

Abstract
Therapy-induced neuroendocrine prostate cancer (NEPC), an extremely aggressive variant of castration-resistant prostate cancer (CRPC), is increasing in incidence with the widespread use of highly potent androgen receptor (AR)-pathway inhibitors (APIs) such as Enzalutamide (ENZ) and Abiraterone and arises via a reversible trans-differentiation process, referred to as neuroendocrine differentiation (NED). The molecular basis of NED is not completely understood leading to a lack of effective molecular markers for its diagnosis. Here, we demonstrate for the first time, that lineage switching to NE states is accompanied by key miRNA alterations including downregulation of miR-106a~363 cluster and upregulation of miR-301a and miR-375. To systematically investigate the key miRNAs alterations driving therapy induced NED, we performed small RNA-NGS in a retrospective cohort of human metastatic CRPC clinical samples + PDX models with adenocarcinoma features (CRPC-adeno) vs those with neuroendocrine features (CRPC-NE). Further, with the application of machine learning algorithms to sequencing data, we trained a ‘miRNA classifier’ that could robustly classify ‘CRPC-NE’ from ‘CRPC-Adeno’ cases. The performance of classifier was validated in an additional cohort of mCRPC patients and publicly available PCa cohorts. Importantly, we demonstrate that miR-106a~363 cluster pleiotropically regulate cardinal nodal proteins instrumental in driving NEPC including Aurora Kinase A, N-Myc, E2F1 and STAT3. Our study has important clinical implications and transformative potential as our ‘miRNA classifier’ can be used as a molecular tool to stratify mCRPC patients into those with/without NED and guide treatment decisions. Further, we identify novel miRNA NED drivers that can be exploited for NEPC therapeutic targeting.
Keywords: MicroRNAs, miR-106a~363 cluster, miR-375, miR-301a, neuroendocrine differentiation, castration-resistant prostate cancer
INTRODUCTION
Prostate cancer (PCa) is fueled by androgens acting via androgen receptor (AR) signaling. Therefore, AR signaling ablation via Androgen deprivation therapy (ADT) is the goal of first-line therapy (1) that results in cancer regression initially. However, 2–3 years post-ADT, the disease develops into castration-resistant prostate cancer (CRPC) (2) with poor survival rates. Next generation of AR pathway inhibitors (API) such as Enzalutamide (ENZ) and Abiraterone (ABI) are used for treatment of men with non-metastatic and metastatic CRPC (3, 4) (1, 3–6) that are effective initially. However, about 30% of patients develop API resistance owing to heterologous mechanisms such as restored/bypass AR signaling or evolution to an AR independent state, called neuroendocrine prostate cancer (NEPC). NEPC is highly aggressive and present a significant clinical challenge (7, 8). It arises via a reversible trans-differentiation process, referred to as neuroendocrine differentiation (NED) (7, 8), wherein cells undergo a lineage switch and exhibit neuroendocrine features, characterized by expression of neuronal markers including enolase 2 (ENO2), chromogranin A (CHGA) and synaptophysin (SYP) (7, 8). Aggarwal et al reported the presence of treatment emergent small cell neuroendocrine prostate cancer in 17% of mCRPC patients with disease progression on abiraterone or enzalutamide (9). Clinically, NEPC manifests as the presence of visceral metastatic disease including metastasis to liver, lung, central nervous system or bone despite low serum PSA (8). Histopathological assessment combined with immunohistochemical detection in PCa tissues/serum levels of neuronal markers including SYP, NSE, CHGA and CD56 is currently used to monitor NED (10, 11). However, these markers are not sufficiently specific (8), highlighting the urgent need of novel molecular markers to assess NED and predict API resistance.
It has been realized that therapy-induced NED represents a continuum of treatment-induced changes at phenotypic and molecular level resulting from a series of genetic and epigenetic alterations (12). The genetic and epigenetic changes underlying NEPC have been investigated by several groups recently (9, 12–17) that show that these states are derived via clonal evolution from adenocarcinomas (13). The key genetic events driving this transition include frequent TMPRSS2-ERG rearrangements (16), loss of retinoblastoma (RB1) and tumor protein (TP53), amplifications of Aurora kinase A (AURKA), NMYC, EZH2 (13–15, 17), BRN2 (18) and BRN4 (19). Though these studies have characterized the genomic and epigenetic alterations driving NED, we are still far from complete understanding of the key molecular alterations. With a goal of understanding molecular mechanisms underlying progression of PCa to NEPC and resulting API resistance, the focused objective of our study was to systematically investigate the key microRNA (miRNAs) alterations that are associated with progression of advanced CRPC with adenocarcinoma characteristics (CRPC-Adeno) to therapy-induced, androgen-independent NE (CRPC-NE) states. miRNA alterations associated with NEPC and their associated roles have not been thoroughly investigated though there are reports on a few miRNAs (20–24). We hypothesized that this lineage switch is associated with significant miRNA alterations that drive gene expression changes leading to NEPC induction. Here, we identify for the first time, that a characteristic set of miRNA alterations, including downregulation of miR-106a~363 cluster of miRNAs and upregulation of miR-375 and miR-301a, promote plasticity of advanced prostate adenocarcinomas to NEPC. Importantly, we develop a ‘novel miRNA classifier’ that can robustly stratify CRPC-NE tumors from CRPC-Adenocarcinomas and has translational potential for diagnosing therapy-induced NED in mCRPC patients and predicting API responsiveness. Further, we demonstrate that miR-106a~363 cluster pleiotropically regulate cardinal nodal proteins instrumental in driving NEPC including Aurora Kinase A, N-Myc (14, 15, 25), E2F1 and STAT3. Aurora Kinase A inhibitor, Alisertib that disrupts N-Myc/AURKA interaction is currently in clinical trials as a targeted NEPC therapy (26). In view of our results, we propose restoration of miR-106a~363 cluster as a novel NEPC therapeutic modality.
RESULTS
MicroRNAs are dysregulated upon transition from CRPC-Adeno to CRPC-NE states
To test our hypothesis that lineage switching from prostate adenocarcinomas to NE states is driven by miRNAs, we performed small RNA-NGS in a retrospective cohort of human mCRPC clinical samples with no evidence of NED+ PDX models with adenocarcinoma features (CRPC-adeno) (n=25) vs those with therapy-induced NED (CRPC-NE) (n=9) (Fig. S1). Clinicopathological characteristics of samples are represented in Table S1. PDX models included CRPC-Adenocarcinoma models LuCaP 70, 78, 81, 92 and CRPC-NE models LuCaP 49, 145.1 and 145.2. These models have been previously comprehensively characterized by Nguyen et al. (27) and NE PDXs LuCaP 49, 145.1, 145.2 are reported to be AR negative. As a positive control, we included NEPC cell line, NCI-H660 (28). Libraries were generated using an Illumina® TruSeq® small RNA library prep kit and sequenced on Illumina NextSeq 500 (Fig. 1). We obtained an average of ~9 million raw reads/library. Of these, 74–97% reads were mapped to known RNA species and the human genome (hg38). Among mapped reads, miRNA reads included 30–65% (~48% average) of known mature miRNAs and 0.1–0.5% (0.3% average) novel mature miRNAs (Fig. S2). Interestingly, we observed 30–70% abundance of known isomiRs (50% average) and 1–5% novel isomiRs (3% average). Raw sequencing data is deposited in the Sequence Read Archive at NCBI (accession no. SUB6292342). Significant miRNA dysregulation was observed between CRPC-Adeno vs CRPC-NE cases (Fig. 1A). Using a cutoff false discovery ratio (FDR) of 5%, adjusted P-value <0.05, a total of 397 known and 42 novel miRNAs were found to be differentially expressed (Table S2). When miRNAs with fold change >2 were considered and iso-miRs were excluded, a set of 62 miRNAs (58 known + 4 novel miRs) were found to be differentially expressed in CRPC-NE vs CRPC-Adeno cases. These miRNAs could cluster CRPC-NEs distinctly from CRPC-Adenocarcinomas. Top significantly downregulated miRs included miRs-363–3p, −20b-5p, −106a-5p, −148–3p, −582–5p, −203a-3p while top significantly upregulated miRNAs included miRs-4662a-5p, 138–5p, 708–5p, 181d-5p, 301b-3p, 10a-5p, 181c-5p, −375 and −301a-3p. The differentially expressed miRNAs fall into 50 miRNA families (Table S3), with significantly downregulated miRNA families including miR-106a~363 cluster, miR-1248, miR-148 families and top upregulated families including miR-873, miR-4662 and miR-4421 families.
