

Abstract
Through the National Ambient Air Quality Standards (NAAQS), the Clean Air Act of the United States outlines acceptable levels of six different air pollutants considered harmful to humans and the environment. Included in this list is ozone (O3), a highly reactive oxidant gas, respiratory health hazard, and common environmental air pollutant at ground level. The respiratory health effects due to O3 exposure are often associated with molecular and cellular perturbations in the respiratory tract. Periodic review of NAAQS requires comprehensive scientific evaluation of the public health effects of these pollutants, which is formulated through integrated science assessment (ISA) of the most policy-relevant scientific literature. This review focuses on the protective and pathogenic effects of macrophages in the O3-exposed respiratory tract, with emphasis on mouse model-based toxicological studies. Critical findings from 39 studies containing the words O3, macrophage, mice, and lung within the full text were assessed. While some of these studies highlight the presence of disease-relevant pathogenic macrophages in the airspaces, others emphasize a protective role for macrophages in O3-induced lung diseases. Moreover, a comprehensive list of currently known macrophage-specific roles in O3-induced lung diseases is included in this review and the significant knowledge gaps that still exist in the field are outlined. In conclusion, there is a vital need in this field for additional policy-relevant scientific information, including mechanistic studies to further define the role of macrophages in response to O3.
Keywords: Ozone, macrophages, airspaces, lung, epithelial lining fluid
Ozone: a respiratory health hazard
Ozone (O3), a highly reactive oxidant gas, is a respiratory health hazard and one of the most common environmental air pollutants at ground level. Approximately, 90% of Earth’s O3 is confined to the stratosphere where it is generated when oxygen is exposed to ultraviolet radiation, electrical discharges, and heat (Aucamp 2007). In contrast, ground level (tropospheric) O3 is formed during a photochemical reaction between oxides of nitrogen and volatile organic hydrocarbons that originate from automobile emissions and other high-heat combustion-based industrial processes (Moller 2004). While stratospheric O3 is beneficial to life as it prevents the entry of harmful ultraviolet radiation into the earth’s atmosphere, tropospheric O3 causes serious health problems.
Elevated levels of ground-level O3 contribute to a significant increase in hospitalization resulting in substantial health and economic burdens (Schwartz 1994; Medina-Ramon et al. 2006; Lin et al. 2008; Moore et al. 2008). Increases in tropospheric O3 levels are associated with reduced pulmonary function, exacerbations of asthma and chronic obstructive pulmonary disease (COPD), and mortality (Halonen et al. 2010; Kariisa et al. 2015). Children, the elderly, and individuals with preexisting respiratory diseases are particularly vulnerable to elevated levels of ambient O3 (Burnett et al. 2001; Gent et al. 2003; Bell et al. 2004; Gryparis et al. 2004; Ito et al. 2005). According to one estimate, a 25-ppb increase in the levels of ambient O3 causes an approximately 4% increase in mortality (Parodi et al. 2005). Further, the unto-ward effects of O3 are worsened when other pollutants, such as particulate matter, allergens, and noxious gases are simultaneously inhaled (Jakab and Hemenway 1994; Linn and Gong 1999; Bernard et al. 2001; Schelegle et al. 2003; Wong et al. 2018).
Risk-assessment paradigm for ozone
O3 is one of six air pollutants, the others being carbon monoxide, lead, particulate matter, nitrogen dioxide, and sulfur dioxide identified under Section 109 of the Clean Air Act. The Clean Air Act requires the Environmental Protection Agency (EPA) to set National Ambient Air Quality Standards (NAAQS) for all criteria air pollutants, including O3. In the USA, the atmospheric levels of these air pollutants are closely monitored to attain NAAQS status. According to 2015 NAAQS, a level of O3 below 70 ppb (annual 4th highest daily maximum 8-h concentration averaged over 3 years) is required to achieve NAAQS attainment status (McClellan et al. 2009).
The exposure and risk assessment paradigm for O3 is employed by EPA administrators to make informed revisions of NAAQS (Figure 1). Studies focused on the toxicodynamic aspects of O3 toxicity are critical components of risk assessment and as a result, risk management. In particular, studies employing rodent models have continuously increased the mechanistic understanding of O3 toxicity. Here, we review critical advancements in understanding of the contribution of macrophages to O3-induced airway diseases.
Figure 1.
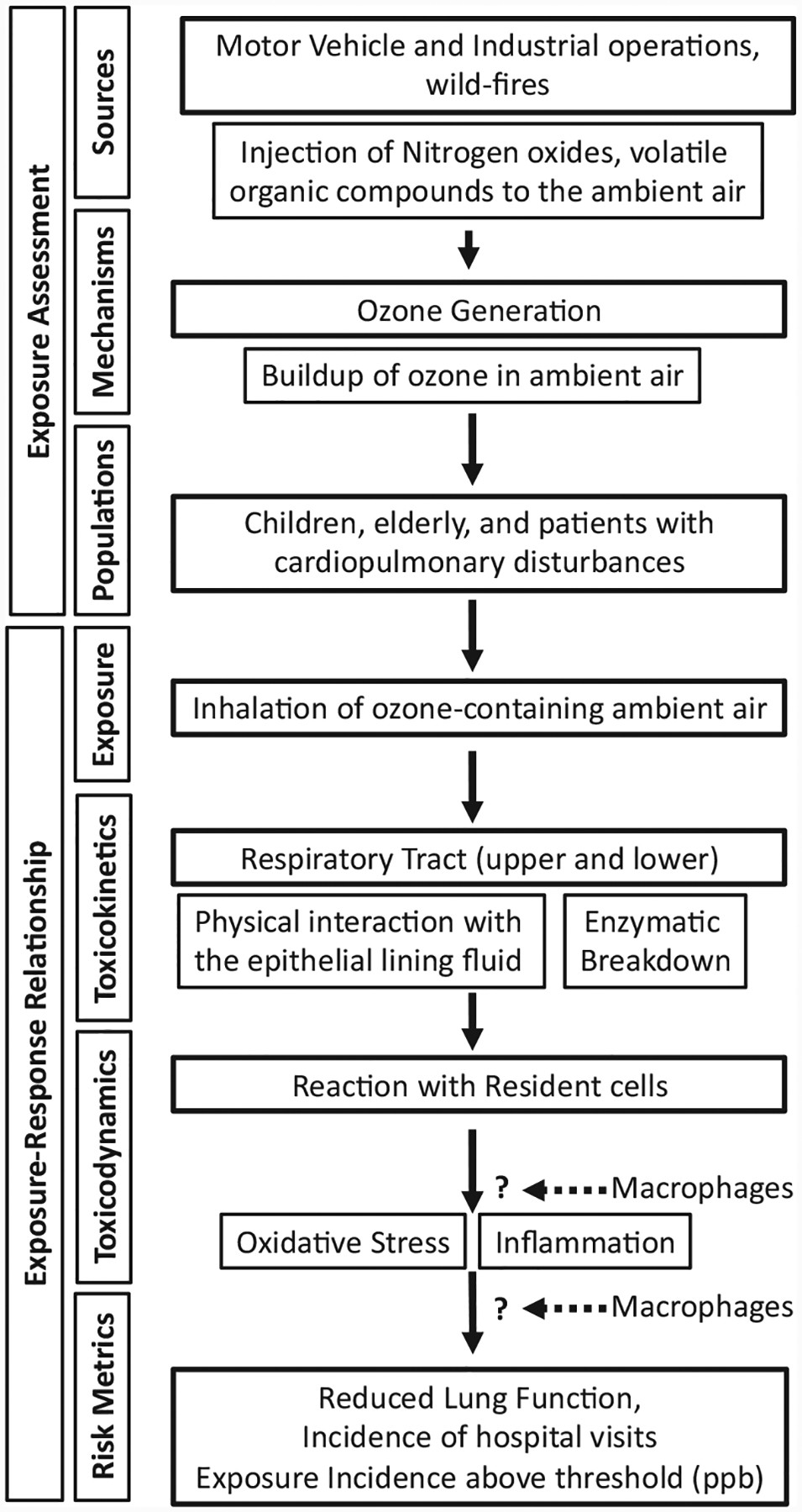
Risk assessment paradigm for ozone.
Lifecycle of ozone in the respiratory tract
Luminal surfaces of the airways and alveolar spaces are lined with an aqueous epithelial lining fluid (ELF) layer. The ELF layer acts as a physical barrier that prevents the direct onslaught of epithelial cells by inhaled entities including O3. Thus, before interacting with the resident cells in airspaces, the inhaled O3 interacts with the ELF layer of the conducting airways and alveolar spaces (Pryor 1992; Gerrity et al. 1995). The low solubility of O3 in the ELF layer limits its rate of diffusion toward the apical surfaces of the epithelial cells (Gerrity et al. 1995). During its diffusion toward the epithelial cell surfaces, a portion of the dissolved O3 is detoxified by antioxidants, including urate, ascorbate, vitamin E, and reduced glutathione, normally found in the ELF (DeLucia et al. 1975; Housley et al. 1995; Duan et al. 1996; Kelly et al. 1996; Mudway et al. 1996; Mudway and Kelly 1998; Mudway et al. 1999). Kelly et al. (1995) previously provided a detailed overview of the antioxidant defense system against O3.
While the ELF layer serves to protect the mucosal surfaces of the respiratory tract from inhaled agents, molecular interactions between O3 and constituents of the ELF layer result in the generation of harmful ozonation products. Studies employing radioactive oxygen (18O) labeled O3 revealed a higher concentration of 18O in the ELF suggesting its incorporation into ELF biomolecules (Hatch et al. 1994). The incorporation of O3 into the unsaturated carbon backbone of ELF constituents, including phospholipids, cholesterol, epoxy cholesterol, proteins, and hyaluronic acids (HA), leads to a rapid drop in its levels within the ELF layer before it reaches the epithelial cells (Johnson 1980; Gordon et al. 1981; Sharman and Mudd 1981; Friedman et al. 1985; Madden et al. 1987). Through the Criegee reaction, ozonation of unsaturated fatty acids yields aldehydes, such as hexanal, nonanal, heptanal, and carbonyl oxide. These aldehydes have been shown to be elevated in the BALF of O3-exposed humans and rats (Pryor et al. 1996; Frampton et al. 1999a, 1999b). Among these, carbonyl oxide, being a reactive species, further combines either with aldehydes to form ozonide (incorporation of O3 in carbon–carbon chain) or with water to form hydroxy hydroperoxide in aqueous environments (Santrock et al. 1992).
