

Abstract
Chemoprevention trials for prostate cancer (PCa) by androgen receptor or androgen synthesis inhibition have proven ineffective. Recently, it has been demonstrated that the histone methlytransferase, EZH2 is de-regulated in mouse and human high-grade prostatic intraepithelial neoplasia (HG-PIN). Using pre-clinical mouse and human models of PCa, we demonstrate that genetic and chemical disruption of EZH2 expression and catalytic activity reversed the HG-PIN phenotype. Further, inhibition of EZH2 function was associated with loss of cellular proliferation and induction of Tp53 dependent senescence. Together, these data provide provocative evidence for EZH2 as an actionable therapeutic target towards prevention of prostate cancer.
Introduction
Prostate cancer is the currently the most diagnosed malignancy and second in the number of cancer associated deaths amongst men in the United States (1). Because of its relative latency in disease onset and progression, prostate cancer should be an ideal target for chemoprevention strategies. This idea is further strengthened because of an established hormonal dependence underlying prostate cancer initiation and progression. This knowledge led to 2 landmark randomized, placebo controlled prostate cancer chemoprevention trials: the Reduction by Dutaseride of Prostate Cancer Events (REDUCE) trial and the Prostate Cancer Prevention Trial (PCPT) with Finasteride (2,3). Both agents target 5α-reductase, which is important for the production of dihydrotesterone (DHT) and androgen receptor function. While reduction in overall risk of low-grade prostate cancer occurred, the cumulative risk of high-grade prostate cancers at the conclusion of both trials generated great concern regarding the targeting of the androgen axis.
The epigenome has exciting potential as a target for prostate cancer prevention. Increasing evidence that the epigenome becomes corrupted before prostate cancer is invasive, suggests the role of epigenetic mechanisms driving prostate cancer initiation. An example involves promoter hypermethylation of GSTP1, which is observed in more than 75% of HG-PIN and 95% of prostate cancers in humans (4). Decitabine, a DNA methyl-transferase inhibitor, demonstrated chemo preventative action in TRAMP mice, delaying tumor progression and improving overall survival (5). Moreover, the histone methlytransferase EZH2 is over-expressed in localized prostate cancers and associated with high-risk of recurrence following prostatectomy (6). EZH2 is regulated transcriptionally and post transcriptionally by the oncogene c-MYC (MYC), a known oncogenic driver of prostate cancer (7–10). Additionally, concurrent over expression of MYC and EZH2 has been observed in human and mouse high-grade prostatic intraepithelial neoplasia (HG-PIN) (7,9). These data highlight an important question that has not yet been addressed; would targeting epigenetic dysfunction, specifically EZH2, inhibit HG-PIN (a PCa precursor lesion) progressing to PCa?
To interrogate the potential of EZH2 as a targetable action for prostate cancer prevention, we carried out genetic and chemical inhibition of EZH2 activity using preclinical mouse and human models of PCa. Loss of EZH2 histone methyltransferase activity inhibits clonogenic survival in vitro. In addition, attenuation of EZH2 activity in vivo reverses HG-PIN with associated induction of cellular senescence and loss of cell proliferation. Hence, targeting EZH2 early in patients with HG-PIN may reduce the incidence of progression to aggressive prostate cancer.
Methods and Methods
Mice:
The Institute of Animal Care and Use Committee (IACUC) at Dana-Farber Cancer Institute approved all mouse procedures. The ARR2Probasin-MYC transgenic line (Pb-Hi-MYC) (10), expresses human c-MYC under the control of a modified rat probasin promoter. EZH2fl/fl (11) mice possess LoxP sites flanking exons 14–15 of the enhancer of zeste homolog 2 (EZH2) gene. The PSA-Cre(ERT2) line (12), has Cre recombinase under the control of the prostate specific antigen promoter. Cre recombinase is fused to a mutant estrogen receptor (ERT2) making activation of Cre inducible by administration of tamoxifen (Tam) (Supplement Fig. 1A). To generate mice with conditional over expression of MYC and ablation of EZH2 in the prostate, Pb-Hi-MYCpos;EZH2fl/fl and PSA-Cre(ERT2)pos;EZH2fl/fl were mated to generate Pb-Hi-MYCpos;PSA-Cre(ERT2)pos;EZH2fl/fl mice (EMC). Mice lacking expression of PSA-Cre(ERT2) transgene (EM) were used for control toward Tam treatment. Tam administration (daily 1mg i.p. injection for 5 days [d1-d5]) was performed with 8-week- old male mice. All mice were mixed background consisting of FVBN and C57Bl/6J. To assess genetic ablation of EZH2 effect of MYC driven prostate cancer, EMC and EM mice were treated with Tam. At 12 weeks (2 months post initiation of Tam treatment) of age all mice were sacrificed and prostate tissue was assessed by histopathology and immunohistochemical procedures.
PCR Analysis:
Genotyping was confirmed by using genomic DNA extracted from mouse-tails. Primers used were PSA-Cre(ERT2): FOR 5’ -ATC CGA AAA GAA AAC GTT GA - 3’ and REV 5’ - ATC CAG GTT ACG GAT ATA GT - 3’, MYC: FOR 5’ - AAA CAT GAT GAC TAC CAA GCT TGG - 3’ and REV 5’ - ATG ATA GCA TCT TGT TCT TAG TCT TTT TCT TAA TAG GG - 3’, EZH2: FOR 5’ - CAT GTG CAG CTT TCT GTT CA - 3’ and REV 5’ - CAC AGC CTT TCT GCT CAC TG - 3’.
In vivo Therapy Experiments:
EM mice were used for all described studies. To evaluate DZNep, EM mice were treated with either DZNep 5mg/kg daily or 5% DMSO/PBS daily by i.p. administration. For EPZ0011989 studies, EM mice were treated with EPZ0011989 312.5 mg/kg or 0.5% carbomethylcellulose (CMC) by oral gavage twice daily (PO, BID). Weighing mice twice weekly monitored therapy toxicity. Following treatment of 4 weeks (8 weeks-of-age to 12 weeks-of-age) mice were sacrificed and gross genitourinary and prostate weights were measured. Prostate tissue was further assessed by histopathology and immunohistochemical procedures.
Immunohistochemical Staining and Quantitation:
Genitourinary tract and mouse prostates were weighed and collected at appropriate time points. Dorsal, lateral, and ventral lobes were separated under a dissection microscopy (Zeiss). Tissues were fixed in 10% neutral buffered formalin solution (Sigma) overnight and then transferred to 70% ethanol for an additional 24 hours. Four-micron thick sections were cut from paraffin- embedded blocks for H&E and immunohistochemistry analysis. For immunohistochemistry, slides were first deparaffinized in xylene solutions and then rehydrated in graded ethanol. Slides were boiled in 10mM sodium citrate solution (pH 6) in a microwave for 10 minutes. The slides were then blocked in 2.5% normal horse serum (Vector Laboratories) for 30 minutes at room temperature and subsequently incubated with primary antibodies diluted in Tris-buffered saline containing 1.25% horse serum in a humidified chamber for overnight. The slides were then washed by Tris-buffered saline containing 0.1% Tween-20 (Sigma) following the incubation with secondary antibody conjugated with peroxidase (Vector Laboratories). Slides were developed by DAB (Dako) and subsequently counterstained with hematoxylin. For senescence associated® - galactosidase staining, tissue samples were embedded in OCT and cryosections prepared at 5∝M depth. Sections were fixed and stained as recommended using a senescence detection kit (Calbiochem). Quantitation of IHC: At least 7 representative images were taken using light microscope. Staining intensity was scored by Aperio ImageScope software.
