

Abstract
Cystic fibrosis (CF) is characterized by dehydration of the airway surface liquid (ASL) layer with persistent mucus obstruction. T-helper 2 (Th2) immune responses are often manifested as increased mucous cell density [mucous cell metaplasia (MCM)] associated with mucus obstruction. Interleukin 33 (IL33) is a known inducer of Th2 immune responses but its roles in mucus obstruction and related phenotypes in a CF-like lung disease model, i.e., Scnn1b-Tg+ (Tg+) mouse, remains unclear. Accordingly, IL33-knockout (IL33KO) Tg+ mice were examined and compared with IL33-heterozygous (IL33HET) Tg+ mice. As compared to IL33HET/Tg+ mice, the IL33KO/Tg+ mice had complete absence of BALF eosinophilia, accompanied with significant reduction in BALF concentration of IL5, a cytokine associated with eosinophil differentiation and recruitment, and IL4, a major Th2 cytokine. As compared to IL33HET/Tg+ mice, IL33KO/Tg+ mice had significantly reduced levels of Th2-associated gene signatures (Slc26a4, Clca1, Retnla, and Chi3l4), along with complete loss of intracellular muco-polysaccharide staining in the airway epithelium. As compared to IL33HET/Tg+ mice, while the IL33KO/Tg+ mice had significantly reduced levels of MUC5AC protein expression, they showed no reduction in the degree of mucus obstruction, MUC5B protein expression, bacterial burden, and neonatal mortality. Interestingly, the histological features including sub-epithelial airway inflammation and alveolar space enlargement were somewhat exaggerated in IL33KO/Tg+ mice compared to IL33HET/Tg+ mice. Taken together, our data indicate that while IL33 modulates Th2 inflammatory responses and MUC5AC protein production, mucus obstruction is not dependent on IL33.
Keywords: Interleukin 33, Scnn1b, airway surface liquid dehydration, mucus obstruction
Introduction:
Airway mucosal surfaces are lined by a thin layer of liquid, i.e., the airway surface liquid (ASL) layer, consisting of water, ions, proteins, and macromolecules including mucin glycoproteins (1). Optimal solid contents of ASL layer are essential for the proper functioning of the mucociliary apparatus that propels the mucus layer with entrapped airborne particles towards the epiglottis (1). The dysfunction of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) in CF airways induces ionic imbalance which results in the dehydration of ASL layer and associated increase in solid contents in ASL layer and consistent mucus obstruction (2, 3). In addition to ASL dehydration, muco-inflammatory responses, i.e., excessive mucin production accompanied with increased inflammatory cell density and cellular debris, may also result in increased ASL solid contents, and ultimately, contribute to the mucus obstruction. Therefore, it is unclear whether the CF-like muco-obstructive phenotype is due to ASL dehydration alone or muco-inflammatory responses or a combination of both. While investigating these scenarios is challenging in human CF patients, the Scnn1b Transgenic (Tg+) mouse is an ideal model for experimental testing as it results in CF-like lung disease.
The Scnn1b-Tg+ (Tg+) mouse model overexpresses the sodium channel non-voltage gated 1 β subunit (Scnn1b) in the airway epithelial cells that induces epithelial water hyperabsorption, resulting in a phenocopy of CF-like ASL dehydration (4)(5). The Tg+ mice exhibit a robust T-helper 2 (Th2) muco-inflammatory airway pathology with marked mucous cell metaplasia (MCM) and increased molecular markers of Th2 inflammation (6, 7). These responses are associated with mucus obstruction that provides a fertile nidus for bacterial colonization (5). Interleukin 33 (IL33) has been shown to be a potent stimulator of Th2 inflammation (8). Our recent report identified an intriguing association between reduced IL33 expression and diminished Th2 muco-inflammatory responses, i.e., MCM, mucus obstruction, and molecular markers of Th2 inflammation (9). However, it remains to be mechanically tested whether the germ-line deletion of IL33 prevents Th2 responses and mucus obstruction in Tg+ mice.
In this study, we hypothesized that IL33 is essential for Th2 inflammatory responses that contribute to the mucus obstruction in the airways of Tg+ mice. In order to test this hypothesis, IL33-knockout Tg+ (IL33KO/Tg+) mice were generated and compared with IL33-heterozygous Tg+ (IL33HET/Tg+) for various pathological manifestations of CF-like muco-obstructive lung disease. Specifically, the effects of IL33 deletion on a) inflammatory cell recruitment, b) levels of inflammatory mediators, c) clearance of spontaneous bacterial infection, d) neonatal mortality, e) presence of mucous secretory cells, and f) mucus obstruction were determined. Importantly, we also determined the effect of IL33 deletion on the mRNA and protein expression levels of two major gel-forming mucins, i.e., MUC5B and MUC5AC. The results from this study delineate the contribution of IL33 to the muco-inflammatory responses in the CF-like lung disease with dehydrated ASL layer.
Methods:
Generation of transgenic mice and animal husbandry:
Scnn1b-Tg+ (Tg+) mice (Tg(Scgb1a1-Scnn1b)6608Bouc/J) were procured from Jackson Laboratory (Bar Harbor, ME) and Interleukin 33 knockout (IL33KO) mice were generously provided by Dr. Susumu Nakae (University of Tokyo, Tokyo, Japan) (10). Both of these mice strains were on C57BL/6 background. These two strains were crossbred to generate IL33 knockout Tg+ (IL33KO/Tg+) and IL33 heterozygous Tg+ (IL33HET/Tg+) mice and their Tg- (WT) counterparts. Various genetic combinations, i.e., IL33HET/WT, IL33KO/WT, IL33HET/Tg+, IL33KO/Tg+, were generated by reciprocal crosses between IL33HET/Tg+ and IL33KO/WT mice, and, in parallel, by reciprocal crosses between IL33HET/WT and IL33KO/Tg+ mice. Genotyping of experimental mice was performed by polymerase chain reaction (PCR) as previously described for the presence of Scnn1b-Tg and Il33 (Supplemental Figure 1) (5, 10). Mice were maintained in individually ventilated, hot-washed cages at the Division of Laboratory Animal Medicine (DLAM; Louisiana State University, Baton Rouge) on a 12-hour dark/light cycle. Mice were fed regular diet and water ad libitum. All animal use procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Louisiana State University.
Bronchoalveolar lavage fluid (BALF) Analyses:
Aseptically harvested BALF was serially diluted onto Columbia Blood Agar (CBA) plates (Hardy Diagnostics, Santa Maria, CA) as previously described (9, 11, 12). The CBA plates were incubated in an anaerobic candle jar for 48 hours at 37°C. Bacterial burden was determined by enumerating colony-forming units (CFUs). Remaining BALF was centrifuged at 300 x g for 5 minutes. The supernatant was removed and stored at −80°C for cytokine analyses. The cellular pellet was resuspended in 500 μl of PBS. Total cell counts were determined using a hemocytometer (Brightline, Horsham, PA). Cytospins (Cytospin 3; Thermo Shandon, Pittsburgh, PA) were prepared with 200 μl cell suspension and differentially stained (Modified Giemsa kit; Newcomer Supply, Middleton, WI).
Analyses of BALF for Cytokines:
Cell-free BALF was assayed for various cytokines (Table 1) using a Luminex XMAP-based assay (MCYTOMAG-70K), according to manufacturer’s instructions (EMD Millipore, Billerica, MA). To estimate IL33 concentration, cell-free BALF supernatant or lung homogenate supernatant was assayed with a Mouse/Rat IL33 ELISA Kit (M3300) according to manufacturer’s instructions (R&D Systems, Minneapolis, MN). In case of tissue IL33 estimation assay, right middle lobe was homogenized in 500μl of radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors.
Table 1.
