6s mouse. (A2) Illustration of the grid of regions of interest (ROIs) used in event detection superimposed on a portion of the GCaMP6s fluorescence image. (A3) ΔF/F peaks with a prominence greater than 15% were identified in each ROI.
(B1) Maximum event prominence plotted as a function of width at half-maximum for WT-G6s and Kcnt1m/m-G6s mice. Kcnt1m/m-G6s mice generate events (narrow, high prominence) not seen in WT-G6s. Lines at 875 ms and 30% ΔF/F indicate our criteria for classifying events as IEDs. (B2) Overlaid ΔF/F traces of all IEDs in all Kcnt1m/m-G6s mice with the mean waveform in red. The Kcnt1m/m plot was cropped to highlight the region containing IEDs, which obscures one large event (0.52, 2.17).
(C) To investigate the cortical localization of spike-like events, we generated a maximum projection across all event frames for each IED. The average of these is shown for each Kcnt1m/m-G6s mouse, showing consistent peak localized to the anterior medial cortex, mostly within primary and secondary motor cortices. Boundaries indicate cortical regions demarcated by aligning brains to the Allen Common Coordinate Framework (CCF) (see STAR Methods).
In Vivo Electrocorticography Shows Early-Onset Cortical Network Hyperactivity in Kcnt1m/m Mouse Pups
KCNT1 epileptic encephalopathy is a childhood genetic disease; therefore, we next tested for evidence of epileptiform activity in mouse pups by performing acute, head-fixed, unanesthetized, in vivo electrocorticography on a cohort of Kcnt1m/m (n = 8) and WT (n = 5) mice at postnatal day 13 (P13) to P15. Of the eight Kcnt1m/m pups, three showed definite seizure activity (Figure 3A), and all eight exhibited waveforms consistent with IEDs (Figure 3C). Electrographic seizure patterns consisted of evolving patterns of spike and wave discharges that had restricted spatial distribution across the cortical surface (Figure 3A). These seizures lasted from 10 to 280 s and stopped spontaneously in all cases (Figure 3B). No WT pups exhibited electrographic seizure activity. Identical offline IED detection algorithms were applied to Kcnt1m/m and WT pups, with all Kcnt1m/m pups demonstrating a higher occurrence rate of IEDs (Figure 3D). These IEDs were focally distributed over the surface of the cortex across Kcnt1m/m pups and, similar to the results from widefield Ca2+ imaging, were more concentrated in anterior regions of the somatomotor cortex (Figure 3E).
Figure 3. Kcnt1m/m Mice Have Epileptic Activity In Vivo at 2 Weeks of Age.
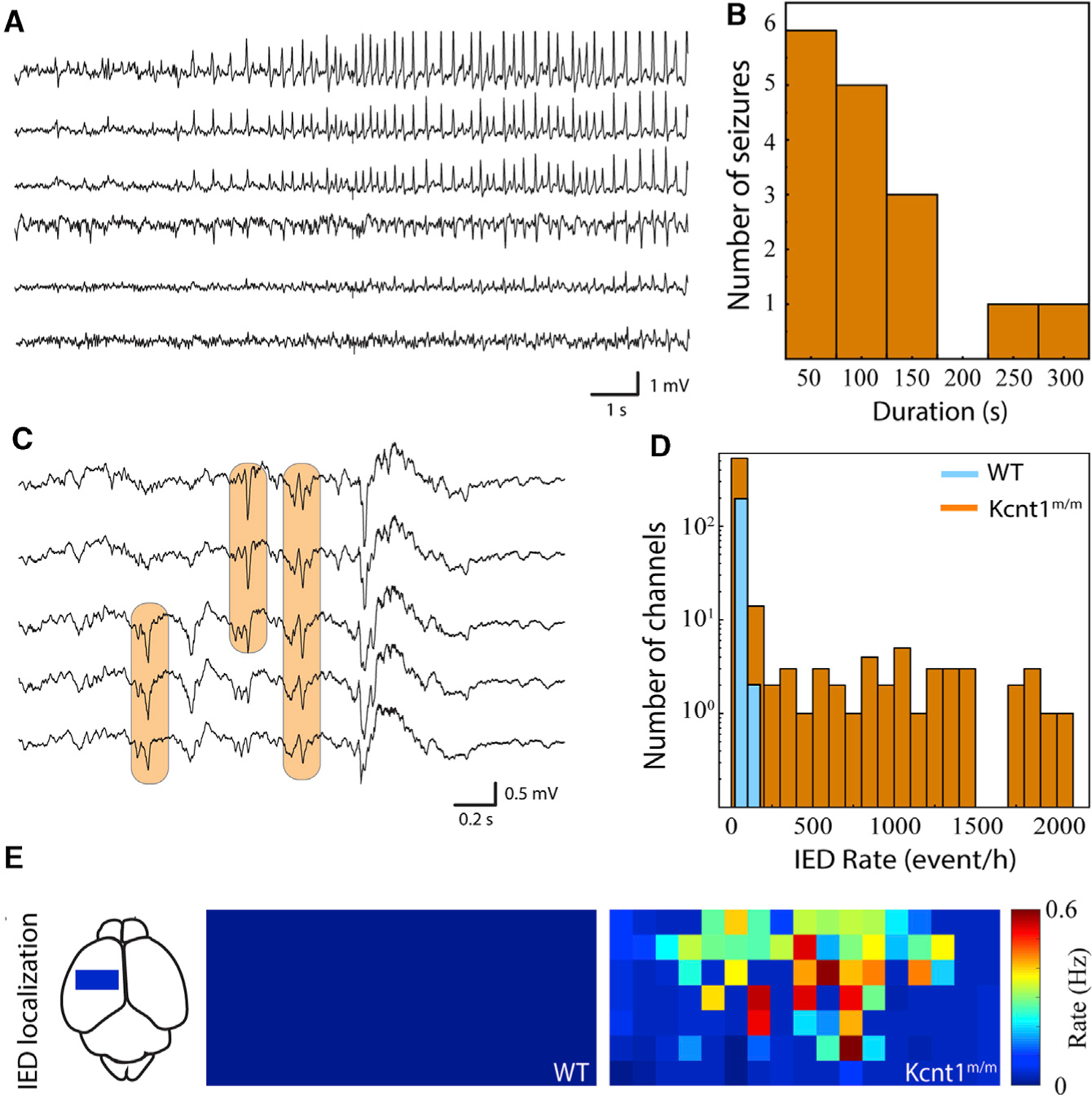
(A) Sample raw traces from electrocorticography array demonstrate focal, evolving ictal pattern in a head-fixed pup.
(B) Histogram of seizure duration across Kcnt1m/m pups. Three out of eight Kcnt1m/m pups had definite seizures, and none of the five WT pups had seizures.
(C) Sample raw traces from surface electrocorticography array demonstrate populations of interictal epileptiform discharges (IEDs; orange boxes).
(D) Histogram of IED occurrence across all functioning electrocorticography channels in Kcnt1m/m (orange) and WT (blue) pups.
(E) IED occurrence rate localized anatomically across the electrocorticography array and pooled across pups reveals focal distribution in mutants. Each square represents one electrocorticography electrode. The approximate location and scale of the arrays on the dorsal cortical surface is shown at left. Color map is normalized across Kcnt1m/m and WT pups.
The Kcnt1-Y777H Variant Impairs Excitability and AP Generation in Cortical GABAergic, but Not Glutamatergic, Neurons
Video-EEG, widefield Ca2+ imaging, and in vivo electrocorticography all demonstrated hyperexcitable cortical networks in Kcnt1m/m mice, starting as early as the second postnatal week. What are the origins of this hyperexcitability? KCNT1 and the associated KNa current have several proposed roles in the regulation of membrane excitability and AP generation, and KCNT1 is broadly expressed in neurons (Bhattacharjee et al., 2002, 2005; Rizzi et al., 2016), including multiple subtypes of glutamatergic and GABAergic neurons from both human and mouse cortices (Figure S3). Thus, it has been hypothesized that KCNT1 GOF enhances the intrinsic excitability of glutamatergic neurons, or reduces that of GABAergic neurons, to generate a hyperexcitable network. To test these hypotheses, we assessed neuron-subtype-specific effects of KCNT1 GOF on membrane properties and AP firing by performing whole-cell current-clamp analysis of cultured cortical neurons during the time frame of network hyperexcitability onset (days in vitro 13 [DIV13] to DIV17). The recorded neurons were classified as glutamatergic, fast-spiking (FS) GABAergic, or non-fast-spiking (NFS) GABAergic, based on expression of GFP driven by the CaMKII promoter, AP parameters, and evoked synaptic responses (see STAR Methods).
First, we assessed the membrane excitability of Kcnt1m/m glutamatergic neurons. If the KNa current activated at subthreshold voltages in Kcnt1m/m neurons, KCNT1 GOF would be expected to alter the resting membrane potential (Vrest), input resistance (Rin), AP threshold, or the minimum amount of current needed to trigger an AP (rheobase); however, none of these parameters were significantly affected by the Y777H variant in glutamatergic neurons (Table S2; Figures 4A1 and 4A2). Alternatively, it has been hypothesized that the KNa current activates in response to AP firing and that KCNT1 GOF increases the rate of AP repolarization in glutamatergic neurons, leading to an increase in AP firing frequency. In this case, KCNT1 GOF would be expected to alter the AP shape; however, the AP half-width, AP repolarization rate, and AHP were also not significantly affected by the Y777H variant (Table S2; Figure 4A3). Accordingly, the AP firing responses to incremental current injections were also indistinguishable between WT and Kcnt1m/m glutamatergic neurons (Figure 4A4). Thus, despite the reported expression of KCNT1 and KNa currents in cortical glutamatergic neurons (Budelli et al., 2009, Rizzi et al., 2016), there were no significant alterations in any of the electrophysiological parameters measured in this neuron type.
Figure 4. The Kcnt1 Y777H Variant Causes a Reduction in Membrane Excitability and AP Generation in GABAergic, but Not Glutamatergic, Cortical Neurons.
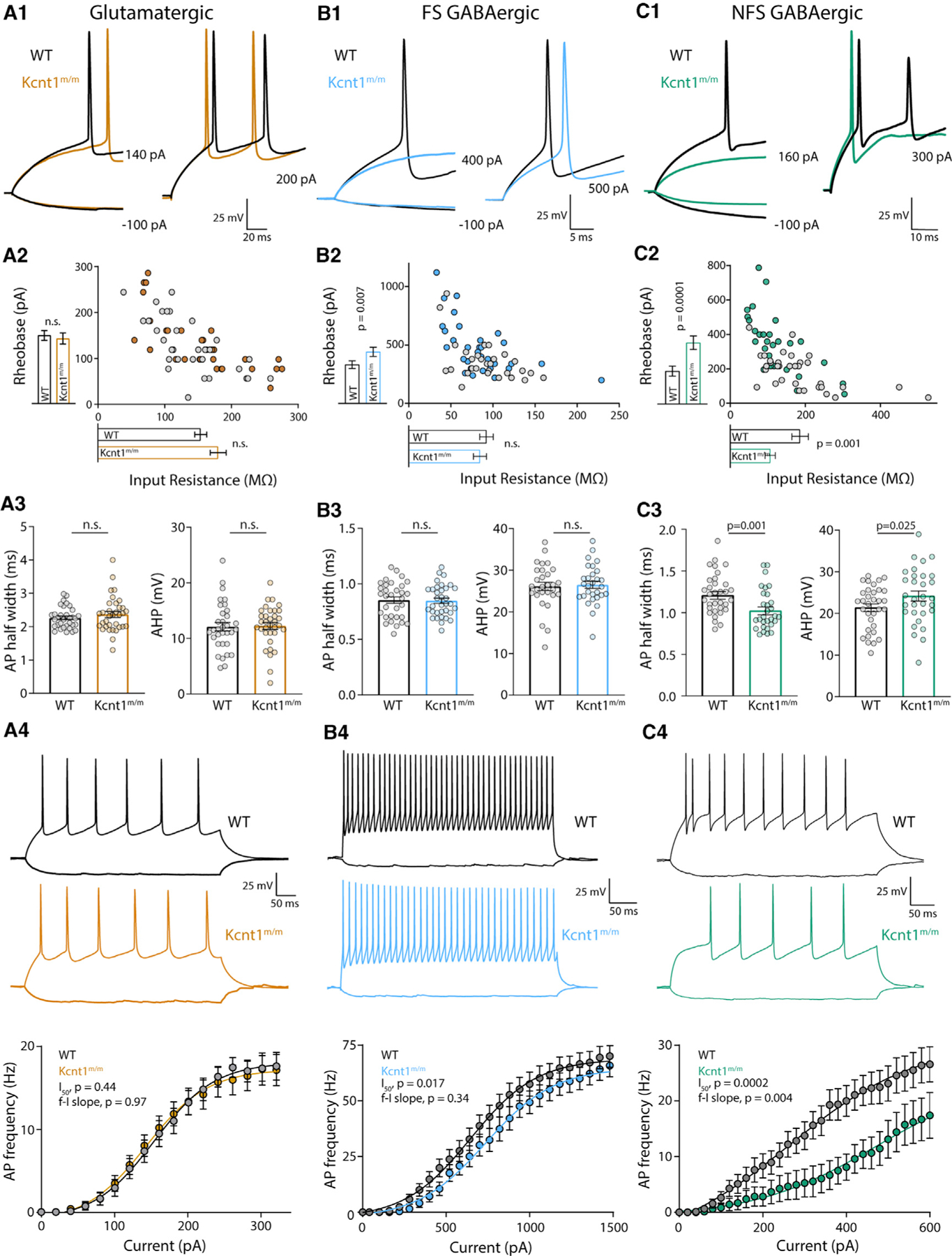
(A1–C1) Representative membrane voltage traces of WT (black) and Kcnt1m/m (colors) neurons of the indicated type in response to current injections illustrate differences in the input resistance (Rin) and/or rheobase between WT and Kcnt1m/m GABAergic neurons.
