

Abstract
Objective
To determine genotype–phenotype correlation in succinic semialdehyde dehydrogenase (SSADH) deficiency.
Methods
ALDH5A1 variants were studied with phenotype correlation in the SSADH natural history study. Assignment of gene variant pathogenicity was based on in silico testing and in vitro enzyme activity after site-directed mutagenesis and expression in HEK293 cells. Phenotypic scoring used a Clinical Severity Score (CSS) designed for the natural history study.
Results
Twenty-four patients were enrolled (10 male, 14 female, median age 8.2 years). There were 24 ALDH5A1 variants, including 7 novel pathogenic variants: 2 missense, 3 splice site, and 2 frameshift. Four previously reported variants were identified in >5% of unrelated families. There was a correlation with age and presence (p = 0.003) and severity (p = 0.002) of epilepsy and with obsessive-compulsive disorder (OCD) (p = 0.016). The median IQ score was 53 (Q25–Q75, 49–61). There was no overall correlation between the gene variants and the CSS, although a novel missense variant was associated with the mildest phenotype by CSS in the only patient with a normal IQ, whereas a previously reported variant was consistently associated with the most severe phenotype.
Conclusions
Seven novel pathogenic and one previously unpublished benign ALDH5A1 variants were detected. There is an age-dependent association with worsening of epilepsy and presence of OCD in SSADH deficiency. Overall, there does not appear to be a correlation between genotype and phenotypic severity in this cohort of 24 patients. We did find a suspected correlation between a novel pathogenic missense variant and high functionality, and a previously reported pathogenic missense variant and maximal severity.
Succinic semialdehyde dehydrogenase (SSADH) deficiency is a rare autosomal recessive disease that interferes with the catabolism of the major inhibitory neurotransmitter γ-aminobutyric acid (GABA) and leads to accumulation of various potential toxic metabolites, most prominently γ-hydroxybutyric acid (GHB).
Increased endogenous levels of both GABA and GHB have been associated with protean neurologic manifestations, which typically include early life developmental impairment with cognitive deficiency and severe limitation in expressive language. Phenotypic variation is broad, and no genotype–phenotype relationship has been discovered to date.1 Recent longitudinal studies have demonstrated changes in GABA and GHB levels with age, with highest levels seen in the first years of life.2,3 Clinical presentation is typically hypotonia and developmental delay. Nonprogressive ataxia, hyporeflexia, behavioral dysregulation, obsessive-compulsive disorder, anxiety disorder, and attention-deficit/hyperactivity disorder are additional signs and symptoms with high prevalence.4–7 Progressive worsening seizures over time are common in the adolescent/adult cohort, as well as a high sudden unexpected death in epilepsy (SUDEP) rate.8,9 MRI reveals an abnormal intensity in the globus pallidus, subthalamic nucleus, and dentate nucleus of the cerebellum on T2-weighted images, typically bilateral, but it can be unilateral.10
SSADH deficiency has been identified in hundreds of patients worldwide, and sequencing reveals homozygous or compound heterozygous variants in the ALDH5A1 gene, which consists of 10 exons at locus 6p22. SSADH deficiency may be underdiagnosed due to the highly variable nonspecific neurologic manifestations. Over 45 pathologic variants have been identified, with the majority being missense. Of note, only a handful of splice site, deletion, and insertion variants have been identified.11–17
Methods
Standard protocol approvals, registrations, and patient consents
The Natural History Study of Patients with SSADH Deficiency was approved by the Boston Children's Hospital (BCH) Institutional Review Board (P-00029917). Written informed consent was obtained for all participants. A standard of care (SOC) cohort was included for patients unable to travel to BCH and evaluated through medical records, questionnaire surveys, and laboratory samples.
Variant grouping and assignment
All participants have confirmed SSADH deficiency based on clinical and biochemical phenotyping in addition to gene sequencing (ALDH5A1, NM_001080.3 transcript). The variants found are reported following the nomenclature established in the American College of Medical Genetics recommendations.18 Variants were placed in groups as missense, truncating (nonsense or frameshift), or intronic splice site variants for analysis.