BALF Cytokine Levels (pg/ml)
| Cytokine | IL33HET/WT | IL33KO/WT | IL33HET/Tg+ | IL33KO/Tg+ | LOD |
|---|---|---|---|---|---|
| IL4 | 10.4 ± 0.18 # | 10.5 ± 0.46 π | 22.17 ± 4.96 # π | 11.56 ± 0.50 | 3.2 |
| IL5 | 13.88 ± 0.2# | 14.1 ± 0.4 π | 42.4 ± 16.0 # π | 14.4 ± 0.3 | 3.2 |
| RANTES | 26.3 ± 0.5# | 24.9 ± 0.7 π | 30.1 ± 1.1# π | 26.6 ± 0.8 | 3.2 |
| MIP1α | 9.25 ± 0.3 # Φ | 10.1 ± 0.3 π θ | 46.9 ± 8.9 # π | 26.7 ± 2.7 Φ θ | 3.2 |
| IP10 | 34.6 ± 1.9 # Φ | 38.1 ± 2.1 π θ | 86.4 ± 15.2 # π | 52.5 ± 5.4 Φ θ | 3.2 |
| MIP1β | 31.0 ± 0.9 # Φ | 31.2 ± 0.8 π θ | 85.0 ± 13.6 # π | 64.6 ± 6.5 Φ θ | 3.2 |
| IL12 (P70) | 26.6 ± 1.0 # | 23.3 ± 0.9 π | 25.4 ± 1.0 # π | 23.3 ± 0.9 | 3.2 |
| KC | 16.6 ± 0.8 # Φ | 18.3 ± 0.9 π θ | 844.7 ± 148.0 # π | 623.6 ± 170.1 Φ θ | 3.2 |
| IL6 | 11.8 ± 0.4 # | 12.1 ± 0.5 π θ | 53.8 ± 32.6 # π | 29.1 ± 8.7 θ | 3.2 |
| TNFα | 15.3 ± 0.4# Φ | 15.3 ± 0.3 π θ | 28.1 ± 2.4# π | 23.6 ± 1.4 Φ θ | 3.2 |
| MIP2 | 14.6 ± 0.4# Φ | 14.8 ± 0.2 π θ | 1282.0 ± 665.8# π | 893.3 ± 495.3 Φ θ | 3.2 |
| G-CSF | 16.9 ± 0.5 # Φ | 14.6 ± 0.5 π θ | 1166.0 ± 507.6 # π | 727.1 ± 171.1 Φ θ | 3.2 |
| IL17 | 16.4 ± 0.3 # Φ | 15.3 ± 0.5 π θ | 22.4 ± 2.8 # π | 18.0 ± 0.6 Φ θ | 3.2 |
| IL10 | 16.4 ± 0.8 # | 15.5 ± 0.5 | 15.1 ± 0.5 | 14.0 ± 0.6 # | 3.2 |
| MCP1 | 10.2 ± 0.4 # | 10.2 ± 0.3 π | 11.61 ± 0.5 # π | 11.4 ± 0.3 | 3.2 |
| IL2 | 18.9 ± 1.1 # Φ | 20.2 ± 1.9 π θ | 14.3 ± 1.1 # π | 13.2 ± 0.9 Φ θ | 3.2 |
| IL9 | 19.5 ± 1.0 # Φ | 20.5 ± 1.6 π θ | 16.4 ± 1.2 # π | 15.2 ± 0.5 Φ θ | 3.2 |
| IL1α | 24.9 ± 2.3 | 26.5 ± 2.8 | 34.2 ± 4.4 | 27.5 ± 2.6 | 3.2 |
| IL1β | 9.4 ± 0.3 | 9.5 ± 0.3 | 10.8 ± 0.6 | 9.8 ± 0.2 | 3.2 |
| IFNγ | 24.3 ± 0.5 | 22.6 ± 0.6 | 25.0 ± 1.4 | 22.7 ± 0.8 | 3.2 |
| IL7 | 21.3 ± 0.7 | 18.9 ± 0.6 | 20.0 ± 0.8 | 20.2 ± 1.1 | 3.2 |
| IL12 (P40) | 26.6 ± 1.0 | 23.3 ± 0.9 | 25.4 ± 1.0 | 23.3 ± 0.9 | 3.2 |
Histopathological analyses:
Left lung lobes were formalin-fixed, paraffin-embedded, sectioned, and stained for histopathological analyses. Histopathological parameters were compared by blinded board-certified pathologists (SP, NW), using a previously described semi-quantitative grading strategy (13). Briefly, histological sections stained with hematoxylin and eosin (H&E) were used to evaluate histopathological changes. To evaluate mucus obstruction and MCM, histological slides were stained with Alcian Blue-Periodic Acid Schiff (AB-PAS). Mucous cell density (number of mucous cells per millimeter (mm) of basement membrane) was used as a quantitative parameter to evaluate Mucous Cell Metaplasia, using 40X objective of Nikon Eclipse Ci light microscope (Nikon, Japan) with camera attachment (Nikon Digital Sight DSFi-2). Airway epithelial cells (at least 50 cells) and mucous cells, distinguished by presence of AB-PAS staining, were counted.
Gene expression analyses:
Total RNA was extracted and analyzed for quantity and purity as described previously (9). cDNA generation and RT-PCR were done to assess the gene expression signals of selected genes Slc26a4, Clca1, Retnla, Chi3l4, Muc5b, and Muc5ac, as described previously (9).
Mucins agarose gel electrophoresis and western blotting:
BALF mucin electrophoresis and western blotting was performed using a slightly modified protocol as described previously (14). Briefly, BALF mucins were denatured with urea (6M). Equal volumes (50μl per well) of BALF samples were electrophoresed on 1% agarose gel (at 80V for 90 min) and vacuum-blotted onto nitrocellulose membranes with 4x sodium saline citrate buffer (SSC) for two hours at −50 mBar pressure. After transfer, nitrocellulose membrane was blocked with blocking buffer (LI-COR Biosciences, Lincoln, NE) for one hour at room temperature, followed by primary antibody incubation (overnight at 4°C) with rabbit polyclonal antibodies against MUC5B (UNC223, University of North Carolina, Chapel Hill, NC) and mouse monoclonal antibodies against MUC5AC (AB3649, Abcam, Cambridge, MA). After washing, the membrane was probed with IRDye 680RD Goat anti-rabbit and IRDye 800CW Goat anti-mouse IgG (LI-COR Biosciences, Lincoln, NE) secondary antibodies, each diluted to 1:5,000 in blocking buffer. After one hour of incubation at room temperature, the membrane was analyzed by Odyssey CLx infrared imaging system (LI-COR Biosciences, Lincoln, NE). The band intensity was measured using Image J software (NIH).
RNAScope (RNA in situ hybridization):
To access the distribution and relative abundance of Muc5b and Muc5ac transcripts, in situ hybridization was performed on paraffin embedded airway sections using Advanced Cell Diagnostics (ACD) proprietary RNAScope® technology (ACD, Newark, CA). Predesigned transcript-specific probes were used to hybridize Muc5b and Muc5ac transcripts in the airway sections and RNAScope 2.5 HD Duplex Assay Kit (ACD, Newark, CA) was used to amplify the transcript signals. Muc5b and Muc5ac amplification signals were detected as green and red staining, respectively.
Mean linear intercept measurements:
Briefly, two different lung sections per mouse were imaged at 10X magnification using Nikon scope (Nikon, Japan) with camera attachment (Nikon Digital Sight DSFi-2). Images were loaded onto Image J software and converted to binary. Horizontal grid lines were superimposed on lung section image. Using line drawing software, lines were drawn along gridlines and measured. Intercepts were considered as points of contact between drawn line and alveolar septi. Mean Linear Intercepts (MLI) were determined by dividing the sum of the length of all drawn lines (μm) by the sum of the total intercepts between alveolar septi and drawn lines (15).
Statistical analyses:
One-way Analysis of Variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons was used to determine significant differences among groups. Measurements from two groups were compared using Student’s t test assuming unequal variance. All data were expressed as mean ± standard error of the mean (SEM). A p value <0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA).
Results:
IL33 contents were elevated in the BALF and lung tissue of Scnn1b-Tg+ mice:
To identify the age at which IL33 is released into the airspaces of Tg+ mice, IL33 contents were estimated in BALF from wild-type (WT) and Tg+ neonates (PND 7) and juveniles (PND 21). IL33 protein was undetectable in BALF from WT as well as Tg+ neonates (Figure 1a, left panel). While IL33 protein was undetectable in BALF from WT juveniles, the Tg+ juveniles had significantly elevated levels of IL33 (Figure 1a, right panel).
Figure 1: IL33 protein contents are elevated in the Scnn1b-Tg+ lungs.