Ozonation of proteins at aliphatic amino acids results in the formation of nitrates, ammonia, carbonyl, and carboxyl byproducts (Mudd et al. 1969; Pryor and Uppu 1993; Kotiaho et al. 2000; Kelly and Mudway 2003). In addition, ozonation of proteins at aromatic amino acid residues (tyrosine, tryptophan, and phenylalanine), also results in alteration of the folding abilities of proteins, which in turn alters their tertiary structures and biologic activities (Berlett et al. 1996). For instance, O3 is known to reduce α1-proteinase activity (Johnson, 1980, 1987; Nadziejko et al. 1995), presumably through misfolding. The enzyme α1-proteinase is an endogenous inhibitor of neutrophil elastase, an enzyme that causes alveolar wall destruction resulting in pulmonary emphysema (Johnson 1980, 1987). The negative effect of O3 on α1-proteinase activity suggests that O3 may contribute to the alveolar space enlargement by negatively affecting α1-proteinase activity.
Given the poor solubility of O3 in the ELF layer, its significant detoxification by ELF antioxidants, and the molecular reactions of O3 with the constituents of the ELF layer, it is highly likely that for the most part, ozonation products produced within the ELF layer, rather than the O3 itself, interact directly with epithelial and immune cells, particularly macrophages. Since resident macrophages patrol the ELF layer, these cells are likely the first to respond to both dissolved O3 and ozonation products produced within the ELF (Figure 2). In the following sections, we will focus on the current understanding of lung macrophages and their responses to O3 exposure.
Figure 2.
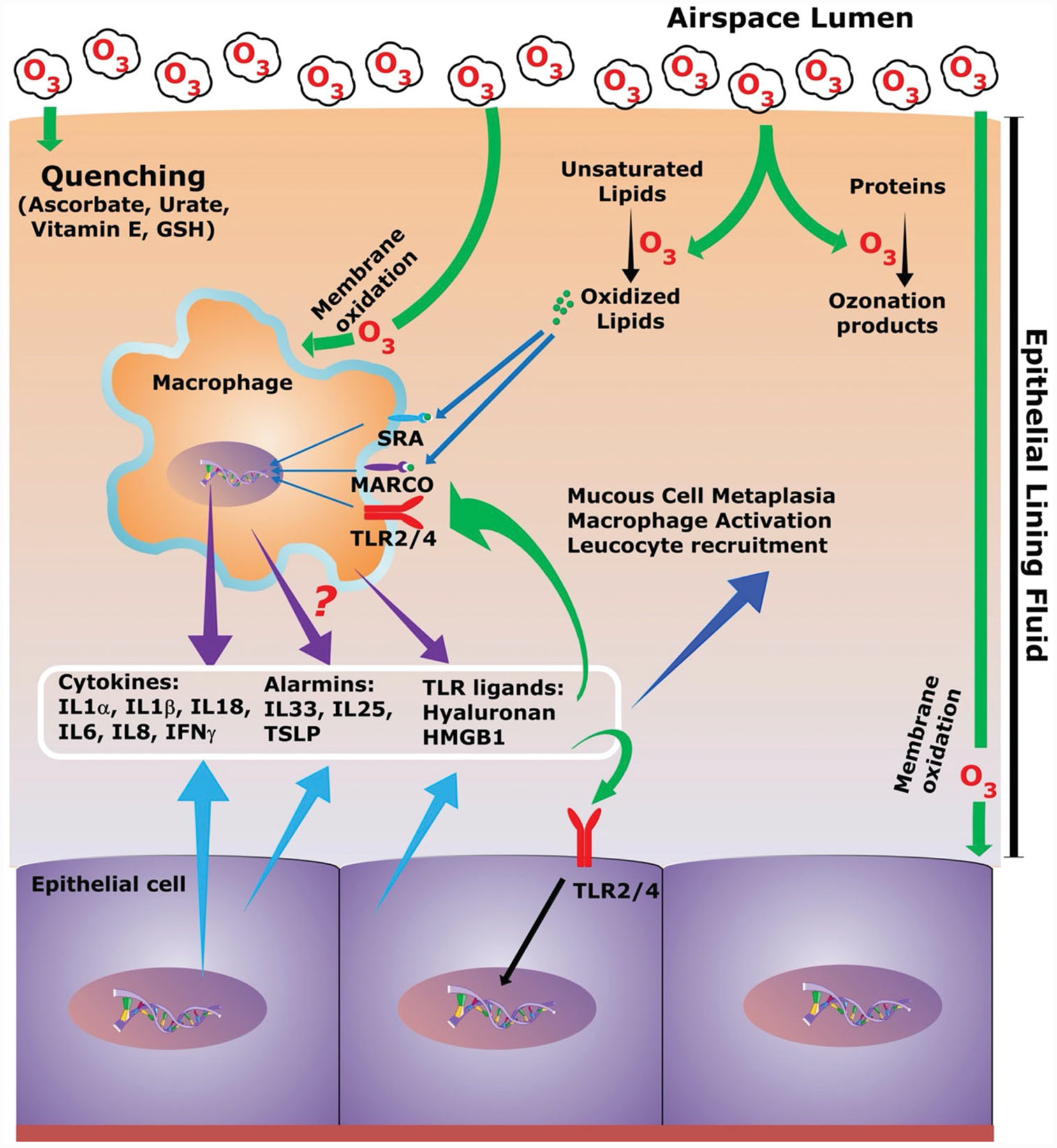
Schematic of the ozone life cycle in airspaces and the potential effects on airspace cells. Inhaled ozone molecules interact with the epithelial lining fluid (ELF) in the airway and alveolar compartments. Soluble ozone is either quenched by the antioxidant system of the ELF or reacts with lipids and proteins to form ozonation products. Ozone molecules also oxidize membrane proteins and lipids of epithelial cells and macrophages. Surface receptors such as TLRs, MARCO, and SR-A on macrophages are known to bind oxidized lipids. Resident cells, macrophages, and epithelial cells respond to the presence of ozone and ozonation products through the release of cytokines, alarmins, and Toll-like receptors (TLRs) ligands, which leads to multiple outcomes including immune cell recruitment, macrophage activation, and epithelial remodeling.
Lung macrophages
Macrophages are present in all vertebrate tissues where they play critical roles in tissue development, homeostasis, and pathogenesis of diseases (Epelman et al. 2014). Based on their anatomic locations, macrophages have been assigned organ-specific nomenclature including lung alveolar macrophages (AM), liver Kupffer cells, bone osteoclasts, and brain microglial cells (Davies et al. 2013; Hoeffel and Ginhoux 2015). Despite their distinct anatomical locations, macrophages throughout the vertebrate body act as professional phagocytes to clear pathogens, abiotic entities, and dead host cells. In addition, fine-tuned by their distinct tissue microenvironment, macrophages also possess varied functions including antigen-presentation, pathogen killing, tissue repair, tissue destruction, and scavenging (Wynn and Vannella 2016).
Due to their ability to sense molecular cues in the extra-cellular microenvironment and mount a wide array of functional responses, macrophages are regarded as a remarkably plastic cell type. In addition, macrophage populations within a tissue are heterogenous. While plasticity refers to the capability of a cell to acquire a distinct molecular and functional phenotype, macrophage heterogeneity refers to the ability of a macrophage population to exhibit a wide range of diverse molecular and functional phenotypes. This heterogeneity arises either due to the presence of cells that are in various stages of differentiation or due to differential responses to diverse extrinsic cues present in the immediate vicinity of the cells. Due to the presence of a multitude of surface receptors, including Toll-like receptors (TLRs), scavenger receptors, C-type lectin receptors, and cytokine receptors, macrophages possess extraordinary abilities to sense the presence of aberrant entities of both biotic and abiotic nature in the tissue microenvironment (Taylor et al. 2005; Ley et al. 2016).
Lung macrophages patrol the epithelial surfaces of the pulmonary airway and alveolar spaces and, thus, face a continued onslaught from a plethora of inhaled environmental insults (Hussell and Bell 2014). Alongside epithelial cells, lung macrophages constitute the first line of cellular host defense against inhalational exposure to aberrant biotic and abiotic agents. According to the nature and degree of the insult, macrophages respond in a variety of ways including functional modulation (activation), release of chemical mediators of inflammation that dictate recruitment of immune cells, and modulation of epithelial responses (Mosser and Edwards 2008). Macrophage-derived inflammatory mediators communicate with neighboring cells including neighboring naïve macrophages, other immune cells, and epithelial cells. The net overall response generally encompasses a wide variety of responses ranging from localized activation of macrophages, in the case of mild exposure, to large-scale systemic responses, in the case of severe exposure where increased macrophage recruitment and proinflammatory classical macrophage activation responses play critical roles in conjunction with epithelial- and other immune cell-driven responses.
Macrophage types in the lung
Broadly, lung macrophages are classified into two major categories, alveolar macrophages (AM) and interstitial macrophages (IM). AM reside in the two anatomically distinct airspace compartments, alveolar airspaces, and the airway lumen. IM are localized to the interstitial spaces between the alveolar epithelium and the endothelium of septal blood vessels. These two cell types are distinguished by the differential expression of surface receptors including CD11b, CD11c, and sialic acid-binding immunoglobulin-like lectin F (SIGLEC-F) (Misharin et al. 2013; Zaynagetdinov et al. 2013). AM and IM are defined as CD11b−CD11chighSIGLEC-Fhigh and CD11bintCD11c−SIGLEC-F−, respectively. In addition, AM express CD206 and CD204R at higher levels than IM (Misharin et al. 2013; Zaynagetdinov et al. 2013).
Origins of lung macrophages under steady state
At embryonic stage E18.5 (day of observance of vaginal plug is set at E0.5) in mice, fetal liver-derived monocytes start differentiating into the pre-alveolar macrophages (preAM) within the alveolar septa. At birth, these preAMs start migrating to the alveolar spaces as immature macrophages, where, under the action of granulocyte-macrophage colony-stimulating factors (GM-CSF), they develop into long-lived AM. This process carries on during the first week of postnatal life (Guilliams et al. 2013). Tan and Krasnow (2016) carried out detailed studies and identified three distinct waves of macrophage development in the lung. At E10.5 the first wave originates from the yolk sac resulting in a population of F4/80+ macrophages in the interstitial spaces. At E12.5, a second wave originates from the fetal liver and populates interstitial spaces with Mac2+macrophages. These macrophages then enter the alveolar spaces at the time of birth and give rise to self-renewing long-lived AM (Guilliams et al. 2013). Finally, the third wave starts with the recruitment of circulating monocytes into the interstitial spaces to give rise to F4/80+definitive IM (Tan and Krasnow 2016). Thus, these findings indicate that the AM in healthy lungs are a self-renewing population of macrophages originated from the fetal liver.
Origin of lung macrophages during inflammation and stress
Inhalation of aberrant entities into airspaces causes varied macrophage responses including pro-inflammatory, anti-inflammatory, and phagocytic responses as well as proliferation, apoptosis, and death. These responses likely contribute to the heterogeneity of alveolar macrophage populations via perturbed self-renewal and recruitment responses. In macrophage depletion models, circulating monocytes have been regarded as a central source of macrophages in the alveolar spaces. However, Landsman et al. (2007) showed that Gr1lowCX3CR1highCCR2− blood monocytes are the precursors of interstitial lung macrophages. These IM were later identified as precursors of AM (Landsman and Jung 2007). Using fate mapping and parabiosis studies in both steady state and macrophage depletion models, circulating monocytes were later ruled out as precursors for AM and a self-renewal mechanism was found to be responsible for the maintenance of resident alveolar macrophages (Hashimoto et al. 2013).
