

Abstract
The NF-E2-related factor 2 (referred to as NRF2) transcription factor binds antioxidant responsive elements within the promoters of cytoprotective genes to induce their expression. Next-generation sequencing studies in lung cancer have shown a significant number of activating mutations within the NRF2 signaling pathway. Mutations in components of the SWI/SNF chromatin-remodeling complex, a general regulator of transcription employing either BRG1 or BRM as the catalytic subunit, also frequently occur in lung cancers. Importantly, low BRG1 expression levels in primary human NSCLC correlated with increased NRF2-target gene expression. Here, we show that loss of SWI/SNF complex function activated a subset of NRF2-mediated transcriptional targets. Using a series of isogenic NSCLC lines with reduced or depleted BRG1 and/or BRM expression, we observed significantly increased expression of the NRF2-target genes HMOX1 and GSTM4. In contrast, expression of the NRF2 target genes NQO1 and GCLM modestly increased following BRM reduction. Chromatin immunoprecipitation showed that BRG1 knockdown led to increased NRF2 binding at its respective ARE sites in the HMOX1 promoter but not in NQO1 and GCLM. Our data demonstrate that loss of BRG1 or BRM in lung cancer results in activation of the NRF2/KEAP1 pathway and HMOX1 expression. Therefore, we provide an additional molecular explanation for why patients harboring BRG1 or BRM mutations show poor prognoses. A better understanding of this mechanism may yield novel insights into the design of targeted treatment modalities.
Keywords: SWI/SNF, KEAP1, NRF2, BRG1/SMARCA4, Lung cancer
INTRODUCTION
Lung cancer remains the leading cause of cancer death worldwide(1). The World Health Organization has divided these tumors into two major groups based on their biology and treatment: small cell lung cancer and non-small cell lung cancer (NSCLC), the latter accounting for more than 85% of all cases. Despite advances in early detection and treatment, NSCLC patients still have a poor five year overall survival. To improve upon this dismal prognosis, identification and characterization of the key molecular events that fuel initiation and spread of this disease is imperative. To address this need, recent next-generation sequencing (NGS) studies in lung cancer have shown a significant number of activating mutations in the KEAP1/NRF2 signaling pathway (2,3) as well as inactivating mutations in key members of the SWI/SNF complex(4,5).
The human SWI/SNF complex, a 1.5- to 2-MDa multisubunit complex employing either BRG1 or BRM as the catalytic subunit, alters nucleosome arrangement along DNA in an ATP-dependent manner(6). Previous studies have indicated that the SWI/SNF chromatin remodeling complex participates in various biological processes, including gene transcription(7), cell cycle regulation (8) and cell differentiation(7,9). Mutations and deletions of the BRG1 gene, also known as SMARCA4, occur frequently in a variety of human cancer cell lines, especially in ~35% of those derived from non-small cell lung carcinoma(10,11). Loss of heterozygosity (LOH) on the short arm of chromosome 19p (50% in negative BRG1 immunostaining tumors) (12) and loss of BRG1 protein expression (~10%) (4,13) are present in primary lung cancers as well.
Oxidative stress-response signaling contributes to the development of many human cancers including NSCLC (14,15). The NF-E2-related factor 2 (NFE2L2, referred to as NRF2) transcription factor induces the expression of ~200 cytoprotective genes which collectively combat oxidative stress, including HMOX1 (heme oxygenase (decycling) 1), NQO1 (NAD(P)H dehydrogenase, quinone 1), and GCLM (Glutamate--cysteine ligase regulatory subunit). NRF2 activates these genes via binding to the antioxidant responsive element (ARE) sequence. Under a quiescent state, KEAP1/CUL3/RBX1, an E3 ubiquitin ligase, ubiquitylates NRF2 in the cytoplasm, thus facilitating its degradation by the 26S proteasome. In the presence of oxidative stress, KEAP1 undergoes a conformational change that results in NRF2 stabilization, NRF2 nuclear translocation and transcriptional activation of NRF2 target genes like HMOX1 and GSTM4.
HMOX1 is a rate-limiting enzyme in the catabolism of heme into biliverdin with anti-inflammatory, anti-apoptotic, and anti-oxidative effects (16). Several groups have reported that HMOX1 is elevated in a variety of human cancers, including prostate (17,18), pancreas (19,20), and liver (21). High expression of HMOX-1 is also associated with tumor invasiveness and poor prognosis in non-small cell lung cancer patients (22). In addition, HMOX1 expression correlates with advanced stages and lymph node involvement (23).
Previous reports have demonstrated a functional interaction between the SWI/SNF complex and the KEAP1/NRF2/ARE pathway using colorectal carcinoma and immortalized embryonic kidney cell lines (24,25). However, functional relationships between chromatin remodeling and NRF2-dependent transcription have not been examined in lung cancer. We hypothesized that BRG1 and/or BRM mutations might promote NSCLC progression in part through regulation of NRF2 target genes. Collectively, our data suggest that loss of SWI/SNF chromatin remodeling complex activity activates the NRF2 pathway in NSCLC, providing a mechanism for its role in NSCLC development. Thus, therapeutic approaches that inhibit NRF2 or NRF2 target genes might benefit BRG1 deficient NSCLC patients.
MATERIAL AND METHODS
Cell culture and chemicals
The H358, H441, H520, H522, and A427 cell lines were obtained from ATCC. All cell lines were grown in RPMI 1640 with 10% FBS (Gibco, Life Technologies, Grand Island, NY). All experiments were performed with cell lines within 20 passages of receipt (<3 months) to ensure the identity of each cell line. Cell lines containing stable BRG1 or BRM knockdown were maintained in RPMI1640 supplemented with 1 μg/ml puromycin or 0.4mg/ml neomycin, respectively. Stable BRG1 and BRM double knockdown cell lines were maintained in RPMI1640 supplemented with 1 μg/ml puromycin and 0.4mg/ml neomycin. tert-Butylhydroquinone (tBHQ) was purchased from Sigma (St. Louis, MO). All cell lines were routinely tested for mycoplasma contamination by the UNC Tissue Culture Facility using a co-culture/DNA staining technique. All cell lines were found to be negative.
Generation of stable RNAi clones
Two methods are used to establish stable BRG1 knockdown cell lines. (1) H358 Control and H358 Brg1i.1 were generated using RNAi expression vectors: pHTP empty vector and pHTP-BRG1i, respectively (26,27). H358 parental cells were transfected with each vector using FuGENE 6 (Promega, Madison, WI). Twenty-four hours post-transfection, cells were selected with 1mg/ml puromycin. (2) To generate lentiviral particles for infection of H358 cells, DNA for the MISSION BRG1 shRNA (TRCN0000015549) from Sigma (St. Louis, MO) was co-transfected with packaging plasmids (ΔNRF, PMDK64) into 293FT cells by the calcium phosphate method as previously described (28). H358 cells were incubated with 4μg/ml polybrene for 30 minutes before being exposed to lentiviral particles, and, 24 hours later, selected with 1mg/ml puromycin. We designated the BRG1 knockdown cell lines as H358 Brg1i.2 (TRCN0000015549).
To generate stable H358 Brmi knockdown cell line, H358 parental cells were transfected with pSR-BRMi using FuGENE 6 (Promega, Madison, WI) (26). Twenty-four hours post-transfection, cells were selected with 0.4mg/ml neomycin.
To generate stable BRG1 and BRM double knockdowns, puromycin resistant BRG1 knockdown cell lines H358 Brg1i.1.25 and H358 Brg1i.2.4 were infected with pSR-BRMi retroviral particles. Retroviral particles were produced using Phoenix cells by the calcium phosphate method (29). Twenty-four hours post-infection, cells were selected with 1μg/ml puromycin and 0.4mg/ml neomycin. Stably infected or transfected clones were isolated, expanded and screened for BRG1 and BRM expression by Q-PCR and western blotting. At least three clones with the lowest BRG1 and/or BRM expression were combined to generate pooled cell lines for further analysis. We confirmed reduced BRG1 and/or BRM protein and mRNA levels in all cell lines by western blotting and qPCR, respectively (Supplemental Figure 1 and Figure 3).
Figure 3. BRG1 knockdown leads to a significant increase in NRF2 binding to the HMOX1 promoter.