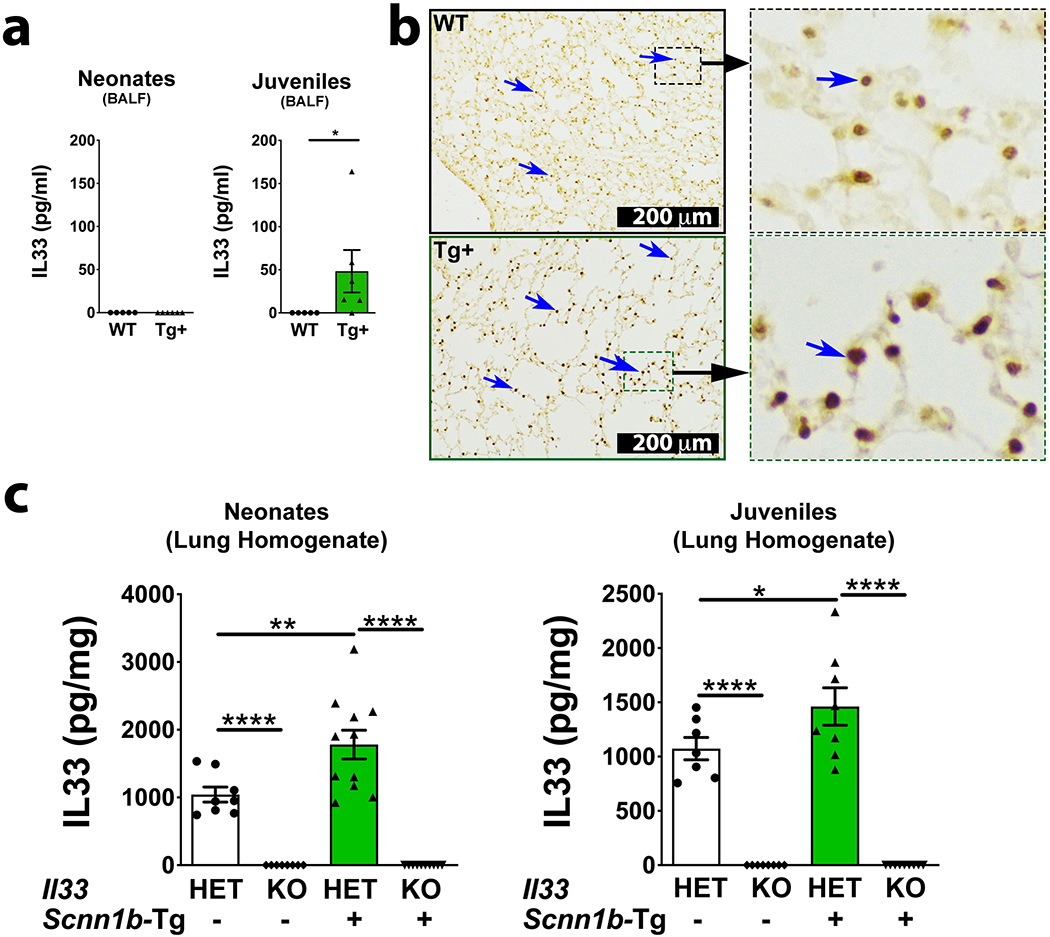
IL33 concentration in cell-free BALF from WT (white bar) and Scnn1b-Tg+ (green bar) neonates (a, left panel) and juveniles (a, right panel) (n=5–6 per group). For IL33 estimation in BALF, since equal volumes of BALF samples were used, the estimated IL33 values (picograms per milliliter, pg/ml) were used directly. Error bars represent SEM. *p<0.05, using Student’s t test. Representative photomicrographs from lung sections showing immuno-localization for IL33 protein (blue arrows) in WT [b, top left and top right (higher magnification)] and Scnn1b-Tg+ (bottom) juveniles [b, bottom left and bottom right (higher magnification)]. IL33 concentration (picogram per milligram; pg/mg) in lung homogenates from WT (white bar) and Scnn1b-Tg+ (green bar) neonates (c, left) and juveniles (c, right) (n=8–11 per group). The estimated IL33 values were normalized to the total lung homogenate protein content. Error bars represent SEM. *p<0.05, **p<0.01, ****p<0.0001 using ANOVA followed by Tukey’s multiple comparison post hoc test.
To identify the cellular source of IL33 in Tg+ lungs, we performed immuno-histochemical localization for IL33 in lung sections from WT and Tg+ juveniles. While IL33 was found to be localized in the alveolar epithelial cells of both WT and Tg+ juveniles, the IL33 staining was remarkably intense in alveolar epithelial cells from Tg+ juveniles (Figure 1b). Further, the level of IL33 protein in the lung homogenates from WT and Tg+ mice on IL33Het or IL33KO background were assessed (Figure 1c). As compared with IL33Het/WT neonates and IL33Het/WT juveniles, the total IL33 contents were significantly higher in both IL33Het/Tg+ neonates (Figure 1c, left panel) and IL33Het/Tg+ juveniles (Figure 1c, right panel). As expected, IL33 was undetected in the lung homogenates from both IL33KO/WT and IL33KO/Tg+ neonates (Figure 1c, left panel) and juveniles (Figure 1c, right panel).
IL33 deletion modulates immune cell recruitment in Scnn1b-Tg+ airways:
To determine the effect of IL33 deletion on immune cell recruitment into the Tg+ airspaces, BALF samples were analyzed for alterations in the inflammatory cell counts and soluble inflammatory mediators. The IL33KO/WT and IL33HET/WT mice had comparable number of total immune cells in their BALF (Figure 2a). The proportion of individual immune cells including macrophages, neutrophils, eosinophils, and lymphocytes were also comparable among IL33KO/WT and IL33HET/WT mice (Figure 2b–c). The IL33HET/Tg+ mice had significant increase in number of total BALF cells as a result of an increase in three cell types, i.e., neutrophils, eosinophils, and lymphocytes (Figure 2b–c). The total cell, macrophage, neutrophil, and lymphocyte counts in IL33KO/Tg+ BALF were comparable to BALF cell counts obtained from IL33HET/Tg+ littermates (Figure 2b–c). Nonetheless, the eosinophilic counts were significantly lower in the IL33KO/Tg+ BALF as compared to IL33HET/Tg+ BALF (Figure 2b–c).
Figure 2: IL33 deficiency alters immune cell recruitment.
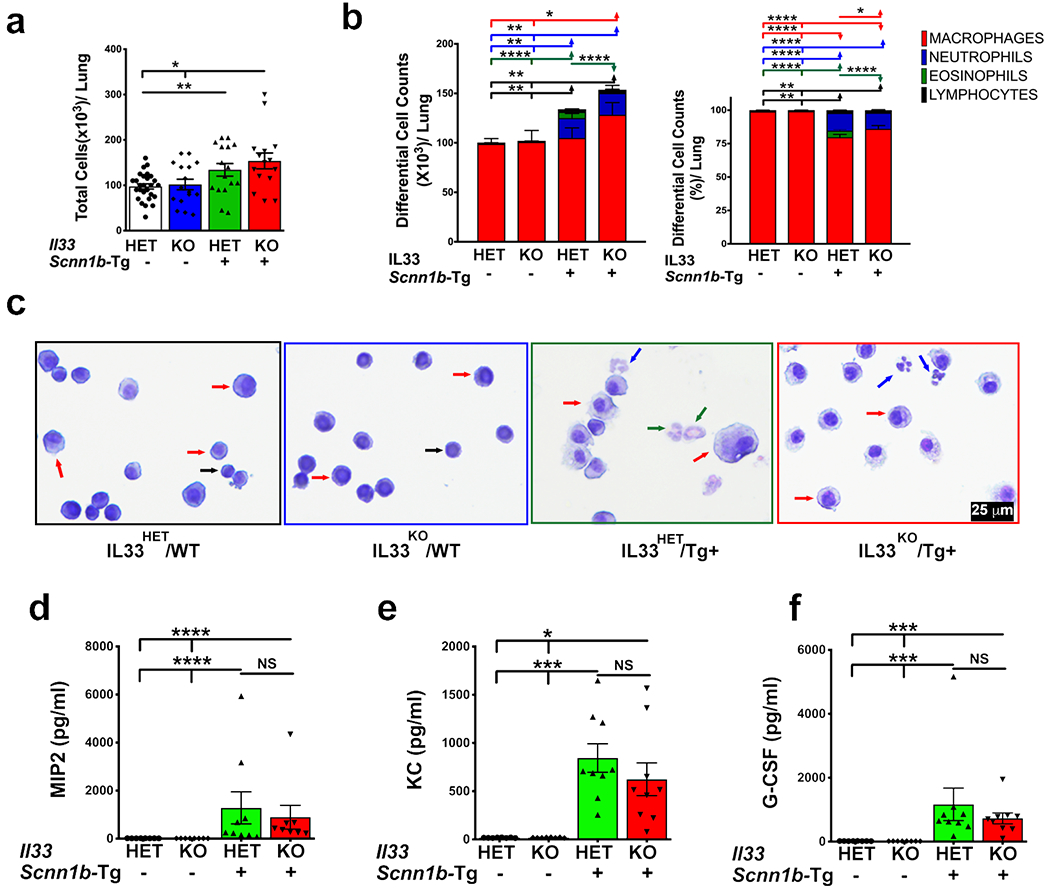
(a) Total cell counts in BALF from IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), and IL33KO/Tg+ (red bar) (n=15-26 per group). (b) Differential cell counts (left) and relative percentages (right) are shown as stacked bar graph [macrophages (red), neutrophils (blue), eosinophils (green), and lymphocytes (black)]. Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 using ANOVA followed by Tukey’s multiple comparison post hoc test. Significant difference in numbers or proportions of different cell types is indicated by horizontal lines of cell-specific colors, i.e., macrophages (red line), neutrophils (blue line), eosinophils (green line), and lymphocytes (black line) (c) Representative photomicrographs of BALF cytospins from IL33HET/WT, IL33KO/WT, IL33HET/Tg+ IL33KO/Tg+. Macrophages (red arrow), neutrophils (blue arrow), eosinophils (green arrow), and lymphocytes (black arrow). BALF cytokine levels (pg/ml) of MIP2 (d), KC (e), and G-CSF (f), in cell-free BALF from IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar). Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 using ANOVA followed by Tukey’s multiple comparison post hoc test. NS, non-significant; IL33, Interleukin 33; MIP2, Macrophage inflammatory protein-2; KC, Keratinocyte chemoattractant; G-CSF, Granulocyte-colony stimulating factor.