Assignment of novel coding variant pathogenicity was based on in silico testing using the software programs integrated into the ALAMUT VISUAL software, version 2.9: SIFT, MutationTaster, and PolyPhen-2 for missense variants, and NNSplice Splice Site Predictor, MaxEntScan, and Human Splicing Finder for splice site variants. The effect of the novel missense variants on SSADH enzyme activity was determined using overexpression in HEK293 cells. The 2 novel missense variants were introduced in the pAD1/RSV vector containing the wild-type ALDH5A1 cDNA,19 using site-directed mutagenesis. Transfections of the ALDH5A1 recombinant constructs were performed in triplicate using Fugene (Promega, Madison, WI), following the manufacturer’s protocol. Cells were harvested 48 hours after transfection and the SSADH activity was evaluated using the method of Gibson et al.,20 with minor modifications. Western blot (ALDH5A1 mouse polyclonal antibody [B01P, Abnova Corporation, Taipei, Taiwan]) and RNA studies using standard molecular techniques were performed on the SSADH transfectants to confirm transfection efficiency.
Clinical severity determination
Disease severity was assessed using a Clinical Severity Score (CSS) devised for the study based on clinical and published experience with this patient population. A composite score ranging from 5 (profound impairment) to 25 (no impairment) was calculated from 5 clinical subdomains (cognition, communication, motor, psychiatric, and epilepsy), each scored from 1 (worse) to 5 (no impairment).
The cognitive subdomain was assessed using neuropsychological evaluation of age-appropriate cognitive testing (Mullen Scales of Early Learning, Differential Ability Scale, Second Edition [DAS-II], or Wechsler Abbreviated Scale of Intelligence, Second Edition). If participants scored below the floor value on cognitive testing, the floor value was assigned as the score. The motor subdomain was assessed using the Movement Assessment Battery for Children, Second Edition, and clinical evaluation. The psychiatric and communication subdomains were assessed using the Vineland, Third Edition, Child/Adult Behavioral Checklist, Behavior and Sensory Interests Questionnaire, Receptive-Expressive Emergent Language Test, Third Edition, and by clinical evaluation and medical history review. In addition, symptoms of autism spectrum disorder (ASD) were measured using the Autism Diagnostic Observation Scale (ADOS) or the Autism Observation Scale for Infants (AOSI), depending on age/functional level. The subdomain of epilepsy was assessed by severity of epilepsy based on the presence of status epilepticus, seizure frequency, and whether there were convulsive episodes.
Statistical analysis
Given the recognized variable clinical presentation depending on age,9 the analysis of clinical severity was undertaken for the whole population and stratified as above and below 12 years of age. One participant was excluded from the analysis due to the presence of a neurodegenerative disorder in addition to SSADH deficiency. Qualitative variables are expressed in percentages and relationship contrasts were analyzed using the χ2 or Fisher exact test. Statistical analyses were conducted with SPSS, version 24 (released 2016; IBM SPSS Statistics for Windows version 24.0, Armonk, NY).
Data availability
Anonymized data not provided in the article will be shared by request from any qualified investigator for purposes of replicating procedures and results.
Results
The natural history study consisted of 24 patients with confirmed SSAHD from 22 unrelated families. The study group consisted of 21 evaluated at BCH and 3 in the SOC cohort. There were 23 different pathologic ALDH5A1 variants. The majority of patients were compound heterozygotes, with 3 patients having homozygous missense variants. Table 1 contains information about the clinical characteristics of the study population, and a breakdown by age (above or below 12 years). Epilepsy was more prevalent in the adolescent/adult cohort: 83% (5/6) >12 vs 11% (2/18) <12 (p = 0.003). Antiseizure medications (ASMs) used in the cohort were carbamazepine, clobazam, lamotrigine, levetiracetam, oxcarbazepine, phenobarbital, topiramate, and valproate. One patient had daily seizures and takes 3 ASMs; all others had a seizure frequency of 2 per year or less and are on 1–2 ASMs. One patient (age 10) was recently diagnosed with epilepsy, and was not on daily ASMs. The most common EEG abnormality was diffuse background slowing in 63% (15/24). Other findings included focal spikes in 2 patients, sharp waves in 2 patients, and generalized spike and wave in 1 patient. Focal spikes were seen in 2 patients who do not have a history of seizures.
Table 1.