In silico Gene Set Enrichment Analysis:
The gene expression profiles of mouse models of PCa at PIN stage and wild type (WT) mouse were downloaded from National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) and UCLA DOE Institute. The gene expression profiles for the Hi-MYC and wild type mice gene expression data were downloaded from (http://doembi.ucla.edu/myc_driven_prostate_cancer/). The gene expression profiles for Pb-Cre4; Ptenf/f (Pten null) were downloaded from NCBI GEO (GSE47220). The EZH2 and/or H3K27me3 associated molecular signatures were downloaded from Molecular Signature Database. The 49 Molecular signatures were identified and downloaded. The Gene Set Enrichment Analysis (GSEA) tool (http://www.broadinstitute.org/-gsea) was used to analyze relationship of gene expression profiles from mouse models in EHZ2 and/or H3K27me3 associated molecular signatures. The Signal2Noise metric for gene ranking and 1000 number of permutations were used for GSEA analysis.
Cell lines and Reagents:
The AR expressing, via lentiviral infection of LV-AR, hTERT immortalized human prostate epithelial 957E/hTERT cells (PrEC-AR) and c-Myc transformed PrEC-AR cells (PrEC-AR-c-Myc) were from Dr. John T. Issacs (Johns Hopkins University). Hi-MYC cell lines were purchased from AACR and cultured in DMEM supplemented with 10% FBS. PrEC-AR and PrEC-AR-c-Myc cell lines were cultured in Keratinocyte Serum Free Media supplemented with supplemented with bovine pituitary extract and recombinant epidermal growth factor (ThermoFisher) and 1% penicillin/streptomycin in 5% CO2 environment at 37°C. For in vitro experiments DZNep, GSK126, and EPZ6438 were all purchased from Xcessbio. For in vivo experiments DZNep was purchased from Sigma, and Epizyme Pharmaceuticals generously supplied EPZ011989. Antibodies used were anti-P53 (D2H9O, Cell Signaling), anti-EZH2 (D2C9, Cell Signaling), anti-H3K27me3 (C36B11, Cell Signaling), and anti-Ki67 (Sp6, Thermo Fisher). Mycoplasma testing is conducted every 3 months using standard PCR protocol from Sigma: Venor GeM Mycoplasma Detection Kit. Cell lines are authenticated using standard STR profiling.
3-dimensional organoid models:
All 3D organoid models were generated using previously described methodology and maintained in accordance as previously published (13). Murine EMC 3D organoids were generated from the dorsolateral prostates of 8-week-old GEMMs. Mouse 3D organoids were validated by genotyping and recombination PCRs prior to use in subsequent studies.
MTT Growth Assay with EZH2 Inhibitors:
Briefly, 2.5x103 cells of PrEC-AR-c-Myc were plated in 96 well plate. Following day, 3–4,5-dimethylthiazol-2-yl-5–3- carboxymethoxyphenyl-2–4-sulfophenyl-2H-tetrazolium (MTT) were added and incubated for 4 hours. After 4 hours Day 0 was read at 490nm on microplate reader. Nine point 3-fold dilution (2nM-15mM) of DZNep made and added to new wells. For GSK126, a nine point 1.5-fold dilution (0.975mM-25mM) of GSK126 made and added to new wells. Fro EPZ6438, a nine point 1.5-fold dilution (0.975mM-25mM) of EPZ6438 made and added to new wells. The end point readout was done at 72 hours for DZNep and GSK126 and 96 hours for EPZ6438 at 490nm on microplate reader. Emax50 Calculation: Day0 readouts were subtracted from end point readouts. Results were normalized to DMSO control and Log10 transformed. To calculate Emax50, Nonlinear regression Find EC anything algorithm in GraphPad Prism was used with F=50.
Histone Activity Assay:
Adherent cells were harvested using Trypsin-EDTA and centrifuged at 1600rpm for 5 minutes. Cell pellets were washed with PBS. Histones were extracted according to Epigentek manufacturer instruction using EpiQuik Total Histone extraction kit. Histone protein concentration was measured by Bradford protein assay. To assess H3K27me3 levels, ELISA was performed according to Epigentek manufacturer instruction using EpiQuik Global Histone H3K27 Tri-Methylation Quantification Kit. 50ml of C2 buffer added and 50mg of histone extracts were added into each well and incubated for 2 hours at RT. Wells were aspirated and washed 3 times with 150ml diluted (1:9 ratio with water) C1 buffer. Followed by adding 50ml diluted (1:1000 ratio with C2 buffer) C3 to each well and incubating for 1 hour at RT on orbital shaker at 100rpm. Wells were aspirated and washed 6 times with 150ml diluted (1:9 ratio with water) C1 buffer. Then 100ml of C4 added into each well and incubated at RT for color development. After 1 minute when color turned blue, 50ml of C5 added to each well to stop enzyme reaction. Absorbance was read at 450nm on mircoplate reader. Relative H3K27me3 were calculated by normalizing to control/parental cell.
In Vitro Senescence Analysis:
Fifty thousand (5x104) cells of PrEC-AR-c-Myc were plated in 6 well plate. Following day, cells were treated with Emax50 concentration of EZH2 inhibitor for 72–96 hours (DZNep and GSK126 for 72 hours and EPZ6438 for 96 hours) in the presence of absence of 10nM R1881. Post EZH2 inhibitor treatment media was changed and refreshed with or without 10nM R1881 every 72 hours. Senescence Detection was performed as per BioVision manufacturer instruction using Senescence Detection Kit. On Day 6, media was removed, and cells were washed with 1x PBS. Cells were fixed with fixative solution for 15 minutes at RT. Following fixation cells were washed with 1x PBS and 0.5ml Staining Solution (containing 470 μl of Staining Solution, 5 μl of Staining Supplement, and 25 μl of 20 mg/ml X-gal) were added and incubated overnight at 37oC. Cells were then stained with 1x DAPI staining. Cells were washed with 1x PBS and then counterstained with DAPI fro 15 minutes at RT. Images of at least 7 separate fields of each well were taken with light and fluorescent microscope. Images were analyzed using ImageJ software. To generate senescence index, number of X-gal positive cells were divided by number of DAPI positive nuclei.
Adherent Clonogenics:
PrEC-AR-c-Myc (1x103 cells/well) were plated in 6 well plate. Following day, cells were treated with Emax50 concentration of EZH2 inhibitor for 72–96 hours (DZNep and GSK126 for 72 hours and EPZ6438 for 96 hours) in the presence of absence of 10nM R1881. Post EZH2 inhibitor treatment media was changed and refreshed with or without 10nM R1881 every 72 hours.