To determine the effect of IL33 deletion on chemokine levels in Tg+ airspaces, BALF was assayed for the presence of cell-specific chemokines. Neutrophil chemoattractants including Keratinocyte chemoattractant (KC), Macrophage inflammatory protein-2 (MIP2), Granulocyte-colony stimulating factor (G-CSF), Macrophage inflammatory protein-1 alpha (MIP1α), and Monocyte chemoattractant protein-1 (MCP-1) were below detection levels in IL33HET/WT and IL33KO/WT mice (Figure 2d–f; Table 1). All of these cytokines were significantly elevated, but comparable to one another, in BALF from IL33HET/Tg+ and IL33KO/Tg+ mice (Figure 2d–f; Table 1). Eosinophil chemoattractants, Interleukin 5 (IL5) (Figure 3a) and Regulated on Activation, Normal T cell Expressed and Secreted (RANTES) (Table 1) were below detection levels in IL33HET/WT and IL33KO/WT mice. Both of these mediators were significantly elevated in BALF from IL33HET/Tg+ mice. However, IL5 and RANTES were significantly reduced to the basal level in IL33KO/Tg+ mice (Figure 3a; Table 1).
Figure 3: IL33 deficiency suppresses molecular markers of Th2 inflammation in Scnn1b-Tg+ lungs.
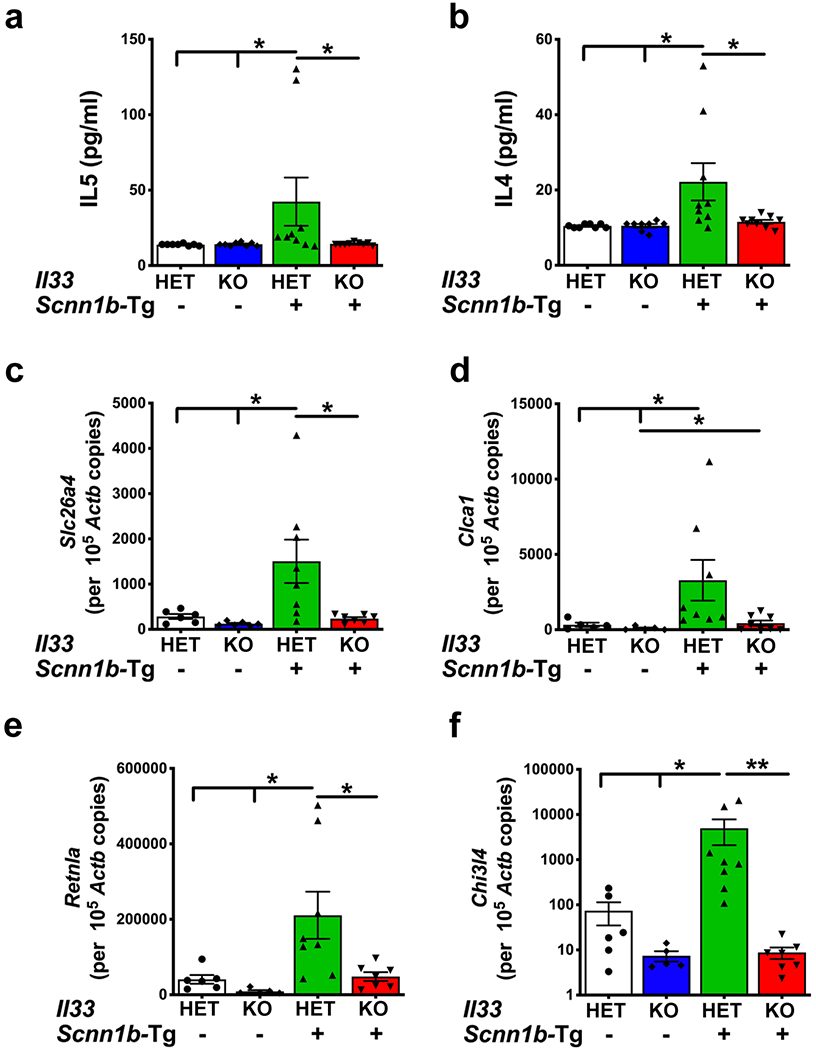
Cytokine levels (pg/ml) of IL5 (a) and IL4 (b) in cell-free BALF from IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar). Error bars represent SEM. *p<0.05 using ANOVA followed by Tukey’s multiple comparison post hoc test. Absolute quantification (mRNA copied per 105 copies of Actb mRNA) of mRNA for Slc26a4 (c), Clca1 (d), Retnla (e), and Chi3l4 (f) in lung homogenate of IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar) mice. Error bars represent SEM. *p<0.05, **p<0.01 using ANOVA followed by Tukey’s multiple comparison post hoc test.
Deletion of IL33 results in diminished Th2 inflammation markers in Scnn1b-Tg+ airways:
The Scnn1b-Tg+ mice predominantly exhibit Th2 inflammation characterized by elevated levels of eosinophils in BALF, elevated levels of soluble mediators, i.e., IL4 and IL5, and elevated levels of Th2 gene signatures, i.e., Slc26a4 (pendrin), Clca1 (Gob5), Retnla (Fizz1), and Chi3l4 (YM2) (9, 16). In agreement, as presented in the previous section, IL33HET/Tg+ mice had significant BALF eosinophilia (Figure 2b–c) and concomitant increase in BALF IL5 levels (Figure 3a). Both of these features were completely abolished in IL33KO/Tg+ pups (Figure 2b–c, 3a). While levels of IL4 were at baseline in IL33HET/WT and IL33KO/WT pups, the IL33HET/Tg+ pups had significantly elevated BALF IL4 levels (Figure 3b). The BALF from IL33KO/Tg+ pups had significant reduction in IL4 contents to the baseline level (Figure 3b).
Th2 inflammation-associated gene signatures were assessed in the total lung RNA preparations. In the absence of Scnn1b transgene, the IL33HET as well as IL33KO mice had baseline levels of expression of four Th2 inflammation-associated gene, i.e., Slc26a4 (Figure 3c), Clca1 (Figure 3d), Retnla (Figure 3e), and Chi3l4 (Figure 3f). While the levels of all the four genes were significantly elevated in IL33HET/Tg+ lungs, their expression levels were significantly reduced to the baseline level in IL33KO/Tg+ mice (Figure 3c–f).
IL33 deletion results in significant reduction in airway mucous cell density but without amelioration of muco-obstructive phenotype of Scnn1b-Tg+ mice:
To determine the effect of IL33 deletion on mucus obstruction, we analyzed the airway luminal contents in AB-PAS-stained lung sections from WT and Tg+ mice on IL33HET and IL33KO background. While there was no sign of mucus plugging in IL33HET/WT as well as IL33KO/WT mice, the IL33HET/Tg+ mice had marked mucus obstruction (Figure 4a). Contrary to our expectation, the degree of mucus obstruction was significantly increased in IL33KO/Tg+ mice versus IL33HET/Tg+ mice (Figure 4a). This quantitative scoring was consistent with our visual observations, made while performing BALF collection, that IL33KO/Tg+ mice contained relatively larger mucus plugs (data not shown).
Figure 4: IL33 deletion does not ameliorate mucus obstruction despite significant suppression of mucous cell density in Scnn1b-Tg+ airways.