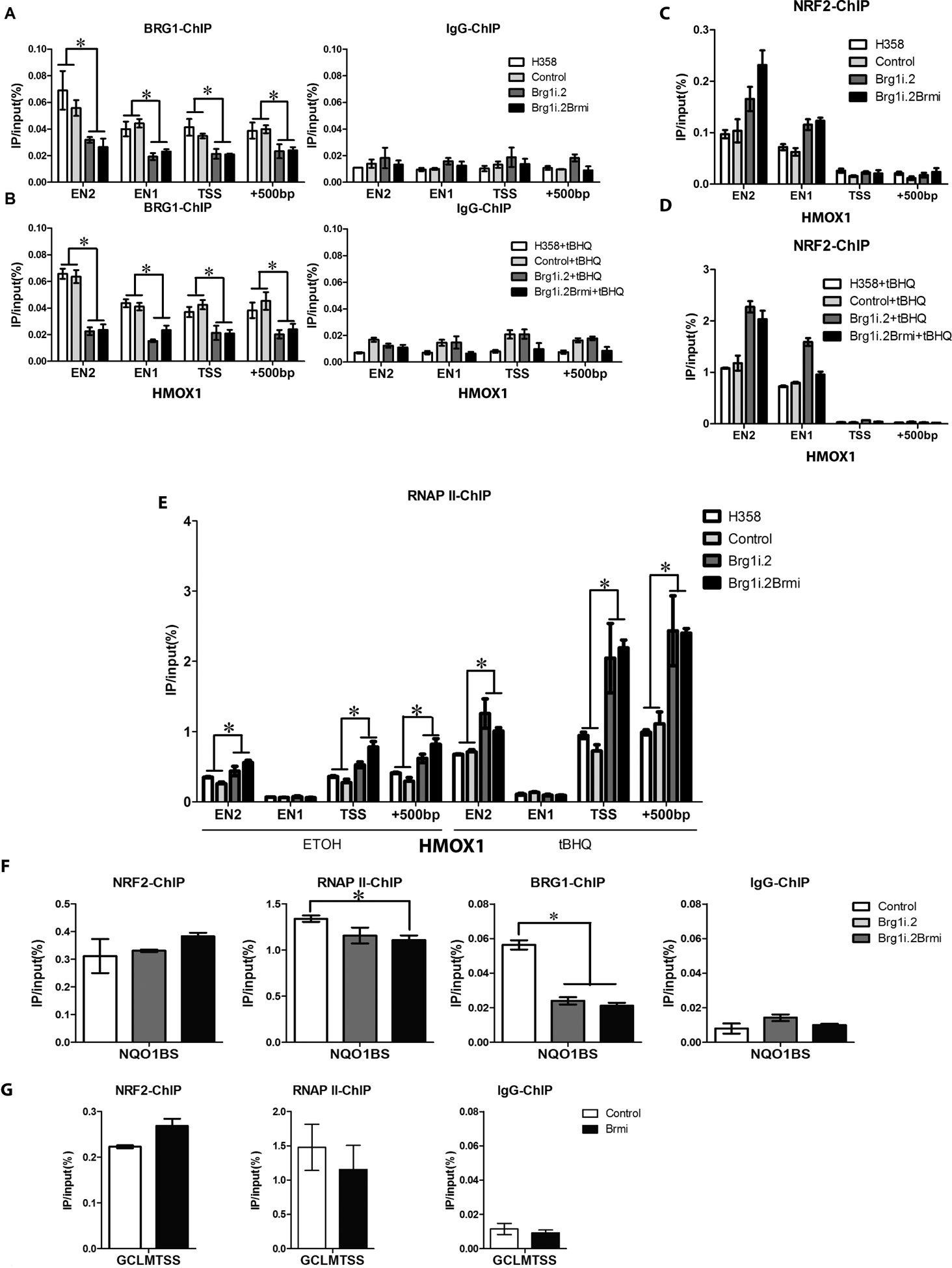
ChIP assays were carried out using cross-linked chromatin from H358, H358 control, H358 BRG1 knockdown (H358 Brg1.2) and H358 BRG1 and BRM knockdown (H358Brg1i.2/Brmi) cells treated with 100% ethanol (vehicle control) or tBHQ (75μM) for 6h. Nucleoprotein complexes were immunoprecipitated with antibodies against BRG1, NRF2, RNAP II, or a rabbit IgG control and the amount of precipitated DNA was determined by qPCR with oligonucleotide primers complimentary to the ARE, TSS and +500bp sequences of HMOX1 promoter region and the NRF2 ARE binding sites (BS) of the NQO1 and GCLM promoter regions. (A) BRG1 binding is reduced to the 4 sites within the HMOX1 promoter region after knockdown in the H358 Brg1.2 and H358Brg1i.2/Brmi cell lines (B) tBHQ treatment (75μM) for 6h does not affect BRG1 association with 4 sites within the HMOX1 promoter region (C) Differential NRF2 binding to its regulatory regions in the HMOX1 promoter among the H358, H358 control, H358 BRG1 knockdown and H358 BRG1/BRM knockdown cell lines. (D) In response to tBHQ treatment, NRF2 binding was increased ~10 fold to its regulatory regions in the HMOX1 promoter in the H358, H358 control, H358 BRG1 knockdown and H358 BRG1/BRM knockdown cell lines. (E) The recruitment of RNA polymerase II to EN2, TSS, and +500bp was increased in H358 cells after BRG1 loss and/or tBHQ treatment. (F) and (G) Nucleoprotein complexes were immunoprecipitated with antibodies against NRF2, RNAP II, BRG1 or a rabbit IgG control and the amount of precipitated DNA was determined by qPCR with oligonucleotide primers complimentary to the ARE sequences of NQO1 (F) and GCLM (G). Data are representative of one experiment (NRF2-ChIP) and two independent experiments (RNAPII-ChIP, BRG1-ChIP and IgG-ChIP) and assayed twice. *P-value < 0.05, (Student T test) and error bars represent ± SEM.
Generation of stable CRISPR clones
The single guide RNAs (sgRNA) targeting the BRG1 ATPase domain and the BRM ATPase domain were designed using an online CRISPR design tool (http://crispr.mit.edu/). LentiCRISPR (pXPR_001) plasmid was used to generate stable CRISPR clones. The target guide sequence cloning protocol is adopted based on the protocol developed by Feng Zhang’s group (30,31). Briefly, it was digested with BsmBI enzyme (FD0454, Fermentas) and gel-purified using QIAquick Gel Extraction Kit (28704, Qiagen, Ipswich, MA). In order to clone the target sequence into the pXPR_001 backbone, a pair of oligos for BRG1 (oligo1: CACCGAGGTACGTGATGAGCGCGA, oligo2: AAACTCGCGCTCATCACGTACCTC) and for BRM (oligo1: GAATCTTAGCCGATGAAATGGGG, oligo2: AAACATTTCATCGGCTAAGATTC) were synthesized by IDT (San Jose, CA) and annealed in our lab. The pXPR_001 backbone and the oligo duplexes were ligated using 2X Quick Ligase Buffer (M2200, NEB, Ipswich, MA). H358 parental cells were transfected with each ligated vector using FuGENE 6 (Promega, Madison, WI). Twenty-four hours post-transfection, cells were selected with 1mg/ml puromycin for three days. Single clones were isolated, expanded and screened for BRG1 and BRM expression by western blotting. Three clones of H358 with no BRG1 expression or no BRM expression were combined to generate pooled cell line for further analysis. We confirmed reduced BRG1 and/or BRM protein levels by western blotting, respectively (Figure 3).
Protein extracts and immunoblotting
Cell lysates were prepared by whole cell extract buffer (20 mM Tris pH 7.5, 400 mM NaCl, 10% Glycerol, 0.5% NP40, 0.1% SDS) and were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4%−12% Bis-Tris gel) and transferred to PVDF membrane or nitrocellulose (Figure 2). Protein concentrations were determined by Bradford or BCA protein assay reagent (Pierce, Rockford, IL). The PVDF membrane was incubated in a blocking buffer for 1 hour and then with a primary antibody (1:1000 dilution) overnight at 4 °C, followed by a HRP-conjugated secondary antibody (1:1000 dilution in blocking buffer). Proteins of interest were visualized with ECL western blotting substrate according to manufacturer’s instructions (GE Healthcare, Uppsala, Sweden). The following antibodies were used: KEAP1 (10503–2-AP, Protein Tech, Chicago, IL), NRF2 (2178–1, Epitomics, Burlingame, CA), HMOX1 (ab13243, Abcam, Cambridge, England), NQO1 (11451, Protein Tech, Chicago, IL), GCLM (sc22754, Santa Cruz Biotechnology, Dallas, TX), BRG1 (G7, sc17796, Santa Cruz Biotechnology, Dallas TX), BRM (ab15597, Abcam, Cambridge, England) and β-ACTIN (A2066, Sigma, St. Louis, MO).