Along similar lines, local proliferation, rather than recruitment from blood, was also reported to be the primary mechanism responsible for the increased number of alternatively activated (M2) macrophages in the pleural cavity of helminth-infected mice (Jenkins et al. 2011). In allergen-exposed mice, the self-renewal mechanism was also found to be responsible for maintenance of the resident AM pool during the early stages of inflammation (Zasłona et al. 2014). Misharin et al. (2017) reported that monocyte-derived macrophages are recruited during various lung fibrotic stages, and that expression of caspase-8, a cysteine protease, on these macrophages contributes to the worsening of lung pathology. Acute exposure to lipopolysaccharide (LPS), a potent inflammatory insult, results in a drastic reduction in the number of AM, which are restored via self-renewal mechanisms (Dong et al. 2018). Simultaneously, monocyte-derived macrophages also appear in the alveolar spaces, albeit at a slower rate and to a relatively lesser extent than occurs with self-renewal mechanisms (Dong et al. 2018). These studies point to the involvement of insult-dependent mechanisms for the maintenance and turnover of macrophages in the lungs.
Macrophages in ozone-exposed lungs
To review the current literature relevant to lung macrophages in mouse model-based O3 toxicity studies, we identified relevant studies published in the PubMed database using the EndNote citation manager. Our scheme for this literature search is outlined in Figure 3. We retrieved 142 references containing four keywords, O3, lung, mice, and macrophage, within any field of reference. Through manual screening, we selected 39 references that reported macrophage-relevant findings in O3-exposed mouse lungs. Most of the studies excluded used the term “macrophage” only in reference to increased levels of macrophage inflammatory protein 2 (MIP2/CXCL2) chemokines or increased numbers of macrophages in O3-exposed mice. Select studies are discussed in this review, while the exposure conditions and salient findings from all 39 studies are presented in Table 1.
Figure 3.

Screening strategy for review of literature.
Table 1.
Selected mouse model-based studies with focus on macrophage responses to ozone exposure.
| Dose (ppm) | Duration | Endpoint (h/d post exposure) | Findings | References | |
|---|---|---|---|---|---|
| 1 | 0.1–3 | 3 h | 0h | Ozone exposure decreases superoxide anion radical production in mouse alveolar macrophages | Ryer-Powder et al. (1988) |
| 2 | 0.4–0.8 | 3 h | 20 d | Alveolar macrophages from ozone-exposed mice exhibit poor clearance of Streptococci | Gilmour et al. (1993), Gilmour and Selgrade (1993) |
| 3 | 0.1–2 | 3 h | 6h | Ozone exposure induces expression of MIP2 in macrophages | Driscoll et al. (1993) |
| 4 | 1.5 | 4 h | 24 h | Ozone suppresses phagocytic responses of macrophages | Jakab and Hemenway (1994) |
| 5 | 2 | 3 h | 4–72 h | Monocyte influx in ozone-exposed airways is mediated through MCP1 | Zhao et al. (1998) |
| 6 | 0.8 | 3h | 24–48 h | Macrophages from ozone-exposed mice with iNOS deficiency do not produce nitric oxide or nitrotyrosine in lungs | Laskin et al. (2001) |
| 7 | 1 | 6 h/d × 3d | 24–48 h | The deficiency of iNOS does not protect against ozone-induced tissue injury | Kenyon et al. (2002) |
| 8 | 0.8 | 3h | 24–48 h | Ozone exposure induces nitric oxide production and iNOS expression in alveolar macrophages | Laskin et al. (2002) |
| 9 | 0.5 | 24 h | 2h | Ozone exposure compromises release of IL6 from LPS-challenged macrophages | Yu et al. (2002) |
| 10 | 0.8 | 3h | 24–48 h | iNOS expression is elevated in alveolar macrophages from ozone-exposed mice and iNOS deficiency protects against ozone-induced tissue injury | Fakhrzadeh, Laskin, et al. (2004) |
| 11 | 0.8 | 3h | 24–48 h | Overexpression of superoxide-dismutase (SOD), an antioxidant enzyme, confers protection against oxidant stress and ozone-induced lung injury | Fakhrzadeh, Laskin, Gardner, et al. (2004) |
| 12 | 0.3 | 48 h | 0h | Scavenger receptors (MARCO and SR-AI/II) on macrophages scavenge oxidized lipids and mitigate inflammatory response to ozone | Dahl et al. (2007) |
| 13 | 2 | 3h | 4h | Alveolar macrophages from ozone-exposed mice show elevated apoptosis and functional response to endotoxin | Hollingsworth, Maruoka, et al. (2007) |
| 14 | 0.8 | 3h | 12 h | TNFα suppresses caveolin 1 expression in alveolar macrophages | Fakhrzadeh et al.(2008) |
| 15 | 2 | 3h | 0h | Ozone exposure reduces phagocytic capabilities of macrophages | Mikerov, Haque, et al. (2008) |
| 16 | 2 | 3h | 0h | Ozone-exposed mice with SP-A deficiency exhibit poor macrophage phagocytic potential in Klebsiella pneumonia infection | Mikerov, Gan, et al. (2008) |
| 17 | 3 | 3h | 21 −24 h | Ozone-induced macrophage recruitment to lungs is attenuated in IL13 and IL4/13 knockout mice | Williams et al. (2008) |
| 18 | 2 | 3h | 24 h | Macrophages from ozone-exposed mice express CD44, a hyaluronan (HA) receptor | Garantziotis et al. (2009) |
| 19 | 2 | 3h | 4h | Surfactant protein A (SP-A) plays a critical role in scavenging ozone-induced reactive oxidants and mitigates oxidative stress | Haque, Umstead, Freeman, et al. (2009) |
| 20 | 2 | 3h | 24 h | Ozone exposure increases TLR4 expression on lung macrophages | Garantziotis et al. (2010) |
| 21 | 1 | 3h | 24 h | Ozone exposure induces TLR4 peripheralization in macrophages and enhances their responsiveness to LPS | Li et al. (2010) |
| 22 | 2 | 3h | 24 h | Short-term exposure of mice to 2 ppm ozone for 3 h induces appearance of a novel subset of resident-intermediate derived macrophages that appear to be protective against ozone-induced pulmonary responses | Tighe et al. (2011) |
| 23 | 0.8 | 3h | 0.5, 12, 24, 48 h | Macrophage activation is compromised in the absence of TLR4 | Connor et al. (2012) |
| 24 | 0.8 | 3h | 72 h | Mice deficient for surfactant protein D (SP-D) exhibit exaggerated macrophage recruitment and activation responses upon ozone exposure | Groves et al. (2012) |
| 25 | 1 | 4h | 24 h | Deletion of CD36, a macrophage scavenger receptor, protects against ozone-induced lung inflammation | Robertson et al. (2013) |
| 26 | 2 | 3h | 4/24 h | Ozone induces expression of TLR1/2/4 on alveolar macrophages | Oakes et al. (2013) |
| 27 | 0.3 | 24–72 h | 24 h | ΓδT cell-derived IL17A induces alternative activation of macrophages that promotes clearance of apoptotic cells and resolution of inflammation | Mathews et al. (2014) |
| 28 | 0.8 | 3h | 24 h | Galectin-3 expressing proinflammatory macrophages accumulate in ozone-exposed lungs and promote lung toxicity | Sunil et al. (2015) |
| 29 | 0.7 | 72 h | 24 h | Alveolar macrophages from ozone-exposed mice exhibit higher levels of mitochondrial ROS, elevated cytosolic mtDNA, increased caspase-1 activation, and increased IL-1β production | Che et al. (2016) |
| 30 | 2 | 3h | 4/24 h | Ozone-induced lung injury is comparable between wildtype and plasminogen activator inhibitor-1 (PAI-1) knockout mice | Elkhidir et al. (2016) |
| 31 | 3 | 3 h/twice a week (6 weeks) | 24 h | Ozone exposure induces macrophage migration inhibitory factor (MIF) release, which promotes lung inflammation | Russell et al. (2016) |
| 32 | 0.8 | 3h | 24 h | Spleen acts as a source of pro-inflammatory macrophages in ozone-exposed lungs, which contribute to inflammation and oxidative stress | Francis, Groves, et al. (2017) |
| 33 | 0.8 | 3h | 24 h | Ozone induces recruitment of macrophages via CCL2/MCP1-CCR2 ligand-receptor interactions, which promotes lung injury and oxidative stress | Francis, Sun, et al. (2017) |
| 34 | 2 | 3h | 4 and 18h | Surfactant protein A2 provides microRNA-mediated protection against oxidative stress in ozone-exposed males, but not females | Noutsios et al. (2017) |
| 35 | 2 | 3h | 24 h | Ozone induces release of free-actin into the ELF, which competitively inhibits microbial interactions with SRA/MARCO receptors | Ordija et al. (2017) |
| 36 | 1 | 1h | 24 h | Acute ozone exposure recruits IL-33 expressing macrophages to the lungs. Absence of IL-33 receptor increases macrophage recruitment to the lungs | Michaudel et al. (2018) |
| 37 | 2 | 3h | 4h | Alveolar macrophages from females are more susceptible to ozone-induced oxidative stress. SP-A provides protection in males via regulation of microRNAs involved in protection against oxidative stress | Thorenoor et al. (2019) |
| 38 | 1 and 2 | 3h | 21 h | Acute ozone exposure alters gene expression in alveolar macrophages | Tovar et al. (2020) |
Effect of ozone on macrophage phagocytosis
Phagocytosis relies on cell surface receptors that have been evolutionarily selected to recognize particles including microbes, apoptotic and necrotic debris, and foreign abiotic substances. For example, antibody/complement-coated (opsonized) bacteria are recognized by Fc-gamma (Fcγ)/complement receptors (Allen and Aderem 1996). Ligand bound Fcγ/complement receptors signal substantial reorganization of the macrophage actin cytoskeleton, which, in turn, triggers the formation of membrane extensions called pseudopodia that surround and finally engulf recognized particles (Allen and Aderem 1996). Critical known receptors in opsonin-dependent phagocytosis include FcγRI (CD64), FcγRIIa (CD32), FcγRIIIa (CD16), FcαR1 (CD89), FcεR1, CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18 complex), CR4 (CD11c/CD18 complex), C5a (CD88), and surfactant protein A (SPA) (Gordon 2016; Rosales and Uribe-Querol 2017). In contrast to bacteria, environmental toxicants devoid of microbial contents are scavenged through scavenger receptors (Palecanda and Kobzik 2001). Mechanistically, these receptors scavenge inflammatory products, such as oxidized lipids, that are also potent neutrophil chemoattractants.
