

Abstract
Extracellular deposits of the amyloid-beta peptide (Aβ) are known as the main pathological hallmark of Alzheimer’s disease. In Alzheimer’s disease, neurons are injured and die throughout the brain, a process in which Aβ neurotoxicity is considered to play an important role. However, the molecular mechanisms underlying Aβ toxicity that lead to neurodegeneration are not clearly established. Here we have elucidated the molecular pathways and networks which are impacted by Aβ in neurons using SH-SY5Y human neuroblastoma cells as a model. These cells were treated with Aβ1–42 peptides to study changes in biochemical networks using tandem mass tag labeled quantitative proteomic technique followed by computational analysis of the data. The molecular impacts of Aβ on cells were evident in a time- and dose-dependent manner, albeit the duration of treatment induced greater differential changes in cellular proteome compared to the effects of concentration. Aβ induced early changes in proteins associated with lysosomes, collagen chain trimerization and extracellular matrix receptor interaction, complement and coagulation cascade, oxidative stress induced senescence, ribosome biogenesis, regulation of insulin-like growth factor transport and uptake by insulin-like growth factor-binding protein. These novel findings provide molecular insights on the effects of Aβ on neurons, with implications for better understanding the impacts of Aβ on early neurodegeneration in Alzheimer’s disease pathology.
Keywords: Alzheimer's disease, amyloid, lysosomes, neurodegeneration, oxidative stress, proteomics, ribosome, SH-SY5Y cells
Chinese Library Classification No. R446; R364; Q2
Introduction
Every three seconds, there is one new case of dementia diagnosed, with over 9.9 million people being affected each year worldwide. It was estimated that 50 million people worldwide were living with dementia in 2018 (Patterson, 2018). Alzheimer’s disease (AD) is the most common form of dementia, and is characterised as neurodegenerative, chronic, and progressive (Hardy, 2002; Gouras et al., 2015). A hallmark pathological feature of AD is the misfolded and accumulated amyloid-beta peptide (Aβ), which leads to the formation of extracellular fibrils and plaques (Murphy and LeVine, 2010; De Strooper and Karran, 2016). Nearly 30 years of studies have produced extensive evidence that accumulated Aβ in the brain is responsible for memory and cognitive modalities, and plays a central role in the progression of the disease. Changes of mitochondrial function, synaptic plasticity in addition to many other processes induced by Aβ have a direct impact on activity-dependent signaling and gene expression (Song et al., 2006; Parihar and Brewer, 2010; Olah et al., 2011; Krishtal et al., 2017). The gradually accumulated Aβ plaques in the brain potentially cause an axonal physical damage, which finally result in cytoskeletal alternations, an important underlying factor in neurofibrillary tangle pathology and neurodegeneration (Vickers et al., 2000).
Aberrant modifications of amyloid precursor protein (APP) processing leads to increased accumulation of Aβ, which has been suggested to be one of the initial steps in the progression of AD, and potentially causes downstream pathological lesions, such as the initiation of neuroinflammatory processes. Aβ pathology expands in a time-dependent manner, and the deposits that play an important role in the disease development are formed progressively throughout the brains of AD patients (Sowade and Jahn, 2017). Several in vivo and in vitro studies have highlighted numerous toxic mechanisms of Aβ, including excitotoxicity, mitochondrial and synaptic dysfunction, oxidative stress and calcium imbalance. However, it is not clear how the amyloid aggregates initiate the death of neuronal cells. The main challenges for AD research currently is to achieve a better understating of the complex cellular reactions underlying the initial stages of the disease, and systematically illustrate the progressive cellular alterations that occur in the cells in response to APP dysregulation and consequent Aβ accumulation (De Strooper and Karran, 2016).
With the “big data” proteomic approaches that have been applied to AD research for more than 15 years, we are increasingly gaining insights into AD pathogenesis and identifying novel molecular players that may play critical roles in various stages of the disease process (Butterfield et al., 2003; Zellner et al., 2009). Human neuroblastoma SH-SY5Y cells are one of the most commonly used cell lines in neuroscience research (Agholme et al., 2010; Omar et al., 2018), and are also known to be a relevant cellular model for biochemical investigations in AD (Krishtal et al., 2017). In our study, Aβ toxicity on SH-SY5Y cells was established with two time points (6 and 24 hours) and two concentrations (5 and 25 μM). Several biochemical pathways responded to treatment in a time- and concentration-dependent manner, such as regulation of insulin-like growth factor (IGF) transport and uptake by IGF-binding proteins (IGFBPs), collagen chain trimerization and extracellular matrix (ECM) receptor interaction, complement and coagulation cascades, lysosome, autophagy, ribosomal biogenesis, and oxidative phosphorylation. These pathways and regulated proteins provided global and comprehensive insights into the impacts of Aβ peptides on human neurons. The findings will advance the pathological understanding of the disease and may lead to the development of new mechanism based therapeutic strategies.
Materials and Methods
Cell culture and Aβ treatments
SH-SY5Y human neuroblastoma cells (American Type Culture Collection, Rockville, MD, USA) were cultured in DMEM culture medium with pH 7.4, supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 U/mL streptomycin, at 37°C, 5% CO2 (Gupta et al., 2010, 2012; You et al., 2014). The old culture medium was discarded and exchanged with new culture medium when cells underwent exponential growth phase to approximately 80% confluence. Aβ1–42 fragments (Sigma, St. Louis, MO, USA) were dissolved in PBS and added into the plates to a final concentration of 5 and 25 μM, respectively (Guivernau et al., 2016; Deng et al., 2019). Cells were incubated in the culture medium at 37°C as reported previously (Deng et al., 2019). Samples were separately collected after cells were treated with Aβ for 6 and 24 hours. Two concentrations and two time points established four treatments, including T1 (5 μM_6 hours), T2 (5 μM_24 hours), T3 (25 μM_6 hours), and T4 (25 μM_24 hours), to mimic the AD progression and assess the effect of Aβ toxicity on neuronal cells. Four biological replicates were considered for each Aβ treatment and five biological replicates for controls (vehicle treated cells).
Protein sample preparation
Cells were lysed by sonication (twice, 30-second intervals, 40 Hz × 15 seconds) after adding cold lysis buffer (50 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 1% NP40, 1 mM EDTA, 0.1% SDS) containing protease inhibitor. After centrifugation (18,000 × g, 15 minutes) at 4°C, the supernatant was collected in a fresh Eppendorf tube while the remaining cell debris was removed (Basavarajappa et al., 2011; Gupta et al., 2014). Dithiothreitol (20 mM) was added into the extracted proteins, and reduction was carried out for 15 minutes at room temperature, followed by alkylation for 30 minutes in dark with 50 mM iodoacetamide. Proteins were precipitated using chloroform and methanol approach for removing the interferents and other contaminants (Deng et al., 2019). Samples were air-dried for evaporation of the remaining methanol. Protein pellets were completely dissolved with 200 μL of urea buffer (8 M urea, 100 mM Tris-HCl, pH 8.5), followed by protein concentration measurement using BCA protein assay kit. An aliquot of 200 μg of proteins was digested using trypsin (protein: enzyme = 50:1) at 37°C overnight. The resultant peptides were then acidified using trifluoroacetic acid. Samples were cleaned by SDB-RPS (3M-Empore, Maplewood, MN, USA) stage tips before drying in speed vacuum. Before tandem mass tag (TMT) labeling, the dried peptides reconstituted with hydroxyethyl piperazine-ethane-sulfonic acid (HEPES) buffer (100 mM HEPES, pH 8.0).
TMT labeling of peptides, liquid chromatography tandem mass spectrometry analysis and peptide to spectrum matching
Two separate 10-plex TMT experiments were designed to accommodate all biological replicates of different treatments and controls, as outlined in previously published studies (Mirzaei et al., 2017b, 2019; Deng et al., 2019). Briefly, 100 μg of peptides from each biological replicate was labeled with TMT reagents (Thermo Fisher, Waltham, MA, USA). The reaction was carried out at room temperature for 1 hour, followed by addition of 8 μL of 5% fresh hydroxylamine to samples for fully quenching the unbounded TMT labels. For each TMT experiment, ten labeled samples were pooled together and dried completely using a speed vacuum. After resuspension with 1% formic acid, labeled samples were desalted using Sep-Pak C18 cartridges (Waters, Milford, MA, USA). Subsequently, peptides were fractionated using high pH reversed phase fractionation (HpH), and pooled into 16 fractions. The fractions were dried and reconstituted in 1% formic acid and desalted again using SDB-RPS stage tips. The liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis of peptides was performed using reversed phase nano-flow liquid chromatography, and identified and quantified using high resolution mass spectrometry on a Q Exactive orbitrap, followed by peptide to spectrum matching using local Mascot search engine against the Homo Sapiens SwissProt database (http://www.ebi.ac.uk/swissprot/) as described extensively in our previous studies (Deng et al., 2019; Mirzaei et al., 2019).
Analysis of multiplexed quantitative proteomics data
In-house TMTPrepPro package, which are deployed on a local GenePattern server, was used to further analyse the identified proteins (Mirzaei et al., 2017a). All protein ratios with respect to the reference (label-126) were extracted and combined across runs. The differentially expressed proteins (DEPs) were identified using two-way analysis of variance (ANOVA) of all proteins identified reproducibly across different treatments including control, in addition to pairwise comparisons (Student’s t-test) of each treatment versus control. Significantly regulated proteins based on two-way ANOVA comparison (log-transformed ratios) were clustered to check whether the four Aβ peptide treatments and controls were well separated from each other. For the pairwise comparisons, the relative quantitation of proteins in Aβ peptide treatments versus control was derived from the ratio of the TMT label signal to noise (S/N) detected in each specific treatment to control. The overall fold changes were calculated as geometric means of the respective ratios. Two criteria were applied to determine significantly regulated proteins: fold change over 1.20 and P-value lower than 0.05 (Margolin et al., 2009; Mirzaei et al., 2017b, 2019; Deng et al., 2019). The proteins with changed expression through the pairwise comparisons were subjected to pathway enrichment analysis using Ingenuity Pathway Analysis (IPA) software (QIAGEN, Venlo, Netherlands). Protein interaction networks and molecular/cellular functions (P-value < 0.05) were identified based on known protein-protein interactions in the Ingenuity knowledge base. Canonical pathway analysis was used to identify the enriched pathways from the up- and down-regulated proteins. These regulated proteins were further classified using gene ontology functional classification in PANTHER (http://www.pantherdb.org) (Mi et al., 2013). Further, the regulated proteins identified from the one-way ANOVA were classified using STRING plugin in Cytoscape (https://cytoscape.org) based on their pathways and biological processes. In addition, we compared this data with our previous study on 661W mouse photoreceptor cells treated with Aβ using the same strategy, treating cells with Aβ at two concentrations (5 and 25 µM) and two time points (6 and 24 hours) (Deng et al., 2019).
