

Abstract
Purpose:
In patients with >1 non-small cell lung carcinoma (NSCLC), the distinction between separate primary lung carcinomas (SPLCs) and intrapulmonary metastases (IPMs) is a common diagnostic dilemma with critical staging implications. Here, we compared the performance of comprehensive next-generation sequencing (NGS) with standard histopathologic approaches for distinguishing NSCLC clonal relationships in clinical practice.
Experimental Design:
We queried 4,119 NSCLCs analyzed by 341–468 gene MSK-IMPACT NGS assay for patients with >1 surgically resected tumor profiled by NGS. Tumor relatedness predicted by prospective histopathologic assessment was contrasted with comparative genomic profiling by subsequent NGS.
Results:
Sixty patients with NGS performed on >1 NSCLCs were identified, yielding 76 tumor pairs. NGS classified tumor pairs into 51 definite SPLCs (median, 14; up to 72 unique somatic mutations per pair), and 25 IPMs (24 definite, one high probability; median, 5; up to 16 shared somatic mutations per pair). Prospective histologic prediction was discordant with NGS in 17 cases (22%), particularly in the prediction of IPMs (44% discordant). Retrospective review highlighted several histologic challenges, including morphologic progression in some IPMs. We subsampled MSK-IMPACT data to model the performance of less comprehensive assays, and identified several clinicopathologic differences between NGS-defined tumor pairs, including increased risk of subsequent recurrence for IPMs.
Conclusions:
Comprehensive NGS allows unambiguous delineation of clonal relationship among NSCLCs. In comparison, standard histopathologic approach is adequate in most cases, but has notable limitations in the recognition of IPMs. Our results support the adoption of broad panel NGS to supplement histology for robust discrimination of NSCLC clonal relationships in clinical practice.
Introduction
Distinguishing between separate primary lung carcinomas (SPLCs) and intrapulmonary metastases (IPMs) is an increasingly common dilemma encountered in clinical practice. In the era of widespread utilization of CT imaging, it is estimated that screening detects more than one tumor nodule in 15%–20% of current/former smokers with non–small cell lung carcinomas (NSCLC; refs. 1, 2). In addition, NSCLC can spread to other parts of pulmonary parenchyma via IPMs—a process for which the underlying pathogenesis is still poorly understood. The distinction between SPLCs and IPMs has major implications for staging: SPLCs are staged individually, whereas IPMs are staged as pT3 if in the same lobe, pT4 if in another ipsilateral lobe, and pM1a if in a contralateral lobe (3). The method for the distinction of SPLCs and IPMs has evolved substantially over the years, beginning with the criteria proposed in 1975 by Martini and Melamed (4), which remained the primary method in clinical practice for many years. Those criteria defined SPLCs on the basis of different general tumor histotype (e.g., squamous cell carcinoma vs. adenocarcinoma) and other clinicopathologic characteristics (presence of carcinoma in situ, involvement of a different lung lobe after a >2-year interval, and lack of nodal or systemic metastases; ref. 4). However, recent studies with molecular data have highlighted the significant limitations of the Martini and Melamed criteria (5). It is now recognized that the distinction between SPLCs and IPMs is complex and requires multidisciplinary correlation of all available clinical, radiologic, pathologic, and molecular information (6–8), but the criteria are not well defined.
In recent years, adenocarcinoma has become the most prevalent lung cancer type in the United States and Asia, and it is the tumor type that frequently presents with multiple separate nodules. Thus, the issue of separating SPLCs from IPMs largely concerns the determination of the relatedness of adenocarcinomas. The hallmark of lung adenocarcinomas is their enormous histologic and molecular heterogeneity (9). The histologic variation manifests in a strikingly diverse combination of architectural and cytologic features in individual tumors, accompanied by the variability in stromal features and associated inflammatory milieu. In combination, such variability yields a distinctive histologic signature for individual tumors. On the basis of this principle, a landmark study has demonstrated the value of comprehensive histologic assessment as a tool to determine whether two tumors are SPLCs versus IPMs (5). This approach has been adopted by the latest 7th and 8th editions of American Joint Committee on Cancer tumor–node–metastasis (TNM) classification (3). However, the accuracy of histologic comparison has not been benchmarked against highly robust molecular approaches.
A variety of molecular methods have been applied over the years to distinguish SPLCs from IPMs. The early methods included microsatellite and loss of heterozygosity analysis (10–14), array comparative genomic hybridization (5, 15), and TP53 gene mutation status (16, 17). More recently, oligo-gene panels for hotspot mutations in two to five major driver genes (18–21) and next-generation sequencing (NGS) for up to 50 cancer genes (22–27) have been applied to the problem of separating SPLCs from IPMs. Although providing valuable information on clonal relationship of lung adenocarcinomas, the limitation of such methods is that they are uninformative in a substantial proportion of cases (see Discussion). Also, recently reported was the application of assays still mainly confined to research settings such as whole-exome sequencing (28, 29) and NGS-based genomic breakpoint analysis (30) as a means to delineate tumor clonal relationships. However, still lacking are large-scale studies using NGS assays with comprehensive coverage of cancer-related genes, which are currently in use at increasing number of institutions for guiding the use of targeted therapies. Large-panel NGS platforms provide an unprecedented opportunity to address the problem of SPLCs versus IPMs using a highly granular molecular gold standard. In this study, we compared the results of prospective histologic prediction with subsequent NGS results using the Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) NGS assay covering up to 468 cancer-related genes for distinguishing SPLCs from IPMs in our clinical practice. Using the clonal relationships established by large panel NGS as the molecular gold standard, we analyzed the accuracy and pitfalls of histologic predictions of SPLCs versus IPMs, and we began to elucidate clinicopathologic and genomic features that may aid in the prediction of NSCLC clonality in clinical practice.
Materials and Methods
Study design
We queried the data on NSCLC specimens from the MSK-IMPACT clinical sequencing cohort in the cBioPortal database (9, 31), and selected patients who underwent surgical resections for >1 NSCLC profiled by NGS. Patients on targeted therapies undergoing resection for known or suspected metastatic disease were excluded. Also excluded were patients with >1 invasive mucinous adenocarcinoma because those tumors commonly present with multifocal, multilobar disease, which will be addressed in a separate study. This study was approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center.
Clinical and histologic review
Clinical information, including patient demographics, smoking, treatment history, and survival outcomes were obtained from hospital electronic medical records. Prospective histologic predictions of SPLCs versus IPMs were obtained from surgical pathology reports, based on the statement of whether the tumors appeared morphologically similar or different—a routine practice at our institution. Histologic assessment of tumor relatedness was performed by experienced thoracic pathologists based on the histologic criteria proposed by Girard and colleagues (5). In all cases the histologic predictions were made prior to comprehensive molecular profiling by MSK-IMPACT. The histologic features were re-reviewed by two thoracic pathologists (J.C. Chang and N. Rekhtman).
Progression-free survival (PFS) was calculated using the Kaplan-Meier approach from the time of most recent procedure to the time of progression or death. Patients were otherwise censored at the time of last clinical follow-up. Survival curves between IPMs and SPLCs were compared using the log-rank tests. Analyses were conducted in R 3.5.3 (R Core Team).
Genomic profiling
The detailed methodology of the MSK-IMPACT assay has been described previously (32). In brief, the MSK-IMPACT assay is a hybridization capture-based NGS platform that sequences the entire coding region and select noncoding regions of 341 (v3), 410 (v4), or 468 (v5) genes and provides data on nonsynonymous somatic mutations, copy number alterations, and structural variants. Synonymous mutations are detected and maintained in database but not clinically reported. DNA extraction was performed using standard methods on formalin-fixed, paraffin-embedded (FFPE) tissue on tumor specimens with matched blood normal control on all cases.
To determine the clonal relationship between two tumors, we compared somatic mutations and structural variants but not copy number alterations, as the latter are heavily influenced by tumor purity (proportion of tumor cells relative to nonneoplastic cells such as inflammatory and stromal cells). Tumor pairs exhibiting entirely nonoverlapping, unique mutations were classified as clonally unrelated (SPLCs). In contrast, tumors sharing multiple (≥2) mutations were regarded as clonally related (IPMs). For tumors sharing a single hotspot mutation, the designation of IPMs versus SPLCs was adjudicated on an individual basis by extended molecular review (see Results). Visual review of the alignment data in integrated genomic viewer (33) was performed to confirm the absence (in SPLCs) or presence (in IPMs) of overlapping mutations below the bioinformatic threshold used for clinical variant calls [<5% variant allele frequency (VAF)]. Tumor pairs sharing translocations and splice-site mutations were manually reviewed to determine whether the breakpoints/splice sites occurred in identical locations.
To determine the probability for a set of observed shared mutations to occur coincidentally, we collected the data on the prevalence of each mutation in our patient population within NSCLCs profiled by MSK-IMPACT (n = 4,119), and manually calculated the odds of their cooccurrence by chance. In addition, all tumors were analyzed by a biostatistical method developed for NGS-based clonality assessment. In brief, for each tumor pair, the number of shared mutations versus unique mutations and prevalence of each mutation in NSCLC were used to calculate the probability of clonality using statistical formulas, where 0 means definitely nonclonal (SPLCs) and 1 means definitely clonal (IPMs). For detailed description, see Supplementary Materials and Methods 1 and prior publications (34, 35).