Figure 2. BRG1 deficiency activates transcription of a subset of NRF2 target genes.
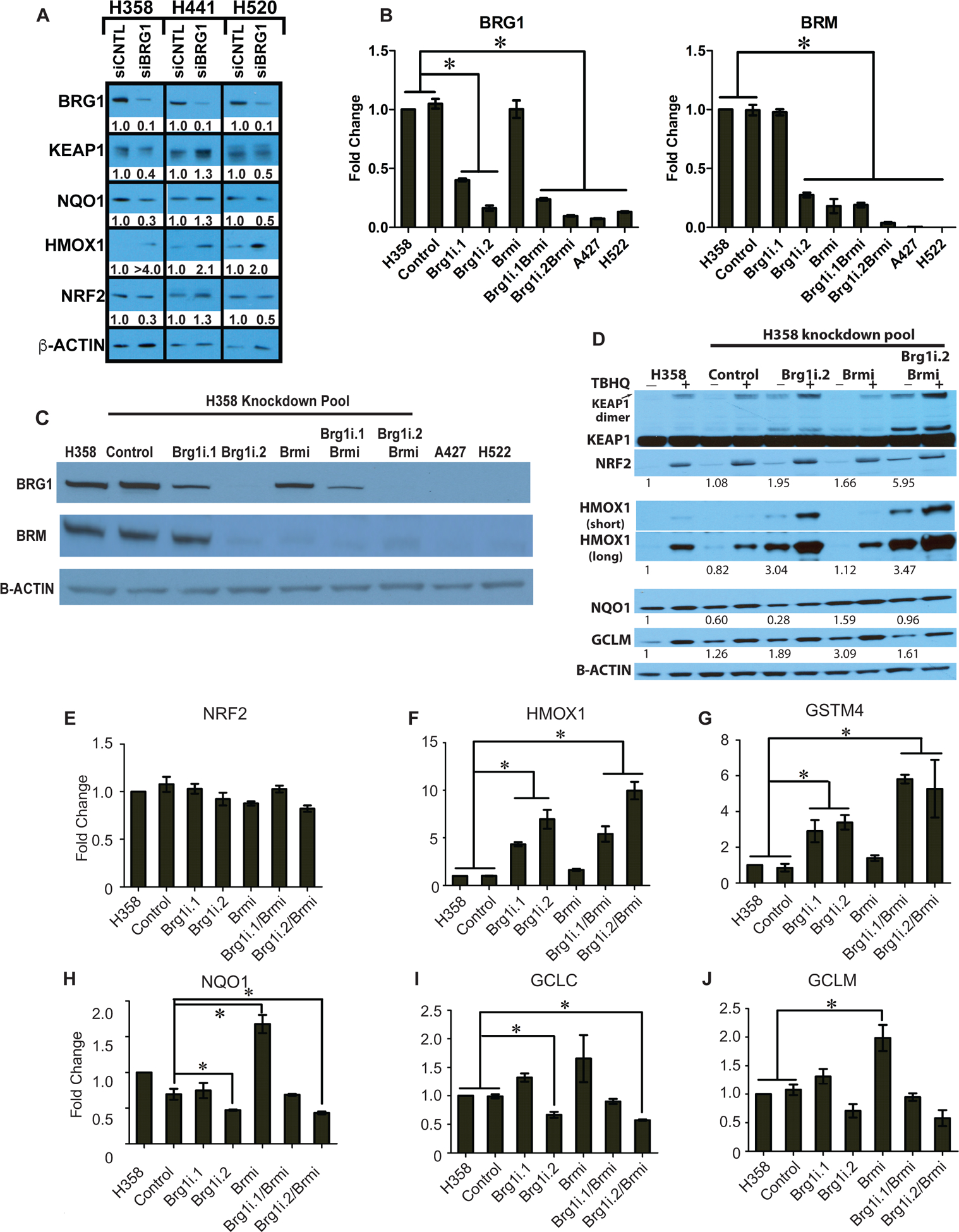
BRG1 and/or BRM were knocked down in H358 cell line using a lentiviral system or transfection to deliver shRNA. The A427 and H522 lung cancer cell lines possessing inactivating mutations of BRG1 and epigenetic silencing of BRM were used as controls. (A) The indicated human NSCLC cell line was transfected with either a non-targeting siRNA (siCONTROL) or a siRNA targeting BRG1 (siBRG1). 72h after transfection, the indicated proteins were detected and quantified by western blot analysis from whole cell lysates. Relative protein abundance was quantified by densitometry from linear-range film exposures. (B) mRNA levels were determined by qPCR for each gene normalized to B-ACTIN. *P-value< 0.05, (Student T test) and error bars represent ± SEM. qPCR results are representative of two independent experiments and assayed twice. (C) Whole cell lysates were separated by 4–12% SDS-PAGE and probed with the indicated antibodies. (D) Clonal H358 cells stably expressing BRG1 and/or BRM shRNAs were treated with tBHQ for 16h before protein extraction and western blot analysis. Protein abundance was quantified by densitometry from linear-range film exposures. B-actin-normalized values are presented. (E-J) Transcript abundance for the indicated mRNAs was determined by qPCR. B-ACTIN was used for normalization. Data represent two independent experiments, assayed in technical duplicate. * P-value < 0.05 (Student T test) and error bars represent ± SEM.
Quantitative RT-PCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Venlo, Limburg) according to manufacturer’s protocol, and was quantified by nanodrop spectrophotometry. 1 μg was used for cDNA synthesis and reverse transcription was performed using M-MLV reverse transcriptase (Invitrogen, Grand Island, NY) with random primers (Invitrogen, Grand Island, NY) and dNTP mix according to manufacturer’s instructions. We determined cDNA levels using the ABI 7900HT sequence detection system (Applied Biosystems, Grand Island, NY) with TaqMan Universal PCR Master Mix reagents. Relative quantification was analyzed by the 2−ΔΔCt method with β-ACTIN serving as an endogenous control. The primers used to detect the expression of β-ACTIN (Hs00357333_g1) GCLC (Hs00155249_m1), GCLM (Hs00157694_m1), GSTM4 (Hs00426432_m1), HMOX1 (Hs01110250_m1), NFE2L2 (Hs00975961_g1), NQO1 (Hs02512143_s1), SMARCA2 (Hs01030846_m1) and SMARCA4 (Hs00946396_m1) were purchased from Life Technologies, (Grand Island, NY).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously (28). Briefly, H358, H358 Control, or H358 Brg1i.2 cells were fixed with 1% formaldehyde in PBS for 15 minutes at room temperature, whereas H358 Brg1i.2Brmi double knockdown cells were fixed for 8 minutes. Crosslinking was quenched by adding glycine to a final concentration of 125mM for at least 10 minutes. Cells were then collected in lysis buffer (1% NP40, 0.1% SDS, 5mM EDTA, 50mM Tris, pH 8.0, 150mM NaCl, protease inhibitors cocktail tablet) and cell lysates were sonicated for 5minutes (for 1 cycle:15 sec on/45 sec off, 20 cycles) at 20% amplitude. ChIP was performed with antibodies specific to NRF2 (H300, Santa Cruz Biotechnology, Dallas, TX), BRG1 (A300–813A, Bethyl Laboratories, Inc., Montgomery, TX), RNA polymerase II (MMS-126-R, Covance, Raleigh, NC) or normal rabbit IgG (SC-2027, Santa Cruz Biotechnology, Dallas, TX). Precipitated DNA was determined by Q-PCR using gene-specific primers as listed in Table 1 and normalized against input DNA.
Table 1.