(A2–C2) The individual values of Rin (x axis) and rheobase (y axis) for WT (gray) and Kcnt1m/m (colors) neurons. The mean and SEM for each group are indicated by the bars next to the corresponding axis.
(A3–C3) Individual values and mean ± SEM for AP half-widths and AHP in WT (black and gray) and Kcnt1m/m (colors) neurons.
(A4–C4) Example traces and summary data showing the number of APs (mean and SEM) per current injection step in WT (black and gray) and Kcnt1m/m (colors) neurons. The lines in the current/AP plots are fits to a Boltzmann sigmoidal curve and were used to determine the half-max current (I50) and the frequency-current (f-I) slope for each group. Statistical significance was tested using generalized linear mixed models.
An alternative hypothesis is that KCNT1 GOF selectively reduces the excitability of GABAergic neurons. Although, like glutamatergic neurons, the Kcnt1m/m FS neurons showed no alterations in Vrest, Rin, or AP threshold, they did show an increase in the rheobase current (Table S2; Figures 4B1 and 4B2), suggesting there may be an increased KNa current near AP threshold levels in this neuron population. Other electrophysiological parameters assessed, including AP half-width, AP repolarization rate, and AHP, were not affected by the Y777H variant in FS neurons (Table S2; Figure 4B3). However, incremental current injections showed a decrease in the number of APs per current step in Kcnt1m/m FS neurons relative to that of WT (Figure 4B4).
In contrast to glutamatergic and FS neurons, the Kcnt1m/m NFS neurons showed drastic alterations in membrane properties and AP generation relative to those of WT. The Kcnt1m/m NFS neurons showed a decrease in Rin, accompanied by a large increase in the rheobase (Figures 4C1 and 4C2), whereas the Vrest and AP threshold were unchanged (Table S2). The membrane capacitance (Cm) was also increased in Kcnt1m/m NFS neurons (Table S2). There were several other alterations to the AP parameters in Kcnt1m/m NFS neurons, including a narrower AP half-width, a faster AP repolarization rate, and a larger AHP (Table S2; Figure 4C3). These data suggest there may be a larger increase in the KNa current in this neuron population or that the KNa current is increased across a wider voltage range. Importantly, incremental current injections showed a strong decrease in the number of APs per current step in Kcnt1m/m NFS neurons relative to that of WT (Figure 4C4). Overall, these data demonstrate that the Y777H variant selectively reduces the intrinsic excitability of FS and, to a greater extent, NFS GABAergic neurons, thus providing a functional answer to the question of how a variant that causes a decrease in excitability could lead to the formation of a hyperexcitable network.
The Y777H Variant Left-Shifts the Voltage-Dependent Activation of KCNT1 and Increases Subthreshold KNa Currents Only in GABAergic Neurons
Why are GABAergic neurons more sensitive to the effects of a variant in a gene that is widely expressed in neurons? To address this question, we recorded KNa currents in cultured cortical neurons by applying voltage steps to voltage-clamped neurons and comparing the delayed outward current before and after the addition of tetrodotoxin (TTX), as KCNT1 channels are thought to be the major contributors to this current (Budelli et al., 2009; Reijntjes et al., 2019; Cervantes et al., 2013). Consistent with KCNT1-mediated currents recorded previously using the same technique, we observed KNa currents in all three WT neuron types, beginning at approximately ~10 mV and increasing with depolarization (Figures 5A1,2–5C1,2). At more negative potentials, the TTX-sensitive current was often net inward (Figures 5A3–5C3), likely due to the persistent Na+ current (INaP), which has been shown to prime KNa for activation in some neuron types (Budelli et al., 2009; Hage and Salkoff, 2012).
Figure 5. The Y777H Variant Causes Cell-Type-Specific Increases in KNa Currents.
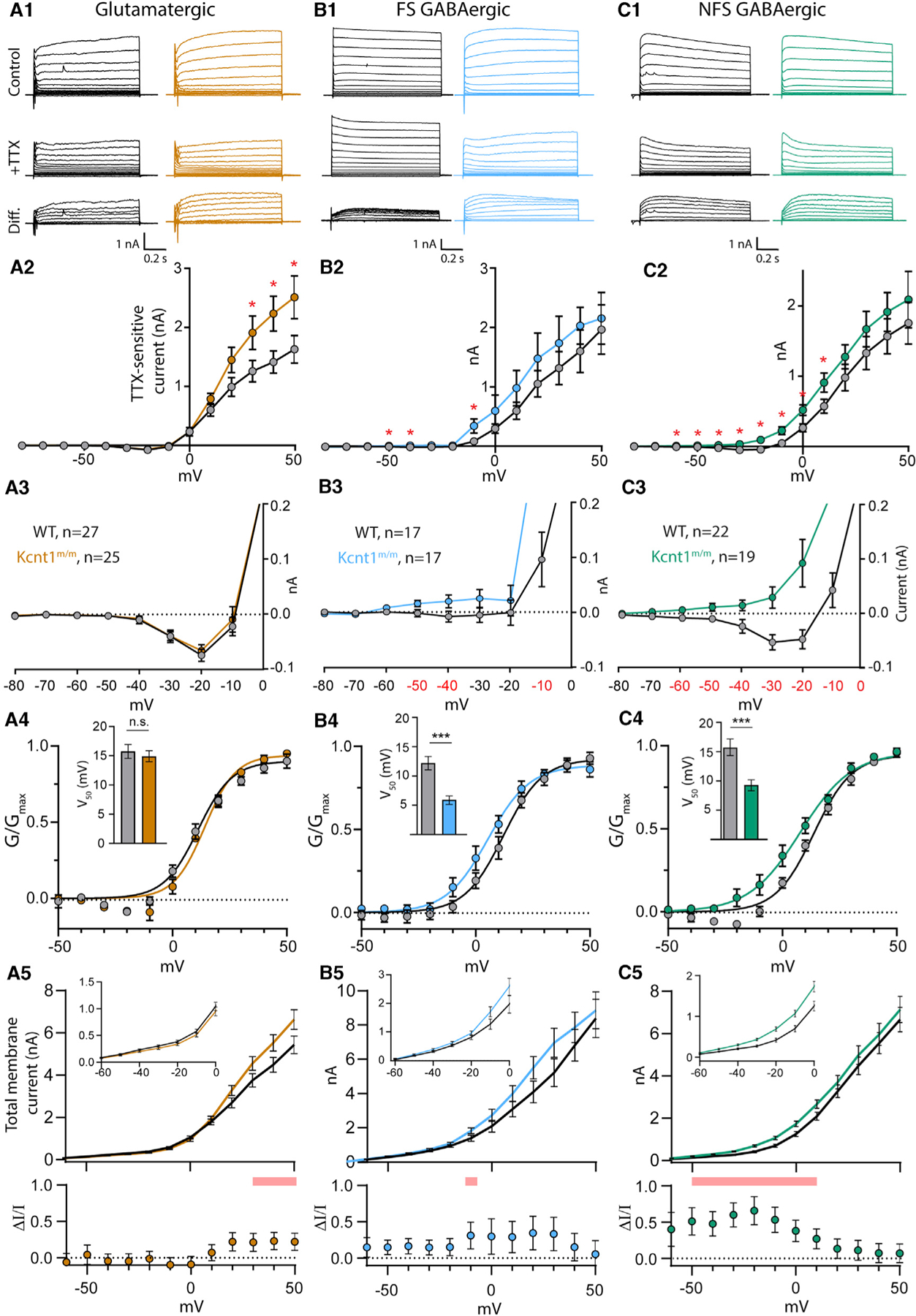
(A1–C1) Representative traces in control external solution (top), 0.5 mM TTX (middle), and the difference current (bottom), which was calculated by subtracting the membrane current response to voltage steps (−80 to +50 mV) in TTX from the response in control, in WT (black) and Kcnt1m/m (colors) neurons.
(A2–C2) Summary data show the KNa current (mean ± SEM) for each voltage step in WT (black and gray) and Kcnt1m/m (colors) neurons.
(A3–C3) Plots of the KNa current (mean ± SEM) for each voltage step from −80 to 0 mV in WT (black and gray) and Kcnt1m/m (colors) neurons to illustrate the values that are too small to be seen on the graphs in A2-C2.
(A4–C4) Normalized Conductance-Voltage (G-V)plots (mean ± SEM) for WT (black and gray) and Kcnt1m/m (colors) neurons with Boltzmann fits. Insets show the Vm at half-maximum (V50; mean ± SEM).
(A5–C5) Plots of the total membrane current (mean and SEM) for each voltage step from −60 to +50 mV in WT (black) and Kcnt1m/m (colors) neurons. Insets show values too small to be seen on the larger graph. Shaded red areas indicate significantly higher currents in Kcnt1m/m neurons. The plots below show the fractional increase in the total membrane current (ΔI/I, mean ± SEM) caused by the Y777H variant. Statistical significance for Current-Voltage (I-V) plots was tested using generalized linear mixed models with genotype and current step as fixed effects followed by pairwise comparisons at each level.
p values < 0.05 are labeled on the plots in A2–C2 as asterisks and on the plots in A3–C3 as red numbers. p values < 0.001 are labeled on the plots in A4–C4 as three asterisks. See also Figures S4 and S5.
The Y777H variant altered KNa currents in ways consistent with it being a GOF variant, but the nature of the changes was unique to each neuron type. In all Kcnt1m/m neuron types, the overall KNa current was increased, as measured by a significant effect of genotype using a linear model, and all showed apparent increases in current at positive potentials compared to those of WT (Figures 5A2–5C2). However, pairwise comparisons at each voltage step showed voltage-dependent differences in current increases among the Kcnt1m/m neuron types. For Kcnt1m/m glutamatergic neurons, significant increases in the KNa current were only at very depolarized voltages, from +30 to +50 mV (Figure 5A2), whereas the currents at 0 mV and below were indistinguishable from those of WT (Figure 5A3). Conversely, in Kcnt1m/m FS neurons, significant increases in KNa currents only occurred at more negative voltage steps, including −50, −40, and −10 mV (Figures 5B2 and 5B3). Kcnt1m/m NFS neurons showed the broadest increase in KNa currents, with significant increases at all voltage steps from −60 to +10 mV (Figures 5C2 and 5C3). Plotting the KNa current densities (pA/pF) of NFS neurons showed similar results (Figure S4), suggesting that the increased Cm of Kcnt1m/m NFS neurons does not contribute to their increased KNa currents.
To better assess neuron-subtype-specific effects of the Y777H variant on KCNT1 channel function, we plotted the mean normalized conductances of WT and Kcnt1m/m neurons as a function of voltage (activation curves). For glutamatergic neurons, the activation curves were not different between the two genotypes, as indicated by their similar membrane potential at half-maximum (V50) values (WT: 16.5 ± 1.0 mV, Kcnt1m/m: 15.8 ± 0.6 mV, p = 0.54; Figure 5A4). These data, together with the overlapping KNa currents observed throughout the subthreshold voltage range in WT and Kcnt1m/m glutamatergic neurons, are consistent with the unaltered membrane properties of Kcnt1m/m glutamatergic neurons. However, for FS and NFS GABAergic neurons, the activation curves were significantly left-shifted by the Y777H variant, as evidenced by a 6.5-mV (WT: 12.1 ± 1.2 mV, Kcnt1m/m: 5.6 ± 1.4 mV, p = 0.0005; Figure 5B4) and 6.3-mV (WT: 15.7 ± 0.8 mV, Kcnt1m/m: 9.4 ± 0.8 mV, p < 0.0001; Figure 5C4) decrease in V50, respectively. These strong GABAergic-specific left shifts in the voltage-dependent activation of the KCNT1-Y777H channel likely contribute to the increases in KNa currents at subthreshold membrane potentials, and the increases in rheobase and decreases in AP firing, that were observed in the FS and NFS neurons.
Glutamatergic neurons showed large increases in the KNa current at depolarized voltages but no effects on membrane excitability, whereas GABAergic neurons, especially NFS, showed smaller absolute increases but large effects on membrane excitability. Reasoning that the effect of the altered KNa current on membrane excitability would depend on how much it increased the total membrane current (Im), we plotted Im across all voltage steps for each neuron type, WT and Kcnt1m/m (Figures 5A5–5C5), as well as the fractional increase in Im caused by the variant (ΔI/I; Figures 5A5–5C5, lower panels). In glutamatergic neurons, the maximal ΔI/I was from +20 to +50 mV, where Kcnt1m/m neurons had a ~20% increase compared to WT. In FS Kcnt1m/m neurons, the maximum ΔI/I was ~30%, which occurred from −10 to +30 mV. In NFS Kcnt1m/m neurons, the ΔI/I was 50%–60% throughout the subthreshold voltages, reaching values as high as 65% at −20 mV. These results are consistent with the observed neuron-subtype-specific effects of the Kcnt1 GOF variant, with the smallest fractional increase in the K+ current occurring in glutamatergic neurons and the largest in NFS neurons; more specifically, the large increase in ΔI/I across subthreshold voltages in Kcnt1m/m NFS neurons predict the robust effects observed on membrane excitability, including the decreased Rin, increased rheobase, and impaired AP firing.
To provide further evidence for the specificity of the KNa current increases and their relationship to membrane excitability, we recorded KNa currents and assessed alterations in membrane excitability in neurons with heterozygous expression of the Y777H variant (Figure S5). Heterozygous expression of Y777H increased KNa currents in the same voltage ranges as homozygous expression in all three neuron types, and with current magnitudes intermediate to those seen in corresponding WT and homozygous neurons, demonstrating a gene dosage effect of the Kcnt1-Y777H variant on KNa current increases. Furthermore, NFS neurons with heterozygous expression of Y777H showed an intermediate increase in rheobase current compared with those of WT and homozygous neurons, suggesting a tight relationship between the increase in KNa current at subthreshold voltages and the deficit in AP generation in NFS neurons.