Goldstein et al. (1974) reported that AM from O3-exposed rats are defective in bacterial clearance. Later, McAllen et al. (1981) and Sherwood et al. (1986) reported compromised macrophage mobility and lysosomal enzyme contents in O3-exposed rats. Further, Pearson and Bhalla (1997) reported that O3-exposed rat macrophages exhibit increased adherence to epithelial cells. O3-exposed rat macrophages have also been found to show diminished capabilities to produce superoxide radicals, which in turn, limits their bactericidal activities (Amoruso et al. 1981). Along these lines, in vitro exposure of cultured human AM to O3 results in reduced phagocytosis (Becker et al. 1991). Together, these studies indicate that there is a negative impact of O3 on macrophage function.
Several mouse-based studies have also linked O3 exposure to poor phagocytic abilities of alveolar macrophages. AM from mice exposed to 2 ppm O3 for 3 h exhibited poor ability to phagocytose Klebsiella pneumonia (Mikerov, Gan, et al. 2008; Mikerov, Haque, et al. 2008). Similarly, in another study, macrophages from O3-exposed mice (1.5 ppm O3 for 4 h) were found to exhibit compromised phagocytosis of carbon black (Jakab and Hemenway 1994). In yet another study, macrophages from mice exposed to 0.4–0.8 ppm for 3 h had reduced ability to engulf bacteria, i.e. Streptococcus zooepidemicus (Gilmour et al. 1993).
The mechanisms by which O3 compromises phagocytosis are, however, not understood. Interestingly, S. zooepidemicus was found to develop a virulence factor, antiphagocytic capsulation, within 3 h of O3 exposure at 0.4–0.8 ppm that resisted phagocytosis by AM (Gilmour et al. 1993). O3 is known to impair the structure and function of SPA, an opsonin that is important for the phagocytic ability of macrophages (Stringer and Kobzik 1996; Benne et al. 1997; Tenner 1998; Schagat et al. 2001), thus compromising opsonin-mediated phagocytosis in the alveolar spaces (Oosting et al. 1991; Su and Gordon 1996). In fact, O3-exposed SPA-deficient mice exhibit severely compromised clearance of K. pneumonia (Mikerov, Gan, et al. 2008; Mikerov, Haque, et al. 2008) suggesting that SPA acts as a critical modulator of host defense against bacterial infections. Haque and colleagues reported that SPA plays a critical role in scavenging O3-induced reactive oxidants, thus mitigating oxidative stress, a protective response that was found to be absent in SPA knockout mice (Haque, Umstead, Ahn, et al. 2009). However, the effects of O3 on other opsonins and phagocytosis-related receptors remain largely unknown.
Effect of ozone on the production of reactive species in macrophages
Macrophages recognize, engulf, and kill bacteria inside phagolysosomes where NADPH-dependent oxidases produce reactive superoxides, which are further converted to hydrogen peroxide (H2O2) by the enzyme superoxide dismutase (Bedard and Krause 2007). Earlier studies conducted on mice exposed to 0.1 ppm O3 for 3 h revealed that AM from these mice are poor producers of superoxide anions, substrates required for the production of bactericidal H2O2 (Ryer-Powder et al. 1988). Superoxide and nitric oxide (NO) species also react to produce highly reactive peroxynitrite (ONOO−). While superoxides, NO, and ONOO− are essential for creation of the bactericidal microenvironment of lysosomes, they have also been implicated in causing tissue injury (Bedard and Krause 2007).
AM from O3-exposed mice (0.8 ppm O3 for 3 h) exhibit higher expression of inducible nitric oxide synthase (iNOS/NOS2) and elevated production of NO and ONOO− compared to control mice (Laskin et al. 2002). In contrast, O3-exposed iNOS-deficient mice exhibit attenuated epithelial injury, and AM from these mice do not produce NO or ONOO− (Laskin et al. 2001). In a more long-term study, iNOS-deficient mice exposed to 1 ppm O3 for 8 h per night for three consecutive nights had elevated lung injury as compared to O3-exposed wild-type mice (Kenyon et al. 2002). The difference in the outcomes of iNOS deletion in the two studies is most likely due to differences in the O3 exposure paradigm. Furthermore, overexpression of superoxide-dismutase (SOD), an antioxidant enzyme, was shown to confer protection against oxidative stress and O3-induced lung injury (Fakhrzadeh, Laskin, Gardner, et al. 2004). Overexpression of SOD also reduced the production of NO and ONOO− and expression levels of iNOS (Fakhrzadeh, Laskin, Gardner, et al. 2004).
While the elevated production of reactive nitrogen species in macrophages from O3-exposed mice was found to be associated with increased lung injury, its impact on the bactericidal potential of macrophages following O3/bacteria co-exposure is not known. While earlier studies reported that the worsening of bacterial infection in O3-bacteria co-exposure models is attributed to the poor phagocytic ability of macrophages (Gilmour et al. 1993; Mikerov, Gan, et al. 2008; Mikerov, Haque, et al. 2008), in depth analyses of the effects of O3 on iNOS and SOD enzyme activity and generation of their products is still awaited.
Effect of ozone on macrophage activation
O3 exposure leads to activation, i.e. enhanced function, of macrophages. Macrophages have also been suggested to react to products released from epithelial cells injured by O3 resulting in classical macrophage activation and release of products such as reactive oxygen species (ROS) and TNFα, which further promote lung injury (Pendino et al. 1995; Cho et al. 2001; Fakhrzadeh et al. 2002; Toward and Broadley 2002; Fakhrzadeh, Laskin, et al. 2004; Fakhrzadeh, Laskin, Gardner, et al. 2004). AM from O3-exposed mice show elevated expression of iNOS (Fakhrzadeh et al. 2002), a bonafide marker of M1 macrophages. Further, iNOS deficiency is protective against O3-induced (0.8 ppm for 3 h) tissue injury implicating M1 activation as a factor responsible for tissue injury (Fakhrzadeh et al. 2002). These reports clearly highlight the role of classically activated macrophage-derived nitrosative and oxidative stress as a promoter of acute O3-induced lung injury (Fakhrzadeh et al. 2002; Fakhrzadeh, Laskin, Gardner, et al. 2004; Hollingsworth, Kleeberger, et al. 2007; Laskin et al. 2011).
Alternatively activated (M2) macrophages are known to release anti-inflammatory mediators and actively participate in wound repair, resolution of inflammation (Reinhart et al. 1999; Backus et al. 2010), and clearance of apoptotic/necrotic cells (Ishii et al. 1998; Dahl et al. 2007). Mathew and colleagues showed that γδT cell-derived IL17A induces alternative activation of macrophages, which promotes clearance of apoptotic cells and resolution of O3-induced lung inflammation (Mathews et al. 2015).
Effect of ozone on macrophage survival
O3 is known to induce cell death via autophagy, apoptosis, as well as necroptosis (Sunil et al. 2012). Macrophages from O3 exposed lungs were reported to undergo cell death as indicated by induction of markers of apoptosis (cleaved caspase-9) and autophagy (beclin-1) (Sunil et al. 2012). ROS-induced apoptosis-like cell death (oxeiptosis) has been reported in O3-exposed mice lungs (Holze et al. 2018). Further, it was reported that the abolition of mitochondrial, serine/threonine protein phosphatase (PGAM5), a major player in oxeiptosis pathway, resulted in exaggerated oxidative stress and inflammation. While oxeiptosis have been observed in a wide range of cell types, macrophages appear to be insensitive to oxeiptotic response (Moriwaki et al. 2016). Since activated macrophages express a variety of heat-shock proteins (HSP), including HSP60, the γδT cells in O3-exposed airspaces most likely recognize these HSPs and target macrophages for apoptotic cell death (Hirsh and Junger 2008; Mathews et al. 2015). Despite a growing list of cell death pathways, our current understanding of macrophage cell death in O3-exposed lungs is still limited.
Cell surface receptors involved in the recognition of ozonation products
Toll-like receptors (TLRs)
TLRs, including TLR2 and 4, are expressed on AM (Fan et al. 2002; Droemann et al. 2003), and their expression increases following O3 exposure (Oakes et al. 2013). The increased expression of TLR4 on AM likely occurs through an increase in lipid raft colocalization and peripheralization (Garantziotis et al. 2010; Li et al. 2010). O3-induced inflammatory responses in TLR4 knockout mice have been investigated in multiple studies. Using C3H/HeJ mice with a missense mutation in the Tlr4 gene, Kleeberger et al. (2000) and Kleeberger et al. (2001) showed that TLR4 mediates Nos2 mRNA expression, which causes ozone-induced epithelial injury and TNFα expression. Another study revealed that knockdown of TLR4, TLR2 or the intracellular adaptor protein, MyD88, abrogates O3-induced (3 ppm for 3 h) airway hyper-responsiveness in mice (Williams et al. 2007). Interestingly, the absence of Myd88, but not TLR2 or TLR4 individually, inhibited O3-induced lung neutrophilia (Williams et al. 2007) suggesting two possibilities: 1) Redundancy of TLR2 and TLR4, 2) Presence of additional triggers, such as IL33 or IL1α/β, that also work through MyD88 signaling (Griesenauer and Paczesny 2017). A second study revealed that although the absence of TLR4 on the C57BL6 background did not provide protection against O3-induced (0.8 ppm for 3 h) lung injury, it did confer partial protection against airway hyperresponsiveness (Garantziotis et al. 2010). In contrast, Connor et al. (2012) reported that mutation of TLR4 in C3H/HeJ mice results in protection against O3-induced (2 ppm for 3 h) inflammation, oxidative stress, and classical macrophage activation. The discrepancy in these findings may be attributed to differences in strain backgrounds, O3 concentrations, or both.
Both TLR2 and TLR4 bind to multiple endogenous ligands including HMGB1, HSP60, HSP70, hyaluronan, monosodium urate crystals, and biglycans (Erridge 2010). In addition, other endogenous molecules, including β-defensin, fibronectin, S100 protein, and SP-A, specifically bind only TLR4. Garantziotis et al. (2009) found increased levels of hyaluronan (HA) in BALF from O3-exposed mice and mice deficient in CD44, a major receptor for HA, were protected from O3-induced airway hyper-responsiveness (AHR). Li and colleagues reported that O3 causes the conversion of HA, a glycosamino-glycan-rich macromolecule found in lung epithelial cell basement membranes, to low molecular weight (LMW)-HA. LMW-HA has been shown to interact with macrophage surface receptors, CD44 and TLR4, resulting in proinflammatory cytokine production (Li et al. 2010, 2016; Li et al. 2011). Further, HSP70, another endogenous ligand for TLR4, was elevated in the lungs of O3-exposed (0.3 ppm for 6–72 h) mice both at the level of mRNA and protein, and HSP70-deficient mice were protected from O3-induced inflammation (Bauer et al. 2011). Whether other TLR ligands are elevated in O3-exposed lungs remains unexplored.