List of PCR primers used to determine ChIP enrichment of ARE sites in HMOX1, NQO1 GCLM.
| Q-PCR Primers used in ChIP | ||
|---|---|---|
| Gene: Position | sequence | |
| HMOX1:EN2 | F: TCGCTAAGTCACCGCCCCGA R: AGCGAAAACAGACACCGGGACC |
|
| HMOX1: EN1 | F: TGCGTCATGTTTGGGAGGGGG R: AGCTGAGGAGGCACTGGTGA |
|
| HMOX1: TSS | F: CCAGAAAGTGGGCATCAGCT R: GTCACATTTATGCTCGGCGG |
|
| HMOX1: +500 | F: GTCCGCAACCCGACAGGCAA R: GGTGGGGCTAGGACGCAAGC |
|
| NQO1BS | F: CAGGATTCAGGCGTTGGGT R: ATATATCCTGTCCGGCCCGT |
|
| GCLMTSS | F: TCTCGGCTACGATTTCTGCT R: GCGGGAGAGCTGATTCCAAA |
|
For each primer set, the forward (F) and reverse (R) primers are indicated in the center column.
Expression Analyses of Primary Human Lung Adenocarcinomas
Data from the TCGA lung adenocarcinoma project (32) was analyzed in an effort to determine whether KEAP1-NRF2 signaling activity was associated with low SMARCA4/BRG1 expression status (33). Samples with non-silent mutations of KEAP1, NFE2L2, and SMARCA4/BRG1 were identified. Starting with raw read-counts per gene available through the Broad Firehose Portal, gene expression measurements were computed: First, normalization was applied using the EdgeR R package CalcNormFactors() function, applying the “TMM” method. Gene expression (LCPM) was then calculated by gene specific read-counts per million total normalized reads, then applying log2 transformation. We defined a threshold for low SMARCA4/BRG1 expression to be the highest SMARCA4/BRG1 expression value observed in any of the samples with BRG1 nonsense or frameshift mutations. Relevant NRF2 target genes for LUAD were defined using differential expression analysis. Briefly, tumors with KEAP1 or NRF2 mutations were compared to those without any alterations in the NRF2 pathway (mutations or copy number variants effecting KEAP1, NRF2, or CUL3). Mutations (Varscan) and copy number variants (GISTIC) were accessed through the Broad Firehose Portal. Expression data was prepared as above and analyzed for differential expression using the R Limma package as described in (34). A two-sided Wilcoxon rank sum test was applied to test the null hypothesis that there is association between NRF2 gene expression and low SMARCA4/BRG1 expression status. R 2.15.1 was used to perform statistical analysis and generate figures (35).
Gene Sequencing
KEAP1 and NFE2L2 genotype was validated using targeted capture next generation sequencing using the Agilent SureSelect Targeted capture protocol and illumina hiSeq 2500 sequencer and chemistry (36).
RNA Sequencing
RNA was extracted using a Zymo QuickRNA Mini Prep. Samples were submitted to Novogene Corporation, Inc (Sacramento, CA) for library preparation and sequencing using an Illumina NovaSeq 6000 at a depth of 40 million read pairs. Fastq files were aligned to the genome (hg38) using STAR version 2.6.0a. BAM files were quantified using salmon version 0.12.0. Differential expression was performed the DEseq2 package in R version 3.6.
RESULTS
Low BRG1 expression levels in primary human NSCLC are associated with increased KEAP1-NRF2 signaling –
We evaluated relationships between the expression of NRF2 target genes and mutations within KEAP1, NRF2 and SMARCA4/BRG1 in 513 primary human adenocarcinoma samples investigated by TCGA. The LUAD cohort contained 90 (17.6%) samples with KEAP1 mutations and 10 samples (2%) with NFE2L2 mutations. Samples (n=20) with inactivating mutations (frame shift, nonsense or splice site) in SMARCA4/BRG1 were used to define a threshold for low SMARCA4/BRG1 expression (Figure 1A). As expected, the 19 tumors samples harboring missense mutations in SMARCA4/BRG1 did not show decreased expression (Figure 1A). Interestingly, tumors with low SMARCA4/BRG1 expression displayed relatively higher NRF2 target gene expression than wildtype tumors, including tumors without mutations in KEAP1 and/or NRF2 (Figure 1B). Thus, we conclude that decreased SMARCA4/BRG1 expression positively correlates with increased NRF2 target gene expression in human LUAD tumors.
Figure 1. Analysis of TCGA Lung Adenocarcinoma Cohort.
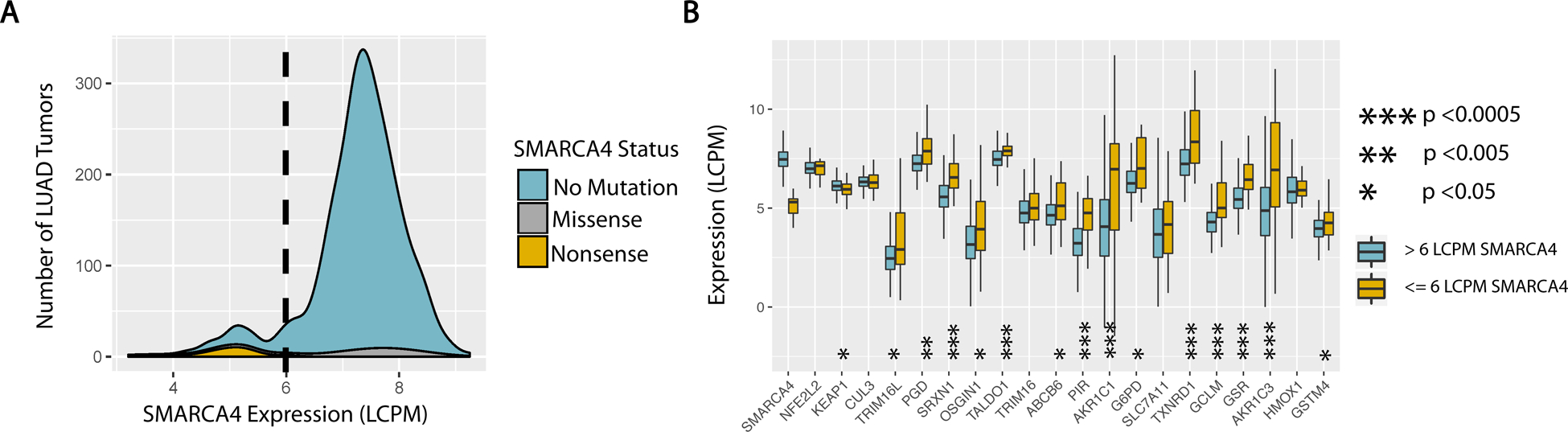
(A) Density plot of SMARCA4 expression amongst LUAD tumors in TCGA. Contributions of tumors with mutated vs. unaltered SMARCA4 loci are color coded. blue - no alteration. grey - missense or splice site mutation. orange - nonsene or frame shift deletion. dashed line - highest expression of SMARCA4 in a LUAD tumor with SMARCA4 nonsense mutation (6 LCPM). (B) Expression of NRF2 regulatory and target genes by SMARCA4 status. Displayed target genes include the top 15 (by p-value) differentially expressed genes, comparing LUAD tumors with and without NRF2/KEAP1 mutations. Standard NRF2 target genes HMOX1 and GSTM4 were also added for comparison with experimental data. orange - low SMARCA4 expression as defined by range of expression seen in tumors with non-sense mutations in SMARCA4, below 6 LCPM. blue - normal range of SMARCA4 expression, above 6 LCPM.
Reduced SWI/SNF complex activity alters NRF2 signaling-
To validate the correlations observed in human tumors, we tested whether reduced SWI/SNF complex activity affected KEAP1-NRF2 signaling in cultured human lung cancer cell lines. We chose 2 cell lines derived from lung adenocarcinomas, H358 and H441, and 1 cell line derived from a squamous cell carcinoma of the lung, H520. These cell lines possess wild-type BRG1, NRF2 and KEAP1 genes by exome sequencing (10,36). Two approaches were used to suppress SWI/SNF function: siRNA-based transient transfection and shRNA-based stable suppression. First, we measured the effects of transient BRG1 knockdown on KEAP1-NRF2 signaling in each cell by siRNA-based silencing and western blot. As shown in Figure 2A, we reduced BRG1 expression by approximately 90% in each cell line as compared to the non-targeting control. Expression of the consensus NRF2 target, HMOX1, increased in each cell line after BRG1 reduction. In contrast, protein levels for KEAP1, NRF2 and NQO1, another consensus NRF2 target, did not increase following BRG1 loss. Therefore, transient repression of BRG1 expression increased the expression of one key NRF2 target gene, HMOX1, a recently described driver of lung tumor metastasis (37).