Clinical characteristics of study participants

Novel and common variants
Seven novel pathologic variants were identified: 2 missense, 3 splice site, and 2 frameshift variants (table 2). The missense variants were c.416C>A (p.Ala139Asp) and c.1321G>A (p.Gly441Arg); splice site variants were c.870+3delA, c.1015-3C>G, and c.1402+2T>C. The novel frameshift variants were c.1253del (p.Asn418llefs*39) and c.967_968dupCA (p.Gln323Hisfs*4).
Table 2.
Novel pathologic ALDH5A1 variants (NM_001080.3)

Triplicate overexpression experiments of the 2 novel missense variants showed no residual enzymatic activity, demonstrating their pathogenic nature. Western blot and RNA expression data confirmed the overexpression of the SSADH constructs, validating the enzyme activity results (figure 1). The in silico pathogenicity predictions of the missense variants are in line with the functional data. The 3 intronic variants are localized in the canonical splice sites and are predicted to be pathogenic. The deletion/duplication variants are predicted to result in frameshifts and should be also considered pathogenic.
Figure 1. Confirmation of successful transfection of the 2 missense succinic semialdehyde dehydrogenase (SSADH) alleles in HEK293 by Western blot and reverse transcription PCR (RT-PCR).
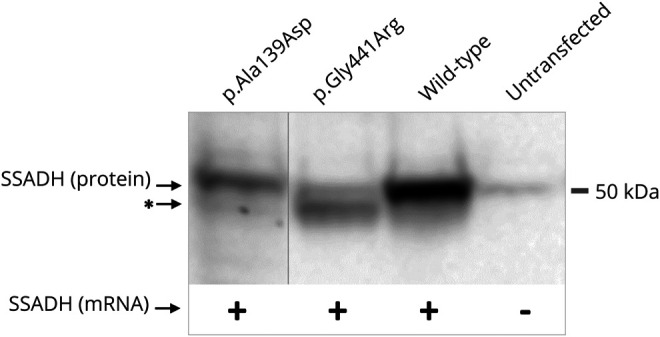
Cell pellets were resuspended in urea lysis buffer (10 mmol/L Tris HCl, 8 mol/L urea, 100 mmol/L NaCl, pH 8.0) and the protein content was determined using the bicinchoninic acid method (Sigma-Aldrich, St Louis, MO). Thirty micrograms of total protein was size-separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using a 12% stain‐free TGX gel (Bio‐Rad Laboratories, Hercules, CA). Proteins were transferred to a polyvinylidene fluoride membrane using the Trans‐Blot Turbo Transfer System (Bio‐Rad). Immunodetection was performed using the ALDH5A1 mouse polyclonal antibody (B01P; Abnova Corporation, Taipei, Taiwan), anti-mouse secondary antibody (A9044; Sigma-Aldrich, St Louis, MO), and enhanced chemiluminescent substrates (Lumi-Light Plus Western Blotting Substrate; Roche Applied Science, Indianapolis, IN). Images were acquired and analyzed with the ChemiDoc MP imager and Image Lab software (Bio-Rad). Total RNA was isolated from the HEK293 transfectants and cDNA was synthesized using standard molecular biology techniques. mRNA expression of the transfected constructs is confirmed by RT-PCR amplification of the full length cloned ALDH5A1 open reading frame, with the forward primer specific for the pAD1/RSV ALDH5A1 cDNA sequence.19 *Lower molecular weight protein band detected for the p. Gly441Arg construct, which could be due to the instability and partial degradation of the mutated protein, but alterations in posttranslational protein modification cannot be excluded.
An additional missense variant c.1277G>A (p.Cys426Tyr) was discovered from a patient who was screened. This variant was predicted to be possibly damaging by PolyPhen 2 (score of 0.815), deleterious by SIFT (score of 0.02), and a polymorphism by Mutation Taster (p value 0.994). Despite a clinical presentation that included ASD and global developmental delay, this patient had no abnormal levels of GHB detected in 2 samples of urine organic acid testing by CLIA-approved testing (1 qualitative and 1 quantitative). In addition, a blood test did not reveal abnormal levels of GHB. Given that the diagnosis requires biochemical (4-hydroxybutyric aciduria) and molecular confirmation, this variant was classified as nonpathogenic and led to exclusion of the patient from enrollment.