Fig. 1. Small RNA sequencing analyses of CRPC-Adeno and CRPC-NE clinical samples, PDX and cellular models.
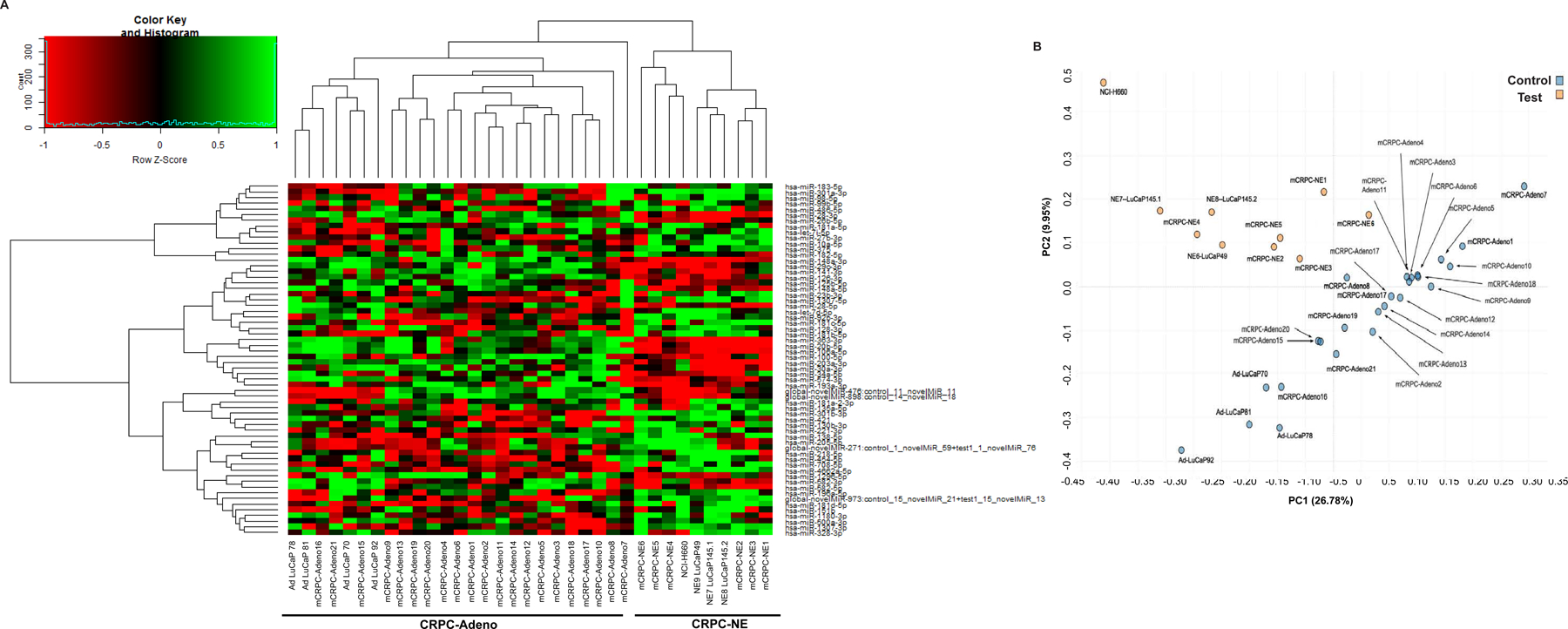
A. Heat map showing differentially expressed mature miRNAs in CRPC-adeno cases (n=25; clinical tissues, n=21 + PDX models AdLuCaP-70, −78, −81) as compared to CRPC-NE (n=10; clinical tissues, n=6 + PDX models LuCaP-49, −145.1, −145.2 + NCI-H660 cell line).
B. Based on differential expression of miRNA families, unsupervised principal component analyses (PCA) was performed that revealed separate clustering of the CRPC-NE tumors/models from CRPC-Adeno.
Distinct clustering of CRPC-NE tumors from CRPC-Adenocarcinomas based on miRNA families
Further, based on miRNA families, unsupervised analysis was performed using principal component analyses (PCA) (Fig. 1B) that revealed separate clustering of the CRPC-NE tumors from CRPC-Adenocarcinomas. These data suggest that specific miRNAs are dysregulated in induction of NE states and that miRNA expression patterns can distinguish between CRPC-Adeno and CRPC-NE tumors. Interestingly, this ability of miRNAs was very specific as a similar analysis based on differential piRNA expression revealed a non-distinct clustering of CRPC-NE from CRPC-Adeno tumors (Fig. S3).
A microRNA classifier of neuroendocrine differentiation in CRPC
We next explored if the observed differentially expressed miRNAs can be used as a ‘molecular classifier’ to differentiate between CRPC-NE and CRPC-Adenocarcinomas. To this end, we employed random forest machine learning technique with leave-pair-out cross validation (LPOCV) to the NGS dataset. For development of the classifier, miRNAs were filtered to exclude miRNA isoforms. Interestingly, a set of 43 miRNAs formed a ‘miRNA classifier’ (Fig. 2A). Fig. 2A shows the miRNA classifier with the miRNAs listed in the order of importance as determined by LPOCV. Top ten features of the classifier included miRs-301a-3p, −375, −1307–3p, −181c-5p, −106a-5p, −29a-3p, −218–5p, −106b-5p, miR-363–3p and miR-20b-5p. Interestingly, miR-363–3p, miR-20b-5p and miR-106a-5p, members of miR-106~363 cluster located on chrX, constituted important features of the classifier (Fig. 2A). This miRNA classifier could distinguish CRPC-NEs from CRPC-Adeno with an AUC of 0.98 (Fig. 2B).
Fig. 2. A microRNA classifier to distinguish between CRPC-NE and CRPC Adenocarcinoma cases.