Results
Differential proteome profile of human neuronal cells in response to Aβ treatment
We identified a total of 7525 proteins from SH-SY5Y human neuronal cells treated with two doses of Aβ (5 and 25 µM) at two time points (6 and 24 hours) (Additional Table 1 (2.5MB, pdf) ). We identified 473 DEPs under different conditions using two-way ANOVA (Additional Table 2 (1.1MB, pdf) ). The expression patterns of differentially regulated proteins demonstrated that the consistency of up- or down-regulation proteins within 6 hours (5 and 25 µM) and 24 hours (5 and 25 µM) treatments (Figure 1). The data clearly indicates the changes in regulation of SH-SY5Y human neuron cells proteome in regard to both the duration and concentrations of Aβ treatment.
Figure 1.
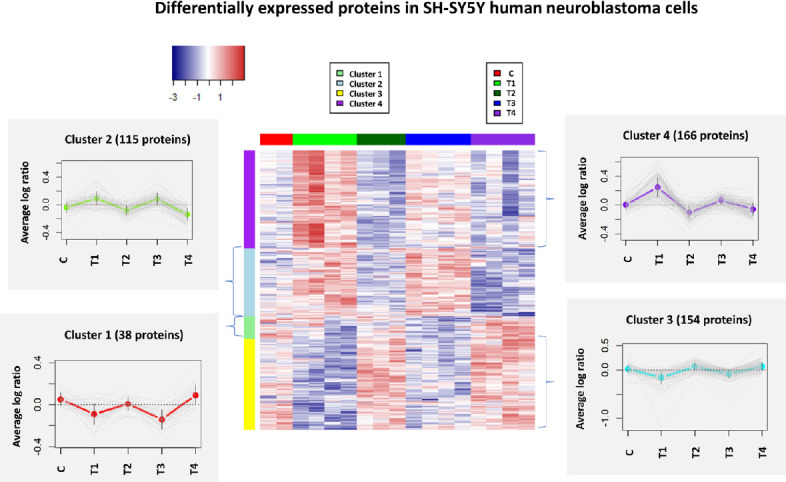
Heatmap (hierarchical clustering) of the log-transformed ratios of differentially expressed proteins in SH-SY5Y human neuroblastoma cells after β-amyloid peptide treatments.
Differences among all experimental conditions via two-way analysis of variance are found (P < 0.05); row clustering only. Column colors indicate treatment type, and the cluster patterns are detailed on the side plots (clusters 1–4). C, T1, T2, T3 and T4 represent control and four specific conditions as described in the methods.
Additionally, four comparative analyses comparing protein expression using Student’s t-tests were applied to identify the significantly regulated proteins between control and four treatment groups (Figure 2). 110 and 75 proteins were identified as up- and down-regulated in T1 (5 µM_6 hours) compared to the control condition, respectively (Additional Table 3 (956.2KB, pdf) ). In contrast, fewer differentially regulated proteins were identified in the other three treatments, among which T2 had the minimum number of DEPs with 17 up- and 25 down-regulated proteins. Although similar patterns and proteome profiles were shown between T1 (5 µM_6 hours) and T3 (25 µM_6 hours) (Figure 1), T1, with lower concentration and shorter exposure time, was the treatment that elicited the most significant changes in the proteome (Figure 2). The number of differentially regulated proteins in T3 (25 µM_6 hours) and T4 (25 µM_24 hours) were quite similar, with 37 up- and 40 down-regulated proteins being identified in T3 while 34 up- and 38 down-regulated proteins being found in T4. The top 10 regulated proteins in four treatments, sorted by fold changes, are shown in Table 1. Fold changes of DEPs varied from –4.718 (down-regulated) to 3.128 (up-regulated). Two up-regulated proteins, COPS9 and HMOX1, and two down-regulated proteins, MT1G and GM2A, overlapped between T1 and T3 at 6 hours with either 5 or 25 µM. Two proteins, TNFAIP8 and TAF1, were commonly up-regulated in T2 and T4 at 24 hours of time point with either 5 or 25 µM concentration. One down-regulated protein, JPT1, was identified in both T3 and T4 with 25 µM Aβ1–42 at either 6 or 24 hours.
Figure 2.
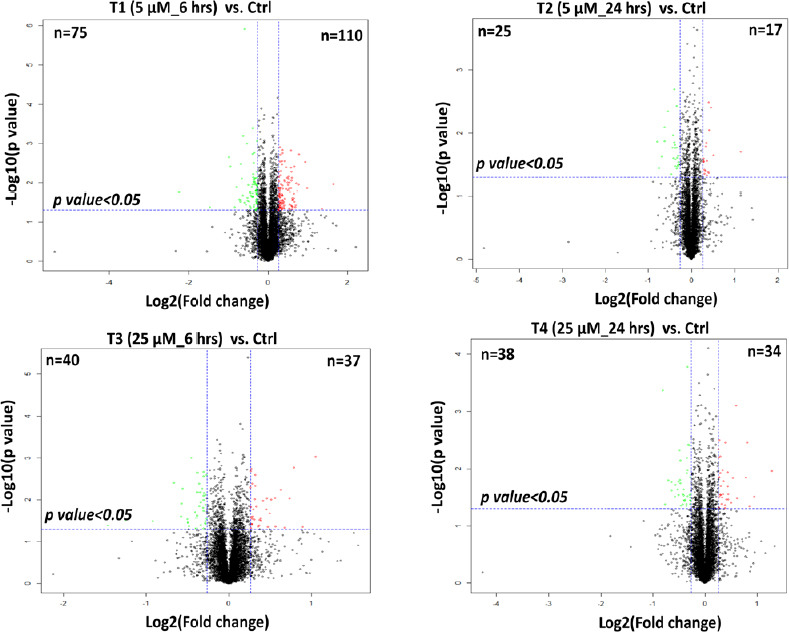
Volcano plots demonstrating the dual thresholds for differentially regulated proteins after four comparative analyses with control using Student’s t-test.
Proteins within the upper and outer quadrants meet both the fold change and P-value cut-off and are therefore considered as differentially regulated. Red and green represent the up- and down-regulated proteins in each treatment respectively.
Table 1.
Top 10 regulated proteins in four treatments with P-value lower than 0.05
| T1 (5 μM_6 h) vs Ctrl |
T2 (5 μM_6 h) vs Ctrl |
T3 (25 μM_6 h) vs Ctrl |
T4 (25 μM_6 h) vs Ctrl |
|||||
|---|---|---|---|---|---|---|---|---|
| Gene ID | FC | Gene ID | FC | Gene ID | FC | Gene ID | FC | |
| Up-regulated (Treatment vs. Ctrl) | UBE2W | 3.128 | TNFAIP8 | 2.218 | COPS9 | 2.079 | RFXANK | 2.433 |
| HIST1H1E | 2.574 | RNF121 | 1.419 | FLG2 | 1.864 | PHOSPHO2 | 1.924 | |
| COPS9 | 1.976 | SET | 1.369 | RAD51D | 1.732 | CALR3 | 1.804 | |
| HMOX1 | 1.924 | UQCR11 | 1.337 | HMOX1 | 1.671 | NEDD4L | 1.748 | |
| CDH6 | 1.716 | TAF1 | 1.322 | ZNF384 | 1.604 | TNFAIP8 | 1.724 | |
| CCDC138 | 1.711 | SERBP1 | 1.319 | KRT17 | 1.482 | TAF8 | 1.536 | |
| C15orf41 | 1.667 | GPC6 | 1.314 | SLC35F6 | 1.479 | CALU | 1.510 | |
| C8B | 1.640 | NDUFAF2 | 1.288 | BRD9 | 1.427 | KRT6A | 1.445 | |
| C9 | 1.621 | APOO | 1.283 | OGFOD2 | 1.410 | TAF1 | 1.416 | |
| KLC1 | 1.616 | ATG4A | 1.276 | ADGRE5 | 1.390 | SGO1 | 1.362 | |
| Down-regulated (Treatment vs. Ctrl) | MT1G | –4.718 | RBX1 | –1.735 | MT1G | –2.752 | CFB | –1.742 |
| ING1 | –2.757 | CPNE4 | –1.683 | JPT1 | –2.368 | JPT1 | –1.687 | |
| NPC2 | –1.975 | NOTCH3 | –1.599 | POLL | –1.887 | XRCC2 | –1.617 | |
| HPCAL1 | –1.935 | CFI | –1.533 | PCNP | –1.579 | RAP2A | –1.554 | |
| DECR1 | –1.787 | LTF | –1.530 | GM2A | –1.487 | HSP90AB4P | –1.554 | |
| GM2A | –1.735 | AHSG | –1.452 | CYCS | –1.478 | DLX2 | –1.491 | |
| SMIMI5 | –1.643 | COL1A1 | –1.380 | FUBP1 | –1.471 | MMGT1 | –1.474 | |
| CELF2 | –1.625 | CHIA | –1.348 | BOLA1 | –1.410 | B4GALT7 | –1.435 | |
| GPR155 | –1.614 | PLXDC2 | –1.338 | DEPTOR | –1.406 | ZFP36L2 | –1.418 | |
| TXN | –1.602 | PLEKHA8 | –1.331 | SERGEF | –1.403 | HNRNPAB | –1.416 | |
The red and blue colors represent the top ten up- and down-regulated proteins respectively. Ctrl: Control; FC: fold change.