CRISPR-Cas9-mediated Gene Disruption:
Disruption of Tp53 in Hi-MYC cell lines was performed in collaboration with the Genome Engineering and iPSC Center (GEiC) at Washington University. Guide RNA (gRNA) sequences were designed for Tp53 and validated in Neuro2a cells by GEiC. Hi-MYC cells were co-transfected with separate gRNA and Cas9 expression plasmids and selected by puromycin and blasticidin resistance. Tp53 expression was examined by western blot analysis.
Western Blot
Whole Cell Lysate Preparation (WCL):
Cells were harvested and lysed with RIPA buffer (Sigma Aldrich, USA) + 1X P-STOP + 1X PIC (Roche) for 30 minutes on ice. Eppendorf tubes were vortexed every ten minutes for 10 seconds. After cell lysis tubes were centrifuged at 13,000 rpm for 15 minutes at 4°C. Supernatant of each tube was collected and transferred to a new tube. Protein concentrations of whole cell lysates (WCL) and histone extractions were measured by the bradford protein assay (Bio-Rad laboratories). Protein lysates (50μg WCL, 5μg Histone Extraction) where separation using 4–15% by SDS-PAGE gels (Bio Rad). The proteins were transferred from the SDS-PAGE gel onto nitrocellulose membrane (0.2 μm) (Bio-Rad, Hercules, CA) via the semi-dry method (Bio-Rad, Hercules, CA) for 35 minutes at 15V. Membranes were blocked in either 5% skim milk or BSA in 0.1% tween-PBS (tPBS) for 1-hour at RT. Membranes were washed briefly 3x with tPBS prior to primary antibody incubation at 4°C over night. Membranes were then washed 3×10 minutes before the addition of secondary horseradish peroxidase (HRP) conjugated antibodies (Bio-Rad, Hercules, CA) diluted in tPBS. After incubation at RT for 1-hour with agitation the membranes were washed 3×10 minutes in tPBS. The immunoreactive bands were visualized by enhanced chemiluminescence with ECL detection reagents (GE Healthcare Life Sciences, UK). The blots were exposed to Bio film for 1 second-10 minutes. The films were then developed in a Kodak film developer. To estimate molecular weight of bands a pre-stained protein ladder was used (Bio-Rad, Hercules, CA).
Flow Cytometry – Organoid Senescence Analysis
Our Senescence Detection Kit was used to measure organoid cellular senescence synthesis according to manufacturer’s instructions. Cultures were drug treated for 72 hours. Organoid discs were dislodged by pipetting, then digested to a single cell suspension by treatment with TrypLE (Gibco), which was in turn deactivated by resuspension in DMEM (Gibco) supplemented with 10% FBS (Sigma). Cells were washed with PBS (Gibco) by centrifugation at 500g at 4˚C and fixed with 4% paraformaldehyde for 15 minutes. Following another PBS wash, cells were permeabilized by the addition of ice-cold methanol to a final concentration of 90% methanol. This suspension was incubated for 30 minutes on ice. Following 2 washes with FACS buffer (PBS supplemented with 10% FBS), cells were resuspended in FACS buffer These cell suspensions were incubated overnight in the dark at 4˚C or for 1 hour at room temperature. Cells were washed two additional times in FACS buffer. H3K27me3 and Edu was analyzed using an Amnis ImageStream Mark II (Luminex) and dsRNA and PD-L1 with a BD LSRFortessa (BD Biosciences).
Results
EZH2 catalytic activity is enriched during MYC driven prostate cancer initiation and progression.
We first performed in silico GSEA of 49 EZH2 and/or H3K27me3 associated molecular signatures showed positive enrichment in independent EMC GEMM studies (14) (Fig. 1A and Supplement Table 1). We validated enriched expression of EZH2 protein and activity as indicated by tri-methylation of histone H3 lysine 27 (H3K27me3) in dorso-lateral prostate lobes of EMC GEMMs that had progressed to HG-PIN (Fig. 1B). We next performed longitudinal immunohistochemical (IHC) analysis for H3K27me3 in EMC mice (Fig. 1C). Our data indicates that H3K27me3 levels remain similar in wild type and EMC mice at 1 month of age. This is expected due to the well-documented role of EZH2 during differentiation, cell fate and maintenance of developmental genes (15–17). With this, H3K27me3 staining intensity decreased over time in wild type mice, which can be attributed to prostate luminal epithelium differentiation. Conversely, in luminal epithelium over-expressing MYC maintained high H3K27me3 expression during HG-PIN through progression to prostate cancer (Fig. 1C). Collectively, this data suggests that enriched EZH2 expression and activity are early molecular events important for HG-PIN progression to primary prostate cancer.
Figure 1. EZH2 expression and activity is increased in mouse HG-PIN and maintained during prostate cancer initiation and progression.
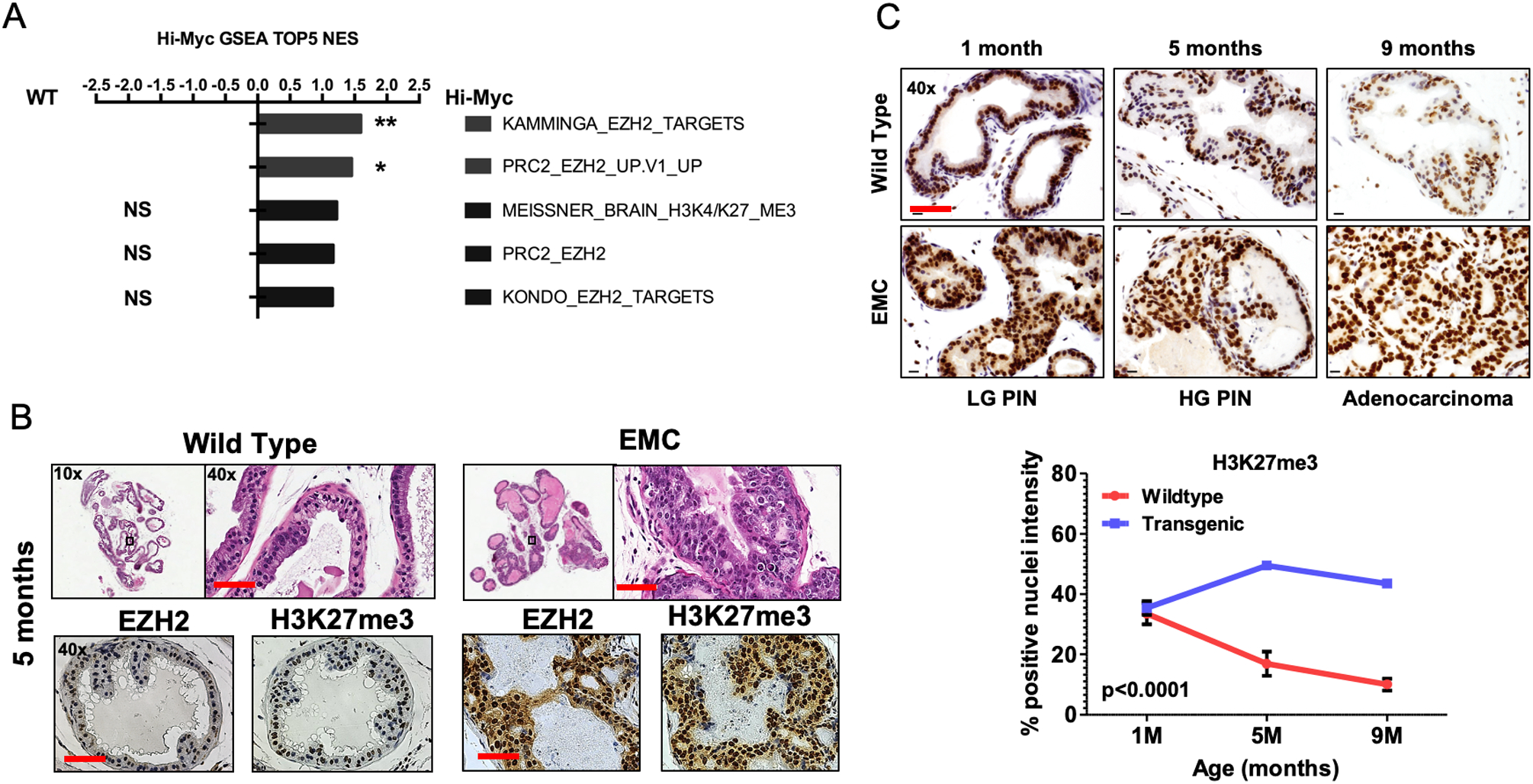
(A) Gene set enrichment analysis (GSEA) shows that EHZ2 and/or H3K27me3 associated molecular signatures are positively enriched in Hi-MYC murine PCa tumors when compared to wild-type prostate tissue. * p<0.05. (B) Representative H&E and IHC images from indicated mouse genotypes at 5 months of age (HG-PIN). It is observed that EZH2 and H3K27me3 is increased in PCa mouse models compared to age matched wild-type controls. (C) IHC images and quantitation from Hi-MYC mice indicating H3K27me3 positivity remains significantly elevated during HG-PIN through progression to PCa. For B-C, a total of 5 mice/genotype were analyzed. Scale bar = 50μm.