To compare the performance of MSK-IMPACT with other molecular platforms commonly used in clinical practice at other institutions, we modeled the results of clonality assessment by other molecular platforms by subsampling of the MSK-IMPACT data on the same cases (see Supplementary Materials and Methods 2 for details).
Results
Patient characteristics
Between 2014 and 2018, 4,119 NSCLC specimens underwent genomic profiling by MSK-IMPACT, from which we identified a total of 60 patients with NGS performed on >1 resected NSCLC. Fifty-two patients had two tumors, and eight patients had three tumors, for a total of 128 individual tumors. Considering that patients with three tumors each represented three tumor pairs, this yielded a total of 76 tumor pair comparisons. On clinical grounds, all patients were considered to have separate primary tumors or the relationship of the tumors was uncertain at the time of surgery; none of the patients were known to have IPMs prior to surgery.
The cohort comprised paired adenocarcinomas (AD vs. AD; n = 70), paired squamous cell carcinomas (SQ vs. SQ; n = 2), and pairs with different histotypes: adenocarcinoma versus pleomorphic carcinoma (n = 3), and adenocarcinoma versus large-cell neuroendocrine carcinoma (n = 1). Tumors were synchronous in 54 and metachronous in 22 tumor pairs.
Tumor relatedness based on prospective histologic prediction
Of 76 tumor pairs, prospective histologic comparison predicted that 20 tumor pairs (26%) were morphologically similar representing IPMs, whereas 56 tumor pairs (74%) were morphologically different representing SPLCs (Fig. 1C). Specifically, within AD-AD pairs (n = 70), 19 were predicted to represent IPMs and 51 SPLCs. In 23 cases, a histologic prediction was rendered, but the pathologist recommended molecular testing to confirm the clonal relationships between the tumors (Fig. 1D).
Figure 1.
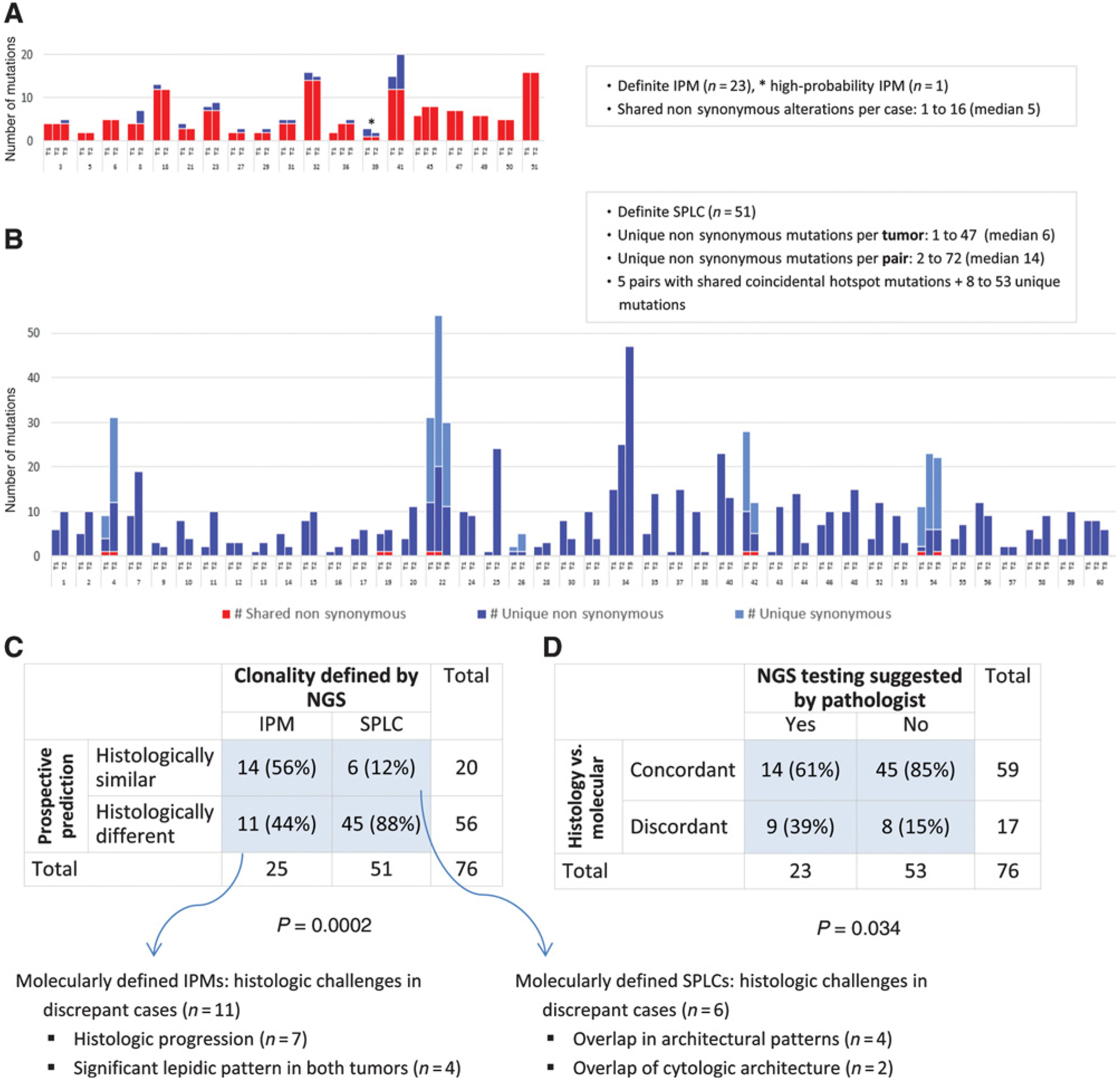
Overall distribution of molecular alterations and histologic/molecular correlation of tumor clonality typing. A, Shared and unique mutations in NGS-defined IPMs. B, Shared and unique mutations in NGS-defined SPLCs. C, Comparison of prospective histologic prediction with clonality defined by NGS. D, Comparison of histology-molecular concordance rate when NGS testing was suggested versus not suggested by the pathologist.
Clonality assessment based on MSK-IMPACT results
Genomic profiling by MSK-IMPACT of the 128 tumors yielded a median of eight somatic alterations per tumor (range, 1–47) with a mean coverage of 704× (range, 186×−1,132×; Supplemental Table S1). A major oncogenic driver alteration (e.g., EGFR, KRAS, ALK, ROS1, and MET exon 14) was identified in 107 tumors (84%). As shown in Figs. 1 and 2, NGS classified tumor pairs as 25 IPMs (24 definite and one high probability) and 51 definite SPLCs as detailed below.
Figure 2.
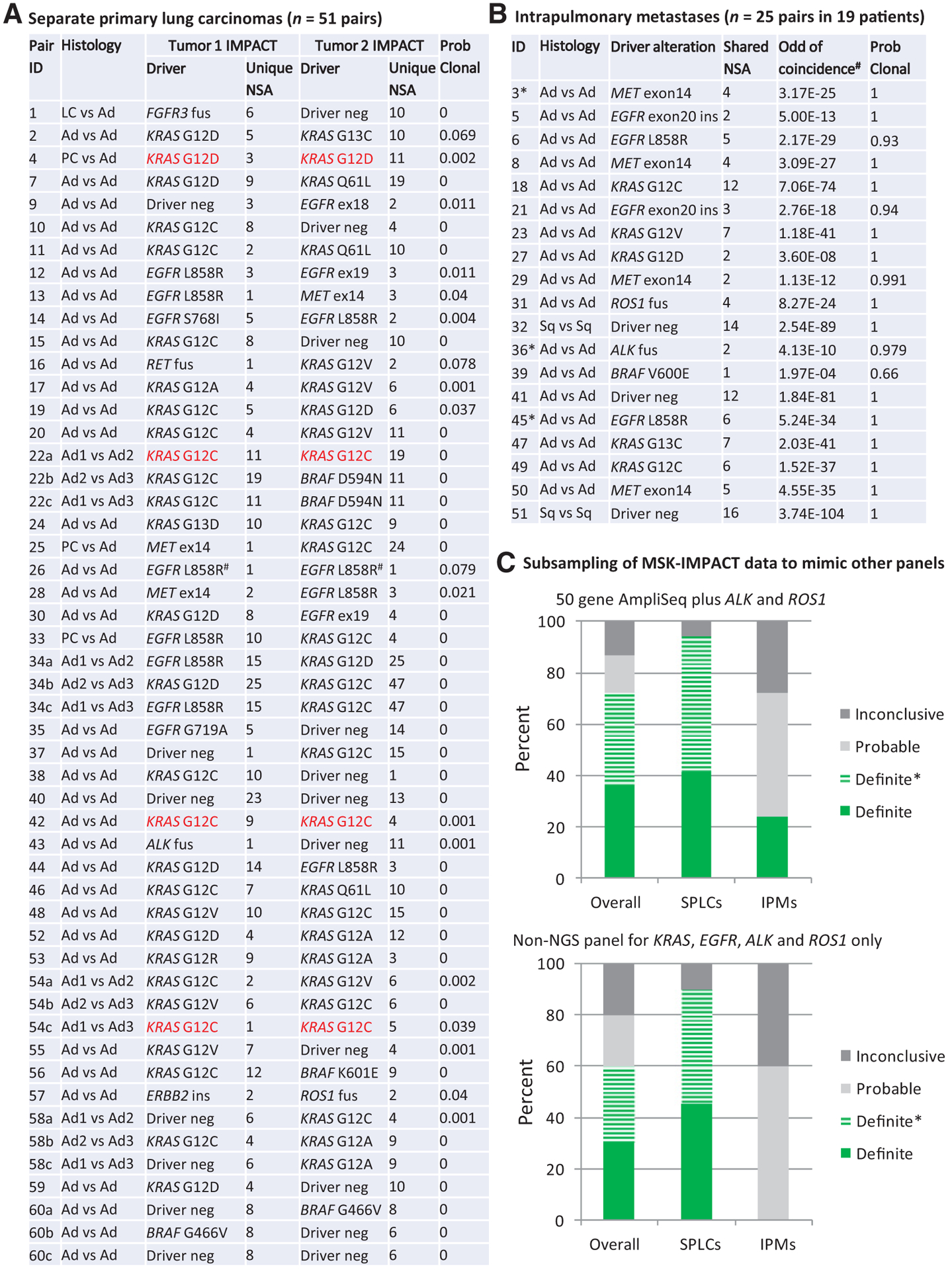
Distribution of driver alterations and subsampling to mimic more limited platforms. A, Driver alterations in 51 SPLC tumor pairs from 41 patients. Highlighted in red are SPLCs with coincidentally shared hotspot driver mutations that could be misclassified as favoring IPMs by other platforms. #, shared EGFR L858R in case 26 represents tumor pairs with different nucleotide changes in EGFR gene resulting in the same amino acid substitution. B, Driver alterations in 25 IPM tumor pairs from 19 patients.*, three IPM cases with asterisks (3, 36, and 45) denote patients with three tumors. C, Subsampling of MSK-IMPACT data to mimic performances of noncomprehensive molecular platforms. Definite SPLC tumor pairs showing different driver alterations in each tumor. Definite SPLC*, one tumor shows a driver alteration while the other lacks that alteration (See Supplementary Materials and Method 2). Definite IPM, ≥2 shared somatic alterations. Probable IPM, one shared somatic alteration. Inconclusive, neither tumor shows a driver alteration. Ad, adenocarcinoma; amp, amplification; fus, fusion; ins, insertion; LC, large-cell neuroendocrine carcinoma; NSA, nonsynonymous alterations; PC, pleomorphic carcinoma; Sq, squamous cell carcinoma.