A clear visual representation of differentially regulated proteins in four treatments are presented as Venn diagrams in Figure 3. Initially, we assessed the effects of exposure time of neuronal cells to Aβ on protein expression. The result (Figure 3A) indicated that a greater number of proteins were regulated in lower concentration of Aβ1–42 (5 µM) in response to 6-hour treatment (T1) compared with 24 hours (T2). Besides, although a similar number of regulated proteins responded to the higher concentration of Aβ treatments (25 µM), only a small overlap of four DEPs were identified between two treatments at 6 (T3) and 24 hours (T4) (Figure 3B) and lower number of proteins identified as deferentially expressed at higher concentration of Aβ1–42 (25 μM) (Figure 3C). Further, we found more protein changes in 25 µM of Aβ treatment when compared to the treatment with lower concentration in response to 24-hour treatment (Figure 3D). In a summary, more proteins were differentially expressed with 6 hours of Aβ treatments (T1 and T3) compared to 24 hours of Aβ treatments (T2 and T4) (Figure 3E). These results indicated that 6 hours of Aβ treatment more severely affected the SY-SY5Y human neuronal cells, especially T1 (5 µM_6 hours) when compared with the other three treatments with either higher concentrations or longer treatment period.
Figure 3.
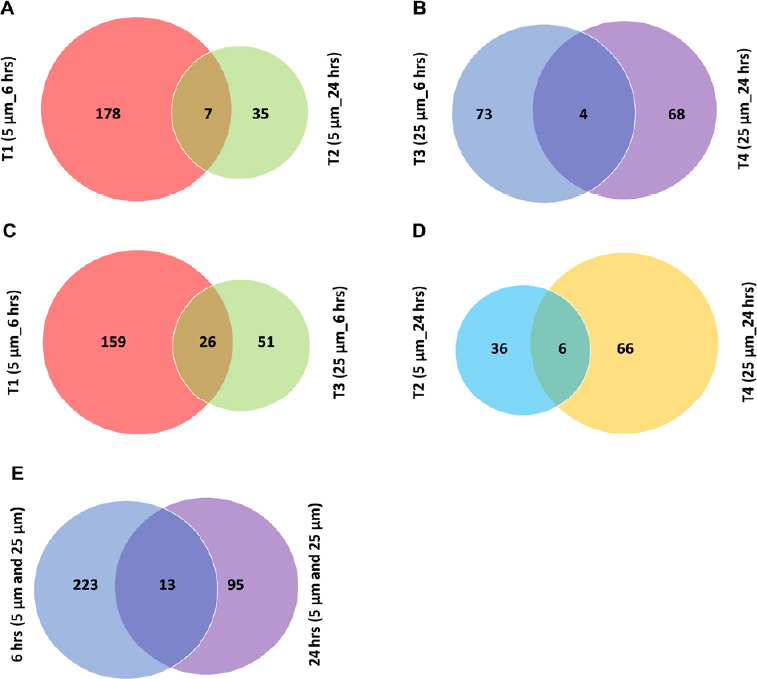
Venn diagram analysis of regulated proteins identified under different conditions.
(A) Venn diagram indicating the overlap between the differentially expressed proteins identified and quantified in T1 and T2 with 5 µM of β-amyloid peptide (Aβ) at 6 and 24 hours, respectively (1% false discovery rate (FDR). (B) Venn diagram indicating the overlap between the differentially expressed proteins identified and quantified in T3 and T4 with 25 µM of Aβ at 6 and 24 hours, respectively (1% FDR). (C) Venn diagram indicating the overlap between the differentially expressed proteins identified and quantified in T1 and T3 at 6 hours with 5 µM and 25 µM of Aβ, respectively (1% FDR). (D) Venn diagram indicating the overlap between the differentially expressed proteins identified and quantified in T2 and T4 at 24 hours with 5 and 25 µM of Aβ, respectively (1% FDR). (E) Venn diagram indicating the overlap between the differentially expressed proteins identified and quantified at 6 hours with two concentrations (T1 and T3) and at 24 hours with two concentrations (T2 and T4), respectively (1% FDR).
Pathway classification of the DEPs
To identify the molecular mechanisms and biological processes impacted by Aβ in neuronal cells, biochemical pathway analysis was applied using IPA analysis to characterize the DEPs identified after exposure to the four treatments (Figure 4). The acute phase response signaling pathway, complement and coagulation system, and autophagy were among the most affected pathways at 5 µM at 6 and 24 hours while the acute phase response signaling pathway was mostly affected at 6 hours. In contrast, proteins involved in oxidative phosphorylation were more impacted at 6 hours at both concentrations. Liver X receptor/retinoid X receptor (LXR/RXR) activation and farnesoid X receptor/retinoid X receptor (FXR/RXR) were also impacted by Aβ, but this impact was not apparent in T4 with 25 µM of Aβ at 24 hours. Sirtuin signaling pathway was enriched in T2 with 5 µM of Aβ at 24 hours, while mammalian target of rapamycin (mTOR) signaling pathway was affected in T1 (5 µM_6 hours) and T4 (25 µM_24 hours). Interestingly, nuclear factor erythroid 2-related factor 2-mediated oxidative stress response pathway was altered in abundance to a much greater extent in T4 (25 µM_24 hours).
Figure 4.

Comparison of the top canonical pathways enriched from ingenuity pathway analysis of differentially regulated proteins (treatments vs. control) in four treatments.
The significance of functional enrichment is highlighted with red.
To deeper characterize the data, DEPs were also categorized using PANTHER tool, based on their gene ontology terms ‘biological process, molecular process and cellular component’ (Mi et al., 2013). The percentage of proteins in specific processes and components were calculated, as shown in Figure 5. More biological processes were generated in the initial treatment T1 (5 µM_6 hours), which corresponds to the identification of more DEPs (Figure 5A). A higher percentage of proteins were related to “cellular component organization or biogenesis” at 6 hours at both concentrations (T1 and T3), while a larger proportion of proteins was involved in ‘biological regulation’ at 24 hours (T2 and T4). Interestingly, the highest ratio of proteins in two processes, ‘response to stimulus’ and ‘immune system process’, were detected at T2 and T4 at 24 hours, respectively. Additionally, more proteins with ‘transporter activity’ were found at 25 µM of Aβ at 24 hours (T4) (Figure 5B).
Figure 5.
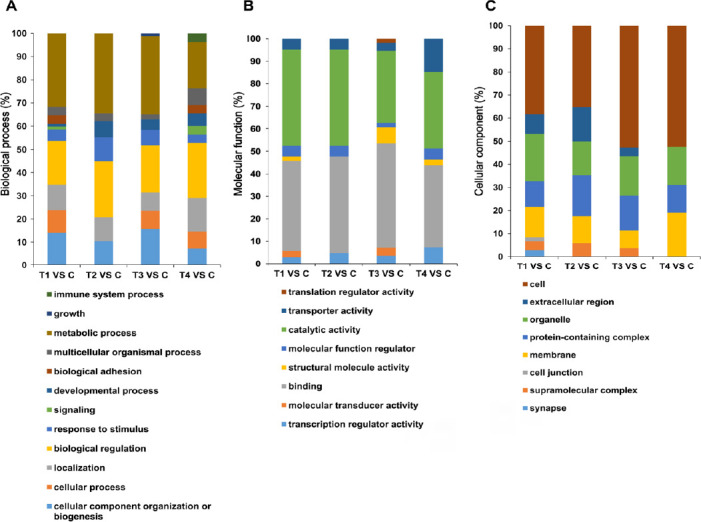
Categorization of the differentially expressed proteins in four treatments with Student’s t-test according to their involvements in different gene ontology terms “biological process, molecular process and cellular component”.
Percentage of proteins in specific terms were calculated and illustrated. C, T1, T2, T3 and T4 represent control and four specific conditions as described in the methods. (A) Biological process; (B) molecular process; (C) cellular component.
Besides, an interaction network using the STRING Cytoscape plugin was built with DEPs identified with two-way ANOVA. Pathways related to Alzheimer’s disease were enriched and illustrated (Figure 6), and the expression data of proteins was shown using a heatmap (Figure 7). Several pathways enriched with IPA analysis were also introduced using the STRING, such as mTOR signaling, autophagy, oxidative phosphorylation, complement, and coagulation. In addition, other interaction networks illustrating changes in lysosomal proteins, collagen chain trimerization and ECM receptor interaction, oxidative stress induced senescence, ribosome biogenesis, and regulation of insulin-like growth factor transport and uptake by IGFBPs were also identified. Importantly, inverse protein regulations were obvious between the two concentrations at 6 hours (T1 and T2) but with pathways being more significantly impacted initially (5 µM_6 hours), which may illustrate the molecular response during the early AD. Additionally, similar regulation patterns were detected at 6 or 24 hours, indicating the impact of Aβ on neuronal cells was more time-dependent (6 and 24 hours) rather than concentration-dependent (5 and 25 µM).
Figure 6.
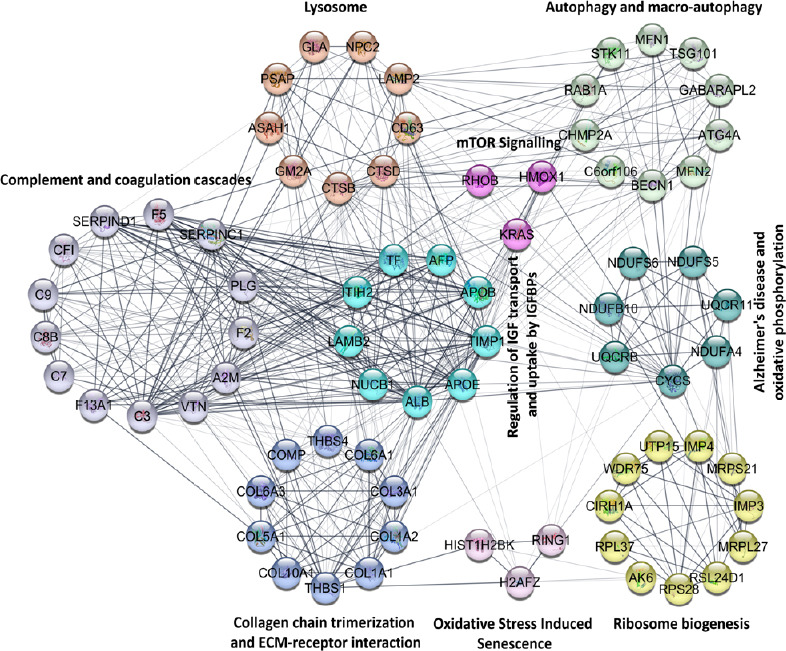
Functional interaction network analyzed by String Cytoscape plugin.