Elevated in vitro and in vivo MYC expression elevates EZH2 expression and activity increasing sensitivity to EZH2 inhibition.
To evaluate the effect of EZH2 inhibition in a human model of primary PCa, we utilized the human 957E/hTERT (957E/T) immortalized prostate epithelial cell line (18) that was further engineered to express human wild-type AR and cMYC (19). It was previously demonstrated that addition of cMYC inhibited AR induced growth in 957E/T cells, indicating this model ideal to investigate PCa initiation (19). Our analysis shows that addition of cMYC does increase EZH2 protein expression in 957E/T cells (Fig. 2A), and these cells are responsive to EZH2 inhibitors DZNep (20), GSK126 (21), and EPZ6438 (22) (Fig. 2B–C). All inhibitors displayed inhibition of cell proliferation which was associated with a loss of H3K27me3 positivity. Collectively, all inhibitors significantly repressed clonogenic survival of 957E/T cells (Fig. 2D).
Figure 2. MYC expression in prostate epithelial cells increases EZH2 expression and sensitizes to EZH2 inhibition.
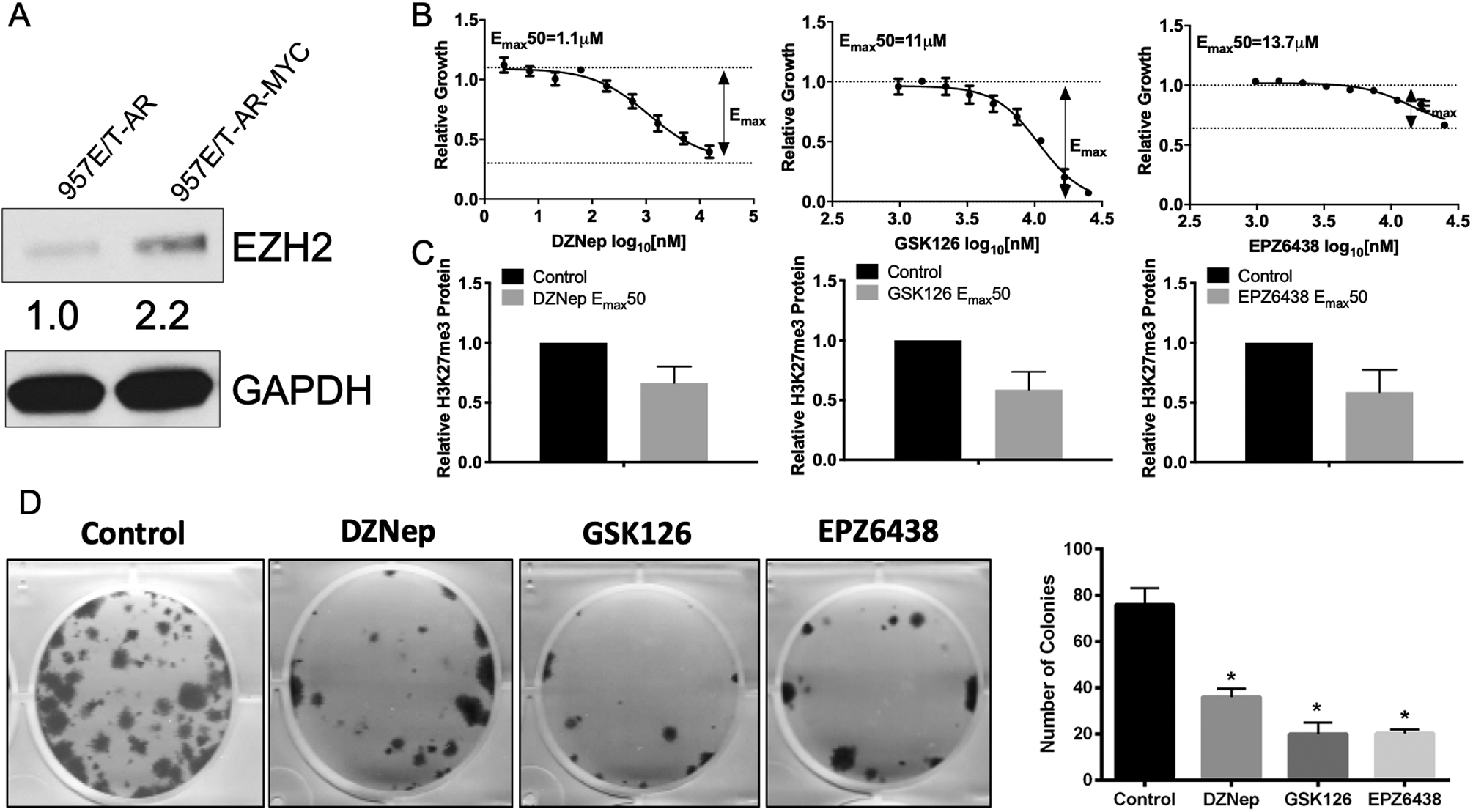
(A) Western blot analysis indicates over-expression of MYC increases EZH2 expression in 957E/T-AR cells. (B) EZH2 inhibitor response curves indicate 957E/T-AR-MYC cells respond in a dose-dependent inhibition of cell growth following 3 days treatment. (C) Calculated Emax50 concentrations from (B) were used to treat cells for 3 days and show reduction of H3K27me3 levels in 957E/T-AR-MYC cells. (D) Emax50 concentrations were used to treat for 3 days prior to drug wash out to assess clonogenic potential in 957E/T-AR-MYC cells.