A total of 24 tumor pairs (32%) shared multiple (≥2) nonsynonymous somatic alterations (mean, 6.3; up to 16). In such pairs, the odds of coincidence for all shared alterations was <1E-06 in all cases (median, 2.17E-29; range, 3.60E-08–3.74E-104) based on the prevalence of individual mutations in our patient population (Fig. 2B). In addition, tumors sharing translocations (ALK and ROS1) and splice-site mutations in MET exon 14 were confirmed to have identical breakpoints/splice sites. These pairs were thus classified as definite IPMs.
Compared with the number of shared mutations, the number of unique mutations in IPMs was substantially lower (mean 0.7; range, 0–11). There were no unique mutations in 10 IPMs, one to two unique mutations per tumor in 12 IPMs, and >2 unique mutations per tumor in only two IPMs. On average, unique mutations in IPMs represented only 9% of total mutations per tumor (range, 0%–42%).
Conversely, 46 other tumor pairs (61%) exhibited entirely unique mutation profiles in each tumor. These pairs harbored a median of 14 unique nonsynonymous somatic mutations per pair (range, 2–72). On manual review, no overlapping mutations were detected in the paired tumors even at subthreshold level (VAF < 5%). These pairs were classified as definite SPLCs.
A total of six cases shared a single hotspot mutation; their classification was adjudicated individually by extended molecular review that included review of synonymous mutations and chromosome arm level gains and losses (Supplementary Table S2). Of those, four tumor pairs shared a single KRAS hotspot mutation (p.G12C in three and p.G12D in one); each tumor also harbored an abundance of unique mutations (both nonsynonymous and synonymous), ranging from eight to 53 mutations per tumor, with no shared mutations even at subthreshold level, and no shared chromosome arm level gains or losses. Classification of those tumors as SPLCs were supported by (i) fair probability of coincidentally shared driver; in particular, given the prevalence of KRAS p.G12C mutation in our population of approximately 15% (~24% in smokers), the odds of coincidental occurrence of this mutation in two unrelated tumors is approximately 1 in 44 (~1/17 in smokers; ref. 36) and (ii) substantially higher unique/total mutation ratio (>90%) compared with definite IPMs in our series. All 4 patients were indeed smokers with a 15–70 pack-year smoking history.
One other tumor pair shared a single U2AF1 p.S34F hotspot mutation. These tumors also harbored distinct KRAS-driver muta tions (p.G12C vs. p.G12D) plus multiple unique nonsynonymous and synonymous mutations in each tumor, resulting in an unambiguous classification of SPLCs with coincidental U2AF1 hotspot mutation.
Finally, one tumor pair shared a single BRAF p.V600E mutation with only one and two unique mutations per tumor. On manual review, all mutations had low VAF (<15%), and no synonymous mutations were detected in either tumor. Such findings indicate low tumor purity and high likelihood of incomplete detection of mutations. Low tumor purity was in-line with marked admixed inflammatory infiltrate in corresponding hematoxylin and eosin sections. On the basis of the low prevalence of BRAF p.V600E in NSCLC (1.4%), the odds of coincidental occurrence of this mutation in two independent tumors is very low (1.97E-04); thus, the tumor was classified as high-probability IPM.
Overall, of 76 tumor pairs, five (6%) had no driver alteration in either tumor, and 14 SPLCs (18%) had a driver in only one of the tumors. In the entire cohort, nine tumor pairs shared KRAS mutations, of which four (44%) represented coincidental cooccurrence in SPLCs on the basis of the aforementioned analysis.
Biostatistical analysis was fully concordant with the above manual classification of tumors as SPLCs versus IPMs, revealing probability of clonality of 0–0.079 for SPLCs, and 0.93–1 for IPMs (Fig. 2). In particular, this method supported the manual classification of pairs with a single shared alteration (Supplementary Table S2).
Modeling of clonality assessment by less comprehensive molecular platforms by subsampling of MSK-IMPACT data
Because many institutions do not currently have access to comprehensive NGS panels, we modeled how smaller NGS panels and standalone assays for major NSCLC drivers would be predicted to perform with the cases in this study by subsampling the MSK-IMPACT data to mimic more limited panels, which are widely used in clinical practice.
As summarized in Fig. 2C and Supplementary Materials and Methods 2, subsampling for four major drivers (EGFR, KRAS, ALK, and ROS1) would distinguish SPLCs versus IPMs in 60% of cases, and subsampling for 50-gene AmpliSeq plus ALK and ROS1 would distinguish 72% of cases. Notably, because >90% of SPLCs were characterized by distinct drivers or presence versus absence of driver in paired tumors, most SPLCs would be readily identified by noncomprehensive panels. Conversely, noncomprehensive panels would have a limited ability to definitively confirm that tumors are clonally related (i.e., IPMs) because shared single-hotspot alteration can occur in unrelated tumors (see Discussion and Supplementary Materials and Methods 2 for details), and detection of secondary mutations would be feasible only by AmpliSeq in a minority cases. To model the number of genes needed to achieve high probability of confirming clonal relatedness of NSCLC pairs, we utilized a computational approach (Supplementary Materials and Methods 3), which estimated that at least 100 commonly mutated genes would be needed to allow confirmation of clonal relatedness in >95% of lung adenocarcinomas (Supplementary Table S3).
Comparison of histologic prediction to NGS classification
Figure 1C shows the comparison of prospective histologic prediction with NGS classification. Overall, 17 of 76 tumor pairs (22%) showed discordant results between prospective histologic prediction and final molecular classification. The discordance rate was significantly higher for IPMs (11/25, 44%) than SPLCs (6/51, 12%), P = 0.001. As shown in Fig. 1D, the discordance rate was significantly higher when histologic prediction was regarded as potentially equivocal by a pathologist and confirmation by NGS was suggested (P = 0.02).
Retrospective review of cases with challenging histologic prediction of tumor relatedness
The 17 cases in which histologic prediction of tumor relatedness was discordant with NGS results comprised six SPLCs and 11 IPMs, as defined by NGS (Fig. 1C; detailed summary in Supplementary Table S4). On re-review, of six SPLCs that were misclassified histologically as IPMs, four adenocarcinoma pairs indeed shared architectural similarity (similar growth patterns); however, they showed subtle differences in cytologic features. Two other discordant adenocarcinoma pairs exhibited overlap in cytologic features, while they differed in architectural patterns. All these cases were regarded as challenging on prospective evaluation, and molecular testing to confirm tumor relationship was recommended in pathology reports.
Of 11 tumor pairs classified as IPMs by NGS but thought to be histologically different, four adenocarcinoma pairs harbored 5%–30% lepidic component in both tumors (Fig. 3), resulting in erroneous conclusions of separate primary tumors, given the current concept that non-mucinous lepidic pattern represents “in situ” (precursor) lesion (See Discussion). Seven other tumor pairs showed histologic progression: six adenocarcinoma pairs were characterized by significantly increased proportions of high grade (solid or micropapillary) patterns in the subsequent tumor (Fig. 4), and one squamous cell carcinoma pair showed strikingly distinct levels of differentiation (Supplementary Fig. S1).