The Kcnt1-Y777H Variant Increases Homotypic Synaptic Connectivity and Induces a Hyperexcitable and Hypersynchronous Network
Our finding that KCNT1 GOF caused a selective reduction in GABAergic neuron excitability is not mutually exclusive with the third proposed hypothesis, which is that KCNT1 GOF variants cause network excitability because increases in K+ current during development alter normal patterns of synaptic connections (Kim and Kaczmarek, 2014). Thus, we tested for altered synaptic connectivity by performing paired recordings of glutamatergic (excitatory [E]) and GABAergic (inhibitory [I]) neurons and alternatively stimulating each neuron at 0.1 Hz to test baseline connection probability (Pc) and strength at the four possible motifs (I-I, I-E, E-I, and E-E). Pc at the I-E and E-I motifs was not altered in Kcnt1m/m networks (Figures 6B and 6C), indicating grossly normal synaptic interactions between glutamatergic and GABAergic neurons. However, Pc between both I-I and E-E pairs was higher in Kcnt1m/m networks than in those of WT (Figures 6A and 6D). The amplitudes of the evoked postsynaptic currents (ePSCs) between connected neurons was not significantly different between genotypes for any of the four connection types (Figures 6A–6D). Thus, in addition to impairing the excitability of GABAergic neurons, the Y777H variant increased synaptic connectivity in the network in ways that should, theoretically, further enhance excitation (increased E-E connections) and reduce inhibition (increased I-I connections).
Figure 6. Increases in Synaptic Connectivity in Kcnt1m/m Networks Contribute to Hyperexcitability and Hypersynchrony.
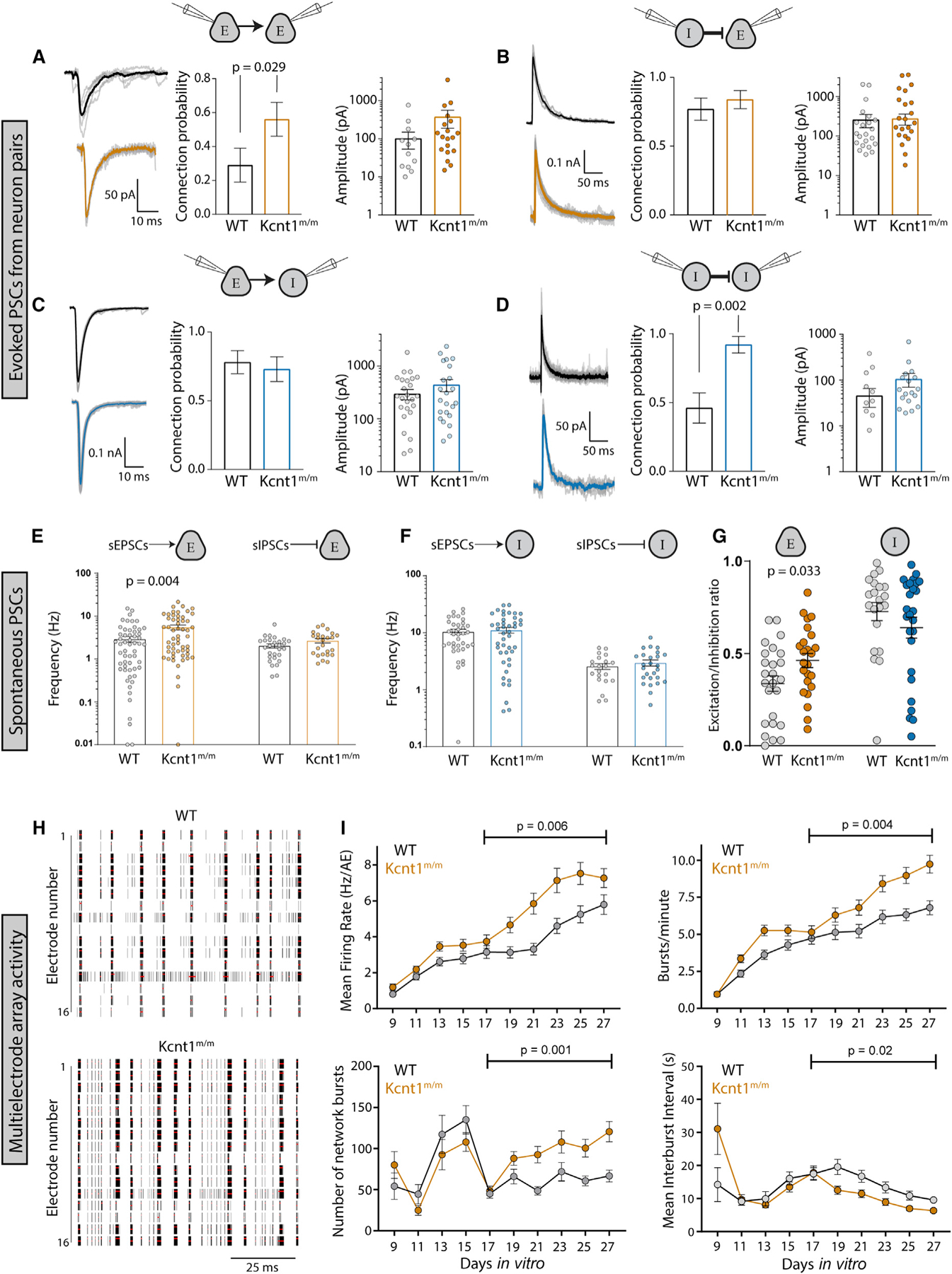
(A–D) Evoked postsynaptic currents (PSCs) recorded from neuron pairs (glutamatergic [excitatory (E)] and GABAergic [inhibitory (I)]) by stimulating the neuron type indicated on the left and recording the response in the neuron type indicated on the right (WT, black; Kcnt1m/m E, orange; Kcnt1m/m I, blue). Left: example traces of evoked ePSCs. Middle: summary data (mean ± SEM) of the connection probability (Pc) between motifs. Right: individual pair values and mean ± SEM of peak evoked PSC amplitudes of connected pairs.
(E) Individual values and mean ± SEM of the spontaneous excitatory postsynaptic current (sEPSC) or inhibitory postsynaptic current (sIPSC) frequency onto excitatory neurons (WT, black; Kcnt1m/m, orange).
(F) Individual values and mean ± SEM of the sEPSC or sIPSC frequency onto inhibitory neurons (WT, black; Kcnt1m/m, blue).
(G) The E/I ratio onto excitatory neurons, but not inhibitory neurons, is increased. Individual values and mean ± SEM of the E/I ratios are shown.
(H) Raster plots of multi-electrode array (MEA) data showing WT and Kcnt1m/m cortical network firing across the 16 electrodes of a well of a representative plate at DIV23. The black bars indicate spikes, and the red bars indicate bursts.
(I) Graphs plotting measures of spontaneous activity as a function of DIV on MEAs of WT (gray, n = 65 wells, 5 mice) and Kcnt1m/m (orange, n = 66 wells, 5 mice) cortical neurons. Mean firing rate (MFR) per active electrodes, number of bursts per minute, number of network bursts, and interburst interval are each shown as the mean ± SEM. Permutated p values for mature DIV17 to DIV27 neurons calculated with a Mann-Whitney U test followed by 1,000 permutations are indicated on the graphs.
To determine whether the increases in synaptic connectivity were accompanied by alterations in synaptic activity, we recorded spontaneous postsynaptic currents (sPSCs; spontaneous excitatory postsynaptic currents [sEPSCs] and spontaneous inhibitory postsynaptic currents [sIPSCs]) onto voltage-clamped glutamatergic and GABAergic neurons. Similar to the increase in E-E synaptic connectivity, we observed an increase in sEPSC frequency onto Kcnt1m/m glutamatergic neurons (Figure 6E). Interestingly, although sEPSC frequency was not altered onto the Kcnt1m/m GABAergic neuron population as a whole, it was increased onto those with an NFS phenotype, suggesting there may be a compensatory increase in excitatory drive onto Kcnt1m/m NFS neurons (Figure S6). This increased excitatory drive may offset the effects of the decreased membrane excitability of these neurons, as sIPSCs were not significantly affected onto Kcnt1m/m glutamatergic or GABAergic neurons (Figure 6F). Moreover, in agreement with the ePSC measurements, the amplitudes and kinetics of the sPSCs were not different between the genotypes onto either neuron group (Table S3). Finally, to assess the net effect of altered sPSC activity onto Kcnt1m/m neurons, we calculated the E/I ratio, taking into account the relative frequency and size of the sPSCs. E/I ratio onto Kcnt1m/m glutamatergic neurons was significantly higher than that onto WT, whereas the E/I ratio onto GABAergic neurons was similar between the two groups (Figure 6G). Thus, the overall balance of excitation and inhibition in in vitro Kcnt1m/m networks is shifted toward excitation.
Finally, we tested whether in vitro networks of Kcnt1m/m neurons exhibit hyperexcitable and/or hypersynchronous behavior, effects that would be predicted downstream of the GABAergic impairments, increased homotypic synaptic connectivity, and altered E/I ratio. To assess these network behaviors, we made primary cultures of cortical neurons from Kcnt1m/m and WT P0 pups and recorded spontaneous spiking activity using multi-electrode array (MEA) analysis. We analyzed a variety of spiking and bursting features between DIV9 and DIV27 (see STAR Methods). Networks of Kcnt1m/m and WT neurons matured at a similar rate, as measured by the increase in the number of active electrodes (nAEs) per well, which reached a maximum by DIV17 (not shown), and displayed a variety of activity patterns (Figure 6H). In networks of Kcnt1m/m neurons, we observed a hyperexcitability phenotype, characterized by an increased mean firing rate (MFR) and an increased bursting frequency (increase in bursts/minute and decrease in interburst interval) (Figure 6I). We also observed an increase in synchrony at the network level, characterized by an increased number of network bursts (Figure 6I). Together, these data suggest that KCNT1 GOF alters intrinsic GABAergic excitability and synaptic connectivity to promote a hyperexcitable and hypersynchronous network.
Acute Cortical Slice Recordings Confirm the Reduced Intrinsic Excitability of Kcnt1m/m NFS GABAergic Neurons
Having identified a brain region (Figures 2 and 3) and a neuron type (Figures 4 and 5) that were particularly sensitive to the Kcnt1 variant, we next wanted to test whether the physiological changes observed in cortical neuron cultures are also present in acute slice, an experimental preparation with a more organized network. We first performed Nissl staining of multiple, coronal 40-mm sections, each containing portions of motor cortex (Figure 7A), from P28 WT and Kcnt1m/m littermates to examine potential effects of the Kcnt1 GOF variant on general cortical structure and lamination. Overall, the brain structures between WT and Kcnt1m/m mice appeared highly similar, and the cortical regions in brains of Kcnt1m/m mice showed grossly normal cortical thickness and lamination.
Figure 7. NFS GABAergic Neurons in Layer 2/3 of Acute Slices from Motor Cortex of Kcnt1m/m Mice Show a Strong Impairment in AP Generation.
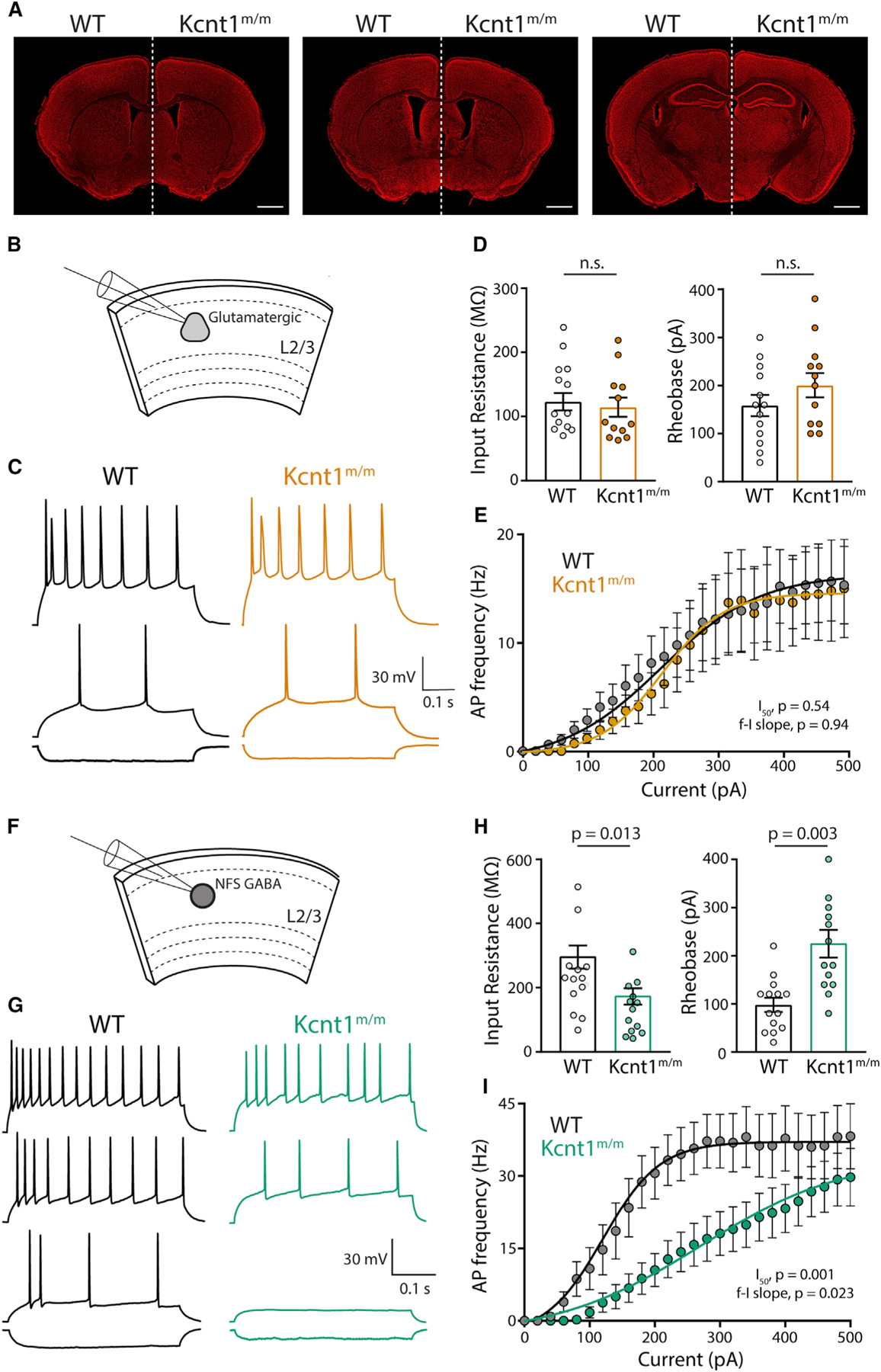
(A) Representative images of 10-week-old, Nissl-stained coronal sections at three matched bregma levels from WT (left) and Kcnt1m/m (right) mice. Scale bar, 1 mm.