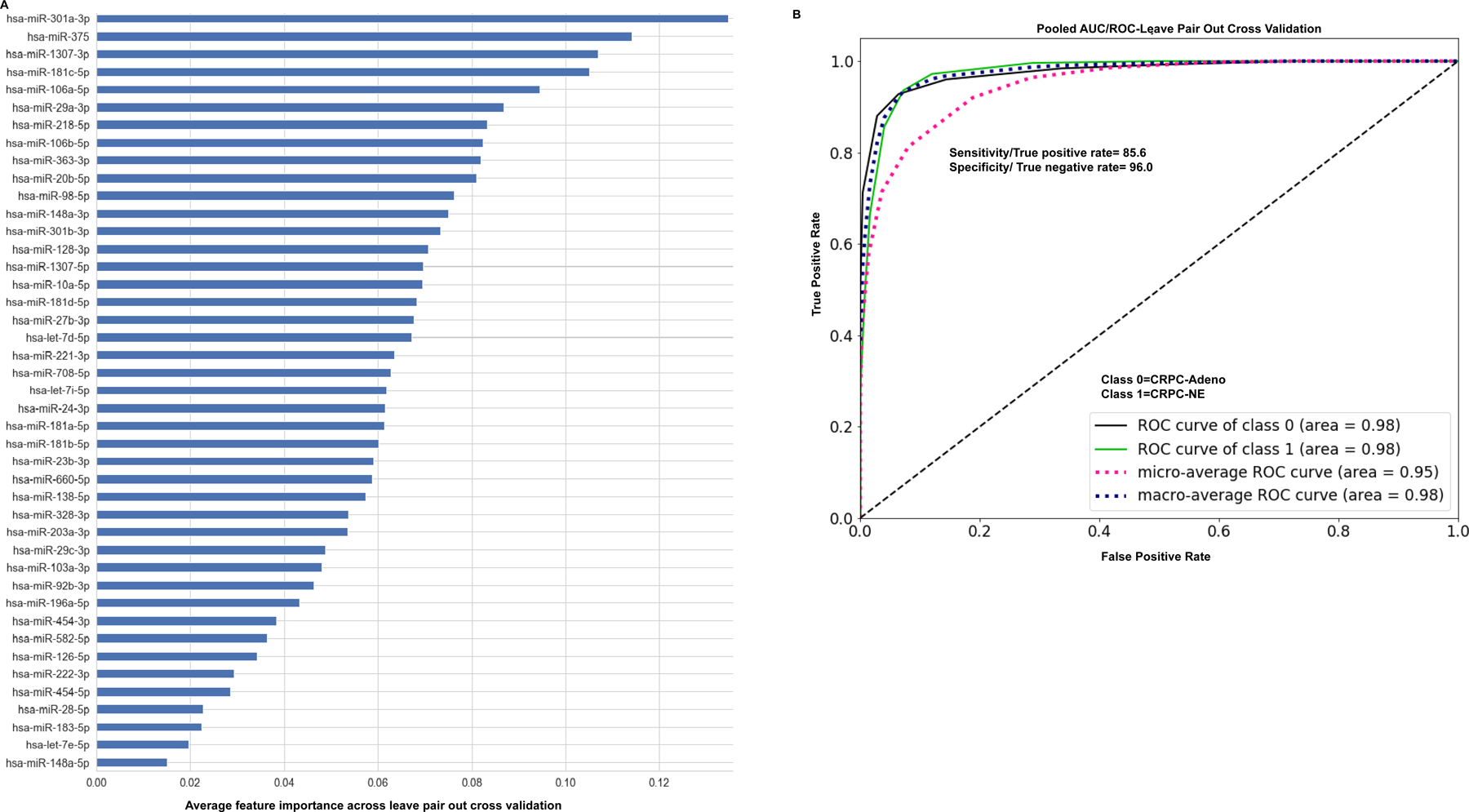
A. Application of machine learning methods (random forest machine learning technique with leave-pair-out cross validation) to the NGS dataset of analyzed NE tissues + PDX models+ NCI-H660 cell line (CRPC-NE, n=10) vs those with adenocarcinoma features (CRPC-Adeno, n=25) yielded a ‘43 miRNA classifier’. miRNAs are listed in the order of feature importance as determined by these methods.
B. ROC curve analyses showing the ability of ‘miRNA classifier’ to distinguish between class 0 (CRPC-Adeno) and class 1 (CRPC-NE).
Predominant differential expression of microRNA isoforms (iso-miRs) is associated with PCa NED
Of the 397 known and 42 novel miRNAs differentially expressed in CRPC-NE vs CRPC-Adenocarcinomas, we observed a predominant representation of iso-miRs (Fig. 3). Using a cutoff FDR of 5%, P-value<0.05 and fold change >2, a total of 337 iso-miRs (299 known and 38 novel) were found to be differentially expressed (Fig. 3A). Iso-miRs were found to range in length from 14–45 nucleotides (nt) (Fig. S4A), with the majority of iso-miRs at 22–23nt, a length typical of mature miRNAs. Fig. 3B represents the miRNA loci producing ≥5 differentially expressed iso-miRs with the predominance of miR-92b, miR-363, miR-10a, miR-148a and miR-375 iso-miRs. Top ten downregulated iso-miRs included 9 isoforms of miR-363 and 1 of miR-148a. Isoforms of other members of miR-106a~363 cluster, including one each of miR-106a and miR-20b were also downregulated in CRPC-NE tissues. Among top ten upregulated, isoforms of miR-375, miR-181c, miR-10a, miR-205, miR-27b and miR-95 were represented. In view of observed significant iso-miR dysregulation, we examined their potential to classify NED in CRPC by performing second machine learning method with iso-miRs included that identified a ‘126- features miRNA classifier’ (Fig. S4B and Table S4). Top features of the classifier included one isoform each of miRs-19b-1, −95, −103a-1, −103a-2, −126, two isoforms of miR-363–3p apart from miRNAs miR-301a-3p, miR-203a-3p and miR-181a-5p. This ‘126-features miRNA classifier’ could distinguish CRPC-NE from CRPC-Adeno with an AUC of 0.96 (Fig. 3C) suggesting that while iso-miRs are significantly dysregulated in NEPC, incorporating iso-miRs in the ‘classifier’ does not significantly improve its performance.
Fig. 3. Predominant differential expression of microRNA isoforms (iso-miRs) is associated with neuroendocrine differentiation in CRPC.