Figure 3.
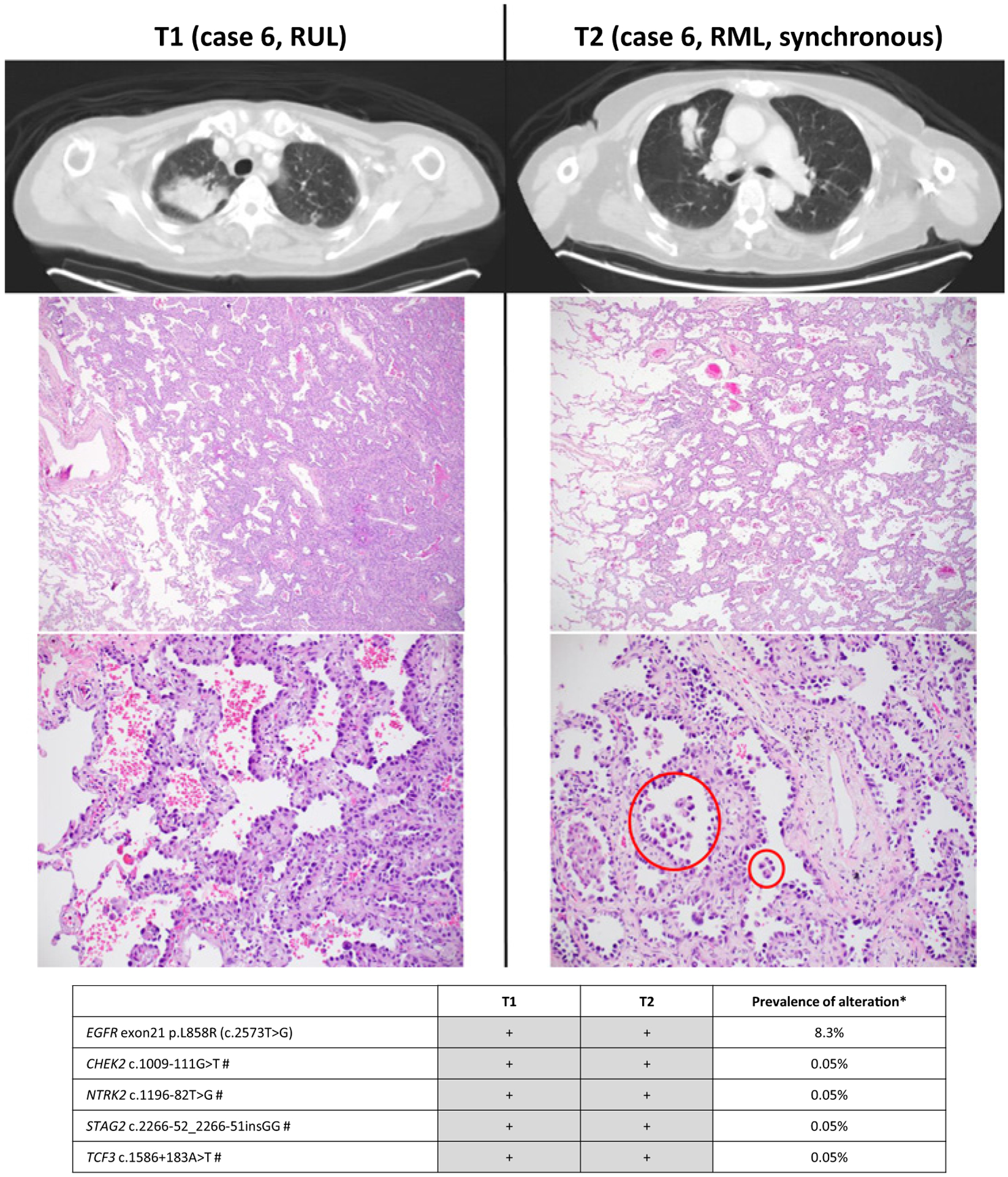
Radiologic and pathologic appearances and NGS profile of an intrapulmonary metastasis showing prominent lepidic pattern. Focal micropapillary pattern was present elsewhere in the tumor (right, bottom; circled). #, synonymous mutations.*, prevalence is based on MSK-IMPACT results for 4,119 NSCLCs in cBioPortal database. On the basis of combined prevalence of each mutation the odds of coincidental cooccurrence is 2.17E-29.
Figure 4.
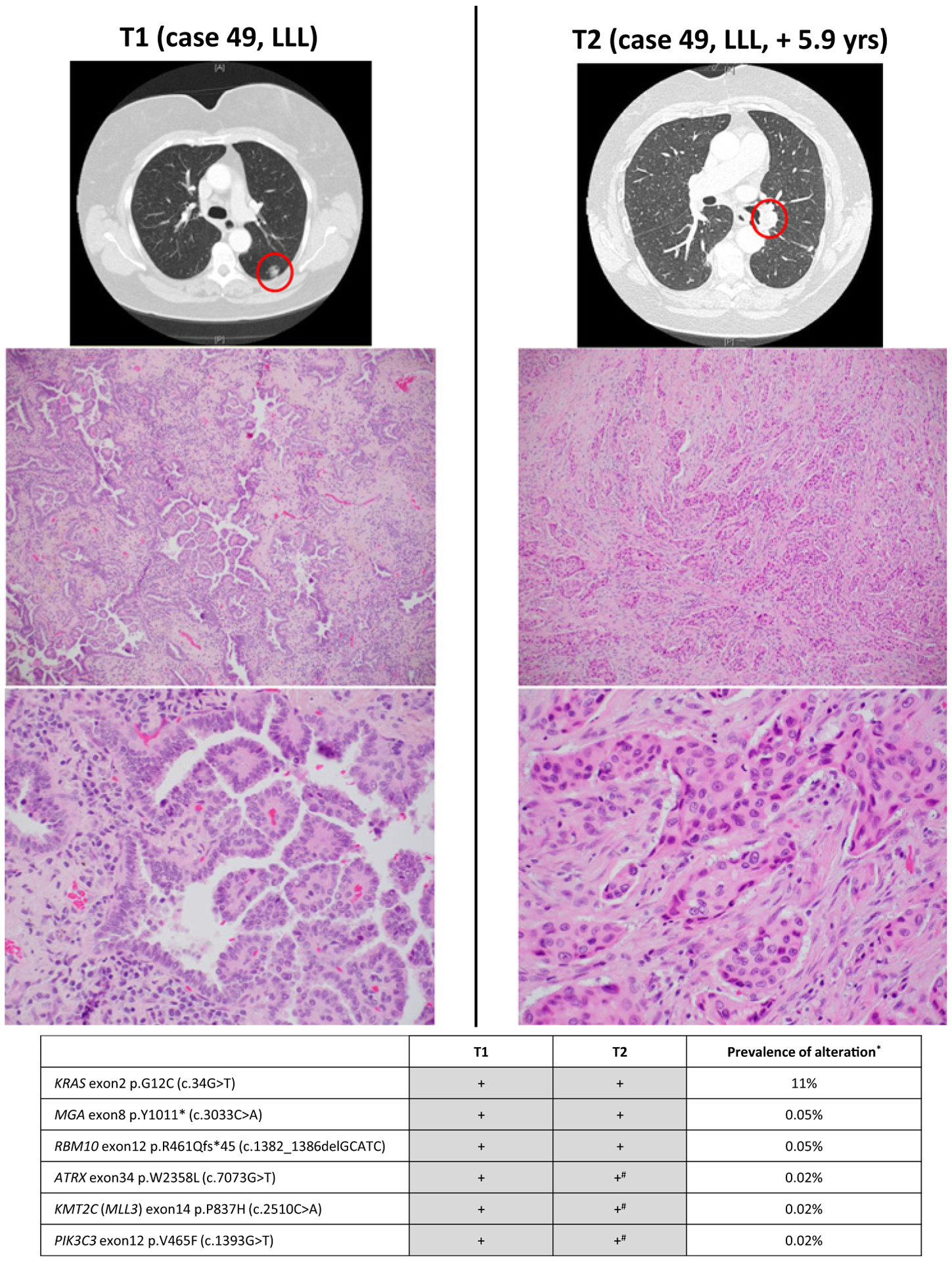
Radiologic and pathologic appearances and NGS profile of an intrapulmonary metastasis showing histologic progression consisting of entirely solid pattern. #, mutations present (subthreshold) in T2 on manual review.*, prevalence is based on MSK-IMPACT results for 4,119 NSCLCs in cBioPortal database. On the basis of combined prevalence of each mutation the odds of coincidental cooccurrence is 1.52E-37.
Clinicopathologic comparison of adenocarcinomas classified as IPMs versus SPLCs
Table 1 summarizes the clinicopathologic characteristics of patients with adenocarcinomas classified by NGS as IPMs versus SPLCs. Compared with SPLCs, IPMs showed greater propensity for metachronous presentation (61% vs. 15%; P = 0.0002), association with never smoker status (41% vs. 14%; P = 0.035), and overall lower pack-year smoking history (mean 14 vs. 34; P = 0.007). Genomically, IPMs showed a tendency for lower rate of KRAS mutations than SPLCs (30% vs. 54%; P 0.11), and were instead significantly enriched in non-KRAS-driver alterations (EGFR, ALK, ROS1, MET Exon 14 splicing, and BRAF V600E) with a combined rate of 65% versus 22% in IPMs versus SPLCs, respectively (P = 0.0009). In particular, the prevalence of MET Exon 14 was significantly higher in IPMs compared with SPLCs (23% vs. 3%, respectively; P = 0.0085). In addition, IPMs showed overall lower tumor mutation burden compared with SPLCs (mean 4.5 vs. 7.0 per Mb; P 0.025).