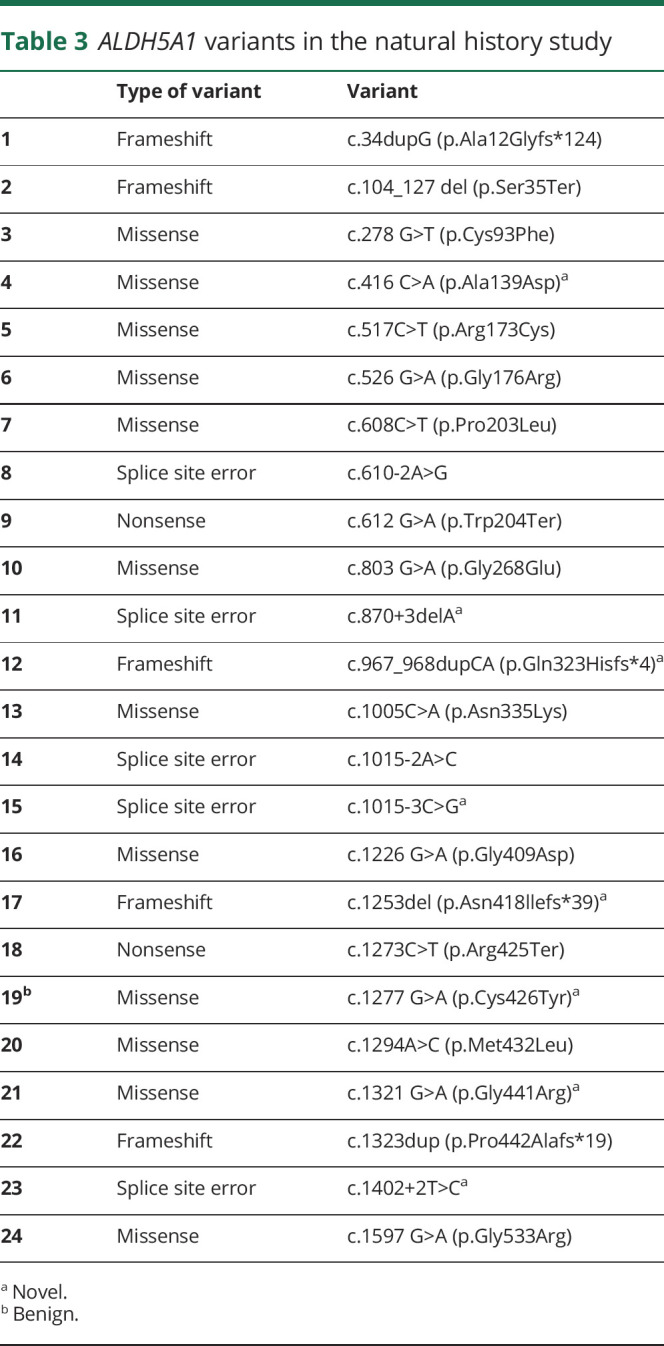
A number of variants were present in multiple families (figure 2). Four previously reported variants were identified in >5% unrelated families: c.612G>A (p.Trp204Ter) (11 participants, 1 homozygous), c.1226G>A (p.Gly409Asp) (3 subjects, 2 homozygous), c.803G>A (p.Gly268Glu) (3 participants), and c.1015-2A>C (8 participants). Table 3 contains the variants present in the cohort.
Figure 2. ALDH5A1 variants in the study population.
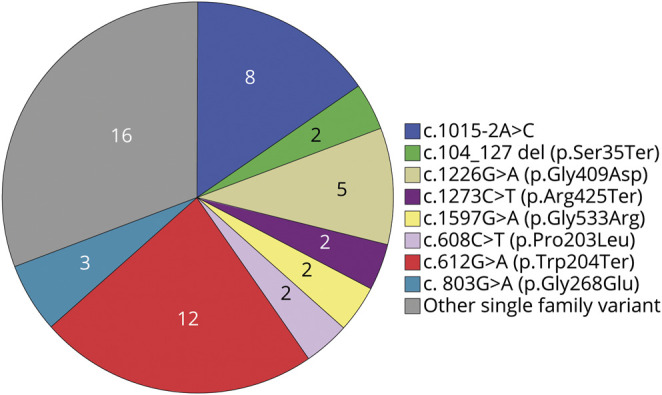
Table 3.