(B) Schematic shows the experimental approach of recording glutamatergic neurons in layer 2/3 of the motor cortex.
(C) Representative membrane voltage traces of glutamatergic WT (black) and Kcnt1m/m (orange) neurons in response to −100, +140, and +240 pA current injections.
(D) Individual neuron values, mean, and SEM of the Rin and rheobase of WT and Kcnt1m/m glutamatergic neurons.
(E) Number of APs (mean ± SEM) per current injection step in WT and Kcnt1m/m glutamatergic neurons.
(F) Schematic shows the experimental approach of recording NFS GABAergic neurons in layer 2/3 of the motor cortex.
(G) Representative membrane voltage traces of NFS GABAergic WT (black) and Kcnt1m/m (green) neurons in response to −100, +100, +180, and +300 pA current injections.
(H) Individual neuron values, mean, and SEM of the Rin and rheobase of WT and Kcnt1m/m NFS GABAergic neurons.
(I) Number of APs (mean ± SEM) per current injection step in WT and Kcnt1m/m NFS GABAergic neurons. The lines in the current/AP plots are fits to a Boltzmann sigmoidal curve that were used to determine the I50. Statistical significance was tested using generalized linear mixed models.
See also Table S4.
We next prepared acute slices containing the anterior motor cortex from P20 to P29 WT and Kcnt1m/m mice and performed whole-cell recordings from pyramidal neurons (Figure 7B) and GABAergic neurons (Figure 7F) in the medial portion of the slice. Because more superficial cortical layers are enriched for NFS neurons (Tremblay et al., 2016; Lee et al., 2010) and have been reported to show higher levels of KCNT1 expression (Rizzi et al., 2016), we current-clamped neurons in layer 2/3 and injected steps to elicit APs (Figures 7C and 7G). Similar to observations in cortical neuron cultures, Kcnt1m/m glutamatergic neurons showed no changes in Rin or rheobase relative to those of WT (Figure 7D). They did, however, have a more depolarized AP threshold (Table S4), an effect that is notably opposite of that previously reported from Kcnt1 knockout mouse models (Martinez-Espinosa et al., 2015; Reijntjes et al., 2019). Despite their depolarized AP threshold, Kcnt1m/m glutamatergic neurons showed AP firing patterns similar to those of WT glutamatergic neurons (Figure 7E).
Importantly, Kcnt1m/m NFS neurons showed a significant reduction in Rin and large increases in rheobase (Figure 7H) and Cm (Table S4), all of which were changed in the same manner in cultured neurons, although the AP half-width and AHP were unchanged (Table S4). Moreover, incremental current injections demonstrated significant impairments in AP generation in NFS neurons expressing the Y777H variant (Figure 7I). Thus, the main finding from culture, that NFS neurons are most strongly affected by the Kcnt1-Y777H variant, was verified in acute slices made from a brain area with demonstrated pathology.
DISCUSSION
Classic and recent human genetic studies have provided key insights into the causes of neurodevelopmental disease and severe epilepsies, revealing that genetic variants in ion channels, both ligand and voltage gated, comprise approximately one-third of known monogenic causes of seizure disorders (Noebels, 2017). Despite the fact that molecular dysfunction of these channel variants is relatively well modeled and correctable in heterologous systems, translating these findings to therapies has faced unexpected challenges, likely due to the developmental, cellular, and synaptic complexity of the brain. Progress in tracing the critical steps between ion channel biophysics and pathological synchronization of cortical neurons thus requires precision genetic disease models and multi-level interrogations of their effects (Oyrer et al., 2018; Farrell et al., 2019).
To address this, we created a mouse model with a missense mutation in a KNa channel gene orthologous to a human missense mutation that causes an early-onset seizure disorder (ADNFLE). Homozygous expression of this GOF variant caused an epileptic phenotype with strong parallels to the human disease, and electrocorticography analysis showed epileptiform activity in Kcnt1m/m mice by P14. Early childhood seizures are a common feature of KCNT1-associated (and numerous other) genetic epilepsies, but seizure phenotyping is rarely done at this age in rodents, even though early phenotyping has the potential to uncover seizure phenotypes not present in adults (Lozovaya et al., 2014) and improve our understanding of how well genetic models recapitulate human diseases. Likewise, widefield Ca2+ imaging pinpointed the M2 region as the cortical source of IEDs. These results do not rule out the involvement of other cortical areas in seizure generation or speak to the strong possibility that cortical interactions with subcortical structures are critical for seizure generation. In fact, the finding that seizures in our model are most likely to happen during NREM sleep, coupled with the fact that ADNFLE is also caused by variants in genes that encode nicotinic acetylcholine receptor subunits (Phillips et al., 1995; Sutor and Zolles, 2001), suggests that thalamic input or cholinergic transmission may play a key role in seizure generation (Klaassen et al., 2006).
The multiplatform, in vivo phenotyping of the KCNT1 GOF mouse model focused our cellular studies on a discrete time and place. Results from these in vitro and ex vivo studies support a model in which the Kcnt1-Y777H variant increases the KNa current over different voltage ranges in the three major neuron types of the cerebral cortex; glutamatergic, FS GABAergic, and NFS GABAergic. In GABAergic neurons, because it occurs at subthreshold voltages, the KNa current increase impairs membrane excitability and AP generation, changes that are especially strong in the NFS neurons. The reduced excitability of GABAergic neurons is accompanied by increases in E-E and I-I synaptic connectivity, and together, these alterations likely interact to promote both hyperactivity and hypersynchrony in the cortical circuits, especially in cortical region M2, which leads to epileptiform discharges and seizures. This model provides a strong framework for understanding how GOF variants in KCNT1 lead to epilepsy; however, it remains to be seen if this model will generalize to other GOF variants in K+ channels, or even other variants within the KCNT1 gene.
Prior to this study, two other studies were published characterizing KCNT1 GOF variants, both of which are associated with MMPSI, an earlier onset and more severe form of epilepsy than ADNFLE. Using a knockin mouse model, Quraishi et al. showed that heterozygous expression of the Kcnt1-R455H variant results in spontaneous seizure activity in four out of seven mice (Quraishi et al., 2020). However, there were no reports of the effects of the R455H variant on KNa currents or neuronal physiology, making it difficult to compare with mechanistic data from our model. In the other study, homozygous expression of the P942L variant in human induced pluripotent stem cell (iPSC)-derived neurons increases the KNa current, but only at voltages above +40 mV (Quraishi et al., 2019). Similar to our data, they reported a decrease in AP width and increase in AHP; however, in contrast to our data, the P942L variant increases the peak AP firing rate in what are presumably immature glutamatergic neurons. This discrepancy in AP generation identified downstream of the Y796H and P942L variants may be because the KCNT1 GOF mechanism is variant and/or disease dependent. Alternatively, we also observed an increased KNa current at suprathreshold voltages in Kcnt1m/m glutamatergic neurons. Thus, it is possible that early effects in cortical glutamatergic neuron excitability have resolved by the time we performed our electrophysiology on more mature glutamatergic neurons but could contribute to our observed increase in E-E connectivity (Figure 6A). Future mechanistic studies using knockin mouse models of additional KCNT1 GOF variants should help resolve these discrepancies and clarify the generality of our findings.
A caveat of our mouse model is that although a heterozygous GOF mutation in KCNT1 is sufficient to cause ADNFLE in humans, we only detected seizures in homozygous mice. Unfortunately, this caveat is not unique to our model, as several attempts to generate mouse models of heterozygous LOF mutations in K+ channel genes associated with epilepsy in humans have failed to result in a seizure phenotype in the heterozygous state in mice (Oyrer et al., 2018). Nevertheless, some of these models in the homozygous state, such as Kcna1 null mice and knockin LOF Kcnq2 and Kcnq3 mice, display seizure phenotypes highly similar to those of human patients (Smart et al., 1998; Glasscock et al., 2010; Singh et al., 2008) and have been used successfully to model the human disease and initiate therapeutic studies (Fenoglio-Simeone et al., 2009; Roundtree et al., 2016). Similarly, the strong parallels observed in both the behavior and seizure phenotypes between the homozygous Y777H mouse model and the Y796H-associated disease suggest analogous underlying mechanisms and indicate that the homozygous Y777H mice are a valid preclinical model. Moreover, during the preparation and review of this article, we observed seizures in two Kcnt1-Y777H heterozygous mice, one during a widefield Ca2+ imaging experiment and the other during a video-EEG experiment. Thus, heterozygous expression can result in seizures in a rodent model, but apparently at a much lower frequency than that observed with homozygous expression, and studies are ongoing to further characterize the heterozygous Kcnt1-Y777H phenotype.
A somewhat perplexing finding from our study was that the decreased excitability of Kcnt1m/m GABAergic neurons did not lead to decreased sIPSC frequency onto cultured cortical glutamatergic or GABAergic neurons; however, there are several potential explanations for this inconsistency. First, the large literature on Dravet syndrome (DS), another childhood epilepsy disorder, has demonstrated that LOF mutations in the Na+ channel gene Scn1a decrease the excitability of inhibitory neurons. Despite this consensus, electrophysiological recordings in slices have sometimes failed to find reductions in sIPSC frequencies (De Stasi et al., 2016), especially when Scn1a deletion is restricted to NFS GABAergic neurons (Rubinstein et al., 2015), which is likely due to the fact that a large portion of NFS GABAergic neurons target the distal dendrites of neurons, far from where sIPSCs are measured at the soma. Moreover, in vivo studies of DS models have not shown a decrease in GABAergic neuron firing rates (De Stasi et al., 2016; Tran et al., 2020), even though SCN1A loss causes a more severe phenotype in mice than KCNT1 GOF in our model. Finally, in this study, we also provided experimental evidence that sEPSC frequency onto Kcnt1m/m NFS GABAergic neurons is increased (Figure 6H), which likely compensates for their reduced membrane excitability and normalizes sIPSC frequency. Thus, the lack of a decrease in sIPSC frequency is not strong evidence against the hypothesis that inhibitory neuron dysfunction due to KCNT1 GOF contributes to epilepsy.
Kcnt1 mRNA and protein expression in the CNS are thought to be widespread and present in both glutamatergic and GABAergic neurons, indicating that the differential effects we found in these neuron populations are likely not due to selective expression of KCNT1 (Figure S3). In support of this, we found that the Y777H variant increases KNa current in all neuron types analyzed; however, the voltage dependence of these increased currents in each neuron type resulted in drastically different effects on neuronal physiology. In cortical glutamatergic neurons, the KNa current was increased in the +30 to +50 mV range, but the effects of this increase on physiology were extremely limited. This lack of an effect may be due to the fact that in normal physiology, neurons only reach these voltages (30–50 mV) at the peak of the AP and remain there for less than a millisecond, which may not allow for activation of KNa, as the time constant for full activation of KCNT1 channels has been estimated at 25–60 ms (Joiner et al., 1998; Chen et al., 2009). Moreover, the KNa current was increased at negative potentials in both Kcnt1m/m GABAergic neuron types, but not in Kcnt1m/m glutamatergic neurons. The reasons for the lack of a KNa current increase at subthreshold voltages in glutamatergic neurons will require further study; however, candidate mechanisms include glutamatergic-specific expression of alternative splice forms of KCNT1, which have been shown to have different activation kinetics, and coexpression of other channels, such as KCNT2, which can form heteromers with KCNT1 and alter its biophysical properties (Chen et al., 2009; Joiner et al., 1998), thus protecting glutamatergic neurons from the more severe effects of KCNT1 GOF.
In GABAergic neurons, we observed the strongest effects of the Y777H variant on KNa currents at voltages between −60 and +10 mV. The role of the KNa current in neurons, at subthreshold voltages in particular, has been controversial, largely because early studies suggested that the Na+ concentration needed to activate KNa was higher than that normally found in neurons. However, recent studies have shown that KNa can be activated by a persistent Na+ (NaP) current present at voltages as low as −90 mV (Hage and Salkoff, 2012). Consistent with this subthreshold role for KCNT1, multiple studies have shown that mouse sensory neurons lacking a KNa current, due to Kcnt1 knockout, have no alterations in AHP or AP repolarization but instead show decreases in AP threshold and rheobase and increases in AP firing (Martinez-Espinosa et al., 2015; Reijntjes et al., 2019; Lu et al., 2015). These KCNT1 LOF effects on neuronal physiology nicely complement our GOF effects, and taken together, these data support a central role for KCNT1-mediated currents in regulating neuronal excitability at subthreshold potentials.