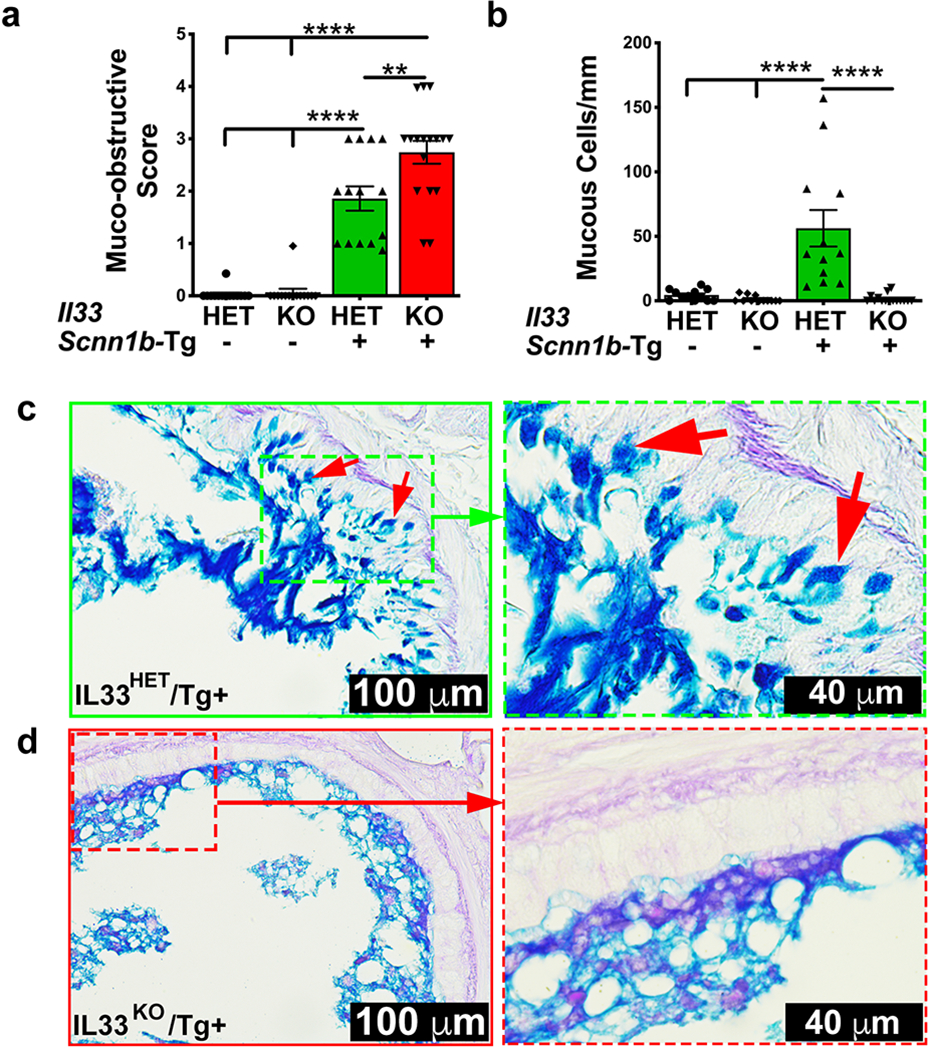
(a) Semi-quantitative histological scoring for airway mucus obstruction IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar). (b) Number of mucous cells per mm of basement membrane, IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar). (c) Representative photomicrographs from AB-PAS-stained left lung sections from IL33HET/Tg+ (higher magnification of inset is shown as dotted green margin). Intracellular AB-PAS staining of mucous cells is indicated by red arrows. (d) Representative photomicrographs from AB-PAS-stained left lung sections from IL33KO/Tg+ (higher magnification of inset is shown as dotted red margin). Error bars represent SEM. **p<0.01, ****p<0.0001 using ANOVA followed by Tukey’s multiple comparison post hoc test.
Increased mucous cell density, i.e., mucous cell metaplasia (MCM), is a hallmark feature of Scnn1b-Tg+ lung disease(15). To explore whether the sustained mucus obstruction in IL33KO/Tg+ mice is resulting due to increased mucous cell density, AB-PAS stained lung sections were examined for intracellular muco-polysaccharide contents. AB-PAS positive airway epithelial cells were sporadically present in the airways of IL33HET/WT but not in IL33KO/WT mice (Figure 4b). While the IL33HET/Tg+ airway epithelium had significantly higher number of AB-PAS positive cells (Figure 4b, 4c), the proportion of mucous cells was significantly reduced in IL33KO/Tg+ mice (Figure 4b, 4d).
Absence of IL33 has differential effect on the gene and protein expression of gel-forming mucins:
To determine the effect of IL33 deletion on gene expression of two major gel-forming mucins, i.e., Muc5b and Muc5ac, gene expression analyses was performed on total RNA from lungs. As reported previously (16), the Muc5b gene signatures were elevated in the IL33HET/Tg+ lungs (Figure 5a). However, there was no significant difference in the Muc5b gene signatures in IL33HET/Tg+ versus IL33KO/Tg+ mice (Figure 5a). As compared to WT counterparts, IL33HET/Tg+ and IL33KO/Tg+ mice had significantly elevated Muc5ac gene signatures (Figure 5b). Although trending towards lower expression, the reduction in Muc5ac gene signature in IL33KO/Tg+ mice did not reach statistical significance (Figure 5b). In addition, we performed in situ hybridization for the airway epithelium-specific expression patterns of Muc5b and Muc5ac transcripts. Both Muc5b and Muc5ac transcripts were almost equally abundant in the epithelial cells of large airways of IL33HET/Tg+ (Figure 5c) as well as IL33KO/Tg+ (Figure 5d) mice.
Figure 5: IL33 deletion results in reduced MUC5AC, not MUC5B, protein contents in Scnn1b-Tg+ BALF.
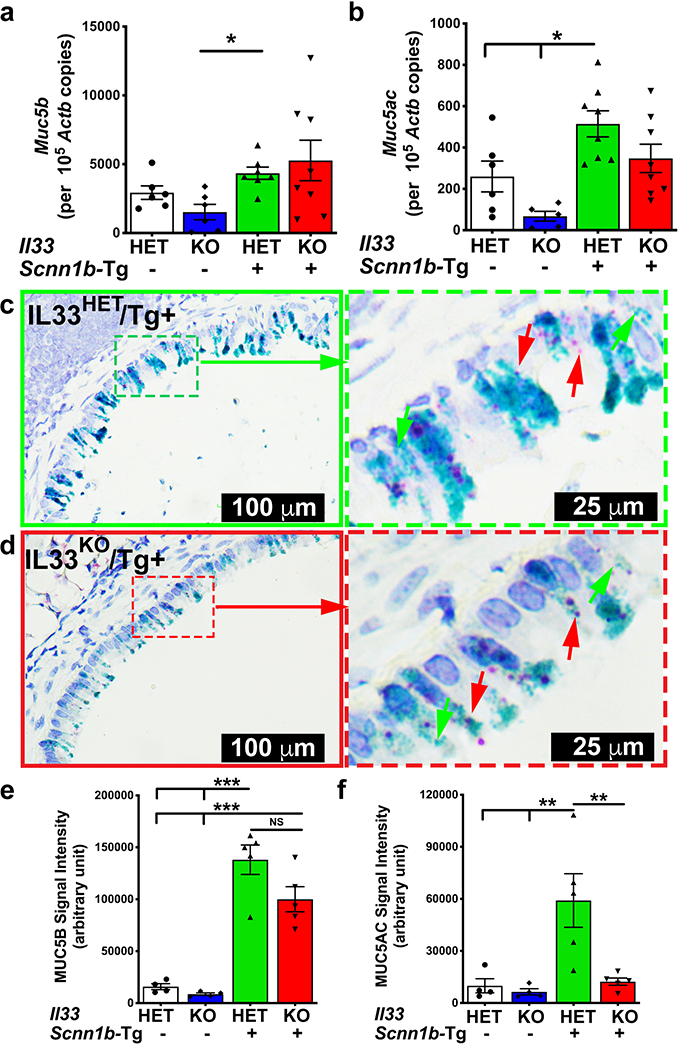
Absolute quantification (mRNA copied per 105 copies of Actb mRNA) of mRNA for Muc5b (a) and Muc5ac (b) in lung homogenate of IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar) mice. Error bars represent SEM. *p<0.05 using ANOVA followed by Tukey’s multiple comparison post hoc test. Representative photomicrograph from in situ mRNA hybridization (RNAScope) analyses of Muc5b (green stain) and Muc5ac (red stain) transcripts in lung sections from IL33HET/Tg+ (c; higher magnification of inset is shown as dotted green margin) and IL33KO/Tg+ (d; higher magnification of inset is shown as dotted red margin) mice. Western blot analyses for MUC5B (e) and MUC5AC (f) in the BALF from IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar) mice. Error bars represent SEM. **p<0.01, ***p<0.001 using ANOVA followed by Tukey’s multiple comparison post hoc test. Images used for the quantification of agarose western blot for MUC5B and MUC5AC are supplied as Supplemental Figure 2.