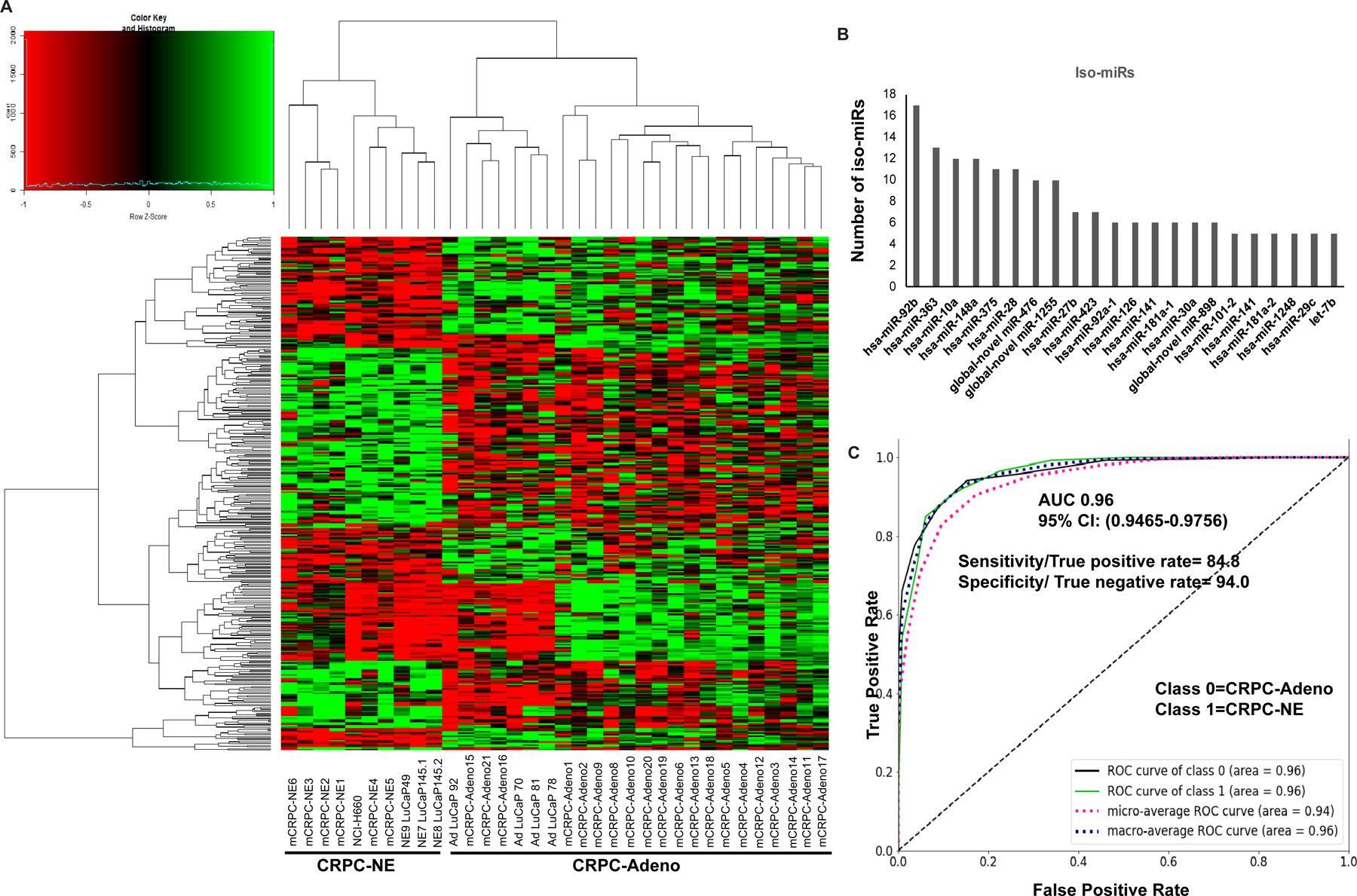
A. Heat map showing differentially expressed iso-miRs between CRPC-adeno cases (n=25; clinical tissues, n=21 + PDX models AdLuCaP-70, −78, −81) as compared to CRPC-NE (n=10; clinical tissues, n=6 + PDX models LuCaP-49, −145.1, −145.2 + NCI-H660 cell line).
B. miRNA loci producing ≥5 differentially expressed iso-miRs plotted as a function of number of observed iso-miRs.
C. ROC curve analyses showing the ability of ‘miRNA classifier including iso-miRs’ to distinguish between class 0 (CRPC-Adeno) and class 1 (CRPC-NE).
‘miRNA classifier’ is robust in differentiating between CRPC-Adenocarcinoma and CRPC-NE states in validation cohort of PCa
In order to rigorously validate the robustness of our miRNA classifier, we performed small RNA-NGS in a validation cohort (cohort 2) of human mCRPC clinical samples (n=20) (Fig. 4) that included mCRPC-adeno (n=18) (denoted CRPC-adeno 22–39) and mCRPC-NE (n=2) (denoted CRPC-NE 10–11). NGS analyses of this cohort confirmed key miRNA alterations such as miR-106a~363 cluster downregulation apart from −148–3p, −203a-3p downregulation that were also observed in training cohort (Table S5). Common significantly upregulated miRNAs included miRs-138–5p, 181d-5p, 301b-3p, −301a-3p, 181c-5p, 181d-5p, miR-182 and miR-183. Importantly, miRNA analyses in validation cohort confirmed key miRNA alterations and also confirmed the ability of miRNA alterations to distinctly stratify CRPC-NEs from CRPC-adenocarcinomas as shown in PCA plot (Fig. 4A and S5A). We further examined if the miRNAs included in our ‘miRNA classifier’ could distinguish/classify NEs from adenocarcinomas in this cohort. Towards this, we applied the ‘classifier data’ obtained from cohort 1 to validation cohort (cohort 2) by employing single model prediction algorithm. This modeling validated a set of 24 miRNAs of the classifier to be important in distinguishing between CRPC-Adeno vs CRPC-NE (Fig. 4B). Importantly, modeling in cohort 2 preserved the top features of the classifier including miRs-301a-3p, −375, −106a-5p, −181c-5p, miR-363–3p and miR-20b-5p. Interestingly, miR-301a-3p, miR-375 and miR-106a~363 cluster of miRNAs (including miR-363–3p, miR-20b-5p and miR-106a-5p) formed top features of the validated classifier. Fig. 4C shows the prediction probabilities of a clinical sample to be ‘adenocarcinomas’ (grey) or ‘NE’ (red) based on validated miRNA classifier. For mCRPC-Adeno22–39 cases, the prediction probabilities for adenocarcinomas ranged from 0.872 to 1 with concomitant low NE probabilities. However, mCRPC-Adeno38 showed ‘adeno’ probability of 0.685 and ‘NE’ probability at 0.315. Examination of clinical information showed that this patient had low final serum PSA suggesting that this case likely was progressing towards NE. The two NE cases examined showed predominant NE probability (0.81–0.873). These data validate the robustness of our miRNA classifier to distinguish between ‘Adeno’ vs ‘NE’ states. The performance of classifier was measured using ROC analyses that showed AUC =1 showing high overall prediction accuracy (Fig. S5B). We also performed logarithmic loss performance metric on validation cohort to test the performance of our classifier. This metric measures the performance of a supervised learning classification model and the predicted probabilities from the model, with lower score (0) being a perfect score. Interestingly, the validated set of ‘miRNA classifier’ showed a log loss score of 0.09258, demonstrating its robustness.
Fig. 4. ‘miRNA classifier’ is robust in differentiating between CRPC-Adenocarcinoma and CRPC-NE states in validation cohorts of prostate adenocarcinomas.