Scavenger receptors
O3 has also been implicated in the production of unique ozonation products including 1-palmitoyl-2-(9′-oxo-nonanoyl)-glycerophosphocholine (PON-GPC) as well as nonspecific auto-oxidation products such as 5β, 6β-epoxy-cholesterol (β-epoxide) and secosterols (Seco A and B) (Almstrand et al. 2015). PON-GPC is known to compromise macrophage viability (Uhlson et al. 2002) and induce release of proinflammatory mediators via activation of the 5-lipoxygenase pathway (Zemski Berry and Murphy 2016). Macrophage receptors of the SRA family (scavenger receptor A-I/II (SRA-I/II) and MARCO) have been implicated in the scavenging of oxidized lipids. Dahl and colleagues mechanistically established that β-epoxide and PON-GPC fail to induce neutrophil recruitment in MARCO-deficient mice implicating MARCO as a likely receptor for β-epoxide and PON-GPC. They further reported that mice deficient in MARCO as well as SRA-I/II had more robust O3-induced lung injury suggesting a protective role for MARCO and SRA-I/II in O3-induced lung injury (Dahl et al. 2007). CD36, a class B macrophage scavenger receptor, is known to bind to a number of oxidized lipoproteins and phospholipids, and CD36 expression is elevated in mice upon O3 exposure (Valacchi et al. 2007). Moreover, genetic deletion of CD36 protects against lung inflammation (Robertson et al. 2013; Mumaw et al. 2016).
Ozone and ozonation products: effect on macrophage population size
O3 exposure results in an increase in total numbers of airspace macrophages; however, the source of this increase is not fully known. Francis and colleagues reported that O3 exposure induces the recruitment of pro- as well as anti-inflammatory macrophages via CCL2(MCP1)-CCR2 ligand-receptor interactions. The recruited pro-inflammatory macrophages promoted lung injury and oxidative stress (Francis, Groves, et al. 2017). In a subsequent study, Francis and colleagues identified the spleen as a source of pro-inflammatory macrophages in mice exposed to 0.8 ppm O3 for 3 h (Francis, Sun, et al. 2017). These splenic macrophage populations appeared to contribute to O3-induced inflammation and oxidative stress. In contrast, Tighe and colleagues reported that exposure of mice to 2 ppm O3 for 3 h did not induce recruitment of either CD11b + exudative macrophages (ExMacs) or CCR2-dependent macrophages (Tighe et al. 2011). Instead, a novel subset of cells derived from CX3CR1-dependent resident macrophages appeared to be protective against O3-induced pulmonary responses. Thus, the authors concluded that a higher dose of O3 causes proliferation of local macrophages rather than extrapulmonary mobilization into the lung airspaces. Therefore, further dose-response and time-course studies are required to fully understand macrophage recruitment in response to O3.
Macrophage response to ozone and ozonation products
Recently, Ordija et al. (2017) reported that free actin, possibly a necrotic product, is elevated in the BALF of O3-exposed mice. These authors went on to show that this free actin blocks recognition of bacterial products, likely, via competitive binding to SRA-I/II and MARCO on macrophages. Although not yet fully established, it is likely that ozonation/oxidation-induced disruption of multiple surface receptors accounts for poor bacterial clearance.
Cell–cell interactions in ozone-exposed airspaces
Studies using macrophage/epithelial co-culture models are critical to our understanding of how O3-induced stress is communicated between various cell-types in airspaces and then translated to airway disease. Earlier studies in mice exposed to O3 (2 ppm for 3 h) resulted in increased expression of MIP2, a potent neutrophil chemoattractant, and CCL2 (MCP1), a potent monocyte chemoattractant, in macrophages (Driscoll et al. 1993; Zhao et al. 1998).
Manzer et al. (2008) studied the effect of mediators derived from O3-exposed macrophages on epithelial cells. In vitro exposure of macrophages to O3 (100 ppb for 1 h) induces release of IL1α, IL1β, IL6, IL18, and GM-CSF into the culture medium. Moreover, an alveolar epithelial cell culture stimulated with macrophage-conditioned media for 24 h was shown to release CCL2 (MCP1) and CXCL1 (KC), key chemoattractants for monocytes and neutrophils, respectively. These in vitro and in vivo studies suggest there is a flow of information between O3-exposed macrophages and epithelial cells; however, whether these interactions are macrophage→epithelium (unidirectional) or macrophage←→epithelium (bidirectional) is not clear. Further, it remains to be tested whether macrophage-derived inflammatory mediators communicate in an autocrine or paracrine manner with neighboring cells including neighboring naïve macrophages, other myeloid cells, and epithelial cells.
Bauer and colleagues studied the effect of O3-exposed epithelial-derived mediators on macrophages (Bauer et al. 2015). This study revealed an interesting interaction between epithelial cells and macrophages involving the release of hyaluronic acid (HA; also known as hyaluronan), a collagen synthesis marker, shown to be elevated in the BALF from volunteers exposed to O3 as well as O3-exposed mice (Garantziotis et al. 2009, 2016; Garantziotis et al. 2010). While the levels of HA did not increase in O3-exposed epithelial monocultures, HA levels were significantly elevated in O3-exposed epithelial/macrophage co-cultures. The study also revealed that, in contrast with macrophages in monoculture, macrophages co-cultured with airway epithelial cells had increased alternative activation, poor phagocytosis, and enhanced cytotoxicity to O3 (Bauer et al. 2015). Further, comprehensive profiling of macrophage- and epithelium- (airway versus alveolar) derived mediators in in vivo models is critical to understand the roles of macrophages and the epithelium in modulating airspace responses to O3. Since paracrine, autocrine, and contact-dependent cell–cell interactions likely orchestrate O3-induced airspace perturbations, the co-culture model should be customized to identify the nature of the cell–cell interactions in airspaces.
Although airspace macrophage populations are composed primarily of motile subpopulations, a subpopulation of sessile macrophages bound to alveolar epithelium has also been reported (Westphalen et al. 2014; Bhattacharya and Westphalen 2016). Using an LPS-induced lung injury model, Westphalen and colleagues identified connexin 43 (Cx43)-mediated syncytial communication channels between sessile macrophages and alveolar epithelial cells that suppress the release of proinflammatory cytokines (Westphalen et al. 2014; Bhattacharya and Westphalen 2016). Whether such interactions play a role in O3-induced airway pathogenesis is currently unclear.
Conclusions
While major cellular and molecular players in O3-induced airway diseases have been identified, little is known about the exact sequence in which they are involved in airway disease pathogenesis. Between the two primary responders in airspaces, epithelial cells and macrophages, the exact cascade of events resulting in pulmonary pathology is poorly understood. Since diffusion of O3 across the ELF towards epithelial cells is a rate-limiting step, and because macrophages are motile cells, it is highly likely that macrophages encounter O3 and/or ozonation/oxidation products well before epithelial cells do; however, this notion remains to be tested.
It is fairly well understood that macrophage populations in airspaces are highly plastic and manifest remarkable heterogeneity in the inflammatory microenvironment. The macrophages localized to the airspace compartment with the highest concentration of dissolved O3, and therefore the highest concentration of ozonation products, most likely differ from the macrophages localized in airspace compartments with lower levels of O3 deposition. This heterogeneity is further compounded during airspace inflammation when macrophage precursors in various stages of differentiation populate the airspaces. Thus, further studies to understand the anatomic distribution of O3 and its products in airspaces (trachea, airways, and alveoli) may aid in our understanding of macrophage responses. Comprehensive studies, including acute versus chronic and low-dose versus high-dose of O3 exposure, should be conducted to understand macrophage recruitment or localized proliferative responses following O3 exposure.
Future studies with an intense focus on the cell-specific roles of receptors, cytokines, and inflammatory mediators will provide further information on the initiation, progression, and resolution of O3-induced airway diseases. Controlled experimental models, such as co-culture models and gene-knockout mice, may be employed to further our understanding of the complex interactions between O3 and lung airspaces. Together, these studies will have a transformative impact on the development of therapeutic strategies against O3-induced airway diseases.
Acknowledgments
The authors are grateful for the funding provided by the National Institute of Environmental Health Sciences (NIEHS) Grant R01ES030125. The authors gratefully acknowledge the extensive critiques suggested by the three reviewers that were selected by the Editor and presented anonymously to the authors. The authors also gratefully acknowledge valuable feedback received from Dr. Roger O McClellan. These critiques were extremely helpful in revising the manuscript.
Abbreviations:
- O3
Ozone
- NAAQS
National Ambient Air Quality Standards
- ppm
parts per million
- COPD
chronic obstructive pulmonary disease
- AM
alveolar macrophage
- IM
interstitial macrophage
- ELF
epithelial lining fluid
- iNOS
inducible nitric oxide synthase
- SOD
superoxide dismutase
- ROS
reactive oxygen specie
Footnotes
Declaration of interest
The employment affiliation of both the authors is included in the cover page. This work was funded by National Institute of Environmental Health Sciences (NIEHS). Authors involved in the preparation of this manuscript did not receive any compensation from any source and declare that they have no potential conflict of interest. The preparation of this review was conducted during normal course of author’s employment. Both the authors conducted literature search, planned the outline of the manuscript, wrote, and edited the manuscript. The authors have the sole responsibility for the writing and content of the manuscript. None of the authors have been previously involved in any litigation, advocacy or regulatory activities related to the contents of this review.