To explore the effect of IL33 deletion on protein contents of both MUC5B and MUC5AC, their levels were determined in BALF. MUC5B levels were very low and comparable between IL33HET/WT and IL33KO/WT mice. As compared to their WT counterparts, the IL33HET/Tg+ and IL33KO/Tg+ mice had ~ 9 and ~12-fold higher MUC5B content, respectively. Although trending towards lower levels, the MUC5B contents in IL33KO/Tg+ mice were not significantly lower than IL33HET/Tg+ mice (Figure 5e, left panel; Supplemental Figure 2a). MUC5AC BALF contents were present at very low levels in the IL33HET/WT as well as IL33KO/WT mice but were found to be significantly elevated in IL33HET/Tg+ mice (Figure 5f, right panel; Supplemental Figure 2b). Importantly, as compared to IL33HET/Tg+ mice, the MUC5AC levels were significantly reduced to the baseline levels in IL33KO/Tg+ mice (Figure 5f, right panel; Supplemental Figure 2b).
Absence of IL33 does not alter postnatal mortality, body weight, and prevalence of spontaneous bacterial infections in Scnn1b-Tg+ offspring:
The Scnn1b-Tg+ mice on C57BL/6 background exhibit mortality within first three weeks of postnatal life (13). To explore the contribution of IL33 towards postnatal mortality in Scnn1b-Tg+ mice, we observed pups from various breeding strategies (see Methods section) for any postnatal distress and mortality. Pups of all the four genotypes were born at expected Mendelian ratios, and, as indicated by their body weights at the age of three weeks, none of the offspring exhibited any signs of distress due to IL33 deficiency (Supplemental Figure 3a). While ~19 % IL33HET/Tg+ pups did not survive beyond three weeks of age, IL33KO/Tg+ pups exhibited non-significant increase (~23%) in mortality (Supplemental Figure 3b). The status of IL33 deficiency or Tg+ presence in parents undergoing reciprocal crossing had no obvious effect on the postnatal survival or body weights.
To determine the effect of IL33-deletion on bacterial clearance, we analyzed bacterial burden in aseptically collected BALF from neonates (PND 7) and juveniles (PND 21). In the absence of Scnn1b transgene, only 1 out of 10 IL33Het/WT (CFU ~3500) and 3 out of 16 IL33KO/WT (mean CFU ~125.9 ± 85.3) neonates had bacterial infection (Supplemental Figure 3c). As compared to their WT counterparts, IL33Het/Tg+ and IL33KO/Tg+ neonates had significantly higher degree of bacterial infection. Only 1 out of 12 IL33HET/Tg+ and 4 out of 14 IL33KO/Tg+ neonates had no signs of bacterial infection. Mean CFU counts in IL33HET/Tg+ (mean CFU ~6.9 x104 ± 3.7 x104) were not significantly different from IL33KO/Tg+ neonates (mean CFU ~6.1 x104 ± 2.9 x104) (Supplemental Figure 3c).
Scnn1b-Tg+ mice are known to clear airspace infection by the age of three weeks(11). Only mild bacterial infection was detected in 7 out of 29 IL33HET/WT (mean CFU ~16.6 ± 8.5) and 7 out of 16 IL33KO/WT (mean CFU ~11.2 ± 3.8) juveniles (Supplemental Figure 3d). In contrast, bacterial burden was significantly elevated in both IL33HET/Tg+ (mean CFU ~443.2 ± 231.4) and IL33KO/Tg+ (mean CFU ~59.4 ± 24.9) juveniles (Supplemental Figure 3d). While mean CFU counts were trending toward lower in IL33KO/Tg+ juveniles, the differences between IL33KO/Tg+ and IL33HET/Tg+ juveniles were statistically non-significant (p=0.12) (Supplemental Figure 3d).
IL33 deletion modulates pulmonary pathology in Scnn1b-Tg+ mice:
The effects of IL33 deletion on Scnn1b-Tg+ pulmonary pathology including airway inflammation, alveolar space enlargement, and lymphoid hyperplasia, were examined. Sub-epithelial/peri-bronchiolar airway inflammation with marked leukocytic infiltration is associated with the Scnn1b-Tg+ lung pathology(15). The airway inflammation was not evident in IL33HET/WT as well as IL33KO/WT groups. As previously reported(15), the peri-bronchiolar inflammation was present in IL33HET/Tg+ lungs (Figure 6a). The inflammatory cellular infiltrates were an admixture of neutrophils, eosinophils, and lymphocytes (Figure 6a). In contrast, the degree of neutrophilic and lymphocytic infiltration was significantly higher in IL33KO/Tg+ lungs (Figure 6a). As reflected by the absence of eosinophils in IL33KO/Tg+ BALF, the peri-bronchiolar inflammation had negligible presence of eosinophils (Figure 6a).
Figure 6: IL33 deficiency modulates pulmonary pathology in Scnn1b-Tg+ airways.
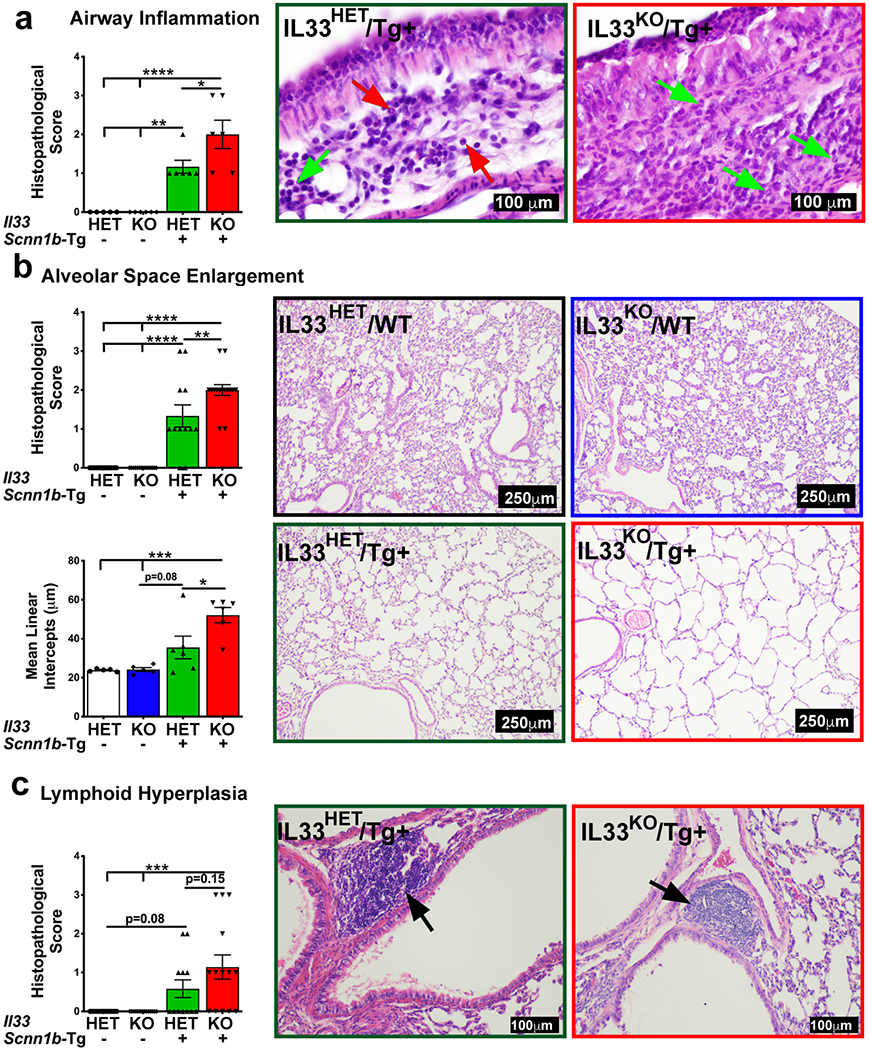
(a) Semi-quantitative histological scoring for airway inflammation for IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), and IL33KO/Tg+ (red bar). Representative photomicrographs from H&E-stained left lung lobe sections showing peri-bronchiolar inflammation in IL33HET/Tg+ (left panel) and IL33KO/Tg+ (right panel). Green and red arrows point to neutrophils and eosinophils, respectively. (b) Semi-quantitative histological scoring (Top bar graph) for alveolar space enlargement for IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), and IL33KO/Tg+ (red bar). Quantitative histological scoring (Bottom bar graph) for mean linear intercepts for IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), and IL33KO/Tg+ (red bar). Representative photomicrographs from H&E-stained left lung lobe sections for IL33HET/WT (black margin), IL33KO/WT (blue margin), IL33HET/Tg+ (green margin) and IL33KO/Tg+ (red margin). (c) Semi-quantitative histological scoring for lymphoid hyperplasia for IL33HET/WT (white bar), IL33KO/WT (blue bar), IL33HET/Tg+ (green bar), IL33KO/Tg+ (red bar). Representative photomicrographs from H&E-stained left lung lobe sections showing lymphoid hyperplasia (black arrow) in IL33HET/Tg+ (left panel) and IL33KO/Tg+ (right panel). Error bars represent SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 using ANOVA followed by Tukey’s multiple comparison post hoc test. IL33, Interleukin 33; WT, Wild-type.