Among GABAergic neurons, we identified strong KCNT1 GOF effects on the NFS subtype, which are still electrically, molecularly, and morphologically diverse, with subclasses performing distinct roles in circuit function (Markram et al., 2015). The similar, yet more subtle, effects on the FS subtype suggest that there is likely not one particular subclass of inhibitory neurons that is uniquely affected. What cell type differences could account for the stronger effects of KCNT1 GOF on NFS than FS GABAergic neurons? First, as proposed for the differential effect of the variant on glutamatergic neurons, NFS and FS GABAergic neurons may express different KCNT1 isoforms and/or interacting channels and proteins that alter the functional effects of the variant. Alternatively, FS neurons already show a large K+ conductance at subthreshold voltages. This is reflected in their low Rin, fast time constant, and high rheobase and caused by high expression of K+ leak channels (Okaty et al., 2009). The relatively small effect of KCNT1 GOF on FS neurons may simply reflect the fact that adding a KCNT1-mediated K+ conductance on top of an already large K+ conductance may have little effect.
KCNT1 and other K+ channel GOF epilepsies are typically treatment resistant, and the use of quinidine, which inhibits most KCNT1 GOF variant currents in heterologous neurons, has shown limited success in treating KCNT1-related epilepsy in humans. Here, we show that Kcnt1m/m neurons, networks, and mice all display clear pathology at 2 weeks of age. In addition to the changes in membrane excitability, which may be correctable by blocking KCNT1 or other ion channel currents, we also found increases in Cm and synaptic Pc at multiple synaptic motifs, which likely play a role in the generation of abnormal network activity. These structural changes would be more difficult to reverse by modulating ion channel currents, unless perhaps the modulation occurred prior to their occurrence, which would argue for earlier treatment with quinidine (Dilena et al., 2018). Alternatively, if the NaP current activates the increased KNa current and results in reduced interneuron excitability, drugs that inhibit the NaP current could have the dually useful effect of mitigating aberrant activation of the KNa current in NFS GABAergic neurons while still providing general dampening of membrane excitability in glutamatergic neurons. NaP current inhibitors are currently available and have been used with success in preclinical models of Na+ channel dysfunction (Baker et al., 2018; Anderson et al., 2014). Our data suggest that the way forward to designing optimal treatment strategies for genetic epilepsies is a better understanding of the complex interactions among neuron-subtype-specific membrane currents, circuit development, and synaptic connections.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew C. Weston (mcweston@uvm.edu).
Materials Availability
The Kcnt1Y777H mouse line is available from the Lead Contact, Matthew C. Weston (mcweston@uvm.edu), with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were bred, and procedures were conducted at the Jackson Laboratory, at Columbia University Irving Medical Center, or at the University of Vermont. Each institution is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and all protocols were approved by their respective Institutional Animal Care and Use Committees. All experiments were performed in accordance with respective state and federal Animal Welfare Acts and the policies of the Public Health Service. The animal protocol numbers for Columbia University were: AC-AAAU8484, AC-AAAZ8450, AC-AAAR4414, AC-AAAU8476, and AC-AAAT6474, and the University of Vermont were: 16-001 and 19-034.
Kcnt1Y777H knockin mice (formal gene and allele symbol: Kcnt1em1(Y777H)Frk) were generated in the C57BL/6NJ (B6NJ) mouse strain (The Jackson Laboratory, JAX: 005304) using CRISPR/Cas9 and an oligonucleotide donor sequence as part of the Jackson Laboratory Center for Precision Genetics program (JCPG, https://www.jax.org/research-and-faculty/research-centers/precision-genetics-center), and maintained by backcrossing heterozygous males to wild-type B6NJ females. Kcnt1Y777H mice were genotyped using PCR amplification primers (Kcnt1 forward primer: 5′-CTAGGGCTGCAAACACAACA-3′; Kcnt1 reverse primer: 5′-TCAAGCAGCAACACGATAGG-3′) with standard thermocycler amplification conditions using MyTaq master mix (Bioline, BIO-25041), and the annealing temperature set at 58°C. Following amplification, a restriction cut was performed with the enzyme NlaIII (NEB, R0125S) to distinguish mutant (127 and 44 bp products after cut) from wild-type alleles (171 bp product). For some experiments, as noted in the text, male mutant mice were crossed with FVB.129P2-Pde6b+ Tyrc-ch/AntJ (The Jackson Laboratory, JAX: 004828) dams to generate cohorts of N2 or F2 hybrid mice.
The following additional mouse lines and strains were used for these studies: Snap25-2A-GCaMP6s-D knockin (B6.Cg-Snap25tm3.1Hze/J, The Jackson Laboratory, JAX: 025111) and C57BL/6J (The Jackson Laboratory, JAX: 000664). Mice were maintained in ventilated cages at controlled temperature (22–23°C), humidity ~60%, and 12-h light: 12-h dark cycles (lights on at 7:00 AM, off 7:00 PM). Mice had access to regular chow and water, ad libitum. For all experiments, male and female littermates were used for each genotype. The ages of the mice for each experiment are indicated in the Method Details section.
Primary Astrocyte Feeder Layer Culture
Astrocyte feeder layers, to support the growth and maintenance of primary neurons, were generated as previously described (Barrows et al., 2017). Briefly, cortices were dissected from P0-1 WT C57BL/6J mice of either sex. The cortices were incubated in 0.05% trypsin-EDTA (GIBCO, 25300-054) for 15 min at 37°C in a Thermomixer (Eppendorf) with gentle agitation (800 rpm). Then, the cortices were mechanically dissociated with a 1-mL pipette tip, and the cells were plated into T-75 flasks containing filter-sterilized astrocyte media [DMEM media supplemented with glutamine (GIBCO, 10569-010), 10% fetal bovine serum (FBS, GE Healthcare, SH3008803), 1X MITO+ Serum Extender (Corning, 355006), and 0.2X penicillin/streptomycin (GIBCO, 15140-122)]. After the astrocytes reached confluency, they were washed with PBS and incubated for 5 min in 0.05% trypsin-EDTA at 37°C, washed, and then resuspended in astrocyte media. Astrocytes were added to 6-well plates containing 25-mm coverslips (Carolina Biological, 633037) precoated with coating mixture [0.7 mg/ml collagen I (Corning, 354236) and 0.1 mg/ml poly-D-lysine (Sigma-Aldrich, P6407) in 10 mM acetic acid].
Primary Cortical Neuron Culture for Electrophysiology
For the primary neuron culture, the dorsomedial cortices from P0-1 WT and Kcnt1m/m mice of either sex were dissected in cold HBSS. The tissue was then digested with papain (Worthington Biochemical, LS003126) for 60–75 min and treated with an ovomucoid inhibitor solution (Worthington Biochemical, LS003085) for 10 min, both while shaking at 800 rpm at 37°C in a Thermomixer (Eppendorf). The cells were then mechanically dissociated and counted. The dissociated cells were added at 200,000 cells/well to 6-well plates containing astrocyte-coated coverslips in filter-sterilized NBA plus [Neurobasal-A medium (GIBCO, 10888-022) supplemented with 1X GlutaMAX (GIBCO, 35050-061), 1X B27 supplement (GIBCO, 17504-044), and 0.2X penicillin/streptomycin (GIBCO, 15140-122)]. After plating (12–24 h), approximately 4 × 1010 genome copies (GC) of AAV8-CaMKIIa-GFP, serotype 8 (UNC Gene Therapy Center - Vector Core) was added to each well to fluorescently label glutamatergic neurons. Every 3–4 days, 20%–40% of the media was replaced with fresh NBA plus.
METHOD DETAILS
Western Blotting
For crude membrane/cytosol fractioning, P0 cortices of either sex were homogenized in the following homogenization buffer: 0.32 M sucrose, 10 mM HEPES pH 7.4, 2 mM EDTA in H2O, containing a protease, and a phosphatase, inhibitor cocktail. The samples were homogenized with a motor-driven homogenizer and centrifuged at 1000 × g for 15 min at 4°C to remove the pelleted nuclear fraction. The supernatant was further centrifuged at 16000 rpm for 20 min at 4°C to yield the cytosolic fraction in the supernatant and the pelleted membrane fraction. The pellet was resuspended in homogenization buffer. Protein concentrations were determined with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225) using BSA as a standard. Sample protein (10 μg) was mixed with 4X LDS Sample Buffer (NuPAGE, NP0007), and 10X Sample Reducing Agent (NuPAGE, NP0009), heated for 10 min at 70°C, separated on a 4%–12% Bis-Tris Mini Gel (NuPAGE, NP0321), and transferred to a PVDF membrane (Millipore, ISEQ00010). Nonspecific binding was blocked for 1 h at RT with 5% BLOTTO (Bio-Rad, 1706404) in Tris-buffered saline with 0.1% Tween (TBST). Membranes were incubated overnight at 4°C with the primary antibody mouse anti-KCNT1 (1:1000, Abcam, ab94578), washed 3 × 10 min with TBST, incubated with the secondary antibody HRP-conjugated goat anti-mouse (1:10000, Santa Cruz Biotechnology, sc-2005) in 5% BLOTTO for 1 h at RT, and washed again 3 × 10 min with TBST. Blots were incubated for 5 min with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32106) and developed with a Kodak X-OMAT 2000A Processor. Blots were stripped for 15 min at RT in ReBlot Plus Strong Solution (Millipore. 2504), blocked for 30 min in 5% BLOTTO, incubated for 1 h at RT with an HRP-conjugated mouse anti-β-actin antibody (1:5000, Santa Cruz Biotechnology, sc-47778) in 5% BLOTTO, and developed as previously described. β-actin was used as a loading control.
Mouse Brain Morphology and Histology
WT, Kcnt1m/+, and Kcnt1m/m littermate mice of either sex were anesthetized with 1.5%–2.5% isoflurane and oxygen at P28, or 10–12 weeks of age, and transcardially perfused with PBS, followed by 4% paraformaldehyde in PBS (pH 7.4). Brains were dissected and then post-fixed overnight in 4% paraformaldehyde at 4°C. Next, brains from 10–12-week-old mice were washed with PBS, weighed, and imaged to assess gross morphology. Then, all brains were cryoprotected with 30% sucrose until sectioning. To section, brains were frozen in Tissue-Plus O.C.T. Compound (Fisher, 23-730-571) and sectioned into 40-mm coronal slices using a cryostat. Brain sections were stored in a cryoprotectant solution with 30% ethylene glycol, 20% glycerol, and 50% 1X PBS until use.
Brain sections from P28 mice were stained with a NeuroTrace fluorescent Nissl stain (1:50, Molecular Probes, N-21482), according to the manufacturer’s instructions, and mounted in Fluoromount-G Mounting Medium, with DAPI (Fisher, 00-4959-52). Nissl-stained, whole coronal section images were continuously captured with a motorized stage and automatically stitched using a 4X objective on a Nikon-A1R-ER confocal laser microscope with NIS Elements software (eight images, 10% overlap/section).
Mouse Behavioral Tasks – Open Field Exploration
Each mouse was gently placed in the center of a clear Plexiglas arena (7.31 × 27.31 × 20.32 cm, Med Associates, ENV-510) lit with dim light (~5 lux), and allowed to ambulate freely for 60 min. Infrared (IR) beams embedded along the X, Y, Z axes of the arena automatically tracked distance moved, horizontal movement, vertical movement, stereotypies, and time spent in center zone. At the end of the test, the mouse was returned to the home cage and the arena was cleaned with 70% ethanol followed by water drying.
Mouse Behavioral Tasks – Elevated Plus Maze
The elevated plus maze test was conducted as described previously (Yang et al., 2012). The elevated plus maze consisted of two open arms (30 × 5 cm) and two closed arms (30 × 5 × 15 cm) extending from a central area (5 × 5 cm). Photo beams embedded at arm entrances registered movements. Room illumination was approximately 5 lux. The test began by placing the subject mouse in the center, facing a closed arm. The mouse was allowed to freely explore the maze for 5 min. Time spent in the open arms and closed arms, the junction, and number of entries into the open arms and closed arms, were automatically scored by the MED-PC V 64-bit software (Med Associates, SOF-736). At the end of the test, the mouse was gently removed from the maze and returned to its home cage. The maze was cleaned with 70% ethanol and wiped dry between subjects.
Mouse Behavioral Tasks – Acoustic Startle Response
Acoustic startle response was tested using the SR-LAB Startle Response System with SR-LAB software (San Diego Instruments, San Diego, CA) as described previously (Yang et al., 2012). Test sessions began by placing the mouse in the Plexiglas holding cylinder for a 5-min acclimation period. For the next 8 min, mice were presented with each of six trial types across six discrete blocks of trials, for a total of 36 trials. The intertrial interval was 10–20 s. One trial type measured the response to no stimulus (baseline movement). The other five trial types measured startle responses to 40 ms sound bursts of 80, 90, 100, 110, or 120 dB. The six trial types were presented in pseudorandom order such that each trial type was presented once within a block of six trials. Startle amplitude was measured every 1 ms over a 65 ms period beginning at the onset of the startle stimulus. The maximum startle amplitude over this sampling period was taken as the dependent variable. A background noise level of 70 dB was maintained over the duration of the test session.
Mouse Behavioral Tasks – Fear Conditioning
Training and conditioning tests were conducted in two identical chambers (Med Associates) that were calibrated to deliver identical footshocks. Each chamber was 30 × 24 × 21 cm with a clear polycarbonate front wall, two stainless side walls, and a white opaque back wall. The bottom of the chamber consisted of a removable grid floor with a waste pan underneath. When placed in the chamber, the grid floor connected with a circuit board for delivery of scrambled electric shock. Each conditioning chamber was inside a sound-attenuating environmental chamber (Med Associates). A camera mounted on the front door of the environmental chamber recorded test sessions, which were later scored automatically using the Video Freeze software (Med Associates, SOF-843). For the training session, each chamber was illuminated with a white house light. An olfactory cue was added by dabbing a drop of imitation almond flavoring solution (1:100 dilution in water) on the metal tray beneath the grid floor. The mouse was placed in the test chamber and allowed to explore freely for 2 min. A pure tone (5 kHz, 80 dB), which serves as the conditioned stimulus (CS), was played for 30 s. During the last 2 s of the tone, a footshock (0.5 mA) was delivered as the unconditioned stimulus (US). Each mouse received three CS-US pairings, separated by 90 s intervals. After the last CS-US pairing, the mouse remained in the chamber for another 120 s, during which freezing behavior was scored by the Video Freeze software. The mouse was then returned to its home cage. Contextual conditioning was tested 24 h later in the same chamber, with the same illumination and olfactory cue present but without footshock. Each mouse was placed in the chamber for 5 min, in the absence of CS and US, during which freezing was scored, and then returned to its home cage. Cued conditioning was conducted 48 h after training. Contextual cues were altered by covering the grid floor with a smooth white plastic sheet, inserting a piece of black plastic sheet bent to form a vaulted ceiling, using near infrared light instead of white light, and dabbing vanilla instead of banana odor on the floor. The session consisted of a 3 min free exploration period followed by 3 min of the identical CS tone (5 kHz, 80 dB). Freezing was scored during both 3 min segments. The mouse was then returned to its home cage, and the chamber thoroughly cleaned of odors between sessions using 70% ethanol and water.