Network nodes are labeled with gene symbols. Different colors represent proteins classified into specific pathways which are highlighted beside the nodes. The lines indicate the interactions among proteins.
Figure 7.
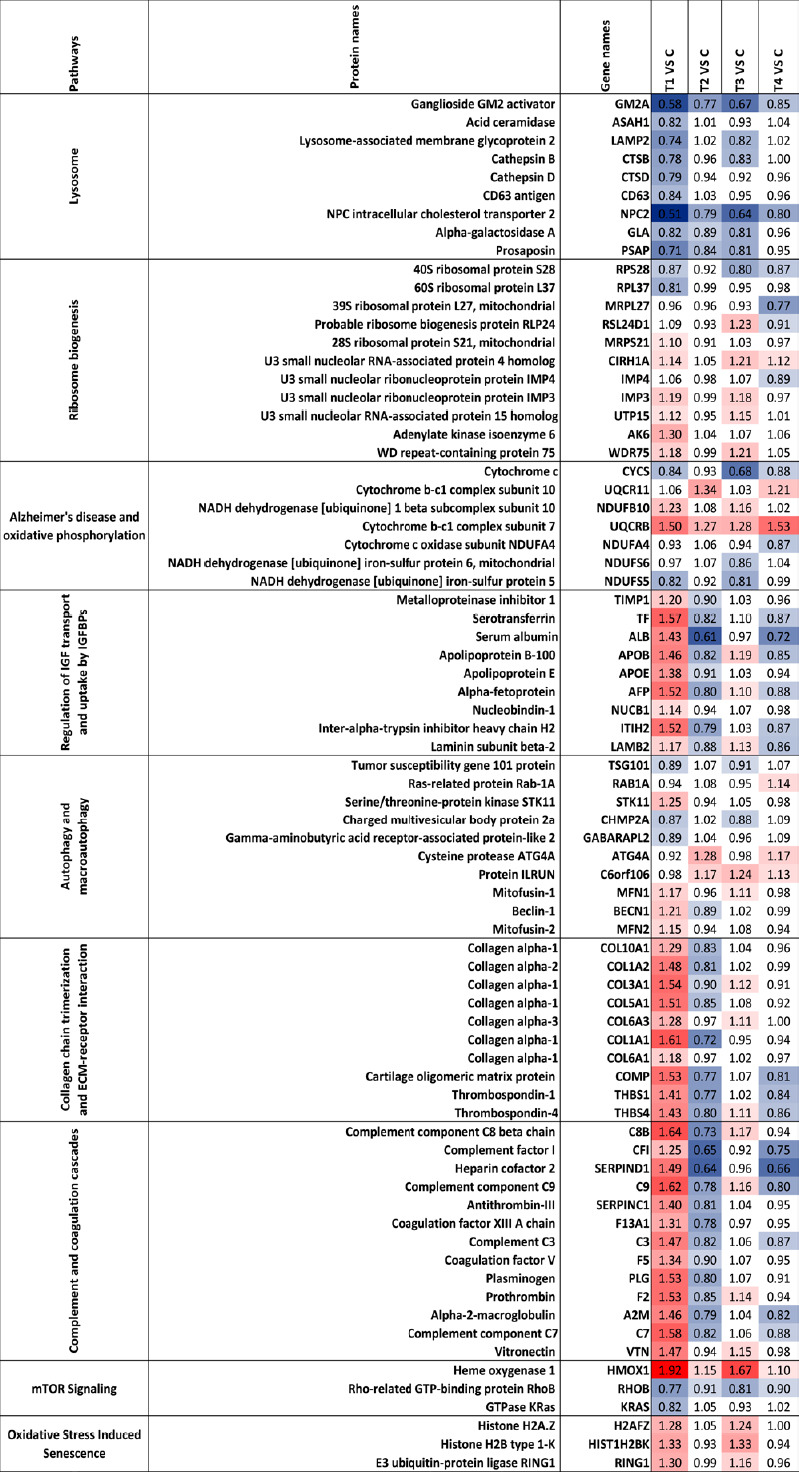
Heatmap of the regulated proteins in pathways related to Alzheimer’s disease.
Red and blue indicate relative increase or decrease in protein abundance, respectively. C, T1, T2, T3 and T4 represent control and four specific conditions as described in the Methods.
Lysosomal dysfunction induced by Aβ toxicity
There were nine proteins associated with lysosomal regulation that were differentially modulated across four treatment groups (Figure 7) with all of them down-regulated in lower concentration at early time point (5 µM_6 hours). GM2A and NPC2 were down-regulated across all the treatment groups suggesting that these proteins were primarily affected upon Aβ exposure, irrespective of concentration and timing of treatment. Most of the lysosomal proteins, in general, were less affected when cells were exposed to Aβ for longer periods (24 hours), suggesting that initial down-regulation of protein expression was transient, and expression bounced back to normal levels with time. At higher concentration (25 µM), the perturbations in lysosomal associated proteins were more subtle compared to 5 µM of Aβ which caused more robust changes in the expression levels of proteins. ASAH1, CD63 and CTSD proteins, in particular, exhibited a significant change at 5 µM, but no significant alteration was observed when the cells were exposed to 25 µM of Aβ.
ECM proteins are up-regulated early upon Aβ treatment
ECM associated proteins including thrombospondin-1 (THBS1), thrombospondin-4 (THBS4), cartilage oligomeric matrix protein (COMP) and seven collagen proteins (collagen type X alpha 1 chain (COL10A1), COL1A2, COL3A1, COL5A1, COL6A3, COL1A1 and COL6A1) were observed to be up-regulated in the early stage at 5 µM at 6 hours (T1). Interestingly, most of these were down-regulated at 24 hours (except COL6A3 and COL6A1) (Figure 7). A similar pattern of regulation was also found at 6 hours with 5 and 25 µM of Aβ (T1 and T3), but with fewer proteins differentially expressed when cells were treated with higher concentration (25 µM). There were only two collagen proteins (COL3A1 and COL6A3) slightly up-regulated at 25 µM at 6 hours (T3), while the expression of all collagen proteins reflected no significant change at this concentration at 24 hours (T4). However, THBS1, THBS4 and COMP were down-regulated at both concentrations at 24 hours (T2 and T4).
Differential modulation of ribosomal biogenesis proteins upon Aβ exposure
A total of 11 ribosomal biogenesis related proteins were induced by Aβ across the four data sets (Figure 7). The majority of these DEPs were identified at 6 hours of treatment regardless of the concentration (T1 and T3), while the levels bounced back closer to original levels at 24 hours of exposure time, especially in T2 (5 µm_24 hours) with no such protein changes detected. 40S ribosomal protein S28 (RPS28) and 60S ribosomal protein L37 (RPL37) were down-regulated initially (5 µM_6 hours), while mitochondrial 39S ribosomal protein L27 (MRPL27) was down-regulated only in T4 (25 µM_24 hours). A majority of proteins, including U3 small nucleolar RNA-associated protein 4 homolog (CIRH1A), U3 small nucleolar ribonucleoprotein protein (IMP3), U3 small nucleolar RNA-associated protein 15 homolog (UTP15), WD repeat-containing protein 75 (WDR75) and 40S ribosomal protein S28 (RPS28), were similarly affected at 6 hours Aβ exposure time irrespective of the concentration (T1 and T3).
Differential effects on oxidative phosphorylation and autophagy related proteins
We identified seven DEPs that have been associated with AD pathogenesis and oxidative phosphorylation, including four NADH dehydrogenases that were differentially regulated across four treatments (NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 10 (NDUFB10), Cytochrome c oxidase subunit NDUFA4, mitochondrial NADH dehydrogenase [ubiquinone] iron-sulfur protein 6 (NDUFS6) and NADH dehydrogenase [ubiquinone] iron-sulfur protein 5 (NDUFS5)) (Figure 7). Ubiquinol-cytochrome c reductase binding protein was identified to be consistently up-regulated upon exposure to Aβ across all the treatment groups. In addition to the mitochondrial proteins, autophagy pathway was impacted with ten proteins differentially modulated in specific conditions, indicating that clearance of the proteins might be affected in the neuronal cells upon Aβ exposure. Corresponding to impaired oxidative phosphorylation and autophagy pathways in cells, we identified changes in oxidative stress and senescence associated proteins histone H2A.Z (H2AFZ), histone H2B type 1-K (HIST1H2BK) and E3 ubiquitin-protein ligase RING1 (RING1). These were enriched at 6 hours (5 and 25 µM) but no changes were observed at 24 hours (5 and 25 µM) (Figure 7). Another pathway regulated by oxidative stress is the mTOR signaling. Three proteins grouped in the mTOR signaling pathway were affected; heme oxygenase 1 (HMOX1), Rho-related GTP-binding protein RHOB, and GTPase KRas (KRAS). The HMOX1 level was elevated while RHOB and KRAS were reduced, particularly at the initial time point of 6 hours and 5 µM Aβ concentration.