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- Allen LA, Aderem A. 1996. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J Exp Med. 184(2):627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almstrand AC, Voelker D, Murphy RC. 2015. Identification of oxidized phospholipids in bronchoalveolar lavage exposed to low ozone levels using multivariate analysis. Anal Biochem. 474:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoruso MA, Witz G, Goldstein BD. 1981. Decreased superoxide anion radical production by rat alveolar macrophages following inhalation of ozone or nitrogen dioxide. Life Sci. 28(20):2215–2221. [DOI] [PubMed] [Google Scholar]
- Aucamp PJ. 2007. Questions and answers about the effects of the depletion of the ozone layer on humans and the environment. Photochem Photobiol. 6:319–330. [DOI] [PubMed] [Google Scholar]
- Backus GS, Howden R, Fostel J, Bauer AK, Cho HY, Marzec J, Peden DB, Kleeberger SR. 2010. Protective role of interleukin-10 in ozone-induced pulmonary inflammation. Environ Health Perspect. 118(12):1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer RN, Muller L, Brighton LE, Duncan KE, Jaspers I. 2015. Interaction with epithelial cells modifies airway macrophage response to ozone. Am J Respir Cell Mol Biol. 52(3):285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AK, Rondini EA, Hummel KA, Degraff LM, Walker C, Jedlicka AE, Kleeberger SR. 2011. Identification of candidate genes downstream of TLR4 signaling after ozone exposure in mice: a role for heat-shock protein 70. Environ Health Perspect. 119(8):1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S, Madden MC, Newman SL, Devlin RB, Koren HS. 1991. Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol Appl Pharmacol. 110(3):403–415. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 87(1):245–313. [DOI] [PubMed] [Google Scholar]
- Bell ML, McDermott A, Zeger SL, Samet JM, Dominici F. 2004. Ozone and short-term mortality in 95 US urban communities, 1987–2000. JAMA. 292(19):2372–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne CA, Benaissa-Trouw B, van Strijp JA, Kraaijeveld CA, van Iwaarden JF. 1997. Surfactant protein A, but not surfactant protein D, is an opsonin for influenza A virus phagocytosis by rat alveolar macrophages. Eur J Immunol. 27(4):886–890. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Levine RL, Stadtman ER. 1996. Comparison of the effects of ozone on the modification of amino acid residues in glutamine synthetase and bovine serum albumin. J Biol Chem. 271(8):4177–4182. [DOI] [PubMed] [Google Scholar]
- Bernard SM, Samet JM, Grambsch A, Ebi KL, Romieu I. 2001. The potential impacts of climate variability and change on air pollution-related health effects in the United States. Environ Health Perspect. 109(2): 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya J, Westphalen K. 2016. Macrophage-epithelial interactions in pulmonary alveoli. Semin Immunopathol. 38(4):461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett RT, Smith-Doiron M, Stieb D, Raizenne ME, Brook JR, Dales RE, Leech JA, Cakmak S, Krewski D. 2001. Association between ozone and hospitalization for acute respiratory diseases in children less than 2 years of age. Am J Epidemiol. 153(5):444–452. [DOI] [PubMed] [Google Scholar]
- Che L, Jin Y, Zhang C, Lai T, Zhou H, Xia L, Tian B, Zhao Y, Liu J, Wu Y, et al. 2016. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci Rep. 6(1):18680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Zhang LY, Kleeberger SR. 2001. Ozone-induced lung inflammation and hyperreactivity are mediated via tumor necrosis factor-alpha receptors. Am J Physiol Lung Cell Mol Physiol. 280(3):L537–46. [DOI] [PubMed] [Google Scholar]
- Connor AJ, Laskin JD, Laskin DL. 2012. Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp Mol Pathol. 92(2): 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, Kobzik L. 2007. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest. 117(3):757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, Taylor PR. 2013. Tissue-resident macrophages. Nat Immunol. 14(10):986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLucia AJ, Mustafa MG, Hussain MZ, Cross CE. 1975. Ozone interaction with rodent lung. III. Oxidation of reduced glutathione and formation of mixed disulfides between protein and nonprotein sulfhydryls. J Clin Invest. 55(4):794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Poon GFT, Arif AA, Lee-Sayer SSM, Dosanjh M, Johnson P. 2018. The survival of fetal and bone marrow monocyte-derived alveolar macrophages is promoted by CD44 and its interaction with hyaluronan. Mucosal Immunol. 11(3):601–614. [DOI] [PubMed] [Google Scholar]
- Driscoll KE, Simpson L, Carter J, Hassenbein D, Leikauf GD. 1993. Ozone inhalation stimulates expression of a neutrophil chemotactic protein, macrophage inflammatory protein 2. Toxicol Appl Pharmacol. 119(2): 306–309. [DOI] [PubMed] [Google Scholar]
- Droemann D, Goldmann T, Branscheid D, Clark R, Dalhoff K, Zabel P, Vollmer E. 2003. Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol. 119(2):103–108. [DOI] [PubMed] [Google Scholar]
- Duan X, Buckpitt AR, Pinkerton KE, Ji C, Plopper CG. 1996. Ozone-induced alterations in glutathione in lung subcompartments of rats and monkeys. Am J Respir Cell Mol Biol. 14(1):70–75. [DOI] [PubMed] [Google Scholar]
- Elkhidir HS, Richards JB, Cromar KR, Bell CS, Price RE, Atkins CL, Spencer CY, Malik F, Alexander AL, Cockerill KJ, et al. 2016. Plasminogen activator inhibitor-1 does not contribute to the pulmonary pathology induced by acute exposure to ozone. Physiol Rep. 4(18):e12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Randolph GJ. 2014. Origin and functions of tissue macrophages. Immunity. 41(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C 2010. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 87(6):989–999. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Gardner CR, Laskin DL. 2004. Superoxide dismutase-overexpressing mice are resistant to ozone-induced tissue injury and increases in nitric oxide and tumor necrosis factor-alpha. Am J Respir Cell Mol Biol. 30(3):280–287. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Laskin DL. 2002. Deficiency in inducible nitric oxide synthase protects mice from ozone-induced lung inflammation and tissue injury. Am J Respir Cell Mol Biol. 26(4):413–419. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Laskin DL. 2004. Ozone-induced production of nitric oxide and TNF-alpha and tissue injury are dependent on NF-kappaB p50. Am J Physiol Lung Cell Mol Physiol. 287(2):L279–L285. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Laskin DL. 2008. Regulation of caveolin-1 expression, nitric oxide production and tissue injury by tumor necrosis factor-alpha following ozone inhalation. Toxicol Appl Pharmacol. 227(3):380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kapus A, Marsden PA, Li YH, Oreopoulos G, Marshall JC, Frantz S, Kelly RA, Medzhitov R, Rotstein OD. 2002. Regulation of Toll-like receptor 4 expression in the lung following hemorrhagic shock and lipopolysaccharide. J Immunol. 168(10):5252–5259. [DOI] [PubMed] [Google Scholar]
- Frampton MW, Pryor WA, Cueto R, Cox C, Morrow PE, Utell MJ. 1999a. Ozone exposure increases aldehydes in epithelial lining fluid in human lung. Am J Respir Crit Care Med. 159(4):1134–1137. [DOI] [PubMed] [Google Scholar]
- Frampton MW, Pryor WA, Cueto R, Cox C, Morrow PE, Utell MJ. 1999b. Aldehydes (nonanal and hexanal) in rat and human bronchoalveolar lavage fluid after ozone exposure. Res Rep Health Eff Inst. (90):1–15. [PubMed] [Google Scholar]
- Francis M, Groves AM, Sun R, Cervelli JA, Choi H, Laskin JD, Laskin DL. 2017. Editor’s highlight: CCR2 regulates inflammatory cell accumulation in the lung and tissue injury following Ozone exposure. Toxicol Sci. 155(2):474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis M, Sun R, Cervelli JA, Choi H, Mandal M, Abramova EV, Gow AJ, Laskin JD, Laskin DL. 2017. Editor’s highlight: role of spleen-derived macrophages in ozone-induced lung inflammation and injury. Toxicol Sci. 155(1):182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M, Madden MC, Saunders DS, Gammon K, White GC 2nd, Kwock L. 1985. Ozone inhibits prostacyclin synthesis in pulmonary endothelium. Prostaglandins. 30(6):1069–1083. [DOI] [PubMed] [Google Scholar]
- Garantziotis S, Li Z, Potts EN, Kimata K, Zhuo L, Morgan DL, Savani RC, Noble PW, Foster WM, Schwartz DA, et al. 2009. Hyaluronan mediates ozone-induced airway hyperresponsiveness in mice. J Biol Chem. 284(17):11309–11317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Garantziotis S, Li Z, Potts EN, Kimata K, Zhuo L, Morgan DL, Savani RC, Noble PW, Foster WM, Schwartz DA, et al. 2016. Hyaluronan mediates ozone-induced airway hyperresponsiveness in mice. J Biol Chem. 291(37):19257–19258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garantziotis S, Li Z, Potts EN, Lindsey JY, Stober VP, Polosukhin VV, Blackwell TS, Schwartz DA, Foster WM, Hollingsworth JW. 2010. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J Respir Crit Care Med. 181(7):666–675. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Gent JF, Triche EW, Holford TR, Belanger K, Bracken MB, Beckett WS, Leaderer BP. 2003. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA. 290(14): 1859–1867. [DOI] [PubMed] [Google Scholar]
- Gerrity TR, Biscardi F, Strong A, Garlington AR, Brown JS, Bromberg PA. 1995. Bronchoscopic determination of ozone uptake in humans. J Appl Physiol. 79(3):852–860. [DOI] [PubMed] [Google Scholar]
- Gilmour MI, Park P, Selgrade MK. 1993. Ozone-enhanced pulmonary infection with Streptococcus zooepidemicus in mice. The role of alveolar macrophage function and capsular virulence factors. Am Rev Respir Dis. 147(3):753–760. [DOI] [PubMed] [Google Scholar]
- Gilmour MI, Selgrade MK. 1993. A comparison of the pulmonary defenses against streptococcal infection in rats and mice following O3 exposure: differences in disease susceptibility and neutrophil recruitment. Toxicol Appl Pharmacol. 123(2):211–218. [DOI] [PubMed] [Google Scholar]
- Goldstein E, Lippert W, Warshauer D. 1974. Pulmonary alveolar macrophage. Defender against bacterial infection of the lung. J Clin Invest. 54(3):519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S 2016. Phagocytosis: the legacy of metchnikoff. Cell. 166(5): 1065–1068. [DOI] [PubMed] [Google Scholar]
- Gordon T, Taylor BF, Amdur MO. 1981. Ozone inhibition of tissue cholinesterase in guinea pigs. Arch Environ Health. 36(6):284–288. [DOI] [PubMed] [Google Scholar]
- Griesenauer B, Paczesny S. 2017. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front Immunol. 8:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves AM, Gow AJ, Massa CB, Laskin JD, Laskin DL. 2012. Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am J Respir Cell Mol Biol. 47(6):776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryparis A, Forsberg B, Katsouyanni K, Analitis A, Touloumi G, Schwartz J, Samoli E, Medina S, Anderson HR, Niciu EM, et al. 2004. Acute effects of ozone on mortality from the “air pollution and health: a European approach” project. Am J Respir Crit Care Med. 170(10):1080–1087. [DOI] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. 2013. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 210(10):1977–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen JI, Lanki T, Tiittanen P, Niemi JV, Loh M, Pekkanen J. 2010. Ozone and cause-specific cardiorespiratory morbidity and mortality. J Epidemiol Community Health. 64(9):814–820. [DOI] [PubMed] [Google Scholar]