ALDH5A1 variants in the natural history study
Clinical severity
The composite CSS did not show significant correlation for either the gene variant group (p = 0.519) or age (p = 0.115). There was also no correlation between the gene variant group and the independent subdomains of the CSS. The clinical severity of the epilepsy domain did correlate with age, being higher in the older cohort (p = 0.002) (figure 3). SUDEP occurred in a single patient in this cohort, a boy age 14 years with compound heterozygosity for ALDH5A1 variants c.1015-2A>C and c.1005C>A (p.Asn335Lys). Prior to this event, epilepsy was well-controlled on 2 ASMs. There was also an increased incidence of obsessive-compulsive disorder in the older cohort (p = 0.016) and a tendency for more severe psychiatric severity in the older cohort (p = 0.09).
Figure 3. Epilepsy severity with age, showing overall worse severity in the older age group (p = 0.002).

Neuropsychological evaluation was obtained on 21 of 24 patients (3 were in the SOC cohort). Eight (38%) scored below the floor values for the cognitive assessment. The median IQ scores were full scale 53 (Q25–Q75 49–61, range 30 [floor value] to 87), verbal 56 [46–65], range 30 [floor value] to 95), and nonverbal 49 (47–62; range 30 [floor value] to 84). Overall, there was no correlation between the gene variant group and degree of intellectual disability (p = 0.370). Autism screening was obtained on 20/21 patients; one patient fell during the study and could not complete this. The ADOS was done on 18 patients, with 6 patients' scores consistent with autism, 4 on the autism spectrum, and 8 nonspectrum. Of note, all the patients who scored as nonspectrum were under 12 years of age (p = 0.083). There was no correlation between the gene variant and ADOS score (p = 0.414). The AOSI was completed on 2 patients who both scored below the ASD cutoff score, and were classified as non-ASD.
A single patient, a 9-year-old boy, scored in the normal IQ range (full-scale IQ 87 on the DAS-II). In addition, he was the only patient to score above the first %ile on the Movement Assessment Battery (total score = 5th %ile), and had the least severity on the CSS (20 of 25 maximum; overall median 15, interquartile range 25–75 12.75–17). He had a novel variant c.1321G>A (p.Gly441Arg) and a known variant present in over 5% of the patients: c.612G>A (p.Trp204Ter). He initially presented at 16 months with an expressive language deficit and hypotonia, and was diagnosed with autism prior to the SSADH diagnosis at 2.5 years old. No seizures have been reported, and the EEG showed mild diffuse background slowing without epileptiform features.
A single patient had the maximum severity level on the CSS (5 of a possible 25), an 8-year-old girl, who presented with neonatal hypotonia followed by infantile spasms, hypsarrhythmia on EEG, and later status epilepticus at age 14 months followed by neurologic regression. She experiences 1–2 daily seizures on 3 ASMs. Her genotype was compound heterozygous c.1294A>C (p.Met432Leu); c.610-2A>G.
Discussion
SSADH deficiency is a lifelong neurometabolic disorder with onset in early childhood and evolving features over time. Infantile onset and a severe phenotype presents in a minority of cases.21 We analyzed the genotype variants of patients participating in our natural history study with confirmed clinical and biochemical diagnosis of SSADH deficiency. Novel pathologic variants are described, including missense, splice site, and frameshifts. Four previously published variants were identified with high incidence (>5% of patients).11,13 The pathogenic nature of the novel missense variants was confirmed using overexpression experiments followed by SSADH activity measurements. The novel pathologic variants caused clinical disease in the patients confirmed by consistently increased urine GHB and clinical phenotyping.
Epilepsy is comorbid in half of patients with SSADH deficiency and increases in incidence during adolescence and adulthood, with a SUDEP incidence of nearly 15% in affected adults.9 Correlations between clinical features and age in the current study show an increased presence of epilepsy and obsessive-compulsive disorder, and epilepsy severity, with increased age. The CSS appears to be more applicable when assessing individual domains instead of a global score.
ALDH5A1 variants reported in 3 SUDEP cases were c.1015-2A>C; c.1005C>A (p.Asn335Lys), c.1226G>A (p.Gly409Asp), and c.608C>G (p.Pro203Arg).8,22 These variants were also seen in 13 of 24 participants in the present study, including the 1 patient who died of SUDEP. There was no statistical relationship between the occurrence of SUDEP and genotype. Each adolescent and adult with these variants had epilepsy (figure 4).