Mouse Behavioral Tasks – Nesting
The nesting test was performed as previously described (Deacon, 2006). Briefly, mice were placed in individual cages at 6 pm, with a piece of nestlet weighing 2.5 g. The next morning, the remaining unshredded nestlet was weighed and a nesting score between 1 (no nest) and 5 (perfect nest) was attributed depending on nest quality.
Video-Electroencephalogram
Electrode implantation of adult (> 7 weeks of age) male and female mice was performed surgically as recently described (Asinof et al., 2016). Mice were anesthetized with tribromoethanol (250 mg/kg i.p., Sigma-Aldrich, T48402). Three small burr holes were drilled in the skull (1 mm rostral to the bregma on both sides and 2 mm caudal to the bregma on the left) 2 mm lateral to the midline. One hole was drilled over the cerebellum as a reference. Using four teflon-coated silver wires soldered onto the pins of a microconnector (Mouser electronics, 575-501101), the wires were placed between the dura and the brain and a dental cap was then applied. The mice were given a post-operative analgesic of carprofen (5 mg/kg subcutaneous Rimadyl injectable) and allowed a 48-h recovery period before recordings were taken. To record EEG signals, mice were connected to commutators (Plastics One) with flexible recording cables to allow unrestricted movements within the cage. Signal (200 samples/s) was acquired on a Grael II 48 EEG amplifier (Compumedics), and the data were examined in Profusion EEG 5 software (Compumedics). Differential amplification recordings were recorded pairwise between all three electrodes, as well as referential, providing a montage of six channels for each mouse. Mouse activity was captured simultaneously by video monitoring using a Sony IPELA EP550 model camera, with an infrared light to allow recordings in the dark. We recorded continuously for 24–72 h.
For sleep analysis, we used a custom-written MATLAB program to classify wake, NREM sleep, and REM sleep, and to detect GTCS and TS events. The program first used fast Fourier transform (FFT) to calculate the power spectrum of the EEG using a 5 s sliding window, sequentially shifted by 2 s increments. Then it semi-automatedly classified into different brain states using the following criteria: NREM sleep, high power at low frequencies (1–4 Hz) and low EMG activity; REM sleep, high power at theta frequencies (6–9 Hz) and low EMG activity; GTCS and TS, high power at ‘‘seizure’’ frequencies (19–23 Hz); and wake, the remaining (or default) state. We chose the 19–23 Hz band to detect seizures based on its clear separation from normal brain oscillatory activities. The classification was followed by manual inspection to further refine the scoring. Brain states prior to each GTCS/TS event were used in Table S1.
Widefield Calcium Imaging
Male and female mice that were heterozygous for the Snap25-GCaMP6s construct (Madisen et al., 2015) (Jackson Labs Stock No: 025111) and either homozygous Y777H or WT at the Kcnt1 locus were generated. To gain optical access to the dorsal cortex, the animal’s scalp and the underlying periosteum were removed before coating the dorsal skull with UV cure cyanoacrylate (Loctite 4305) and attaching a custom aluminum headplate to the skull. This headplate framed the glue covered skull and allowed the animal to be secured on a 20-cm diameter Styrofoam treadwheel during imaging sessions. In one homozygous mutant mouse the dorsal skull was removed entirely and replaced with a glass window (Labmaker, Crystal Skull) following a previously described protocol (Kim et al., 2016). Imaging was performed using a custom tandem-lens epifluorescence macroscope built according to a design previously described (Wekselblatt et al., 2016). This macroscope was configured for 2.1X magnification using a Nikon Nikkor 50mm f/1.2 lens paired with a Nikon 105mm f/1.8 lens. During all sessions, images were acquired at 40 Hz with an Andor Zyla 5.5 10-tap sCMOS camera set to 4 × 4-pixel binning. Successive frames were illuminated with blue and green light, and the green reflectance images were used to estimate changes in fluorescence resulting from hemodynamic factors and correct for them in GCaMP images. For blue illumination, we used an X-Cite 120 LED (Lumen Dynamics, XT120L) with a filter set consisting of an ET470/40x excitation filter, a T495lpxr dichroic, and an ET525/50 m emission filter (Set 49002, Chroma Technology, Bellows Falls, VT). Total blue light power delivered to the implant typically ranged from 18–21 mW. For green illumination, we used a 530 nm 6.8 mW Thorlabs LED (M530F2) driven by a Thorlabs tcube (LEDD1B) coupled to a fiber optic obliquely pointed at the brain. To switch illumination sources between frames, we relied on an Arduino Uno R3 (A000066) microcontroller operating on the Zyla ‘expose’ signal. At the beginning of each session, we exposed a dark frame in which no illumination was used. This frame was subtracted from all illuminated frames in the session as the first processing step. Subsequently, reflectance and GCaMP images were deinterleaved and linearly interpolated from 20 to 40 Hz. We calculated ΔF/F for each pixel in the image that covered the brain, as defined by a hand-drawn mask. We estimated F for each pixel as the average of the bottom 20th percentile of fluorescent values over the entire session. Following ΔF/F calculation, we subtracted fractional green fluorescence (ΔF/Fmean) with Fmean calculated over the full session. Finally, we performed Singular Value Decomposition (SVD) on the ΔF/F image stacks and reconstructed images using the first 50 singular values for all subsequent analysis. The SVD code was a modification of that provided by the Cortex Lab group at University College London. To rigidly align area borders derived from the Allen Common Coordinate Framework v3 to each brain, we relied on the landmark-based strategy previously described (Musall et al., 2019).
Mouse Pup Electrocorticography
Eight Kcnt1m/m and five WT pups of either sex at P13-15 were used for in vivo electrophysiology. All experiments were performed in accordance with protocols approved by IACUC at Columbia University Irving Medical Center. Pups were anesthetized using isoflurane and electromyography (EMG) electrodes were placed for real-time monitoring of respiratory rate, heart rate, and muscle activity. The animals were then given systemic and local analgesia, head-fixed, and a craniotomy was made over the dorsal cortical surface of one hemisphere. This craniotomy was centered over somatomotor cortex, spanning 2.5 mm in the mediolateral direction and 3.5 mm in the anterioposterior direction between bregma and lambda. A NeuroGrid (ultra-conformable, biocompatible surface electrocorticography array, 119 electrodes, 177 μm pitch) array was placed on the surface of the dura and covered with a piece of sterile compressed sponge. The pup was transferred to a temperature and humidity-controlled enclosure and allowed to recover for 30 min, after which recording commenced. Signals were amplified, digitized continuously at 20 kHz using a head-stage directly attached to the NeuroGrid (RHD2000, Intan Technology), and stored for offline analysis with 16-bit format. Data were analyzed using MATLAB (MathWorks) and visualized using Neuroscope. At the completion of electrophysiological recording, pups were euthanized.
Neurophysiologic recordings were synchronized with the EMG signals to facilitate identification and elimination of any epochs with artifacts from subsequent analysis. Ictal patterns were identified using a line length algorithm and confirmed with visual screening of the raw data. Interictal epileptiform discharges were detected using previously employed frequency and duration features (Gelinas et al., 2016).
Cortical Neuron Culture Electrophysiology
Whole-cell recordings were performed with patch-clamp amplifiers (MultiClamp 700B; Molecular Devices) under the control of pClamp Clampex 10.3 or 10.5 software (Molecular Devices). Data were acquired at 20 kHz and low-pass filtered at 6 kHz. The series resistance was compensated at 70%, and only cells with series resistances maintained at less than 15 MU were analyzed. Patch electrodes were pulled from 1.5-mm o.d. thin-walled glass capillaries (Sutter Instruments, BF150-86-75) in five stages on a micropipette puller (model P-97; Sutter Instruments). Internal solution contained the following: 136 mM K-gluconate, 17.8 mM HEPES, 1 mM EGTA, 0.6 mM MgCl2, 4 mM ATP, 0.3 mM GTP, 12 mM creatine phosphate, and 50 U/ml phosphocreatine kinase. Alternatively, internal solution contained: 136 mm KCl, 17.8 mm HEPES, 1 mm EGTA, 0.6 mm MgCl2, 4 mm ATP, 0.3 mm GTP, 12 mm creatine phosphate, and 50 U/ml phosphocreatine kinase. The pipette resistance was between 2 and 4 MΩ. Standard extracellular solution contained the following (in mM): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 2 CaCl2 (pH 7.3, 305 mOsm). All experiments were performed at room temperature (22–23°C). Whole-cell recordings were performed on cortical neurons from control and mutant groups in parallel on the same day (day 13–16 in vitro). All experiments were performed by two independent investigators blinded to the genotypes. Electrophysiology data were analyzed offline with AxoGraph X software (AxoGraph Scientific).
For current-clamp experiments, the intrinsic electrophysiological properties of neurons were tested by injecting 500-ms square current pulses incrementing in 20 pA steps, starting at −100 pA. Resting membrane potential (Vm) was calculated from a 50 ms average before current injection. The membrane time constant (Tau, τ) was calculated from an exponential fit of current stimulus offset. Input resistance (RIn) was calculated from the steady state of the voltage responses to the hyperpolarizing current steps. Membrane capacitance (Cm) was calculated by dividing the time constant by the input resistance. Action potentials (APs) were evoked with 0.5 s, 20 pA depolarizing current steps. AP threshold was defined as the membrane potential at the inflection point of the rising phase of the AP. AP amplitude was defined as the difference in membrane potential between the AP peak and threshold, and the afterhyperpolarization was the difference between the AP threshold and the lowest Vm value within 50 ms. The AP half-width was defined as the width of the AP at half-maximal amplitude. The maximum depolarization and repolarization rates were determined by differentiating the AP waveform and finding the peak values. To obtain the neuron’s maximum firing frequency, depolarizing currents in 20-pA steps were injected until the number of APs per stimulus reached a plateau phase. Rheobase was defined as the minimum current required to evoke an AP during the 500 ms of sustained somatic current injections. AP half-width adaptation was determined by dividing the half-width of the last AP of a train at steady state frequency by the first. AP frequency adaptation was determined by dividing the mean interspike interval of the last four APs by the minimum. The membrane potential values were not corrected for the liquid junction potential. GABAergic neurons were classified as fast spiking (FS) if their maximum mean firing rate reached above 60 Hz and their AP half-widths increased by less than 25% during the AP train (Casale et al., 2015; Avermann et al., 2012). All others were considered non-fast spiking (NFS).
For voltage-clamp experiments to measure synaptic currents, two neurons were patched simultaneously and held at −70 mV, except for evoked IPSC measurements, for which postsynaptic neurons were held at 0 mV. PSCs were triggered by a 2 ms somatic depolarization to 0 mV. The shape of the evoked response, the reversal potential and the effect of receptor antagonists [10 μM NBQX (Tocris, 1044) or 20 μM bicuculline (BIC, Hello Bio, HB0893)] were analyzed to verify the glutamatergic or GABAergic identities of the currents. Neurons were stimulated at 0.1 Hz in standard external solution to measure basal-evoked synaptic responses. Spontaneous synaptic potentials were recorded in control solution with either NBQX or bicuculline to isolate EPSCs or IPSCs, respectively. Data were filtered at 1 kHz and analyzed using template-based miniature event detection algorithms implemented in the AxoGraph X. The threshold for detection was set at three times the baseline SD from a template of 0.5 ms rise time and 3 ms decay. The E/I ratio was calculated as the product of the sEPSC frequency and charge over the sum of the sEPSC frequency and charge and the product of the sIPSC frequency and charge.
KNa Current Measurements
For voltage-clamp experiments to measure the sodium-activated K+ current, neurons were held at −70 mV and given 1 s voltage pulses in 10 mV steps over a range of −80 to +50 mV. Recordings were obtained for each cell in standard extracellular solution or extracellular solution containing 0.5 mM Tetrodotoxin (TTX, Abcam, ab120055). To minimize rundown, experiments were started only after test pulses produced stable delayed outward currents. After steps in standard solution, TTX was applied directly on the recorded neuron with a custom-built fast flow perfusion system capable of complete solution exchange in less than 1 s. After 30 −40 s in TTX, the step protocol was repeated. Current traces from the TTX solution were then subtracted from the current traces obtained from the standard solution to obtain the difference current. The difference current over the 100 ms at the end of the voltage pulse was considered the steady state KNa current. Conductance (G) was calculated by dividing the current at each step by the driving force for K+. Activation curves were created by normalizing the G values to the maximum for each cell. The resulting curves were fit with a Boltzmann sigmoid function with the bottom constrained to 0 to obtain the voltage at half max conductance (V50). Unconstrained fits excluding values at voltage steps below 0 mV to minimize the effect of the TTX-sensitive inward current showed the same significant left-shifts in FS and NFS Kcnt1m/m GABAergic neurons while glutamatergic neurons were not different. Slope was not significantly different for any tests.