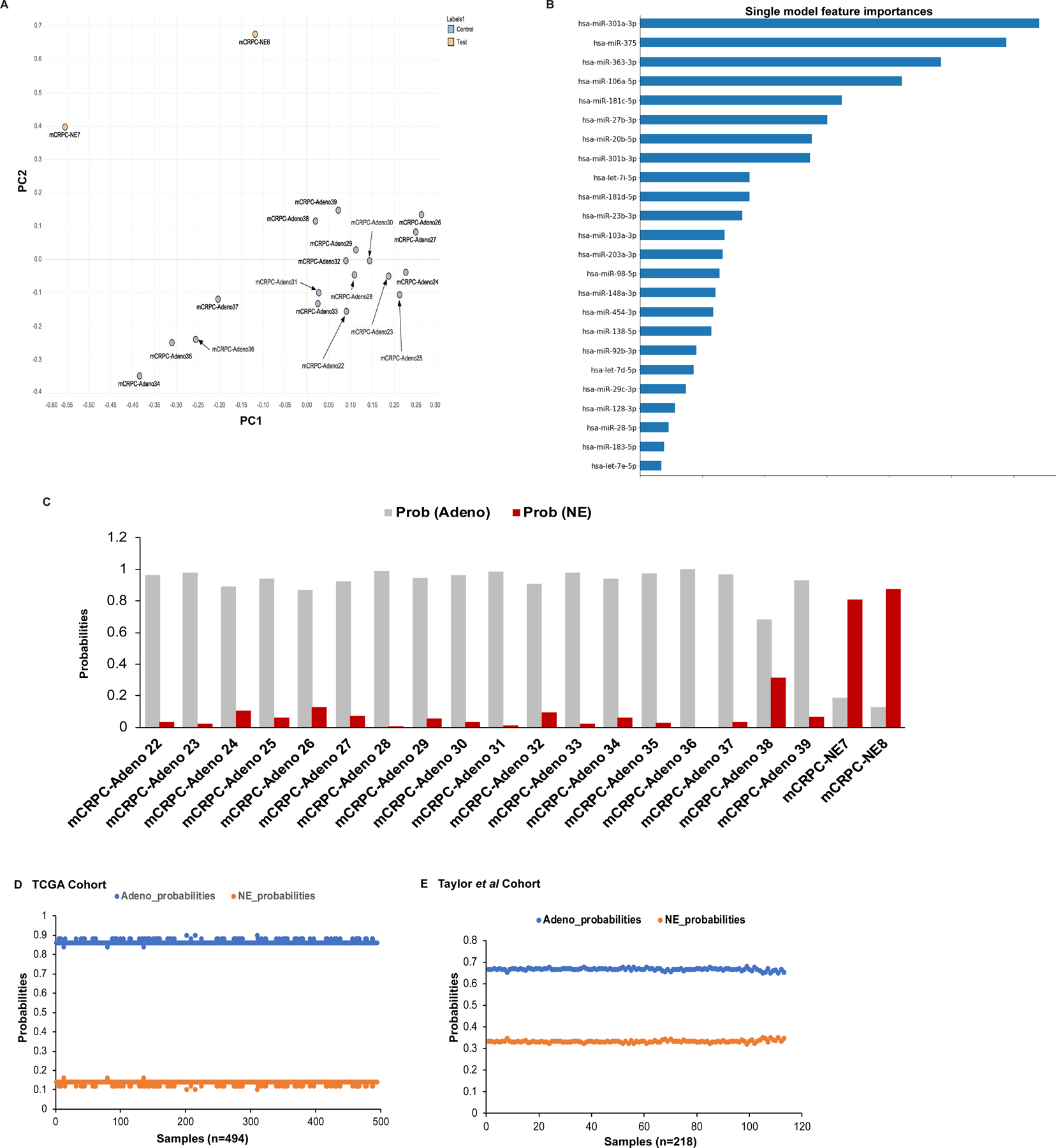
A. Unsupervised principal component analyses (PCA) for validation cohort.
B. Single model prediction algorithm for ‘miRNA classifier data’ from cohort 1 as applied to NGS data for validation cohort,
C. Prediction probabilities for samples in validation cohort to be ‘adenocarcinoma’ (gray) or ‘NE’ (red) histology based on validated miRNA classifier.
D. Application of miRNA classifier to the TCGA database of primary adenocarcinomas (n = 494) [29]. Adeno vs NE prediction probabilities for individual tumors are represented.
E. Prediction probabilities for samples in Taylor et al. [30] cohort to be ‘adenocarcinoma’ (blue) or ‘NE’ (orange) histology based on subsetted miRNA classifier (represented in Fig. S6)
Validation of ‘miRNA classifier’ against existing prostate adenocarcinoma cohorts
Since our classifier is a way of not only diagnosing ‘NE’ tumors, but also a way of ‘calling’ adenocarcinomas, we evaluated the classifier in existing PCa cohorts (Fig. 4D–E). Towards this, we applied it to the TCGA database of primary adenocarcinomas (n=494) (29) (Fig. 4D) and Taylor et al dataset (30) (Fig. 4E). Importantly, it categorized all TCGA PCa cases correctly as ‘adenocarcinomas’ validating its robustness. Taylor et al dataset (30) includes primary and metastatic PCa cases. For application of the classifier to Taylor et al dataset (30) of primary and metastatic prostate cancer cases, we subsetted our classifier as this dataset did not include expression for nine of the miRNAs (Fig. S6A). Table S6 represents list of miRNAs included in our classifier that are missing in Taylor et al dataset. The ‘subsetted classifier’ was then applied to Taylor et al dataset (30) using single model prediction algorithm (Fig. S6B). Fig. 4E shows the prediction probabilities for this dataset (30) using single model prediction based on subsetted miRNA classifier that shows that our classifier correctly categorized all tumors as ‘adenocarcinomas’, supporting its robustness.
Dysregulated miRNAs target signaling pathways implicated in PCa and key genes in ‘NEPC gene signature’ and ‘AR signaling’
Further, we examined the potential target genes affected by dysregulated miRNAs in NEPC by performing Ingenuity Pathway Analysis (IPA 8.0, Qiagen) on our NGS data. Considering only high confidence and experimentally validated targets, 432 mRNAs are potentially targeted by dysregulated miRNAs. Interestingly, significantly miRNA-impacted pathways include glucocorticoid receptor, p53, PTEN, PI3K/Akt, cell cycle G1/S checkpoint regulation, ERK/MAPK, TGFβ, Wnt/β-cat and PCa signaling (Fig. 5A). Recently, Beltran et al. generated a 70-gene NEPC classifier that could distinguish CRPC-NE from CRPC-Adeno, treatment-naïve PCa and BPH (13). Interestingly, we found that 32/70 ‘NE’ genes are potentially targeted by dysregulated miRNAs. Also, dysregulated miRNAs target 17/31 genes within the AR signature (Fig. 5B–C). We employed IPA analyses to further understand the regulatory interplay between the dysregulated miRNAs and NEPC/AR gene signature and identified a set of 134 reciprocal miRNA/mRNA pairings (Table S7). Predominantly, key NE genes such as AURKA, EZH2, CCND1 are potentially controlled by identified miRNAs (Fig 5C). Importantly, RB1 is potentially targeted by identified upregulated miRNAs including miR-98–5p, miR-221–3p and miR-181d-5p, MYCN by downregulated miR-29c-3p, miR-34, miR-20b-5p, miR-19b-3p and AURKA by miR-363–3p. Further, miR-26b downregulation potentially regulates EZH2 and let-7a-5p, miR-92a-3p, miR-421–3p, miR-28–3p regulate AR.