Table 1.
Clinicopathologic and genomic comparison of IPMs versus SPLCs for paired adenocarcinomas
| IPM | SPLCs | P | |
|---|---|---|---|
| Patient characteristics | N total = 17 patients | N total = 37 patients | |
| No. of analyzed tumors per patient | |||
| Two tumors | 14 (82%) | 32 (86%) | 0.70 |
| Three tumors | 3 (18%) | 5 (14%) | |
| Smoking history | |||
| Current/former | 10 (59%) | 32 (86%) | 0.035 |
| Never | 7 (41%) | 5 (14%) | |
| Pack years | |||
| Mean (range) | 14 (0–50) | 34 (0–118) | 0.007 |
| Median | 7.5 | 30 | |
| Background multiple (>1) AAH | 0 | 11 (30%) | 0.011 |
| Staging parameters | |||
| Tumor size mean (range); cm | 2.5 (0.6–9.7) | 2.0 (0.5–9.0) | 0.17 |
| pN1 or pN2 at time of surgery | 8 (47%) | 14 (38%) | 0.56 |
| pM1b (distant metastasis) at time of surgery | 0 | 0 | 1.00 |
| Tumor pair comparisons | N total = 23 tumor pairs | N total = 47 tumor pairs | |
| Time course | |||
| Synchronous | 9 (39%) | 40 (85%) | 0.0002 |
| Metachronous | 14 (61%) | 7 (15%) | |
| Latency for metachronous tumors | |||
| Mean (range); years | 3.6 (0.4–7.6) | 1.6 (0.6–2.7) | 0.038 |
| Intrapulmonary relationship | |||
| Ipsilateral same lobe | 6 (26%) | 20 (43%) | 0.37 |
| Ipsilateral other lobe | 9 (39%) | 16 (34%) | |
| Contralateral lobe | 8 (35%) | 11 (23%) | |
| Histologic pattern in any amount in at least one of the paired tumors | |||
| - Lepidic | 17 (74%) | 41 (87%) | 0.19 |
| - Acinar | 22 (96%) | 47 (100%) | 0.33 |
| - Papillary | 13 (57%) | 23 (49%) | 0.62 |
| - Micropapillary | 21 (91%) | 34 (72%) | 0.12 |
| - Solid | 8 (35%) | 20 (43%) | 0.61 |
| - Either micropapillary or solid | 23 (100%) | 39 (83%) | 0.046 |
| Lepidic in both tumors | 14 (61%) | 25 (53%) | 0.61 |
| Micropapillary in both tumors | 15 (65%) | 15 (32%) | 0.01 |
| Individual tumor comparisons | N total = 17 unique tumors | N total = 79 unique tumors | |
| Driver alterations | |||
| EGFR mutations | 4 (23%) | 13 (16%) | 0.49 |
| ALK fusion | 1 (6%) | 1 (1%) | 0.32 |
| ROS1 fusion | 1 (6%) | 1 (1%) | 0.32 |
| MET exon 14 mutations | 4 (23%) | 2 (3%) | 0.0085 |
| BRAF V600E mutation | 1 (6%) | 0 | 0.18 |
| KRAS mutations | 5 (30%) | 43 (54%) | 0.11 |
| Other driver alterations | 0 | 9 (11%) | 0.35 |
| Unknown driver | 1 (6%) | 10 (13%) | 0.68 |
| Non-KRAS drivers (EGFR, ALK, ROS1, MET, and BRAF) | 11 (65%) | 17 (22%) | 0.0009 |
| TMB | |||
| Mean (range); mt/Mb | 4.5 (0.9–15.8) | 7.0 (0–41.2) | 0.025 |
Abbreviation: TMB, tumor mutation burden.
Importantly, IPMs could not be distinguished from SPLCs by anatomic location, because IPMs were just as likely as SPLCs to present in another ipsilateral or contralateral lobe. Furthermore, IPMs occurred in the absence of nodal involvement in the majority of cases, and no IPMs showed evidence of distant metastases at the time of surgery, similar to SPLCs in this cohort. Also notable was the observation that in metachronous IPMs, the time course between the first and second tumor ranged from 0.4 to 7.6 years (median, 3.1 years), exceeding 2-year latency in eight of 19 patients, and even exceeding 5-year latency in three patients (Supplementary Table S1).
Histologically, high-grade patterns (micropapillary or solid) were invariably present in all 25 IPMs, and the presence of micropapillary pattern in both tumors was significantly associated with IPMs compared with SPLCs (65% vs. 32%, respectively; P = 0.01). The presence of ≥5% lepidic pattern was noted in 61% of paired IPMs. However, none of these IPMs were lepidic predominant, and micropapillary pattern was invariably present in IPMs containing a lepidic component. Multiple (≥2) foci of atypical adenomatous hyperplasia (AAH) were present in 30% of SPLCs but none of IPMs (P = 0.011).
Survival
Exploratory survival analysis with median follow-up of 15 months showed that 2-year PFS was 48% for patients with IPMs, and 74% for patients with SPLCs, with a trend toward worsened PFS for IPMs (P = 0.197) (Supplementary Fig. S2).
Discussion
Large-panel NGS assays are increasingly utilized in clinical practice for guiding the use of targeted therapies. Our results show that such assays also provide a definitive, unambiguous determination of clonal relationships among multiple lung carcinomas as clonally unrelated (SPLCs) versus clonally related (IPMs). While our findings support the overall accuracy of comprehensive histologic assessment for discerning tumor relationships in the majority of NSCLCs, they also highlight its limitations in approximately one-fifth of cases. Our results help to identify specific clinicopathologic settings in which molecular testing should be considered to supplement histologic assessment. In addition, we describe novel clinicopathologic differences between SPLCs and IPMs defined using the highly robust molecular gold standard.
To our knowledge, this is the largest study to date on the performance of comprehensive NGS in distinguishing IPMs from SPLCs. Unlike previously used smaller gene panels (18–24), broad panel NGS provides a way to examine multiple alterations simultaneously, yielding highly granular tumor mutational profiles and a robust discrimination of tumor relatedness. In this series, the tumors harbored a median of eight (up to 47) nonsynonymous somatic alterations per case. In a minority of cases where additional discrimination was needed, those were supplemented with synonymous mutations (median, 16; up to 34 per case); these mutations are excluded for the purposes of predictive testing because they do not lead to protein alterations, but they provide additional useful markers in tumor clonality assessment. Thus, for tumors classified as clonally related (IPMs), multiple shared alterations (median, 5, up to 16) were present. We demonstrate that the odds of a set of such mutations occurring by chance is virtually nil, representing a significant advance over the panels that examine only major drivers or noncomprehensive NGS (see below). We also found that broad panel NGS robustly identified unrelated tumors by demonstrating entirely unique mutational profiles comprising multiple mutations (median, 14; up to 72 per tumor pair). In particular, we found that comprehensive NGS allows clear recognition of SPLCs with coincidentally shared single hotspot mutation, as also discussed below. Finally, this approach permits determination of clonal relationship for cases with driver alterations that are poorly covered by conventional assays (e.g., MET Exon 14) or entirely lacking a known mitogenic driver. Overall, our panel was able to establish definitive tumor clonal relationship in virtually all tumor pairs during the study period.
When comparing the performance of histologic assessment with the definitive NGS classification, we found that histologic prediction was discrepant in 22% of cases overall, similar to the discrepancy rates ranging from 18% to 37% across different platforms in prior studies (5, 19, 24, 26, 27). Difficulties with histologic prediction were substantially more frequent for the recognition of IPMs than SPLCs (44% vs. 12% discordant, respectively). We identified several recurrent challenges limiting the accuracy of histologic prediction of tumor relationships: (i) tumor progression in IPMs leading to histologic dissimilarity, (ii) presence of lepidic pattern in IPMs causing erroneous conclusion of a primary, in situ disease, and (iii) SPLCs with overlapping architectural or cytologic features mimicking morphologic similarity.
Histologic progression in metastatic NSCLC has been noted in prior studies (37). In this study, seven IPMs were incorrectly predicted to be distinct primary tumors because of significant discrepancy in tumor grades. Although in most cases, histologic patterns in the primary and metastatic tumors are preserved, the issue of histologic progression in a subset of cases presents a limitation to histology-based determination of tumor relationship.
Surprisingly, we observed that lepidic pattern was frequently present in IPMs, resulting in an erroneous interpretation of some of such pairs as SPLCs. The current concept of lepidic pattern in non-mucinous lung adenocarcinomas is that it represents noninvasive precursor/in situ disease (38). It may be therefore counterintuitive that tumors molecularly proven to be IPMs can harbor what appears to resemble a pattern equated with in situ disease. We postulate that lepidic pattern in IPMs represents surface colonization rather than a precursor lesion, in line with the recent proposal by Moore and colleagues that some lepidic patterns could represent outgrowth of an invasive tumor.(39) This phenomenon may be analogous to intrapulmonary multilobar spread of invasive mucinous adenocarcinomas, tumors which are well known to exhibit significant lepidic/surface growth pattern during intrapulmonary progression (38), or alveolar surface colonization by metastatic pancreatic carcinomas (40). In contrast, this phenomenon has not been well-described for the intrapulmonary spread of non-mucinous adenocarcinomas, except in isolated case reports (41, 42). Importantly, because lepidic pattern in IPMs was nonpredominant in all cases, lepidic-predominant adenocarcinoma, minimally invasive adenocarcinoma, and adenocarcinoma in situ lesions should not be regarded as potential IPMs.