We next generated BRG1 and/or BRM-depleted clonal cell lines using stable shRNA in the human H358 cell line (Figure 2B–C). We then evaluated KEAP1/NRF2 pathway activity in these cell lines by western blot analysis of NRF2 protein levels, NRF2 target gene expression, and the presence of SDS-resistant dimeric KEAP1 (KEAP1 dimer). Inactive KEAP1 forms a SDS-resistant dimer, thus KEAP1 dimer levels serve as a metric for its activity (38,39). Stable BRG1 reduction resulted in the inactivation of KEAP1 and increased NRF2 and HMOX1 mRNA and protein levels (Figure 2D–F). In contrast, reduction in BRM expression in either parental or BRG1 knockdown cells did not strongly alter NRF2 mRNA levels or NRF2 and KEAP1 dimer protein levels (Figure 2D–E). Similar results were observed in a second BRG1 shRNA clonal cell line, Brg1i.1 (Figure S1). Therefore, stable inhibition of BRG1-containing SWI/SNF complex activity also significantly increased dimeric KEAP1, NRF2 and HMOX1 expression.
The impact of BRG1 and/or BRM suppression on four additional NRF2 target genes, GSTM4, NQO1, GCLM and GCLC, displayed a more complex pattern of expression (40,41). Similar to HMOX1, both BRG1 knockdown cell lines showed a significant increase in GSTM4 transcript levels (Figure 3F). However, the H358 Brg1i.2 cells showed a reduction in mRNA levels for the other three genes (Figure 2H–J). In contrast to BRG1 reduction, cells expressing reduced BRM levels displayed moderately increased NQO1 and GCLM transcript and protein levels. and increased GCLC transcripts levels (Figure 2D, H–J). HMOX1 protein and mRNA levels and GSTM4 mRNA levels remained unchanged following BRM reduction (Figure 2D, F–G). Unexpectedly, double silencing of BRG1 and BRM led to increased dimeric KEAP1, NRF2, HMOX1, and GSTM4 expression but reduced expression of NQO1, GCLM and GCLC, suggesting that BRG1 loss is dominant to BRM loss (Figure 2D–J).
We also examined the effects of reduced BRG1 and/or BRM expression on NRF2-mediated induction of its target genes following tert-butylhydroquinone (tBHQ) treatment, an established KEAP1 antagonist and NRF2 activator (42). NRF2 protein expression increased in all cell lines following tBHQ treatment (Figure 3D). H358 cells lacking either BRG1 alone or both BRG1 and BRM displayed high levels of SDS-resistant KEAP1 dimer; tBHQ treatment did not further increase dimer formation in these cells. HMOX1 protein expression increased in all cell lines after tBHQ treatment, albeit to different levels given the wide-range of baseline expression across the cell lines (Figure 3D). GCLM was similarly responsive to tBHQ treatment, with the notable exception of the BRG1/BRM double knockdown H358 cells. Thus, despite stabilizing NRF2 protein, decreased SWI/SNF complex activity differentially impacts the induction of NRF2 target genes by tBHQ.
Reduced SWI/SNF activity impacts NRF2 binding to its ARE sequences-
To better understand how SWI/SNF chromatin remodeling activity influences NRF2-driven transcription, we performed a series of chromatin immunoprecipitation experiments on the HMOX1, GCLM and NQO1 promoters (43–45). We initially characterized the HMOX1 promoter that harbors two enhancers, E1 and E2, located at approximately 4kb and 10kb upstream from the transcription start site (TSS). These enhancers have been previously shown to contain functional antioxidant response elements (AREs), and bind NRF2. In addition, we looked at binding at the TSS and 500bp upstream of the TSS. ChIP analysis revealed the presence of BRG1 at both enhancers, the TSS and the +500 sites (Figure 3A). As expected, the BRG1 signal was reduced 2–3 fold in the BRG1 knockdown cell lines but not to the level observed with the IgG controls (Figure 3A). Treatment with tBHQ did not impact BRG1 loading onto the any of these sites (Figure 3B).
In contrast to BRG1, recruitment of NRF2 to the regulatory regions of the HMOX1 promoter was greatly increased by tBHQ treatment (Figure 3-compare the lanes in panel C to the corresponding lanes in panel D). In addition, H358 cells deficient for BRG1 or both BRG1 and BRM displayed increased NRF2 on the HMOX1 enhancers (E1 and E2), both in the absence and presence of tBHQ (Figure 3C, D). Consistent with increased NRF2 binding, the recruitment of RNA polymerase II (RNA PolII) to the E2, TSS, and +500bp sites within the HMOX1 gene was also increased significantly by tBHQ and by knockdown of BRG1 or both BRG1 and BRM (Figure 3E). In contrast, we did not observe a significant change in NRF2 binding to the consensus NRF2 binding site in NQO1, consistent with the absence of increased expression following BRG1 loss (Figures 3F and 2D, respectively). However, we did see a slight but statistically significant reduction in RNA PolII binding that mirrored the modest reduction in mRNA levels after BRG1 knockdown (Figures 3F and 2H, respectively). We also examined NRF2 binding at the GCLM promoter where the consensus NRF2 binding site maps ~50bp upstream of the TSS. We did not find any significant differences in NRF2 or RNA PolII binding between the H358 and H358 Brmi cell lines (Figure 3G). Our results demonstrate a correlation between HMOX1 expression and increased NRF2 binding after BRG1 knockdown to its ARE binding sites. These results implicate increased NRF2 binding as a mechanism by which mutations in SWI/SNF complex components contribute to NSCLC development.
Characterization of BRG1 and BRM knockout cell lines
We used CRISPR/Cas9 genome engineering to genetically ablate the BRG1 locus or the BRM locus in H358 lung cancer cells (Figure 4A). Similar to the BRG1i.2 and Brmi shRNA stable clones, BRG1-knockout (BRG1 KO) cells displayed increased levels of dimeric KEAP1 and HMOX1 proteins while BRM-knockout (BRM KO) cells showed no alterations (Figure 4A). In contrast to the stable shRNA cell lines neither the BRG1 KO nor the BRM KO cells displayed altered NQO1 levels (Figure 4A). We then examined global gene expression by RNA-seq to compare the changes in gene expression associated with either BRG1 and BRM knockout (Supplementary Figure 2). As shown in Figure 4B&C, we observed ~ 21% of genes in the BRG1 KO cells and ~17% of genes in the BRM KO cells with a significant change in expression (log2FC <−1 or >1; padj <0.05) compared to the H358 parental cells. Of interest, we saw similar numbers of genes with significantly altered expression (padj <0.05) with increased expression and decreased expression in both cell lines (Figure 4B & C). We also found about 50% of the genes with altered expression overlap in the 2 cell lines (Figure 4D). However, many of these genes changed expression in the opposite direction (Supplementary Table 1). We also carried out GSEA analyses to determine whether NRF2 signaling appeared altered in either knockout cell line. While we did not observe a significant enrichment for NRF2 signaling in the BRG1 KO cell line, we observed a trend towards increased NRF2 signaling in the BRM KO cells (Figure 4E). Finally, we compared expression of genes that changed in the primary LUADs shown in Figure 1B with the same genes in the BRG1 KO and BRM KO cell lines. We found a good correlation between the qPCR data and the RNA-seq for HMOX1, GSTM4 and GCLM gene expression (Figure 3 and Figure 4F). We also saw an increase in gene expression in either the BRG1 KO and/or the BRM KO cell lines for 7 of the 13 genes that showed increased expression in the SMARCA4-low LUADs (Figure 1B and 4F). As with the H358 shRNA knockdown cell lines, we also saw 5 genes with decreased expression in the BRG1 KO and BRM KO cells.
Figure 4. Characterization of H358 BRG1 and BRM knockout cell lines.
