

ABSTRACT
A large number of distal cis-regulatory elements (cREs) have been annotated in the human genome, which plays a central role in orchestrating spatiotemporal gene expression. Since many cREs regulate non-adjacent genes, long-range cRE-promoter interactions are an important factor in the functional characterization of the engaged cREs. In this regard, recent studies have demonstrated that identification of long-range target genes can decipher the effect of genetic mutations residing within cREs on abnormal gene expression. In addition, investigation of altered long-range cREs-promoter interactions induced by chromosomal rearrangements has revealed their critical roles in pathogenic gene expression. In this review, we briefly discuss how the analysis of 3D chromatin structure can help us understand the functional impact of cREs harboring disease-associated genetic variants and how chromosomal rearrangements disrupting topologically associating domains can lead to pathogenic gene expression.
Keywords: cis-regulatory element, long-range chromatin contact, Hi-C, GWAS-SNPs
Introduction
During mammalian development, a fertilized egg gives birth to various cell types and organs throughout the delicate regulation of spatiotemporal gene expression. Systematic investigation of chromatin architecture in various developmental stages and cell-types has shown that such regulation processes are tightly controlled by cis-regulatory elements (cREs) [1–3]. Although cREs refer to all cis-acting noncoding DNA elements that regulate target gene expression such as promoters, enhancers, and silencers, in this review, we mainly focus on enhancer-like cREs. Specific histone modifications such as mono-methylation of histone H3 at lysine 4 (H3K4me1) and co-activator binding patterns are hallmarks of cREs [4]. In addition, DNA methylation and chromatin accessibility are good predictors to identify candidates of regulatory DNA elements [5,6]. Active cREs can be further specified by the enriched acetylation of histone H3 lysine 27 (H3K27ac), bi-directional transcripts (also known as enhancer RNAs), and even tri-methylation of histone H3 lysine 4 (H3K4me3) that is a hallmark of active promoters [7–10]. These chromatin signatures have been utilized to annotate hundreds of thousands of cREs in the mammalian genome through incorporation with machine-learning based computational methods [11–15].
Despite a remarkable achievement in annotating cREs, most of them often regulate non-adjacent genes over large genomic distances, making it challenging to characterize their functional properties [16–19]. Such long-range regulation can take place because chromatin fibers are folded into a higher-order structure, forming DNA loops where regulatory proteins bound at cREs can be in physical closeness to the promoters of their target genes. Thus, target gene identification based on spatial chromatin contacts is crucial in elucidating the regulatory potential of cREs during both normal and pathogenic gene expression control. In this aspect, we will briefly review how the analysis of chromatin interactions is critical to define target gene promoters of cREs and to understanding disease pathogenesis.
Methods to identify target genes based on long-range chromatin interactions
The establishment of Chromosome Conformation Capture (3C) [20] has evolved in the form of high-throughput versions such as 4C, 5C, and Hi-C to detect genome-wide chromatin interactions [21–24]. Through a combination of proximity ligation with next-generation sequencing, these approaches have identified many chromatin contacts of cRE-promoter pairs and provided a comprehensive view of the regulatory interactome [25,26].
To concentrate on well-annotated promoter connecting long-range chromatin interactions, Capture-C and promoter capture Hi-C methods have been developed [17,27–29]. These methods provide high-resolution cRE-promoter interaction maps with a relatively low sequencing cost by capturing ligated DNA fragments containing targeted promoters from 3C or Hi-C libraries. The unprecedented high-resolution cRE-promoter interaction maps have revealed that cell-type specific chromatin interactions often connect cell-type specific promoter and cRE pairs [17,28]. These data strongly support the essential function of long-range chromatin interactions in cell-type specific cRE usage.
To further examine the chromatin interactions bound by a particular protein, Chromatin Interaction Analysis by Paired-End-Tag sequencing (ChIA-PET), HiChIP, and Proximity Ligation-Assisted ChIP-seq (PLAC-seq) have been developed through a combination of ChIP and proximity ligation [30–33]. In ChIA-PET, chromatin immunoprecipitation of a particular protein is performed, followed by proximity ligation to collect chromatin interactions mediated by the target protein. In contrast, HiChIP and PLAC-seq conduct proximity ligation in nuclei prior to immunoprecipitation. Currently, HiChIP and PLAC-seq are widely used owing to their high efficiency and low input requirements.