Complement and coagulation cascade associated proteins are affected upon Aβ treatment
Thirteen proteins associated with complement and coagulation cascade were detected as up-regulated with fold changes over 1.2 at 5 µM at 6 hours (T1), including complement component C8 beta chain (C8B), complement component C9, C3, C7, vitronectin (VTN), alpha-2-macroglobulin (A2M), prothrombin (F2), plasminogen (PLG), coagulation factor V (F5), coagulation factor XIII A chain (F13A1), antithrombin-III (SERPINC1), heparin cofactor 2 (SERPIND1) and complement factor I (CFI) (Figure 7). Interestingly, 24 hours treatment at the same concentration of Aβ (T2) reversed the expression of majority of these proteins. Furthermore, at 25 μM treatment, some of the proteins showed a marginal up-regulation at 6 hours; however, their expression were reversed at the longer time point. These findings indicate that complement and coagulation cascade proteins are an important class that is elevated early upon exposure to neurotoxic Aβ species. At longer time points the cells either adapt or start to give in to the Aβ challenge.
Up-regulation of IGF transport and uptake by IGFBPs early upon exposure to Aβ
Several proteins associated with IGF transport and uptakes by IGFBPs were induced by Aβ peptide, and their expression patterns were also time-dependent (Figure 7). Nine proteins were observed to be increased at 5 µM of Aβ at 6 hours but then decreased upon longer exposure to Aβ for 24 hours at this concentration. A similar expression trend was identified for 25 µM of Aβ at 24 hours but the differential effects were more subtle. Importantly, among the DEPs were apolipoproteins, AOPB and APOE, which have been implicated as risk genes in early onset AD.
STRING analysis was applied to the DEPs at 5 µM of Aβ at both time points (T1 and T2) which were analysed with Student’s t-test, since a greater degree of AD associated protein expression changes were detected at this concentration. In contrast to the results obtained using IPA analysis, some specific pathways were enriched at 5 µM of Aβ and 6 hours. These enriched pathways included exocytosis, regulation of proteolysis, innate immune system, amyloid fiber formation and platelet-derived growth factor (PDGF) signaling (Table 2 and Additional Table 4 (77KB, pdf) ). Some of the differentially regulated proteins had functions in several pathways, such as complement proteins and collagen degradation associated proteins. Interestingly, all six proteins linked to amyloid fiber formation were up-regulated at the early stage (5 µM_6 hours). However, for the 24 hours treatment (T2), fewer enriched pathways were identified with fewer number of proteins found in each pathway. We identified differentially affected proteins in exocytosis, regulation of proteolysis, innate immune system, ECM-receptor interactions, collagen degradation and regulation of IGF transport and uptake by IGBFPs (Table 2 and Additional Table 4 (77KB, pdf) ). Interestingly, at 24 hours, we identified down-regulated proteins falling under ECM-receptor interaction and regulation of IGF transport and uptake by IGFBPs. Superoxide dismutase protein SOD1 was down-regulated and involved in two pathways, exocytosis and immune system process, at 24 hours.
Table 2.
Classification of differentially expressed proteins in T1 (5 μM_6 h) and T2 (5 μM_24 h) based on their enriched biological pathways
| String enriched pathways | Up-regulated proteins under Aβ treatment | Down-regulated proteins under Aβ treatment | |
|---|---|---|---|
| T1 (5 μM_6 h) | Exocytosis | PLG, SEPT5, FN1, NEU1, F5, RAB8B, SPARC | ARSB, ASAH1, GLA, MYH10, CTSD, SERPINB6, CTSB, GAA, RAP1A, LAMP2, SERPINB1, PRDX4, GM2A, NPC2 |
| Regulation of proteolysis | C8B, C9, PZP, C7, F2, ITIH2, RGN, SERPIND1, VTN, CPN1, FN1, COL6A3, SORL1, LAMTOR5 | SUMO2, SERPINB6, SERPINB1 | |
| Regulation of complement activation | C8B, C9, C7, F2, VTN, CPN1 | ||
| Neutrophil degranulation | NEU1 | ARSB, ASAH1, GLA, CTSD, SERPINB6, CTSB, GAA, RAP1A, LAMP2, SERPINB1, PRDX4, GM2A, NPC2 | |
| Lysosome | NEU1, CLTCL1 | ARSB, ASAH1, GLA, CTSD, CTSB, GAA, LAMP2, GM2A, NPC2 | |
| Complement and coagulation cascades | C8B, C9, C7, F2, PLG, SERPIND1, VTN, F5 | ||
| IGF transport and uptake by IGFBPs | F2, PLG, AFP, ITIH2, SERPIND1, APOB, F5 | ||
| ECM proteoglycans | COMP, FMOD, VTN, COL6A3, SPARC | ||
| Amyloid fiber formation | TGFBI, HIST1H2BK, H2AFX, H2AFZ, SORL1 | ||
| Oxidative stress induced senescence | HIST1H2BK, RING1, H2AFX, H2AFZ | TXN | |
| Collagen degradation | COL10A1, COL6A3 | CTSD, CTSB | |
| Signaling by PDGF (platelet-derived growth factor) | THBS4, PLG, COL6A3 | KRAS | |
| Innate immune system | C8B, C9, C7, F2, VTN, APOB, CPN1, NEU1, PDZD11 | ARSB, KRAS, ASAH1, GLA, CTSD, SERPINB6, CTSB, GAA, RAP1A, LAMP2, SERPINB1, ATOX1, PRDX4, TXN, GM2A, NPC2 | |
| T2 (5 μM_24 h) | Exocytosis | FABP5 | SOD1, CTSA, GM2A, THBS1, AHSG, LTF |
| Regulation of proteolysis | TNFAIP8, TAF1 | SERPINC1, ITIH2, THBS1, AHSG, LTF, CFI, RBX1 | |
| Immune system process | FABP5 | SOD1, COL1A2, SERPINC1, CTSA, GM2A, THBS1, CHIA, COL1A1, AHSG, LTF, CFI | |
| ECM receptor interaction | COL1A2, THBS1, COL1A1 | ||
| Collagen degradation | COL1A2, COL1A1 | ||
| Regulation of IGF transport and uptake by IGFBPs | AHSG, ITIH2, SERPINC1 | ||
| Innate immune system | FABP5 | CTSA, GM2A, AHSG, LTF, CFI |
ECM: Extracellular matrix; IGF: insulin-like growth factor; IGFBP: IGF-binding protein; PDGF: platelet-derived growth factor.
Enriched cellular networks in Aβ treated SH-SY5Y cells and comparative analysis with 661W photoreceptor cells
We found 37 DEPs that overlapped between SH-SY5Y cells in this study and our previous report on 661W photoreceptor cells (Deng et al., 2019) using the same treatments. The interaction network (Figure 8A) and their expression changes (Figure 8B) were illustrated. The clustered patterns indicate that the impact of Aβ toxicity on the two different cell lines was, in both cases, more dependent on time rather than Aβ concentration. For example, programmed cell death protein 4 (PDCD4) and PDCD10 from both cell lines were down-regulated at 6 hours (5 and 25 µM), but then up-regulated at 24 hours (5 and 25 µM). Conversely, protein uridine-cytidine kinase 2 (UCK2) was up-regulated at 6 hours (5 and 25 µM), and down-regulated at 24 hours (5 and 25 µM). In contrast, ganglioside GM2 activator (GM2A) was down-regulated across all four treatments in SH-SY5Y neuronal cells, while for 661W photoreceptor cells, decreased expression was only evident at 24 hours (5 and 25 µM). Taken together, these findings suggest similar, yet differential effects of Aβ1–42 on the 661W photoreceptor and SH-SY5Y neuronal cells.
Figure 8.
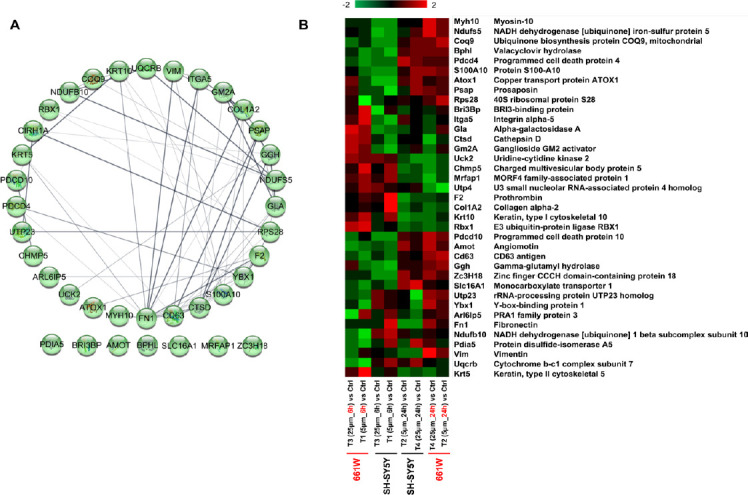
Comparison analysis between 661W mice photoreceptor cells and SH-SY5Y human neuron cells under the β-amyloid peptide toxicity.
(A) Functional interaction networks analyzed by the String Cytoscape plugin with 37 regulated proteins in two cell lines. Network nodes are labeled with gene symbols. (B) Heatmap of the regulated proteins in different treatments on two cell lines. Red and green indicate relative increase or decrease in protein abundance, respectively.
Discussion
Aβ induced toxicity has been identified in ageing brain (Murphy and LeVine, 2010; Allsop and Mayes, 2014; Sadigh-Eteghad et al., 2015; Omar et al., 2018). Its accumulation impairs neuronal function and leads to cell death (Cárdenas-Aguayo et al., 2014; Sengupta et al., 2016; Deng et al., 2019). In this study, Aβ1–42 peptide was used to treat the SY-SY5Y human neuronal cells under culture conditions, and cellular response was monitored using in-depth quantitative proteomic analysis. The study revealed that effect of Aβ induced toxicity on neuronal cells was both time and dose dependent, but the duration of exposure was a more prominent determining factor rather than change in concentration of Aβ alone. These results were consistent with the effects induced by Aβ1–42 on 661 mouse photoreceptor cells, as reported previously (Deng et al., 2019). The major biochemical pathways which were affected by toxic Aβ1-42 in neuronal cells were regulation of lysosomal proteins, collagen chain trimerization and ECM-receptor interaction, ribosomal biogenesis, IGF transport and uptake by IGFBPs, complement and coagulation cascades, autophagy, and oxidative phosphorylation.