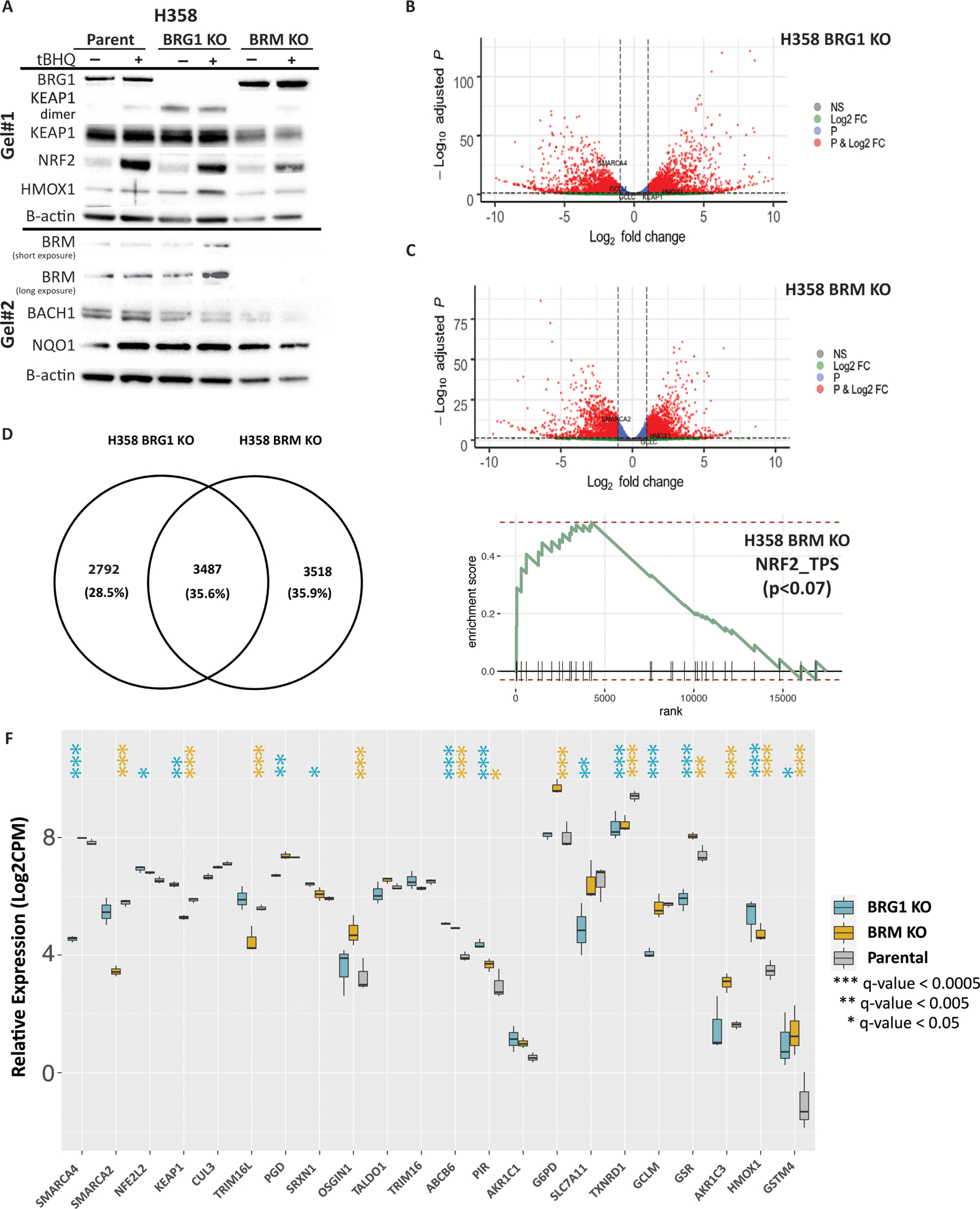
CRISPR/Cas9 engineering was used to delete BRG1 or BRM from H358 cells. (A) Western blots showing the effects on CRISPR knockouts of BRG1 and BRM on NRF2, KEAP1 and downstream targets. (B) Volcano plot results of RNA-seq differential gene expression (H358 BRG1_KO/Parent) for protein coding genes using DESeq2 (n = 19,879 genes total). Significantly upregulated genes (padj < 0.05 and Log2FoldChange > 1) are colored in red (n = 2,082 genes). Significantly downregulated genes (padj < 0.05 & Log2FoldChange < −1) are colored in blue (n = 2010 genes). Non-significant genes are colored in grey (n = 15,787 genes). SMARCA4/BRG1 and NRF2 target genes are identified. (C) Volcano plot results of RNA-seq differential gene expression (H358 BRM_KO/Parent) for protein coding genes using DESeq2 (n = 19,879 genes total). Significantly upregulated genes (padj < 0.05 and Log2FoldChange > 1) are colored in red (n = 1,785 genes). Significantly downregulated genes (padj < 0.05 & Log2FoldChange < −1) are colored in blue (n = 1,531 genes). Non-significant genes are colored in grey (n = 16,653 genes). SMARCA2/BRM and NRF2 target genes are identified. (D) Venn diagram showing the number of genes that change relative to the H358 parental cell line exclusive to the H358 BRG1 KO cell line (n = 2792 genes), exclusive to the H358 BRM KO cell line (n = 3,518 genes) and the number of genes that change in both cell lines (n = 3, 487 genes). (E) Enrichment plot for NRF2 HALLMARK terms in RNA-seq data (LCPM normalized TPMs) using GSEA (MSigDB GO gene set - C5 all v6.0). (F) mRNA expression of NRF2 target genes derived from the H358 parent, H358 BRG1 KO and the H358 BRM KO cell lines. Poly A capture RNAseq was performed in biological triplicate. Raw counts as quantified by Salmon were normalized by the trimmed means method and Log2 transformed. Indicated q-values were estimated using DESeq2, comparing parental H358 to the indicated knock out cell lines.
A recent publication showed that increased HMOX1 expression in a genetically engineered mouse model increased the metastatic properties of lung tumors through stabilization of the BACH1 transcription factor (37). We had previously shown a significant gain of invasive capability in the H358 Brg1i.2 cell line after inoculation in the lungs of nude mice (46). Therefore, we looked at BACH1 expression in the BRG1 and BRM KO cell lines. Despite the increased expression of HMOX1 in these cell lines after BRG1 knockdown or the tBHQ treatment, we did not observe any change in BACH1 protein levels (Figure 4A).
DISCUSSION
Recent next-generation sequencing studies have identified recurrent mutations in key SWI/SNF complex members, including BRG1/SMARCA4, ARID1A, PBM1 and SNF5/INI1 across a broad range of human tumors (47). This observation has proved especially true for human NSCLCs where mutation frequencies for BRG1 and ARID1A have ranged from 10–20% (48,49). Therefore, the significant number of these mutations emphasizes the need to understand the effects of loss of SWI/SNF complex activity during progression of NSCLC.
Multiple studies have established a role for oxidative stress in the development of cancer (50). High levels of ROS are detrimental to normal cells and could lead to tumor development by inducing DNA damage and oncogenic mutations. In a complementary fashion, malignant transformation further increases ROS production. Activation of the KEAP1/NRF2 pathway presumably protects cells against the harmful effects of oxidative stress by inducing the expression of a battery of cytoprotective proteins. Alleviating reactive oxidative species (ROS)-mediated damage by constitutive NRF2 activation should prove beneficial for tumor cells. Our results indicate that mutations in the SWI/SNF complex could also maintain a favorable redox balance in cancer cells and promote their survival by further activating key downstream targets of the KEAP1/NRF2 pathway.
A previous report by Ge and colleagues found BRG1 and NRF2 functionally interacted with each other to regulate hepatic ischaemia/reperfusion injury via propofol post-conditioning (24). They found that NRF2 mediated control HMOX1 expression was BRG1-dependent. Another report (25) by Yamamoto and colleagues demonstrated inhibition of HMOX1 induction in the absence of BRG1 in a human colorectal carcinoma cell line, SW480. Interestingly, they also noted an increase in DEM-inducible NRF2 expression in SW480 cells with stable knockdown of BRG1 using 2 different shRNAs. They also found similar results in that BRG1 loss did not significantly affect NQO1 expression. The differences between this study and our own raise the exciting possibility that BRG1 could play different or even opposing roles in NRF2 signaling in different tissues. Indeed, overexpression of SMARCA4 occurs more frequently in colorectal cancers than deletions or mutations (51).