Fig. 5. Dysregulated miRNAs target signaling pathways implicated in PCa and key genes in ‘NEPC gene signature’ and ‘androgen receptor signaling’.
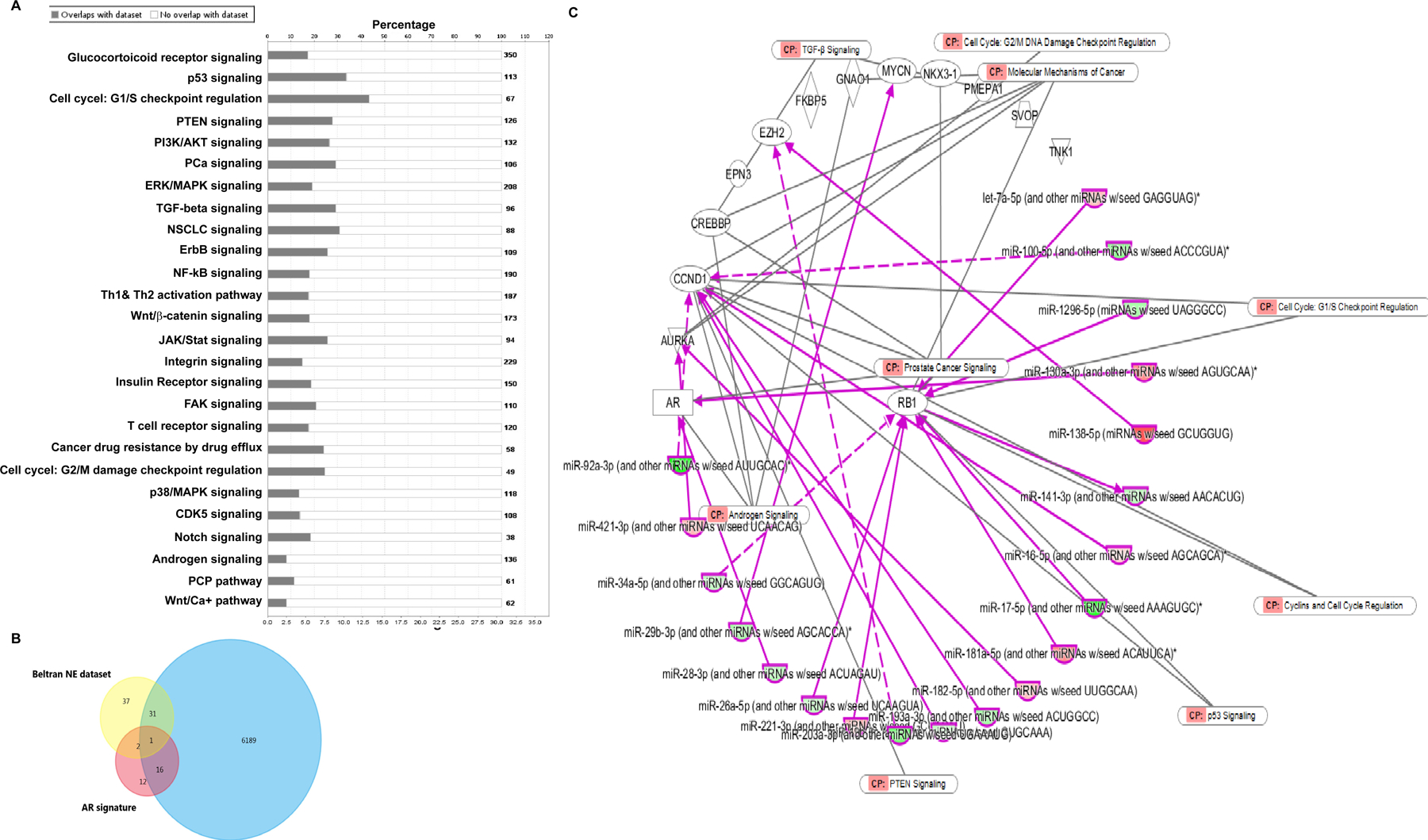
A. Ingenuity Pathway Analysis (IPA 8.0, Qiagen) for potential signaling pathways impacted by dysregulated miRNAs in NEPC. Only high confidence and validated targets were considered for this analysis.
B. Venn diagram showing overlap between potential miRNA targets and NE gene signature and/or AR signaling generated by Beltran et al. (13).
C. IPA analyses to understand the regulatory interplay between the dysregulated miRNAs identified in our present study and NEPC/AR gene signature and identified a set of 134 reciprocal miRNA/mRNA pairings (listed under Table S6). Predominantly, key genes such as AURKA, EZH2, CCND1, MYCN, AR, RB1 were found to be potentially controlled by identified miRNAs.
Downregulation of miR-106a~363 cluster of miRNAs drive NEPC via pleiotropic regulation of multiple NEPC drivers
We further probed the functional significance of top miRNA alterations (downregulation of miR-106a~363 cluster and miR-375 and miR-301a upregulation) that constituted the top features of our ‘miRNA classifier’. First, we validated miR-106a~363 cluster downregulation in NEPC PDX models (Fig. 6A). miR-363, miR-106a and miR-20b were found to be downregulated in CRPC-NE PDX models LuCaP 49, 145.1 and 145.2 (27) vs CRPC-Adeno PDXs LuCaP 70, 78, 81 and 92 (Fig. 6A). Expression analyses in prostate cell lines (Fig. 6B) showed that compared to BPH1 (33), PCa cell lines (PC3, LNCaP) show decreased miRNA expression and these miRNAs were specifically downregulated in ENZ-resistant LNCaP-AR cells as compared to corresponding parental LNCaP-AR cells suggesting the potential association of miRNA downregulation with ENZ resistance. To gauge downstream effects of miR-106a~363 cluster, we employed miRNA sponge technology (31, 32) to simultaneous block multiple miRNAs within this cluster. This technology uses an engineered RNA molecule with miRNA complementary sequence resembling a perfect target that includes a mismatched bulge so the miR-RNA duplex is not degraded (32). To block multiple miRNAs, the sponge construct included multiple bulged miR binding sites, driven by RNA polymerase III-driven H1 promoter (Fig. 6C, upper panel). Following stable transduction of control/ miR-106a~363 sponge constructs in C42B cells, miR-106a, miR-20b and miR-363 expression were examined by real time PCR (Fig. 6C, lower panel) that confirmed miRNA downregulation in miR-106a~363 sponge-treated cells compared to controls. Functional assays showed that miR-106a~363 downregulation led to increased colony formation ability (Fig. 6D) and increased invasiveness of C42B cells as compared to corresponding controls (Fig. 6E). Western blot analyses showed that expression of neuronal markers CHGA, SYP and ENO2 is increased in miR-106a~363 repressed C42B cells as compared to controls (Fig. 6F). Examination of potential miRNA targets showed that repression of this miRNA cluster led to increased STAT3, E2F1 and N-Myc expression as compared to corresponding controls (Fig. 6G). Examination of 3’ UTRs of these genes showed two putative binding sites for miR-106a and miR-20b each in STAT3 (positions 252 and 542) and E2F1 3’ UTRs (positions 387 and 980) and one potential binding site at position 859 within MYCN 3’ UTR (Fig. 6H). To confirm that the observed effects are owing to direct binding of miRNAs to these potential 3’ UTR binding sites, we performed luciferase reporter assays (Fig. 6I). Co-transfection of MYCN, E2F1–1, E2F1–2, STAT3–1 in miR-106a-363 repressed C42B cells led to significant upregulation of luciferase activity as compared to corresponding controls suggesting that these genes are directly targeted by these miRNAs though co-transfection with STAT3–2 did not cause significant alteration in luciferase activity, suggesting that miR-106a~363 acts via primarily binding to site 1 in STAT3 3’UTR. To further validate these genes as direct miR-106a~363 targets, we performed ‘phenocopy experiments’ in C42B cells (Fig. 6J). Overexpression of MYCN and E2F1 ORF clones (that lacks 3’ UTR sites) led to increased invasion of PCa cells as compared to control ORF akin to the effects of 106a~363 sponging (Fig. 6E).