The overall accuracy of comprehensive histologic assessment and the relatively specific scenarios where discrepancies occurred suggest that histologic assessment may be useful in triaging cases that would benefit from molecular confirmation (Fig. 5). In particular, we found that pathologists are generally accurate in determining when a comparison is challenging and requires a molecular confirmation. Given that some of these cases underwent molecular profiling as they were suggested by the pathologists, the cohort may have been enriched in histologically challenging cases, thus elevating the discrepancy rate. Increased familiarity by pathologists with previously underrecognized features in some IPMs (histologic progression and presence of lepidic pattern) should further increase the accuracy of histologic comparison in practice. Overall, retrospective histologic reassessment of cases unambiguously classified by NGS will provide an unprecedented benchmark against which to refine histologic criteria for distinguishing SPLCs and IPMs in future studies.
Figure 5.
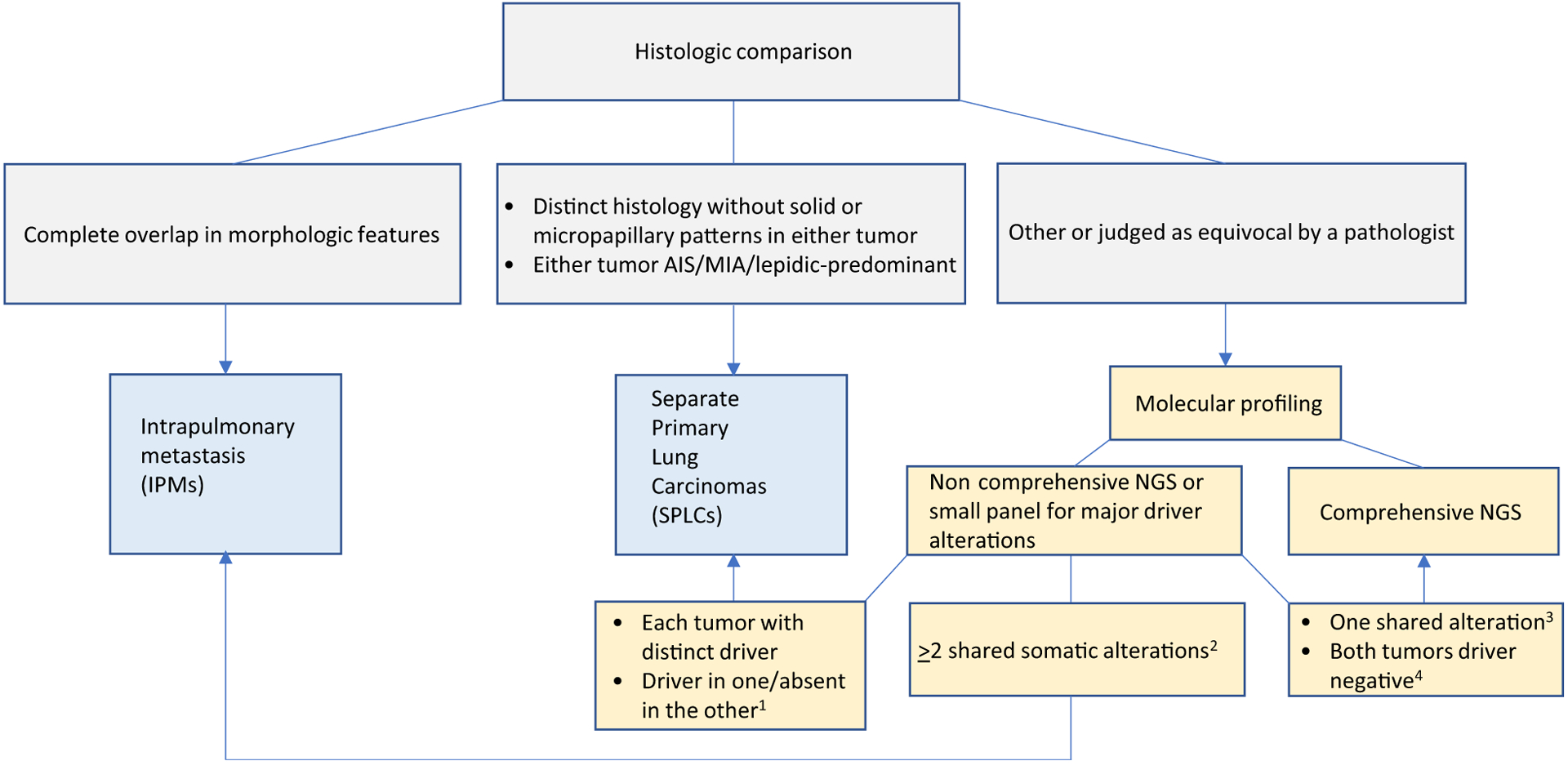
Flowchart for classification of multiple NCSLC using histology*and NGS. *Histologic features may not be sufficiently distinctive for squamous cell carcinoma. Molecular testing should be performed for all cases. 1, this scenario supports SPLCs only if adequate tumor content has been confirmed histologically in the driver-negative tumor (see Supplementary Materials and Method 2 for details). 2, applicable to NGS only but not major driver-only testing. 3, if only a single alteration is shared, the degree to which this supports IPMs should be determined on the basis of prevalence of that alterations in a given population as well as overall clinicoradiologic context (see Supplementary Materials and Method 2 for details). 4, driver negative in both tumors would benefit from broad panel NGS for comprehensive comparison of mutation profiles (see Supplementary Materials and Method 2 for details). AIS, adenocarcinoma in situ; MIA, minimally invasive adenocarcinoma.
We note that histology in this series was dominated by adenocarcinomas, and only two tumor pairs were squamous cell carcinomas, precluding detailed analysis of this subset. Nevertheless, these cases illustrate the effectiveness of NGS in unambiguously establishing tumor relationship in such pairs due to their high tumor mutation burden even in the absence of a driver alteration. Conversely, due to the relative homogeneity of architectural and cytologic features in squamous cell carcinomas (38), histologic features may not be sufficiently distinctive for definitive classification of tumors as related versus unrelated.
Comparison of patient and tumor characteristics associated with SPLCs and IPMs robustly defined by NGS in our series yielded several interesting observations related to the distinct biology underlying these processes. We found that SPLCs were enriched in KRAS mutations (57%), in line with consistent smoking history in those patients (86%) and heavy cigarette exposure (median, 30 pack-years). It is likely that multiple SPLCs arise in the background of generalized cigarette-induced tumor predisposition, as reflected by frequent presence of multifocal precursor lesions (AAH) in such patients in our series and in prior studies (2, 43, 44). Notably, in the Asian populations, SPLCs are dominated by EGFR mutations (28, 45), likely reflecting the overall known geographic differences in genomic profiles of lung adenocarcinomas. In contrast, IPMs were significantly enriched (65%) in non-KRAS–driver alterations, which are not linked to cigarette smoking, including EGFR, ALK, ROS1, and MET Exon 14, in line with the higher rate of never smokers (41%) and lower pack-year exposure (median, 7.5 pack-years). The reason for the predilection of IPMs to tumors with nonsmoking-related driver alterations is less clear, but may be related to a distinct biology of tumors prone to intrapulmonary spread. We note that the propensity for miliary-like intrapulmonary spread is well recognized for EGFR-mutant adenocarcinomas (38, 46); isolated IPMs in this study may represent a limited manifestation of the same phenomenon. We identified a surprising enrichment for MET Exon 14 among IPMs (23%), whereas the rate of this alteration in unselected adenocarcinoma is 3% (9). The distinctive prevalence in KRAS and EGFR mutations in SPLCs and IPMs was recently noted in a study by Mansuet-Lupo and colleagues (27); here we expand on those findings and provide a more comprehensive profile of molecular differences in these tumors.
Another distinctive and previously not emphasized difference was that SPLCs presented synchronously in the vast majority of cases, whereas IPMs were more commonly metachronous. This may reflect the natural history of SPLCs where tumors were likely initiated around similar time frame. In addition, increased screening by low-dose CTs in current/former smokers may contribute to simultaneous detection of SPLCs. Conversely, for IPMs, the detection of a second nodule may be more likely to follow a lag period after the resection of the primary tumor. Given the association of IPMs with never/light smokers, the development of a subsequent lung carcinoma in a never/light smoker is thus statistically more likely to represent IPMs rather than SPLCs, and this could serve as a helpful criterion in clinical practice.