Regulation of IGF transport and uptake by IGFBPs is involved in metabolic regulation, while abnormal metabolism is generally associated with AD pathology. Aβ peptides reduce insulin binding and receptor autophosphorylation, which is observed with AD-related neuronal injury. Proteins APOE, APOB, and TF (transferrin) in this pathway were identified as DEPs in this study. Apolipoproteins are important peripheral biomarkers of Alzheimer’s pathology (Koch et al., 2018). ApoE4, as a driver of multiple impairments seen in AD, negatively affects neuronal insulin signaling (Ozcan et al., 2004; Zhao et al., 2017; Brandon et al., 2018). Transferrin, has been suggested as a potential biomarker of AD, acting as a major protein for iron transportion and also involvement in the generation of free radical, (Hussain et al., 2002). All proteins in this pathway were up-regulated initially (5 µM_6 hours) and then slightly down-regulated at 24 hours (5 and 25 µM). These impacts of Aβ1–42 may be attributed to adaptation of the cells to initial toxicity and stress caused by the peptide.
A series of abnormalities were induced in SH-SY5Y neuronal cells by Aβ in this study, such as alterations in oxidative phosphorylation, ribosomal biogenesis, lysosomal processing, autophagy, and ECM interaction. Oxidative phosphorylation is the most vital process in mitochondria for energy production (Distelmaier et al., 2009; Mirzaei et al., 2018). Abnormalities in cellular bioenergetics have been identified in AD and also in other neurodegenerative diseases (Yao et al., 2009). Aβ and tau have been previously shown to exert synergistical damage to oxidative phosphorylation system in a triple AD transgenic mouse model (Rhein et al., 2009). Ubiquinol-cytochrome c reductase binding protein was shown to undergo enrichment across the four treatment groups in this study. It is one of the components of oxidative phosphorylation complex III, and important for functional stability of this complex, electron transport, cellular oxygen sensing and angiogenesis (Kim et al., 2017). Cytochrome b-c1 complex subunit 10 (UQCR11), which forms one of the regulatory subunits in complex III (Bermejo-Nogales et al., 2015), also demonstrated increased abundance at 24 hours (5 and 25 µM). Regulation of these two proteins may accelerate ATP generation in the neural cells.
Ribosomes are known to be the site of initiation of protein synthesis and its elongation. Both the neuronal growth and maintenance rely on the biogenesis of ribosomes. However, abnormal molecular regulation of ribosomal genesis was induced upon exposure to toxic Aβ in this study. Impaired ribosomal biogenesis has been suggested in numerous studies on neurodegenerative diseases, and impairment of ribosomal biogenesis has been shown to produce neuronal atrophy and synapse loss (Ding et al., 2005; Meier et al., 2016). More perturbations were observed in pathways associated with lysosomal function and autophagy. Optimal lysosomal function is of great importance in neurons, as division of these cells is able to eliminate accumulated toxic molecules and aggregates. In this study, lysosomal proteins were modulated by Aβ especially at lower concentrations (5 µM) at 6 hours. At higher concentrations of Aβ, it is possible that the damage is predominantly caused by some other mechanisms. The abnormal lysosomal function greatly contributes to the degeneration of neurons and the pathogenesis of a variety of neurodegenerative diseases. Lysosomal proteases, in particular, cathepsins, play important functions by regulating the lysosomal death pathway, autophagy, ageing, and other processes such as proteolytic cleavage of APP. Cathepsin B (CTSB) and CTSD are two of the major lysosomal proteases, and were down-regulated at the initial time point (5 µM_6 hours). The reduced abundance of CTSB could lead to lysosomal dysfunction and promote accumulation of misfolded proteins. It has been previously demonstrated that CTSB may contribute to the reduction of Aβ peptides by cleaving Aβ1–42 (Bernstein and Keilhoff, 2018). Similarly, proteolysis process mediated by CTSD is highly vital for eliminating the unfolded/oxidized proteins that are delivered to lysosomes to prevent toxicity. Recent studies have demonstrated that CTSD is involved in processing of amyloid precursor protein, apolipoprotein E, and tau proteins in AD (Di Domenico et al., 2016). It was suggested that enhanced activity or increased expression of cathepsins is important in maintaining the normal function of the lysosomes (Cermak et al., 2016). Autophagy related proteins were differentially modulated across the four treatment groups in our study, however, the effects were more prominent at the initial stages (5 µM_6 hours). Autophagy is also a lysosome-dependent pathway that plays a critical role in maintaining neuronal homeostasis and energy metabolism. Compared to non-neuronal cells, the survival of neurons depends on high basal autophagy. The occurrence of autophagy defects could be identified early in the pathogenesis of AD and promote the deposition of misfolded proteins in the cells leading to endoplasmic reticulum (ER) oxidative stress (Funderburk et al., 2010; Uddin et al., 2018). Enhancement of the autophagy-lysosomal system has been suggested as one of the therapeutic strategies to promote Aβ clearance in AD (Tarasoff-Conway et al., 2015; Zare-Shahabadi et al., 2015).
ECM provides essential physical connections for the cellular constituents and cellular filaments, and also helps the regulation of biochemical and signaling pathways that are necessary for cell differentiation and survival (Frantz et al., 2010; Ucar and Humpel, 2018). Significant ECM changes in laminin, elastin, and collagen proteins are known to occur due to imbalance of proteases and protease inhibitors during the early stages of AD, as well as in other degenerative disorders (Gupta and Gowda, 2008; Gupta et al., 2017). Collagen forms a major protein component of ECM and has been shown to interact with APP (Lepelletier et al., 2017). Several collagen family of proteins were found to be affected by Aβ in this study, among which collagen VI (COL6) has been shown to finely modulate the stiffness and mechanical properties of ECM. Aβ peptides initially increase neuronal expression of COL6 which prevents neurons from Aβ binding, while its reduction augments Aβ neurotoxicity (Cheng et al., 2009). Apart from collagen proteins, other ECM proteins such as thrombospondin proteins THBS1 and THBS4 were also differentially expressed. It has been shown that THBS1 protects against Aβ-induced mitochondrial fragmentation and dysfunction in hippocampal neurons (Kang et al., 2018). THBS1 secreted by human umbilical cord blood-derived mesenchymal stem cells rescued neurons from synaptic dysfunction in AD model (Kim et al., 2018). ECM molecular changes in this study might be acting as a protective mechanism against the Aβ induced neurotoxicity in the initial stages of exposure to peptide. These differentially expressed ECM molecules are also involved in the PI3K-Akt signaling pathway. mTOR is a direct target of PI3K-Akt signaling, and Aβ has been suggested to activate the PI3K/Akt/mTOR axis, which are key cellular survival signaling and autophagy related molecular mechanisms. These results indicate that there might be a link among Aβ, ECM molecules and the PI3K/Akt/mTOR pathway, and provide deeper insights into the relationship between AD pathology and potential relationship with insulin resistance (Tramutola et al., 2015).
Increased expression of the complement pathway has previously been shown to impart beneficial effects to neuronal cells in early stages of AD, and up-regulation of complement proteins in this study suggests that complement pathway related proteins might be activated to restrict the Aβ toxicity and enhance its clearance from the cells (Kolev et al., 2009; Mirzaei et al., 2020). In more chronic phases of AD, however, complement activation appears to contribute to neurotoxicity with subsequent exacerbation of the inflammatory reaction (Morgan, 2018). We observed complement pathway related proteins to be down-regulated at 24 hours, which may highlight the complex biochemical roles played by complement related proteins in neuronal cells. Other protective molecular networks were also activated to improve the resistance of cells to Aβ induced neurotoxicity, such as the nuclear factor erythroid 2-related factor 2-mediated oxidative stress response, liver X receptor/retinoid X receptor activation, and sirtuin signaling pathway. Oxidative stress plays a key role in AD pathogenesis by promoting Aβ deposition, tau hyperphosphorylation, and the subsequent loss of synapses and neurons (Perry et al., 2002; Chen and Zhong, 2014). Abnormalities in cellular metabolism, in turn, could contribute to Aβ production and accumulation, which could potentially promote mitochondrial dysfunction and ROS production, thereby resulting in a vicious cycle of events (Tonnies and Trushina, 2017). Nuclear factor erythroid 2-related factor 2-mediated oxidative stress response pathway imparts resistance to cytotoxicity and protects cells from oxidative damage (Pi et al., 2008; Liu et al., 2010). In this study, we observed a greater abundance of proteins in this pathway in T4 (25 µM, 24 hours), which may indicate enhanced resistance of the cells to oxidative stress induced by Aβ. Another pathway which was activated after exposing the cells to Aβ was LXR/RXR activation. LXR/RXR activation has been suggested to reduce Aβ toxicity and may represent a potential therapeutic approach in AD (Saint-Pol et al., 2013). This pathway was not affected in T4 (25 µM_24 hours), which indicates the complex early and long-term exposure effects of Aβ on neurons. Further, the sirtuin signaling pathway was activated in T2 (5 µM_24 hours). Activated sirtuin signaling pathway has diverse anti-ageing effects, which may offer novel therapeutic strategies to prevent or delay age-related diseases, including AD (Gan, 2009). Interestingly, lysosome dysfunction and abnormal cellular metabolism perturbations were induced in neuronal cells in the initial (5 µM_6 hours) stages, in conjunction with up-regulation of protective ECM molecules and the complement pathway system. However, with the extension of treatment duration to 24 hours, lysosomes apparently recovered their expression and only moderate changes in lysosomal proteins were observed. All these results indicate that neuronal cells were differentially affected by Aβ peptides with multiple molecular networks being regulated in a time dependent manner, and to a lesser extent with concentration of Aβ being a determining factor.