- Haque R, Umstead TM, Ahn K, Phelps DS, Floros J. 2009. Effect of low doses of lipopolysaccharide prior to ozone exposure on bronchoalveolar lavage: differences between wild type and surfactant protein A-deficient mice. Pneumon. 22(2):143–155. [PMC free article] [PubMed] [Google Scholar]
- Haque R, Umstead TM, Freeman WM, Floros J, Phelps DS. 2009. The impact of surfactant protein-A on ozone-induced changes in the mouse bronchoalveolar lavage proteome. Proteome Sci. 7(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. 2013. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 38(4):792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch GE, Slade R, Harris LP, McDonnell WF, Devlin RB, Koren HS, Costa DL, McKee J. 1994. Ozone dose and effect in humans and rats. A comparison using oxygen-18 labeling and bronchoalveolar lavage. Am J Respir Crit Care Med. 150(3):676–683. [DOI] [PubMed] [Google Scholar]
- Hirsh MI, Junger WG. 2008. Roles of heat shock proteins and gamma delta T cells in inflammation. Am J Respir Cell Mol Biol. 39(5):509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Ginhoux F. 2015. Ontogeny of tissue-resident macrophages. Front Immunol. 6:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth JW, Kleeberger SR, Foster WM. 2007. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 4(3):240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth JW, Maruoka S, Li Z, Potts EN, Brass DM, Garantziotis S, Fong A, Foster WM, Schwartz DA. 2007. Ambient ozone primes pulmonary innate immunity in mice. J Immunol. 179(7):4367–4375. [DOI] [PubMed] [Google Scholar]
- Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, Pennemann FL, Schnepf D, Wettmarshausen J, Braun M, et al. 2018. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 19(2):130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley DG, Mudway I, Kelly FJ, Eccles R, Richards RJ. 1995. Depletion of urate in human nasal lavage following in vitro ozone exposure. Int J Biochem Cell Biol. 27(11):1153–1159. [DOI] [PubMed] [Google Scholar]
- Hussell T, Bell TJ. 2014. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 14(2):81–93. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Hashimoto K, Nomura A, Sakamoto T, Uchida Y, Ohtsuka M, Hasegawa S, Sagai M. 1998. Elimination of neutrophils by apoptosis during the resolution of acute pulmonary inflammation in rats. Lung. 176(2):89–98. [DOI] [PubMed] [Google Scholar]
- Ito K, De Leon SF, Lippmann M. 2005. Associations between ozone and daily mortality: analysis and meta-analysis. Epidemiology. 16(4): 446–457. [DOI] [PubMed] [Google Scholar]
- Jakab GJ, Hemenway DR. 1994. Concomitant exposure to carbon black particulates enhances ozone-induced lung inflammation and suppression of alveolar macrophage phagocytosis. J Toxicol Environ Health. 41(2):221–231. [DOI] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. 2011. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 332(6035):1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA. 1980. Ozone inactivation of human alpha 1-proteinase inhibitor. Am Rev Respir Dis. 121(6):1031–1038. [DOI] [PubMed] [Google Scholar]
- Johnson DA. 1987. Effects of ozone and nitrogen dioxide on human lung proteinase inhibitors. Res Rep Health Eff Inst. (11):5–25. [PubMed] [Google Scholar]
- Kariisa M, Foraker R, Pennell M, Buckley T, Diaz P, Criner GJ, Wilkins JR. 2015. Short- and long-term effects of ambient ozone and fine particulate matter on the respiratory health of chronic obstructive pulmonary disease subjects. Arch Environ Occup Health. 70(1):56–62. [DOI] [PubMed] [Google Scholar]
- Kelly FJ, Cotgrove M, Mudway IS. 1996. Respiratory tract lining fluid anti-oxidants: the first line of defence against gaseous pollutants. Cent Eur J Public Health. 4:11–14. [PubMed] [Google Scholar]
- Kelly FJ, Mudway IS. 2003. Protein oxidation at the air-lung interface. Amino Acids. 25(3–4):375–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly FJ, Mudway I, Krishna MT, Holgate ST. 1995. The free radical basis of air pollution: focus on ozone. Respir Med. 89(10):647–656. [DOI] [PubMed] [Google Scholar]
- Kenyon NJ, van der Vliet A, Schock BC, Okamoto T, McGrew GM, Last JA. 2002. Susceptibility to ozone-induced acute lung injury in iNOS-deficient mice. Am J Physiol Lung Cell Mol Physiol. 282(3):L540–5. [DOI] [PubMed] [Google Scholar]
- Kleeberger SR, Reddy SP, Zhang LY, Cho HY, Jedlicka AE. 2001. Toll-like receptor 4 mediates ozone-induced murine lung hyperpermeability via inducible nitric oxide synthase. Am J Physiol Lung Cell Mol Physiol. 280(2):L326–L333. [DOI] [PubMed] [Google Scholar]
- Kleeberger SR, Reddy S, Zhang LY, Jedlicka AE. 2000. Genetic susceptibility to ozone-induced lung hyperpermeability: role of toll-like receptor 4. Am J Respir Cell Mol Biol. 22(5):620–627. [DOI] [PubMed] [Google Scholar]
- Kotiaho T, Eberlin MN, Vainiotalo P, Kostiainen R. 2000. Electrospray mass and tandem mass spectrometry identification of ozone oxidation products of amino acids and small peptides. J Am Soc Mass Spectrom. 11(6):526–535. [DOI] [PubMed] [Google Scholar]
- Landsman L, Jung S. 2007. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. 179(6):3488–3494. [DOI] [PubMed] [Google Scholar]
- Landsman L, Varol C, Jung S. 2007. Distinct differentiation potential of blood monocyte subsets in the lung. J Immunol. 178(4):2000–2007. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Fakhrzadeh L, Heck DE, Gerecke D, Laskin JD. 2002. Upregulation of phosphoinositide 3-kinase and protein kinase B in alveolar macrophages following ozone inhalation. Role of NF-kappaB and STAT-1 in ozone-induced nitric oxide production and toxicity. Mol Cell Biochem. 234–235(1–2):91–98. [PubMed] [Google Scholar]
- Laskin DL, Fakhrzadeh L, Laskin JD. 2001. Nitric oxide and peroxynitrite in ozone-induced lung injury. Adv Exp Med Biol. 500:183–190. [DOI] [PubMed] [Google Scholar]
- Laskin DL, Sunil VR, Gardner CR, Laskin JD. 2011. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 51(1):267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Pramod AB, Croft M, Ravichandran KS, Ting JP. 2016. How mouse macrophages sense what is going on. Front Immunol. 7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Potts EN, Piantadosi CA, Foster WM, Hollingsworth JW. 2010. Hyaluronan fragments contribute to the ozone-primed immune response to lipopolysaccharide. J Immunol. 185(11):6891–6898. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Li Z, Potts EN, Piantadosi CA, Foster WM, Hollingsworth JW. 2016. Correction: hyaluronan fragments contribute to the ozone-primed immune response to lipopolysaccharide. J Immunol. 196(5):2426–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. 2011. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS One. 6(11):e27137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Lin S, Liu X, Le LH, Hwang SA. 2008. Chronic exposure to ambient ozone and asthma hospital admissions among children. Environ Health Perspect. 116(12):1725–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn WS, Gong H Jr. 1999. The 21st century environment and air quality influences on asthma. Curr Opin Pulm Med. 5(1):21–26. [DOI] [PubMed] [Google Scholar]
- Madden MC, Eling TE, Friedman M. 1987. Ozone inhibits endothelial cell cyclooxygenase activity through formation of hydrogen peroxide. Prostaglandins. 34(3):445–463. [DOI] [PubMed] [Google Scholar]
- Manzer R, Dinarello CA, McConville G, Mason RJ. 2008. Ozone exposure of macrophages induces an alveolar epithelial chemokine response through IL-1alpha. Am J Respir Cell Mol Biol. 38(3):318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews JA, Kasahara DI, Ribeiro L, Wurmbrand AP, Ninin FM, Shore SA. 2015. Gammadelta T cells are required for M2 macrophage polarization and resolution of ozone-induced pulmonary inflammation in mice. PLoS One. 10(7):e0131236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews JA, Williams AS, Brand JD, Wurmbrand AP, Chen L, Ninin FM, Si H, Kasahara DI, Shore SA. 2014. Gammadelta T cells are required for pulmonary IL-17A expression after ozone exposure in mice: role of TNFalpha. PLoS One. 9(5):e97707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllen SJ, Chiu SP, Phalen RF, Rasmussen RE. 1981. Effect of in vivo ozone exposure on in vitro pulmonary alveolar macrophage mobility. J Toxicol Environ Health. 7(3–4):373–381. [DOI] [PubMed] [Google Scholar]
- McClellan RO, Frampton MW, Koutrakis P, McDonnell WF, Moolgavkar S, North DW, Smith AE, Smith RL, Utell MJ. 2009. Critical considerations in evaluating scientific evidence of health effects of ambient ozone: a conference report. Inhal Toxicol. 21(2):1–36. [DOI] [PubMed] [Google Scholar]
- Medina-Ramon M, Zanobetti A, Schwartz J. 2006. The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: a national multicity study. Am J Epidemiol. 163(6):579–588. [DOI] [PubMed] [Google Scholar]
- Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis CA, Sokolowska M, Dreher A, Tan HT, Quesniaux VF, Ryffel B, et al. 2018. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol. 142(3):942–958. [DOI] [PubMed] [Google Scholar]
- Mikerov AN, Gan X, Umstead TM, Miller L, Chinchilli VM, Phelps DS, Floros J. 2008. Sex differences in the impact of ozone on survival and alveolar macrophage function of mice after Klebsiella pneumoniae infection. Respir Res. 9(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikerov AN, Haque R, Gan X, Guo X, Phelps DS, Floros J. 2008. Ablation of SP-A has a negative impact on the susceptibility of mice to Klebsiella pneumoniae infection after ozone exposure: sex differences. Respir Res. 9(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. 2013. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 49(4):503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, et al. 2017. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 214(8): 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller D 2004. The tropospheric ozone problem. Arhiv Higijenu Rada Toksikol. 55:11–23. [PubMed] [Google Scholar]
- Moore K, Neugebauer R, Lurmann F, Hall J, Brajer V, Alcorn S, Tager I. 2008. Ambient ozone concentrations cause increased hospitalizations for asthma in children: an 18-year study in Southern California. Environ Health Perspect. 116(8):1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, Bertin J, Welsh RM, Chan FK. 2016. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. 196(1):407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. 2008. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 8(12):958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd JB, Leavitt R, Ongun A, McManus TT. 1969. Reaction of ozone with amino acids and proteins. Atmos Environ. 3(6):669–682. [DOI] [PubMed] [Google Scholar]