Multi-electrode Arrays
One to seven days before isolation of cortical neurons, 48-well MEA plates (Axion Biosystems, M768-KAP-48) were coated with 50 μg/mL poly-D-lysine (Sigma-Aldrich, P0899-50MG) in borate buffer, then washed three times with PBS and stored in PBS at 4°C until use. Prior to use, PBS was aspirated, and plates were dried in a sterilized hood. Cortices were dissected from the brains of P0 C57BL/6NJ WT or Kcnt1m/m mice of either sex. Pups were decapitated, weighed, and genotyped. The entire cerebral cortex was rapidly dissected and cut into small pieces under sterile conditions in cold Hibernate A solution (GIBCO, A1247501). Cortices from two WT or Kcnt1m/m pups were pooled together. The dissected cortices were then enzymatically digested in 20 U mL−1 Papain plus DNase (Worthington Biochemical, LK003178 and LK003172) diluted in Hibernate A for 20 min at 37°C. Cells were pelleted by centrifugation at 300RCF for 5 min, and then the digestion was neutralized by aspirating off the supernatant and adding warm Hibernate A media. Cells were mechanically dissociated by trituration and counted using a hemocytometer with Trypan blue counterstain. Cells were pelleted by centrifugation at 300RCF for 5 min and resuspended at a density of 6,000 cells/ml in warm Neurobasal-A (GIBCO, 10888-022) + 1X B27 supplement (GIBCO, 17504-044) + 1X GlutaMax (GIBCO, 35050-061) + 1% HEPES (GIBCO, 15630-080) + 1% Penicillin/Streptomycin (GIBCO, 15140-122) + 1% FBS (GIBCO, 26140-079) + 5 ug/mL Laminin (Sigma-Aldrich, L2020). 50,000 cells were plated on a pre-coated 48-well MEA plate in a 40 μL drop. The day after plating (DIV1), 100% of the media was removed and replaced with warm Neurobasal-A + 1X B27 supplement + 1X GlutaMax + 1% HEPES + 1% Penicillin/Streptomycin (NBA/B27 medium). Glial growth was not chemically suppressed. Cultures were maintained at 37°C in 5% CO2. Media was 50% changed every other day with fresh, warm NBA/B27 starting on DIV3, after each recording session.
MEA recordings were conducted on media change days prior to media change starting on DIV5. Plates were equilibrated for 5 min then recorded for 15 min/day using an Axion Biosystems Maestro 768 channel amplifier (Axion Biosystems) at 37°C ina CO2 gas-controlled chamber with Axion Integrated Studios (AxIS) software v2.4 (Axion Biosystems). Each well of a 48-well plate is comprised of 16 electrodes on a 4 × 4 grid, with each electrode capturing activity of nearby neurons. A Butterworth band-pass filter (200-3000 Hz) and an adaptive threshold spike detector set at 7X the standard deviation of the noise was used during recordings. Raw data and spike list files were collected. Spike list files were used to extract additional spike, burst, and network features, using a custom MEA analysis software package for interpretation of neuronal activity patterns, meaRtools, based on rigorous permutation statistics that enables enhanced identification of over 70 activity features (Gelfman et al., 2018). Specifically, we analyzed spiking and bursting rates, burst duration, and the time between bursts (i.e., interburst interval, IBI), as well as synchronicity of the network. We determined the parameters for detecting neuronal bursts and network events based on published reports and experimentation (Mack et al., 2014; McConnell et al., 2012). Activity data were inspected to remove inactive electrodes and wells. For an electrode to be considered active, we required that at least five spikes/min were recorded. Wells in which fewer than 4 electrodes were active for > 50% of the days of recording were considered inactive and removed from analyses. For synchronous network events, at least 5 electrodes (> 25% of the total in a well) were required to participate in a network event for the network event to qualify as a network spike or burst. Events with fewer participating electrodes were filtered. Bursts were detected using the maximum interval burst detection algorithm (NeuroExplorer software, Nex Technologies) implemented in the meaRtools package. We required that a burst consist of at least 5 spikes and last at least 0.05 s, and that the maximum duration between two spikes within a burst be 0.1 s at the beginning of a burst and 0.25 s at the end of a burst. Adjacent bursts were further merged if the duration between them was less than 0.8 s. These parameters were chosen based on literature and on in-house experimentation (Mack et al., 2014). To analyze data over time, we performed permuted Mann-Whitney U tests. The values for each well for the chosen DIVs were combined and a Mann-Whitney U (MWU) test was performed. The labels for each well (WT versus Kcnt1m/m) were then shuffled and permuted 1,000 times to create 1,000 datasets that were tested for significance using an MWU test. We report the permuted p values as the rank of the true p value within the distribution of permuted p values. We also report the combined p value of the plates, calculated using an R script developed in-house.
Slice Electrophysiology
P20-29 male and female mice were deeply anesthetized with isoflurane and decapitated. The brain was removed and 350 μm coronal slices of frontal cerebral cortex were cut in ice-cold cutting solution (126 mM NaCl, 25 mM NaHCO3, 10 mM d-glucose, 3.5 mM KCl, 1.5 mM NaH2PO4, 0.5 mM CaCl2, 10.0 mM MgCl2) with a Leica VT1000S. Slices were then transferred to a storage chamber with fresh artificial cerebrospinal fluid (aCSF) containing 126 mM NaCl, 3.5 mM KCl, 1.0 mM MgCl2, 2.0 mM CaCl2, 1.5 mM NaH2PO4, 25 mM NaHCO3, and 10 mM d-glucose, pH 7.3–7.4, and were incubated at 37°C for 30 min. The slices were then incubated at room temperature for at least another 30 min before recording. All solutions were continuously bubbled with 95% O2 and 5% CO2.
Whole-cell current-clamp and recordings were obtained from cortical layer 2/3 pyramidal cells and interneurons at 32°C using a Multiclamp 700B and Clampex 10.5 software (Molecular Devices). Individual slices were transferred to a recording chamber located on an upright microscope (BX51; Olympus) and were perfused with oxygenated aCSF (2 mL/min). Neurons were visualized using IR-differential interference contrast microscopy. Layer 2/3 pyramidal cells were distinguished from interneurons by their triangular morphology, large soma, and pronounced apical dendrite. NFS GABAergic cells were distinguished from pyramidal neurons by their typical ovoid-shaped cell bodies and were finally confirmed by electrophysiological recordings as displaying shorter AP half-widths and higher peak AP frequency (Avermann et al., 2012). Intracellular solution contained (in mM): 136 mM K-gluconate, 17.8 mM HEPES, 1 mM EGTA, 0.6 mM MgCl2, 4 mM ATP, 0.3 mM GTP, 12 mM creatine phosphate, and 50 U/ml phosphocreatine kinase, pH 7.2. When patch electrodes were filled with intracellular solution, their resistance ranged from 4–6 MΩ. Access resistance was monitored continuously for each cell. The protocols and determination of electrophysiological parameters were conducted as described above for the culture recordings. All experiments were performed and analyzed by an investigator blinded to the animal genotypes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism 7 (GraphPad Prism, RRID:SCR_002798) was used to perform statistical tests on adult behavioral tests and to create all graphs. Multiple group comparisons were done using two-tailed t test with correction using the Holm-Sidak method, Mann-Whitney U test, or two-way repeated-measures ANOVA with correction using the Sidak’s test, as indicated. Statistics for MEA data and seizure frequency were performed in R using a Mann-Whitney U test with 1,000 permutations.
To test for statistical significance for all whole cell electrophysiology experiments, we used generalized linear mixed models (GLMM) in SPSS (26.0 Chicago, III (IBM, RRID:SCR_002865), which allows for within-subject correlations and the specification of the most appropriate distribution for the data. Because neurons and animals from the same culture or animal are not independent measurements, culture or litter was used as the subject variable, and animals and neurons were considered within-subject measurements. All data distributions were assessed with the Shapiro-Wilk test. Datasets that were significantly different from the normal distribution (p < 0.05) were fit with models using the gamma distribution and a log link function, except for synaptic connection probability, which was fit with the binomial distribution and probit link. Normal datasets were fit with models using a linear distribution and identity link. We used the model-based estimator for the covariance matrix and goodness of fit was determined using the corrected quasi likelihood under independence model criterion and by the visual assessment of residuals. All values reported in the text, figures, and tables are estimated marginal means ± standard error.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse monoclonal anti-KCNT1 (S3-26) | Abcam | Cat# ab94578; RRID:AB_10674494 |
| goat anti-mouse IgG polyclonal, HRP-conjugated | Santa Cruz Biotechnology | Cat# sc-2005; RRID:AB_631736 |
| mouse monoclonal anti-β-Actin (C4) | Santa Cruz Biotechnology | Cat# sc-47778 HRP; RRID:AB_2714189 |
| Bacterial and Virus Strains | ||
| AAV-CaMKIIa-GFP, serotype 8 | UNC Gene Therapy Center – Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NlaIII | NEB | Cat# R0125S |
| 0.5 % Trypsin-EDTA | GIBCO | Cat# 25300-054 |
| DMEM media supplemented with Glutamine | GIBCO | Cat# 10569-010 |
| Fetal bovine serum | GE Healthcare | Cat# SH3008803 |
| MITO+ Serum Extender | Corning | Cat# 355006 |
| Penicillin/Streptomycin | GIBCO | Cat# 15140-122 |
| Collagen I | Corning | Cat# 354236 |
| Poly-D-lysine | Sigma-Aldrich | Cat# P6407 |
| Papain | Worthington Biochemical | Cat# LS003126 |
| Trypsin inhibitor, Ovomucoid | Worthington Biochemical | Cat# LS003085 |
| Neurobasal-A medium | GIBCO | Cat# 10888-022 |
| GlutaMAX | GIBCO | Cat# 35050-061 |
| B27 supplement | GIBCO | Cat# 17504-044 |
| Tribromoethanol | Sigma-Aldrich | Cat# T48402 |
| NBQX disodium salt | Tocris | Cat# 1044 |
| Bicuculine Methiodide | Hello Bio | Cat# HB0893 |
| Tetrodotoxin Citrate | Abcam | Cat# ab120055 |
| Poly-D-lysine | Sigma-Aldrich | Cat# P0899-50MG |
| Hibernate A solution | GIBCO | Cat# A1247501 |
| Papain-for MEA protocol | Worthington Biochemical | Cat# LK003178 |
| DNase I | Worthington Biochemical | Cat# LK003172 |
| HEPES | GIBCO | Cat# 15630-080 |
| Fetal bovine serum-for MEA protocol | GIBCO | Cat# 26140-079 |
| Laminin | Sigma-Aldrich | Cat# L2020 |
| Critical Commercial Assays | ||
| My Taq | Bioline | Cat# BIO-25041 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| NeuroGrid | Intan Technologies | Cat# RHD2000 |
| Classic MEA 48 | Axion Biosystems | Cat# M768-KAP-48 |
| Experimental Models: Cell Lines | ||
| Primary astrocyte feeder layer | This paper | N/A |
| Primary cortical neurons | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Kcnt1em1(Y777H)Frk | This paper | N/A |
| Mouse: C57BL/6NJ | The Jackson Laboratory | Cat# JAX:005304; RRID:IMSR_JAX:005304 |
| Mouse: FVB.129P2-Pde6b+ Tyrc-ch/AntJ | The Jackson Laboratory | Cat# JAX:004828; RRID:IMSR_JAX:004828 |
| Mouse: B6.Cg-Snap25tm3.1Hze/J | The Jackson Laboratory | Cat# JAX:025111; RRID:IMSR_JAX:025111 |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# JAX:000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| Primer: Kcnt1 Forward: 5′-CTAGGGCTGCAAACACAACA-3′ | This paper | N/A |
| Primer: Kcnt1 Reverse: 5′-TCAAGCAGCAACACGATAGG-3′ | This paper | N/A |
| Software and Algorithms | ||
| NIS Elements Software | Nikon Instruments | RRID:SCR_014329 |
| Med-PC V Software Suite, 64-bit | Med Associates | Cat# SOF-736 |
| SR-LAB Startle Response System Software | San Diego Instruments | N/A |
| Video Freeze Software | Med Associates | Cat# SOF-843; RRID:SCR_014574 |
| Profusion EEG 5 Software | Compumedics | N/A |
| SVD code | University College London – Cortex Lab | https://github.com/cortex-lab/widefield |
| MATLAB | MathWorks | RRID:SCR_001622 |
| pCLAMP Clampex Software (10.3 or 10.5) | Molecular Devices | RRID:SCR_011323 |
| Axograph X Software | Axograph Scientific | RRID:SCR_014284 |
| AxIS Software v2.4 | Axion Biosystems | RRID:SCR_016308 |
| R package - meaRtools | Gelfman et al., 2018 | https://cran.r-project.org/web/packages/meaRtools/ |
| NeuroExplorer Software | Nex Technologies | RRID:SCR_001818 |
| Prism 7 | GraphPad | RRID:SCR_002798 |
| SPSS | IBM | RRID:SCR_002865 |
Highlights.
In humans, KCNT1 GOF variants cause severe epilepsy and intellectual disability
KCNT1 GOF increases subthreshold KNa currents selectively in GABAergic neurons
KCNT1 GOF impairs GABAergic neuron excitability and alters synaptic connectivity
Mice expressing a Kcnt1 GOF variant exhibit seizures and cognitive impairments
ACKNOWLEDGMENTS
This work was supported by NIH/NINDS grants NS087095 (M.C.W.), NS110945 (M.C.W.), NS031348 (W.N.F.), and OD020351 (The Jackson Laboratory Center for Precision Genetics). This project also received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sk1odowska-Curie grant agreement No 799501 (S.D.). The authors would like to acknowledge the support of the Microscopy Imaging Center and the Cellular & Molecular Core at the University of Vermont, especially Dr. Todd Clason, and Dr. Rod Scott for advice on statistical modeling.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108303.
DECLARATION OF INTERESTS
S.C. serves a consultant for Q-State Biosciences. D.B.G. is a founder of and holds equity in Praxis, serves as a consultant to AstraZeneca, and has received research support from Janssen, Gilead, Biogen, AstraZeneca, and UCB. M.J.B. has received research support from Janssen. The remaining authors declare no competing interests.