We previously demonstrated that 10% of lung cancers show concomitant loss of BRG1 and BRM expression with a statistically significant correlation with poorer prognosis (13). The knockdown of both proteins in the H358 cell line also led to the strong induction of the NRF2 targets HMOX1 and GSTM4. While we saw similar results with our BRG1 and BRM knockout cell lines, we could not isolate a double knockout cell line. This failure may reflect a previous report showing a synthetic lethality for BRM in BRG1-deficient cell lines (52). The fact that we could isolate BRG1/BRM double knockdown cell lines supports the notion that cells only require low levels of SWI/SNF complex activity for continued proliferation. It also limits the application of CRISPR technology for characterizing dual loss of the SWI/SNF ATPase subunits in cell lines.
Another group (53) has shown that increased expression of NRF2 and decreased expression of KEAP1 by immunohistochemistry are associated with a poor outcome in NSCLC. In addition, increased expression of HMOX1, one the consensus NRF2 target genes, has been found in various tumor types (54). HMOX-1 up-regulation is implicated in chemoresistance and inhibition of HMOX1 by siRNA or a specific inhibitor ZnPP can sensitize lung cancer cells to cisplatin (55). Therefore, the higher levels of HMOX1 observed in BRG1-deficient NSCLC cell lines, +/− tBHQ, could lead to greater resistance to chemotherapeutic drugs. In addition, a recent report demonstrated a correlation in human LUADs between KEAP1 mutations and high levels of HMOX1 by IHC (37). This report also proposed that LUADs with high expression of HMOX1 may prove more aggressive via upregulation of the BACH1 transcription factor, a known drive of cancer metastasis (37). The increased BACH1 expression results from the high HMOX1 levels that would scavenge free heme and inhibit proteasome-dependent degradation of BACH1. However, we did not observe increased BACH1 expression in our BRG1 KO cell line, even with the high HMOX1 levels induced by tBHQ treatment (Figure 4A). Whether this disparity reflects a difference between mouse and human cells and/or cell lines and genetically-engineered mouse models remains unclear.
We propose that expression of NRF2 target genes after BRG1 loss is regulated by altered nucleosome occupancy at NRF2 binding sites (see model in Figure 5). Our study appears consistent with the recent reports by ourselves and Tolstorukov et al. that inactivation of SWI/SNF leads to reduced nucleosome occupancy at regions surrounding TSS (46,56). Thus, reduced BRG1 expression leads to changes in nucleosome occupancy surrounding NRF2 binding sites. Ultimately, our data suggest that the “relaxed” nucleosome positioning results in increased expression of some NRF2 targets and decreased expression of others, i.e. a significant increase in the levels of HMOX1, GSTM4 and PIR with a concomitant decrease in SLC7A11, GSR and PGD. In contrast, BRM loss led changes in expression of different NRF2 targets including increased G6PD and GSR and decreased TRIM16L. The identification of the mechanisms underlying the differences between the effects of BRG1 and BRM loss on gene expression will require further experiments.
Figure 5. Schematic mechanism for KEAP1-NRF2 activation caused by SWI/SNF chromatin remodeling loss.
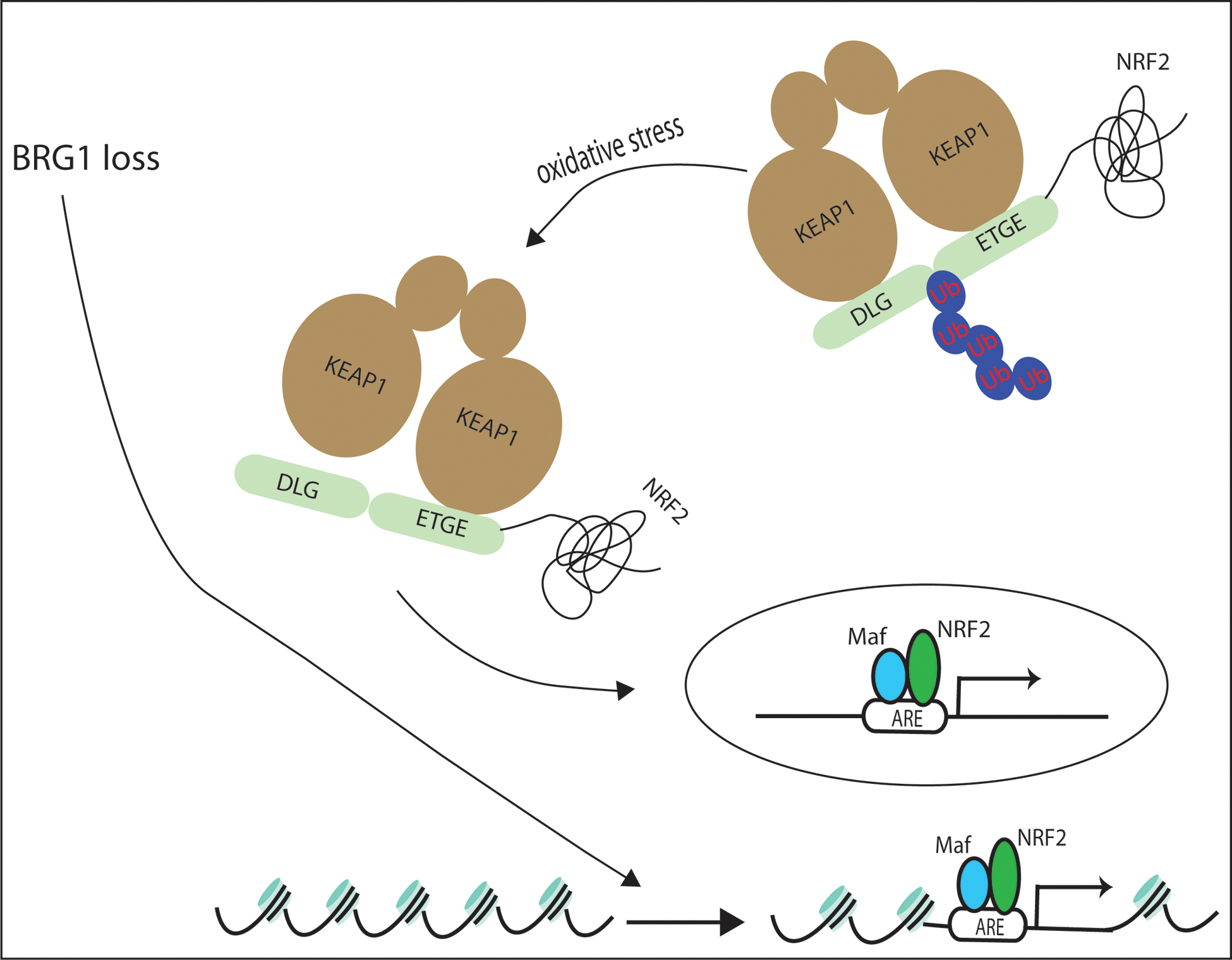
BRG1 loss induced KEAP1-NRF2 activation is shown through ROS production and altered nucleosome occupancy.
Our study establishes a link between SWI/SNF complex function and the KEAP1-NRF2-ARE pathway in NSCLC. Intriguingly, we also found that loss of BRG1 affects KEAP1/NRF2 signaling differently than BRM loss in NSCLC cell lines, further functionally distinguishing these SWI/SNF complexes. Our investigation also emphasizes the importance of personalized medicine: patients with BRG1 and/or BRM deficient NSCLC may need different therapeutic approaches compared to individuals with wild-type expression (57,58). Studies to identify drugs that specifically target SWI/SNF complex loss or inactivate NRF2 signaling will prove critical to the treatment of this population of NSCLC patients.
Supplementary Material
Implication:
Our study identifies a novel mechanism for how mutations in the SMARCA4 gene may drive progression of human lung adenocarcinomas.
ACKNOWLEDGEMENTS
We thank members of Weissman lab and Major lab as well as Dr. William Kim, Dr. James Samet and Dr. Phillip Wages for their support, help and excellent technical input and thoughtful discussions. This study was supported by a grant from National Natural Science Foundation of China (No. 81974434) (Y. Zheng), a grant from the National Institutes of Environmental and Health Sciences (T32ES007126) (D. Wei, V. Nguyen) and grants from the National Cancer Institute (CA138841) (B. Weissman) and (CA216051) (B. Weissman, M. B. Major). M.B. Major was supported by a grant from the American Cancer Society (RSG-14-068-01-TBE) and the Sidney Kimmel Cancer Foundation. These studies were also supported in part by a UNC Lineberger Clinical/Translational Research Award.