Cumulative evidence has shown the impact of AD on the retina, and the accumulation of Aβ in the eye with AD (Goldstein et al., 2003; Koronyo-Hamaoui et al., 2011; Koronyo et al., 2017; Deng et al., 2019). 661W mice photoreceptor cells are a popular in vitro model for studying retinal disorders (Sayyad et al., 2017). A series of AD-associated proteins and pathways were discovered after 661W cells were exposed to Aβ at two concentrations and two time points, as reported previously (Deng et al., 2019). A total of 380 and 473 DEPs were identified from the four treatments from photoreceptor cells and SH-SY5Y neuronal cells, respectively. Some of the main pathways affected by Aβ in both the studies included oxidative phosphorylation, ribosomal dysfunction, lysosomal regulation, mTOR signaling and sirtuin signaling. There was an overlap of 37 DEPs between the two studies, as shown in Figure 8. Interestingly, five of these common proteins (CD63 antigen, prosaposin, cathepsin D, alpha-galactosidase A, and ganglioside GM2 activator) were involved in the regulation of lysosomal function, suggesting this organelle may play a central role in Aβ induced pathology. Our research in these two major cell lines of neuronal origin revealed the molecular effects of Aβ and suggested that this endogenous peptide may have neurotoxic effects on the retinal neurons and SH-SY5Y neuronal cells under disease conditions.
Conclusion
This study revealed several diverse molecular pathways and proteins that are affected in neuronal cells and compared these findings with effects on photoreceptor cells. The data presented in this study serves as a valuable resource for future human and animal studies, which can ultimately provide a better understanding of the disease mechanisms and therapeutic targeting. Candidate neuroprotective agents could be developed to inhibit deleterious molecules in neurons that are potentially triggered upon Aβ exposure. Other potential strategies include designing small molecules that could block Aβ interactions with cell surface and intracellular targets, down-regulating signaling cascades, and reducing the expression of pro-apoptotic proteins in cells (Longo and Massa, 2004).
Additional files:
Additional Tables 1–4 can be available at www.figshare.com under DOI:10.6084/m9.figshare.12017850.
Additional Table 1: (2.5MB, pdf) The combined set of identified proteins from two TMT experiments performed on SH-SY5Y human neuroblastoma cells with four Aβ1–42 treatments and control.
The combined set of identified proteins from two TMT experiments performed on SH-SY5Y human neuroblastoma cells with four Aβ1–42 treatments and control
Additional Table 2: (1.1MB, pdf) Quantitative proteome profiling reveals different protein expressions in four specific treatments on SH-SY5Y human neuroblastoma cells with Aβ1–42 and the control using two-way ANOVA.
Quantitative proteome profiling reveals different protein expressions in four specific treatments on SH-SY5Y human neuroblastoma cells with Aβ1–42 and the control using two-way ANOVA
Additional Table 3: (956.2KB, pdf) The combined four sets of differentially expressed proteins obtained from a student t-test comparison between the specific treatment of Aβ1–42 on SH-SY5Y human neuroblastoma cells and the control.
The combined four sets of differentially expressed proteins obtained from a student t-test comparison between the specific treatment of Aβ1–42 on SH-SY5Y human neuroblastoma cells and the control
Additional Table 4: (77KB, pdf) The cellular networks generated with differentially expressed proteins in T1 (5 µM_6 hours) and T2 (5 µM_24 hours) using String.
The cellular networks generated with differentially expressed proteins in T1 (5 µM_6 hours) and T2 (5 µM_24 hours) using String
Footnotes
Conflicts of interest: The authors have no conflicts of interest to declare.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Li CH; T-Editor: Jia Y
References
- 1.Agholme L, Lindstrom T, Kagedal K, Marcusson J, Hallbeck M. An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J Alzheimers Dis. 2010;20:1069–1082. doi: 10.3233/JAD-2010-091363. [DOI] [PubMed] [Google Scholar]
- 2.Allsop D, Mayes J. Amyloid beta-peptide and Alzheimer’s disease. Essays Biochem. 2014;56:99–110. doi: 10.1042/bse0560099. [DOI] [PubMed] [Google Scholar]
- 3.Basavarajappa DK, Gupta VK, Dighe R, Rajala A, Rajala RV. Phosphorylated Grb14 is an endogenous inhibitor of retinal protein tyrosine phosphatase 1B, and light-dependent activation of Src phosphorylates Grb14. Mol Cell Biol. 2011;31:3975–3987. doi: 10.1128/MCB.05659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bermejo-Nogales A, Calduch-Giner JA, Perez-Sanchez J. Unraveling the molecular signatures of oxidative phosphorylation to cope with the nutritionally changing metabolic capabilities of liver and muscle tissues in farmed fish. PLoS One. 2015;10:e0122889. doi: 10.1371/journal.pone.0122889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein HG, Keilhoff G. Putative roles of cathepsin B in Alzheimer’s disease pathology: the good, the bad, and the ugly in one. Neural Regen Res. 2018;13:2100–2101. doi: 10.4103/1673-5374.241457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandon JA, Farmer BC, Williams HC, Johnson LA. Apoe and Alzheimer’s disease: neuroimaging of metabolic and cerebrovascular dysfunction. Front Aging Neurosci. 2018;10:180. doi: 10.3389/fnagi.2018.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butterfield DA, Boyd-Kimball D, Castegna A. Proteomics in Alzheimer’s disease: insights into potential mechanisms of neurodegeneration. J Neurochem. 2003;86:1313–1327. doi: 10.1046/j.1471-4159.2003.01948.x. [DOI] [PubMed] [Google Scholar]
- 8.Cárdenas-Aguayo MdC, Silva-Lucero MdC, Cortes-Ortiz M, Jiménez-Ramos B, Gómez-Virgilio L, Ramírez-Rodríguez G, Arroyo EV, Fiorentino-Pérez R, García U, Luna-Muñoz J, Meraz-Ríos MA. Physiological role of amyloid beta in neural cells: the cellular trophic activity. In: Heinbockel T, editor. Neurochemistry. London, UK: IntechOpen; pp. 257–281. [Google Scholar]
- 9.Cermak S, Kosicek M, Mladenovic-Djordjevic A, Smiljanic K, Kanazir S, Hecimovic S. Loss of Cathepsin B and l leads to lysosomal dysfunction, NPC-like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS One. 2016;11:e0167428. doi: 10.1371/journal.pone.0167428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. 2014;30:271–281. doi: 10.1007/s12264-013-1423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng JS, Dubal DB, Kim DH, Legleiter J, Cheng IH, Yu GQ, Tesseur I, Wyss-Coray T, Bonaldo P, Mucke L. Collagen VI protects neurons against Abeta toxicity. Nat Neurosci. 2009;12:119–121. doi: 10.1038/nn.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 13.Deng LT, Pushpitha K, Joseph C, Gupta V, Rajput R, Chitranshi N, Dheer Y, Amirkhani A, Kamath K, Pascovici D, Wu JX, Salekdeh GH, Haynes PA, Graham SL, Gupta VK, Mirzaei M. Amyloid β induces early changes in the ribosomal machinery, cytoskeletal organisation and oxidative phosphorylation in retinal photoreceptor cells. Front Mol Neurosci. 2019;12:24. doi: 10.3389/fnmol.2019.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Domenico F, Tramutola A, Perluigi M. Cathepsin D as a therapeutic target in Alzheimer’s disease. Expert Opin Ther Tar. 2016;20:1393–1395. doi: 10.1080/14728222.2016.1252334. [DOI] [PubMed] [Google Scholar]
- 15.Ding QX, Markesbery WR, Chen QH, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH, Smeitink JA. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. doi: 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- 17.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Funderburk SF, Marcellino BK, Yue ZY. Cell “self‐eating”(autophagy) mechanism in Alzheimer’s disease. Mt Sinai J Med. 2010;77:59–68. doi: 10.1002/msj.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gan L. Sirtuin signaling in Alzheimer’s disease. Alzheimers Dement. 2009;5:120. [Google Scholar]
- 20.Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang XD, Mavros C, Coccia JA, Faget KY, Fitch KA, Masters CL, Tanzi RE, Chylack LT, Bush AI. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 21.Gouras GK, Olsson TT, Hansson O. Beta-amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics. 2015;12:3–11. doi: 10.1007/s13311-014-0313-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guivernau B, Bonet J, Valls-Comamala V, Bosch-Morato M, Godoy JA, Inestrosa NC, Peralvarez-Marin A, Fernandez-Busquets X, Andreu D, Oliva B, Munoz FJ. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J Neurosci. 2016;36:11693–11703. doi: 10.1523/JNEUROSCI.1081-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta V, Chitranshi N, You YY, Gupta V, Klistorner A, Graham S. Brain derived neurotrophic factor is involved in the regulation of glycogen synthase kinase 3 beta (GSK3 beta) signalling. Biochem Bioph Res Co. 2014;454:381–386. doi: 10.1016/j.bbrc.2014.10.087. [DOI] [PubMed] [Google Scholar]
- 24.Gupta V, Mirzaei M, Gupta VB, Chitranshi N, Dheer Y, Vander Wall R, Abbasi M, You Y, Chung R, Graham S. Glaucoma is associated with plasmin proteolytic activation mediated through oxidative inactivation of neuroserpin. Sci Rep. 2017;7:8412. doi: 10.1038/s41598-017-08688-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta VK, Gowda LR. Alpha-1-proteinase inhibitor is a heparin binding serpin: molecular interactions with the Lys rich cluster of helix-F domain. Biochimie. 2008;90:749–761. doi: 10.1016/j.biochi.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Gupta VK, Rajala A, Rajala RV. Insulin receptor regulates photoreceptor CNG channel activity. Am J Physiol Endocrinol Metab. 2012;303:E1363-1372. doi: 10.1152/ajpendo.00199.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta VK, Rajala A, Daly RJ, Rajala RV. Growth factor receptor-bound protein 14: a new modulator of photoreceptor-specific cyclic-nucleotide-gated channel. EMBO Rep. 2010;11:861–867. doi: 10.1038/embor.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardy J. Testing times for the “amyloid cascade hypothesis”. Neurobiol Aging. 2002;23:1073–1074. doi: 10.1016/s0197-4580(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 29.Hussain RI, Ballard CG, Edwardson JA, Morris CM. Transferrin gene polymorphism in Alzheimer’s disease and dementia with Lewy bodies in humans. Neurosci Lett. 2002;317:13–16. doi: 10.1016/s0304-3940(01)02403-x. [DOI] [PubMed] [Google Scholar]