To expand this in vitro observation and investigate pharmacological inhibition of EZH2 catalytic activity and its effect on HG-PIN progression in vivo, we treated EMC male mice at 8 weeks of age with vehicle or two EZH2 inhibitors independently over a 4-week period. Mice treated with EPZ0011989 (EPZ) (23) showed by histologically assessment that EPZ treatment resulted in reversal of HG-PIN to where examined dorsal-lateral prostate lobes more resembled normal luminal epithelium architecture compared to the multilayer HG-PIN in the vehicle cohort (Fig. 3A). Also, EPZ treatment reduced overall genitourinary tract and prostate weight. Mechanistically, EPZ treatment significantly reduced proliferation and H3K27me3 expression (Fig. 3A). To further validate our in vivo findings with EPZ, we also treated HG-PIN in EMC mice with DZNep. DZNep treatment showed similar histological response, with normal luminal architecture in dorso-lateral prostate lobes, as well as significant reduction in overall GU and prostate weights (Fig. 3B). DZNep also induced a significant reduction in H3K27me3 staining and cell proliferation compared to vehicle treated animals (Fig. 3B). Importantly, neither drug induced significant toxicity assessed by continual animal body weight measurements (Supplement Fig. 1D).
Figure 3. In vivo progression of MYC driven HG-PIN is attenuated by chemical inhibition of EZH2.
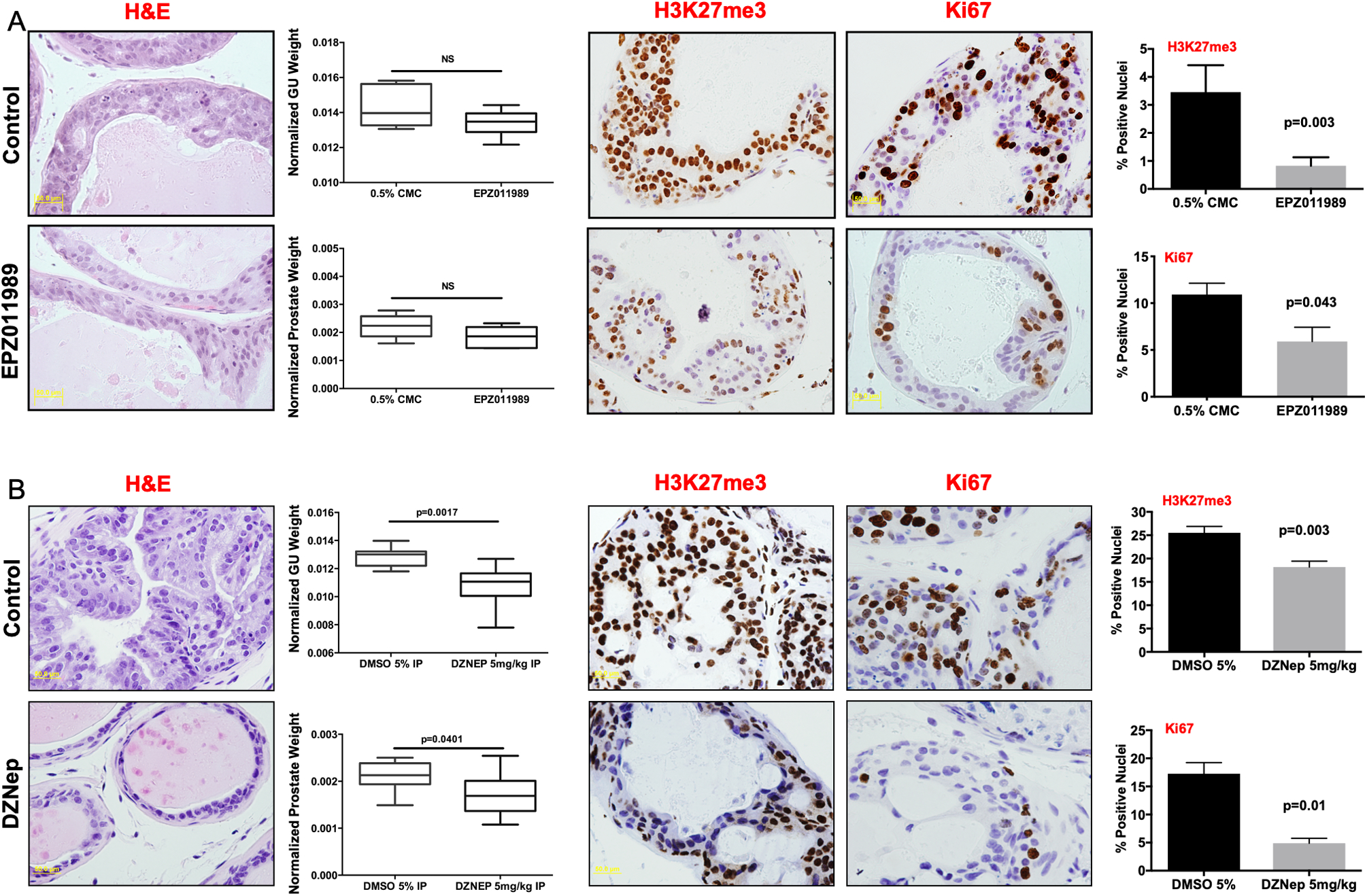
Genetically engineered mice that develop prostate adenocarcinoma driven by MYC overexpression (Hi-MYC) were treated with (A) 0.5% CMC (Control, n=4) or EPZ0011989 (n=4) or (B) 5% DMSO (Control, n=8) or DZNep (n=10). DZNep significantly attenuates progression of MYC driven HG-PIN. EPZ0011989 also attenuated progression but did not reach statistical significance. Immunohistochemical analysis of tumor tissue indicates that both DZNep and EPZ0011989 significantly reduce tumor H3K27me3 levels and tumor proliferation as assessed by Ki67. Scale bar = 50μm.
In order to independently validate the importance of EZH2 catalytic activity underlying HG-PIN progression to prostate cancer, we genetically attenuated EZH2 catalytic function in vivo. The generated EMC experimental mice allow for SET domain deletion from EZH2 under the control of a tamoxifen inducible Cre-recombinase allele (PSA-CreERT2) (Supplement Fig. 1A). Examination of dorso-lateral prostate lobes, following 4 weeks of SET domain deletion, showed similar attenuation of HG-PIN lesions as observed with EZH2 chemical inhibition (Fig. 4A). IHC examination of prostate tissue also showed loss of EZH2 and similar reduction of H3K27me3 positive and cell proliferation (Ki67 staining). (Fig. 4B). We also used our 3D EMC organoid model to further test the dependence for EZH2 towards progression. Using both genetic and chemical attenuation of EZH2 catalytic activity, significant attenuation of organoid formation was observed. These results clearly demonstrate the important of EZH2 catalytic function for the progression of HG-PIN to prostate cancer. EMC mice and organoids without PSA-CreERT2 expression served as tamoxifen treated control and demonstrated that dorso-lateral prostate lobes from Cre-negative mice exhibited no phenotype reversal and prostate tissue remained positive for EZH2 and H3K27me3 expression with increased proliferation (Supplement Fig. 1B–C).