Figure 4. ALDH5A1 variants in the adolescent/adult cohort and association with epilepsy and sudden unexpected death in epilepsy (SUDEP).

We report on neuropsychological evaluation in 21 patients enrolled in our SSADH deficiency natural history study thus far. Nearly 40% (8 of 21) scored so low that floor values had to be applied. While there was a wide range of IQ scores (30–87), the median value of 53 fell in the range of moderate impairment, consistent with our previous studies reporting a baseline average full-scale IQ of 44 (range 34–55).23 While there was no overall correlation between the gene variant group and degree of intellectual disability, we did identify unique genotype correlation in patients at the highest and lowest ends of functionality. The single patient with a normal IQ and highest functioning on the other measures (Movement Assessment Battery and CSS) had a novel missense variant (c.1321G>A [p.Gly441Arg]) that we suspect correlates with a milder phenotype.
The missense variant c.1294A>C (p.Met432Leu) in the patient with the most severe phenotype was also described in an affected boy at age 23 months with severe neurologic deficits with a similar rapidly progressive course presenting in the infantile period with hypotonia, lack of head control, and absent visual fixation, followed by seizure onset and deterioration at 8 months.24 This patient was similarly heterozygous for c.1294A>C (p.Met432Leu), as was our patient with a virtually identical time course and severe residuae. Both of these patients are of Asian descent. We are unaware of other cases with this variant. In addition, the variants present in other published cases with a severe phenotype (c.1501_1503del [p.Glu501del]; c.901A>G [p.Lys301Glu]) were not observed in our cohort.16,17
We report a novel pathogenic variant in a patient with SSADH deficiency with normal cognition, a previously reported pathogenic variant that appears to have a consistent correlation with high phenotypic severity, and a previously unreported benign variant. We found an overall correlation between age and severity of epilepsy and presence of obsessive-compulsive disorder in SSADH deficiency, and suspect a genotype–phenotype correlation in the case of those variants associated with the best and worst outcomes on our CSS. A limitation of our study is the relatively small number of patients, although this is an ultrarare disease.
Glossary
- ADOS
Autism Diagnostic Observation Scale
- AOSI
Autism Observation Scale for Infants
- ASD
autism spectrum disorder
- ASM
antiseizure medication
- BCH
Boston Children's Hospital
- CSS
Clinical Severity Score
- DAS-II
Differential Ability Scale, Second Edition
- GABA
γ-aminobutyric acid
- GHB
γ-hydroxybutyric acid
- SOC
standard of care
- SSADH
succinic semialdehyde dehydrogenase
- SUDEP
sudden unexpected death in epilepsy
Appendix. Authors
Study funding
Funding provided by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, grant 5R01HD091142, and the BCH Intellectual and Developmental Disabilities Research Center (BCH IDDRC, U54HD090255).
Disclosure
M.L. DiBacco is supported in part by a research grant from PTC Therapeutics and NICHD grant 5R01HD091142, both paid to Boston Children's Hospital. Ana Pop, G.S. Salomons, and E. Hanson report no disclosures or conflicts of interest. J.-B. Roullet is funded by NICHD grant 5R01HD091142. K.M. Gibson is funded by NICHD grant 5R01HD091142. P.L. Pearl is supported in part by a research grant from PTC Therapeutics and NICHD grant 5R01HD091142, both paid to Boston Children's Hospital. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000010730 for full disclosures.