- 30.Kang S, Byun J, Son SM, Mook-Jung I. Thrombospondin-1 protects against Abeta-induced mitochondrial fragmentation and dysfunction in hippocampal cells. Cell Death Discov. 2018;4:31. doi: 10.1038/s41420-017-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim DH, Lim H, Lee D, Choi SJ, Oh W, Yang YS, Oh JS, Hwang HH, Jeon HB. Thrombospondin-1 secreted by human umbilical cord blood-derived mesenchymal stem cells rescues neurons from synaptic dysfunction in Alzheimer’s disease model. Sci Rep. 2018;8:354. doi: 10.1038/s41598-017-18542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HC, Chang J, Lee HS, Kwon HJ. Mitochondrial UQCRB as a new molecular prognostic biomarker of human colorectal cancer. Exp Mol Med. 2017;49:e391. doi: 10.1038/emm.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch M, DeKosky ST, Fitzpatrick AL, Furtado JD, Lopez OL, Kuller LH, Mackey RH, Hughes TM, Mukamal KJ, Jensen MK. Apolipoproteins and Alzheimer’s pathophysiology. Alzheimers Dement. 2018;10:545–553. doi: 10.1016/j.dadm.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolev MV, Ruseva MM, Harris CL, Morgan BP, Donev RM. Implication of complement system and its regulators in Alzheimer’s disease. Curr Neuropharmacol. 2009;7:1–8. doi: 10.2174/157015909787602805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koronyo Y, Biggs D, Barron E, Boyer DS, Pearlman JA, Au WJ, Kile SJ, Blanco A, Fuchs DT, Ashfaq A, Frautschy S, Cole GM, Miller CA, Hinton DR, Verdooner SR, Black KL, Koronyo-Hamaoui M. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight. 2017 doi: 10.1172/jci.insight.93621. doi: 101172/jciinsight93621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54 Suppl. 2011;1:S204-217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krishtal J, Bragina O, Metsla K, Palumaa P, Tougu V. In situ fibrillizing amyloid-beta 1-42 induces neurite degeneration and apoptosis of differentiated SH-SY5Y cells. PLoS One. 2017;12:e0186636. doi: 10.1371/journal.pone.0186636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2017;43:167–182. doi: 10.1111/nan.12295. [DOI] [PubMed] [Google Scholar]
- 39.Liu Q, Zhang H, Smeester L, Zou F, Kesic M, Jaspers I, Pi J, Fry RC. The NRF2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the NCI-60 panel. BMC Med Genomics. 2010;3:37. doi: 10.1186/1755-8794-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Longo FM, Massa SM. Neuroprotective strategies in Alzheimer’s disease. NeuroRx. 2004;1:117–127. doi: 10.1602/neurorx.1.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margolin AA, Ong SE, Schenone M, Gould R, Schreiber SL, Carr SA, Golub TR. Empirical Bayes analysis of quantitative proteomics experiments. PLoS One. 2009;4:e7454. doi: 10.1371/journal.pone.0007454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H, Zhu HN, Abisambra JF. Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci. 2016;36:1001–1007. doi: 10.1523/JNEUROSCI.3029-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mi HY, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirzaei M, Gupta V, Gupta V. Retinal changes in Alzheimer’s disease: disease mechanisms to evaluation perspectives. J Neurol Neuromedicine. 2018;3:11–13. [Google Scholar]
- 45.Mirzaei M, Deng L, Gupta VB, Graham S, Gupta V. Complement pathway in Alzheimer’s pathology and retinal neurodegenerative disorders - the road ahead. Neural Regen Res. 2020;15:257–258. doi: 10.4103/1673-5374.265550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mirzaei M, Pascovici D, Wu JX, Chick J, Wu Y, Cooke B, Haynes P, Molloy MP. TMT one-stop shop: from reliable sample preparation to computational analysis platform. In: Keerthikumar S, Mathivanan S, editors. Proteome Bioinformatics, 2016/12/16 Edition. New York, NY: Humana Press; 2017a. pp. 45–66. [DOI] [PubMed] [Google Scholar]
- 47.Mirzaei M, Pushpitha K, Deng L, Chitranshi N, Gupta V, Rajput R, Mangani AB, Dheer Y, Godinez A, McKay MJ, Kamath K, Pascovici D, Wu JX, Salekdeh GH, Karl T, Haynes PA, Graham SL, Gupta VK. Upregulation of proteolytic pathways and altered protein biosynthesis underlie retinal pathology in a mouse model of Alzheimer’s disease. Mol Neurobiol. 2019;56:6017–6034. doi: 10.1007/s12035-019-1479-4. [DOI] [PubMed] [Google Scholar]
- 48.Mirzaei M, Gupta VB, Chick JM, Greco TM, Wu Y, Chitranshi N, Wall RV, Hone E, Deng L, Dheer Y, Abbasi M, Rezaeian M, Braidy N, You Y, Salekdeh GH, Haynes PA, Molloy MP, Martins R, Cristea IM, Gygi SP, et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci Rep. 2017b;7:12685. doi: 10.1038/s41598-017-12858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan BP. Complement in the pathogenesis of Alzheimer’s disease. Semin Immunopathol. 2018;40:113–124. doi: 10.1007/s00281-017-0662-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murphy MP, LeVine H. Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olah J, Vincze O, Virok D, Simon D, Bozso Z, Tokesi N, Horvath I, Hlavanda E, Kovacs J, Magyar A, Szucs M, Orosz F, Penke B, Ovadi J. Interactions of pathological hallmark proteins: tubulin polymerization promoting protein/p25, beta-amyloid, and alpha-synuclein. J Biol Chem. 2011;286:34088–34100. doi: 10.1074/jbc.M111.243907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Omar SH, Scott CJ, Hamlin AS, Obied HK. Olive biophenols reduces Alzheimer’s pathology in SH-SY5Y cells and APPswe mice. Int J Mol Sci. 2018 doi: 10.3390/ijms20010125. doi: 103390/ijms20010125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 54.Parihar MS, Brewer GJ. Amyloid-beta as a modulator of synaptic plasticity. J Alzheimers Dis. 2010;22:741–763. doi: 10.3233/JAD-2010-101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patterson C. World Alzheimer report 2018. London: Alzheimer’s Disease International (ADI); 2018. [Google Scholar]
- 56.Perry G, Cash AD, Smith MA. Alzheimer disease and oxidative stress. Biomed Res Int. 2002;2:120–123. doi: 10.1155/S1110724302203010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pi J, Zhang Q, Woods CG, Wong V, Collins S, Andersen ME. Activation of Nrf2-mediated oxidative stress response in macrophages by hypochlorous acid. Toxicol Appl Pharm. 2008;226:236–243. doi: 10.1016/j.taap.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 58.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 59.Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Drose S, Brandt U, Savaskan E, Czech C, Gotz J, Eckert A. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer’s disease. Med Prin Pract. 2015;24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saint-Pol J, Candela P, Fenart L, Gosselet F. The LXR/RXR approaches in Alzheimer’s disease: is the blood-brain barrier the forgotten partner. J Alzheimers Dis Parkinsonism. 2013;3:4. [Google Scholar]
- 62.Sayyad Z, Sirohi K, Radha V, Swarup G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci Rep. 2017;7:16855. doi: 10.1038/s41598-017-17241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sengupta U, Nilson AN, Kayed R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. Ebiomedicine. 2016;6:42–49. doi: 10.1016/j.ebiom.2016.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song MS, Saavedra L, de Chaves EI. Apoptosis is secondary to non-apoptotic axonal degeneration in neurons exposed to Abeta in distal axons. Neurobiol Aging. 2006;27:1224–1238. doi: 10.1016/j.neurobiolaging.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 65.Sowade RF, Jahn TR. Seed-induced acceleration of amyloid-beta mediated neurotoxicity in vivo. Nat Commun. 2017;8:512. doi: 10.1038/s41467-017-00579-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Menard J, Zetterberg H, Wisniewski T, de Leon MJ. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457–470. doi: 10.1038/nrneurol.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tonnies E, Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis. 2017;57:1105–1121. doi: 10.3233/JAD-161088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739–749. doi: 10.1111/jnc.13037. [DOI] [PubMed] [Google Scholar]
- 69.Ucar B, Humpel C. Collagen for brain repair: therapeutic perspectives. Neural Regen Res. 2018;13:595–598. doi: 10.4103/1673-5374.230273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, Stankiewicz AM. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:04. doi: 10.3389/fnagi.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vickers JC, Dickson TC, Adlard PA, Saunders HL, King CE, McCormack G. The cause of neuronal degeneration in Alzheimer’s disease. Prog Neurobiol. 2000;60:139–165. doi: 10.1016/s0301-0082(99)00023-4. [DOI] [PubMed] [Google Scholar]
- 72.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.You Y, Gupta VK, Li JC, Al-Adawy N, Klistorner A, Graham SL. FTY720 protects retinal ganglion cells in experimental glaucoma. Invest Ophthalmol Vis Sci. 2014;55:3060–3066. doi: 10.1167/iovs.13-13262. [DOI] [PubMed] [Google Scholar]
- 74.Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N. Autophagy in Alzheimer’s disease. Rev Neuroscience. 2015;26:385–395. doi: 10.1515/revneuro-2014-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zellner M, Veitinger M, Umlauf E. The role of proteomics in dementia and Alzheimer’s disease. Acta Neuropathol. 2009;118:181–195. doi: 10.1007/s00401-009-0502-7. [DOI] [PubMed] [Google Scholar]
- 76.Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, Painter MM, Sullivan PM, Bu GJ. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017;96:115–129. doi: 10.1016/j.neuron.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The combined set of identified proteins from two TMT experiments performed on SH-SY5Y human neuroblastoma cells with four Aβ1–42 treatments and control
Quantitative proteome profiling reveals different protein expressions in four specific treatments on SH-SY5Y human neuroblastoma cells with Aβ1–42 and the control using two-way ANOVA
The combined four sets of differentially expressed proteins obtained from a student t-test comparison between the specific treatment of Aβ1–42 on SH-SY5Y human neuroblastoma cells and the control
The cellular networks generated with differentially expressed proteins in T1 (5 µM_6 hours) and T2 (5 µM_24 hours) using String