Alveolar space enlargement is a consistent feature of Scnn1b-Tg+ lung disease(15). We performed semi-quantitative (Figure 6b; top) as well as quantitative (mean linear intercepts) analyses (Figure 6b; bottom) on lung sections. Both IL33HET/WT and IL33KO/WT had comparable alveolar space sizes (Figure 6b). As expected, alveolar space enlargement was a prominent feature of the IL33HET/Tg+ lungs but was widespread and significantly greater in IL33KO/Tg+ lungs (Figure 6b).
Lymphoid hyperplasia, i.e., appearance of lymphoid aggregates/nodules, is another pathological response present in Scnn1b-Tg lungs(17, 18). Both IL33HET/WT and IL33KO/WT were completely devoid of lymphoid aggregates. While 5 out of 12 IL33HET/Tg+ lungs had lymphoid aggregates, 9 out of 14 IL33KO/Tg+ mice had greater incidence of lymphoid aggregates with greater numbers per animals (Figure 6c). However, this increase in incidence of lymphoid aggregates in IL33KO/Tg+ mice was not statistically significant (p=0.15).
Discussion
Our recent report demonstrated a positive correlation between increased levels of IL33 in the muco-obstructive airspaces of Scnn1b-Tg+ mice and robust mucous cell metaplasia (MCM) (9). Both responses were found suppressed in the secondhand smoke (SHS) exposed Scnn1b-Tg+ juveniles (9). Accordingly, we had proposed our conceptual model that the ASL dehydration triggers IL33 release from airway epithelial cells that, via downstream effector cells, orchestrates MCM in Scnn1b-Tg+ airways (9). In addition, we conceptualized that the SHS exposure results in the suppression of IL33 expression in airway epithelial cells that, eventually, leads to the suppression of MCM. This model led us to the next logical step of investigating the effect of germ-line deletion of IL33 on the MCM and MCM-associated pathological features such as mucus obstruction, airway inflammation, and bacterial infection. In the current study, employing IL33-knockout Scnn1b-Tg+ (IL33KO/Tg+) mouse, we have mechanistically tested our central hypothesis that IL33 is essential for Th2 inflammatory responses that contributes to the mucus obstruction in the airways of Tg+ mice.
The Scnn1b-Tg+ lung disease follows a typical age-associated shift in inflammatory milieu in the airspaces that alters activation patterns of macrophages (16). While the M1 activation, a T-helper 1 (Th1)-associated classical macrophage activation pattern, predominates in Scnn1b-Tg+ at PND 3, the M2 activation, Th2-associated alternative macrophage activation, is robust at PND 10 and PND 42 (16). This trend likely suggests that Th1 response that coincides with the onset of spontaneous bacterial infection is switched to Th2 responses during second week of age. The levels of IL33, a primary Th2 response trigger, contents in Scnn1b-Tg+ BALF confirm this correlation, i.e., absence of detectable IL33 at PND 7 and significant presence of IL33 at PND 21 (Figure 1a). These data suggest that release of IL33 into the airspaces, which occurs sometime between PND 7 and PND 21, likely mirrors Th2 responses in Scnn1b-Tg+ mice.
Upon allergen challenge, stressed epithelial cells release IL33, that, along with IL25 and TSLP, initiates Th2-predominated responses (19). The identity of trigger for the release of IL33 in Scnn1b-Tg+ airspaces remains unclear. Although ASL dehydration along with epithelial necrosis is evident well before PND 5 (15), the BALF from 7 days old Scnn1b-Tg+ pups did not show any evidence of IL33 (Figure 1a). It is likely that epithelial necrosis, a response seen during early neonatal stage (15), initiates IL33 release from necrotic cells that achieve measurable concentration somewhere between PND 7 and PND 21. On the other hand, since alveolar epithelial cells in Scnn1b-Tg+ mice stained intensely for nuclear IL33, possibility exists that alveolar epithelial cells are the primary source of BALF IL33 (Figure 1b). Further studies using Scnn1b-Tg+ mice with cell-specific, i.e., airway epithelium, alveolar epithelium, and immune cells ablation of IL33 are required to identify the cellular source of IL33 in Scnn1b-Tg+ mice.
In our recent report, mucus obstruction was significantly reduced in the SHS-exposed Tg+ mice (9). Strong association of this outcome with suppressed IL33 expression and diminished MCM in the SHS-exposed Tg+ mice led to our hypothesis that the mucus obstruction would be significantly reduced in the absence of mucous cells in IL33KO/Tg+ lungs. Counter-intuitively, our data reveal that the complete absence of mucous cells does not ameliorate mucus obstruction from Tg+ airways (Figure 4a–c). In other words, retention of ciliated cell density does not restore normal ciliary clearance function as well. It is, therefore, most likely that the accumulation of normally produced mucopolysaccharides in dehydrated ASL layer of Tg+ mice, rather than increased mucous cell density, contributes to the pathologic mucus obstruction in Tg+ mice.
Our mRNA copy number analyses (RT-PCR based gene expression and in situ RNA hybridization analyses) revealed that IL33HET/Tg+ as well as IL33KO/Tg+ mice had comparable copy number for Muc5b (Figure 5a, c, d). Further, MUC5B protein contents were also not significantly different in the BALF from IL33HET/Tg+ as well as IL33KO/Tg+ mice (Figure 5e). On the other hand, while the Muc5ac copy number was trending lower in IL33KO/Tg+ mice versus IL33HET/Tg+ mice (Figure 5b), the MUC5AC protein contents were significantly lower in IL33KO/Tg+ mice versus IL33HET/Tg+ mice (Figure 5f). The positive correlations between increased MUC5B, not MUC5AC, protein contents and the mucus obstruction have been recently reported (20). In this study, a complete loss of MUC5B (Muc5b−/−/Tg+) resulted in significant reduction in the degree of mucus obstruction in Tg+ mice (20). However, the loss of MUC5AC (Muc5ac−/−/Tg+) did not significantly reduce the mucus obstruction of Tg+ mice(20). Along the the same lines, although the IL33KO/Tg+ mice still had high MUC5B contents (Figure 5e), the mucus obstruction was not ameliorated despite significant reduction in MUC5AC proteins contents (Figure 5f). These data suggest that IL33 does not regulate the transcription or translation of Muc5b mucin. It seems plausible that IL33-independent pathways, including recently implicated interleukin 1 receptor- (IL1R-) mediated pathway (21, 22), may regulate the expression of Muc5b in Scnn1b-Tg+ mice.
IL33 has been shown to increase neutrophil recruitment in response to bacterial infections(23, 24). Here, we report that neutrophil counts and the levels of key neutrophil chemoattractants, i.e., KC, G-CSF and MIP2, remain comparable between IL33KO/Tg+ and IL33HET/Tg+ BALF. These data suggest that IL33-independent mechanisms are self-sufficient in neutrophil recruitment in response to spontaneous bacterial infections in Tg+ mice.
It has been shown that ILC2-derived IL5 and IL13 are essential for eosinophil recruitment, the deletion of IL33 receptor (ST2), however, does not completely inhibit eosinophil recruitment(25). In a recent report, Verma et al. reported the existence of IL13-expressing ILC2 in mice lacking ST2(26). These findings suggest the possible existence of IL33-independent mechanisms for eosinophil recruitment whereby ILC2-derived IL13 may induce eosinophil recruitment. However, eosinophil recruitment into Scnn1b-Tg+ airways was abolished in the absence of IL33, i.e., scarce eosinophils were found only in 3 out of 14 IL33KO/Tg+ mice. These data suggest that the eosinophil recruitment to the Scnn1b-Tg+ airways occurs via IL33-responsive ILC2s.
The ASL dehydration in Scnn1b-Tg+ is strongly associated with the alveolar space enlargement(15). The IL33KO/Tg+ mice had significantly greater alveolar space enlargement suggesting a protective role for IL33 in this outcome. Two distinct cell-specific proteases, i.e., macrophage-specific matrix metalloproteinase 12 (MMP12) and neutrophil-specific neutrophil elastase (NE) have been implicated in the alveolar space enlargement response(27, 28). However, both neutrophil and macrophage counts in IL33HET/Tg+ and IL33KO/Tg+ mice were comparable. Since alveolar epithelial cells in IL33HET/Tg+ have upregulated expression of IL33, it is possible that the loss of IL33 in these cells make them susceptible to the alveolar space enlargement. Further studies are required to elucidate the mechanism by which IL33 provides protection against excessive alveolar space enlargement.