We note that the surgical nature of this cohort may have preselected for a distinctive and unusual patient population with limited intrapulmonary spread of NSCLC in the absence of nodal or extrapulmonary disease (i.e., oligometastatic disease limited to the lung). Our findings suggest that solitary IPMs are substantially underrecognized both clinically and pathologically in current practice. We found that many metachronous IPMs were resected more than 2 years (up to 7.6 years) after the primary tumor resection—a substantially longer latency than the 2-year cutoff suggested for the distinction of IPMs versus SPLCs in the original Martini and Melamed criteria (4), and exceeding a 5-year cutoff suggested in a recent study of molecularly confirmed IPMs (27). Furthermore, we noted frequent spread to different lobes, including the contralateral lung. It is not currently known whether IPMs, especially solitary IPMs, arise from systemic lymphovascular spread or are more likely a manifestation of local spread via bronchoalveolar air spaces or pulmonary vasculature. Greater recognition of this intriguing and poorly understood phenomenon by robust molecular methods will enable more detailed study of its biology, natural history, and long-term clinical outcomes. While our exploratory survival analysis was limited by short follow-up and relatively small number of patients, it did reveal that over half of the patients with resected IPMs developed subsequent disease recurrence, with a trend toward higher recurrence rate compared with patients with SPLCs.
Several interpretative and technical aspects of NGS for clonality assessment are worth highlighting. A significant advantage of comprehensive NGS panels of the type used here is in the ability to discriminate unrelated tumors (SPLCs) that share a single common hotspot mutation by chance. This is especially problematic in smokers with tumors sharing a KRAS p.G12C mutation, or in never-smokers with tumors sharing a EGFR p.L858R mutation, for which the odds of cooccurrence by chance in the respective populations can be as high as one in 17 (see Results). In fact, in our series, shared KRAS mutations were almost as likely to occur coincidentally in SPLC s as in IPMs. The only other instance of coincidentally shared hotspot mutation in our series was a U2AF1 mutation in an otherwise unambiguous SPLCs (see Results). In this study, we illustrate that NGS can readily identify SPLCs despite shared single hotspot mutation by demonstrating numerous additional unique mutations in each of the tumors. We considered the possibility of such cases representing IPMs with early clonal divergence and subsequent acquisition of multiple private mutations. However, arguing against this possibility are our data that in all IPMs the number of shared mutations was consistently substantially higher than that of unique mutations. These data are also in-line with the findings by multiregion and longitudinal NGS of lung adenocarcinomas revealing that the number of early truncal mutations substantially exceeds that of unique subclonal mutations (47, 48).
Clonality assessment by large panel NGS represents a significant advance over less comprehensive gene panels. In this study, we modeled how more limited panels in wide use in clinical practice would perform for typing this set of tumors. In-line with prior studies, we found that such panels can identify clonal relationships in 60%–70% of case (23, 24, 27). On the basis of subsampling of our data, we propose an algorithm for the settings where limited panels are sufficient for interpretation of clonality, and when additional comprehensive approaches would be of value (Fig. 5). In particular, on the basis of the aforementioned data, we caution against concluding that tumors are clonally related solely on the basis of the presence of a single shared hotspot alteration; the degree to which a single shared alteration supports tumor relatedness should be determined on the basis of the prevalence of that alteration in a given population as well as overall clinicoradiologic context.
We also note several advantages of large-panel NGS over nonmutation-based approaches that have been utilized in clonality assessment in research settings. Compared with genomic breakpoint analysis of chromosomal rearrangements, via mate-pair sequencing that requires fresh-frozen tissue (30), MSK-IMPACT and other clinical NGS assays work on routine FFPE material. Array CGH employed in prior studies (5, 49) requires high tumor purity, and this is not always feasible in NSCLC specimens, which commonly show low tumor purity due to abundant admixed inflammatory infiltrate. As shown in this study, without selecting for cases with high tumor purity to preserve a true representation of cases in clinical practice, definitive classification of tumor pairs into IPMs versus SPLCs was possible in virtually all cases.
Another advantage specific to our NGS platform is that tumors are sequenced with matched normal control (patient’s white blood cells), whereas germline (constitutional) variants are filtered out bioinformatically. For large panel NGS performed without normal control, it is difficult to distinguish rare or private germline polymorphisms from shared somatic mutations, which could limit the ability to conclusively address the clonal relationships of the tumors.
The main limitations of large panel NGS platforms include availability, cost, and turnaround time. Because of the cost of implementation and significant requirement of bioinformatic, computational resources, and personnel to analyze and interpret the data, not all institutions currently offer NGS testing. Although the cost of NGS is comparatively lower than sequential testing with multiple standalone single-gene assays (50), clonality assessment using NGS platform requires parallel testing of both tumors, and ideally, matched normal control. Finally, although the turnaround time of NGS assays has improved significantly in the last few years, it still takes substantially longer than pathologic review; therefore, preliminary impression by histologic assessment may still be useful before NGS results are generated.
Although our study cohort by design was limited to surgical resections to facilitate comparison with comprehensive histologic assessment, conclusions regarding clonality assessment by NGS from this study are also applicable to biopsy and cytology specimens, where morphologic assessment of tumor relationship is even more challenging. However, this will require empirical validation in future studies.
In conclusion, to our knowledge, this is the largest study to date that demonstrates the utility of broad panel NGS to accurately establish clonal relationships among NSCLCs in routine clinical practice. Comprehensive NGS highlights select scenarios in which histologic assessment has limitations, and should allow for refinement of histologic criteria for evaluation of tumor relatedness. Overall, our findings suggest that a comprehensive diagnostic approach incorporating histology and molecular analysis is essential to drawing this critical distinction in clinical practice. Molecular staging has the potential to revolutionize the current staging practice in patients with multiple NSCLCs, providing robust confirmation of tumor clonality and information on actionable mutations at the same time.
Supplementary Material
Translational Relevance.
Multiple non-small cell lung carcinomas (NSCLC) may represent either separate primary lung carcinomas (SPLCs) or intrapulmonary metastases (IPMs) of the same carcinoma. To our knowledge, this is the largest study to date to demonstrate the utility of comprehensive next-generation sequencing (NGS) to unambiguously establish clonal relationship among NSCLCs in routine clinical practice. We illustrate the added value of NGS to standard histopathologic assessment to establish tumor relatedness, and elucidate several novel distinctive genomic and clinicopathologic differences between NGS-defined IPMs and SPLCs. Overall, these findings have the potential to revolutionize the approach to multiple NSCLCs in clinical practice, and illustrate the utility of comprehensive NGS not only for identification of targetable alterations but also for assessment of tumor clonal relationships.
Acknowledgments
This work was supported in part by NIH P01 CA129243 (to M. Ladanyi and N. Rekhtman), NIH P30 CA008748 (MSKCC), and the Marie-Jos[notdef]ee and Henry R. Kravis Center for Molecular Oncology at MSKCC
Footnotes
Disclosure of Potential Conflicts of Interest
C.M. Vanderbilt is an employee/paid consultant for Docdoc and Paige.AI. V.W. Rusch reports receiving commercial research grants from Genentech and other remuneration from Intuitive Surgical and NIH TMS Committee. D.R. Jones is an employee/paid consultant for Diffusion Pharmaceuticals, Merck, and AstraZeneca. M.E. Arcila reports receiving speakers bureau honoraria from Invivoscribe and Biocartis. No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Vazquez M, Carter D, Brambilla E, Gazdar A, Noguchi M, Travis WD, et al. Solitary and multiple resected adenocarcinomas after CT screening for lung cancer: Histopathologic features and their prognostic implications. Lung Cancer 2009;64:148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mascalchi M, Comin CE, Bertelli E, Sali L, Maddau C, Zuccherelli S, et al. Screen-detected multiple primary lung cancers in the ITALUNG trial. J Thorac Dis 2018;10:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al. , editors. AJCC cancer staging manual. Switzerland, AG: Springer International Publishing: American Joint Commission on Cancer; 2017. [Google Scholar]
- 4.Martini N, Melamed MR. Multiple primary lung cancers. J Thorac Cardiovasc Surg 1975;70:606–12. [PubMed] [Google Scholar]