Figure 4. Genetic disruption of EZH2 catalytic activity attenuates MYC driven HG-PIN.
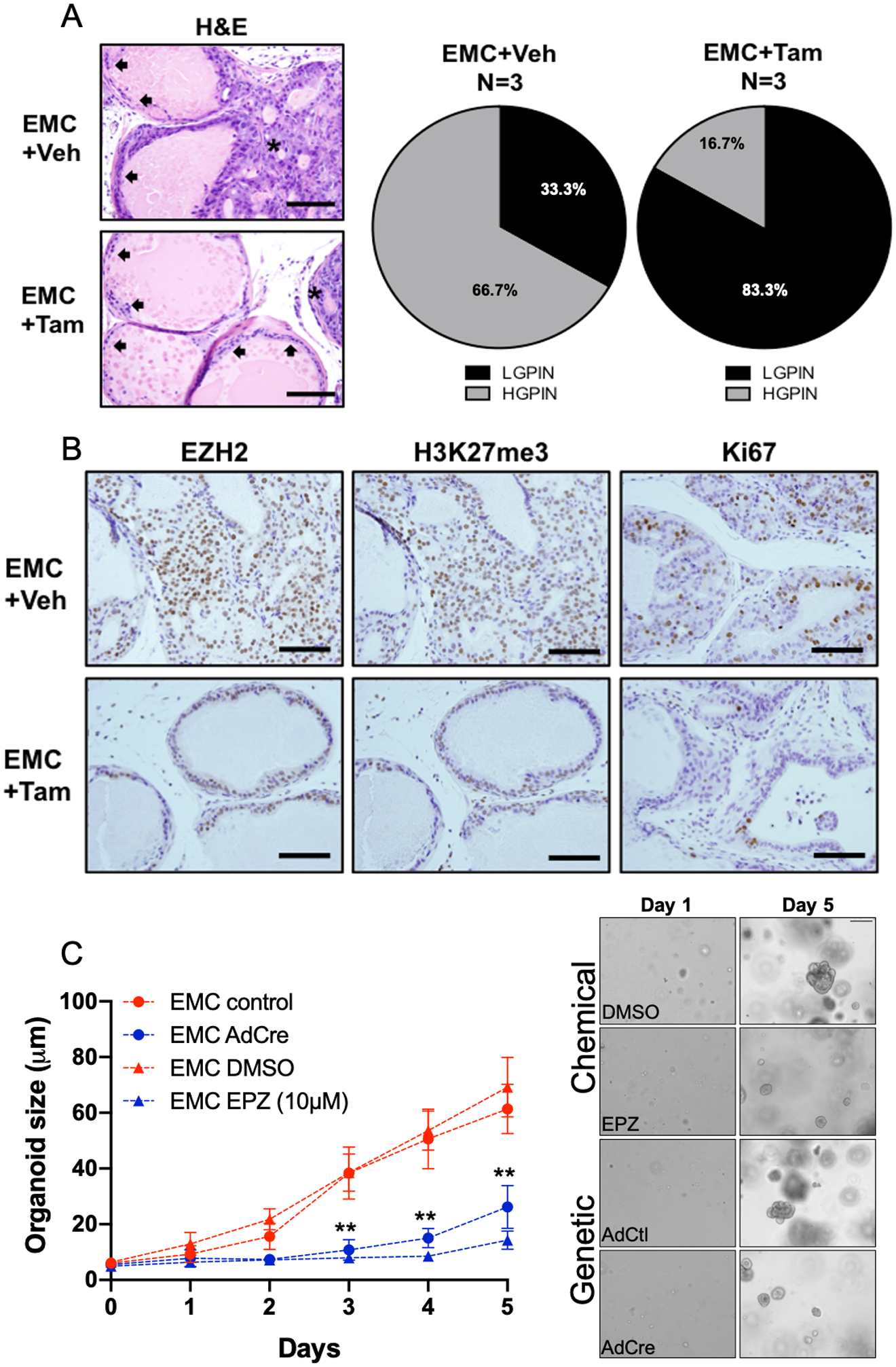
(A) Representative dorso-lateral H&E images and pathological assessment from mice treated with vehicle control or tamoxifen (1mg, days 1–5) attenuates the occurrence of MYC driven HG-PIN. (D) Immunohistochemical staining demonstrates the genetic inhibition of EZH2 catalytic activity reduces tumor positivity for EZH2 and H3K27me3 expression. Tumor cell proliferation is attenuate as indicated by reduction in Ki67 expression. Scale bar = 200μm. (C) Organoids generated from our genetically engineered mouse model had EZH2 catalytic activity inhibited by chemical (DMSO verse EPZ6438, 10μM day 0 and day 3) or genetic (AdCtl or AdCre) methods. Results for (C) represent the mean of 3 independent experiments ± SEM. Multiple T-test analysis demonstrates that EZH2 catalytic inhibition significantly attenuates prostate cancer organoid generation driven by MYC. **p<0.0001.
EZH2 inhibition induces cellular senescence in MYC driven models of prostate cancer.
To further delineate the downstream mechanisms following EZH2 inhibition we analyzed prostate tissue from our mice with EZH2 SET domain deletion and observed an increased level of beta-galactosidase staining (Fig. 5A). This observation indicated that EZH2 inhibition induced cellular senescence and was confirmed by use of matched EMC organoids where both genetic and chemical inhibition of EZH2 resulted in significant increases in cellular senescence (Fig. 5B). This was further validated by showing the ability of the EZH2 inhibitors DZNep and EPZ to significantly induce cellular senescence response in 957E/T-AR-MYC cells (Fig. 5C). Because TP53 is known to be a driver of oncogene-induced senescence we used CRISPR/Cas9 to knock-out Tp53 in murine Hi-MYC prostate cancer cells lines (24). In both cases, treatment of Hi-MYC cells by DZNep or EPZ resulted in the induction of senescence which was drastically decreased by the loss of Tp53 expression and function. These data indicate that induction of senescence by EZH2 inhibition is Tp53 dependent (Fig. 5D).
Figure 5. Inhibition of EZH2 catalytic activity induces tumor cell senescence in MYC prostate cancer.