The rapid accumulation of the rich resource in chromatin interactions enables the development of several computational methods to systematically identify target genes of cREs [34–36]. For example, PSYCHIC annotates several hundred thousand putative cREs and identified their target genes through the analysis of Hi-C data [35]. The development of computational methods in combining various genomic and epigenomic features will be further beneficial for the target gene annotation compared to solely based on chromatin contacts.
Functional annotation of GWAS-SNPs based on long-range chromatin interactions
Many important genetic variants associated with complex human diseases and traits have been identified thanks to the power of genome-wide association studies [37]. Interestingly, more than 95% of GWAS-identified SNPs are in noncoding sequences, and more than three-quarters are associated with open chromatin regions, suggesting their strong association with regulatory elements [38]. Thus, distal target gene information is essential to identify genes they affect. For example, multiple GWAS studies to identify genetic risk factors in obesity had consistently demonstrated variants within the FTO introns as significantly associated variants with the risk of obesity [39–41]. However, there was no clear explanation of how these variants regulate FTO gene expression and potentially cause obesity. This conundrum has been resolved through the analyses of long-range chromatin interactions that have revealed that obesity-associated GWAS-SNPs create cRE activity, a potent preadipocyte super-enhancer, and physically interact with distal promoters of IRX3 and IRX5 (Figure 1) [42,43]. Further experiments found that increased expression of IRX3 and IRX5 genes resulted in changes of body mass index [42,43]. As illustrated in the above example, target gene identification of noncoding associated GWAS-SNPs based on long-range chromatin interactions has been widely applied as a novel strategy to understand gene regulation mechanisms for various complex human diseases.
Figure 1.
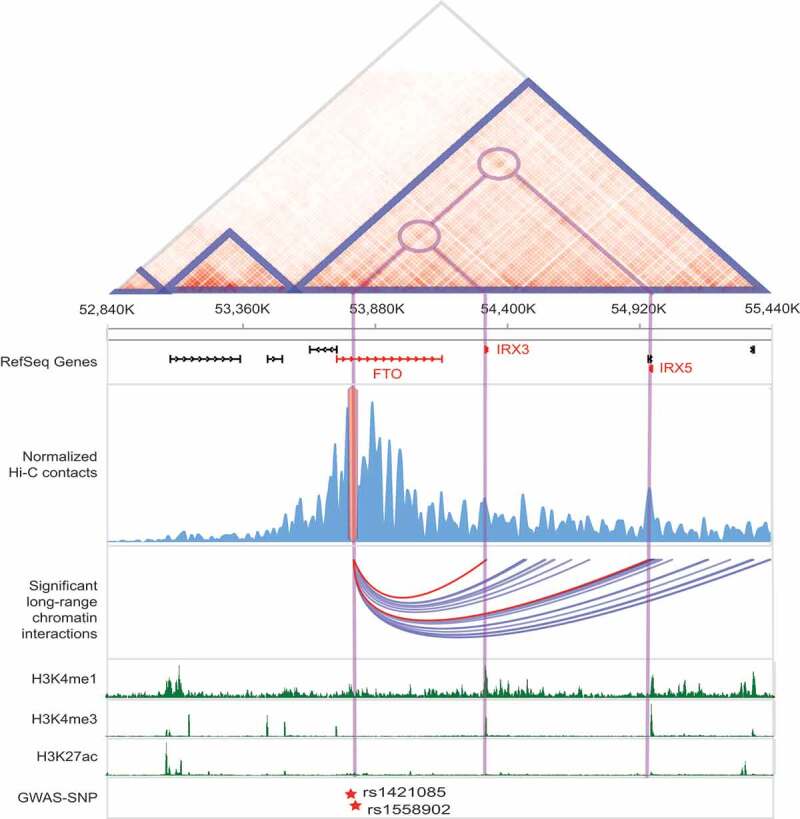
Target gene identification of cREs harboring obesity-associated GWAS-SNPs
Visualization of significant long-range chromatin interactions (arcs) centered on obesity-associated GWAS-SNPs (rs1421085 and rs1558902) located at FTO intron in H1-derived mesenchymal stem cell. From the top, normalized Hi-C contact map with annotation of TADs (blue triangles), RefSeq gene annotation, normalized Hi-C contacts with obesity-associated GWAS-SNPs located in intronic regions of FTO gene, identified significant chromatin interactions, multiple histone modification marks, and GWAS-SNPs are shown together. The long-range chromatin interactions with the promoters of IRX3 and IRX5 are highlighted by red. All data were drawn from 3DIV database [36].