REFERENCES
- Anderson LL, Thompson CH, Hawkins NA, Nath RD, Petersohn AA, Rajamani S, Bush WS, Frankel WN, Vanoye CG, Kearney JA, and George AL Jr. (2014). Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 55, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asinof S, Mahaffey C, Beyer B, Frankel WN, and Boumil R (2016). Dynamin 1 isoform roles in a mouse model of severe childhood epileptic encephalopathy. Neurobiol. Dis 95, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avermann M, Tomm C, Mateo C, Gerstner W, and Petersen CC (2012). Microcircuits of excitatory and inhibitory neurons in layer 2/3 of mouse barrel cortex. J. Neurophysiol 107, 3116–3134. [DOI] [PubMed] [Google Scholar]
- Baker EM, Thompson CH, Hawkins NA, Wagnon JL, Wengert ER, Patel MK, George AL Jr., Meisler MH, and Kearney JA (2018). The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 59, 1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet 44, 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrows CM, McCabe MP, Chen H, Swann JW, and Weston MC (2017). PTEN loss increases the connectivity of fast synaptic motifs and functional connectivity in a developing hippocampal network. J. Neurosci 37, 8595–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, and Goldberg EM (2014). Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol 76, 457–461. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, and Kaczmarek LK (2002). Localization of the Slack potassium channel in the rat central nervous system. J. Comp. Neurol 454, 241–254. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, von Hehn CA, Mei X, and Kaczmarek LK (2005). Localization of the Na+-activated K+ channel Slick in the rat central nervous system. J. Comp. Neurol 484, 80–92. [DOI] [PubMed] [Google Scholar]
- Budelli G, Hage TA, Wei A, Rojas P, Jong YJ, O’Malley K, and Salkoff L (2009). Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat. Neurosci 12, 745–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale AE, Foust AJ, Bal T, and McCormick DA (2015). Cortical interneuron subtypes vary in their axonal action potential properties. J. Neurosci 35, 15555–15567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes B, Vega R, Limón A, and Soto E (2013). Identity, expression and functional role of the sodium-activated potassium current in vestibular ganglion afferent neurons. Neuroscience 240, 163–175. [DOI] [PubMed] [Google Scholar]
- Chen H, Kronengold J, Yan Y, Gazula VR, Brown MR, Ma L, Ferreira G, Yang Y, Bhattacharjee A, Sigworth FJ, et al. (2009). The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J. Neurosci 29, 5654–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong PF, Nakamura R, Saitsu H, Matsumoto N, and Kira R (2016). Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann. Neurol 79, 502–503. [DOI] [PubMed] [Google Scholar]
- De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, Losi G, Mantegazza M, Panzeri S, Carmignoto G, et al. (2016). Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cereb. Cortex 26, 1778–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM (2006). Assessing nest building in mice. Nat. Protoc 1, 1117–1119. [DOI] [PubMed] [Google Scholar]
- Dilena R, DiFrancesco JC, Soldovieri MV, Giacobbe A, Ambrosino P, Mosca I, Galli MA, Guez S, Fumagalli M, Miceli F, et al. (2018). Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 15, 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Lüders HO, Shi J, Cui J, et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet 37, 733–738. [DOI] [PubMed] [Google Scholar]
- Farrell JS, Nguyen QA, and Soltesz I (2019). Resolving the micro-macro disconnect to address core features of seizure networks. Neuron 101, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenoglio-Simeone KA, Wilke JC, Milligan HL, Allen CN, Rho JM, and Maganti RK (2009). Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia 50, 2027–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald MP, Fiannacca M, Smith DM, Gertler TS, Gunning B, Syrbe S, Verbeek N, Stamberger H, Weckhuysen S, Ceulemans B, et al. (2019). Treatment responsiveness in KCNT1-related epilepsy. Neurotherapeutics 16, 848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti S, Lavazza T, Curia G, Aracri P, Panzica F, Sancini G, Avanzini G, and Magistretti J (2003). Na+-activated K+ current contributes to postexcitatory hyperpolarization in neocortical intrinsically bursting neurons. J. Neurophysiol 89, 2101–2111. [DOI] [PubMed] [Google Scholar]
- Gelfman S, Wang Q, Lu YF, Hall D, Bostick CD, Dhindsa R, Halvorsen M, McSweeney KM, Cotterill E, Edinburgh T, et al. (2018). meaRtools: An R package for the analysis of neuronal networks recorded on microelectrode arrays. PLoS Comput. Biol 14, e1006506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Khodagholy D, Thesen T, Devinsky O, and Buzsáki G (2016). Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med 22, 641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler T, Bearden D, Bhattacharjee A, and Carvill G (2018). KCNT1-related epilepsy In GeneReviews, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, and Amemiya A, eds. (University of Washington; ). [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, and Noebels JL (2010). Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J. Neurosci 30, 5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage TA, and Salkoff L (2012). Sodium-activated potassium channels are functionally coupled to persistent sodium currents. J. Neurosci 32, 2714–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, et al. (2012). Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet 44, 1188–1190. [DOI] [PubMed] [Google Scholar]
- Jirkof P (2014). Burrowing and nest building behavior as indicators of well-being in mice. J. Neurosci. Methods 234, 139–146. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, and Kaczmarek LK (1998). Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat. Neurosci 1, 462–469. [DOI] [PubMed] [Google Scholar]
- Kim GE, and Kaczmarek LK (2014). Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell. Neurosci 8, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, and McCormick DA (1998). Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J. Neurophysiol 80, 1222–1235. [DOI] [PubMed] [Google Scholar]
- Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, and Kaczmarek LK (2014). Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Zhang Y, Lecoq J, Jung JC, Li J, Zeng H, Niell CM, and Schnitzer MJ (2016). Long-term optical access to an estimated one million neurons in the live mouse cortex. Cell Rep 17, 3385–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, and Boulter J (2006). Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc. Natl. Acad. Sci. USA 103, 19152–19157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, and Wolfart J (2016). Potassium channels in epilepsy. Cold Spring Harb. Perspect. Med 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hjerling-Leffler J, Zagha E, Fishell G, and Rudy B (2010). The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J. Neurosci 30, 16796–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lin MC, Kornblum HI, Papazian DM, and Nelson SF (2014). Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum. Mol. Genet 23, 3481–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, and Stan Leung L (2004). Sodium-activated potassium conductance participates in the depolarizing afterpotential following a single action potential in rat hippocampal CA1 pyramidal cells. Brain Res 1023, 185–192. [DOI] [PubMed] [Google Scholar]
- Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, Goriounova NA, Buhler E, Watrin F, Shityakov S, et al. (2014). Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat. Commun 5, 4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Bausch AE, Kallenborn-Gerhardt W, Stoetzer C, Debruin N, Ruth P, Geisslinger G, Leffler A, Lukowski R, and Schmidtko A (2015). Slack channels expressed in sensory neurons control neuropathic pain in mice. J. Neurosci 35, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack CM, Lin BJ, Turner JD, Johnstone AF, Burgoon LD, and Shafer TJ (2014). Burst and principal components analyses of MEA data for 16 chemicals describe at least three effects classes. Neurotoxicology 40, 75–85. [DOI] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, Ailamaki A, Alonso-Nanclares L, Antille N, Arsever S, et al. (2015). Reconstruction and simulation of neocortical microcircuitry. Cell 163, 456–492. [DOI] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Wu J, Yang C, Gonzalez-Perez V, Zhou H, Liang H, Xia XM, and Lingle CJ (2015). Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. eLife 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell ER, McClain MA, Ross J, Lefew WR, and Shafer TJ (2012). Evaluation of multi-well microelectrode arrays for neurotoxicity screening using a chemical training set. Neurotoxicology 33, 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, Nair U, Malhotra S, Meyer E, Trump N, Gazina EV, Papandreou A, Ngoh A, Ackermann S, Ambegaonkar G, et al. (2018). Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 90, e55–e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, and Taglialatela M (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci 35, 3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, Milligan CJ, Li M, Grefe A, McConkie A, et al. (2015). Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol 78, 995–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichap JJ, Miceli F, De Maria M, Keator C, Joshi N, Tran B, Soldovieri MV, Ambrosino P, Shashi V, Mikati MA, et al. (2017). Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 58, e10–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, Reid CA, Venkat A, Younkin DP, Dlugos DJ, et al. (2014). KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol 75, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, van Kempen MJ, Klinkenberg S, Andrews I, Kelley K, et al. (2015). Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56, e114–e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen SA, Carney PW, Roten A, Ching M, Lightfoot PA, Churilov L, Nair U, Li M, Berkovic SF, Petrou S, and Scheffer IE (2018). Precision therapy for epilepsy due to KCNT1 mutations: A randomized trial of oral quinidine. Neurology 90, e67–e72. [DOI] [PubMed] [Google Scholar]
- Musall S, Kaufman MT, Juavinett AL, Gluf S, and Churchland AK (2019). Single-trial neural dynamics are dominated by richly varied movements. Nat. Neurosci 22, 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, and Tzingounis AV (2018). Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist 24, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels J (2017). Precision physiology and rescue of brain ion channel disorders. J. Gen. Physiol 149, 533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, Nabatame S, Azuma J, Fujii Y, Hara M, et al. (2015). De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 56, e121–e128. [DOI] [PubMed] [Google Scholar]
- Okaty BW, Miller MN, Sugino K, Hempel CM, and Nelson SB (2009). Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J. Neurosci 29, 7040–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, and Reid CA (2018). Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol. Rev 70, 142–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Sutherland GR, and Mulley JC (1995). Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2. Nat. Genet 10, 117–118. [DOI] [PubMed] [Google Scholar]
- Quraishi IH, Stern S, Mangan KP, Zhang Y, Ali SR, Mercier MR, Marchetto MC, McLachlan MJ, Jones EM, Gage FH, and Kaczmarek LK (2019). An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing Slack KNa currents. J. Neurosci 39, 7438–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quraishi IH, Mercier MR, McClure H, Couture RL, Schwartz ML, Lukowski R, Ruth P, and Kaczmarek LK (2020). Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (KNa1.1) Na+-activated K+ channels. Sci. Rep 10, 3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijntjes DOJ, Lee JH, Park S, Schubert NMA, van Tuinen M, Vijayakumar S, Jones TA, Jones SM, Gratton MA, Xia XM, et al. (2019). Sodium-activated potassium channels shape peripheral auditory function and activity of the primary auditory neurons in mice. Sci. Rep 9, 2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi S, Knaus HG, and Schwarzer C (2016). Differential distribution of the sodium-activated potassium channels Slick and Slack in mouse brain. J. Comp. Neurol 524, 2093–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi LF, Wykes RC, Kullmann DM, and Carandini M (2017). Focal cortical seizures start as standing waves and propagate respecting homotopic connectivity. Nat. Commun. 8, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree HM, Simeone TA, Johnson C, Matthews SA, Samson KK, and Simeone KA (2016). Orexin receptor antagonism improves sleep and reduces seizures in Kcna1-null mice. Sleep (Basel) 39, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, and Catterall WA (2015). Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, and Crill WE (1989). Long-lasting reduction of excitability by a sodium-dependent potassium current in cat neocortical neurons. J. Neurophysiol 61, 233–244. [DOI] [PubMed] [Google Scholar]
- Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, Ru K, Baillie GJ, Alanay Y, Jacquinet A, et al. (2015). Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet 47, 73–77. [DOI] [PubMed] [Google Scholar]
- Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, Noebels JL, White HS, Wilcox KS, and Leppert MF (2008). Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J. Physiol 586, 3405–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, and Tempel BL (1998). Deletion of the K(V) 1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. [DOI] [PubMed] [Google Scholar]
- Steinmetz NA, Buetfering C, Lecoq J, Lee CR, Peters AJ, Jacobs EAK, Coen P, Ollerenshaw DR, Valley MT, de Vries SEJ, et al. (2017). Aberrant cortical activity in multiple GCaMP6-expressing transgenic mouse lines. eNeuro 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutor B, and Zolles G (2001). Neuronal nicotinic acetylcholine receptors and autosomal dominant nocturnal frontal lobe epilepsy: a critical review. Pflugers Arch 442, 642–651. [DOI] [PubMed] [Google Scholar]
- Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS, et al. ; EuroEPINOMICS RES consortium (2015). De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet 47, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QY, Zhang FF, Xu J, Wang R, Chen J, Logothetis DE, and Zhang Z (2016). Epilepsy-related Slack channel mutants lead to channel over-activity by two different mechanisms. Cell Rep 14, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, Murthy P, Muldoon SF, and Goldberg EM (2020). Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J. Neurosci 40, 2764–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa C, and Combi R (2016). Potassium channels and human epileptic phenotypes: an updated overview. Front. Cell. Neurosci 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallén P, Robertson B, Cangiano L, Löw P, Bhattacharjee A, Kaczmarek LK, and Grillner S (2007). Sodium-dependent potassium channels of a Slack-like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. J. Physiol 585, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Quanxin (2020). Cell 10.1016/j.cell.2020.04.007. [DOI] [Google Scholar]
- Wekselblatt JB, Flister ED, Piscopo DM, and Niell CM (2016). Large-scale imaging of cortical dynamics during sensory perception and behavior. J. Neurophysiol 115, 2852–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Desai R, and Kaczmarek LK (2007). Slack and Slick K(Na) channels regulate the accuracy of timing of auditory neurons. J. Neurosci 27, 2617–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wöhr M, Roullet FI, Katz AM, Abrams DN, Kalikhman D, Simon H, Woldeyohannes L, et al. (2012). Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci 32, 6525–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vasylyev DV, Dib-Hajj F, Veeramah KR, Hammer MF, Dib-Hajj SD, and Waxman SG (2013). Multistate structural modeling and voltage-clamp analysis of epilepsy/autism mutation Kv10.2-R327H demonstrate the role of this residue in stabilizing the channel closed state. J. Neurosci 33, 16586–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.