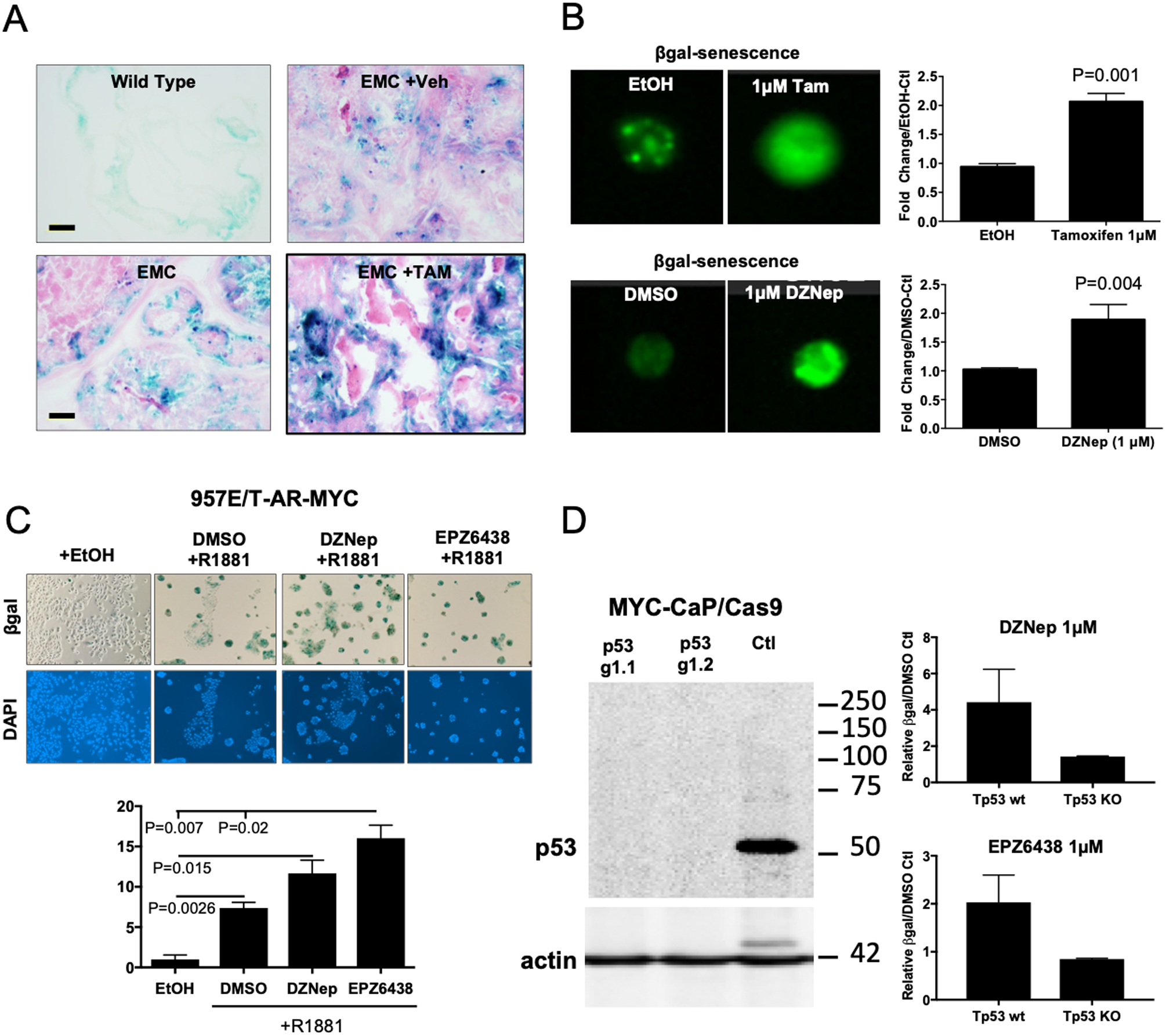
(A) Representative images from 12-week old dorso-lateral prostate samples indicate genetic disruption of EZH2 results in increased tumor cell β-galactosidase positive staining. (B) Organoids generated from our genetically engineered mouse model had EZH2 catalytic activity inhibited by chemical (DMSO verse DZNep, 1μM day 0) or genetic (EtOH or Tam day 0) methods. β-galactosidase staining was assessed 3 days post treatment by flow cytometry. (C) 957E/T-AR-MYC cell lines were treated with EZH2 inhibitors DZNep (1μM) or EPZ6438 (1μM) combined with indicated conditions for 3 days and assessed for β-galactosidase staining. (D) Murine prostate cancer lines, MYC-CaP, were infected Cas9 expressing plasmid followed by control (Tp53 wt) or Tp53 targeting guide-RNA (Tp53 KO). MYC-CaP/Cas9 cells were then treated with DZNep (1μM) or EPZ6438 (1μM) for 3 days and assessed for β-galactosidase staining by flow cytometry. EZH2 inhibitor induction of cellular senescence is Tp53 dependent. For B-D, results represent the mean of 3 independent experiments ± SEM. Unpaired T-tests were used to demonstrate statistical significance.
Discussion
Increased expression of EZH2 mRNA and protein was first reported in human metastatic and clinically localized prostate cancer (6). In addition, increased EZH2 expression was associated with poorer prognosis. Specific to prostate cancer, deregulation of EZH2 expression has been shown to occur via various mechanisms including loss of miR-101, a negative regulator of EZH2 mRNA (25). Alternatively, EZH2 is demonstrated to be a transcriptional target of ERG (26,27) and MYC (7). Koh et al. showed that EZH2 expression was increased in Lo-MYC mice and human pre-cancer (HG-PIN) lesions. To our knowledge, this is the first report to suggest epigenetic targeted therapy to inhibit HG-PIN progression to PCa.
Prevention of PCa has been demonstrated by application of targeted therapy in preclinical PCa mouse models. Using the transgenic adenocarcinoma of the prostate (TRAMP) mouse model, it was previously shown that DNA methyltransferase 1 (DNMT1) mRNA and protein levels were significantly elevated during PIN. Treatment of TRAMP mice in a chemo preventative strategy with the DNMT1 inhibitor, 5-aza-2’-deoxycitodine (5-aza) significantly increased overall survival of treated animals. This was associated with delay in disease progression and the prevention of lymph-node metastasis (5). A more recent study investigated the potential of targeted therapy chemoprevention in a Pten null mouse model of PCa. Jia et al. (28) demonstrated that genetic disruption of the PI3K signaling pathway or treatment of mice with HG-PIN by the dual PI3K-mTORC1 inhibitor BEZ235 significantly reversed HG-PIN induced by Pten loss. Interestingly, Jia et al. also observed that treatment of animals with enzalutamide accelerated disease. This data reinforces the initial results from the PCPT and REDUCE clinical trials, and also highlights potential adverse effects of anti-androgen chemoprevention.
Chemoprevention of PCa is possible and needed. Historically, it has been thought that androgen deprivation therapy (ADT) would inhibit progression of primary disease, however preclinical and clinical data suggest that ADT maybe counter intuitive and accelerate progression of stable disease to more aggressive disease. Therefore, more appropriate targeted therapies, like those highlighted above, need be identified and validated in clinical trials. Towards this, our data and others, show that EZH2 expression and catalytic activity are increased in mouse and human HG-PIN. Genetic and chemical disruption of EZH2 catalytic activity resulted in loss of tumor cell proliferation and increased Tp53 dependent cellular senescence. We speculate this response is due to the ability of EZH2 to suppress the Ink4a/Arf locus that encodes two proteins (p16Ink4a and p19Arf [p14Arf in mice]) critical for a cellular senescence response (29,30). Further our results may be explained by potential reactivation of an AR differentiation transcriptional response due to chromatin remodeling evoked by loss of EZH2 catalytic activity, though further experiments are required to determine this.