In conclusion, this study revealed various findings on the involvement of IL33 in the manifestation of muco-obstructive lung disease in Scnn1b-Tg+ mice. First, IL33 is essential for the recruitment of eosinophils in the airspaces of Tg+ mice. Second, the deletion of IL33 results in the suppression of molecular markers of Th2 inflammation in the Tg+ mice. Third, IL33 is not essential for the transcription of Muc5b and Muc5ac genes, the translation of MUC5AC, however, is suppressed in the absence of IL33. Fourth, while IL33 deletion results in a significant reduction in mucous cell density, the mucus obstruction remains persistent in the Tg+ mice. Consequently, the postnatal mortality and bacterial infection remained persistent in the Tg+ mice. Finally, IL33 provides protection against exaggerated airway inflammation and alveolar space enlargement. These findings indicate that deletion of IL33 suppresses Th2 responses but fails to correct MUC5B hyperconcentration and associated mucus obstruction in Tg+ airways. It remains unclear whether the MUC5B hyperconcentration and mucus obstruction are caused solely by the ASL dehydration or via IL33-independent pathways.
Supplementary Material
Key Points:
IL33 mediates eosinophilic recruitment and Th2 inflammation in Scnn1b-Tg+ airways.
MUC5AC, not MUC5B, is significantly suppressed in the airways of IL33KO/Tg+ mice.
Mucous cell density, not muco-obstruction, is suppressed in IL33KO/Tg+ airways.
Acknowledgments:
We thank Thaya Stoufflet for assistance with multiplex cytokine assays and Sherry Ring for histological tissue processing.
Funding: The work was supported by the Flight Attendant Medical Research Institute (FAMRI) grant (YS), NIGMS Grant # 5P30GM110760 (Pilot Project Funding), and LSU SVM Startup funds (YS).
Footnotes
Disclosures: The authors have no conflicts of interest to disclose.
References
- 1.Knowles MR, and Boucher RC. 2002. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 109: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boucher RC 2007. Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med 261: 5–16. [DOI] [PubMed] [Google Scholar]
- 3.Boucher RC 2007. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58: 157–170. [DOI] [PubMed] [Google Scholar]
- 4.Tarran R 2004. Regulation of airway surface liquid volume and mucus transport by active ion transport. Proc Am Thorac Soc 1: 42–46. [DOI] [PubMed] [Google Scholar]
- 5.Mall M, Grubb BR, Harkema JR, O’Neal WK, and Boucher RC. 2004. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med 10: 487–493. [DOI] [PubMed] [Google Scholar]
- 6.Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, and Nadel JA. 1999. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol 162: 6233–6237. [PubMed] [Google Scholar]
- 7.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, and Elias JA. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest 103: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, and Kastelein RA. 2005. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23: 479–490. [DOI] [PubMed] [Google Scholar]
- 9.Lewis BW, Sultana R, Sharma R, Noel A, Langohr I, Patial S, Penn AL, and Saini Y. 2017. Early Postnatal Secondhand Smoke Exposure Disrupts Bacterial Clearance and Abolishes Immune Responses in Muco-Obstructive Lung Disease. J Immunol 199: 1170–1183. [DOI] [PubMed] [Google Scholar]
- 10.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, Sudo K, Okumura K, Saito H, and Nakae S. 2010. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A 107: 18581–18586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livraghi-Butrico A, Kelly EJ, Klem ER, Dang H, Wolfgang MC, Boucher RC, Randell SH, and O’Neal WK. 2012. Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal Immunol 5: 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saini Y, Wilkinson KJ, Terrell KA, Burns KA, Livraghi-Butrico A, Doerschuk CM, O’Neal WK, and Boucher RC. 2016. Neonatal Pulmonary Macrophage Depletion Coupled to Defective Mucus Clearance Increases Susceptibility to Pneumonia and Alters Pulmonary Immune Responses. Am J Respir Cell Mol Biol 54: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Livraghi A, Grubb BR, Hudson EJ, Wilkinson KJ, Sheehan JK, Mall MA, O’Neal WK, Boucher RC, and Randell SH. 2009. Airway and lung pathology due to mucosal surface dehydration in {beta}-epithelial Na+ channel-overexpressing mice: role of TNF-{alpha} and IL-4R{alpha} signaling, influence of neonatal development, and limited efficacy of glucocorticoid treatment. J Immunol 182: 4357–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsey KA, Rushton ZL, and Ehre C. 2016. Mucin Agarose Gel Electrophoresis: Western Blotting for High-molecular-weight Glycoproteins. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mall MA, Harkema JR, Trojanek JB, Treis D, Livraghi A, Schubert S, Zhou Z, Kreda SM, Tilley SL, Hudson EJ, O’Neal WK, and Boucher RC. 2008. Development of chronic bronchitis and emphysema in beta-epithelial Na+ channel-overexpressing mice. Am J Respir Crit Care Med 177: 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saini Y, Dang H, Livraghi-Butrico A, Kelly EJ, Jones LC, O’Neal WK, and Boucher RC. 2014. Gene expression in whole lung and pulmonary macrophages reflects the dynamic pathology associated with airway surface dehydration. BMC Genomics 15: 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saini Y, Lewis BW, Yu D, Dang H, Livraghi-Butrico A, Del Piero F, O’Neal WK, and Boucher RC. 2018. Effect of LysM+ macrophage depletion on lung pathology in mice with chronic bronchitis. Physiol Rep 6: e13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livraghi-Butrico A, Grubb BR, Kelly EJ, Wilkinson KJ, Yang H, Geiser M, Randell SH, Boucher RC, and O’Neal WK. 2012. Genetically determined heterogeneity of lung disease in a mouse model of airway mucus obstruction. Physiol Genomics 44: 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, and Lambrecht BN. 2009. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med 15: 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livraghi-Butrico A, Grubb BR, Wilkinson KJ, Volmer AS, Burns KA, Evans CM, O’Neal WK, and Boucher RC. 2017. Contribution of mucus concentration and secreted mucins Muc5ac and Muc5b to the pathogenesis of muco-obstructive lung disease. Mucosal Immunol 10: 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fritzsching B, Zhou-Suckow Z, Trojanek JB, Schubert SC, Schatterny J, Hirtz S, Agrawal R, Muley T, Kahn N, Sticht C, Gunkel N, Welte T, Randell SH, Langer F, Schnabel P, Herth FJ, and Mall MA. 2015. Hypoxic epithelial necrosis triggers neutrophilic inflammation via IL-1 receptor signaling in cystic fibrosis lung disease. Am J Respir Crit Care Med 191: 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, Lin JM, Gilmore RC, Batson BD, O’Neal YK, Volmer AS, Dang H, Deng Y, Randell SH, Button B, Livraghi-Butrico A, Kesimer M, Ribeiro CM, O’Neal WK, and Boucher RC. 2019. IL-1beta dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest 129: 4433–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson KM, Ramanan K, Clay ME, McHugh KJ, Rich HE, and Alcorn JF. 2018. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol 11: 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan F, Yuan B, Liu T, Luo X, Huang P, Liu Y, Dai L, and Yin H. 2016. Interleukin-33 facilitates neutrophil recruitment and bacterial clearance in S. aureus-caused peritonitis. Mol Immunol 72: 74–80. [DOI] [PubMed] [Google Scholar]
- 25.Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, and Kita H. 2014. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol 193: 1549–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma M, Liu S, Michalec L, Sripada A, Gorska MM, and Alam R. 2018. Experimental asthma persists in IL-33 receptor knockout mice because of the emergence of thymic stromal lymphopoietin-driven IL-9(+) and IL-13(+) type 2 innate lymphoid cell subpopulations. J Allergy Clin Immunol 142: 793–803 e798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gehrig S, Duerr J, Weitnauer M, Wagner CJ, Graeber SY, Schatterny J, Hirtz S, Belaaouaj A, Dalpke AH, Schultz C, and Mall MA. 2014. Lack of neutrophil elastase reduces inflammation, mucus hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-like lung disease. Am J Respir Crit Care Med 189: 1082–1092. [DOI] [PubMed] [Google Scholar]
- 28.Trojanek JB, Cobos-Correa A, Diemer S, Kormann M, Schubert SC, Zhou-Suckow Z, Agrawal R, Duerr J, Wagner CJ, Schatterny J, Hirtz S, Sommerburg O, Hartl D, Schultz C, and Mall MA. 2014. Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12-dependent emphysema. Am J Respir Cell Mol Biol 51: 709–720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.