- Mudway IS, Blomberg A, Frew AJ, Holgate ST, Sandstrom T, Kelly FJ. 1999. Antioxidant consumption and repletion kinetics in nasal lavage fluid following exposure of healthy human volunteers to ozone. Eur Respir J. 13(6):1429–1438. [DOI] [PubMed] [Google Scholar]
- Mudway IS, Housley D, Eccles R, Richards RJ, Datta AK, Tetley TD, Kelly FJ. 1996. Differential depletion of human respiratory tract antioxidants in response to ozone challenge. Free Radic Res. 25(6):499–513. [DOI] [PubMed] [Google Scholar]
- Mudway IS, Kelly FJ. 1998. Modeling the interactions of ozone with pulmonary epithelial lining fluid antioxidants. Toxicol Appl Pharmacol. 148(1):91–100. [DOI] [PubMed] [Google Scholar]
- Mumaw CL, Levesque S, McGraw C, Robertson S, Lucas S, Stafflinger JE, Campen MJ, Hall P, Norenberg JP, Anderson T, et al. 2016. Microglial priming through the lung-brain axis: the role of air pollution-induced circulating factors. FASEB J. 30(5):1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadziejko C, Finkelstein I, Balmes JR. 1995. Contribution of secretory leukocyte proteinase inhibitor to the antiprotease defense system of the peripheral lung: effect of ozone-induced acute inflammation. Am J Respir Crit Care Med. 152(5):1592–1598. [DOI] [PubMed] [Google Scholar]
- Noutsios GT, Thorenoor N, Zhang X, Phelps DS, Umstead TM, Durrani F, Floros J. 2017. SP-A2 contributes to miRNA-mediated sex differences in response to oxidative stress: pro-inflammatory, anti-apoptotic, and anti-oxidant pathways are involved. Biol Sex Differ. 8(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes JL, O’Connor BP, Warg LA, Burton R, Hock A, Loader J, Laflamme D, Jing J, Hui L, Schwartz DA, et al. 2013. Ozone enhances pulmonary innate immune response to a Toll-like receptor-2 agonist. Am J Respir Cell Mol Biol. 48(1):27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosting RS, van Greevenbroek MM, Verhoef J, van Golde LM, Haagsman HP. 1991. Structural and functional changes of surfactant protein A induced by ozone. Am J Physiol. 261(2):L77–83. [DOI] [PubMed] [Google Scholar]
- Ordija CM, Chiou TT, Yang Z, Deloid GM, de Oliveira Valdo M, Wang Z, Bedugnis A, Noah TL, Jones S, Koziel H, et al. 2017. Free actin impairs macrophage bacterial defenses via scavenger receptor MARCO interaction with reversal by plasma gelsolin. Am J Physiol Lung Cell Mol Physiol. 312(6):L1018–L28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecanda A, Kobzik L. 2001. Receptors for unopsonized particles: the role of alveolar macrophage scavenger receptors. Curr Mol Med. 1(5): 589–595. [DOI] [PubMed] [Google Scholar]
- Parodi S, Vercelli M, Garrone E, Fontana V, Izzotti A. 2005. Ozone air pollution and daily mortality in Genoa, Italy between 1993 and 1996. Public Health. 119(9):844–850. [DOI] [PubMed] [Google Scholar]
- Pearson AC, Bhalla DK. 1997. Effects of ozone on macrophage adhesion in vitro and epithelial and inflammatory responses in vivo: the role of cytokines. J Toxicol Environ Health. 50(2):143–157. [DOI] [PubMed] [Google Scholar]
- Pendino KJ, Meidhof TM, Heck DE, Laskin JD, Laskin DL. 1995. Inhibition of macrophages with gadolinium chloride abrogates ozone-induced pulmonary injury and inflammatory mediator production. Am J Respir Cell Mol Biol. 13(2):125–132. [DOI] [PubMed] [Google Scholar]
- Pryor WA. 1992. How far does ozone penetrate into the pulmonary air/tissue boundary before it reacts? Free Radic Biol Med. 12(1):83–88. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Bermudez E, Cueto R, Squadrito GL. 1996. Detection of aldehydes in bronchoalveolar lavage of rats exposed to ozone. Toxicol Sci. 34(1):148–156. [DOI] [PubMed] [Google Scholar]
- Pryor WA, Uppu RM. 1993. A kinetic model for the competitive reactions of ozone with amino acid residues in proteins in reverse micelles. J Biol Chem. 268(5):3120–3126. [PubMed] [Google Scholar]
- Reinhart PG, Gupta SK, Bhalla DK. 1999. Attenuation of ozone-induced lung injury by interleukin-10. Toxicol Lett. 110(1–2):35–42. [DOI] [PubMed] [Google Scholar]
- Robertson S, Colombo ES, Lucas SN, Hall PR, Febbraio M, Paffett ML, Campen MJ. 2013. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol Sci. 134(2):304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C, Uribe-Querol E. 2017. Phagocytosis: a fundamental process in immunity. BioMed Res Int. 2017:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell KE, Chung KF, Clarke CJ, Durham AL, Mallia P, Footitt J, Johnston SL, Barnes PJ, Hall SR, Simpson KD, et al. 2016. The MIF antagonist ISO-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PLoS One. 11(1):e0146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryer-Powder JE, Amoruso MA, Czerniecki B, Witz G, Goldstein BD. 1988. Inhalation of ozone produces a decrease in superoxide anion radical production in mouse alveolar macrophages. Am Rev Respir Dis. 138(5):1129–1133. [DOI] [PubMed] [Google Scholar]
- Santrock J, Gorski RA, O’Gara JF. 1992. Products and mechanism of the reaction of ozone with phospholipids in unilamellar phospholipid vesicles. Chem Res Toxicol. 5(1):134–141. [DOI] [PubMed] [Google Scholar]
- Schagat TL, Wofford JA, Wright JR. 2001. Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J Immunol. 166(4):2727–2733. [DOI] [PubMed] [Google Scholar]
- Schelegle ES, Miller LA, Gershwin LJ, Fanucchi MV, Van Winkle LS, Gerriets JE, Walby WF, Mitchell V, Tarkington BK, Wong VJ, et al. 2003. Repeated episodes of ozone inhalation amplifies the effects of allergen sensitization and inhalation on airway immune and structural development in Rhesus monkeys. Toxicol Appl Pharmacol. 191(1): 74–85. [DOI] [PubMed] [Google Scholar]
- Schwartz J 1994. PM10, ozone, and hospital admissions for the elderly in Minneapolis-St. Paul, Minnesota. Arch Environ Health. 49(5):366–374. [DOI] [PubMed] [Google Scholar]
- Sharman MC, Mudd JB. 1981. Ozone inactivation of anti-elastase activity of chicken ovoinhibitor and human alpha-1-proteinase inhibitor. Biochem Biophys Res Commun. 102(2):640–645. [DOI] [PubMed] [Google Scholar]
- Sherwood RL, Lippert WE, Goldstein E. 1986. Effect of 0.64 ppm ozone on alveolar macrophage lysozyme levels in rats with chronic pulmonary bacterial infection. Environ Res. 41(2):378–387. [DOI] [PubMed] [Google Scholar]
- Stringer B, Kobzik L. 1996. Alveolar macrophage uptake of the environmental particulate titanium dioxide: role of surfactant components. Am J Respir Cell Mol Biol. 14(2):155–160. [DOI] [PubMed] [Google Scholar]
- Su WY, Gordon T. 1996. Alterations in surfactant protein A after acute exposure to ozone. J Appl Physiol. 80(5):1560–1567. [DOI] [PubMed] [Google Scholar]
- Sunil VR, Francis M, Vayas KN, Cervelli JA, Choi H, Laskin JD, Laskin DL. 2015. Regulation of ozone-induced lung inflammation and injury by the beta-galactoside-binding lectin galectin-3. Toxicol Appl Pharmacol. 284(2):236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil VR, Patel-Vayas K, Shen J, Laskin JD, Laskin DL. 2012. Classical and alternative macrophage activation in the lung following ozone-induced oxidative stress. Toxicol Appl Pharmacol. 263(2):195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SY, Krasnow MA. 2016. Developmental origin of lung macrophage diversity. Development. 143(8):1318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. 2005. Macrophage receptors and immune recognition. Annu Rev Immunol. 23(1):901–944. [DOI] [PubMed] [Google Scholar]
- Tenner AJ. 1998. C1q receptors: regulating specific functions of phagocytic cells. Immunobiology. 199(2):250–264. [DOI] [PubMed] [Google Scholar]
- Thorenoor N, Kawasawa YI, Gandhi CK, Zhang X, Floros J. 2019. Differential impact of co-expressed SP-A1/SP-A2 protein on AM miRNome; sex differences. Front Immunol. 10:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe RM, Li Z, Potts EN, Frush S, Liu N, Gunn MD, Foster WM, Noble PW, Hollingsworth JW. 2011. Ozone inhalation promotes CX3CR1-dependent maturation of resident lung macrophages that limit oxidative stress and inflammation. J Immunol. 187(9):4800–4808. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Tovar A, Smith GJ, Thomas JM, Crouse WL, Harkema JR, Kelada S. 2020. Transcriptional profiling of the murine airway response to acute ozone exposure. Toxicol Sci. 173(1):114–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toward TJ, Broadley KJ. 2002. Airway function, oedema, cell infiltration and nitric oxide generation in conscious ozone-exposed guinea-pigs: effects of dexamethasone and rolipram. Br J Pharmacol. 136(5): 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlson C, Harrison K, Allen CB, Ahmad S, White CW, Murphy RC. 2002. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem Res Toxicol. 15(7):896–906. [DOI] [PubMed] [Google Scholar]
- Valacchi G, Vasu VT, Yokohama W, Corbacho AM, Phung A, Lim Y, Aung HH, Cross CE, Davis PA. 2007. Lung vitamin E transport processes are affected by both age and environmental oxidants in mice. Toxicol Appl Pharmacol. 222(2):227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, Bhattacharya J. 2014. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 506(7489): 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, Chung KF. 2007. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol (1985). 103(4):1189–1195. [DOI] [PubMed] [Google Scholar]
- Williams AS, Nath P, Leung SY, Khorasani N, McKenzie AN, Adcock IM, Chung KF. 2008. Modulation of ozone-induced airway hyperresponsiveness and inflammation by interleukin-13. Eur Respir J. 32(3): 571–578. [DOI] [PubMed] [Google Scholar]
- Wong EM, Walby WF, Wilson DW, Tablin F, Schelegle ES. 2018. Ultrafine particulate matter combined with ozone exacerbates lung injury in mature adult rats with cardiovascular disease. Toxicol Sci. 163(1): 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Vannella KM. 2016. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 44(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Pinkerton KE, Witschi H. 2002. Short-term exposure to aged and diluted sidestream cigarette smoke enhances ozone-induced lung injury in B6C3F1 mice. Toxicol Sci. 65(1):99–106. [DOI] [PubMed] [Google Scholar]
- Zasłona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M. 2014. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 193(8):4245–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaynagetdinov R, Sherrill TP, Kendall PL, Segal BH, Weller KP, Tighe RM, Blackwell TS. 2013. Identification of myeloid cell subsets in murine lungs using flow cytometry. Am J Respir Cell Mol Biol. 49(2):180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemski Berry KA, Murphy RC. 2016. Phospholipid ozonation products activate the 5-lipoxygenase pathway in macrophages. Chem Res Toxicol. 29(8):1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Simpson LG, Driscoll KE, Leikauf GD. 1998. Chemokine regulation of ozone-induced neutrophil and monocyte inflammation. Am J Physiol. 274(1):L39–46. [DOI] [PubMed] [Google Scholar]