Fig. 6. Downregulation of miR-106a~363 cluster of miRNAs (including miR-363–3p, miR-20b-5p and miR-106a-5p) drives NEPC via pleiotropic regulation of multiple NEPC drivers.
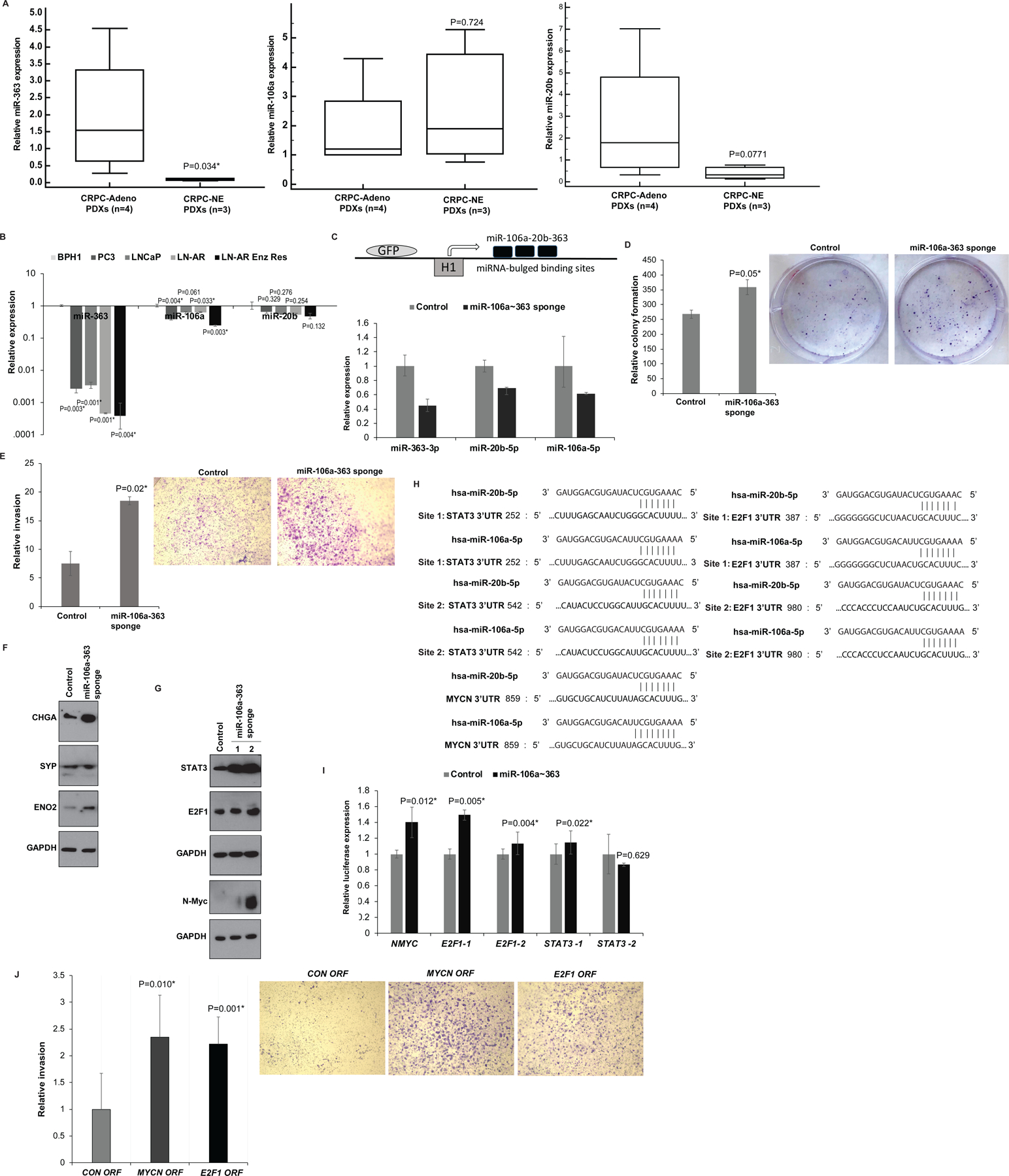
A. Real time PCR analyses of miR-363 (left panel), miR-106a (middle panel) and miR-20b (right panel) in CRPC-NE (LuCaP 49, 145.1 and 145.2) vs CRPC-Adeno PDX models (LuCaP 70, 78, 81 and 92). RNU48 was used as an endogenous control.
B. Real time PCR analyses of miR-363 (left panel), miR-106a (middle panel) and miR-20b (right panel) in benign prostatic hyperplasia cell line (BPH1) and PCa cell lines (PC3, LNCaP, LNCaP-AR, LNCaP-AR-Enzalutamide resistant). RNU48 was used as an endogenous control.
C. Upper panel: Schematic representation of miR-106a~363 sponge construct used for miR-106a~363 downregulation. This construct includes an engineered RNA molecule with multiple bulged miR binding sites, driven by lentiviral pGreen expression system that employs the RNA polymerase III-driven H1 promoter. Lower panel: Real time PCR based expression of miR-106a, miR-363 and miR-20b after stable transduction of control/miR-106a~363 targeting sponge construct in C42B cells.
D. Relative colony formation ability upon control/miR-106a~363 sponge treated-stable C42B cells.
E. In vitro invasion assay in control/ miR-106a~363 sponge-transduced C42B cells.
F. Western blot analyses for CHGA, SYP and ENO2 in control/miR-106a~363 sponge-transduced C42B cells. GAPDH was used as a loading control.
G. Western blot analyses for indicated proteins in control/miR-106a~363 sponge-transduced C42B cells. GAPDH was used as a loading control.
H. Schematic representation of STAT3, E2F1 and MYCN 3’ UTR regions showing potential miR-106a and miR-20b binding sites.
I. Luciferase reporter assays with control 3’ UTR construct or STAT3/E2F1/MYCN 3’ UTR constructs in control/miR-106a~363 sponge-transduced C42B cells. Luciferase activities (Firefly/renilla) were calculated relative to corresponding control 3’UTR constructs and plotted as relative values. Error bars represent SD.