Future directions that may be considered and exciting for PCa prevention is the addition of EZH2 inhibition to standard use of androgen deprivation therapy in current neo-adjuvant clinical trials (31). EZH2 inhibition has been demonstrated to augment DNA damage (32) and recently increased EZH2 expression in PCa diagnostic biopsies was associated with increased risk of postradiotherapy metastatic disease recurrence (33), suggesting that EZH2 inhibition may promote increased radio-sensitivity. Most interesting, is potentiating the induction of senescence by EZH2 inhibition with combination strategies involving immunotherapy. We had recently demonstrated that EZH2 inhibition in PCa models can increase tumor T-cell and M1 tumor associated macrophage trafficking and reversed resistance to PD-1 checkpoint immunotherapy (34). Further, senescent cells are known to be cleared from the body by NK cells (35), and recent data has indicated that EZH2 was shown to be important for NK cell differentiation and EZH2 inhibition mediated eradication of liver cancer cells by NK cells (36,37). In the era of precision medicine, our work suggests that identification of patients with HG-PIN overexpressing EZH2 could be stratified to receive EZH2 inhibitors as a monotherapy or as a combination strategy in future PCa chemo (epi) preventative strategy.
Supplementary Material
Acknowledgements
Dana-Farber Faculty Start-Up Funds, and a Prostate Cancer Foundation Young Investigator Award (awarded to LE) supported research reported in this publication. D.P.L is a Lewis Katz – Young Investigator of the Prostate Cancer Foundation and is a Research Scholar – Junior 1 from The Fonds de Recherche du Québec – Santé funded by a Canadian Institutes of Health Research (CIHR) project grant (PJT-162246). We would like to thank Epizyme Pharmaceuticals for supplying EPZ0011989.
Footnotes
Conflict of Interest Disclosure: No potential conflicts of interest were disclosed.
References:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. The New England journal of medicine 2003;349(3):215–24 doi 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 3.Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, et al. Effect of dutasteride on the risk of prostate cancer. The New England journal of medicine 2010;362(13):1192–202 doi 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 4.Nelson WG, De Marzo AM, Deweese TL, Lin X, Brooks JD, Putzi MJ, et al. Preneoplastic prostate lesions: an opportunity for prostate cancer prevention. Ann N Y Acad Sci 2001;952:135–44. [DOI] [PubMed] [Google Scholar]
- 5.McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer research 2006;66(1):385–92 doi 10.1158/0008-5472.CAN-05-2020. [DOI] [PubMed] [Google Scholar]
- 6.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419(6907):624–9 doi 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 7.Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2011;2(9):669–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwata T, Schultz D, Hicks J, Hubbard GK, Mutton LN, Lotan TL, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PloS one 2010;5(2):e9427 doi 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 2008;21(9):1156–67 doi 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer cell 2003;4(3):223–38. [DOI] [PubMed] [Google Scholar]
- 11.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008;32(4):491–502 doi 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ratnacaram CK, Teletin M, Jiang M, Meng X, Chambon P, Metzger D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 2008;105(7):2521–6 doi 10.1073/pnas.0712021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 2016;11(2):347–58 doi 10.1038/nprot.2016.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer cell 2014;25(5):638–51 doi 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia 2014;28(1):44–9 doi 10.1038/leu.2013.288. [DOI] [PubMed] [Google Scholar]
- 16.Chou RH, Yu YL, Hung MC. The roles of EZH2 in cell lineage commitment. Am J Transl Res 2011;3(3):243–50. [PMC free article] [PubMed] [Google Scholar]
- 17.Yu YL, Chou RH, Chen LT, Shyu WC, Hsieh SC, Wu CS, et al. EZH2 regulates neuronal differentiation of mesenchymal stem cells through PIP5K1C-dependent calcium signaling. J Biol Chem 2011;286(11):9657–67 doi 10.1074/jbc.M110.185124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasunaga Y, Nakamura K, Ewing CM, Isaacs WB, Hukku B, Rhim JS. A novel human cell culture model for the study of familial prostate cancer. Cancer Res 2001;61(16):5969–73. [PubMed] [Google Scholar]
- 19.Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci 2014;10(6):627–42 doi 10.7150/ijbs.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glazer RI, Knode MC, Tseng CK, Haines DR, Marquez VE. 3-Deazaneplanocin A: a new inhibitor of S-adenosylhomocysteine synthesis and its effects in human colon carcinoma cells. Biochem Pharmacol 1986;35(24):4523–7 doi 10.1016/0006-2952(86)90774-4. [DOI] [PubMed] [Google Scholar]
- 21.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492(7427):108–12 doi 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 22.Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Molecular cancer therapeutics 2014;13(4):842–54 doi 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 23.Campbell JE, Kuntz KW, Knutson SK, Warloc NM, Keilhack H, Wigle TJ, et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust In Vivo Activity. ACS Med Chem Lett 2015. doi 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res 2005;65(24):11565–71 doi 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 25.Bohrer LR, Chen S, Hallstrom TC, Huang H. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgen-refractory progression of prostate cancer. Endocrinology 2010;151(11):5136–45 doi 10.1210/en.2010-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer cell 2010;17(5):443–54 doi 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunderfranco P, Mello-Grand M, Cangemi R, Pellini S, Mensah A, Albertini V, et al. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PloS one 2010;5(5):e10547 doi 10.1371/journal.pone.0010547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia S, Gao X, Lee SH, Maira SM, Wu X, Stack EC, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer discovery 2013;3(1):44–51 doi 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, et al. EZH2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res 2011;9(4):418–29 doi 10.1158/1541-7786.MCR-10-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proceedings of the National Academy of Sciences of the United States of America 2009;106(8):2641–6 doi 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery B, Tretiakova MS, Joshua AM, Gleave ME, Fleshner N, Bubley GJ, et al. Neoadjuvant Enzalutamide Prior to Prostatectomy. Clin Cancer Res 2017;23(9):2169–76 doi 10.1158/1078-0432.CCR-16-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep 2018;22(13):3480–92 doi 10.1016/j.celrep.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu X, Scott H, Carlsson SV, Sjoberg DD, Cerundolo L, Lilja H, et al. Increased EZH2 expression in prostate cancer is associated with metastatic recurrence following external beam radiotherapy. Prostate 2019;79(10):1079–89 doi 10.1002/pros.23817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheahan AV, Morel KL, Burkhart DL, Baca SC, Labbé DP, Roehle K, et al. Targeting EZH2 Increases Therapeutic Efficacy of Check-Point Blockade in Models of Prostate Cancer. bioRxiv 2019:730135 doi 10.1101/730135. [DOI] [Google Scholar]
- 35.Antonangeli F, Zingoni A, Soriani A, Santoni A. Senescent cells: Living or dying is a matter of NK cells. J Leukoc Biol 2019;105(6):1275–83 doi 10.1002/JLB.MR0718-299R. [DOI] [PubMed] [Google Scholar]
- 36.Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America 2018;115(15):E3509–E18 doi 10.1073/pnas.1802691115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proceedings of the National Academy of Sciences of the United States of America 2015;112(52):15988–93 doi 10.1073/pnas.1521740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.