- 5.Girard N, Deshpande C, Lau C, Finley D, Rusch V, Pao W, et al. Comprehensive histologic assessment helps to differentiate multiple lung primary nonsmall cell carcinomas from metastases. Am J Surg Pathol 2009;33: 1752–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Detterbeck FC, Franklin WA, Nicholson AG, Girard N, Arenberg DA, Travis WD, et al. The IASLC Lung Cancer Staging Project: background data and proposed criteria to distinguish separate primary lung cancers from metastatic foci in patients with two lung tumors in the forthcoming Eighth Edition of the TNM Classification for lung cancer. J Thorac Oncol 2016;11: 651–65. [DOI] [PubMed] [Google Scholar]
- 7.Detterbeck FC, Marom EM, Arenberg DA, Franklin WA, Nicholson AG, Travis WD, et al. The IASLC Lung Cancer Staging Project: background data and proposals for the application of TNM staging rules to lung cancer presenting as multiple nodules with ground glass or lepidic features or a pneumonic type of involvement in the forthcoming Eighth Edition of the TNM Classification. J Thorac Oncol 2016;11:666–80. [DOI] [PubMed] [Google Scholar]
- 8.Detterbeck FC, Nicholson AG, Franklin WA, Marom EM, Travis WD, Girard N, et al. The IASLC Lung Cancer Staging Project: summary of proposals for revisions of the classification of lung cancers with multiple pulmonary sites of involvement in the forthcoming Eighth Edition of the TNM Classification. J Thorac Oncol 2016;11:639–50. [DOI] [PubMed] [Google Scholar]
- 9.Jordan EJ, Kim HR, Arcila ME, Barron DA, Chakravarty D, Gao J, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov 2017;7:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen C, Wang X, Tian L, Che G. Microsatellite alteration in multiple primary lung cancer. J Thorac Dis 2014;6:1499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimizu S, Yatabe Y, Koshikawa T, Haruki N, Hatooka S, Shinoda M, et al. High frequency of clonally related tumors in cases of multiple synchronous lung cancers as revealed by molecular diagnosis. Clin Cancer Res 2000;6: 3994–9. [PubMed] [Google Scholar]
- 12.Huang J, Behrens C, Wistuba I, Gazdar AF, Jagirdar J. Molecular analysis of synchronous and metachronous tumors of the lung: impact on management and prognosis. Ann Diagn Pathol 2001;5:321–9. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Beltran A, Davidson DD, Abdul-Karim FW, Olobatuyi F, MacLennan GT, Lin H, et al. Evidence for common clonal origin of multifocal lung cancers. J Natl Cancer Inst 2009;101:560–70. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Wang M, MacLennan GT, Abdul-Karim FW, Eble JN, Jones TD, et al. Evidence for common clonal origin of multifocal lung cancers. J Natl Cancer Inst 2009;101:560–70. [DOI] [PubMed] [Google Scholar]
- 15.Arai J, Tsuchiya T, Oikawa M, Mochinaga K, Hayashi T, Yoshiura K, et al. Clinical and molecular analysis of synchronous double lung cancers. Lung Cancer 2012;77:281–7. [DOI] [PubMed] [Google Scholar]
- 16.van Rens MTM, Eijken EJE, Elbers JRJ, Lammers JWJ, Tilanus MGJ, Slootweg PJ. p53 mutation analysis for definite diagnosis of multiple primary lung carcinoma. Cancer 2002;94:188–96. [DOI] [PubMed] [Google Scholar]
- 17.Mitsudomi T, Yatabe Y, Koshikawa T, Hatooka S, Shinoda M, Suyama M, et al. Mutations of the p53 tumor suppressor gene as clonal marker for multiple primary lung cancers. J Thorac Cardiovasc Surg 1997;114: 354–60. [DOI] [PubMed] [Google Scholar]
- 18.Takamochi K, Oh S, Matsuoka J, Suzuki K. Clonality status of multifocal lung adenocarcinomas based on the mutation patterns of EGFR and K-ras. Lung Cancer 2012;75:313–20. [DOI] [PubMed] [Google Scholar]
- 19.Schneider F, Derrick V, Davison JM, Strollo D, Incharoen P, Dacic S. Morphological and molecular approach to synchronous non-small cell lung carcinomas: impact on staging. Mod Pathol 2016;29:735. [DOI] [PubMed] [Google Scholar]
- 20.Asmar R, Sonett JR, Singh G, Mansukhani MM, Borczuk AC. Use of oncogenic driver mutations in staging of multiple primary lung carcinomas: a single-center experience. J Thorac Oncol 2017;12:1524–35. [DOI] [PubMed] [Google Scholar]
- 21.Warth A, Macher-Goeppinger S, Muley T, Thomas M, Hoffmann H, Schnabel PA, et al. Clonality of multifocal nonsmall cell lung cancer: implications for staging and therapy. Eur Respir J 2012;39:1437–42. [DOI] [PubMed] [Google Scholar]
- 22.Patel SB, Kadi W, Walts AE, Marchevsky AM, Pao A, Aguiluz A, et al. Next-generation sequencing: a novel approach to distinguish multifocal primary lung adenocarcinomas from intrapulmonary metastases. J Mol Diagn 2017;19:870–80. [DOI] [PubMed] [Google Scholar]
- 23.Saab J, Zia H, Mathew S, Kluk M, Narula N, Fernandes H. Utility of genomic analysis in differentiating synchronous and metachronous lung adenocarcinomas from primary adenocarcinomas with intrapulmonary metastasis. Transl Oncol 2017;10:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roepman P, Ten Heuvel A, Scheidel KC, Sprong T, Heideman DAM, Seldenrijk KA, et al. Added value of 50-gene panel sequencing to distinguish multiple primary lung cancers from pulmonary metastases: a systematic investigation. J Mol Diagn 2018;20:436–45. [DOI] [PubMed] [Google Scholar]
- 25.Wu C, Zhao C, Yang Y, He Y, Hou L, Li X, et al. High discrepancy of driver mutations in patients with NSCLC and synchronous multiple lung ground-glass nodules. J Thorac Oncol 2015;10:778–83. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi Y, Shien K, Tomida S, Oda S, Matsubara T, Sato H, et al. Comparative mutational evaluation of multiple lung cancers by multiplex oncogene mutation analysis. Cancer Sci 2018;109:3634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mansuet-Lupo A, Barritault M, Alifano M, Janet-Vendroux A, Zarmaev M, Biton J, et al. Proposal for a combined histomolecular algorithm to distinguish multiple primary adenocarcinomas from intrapulmonary metastasis in patients with multiple lung tumors. J Thorac Oncol 2019:14;844–56. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Zhang J, Li L, Yin G, Zhang J, Zheng S, et al. Genomic heterogeneity of multiple synchronous lung cancer. Nat Commun 2016;7:13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma P, Fu Y, Cai MC, Yan Y, Jing Y, Zhang S, et al. Simultaneous evolutionary expansion and constraint of genomic heterogeneity in multifocal lung cancer. Nat Commun 2017;8:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy SJ, Harris FR, Kosari F, Barreto Siqueira Parrilha Terra S, Nasir A, Johnson SH, et al. Using genomics to differentiate multiple primaries from metastatic lung cancer. J Thorac Oncol 2019;14:1567–82. [DOI] [PubMed] [Google Scholar]
- 31.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29: 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostrovnaya I, Seshan VE, Begg CB. Using somatic mutation data to test tumors for clonal relatedness. Ann Appl Stat 2015;9:1533–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mauguen A, Seshan VE, Ostrovnaya I, Begg CB. Estimating the probability of clonal relatedness of pairs of tumors in cancer patients. Biometrics 2018; 74:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res 2012;18:6169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aokage K, Ishii G, Yoshida J, Hishida T, Nishimura M, Nagai K, et al. Histological progression of small intrapulmonary metastatic tumor from primary lung adenocarcinoma. Pathol Int 2010;60:765–73. [DOI] [PubMed] [Google Scholar]
- 38.Travis WD, Brambilla E, Burke A, Marx A, Nicholson AG. WHO classification of tumours of the lung, pleura, thymus and heart. International Agency for Research on Cancer; Lyon, France; 2015. [DOI] [PubMed] [Google Scholar]
- 39.Moore DA, Sereno M, Das M, Baena Acevedo JD, Sinnadurai S, Smith C, et al. In situ growth in early lung adenocarcinoma may represent precursor growth or invasive clone outgrowth-a clinically relevant distinction. Mod Pathol 2019;32:1095–105. [DOI] [PubMed] [Google Scholar]
- 40.Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med 2009;133:413–22. [DOI] [PubMed] [Google Scholar]
- 41.Klempner SJ, Ou SH, Costa DB, VanderLaan PA, Sanford EM, Schrock A, et al. The clinical use of genomic profiling to distinguish intrapulmonary metastases from synchronous primaries in non-small-cell lung cancer: a mini-review. Clin Lung Cancer 2015;16:334–9. [DOI] [PubMed] [Google Scholar]
- 42.Takanashi Y, Tajima S, Tsukui M, Shinmura K, Hayakawa T, Takahashi T, et al. Non-mucinous lepidic predominant adenocarcinoma presenting with extensive aerogenous spread. Rare Tumors 2016;8:6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson BE. Second lung cancers in patients after treatment for an initial lung cancer. J Natl Cancer Inst 1998;90:1335–45. [DOI] [PubMed] [Google Scholar]
- 44.Tucker MA, Murray N, Shaw EG, Ettinger DS, Mabry M, Huber MH, et al. Second primary cancers related to smoking and treatment of small-cell lung cancer. Lung Cancer Working Cadre. J Natl Cancer Inst 1997;89: 1782–8. [DOI] [PubMed] [Google Scholar]
- 45.Park E, Ahn S, Kim H, Park SY, Lim J, Kwon HJ, et al. Targeted sequencing analysis of pulmonary adenocarcinoma with multiple synchronous ground-glass/lepidic nodules. J Thorac Oncol 2018;13:1776–83. [DOI] [PubMed] [Google Scholar]
- 46.Kim HJ, Kang SH, Chung HW, Lee JS, Kim SJ, Yoo KH, et al. Clinical features of lung adenocarcinomas with epidermal growth factor receptor mutations and miliary disseminated carcinomatosis. Thorac Cancer 2015;6:629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014;346:256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol 2014;12:e1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho EK, Tchinda J, Freeman JL, Chung YJ, Cai WW, Lee C. Array-based comparative genomic hybridization and copy number variation in cancer research. Cytogenet Genome Res 2006;115:262–72. [DOI] [PubMed] [Google Scholar]
- 50.Pennell NA, Mutebi A, Zhou Z-Y, Ricculli ML, Tang W, Wang H, et al. Economic impact of next-generation sequencing vs sequential single-gene testing modalities to detect genomic alterations in metastatic non-small cell lung cancer using a decision analytic model. JCO Precis Oncol 2019(3):1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.