References
- 1.Malaspina P, Picklo MJ, Jakobs C, Snead OC, Gibson KM. Comparative genomics of aldehyde dehydrogenase 5a1 (succinate semialdehyde dehydrogenase) and accumulation of gamma-hydroxybutyrate associated with its deficiency. Hum Genomics 2009;3:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jansen EE, Vogel KR, Salomons GS, Pearl PL, Roullet JB, Gibson KM. Correlation of blood biomarkers with age informs pathomechanisms in succinic semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J Inherit Metab Dis 2016;39:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johansen SS, Wang X, Sejer Pedersen D, et al. Gamma-hydroxybutyrate (GHB) content in hair samples correlates negatively with age in succinic semialdehyde dehydrogenase deficiency. JIMD Rep 2017;36:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearl PL, Novotny EJ, Acosta MT, Jakobs C, Gibson KM. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol 2003;54:S73–S80. [DOI] [PubMed] [Google Scholar]
- 5.Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417. [DOI] [PubMed] [Google Scholar]
- 6.Pearl PL, Capp PK, Novotny EJ, Gibson KM. Inherited disorders of neurotransmitters in children and adults. Clin Biochem 2005;38:1051–1058. [DOI] [PubMed] [Google Scholar]
- 7.Gibson KM, Gupta M, Pearl PL, et al. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria). Biol Psychiatry 2003;54:763–768. [DOI] [PubMed] [Google Scholar]
- 8.Lapalme-Remis S, Lewis EC, De Meulemeester C, et al. Natural history of succinic semialdehyde dehydrogenase deficiency. Neurology 2015;85:861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiBacco ML, Roullet JB, Kapur K, et al. Age-related phenotype and biomarker changes in SSADH deficiency. Ann Clin Transl Neurol 2018;6:114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis 2009;32:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akaboshi S, Hogema B, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat 2003;22:442–450. [DOI] [PubMed] [Google Scholar]
- 12.Pearl PL, Parviz M, Vogel K, Schreiber J, Theodore WH, Gibson KM. Inherited disorders of gamma-aminobutyric acid metabolism and advances in ALDH5A1 mutation identification. Dev Med Child Neurol 2015;57:611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leo S, Capo C, Ciminelli BM, et al. SSADH deficiency in an Italian family: a novel ALDH5A1 gene mutation affecting the succinic semialdehyde substrate binding site. Metab Brain Dis 2017;32:1383–1388. [DOI] [PubMed] [Google Scholar]
- 14.Lin CY, Weng WC, Lee WT. A novel mutation of ALDH5A1 gene associated with succinic semialdehyde dehydrogenase deficiency. J Child Neurol 2015;30:486–489. [DOI] [PubMed] [Google Scholar]
- 15.Menduti G, Biamino E, Vittorini R, et al. Succinic semialdehyde dehydrogenase deficiency: the combination of a novel ALDH5A1 gene mutation and a missense SNP strongly affects SSADH enzyme activity and stability. Mol Genet Metab 2018;124:210–215. [DOI] [PubMed] [Google Scholar]
- 16.Puttmann L, Stehr H, Garshasbi M, et al. A novel ALDH5A1 mutation is associated with succinic semialdehyde dehydrogenase deficiency and severe intellectual disability in an Iranian family. Am J Med Genet A 2013;161A:1915–1922. [DOI] [PubMed] [Google Scholar]
- 17.Tay CG, Ariffin H, Yap S, Rahmat K, Sthaneshwar P, Ong LC. Succinic semialdehyde dehydrogenase deficiency in a Chinese boy: a novel ALDH5A1 mutation with severe phenotype. J Child Neurol 2015;30:927–931. [DOI] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blasi P, Boyl PP, Ledda M, et al. Structure of human succinic semialdehyde dehydrogenase gene: identification of promoter region and alternatively processed isoforms. Mol Genet Metab 2002;76:348–362. [DOI] [PubMed] [Google Scholar]
- 20.Gibson KM, Lee CF, Chambliss KL, et al. 4-Hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta 1991;196:219–221. [DOI] [PubMed] [Google Scholar]
- 21.Pearl PL, Acosta MT, Theodore WH, et al. Human SSADH deficiency: phenotype and treatment strategies. In: Hoffmann GF, ed. Disease of Neurotransmission: From Bench to Bed. Heilbronn: SPS Verlagsgesellschaft; 2006:187–198. [Google Scholar]
- 22.Knerr I, Gibson KM, Murdoch G, et al. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr Neurol 2010;42:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber JM, Pearl PL, Dustin I, et al. Biomarkers in a taurine trial for succinic semialdehyde dehydrogenase deficiency. JIMD Rep 2016;30:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamakawa Y, Nakazawa T, Ishida A, et al. A boy with a severe phenotype of succinic semialdehyde dehydrogenase deficiency. Brain Dev 2012;34:107–112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not provided in the article will be shared by request from any qualified investigator for purposes of replicating procedures and results.