Pathogenic long-range chromatin interactions in human diseases
Large-scale chromosomal rearrangements such as structural variants (SVs) can disrupt the organization of topologically associating domains (TADs), thereby inducing aberrant gene expression (Table 1). TADs are megabase-scale local chromatin interaction domains that restrict the interactions between cREs and promoters outside of TAD [44,45]. Thus, changes in the TAD structure can rewire long-range chromatin interactions between cRE and promoter, leading to introducing ectopic cRE-gene interactions called enhancer-hijacking or enhancer-adoption [46–48]. Several studies have shown that TAD fusions, a fused form of multiple TADs due to genomic rearrangements, can activate proto-oncogenes by enhancer-hijacking mechanism [49–55]. Moreover, translocations and inversions spanning TAD boundaries can be responsible for TAD shuffling, which may disrupt originally linked cRE-promoter relationships [56]. Therefore, clarifying target genes of cREs based on rewired chromatin interactions can deeply elucidate the pathological consequence of noncoding-associated genomic rearrangement.
Table 1.
Rewired cRE-promoter interactions in pathogenic gene expression control
| Disease | Type of enhancer usage | Variant type | Cause | Result | Reference |
|---|---|---|---|---|---|
| Limb malformation | Enhancer-hijacking | Inversion, duplication, deletion |
TAD fusion by variants at CTCF-associated boundary domain | Overexpression of WNT or 6IHH or PAX3 | 47 |
| T cell acute lymphoblastic leukemia | Enhancer-hijacking | Deletion | TAD fusion by variants at CTCF-associated boundary domain | Activation of LMO2 proto-oncogene | 50 |
| Medulloblastoma | Enhancer-hijacking | Tandem duplication, deletion, inversion, complex rearrangement | Relocating oncogene proximal to super-enhancer | Activation of GFI1 and GFI1B proto-oncogenes, and overexpression of PRDM6 | 52, 51 |
| Oncogene activation | Enhancer-hijacking | Tandem duplication | neo-TAD appearance | Overexpression of IGF2 | 55 |
| Neuroblastoma | Enhancer-hijacking | Copy number alterations | Relocating oncogene proximal to super-enhancer | Overexpression of TERT | 53, 54 |
| Branchiooculofacial syndrome | Enhancer disconnection | Inversion | TAD shuffling | Monoallelic and reduced TFAP2A expression | 56 |
| Acute myeloid leukemia | Enhancer-hijacking | Inversion | TAD shuffling | Activation of EVI1 proto-oncogene | 49 |
Conclusion
The analysis of chromatin interactions has dramatically advanced our knowledge of understanding disease pathogenesis by identifying target gene information of noncoding located regulatory elements. However, a range of issues remains to be solved. First, the resolution of the 3C based methods to identify chromatin interactions is often too low, generally from kilobases to tens of kilobases. Thus, it hinders their ability to pinpoint the precise location of interacting DNA fragments. Second, more efforts are needed to distinguish the type of cREs interacting with their target promoters. Although we have discussed enhancer-like cREs in this review, especially within a TAD, silencers also act in a cis manner to regulate their target genes similar to enhancers [57]. Recent studies experimentally screened potential silencers, which turns out that strongly silencer-associated DNA elements also pose active chromatin marks that can be misinterpreted as enhancers [57–59]. Thus, discriminating multiple types of regulatory elements is critical in the functional annotation of noncoding genetic variants. Lastly, the identification of target genes at single-cell resolution is ultimately required. Single-cell based Hi-C methods [60–63] and a combination of imaging of the 3D chromatin structure [64,65] will revolutionize our understanding of long-range chromatin interactions and their regulatory potential. Nevertheless, the identification of target genes of cREs based on “C” technologies is highly beneficial in unraveling the regulatory potential of noncoding-associated GWAS-SNPs and large-scale genomic rearrangements, bridging the gap between coding and noncoding sequences.
Acknowledgments
The authors thank to the members of the Jung laboratory for support and critical suggestions throughout the course of this work.