Footnotes
The authors do not declare any conflicts of interests.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67(1):7–30 doi 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489(7417):519–25 doi 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li QK, Singh A, Biswal S, Askin F, Gabrielson E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. Journal of human genetics 2011;56(3):230–4 doi 10.1038/jhg.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herpel E, Rieker RJ, Dienemann H, Muley T, Meister M, Hartmann A, et al. SMARCA4 and SMARCA2 deficiency in non-small cell lung cancer: immunohistochemical survey of 316 consecutive specimens. Ann Diagn Pathol 2017;26:47–51 doi 10.1016/j.anndiagpath.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Manceau G, Letouze E, Guichard C, Didelot A, Cazes A, Corte H, et al. Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. International journal of cancer Journal international du cancer 2013;132(9):2217–21 doi 10.1002/ijc.27900. [DOI] [PubMed] [Google Scholar]
- 6.Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J Mol Biol 1999;293(2):187–98. [DOI] [PubMed] [Google Scholar]
- 7.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene 2009;28(14):1653–68 doi 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 8.Hendricks KB, Shanahan F, Lees E. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol 2004;24(1):362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limpert AS, Bai S, Narayan M, Wu J, Yoon SO, Carter BD, et al. NF-kappaB forms a complex with the chromatin remodeler BRG1 to regulate Schwann cell differentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33(6):2388–97 doi 10.1523/jneurosci.3223-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Human Mutation 2008;29(5):617–22 doi 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 11.Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer research 2000;60(21):6171–7. [PubMed] [Google Scholar]
- 12.Rodriguez-Nieto S, Canada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F, et al. Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Hum Mutat 2011;32(2):E1999–2017 doi 10.1002/humu.21415. [DOI] [PubMed] [Google Scholar]
- 13.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res 2003;63(3):560–6. [PubMed] [Google Scholar]
- 14.Esme H, Cemek M, Sezer M, Saglam H, Demir A, Melek H, et al. High levels of oxidative stress in patients with advanced lung cancer. Respirology 2008;13(1):112–6 doi 10.1111/j.1440-1843.2007.01212.x. [DOI] [PubMed] [Google Scholar]
- 15.Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health 2013;10(9):3886–907 doi 10.3390/ijerph10093886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chau LY. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci 2015;22:22 doi 10.1186/s12929-015-0128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology 1996;47(5):727–33. [DOI] [PubMed] [Google Scholar]
- 18.Nemeth Z, Li M, Csizmadia E, Dome B, Johansson M, Persson JL, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget 2015;6(32):33675–88 doi 10.18632/oncotarget.5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han L, Jiang J, Ma Q, Wu Z, Wang Z. The inhibition of heme oxygenase-1 enhances the chemosensitivity and suppresses the proliferation of pancreatic cancer cells through the SHH signaling pathway. Int J Oncol 2018;52(6):2101–9 doi 10.3892/ijo.2018.4363. [DOI] [PubMed] [Google Scholar]
- 20.Nuhn P, Kunzli BM, Hennig R, Mitkus T, Ramanauskas T, Nobiling R, et al. Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Molecular cancer 2009;8:37 doi 10.1186/1476-4598-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sass G, Leukel P, Schmitz V, Raskopf E, Ocker M, Neureiter D, et al. Inhibition of heme oxygenase 1 expression by small interfering RNA decreases orthotopic tumor growth in livers of mice. International journal of cancer Journal international du cancer 2008;123(6):1269–77 doi 10.1002/ijc.23695. [DOI] [PubMed] [Google Scholar]
- 22.Tsai JR, Wang HM, Liu PL, Chen YH, Yang MC, Chou SH, et al. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cellular oncology (Dordrecht) 2012;35(6):461–71 doi 10.1007/s13402-012-0105-5. [DOI] [PubMed] [Google Scholar]
- 23.Degese MS, Mendizabal JE, Gandini NA, Gutkind JS, Molinolo A, Hewitt SM, et al. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung cancer (Amsterdam, Netherlands) 2012;77(1):168–75 doi 10.1016/j.lungcan.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge M, Chen H, Zhu Q, Cai J, Chen C, Yuan D, et al. Propofol post-conditioning alleviates hepatic ischaemia reperfusion injury via BRG1-mediated Nrf2/HO-1 transcriptional activation in human and mice. J Cell Mol Med 2017;21(12):3693–704 doi 10.1111/jcmm.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Ohta T, Maruyama A, Hosoya T, Nishikawa K, Maher JM, et al. BRG1 Interacts with Nrf2 To Selectively Mediate HO-1 Induction in Response to Oxidative Stress. Molecular and cellular biology 2006;26(21):7942–52 doi 10.1128/MCB.00700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Link KA, Burd CJ, Williams E, Marshall T, Rosson G, Henry E, et al. BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol Cell Biol 2005;25(6):2200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosson GB, Bartlett C, Reed W, Weissman BE. BRG1 loss in MiaPaCa2 cells induces an altered cellular morphology and disruption in the organization of the actin cytoskeleton. J Cell Physiol 2005;205(2):286–94. [DOI] [PubMed] [Google Scholar]
- 28.Kuwahara Y, Charboneau A, Knudsen ES, Weissman BE. Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res 2010;70(5):1854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinsella TM, Nolan GP. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Human gene therapy 1996;7(12):1405–13 doi 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- 30.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. science 2014;343(6166):84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154(6):1380–9 doi 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Network. TCGAR. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature, in press 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law CW, Alhamdoosh M, Su S, Dong X, Tian L, Smyth GK, et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 2016;5 doi 10.12688/f1000research.9005.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R Core Team. 2012. R: A language and environment for statistical computing. <http://www.R-project.org/.>.
- 36.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature biotechnology 2009;27(2):182–9 doi 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019;178(2):316–29 e18 doi 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camp ND, James RG, Dawson DW, Yan F, Davison JM, Houck SA, et al. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. The Journal of biological chemistry 2012;287(9):6539–50 doi 10.1074/jbc.M111.316471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of biological chemistry 2010;285(11):8463–71 doi 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taguchi K, Yamamoto M. The KEAP1-NRF2 System in Cancer. Front Oncol 2017;7:85 doi 10.3389/fonc.2017.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cloer EW, Goldfarb D, Schrank TP, Weissman BE, Major MB. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res 2019;79(5):889–98 doi 10.1158/0008-5472.CAN-18-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang DD. The Nrf2-Keap1-ARE Signaling Pathway: The Regulation and Dual Function of Nrf2 in Cancer. Antioxidants & Redox Signaling 2010;13(11):1623–6 doi 10.1089/ars.2010.3301. [DOI] [PubMed] [Google Scholar]
- 43.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res 2012;40(15):7416–29 doi 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 2016;7:11624 doi 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic acids research 2010;38(17):5718–34 doi 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orvis T, Hepperla A, Walter V, Song S, Simon J, Parker J, et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res 2014;74(22):6486–98 doi 10.1158/0008-5472.CAN-14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45(6):592–601 doi 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150(6):1107–20 doi 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez-Nieto S, Cañada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F, et al. Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Human Mutation 2010;32(2):E1999–E2017 doi 10.1002/humu.21415. [DOI] [PubMed] [Google Scholar]
- 50.Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. Trends Mol Med 2017;23(5):411–29 doi 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. PNAS 2014;111(8):3128–33 doi 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16(14):3743–53 doi 10.1158/1078-0432.ccr-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nitti M, Piras S, Marinari UM, Moretta L, Pronzato MA, Furfaro AL. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants (Basel) 2017;6(2) doi 10.3390/antiox6020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HR, Kim S, Kim EJ, Park JH, Yang SH, Jeong ET, et al. Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung cancer (Amsterdam, Netherlands) 2008;60(1):47–56 doi 10.1016/j.lungcan.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 56.Tolstorukov MY, Sansam CG, Lu P, Koellhoffer EC, Helming KC, Alver BH, et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U S A 2013;110(25):10165–70 doi 10.1073/pnas.1302209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 2015;21(3):231–8 doi 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature 2015;520(7546):239–42 doi 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.