Funding Statement
This work was supported by the Ministry of Science and ICT through the National Research Foundation in Republic of Korea under Grant number NRF-2020R1A2C4001464;
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Consortium EP, Moore JE, Purcaro MJ, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Heintzman ND, Hon GC, Hawkins RD, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hon GC, Rajagopal N, Shen Y, et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45:1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thurman RE, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen K, Chen Z, Wu D, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Henriques T, Scruggs BS, Inouye MO, et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;32:26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Leung D, Jung I, Rajagopal N, et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Firpi HA, Ucar D, Tan K. Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics. 2010;26:1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].He Y, Gorkin DU, Dickel DE, et al. Improved regulatory element prediction based on tissue-specific local epigenomic signatures. Proc Natl Acad Sci U S A. 2017;114:E1633–E1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu F, Li H, Ren C, et al. PEDLA: predicting enhancers with a deep learning-based algorithmic framework. Sci Rep. 2016;6:28517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rajagopal N, Xie W, Li Y, et al. RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput Biol. 2013;9:e1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Amano T, Sagai T, Tanabe H, et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 2009;16:47–57. [DOI] [PubMed] [Google Scholar]
- [17].Jung I, Schmitt A, Diao Y, et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat Genet. 2019;51:1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–257. [DOI] [PubMed] [Google Scholar]
- [19].Sanyal A, Lajoie BR, Jain G, et al. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dekker J, Rippe K, Dekker M, et al. Capturing chromosome conformation. Science. 2002;295:1306–1311. [DOI] [PubMed] [Google Scholar]
- [21].Dostie J, Richmond TA, Arnaout RA, et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 2006;38:1348–1354. [DOI] [PubMed] [Google Scholar]
- [24].Zhao Z, Tavoosidana G, Sjolinder M, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. [DOI] [PubMed] [Google Scholar]
- [25].Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hughes JR, Roberts N, McGowan S, et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat Genet. 2014;46:205–212. [DOI] [PubMed] [Google Scholar]
- [28].Javierre BM, Burren OS, Wilder SP, et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384 e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mifsud B, Tavares-Cadete F, Young AN, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606. [DOI] [PubMed] [Google Scholar]
- [30].Fang R, Yu M, Li G, et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 2016;26:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fullwood MJ, Liu MH, Pan YF, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mumbach MR, Rubin AJ, Flynn RA, et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ay F, Bailey TL, Noble WS. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 2014;24:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ron G, Globerson Y, Moran D, et al. Promoter-enhancer interactions identified from Hi-C data using probabilistic models and hierarchical topological domains. Nat Commun. 2017;8:2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang D, Jang I, Choi J, et al. 3DIV: A 3D-genome Interaction Viewer and database. Nucleic Acids Res. 2018;46:D52–D57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. [DOI] [PubMed] [Google Scholar]
- [40].Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Scuteri A, Sanna S, Chen WM, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Claussnitzer M, Dankel SN, Kim KH, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Smemo S, Tena JJ, Kim KH, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim K, Eom J, Jung I. Characterization of structural variations in the context of 3D chromatin structure. Mol Cells. 2019;42:512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lupianez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Valton AL, Dekker J. TAD disruption as oncogenic driver. Curr Opin Genet Dev. 2016;36:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Groschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. [DOI] [PubMed] [Google Scholar]
- [50].Hnisz D, Weintraub AS, Day DS, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Peifer M, Hertwig F, Roels F, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Valentijn LJ, Koster J, Zwijnenburg DA, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015;47:1411–1414. [DOI] [PubMed] [Google Scholar]
- [55].Weischenfeldt J, Dubash T, Drainas AP, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Laugsch M, Bartusel M, Rehimi R, et al. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell. 2019;24:736–752 e712. [DOI] [PubMed] [Google Scholar]
- [57].Pang B, Snyder MP. Systematic identification of silencers in human cells. Nat Genet. 2020;52:254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Della Rosa M, Spivakov M. Silencers in the spotlight. Nat Genet. 2020;52:244–245. [DOI] [PubMed] [Google Scholar]
- [59].Ngan CY, Wong CH, Tjong H, et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet. 2020;52:264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee DS, Luo C, Zhou J, et al. Simultaneous profiling of 3D genome structure and DNA methylation in single human cells. Nat Methods. 2019;16:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li G, Liu Y, Zhang Y, et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat Methods. 2019;16:991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nagano T, Lubling Y, Stevens TJ, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Ramani V, Deng X, Qiu R, et al. Massively multiplex single-cell Hi-C. Nat Methods. 2017;14:263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ou HD, Phan S, Deerinck TJ, et al. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017;357:6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Su JH, Zheng P, Kinrot SS, et al. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell. 2020;182:1641–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]