J. C42B cells were transiently transfected with control/ MYCN/E2F1 ORF constructs for 72 hours followed by in vitro transwell invasion assay.
miR-363 downregulation is an important NEPC alteration that directly regulates Aurora Kinase A
In view of our data showing significant miR-363 downregulation in NE PDX models, we sought to examine its sole functional role in detail. Towards this, we knocked down its expression in C42B and LNCaP cells followed by functional assays (Fig. 7). Transient transfection of these cell lines with anti-miR-363 inhibitor led to decreased miR-363 expression as confirmed by RT-PCR (Fig. 7A). Decreased miR-363 expression resulted in increased clonogenicity (Fig. 7B) and invasiveness (Fig. 7C) of PCa cell lines as compared to control suggesting that miR-363 loss contributes to increased aggressiveness. Importantly, Western blot analyses showed that miR-363 expression attenuation upregulates AURKA and KLF4 in both cell lines (Fig. 7D). The 3’UTR regions of AURKA possess one while that of KLF4 have two potential miR-363 binding sites, denoted as KLF4–1 and KLF4–2 (Fig. 7E, upper panel). To validate these genes as direct targets, we designed AURKA/ KLF4 3’UTR constructs (Table S8) and performed reporter assays in anti-miR-CON/anti-miR-363 transfected C42B/ LNCaP cells (Fig. 7F). Reporter assays with AURKA/KLF4 3’UTR constructs in miR-363-inhibited C42B/LNCaP cells led to induction of these target genes as compared to corresponding control constructs though induction at site 2 of KLF4 was not statistically significant. To verify that these effects are because of direct anti-miR-363 interaction with the corresponding binding sites, we mutated the putative binding site/sites in AURKA/KLF4 3’UTRs which significantly prevented reporter upregulation upon miR-363 inhibition as compared to corresponding controls (Fig. 7F). In view of these data, we examined the correlation of miR-363 with these target genes in SU2C cohort of PCa (33). Interestingly, we found a significant inverse correlation between miR-363 expression and AURKA mRNA (R=−0.1940, P=0.00095) (Fig. 7G) suggestive of a direct miR-363 regulatory control of AURKA. In addition, we also examined the correlation of miR-363 expression with AURKA in Taylor et al cohort of prostate adenocarcinomas (30) (Fig. 7H) and found a significant inverse correlation, validating miR-363 control of AURKA expression. To further consolidate the interplay of miR-363 with AURKA, we performed ‘phenocopy’ and ‘rescue experiments’. “Phenocopy’ experiments were performed in LNCaP (left panels) and C42B cells (right panels). Overexpression of AURKA ORF in both cell lines led to increased invasiveness similar to the effects of miR-363 inhibition (Fig. 7I). ‘Rescue’ experiments were performed in LNCaP cells wherein cells were transiently transfected with anti-miR-363 construct combined with CON/ AURKA siRNA constructs (Fig. 7J). followed by functional assays (Fig. 7K). As a control, cells were co-transfected with anti-miR-CON alongwith CON siiRNA. AURKA knockdown in miR-363 inhibited cells led to decreased invasiveness of LNCaP cells, validating the role of anti-miR-363 mediated AURKA upregulation in driving invasiveness of PCa cells.
Fig. 7. miR-363 downregulation is an important alteration in NEPC that directly regulates Aurora Kinase A.
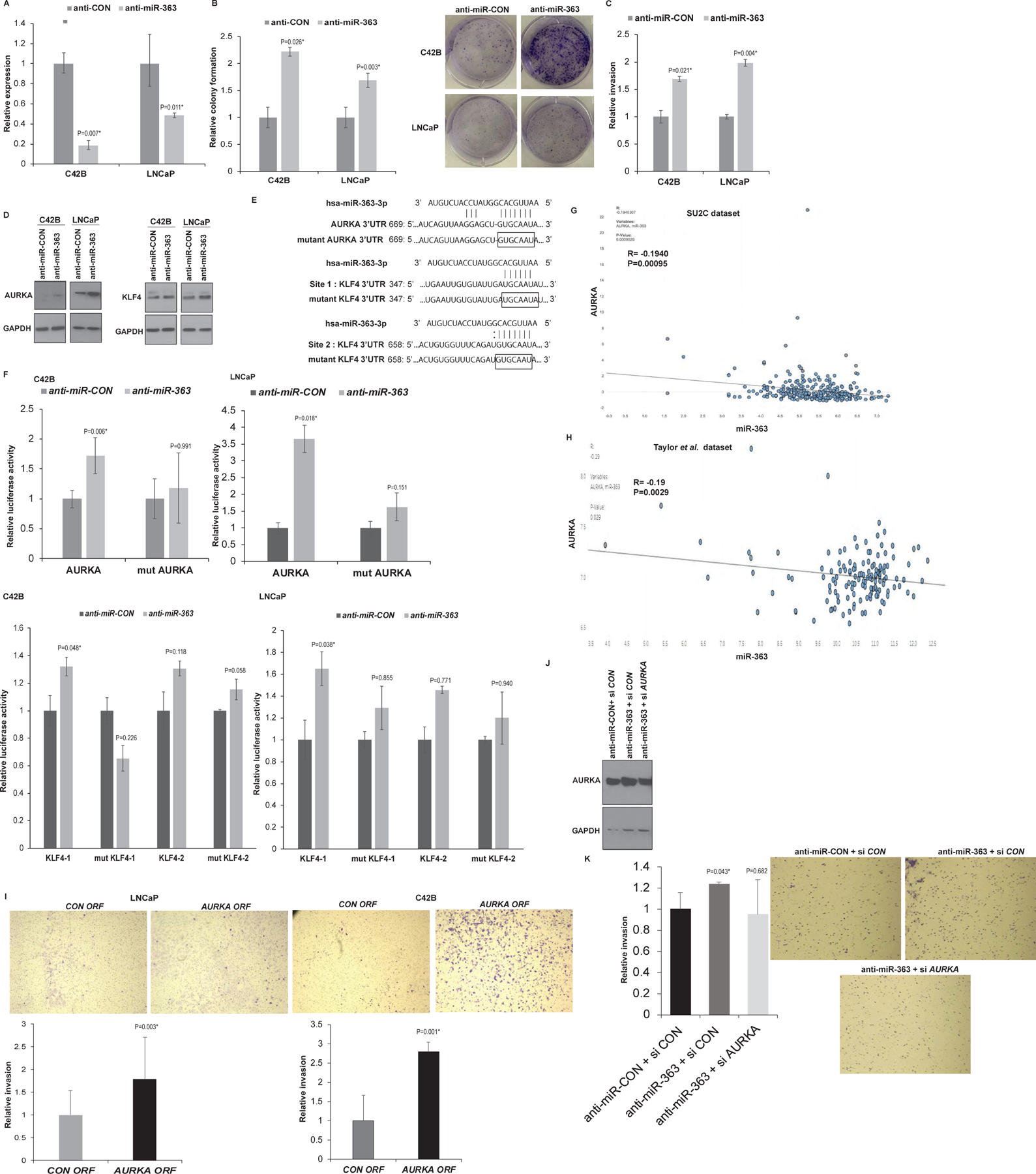
A. Real time PCR analyses of miR-363 expression in C42B and LNCaP cell lines following transient transfection of these cell lines with anti-miR-363 inhibitor/anti-miR-Control inhibitor. RNU48 was used an endogenous control.
B. Colony formation assay and C. in vitro invasion assay upon miR-363 inhibition as compared to control.
D. Western blot analyses for indicated proteins in transfected cells. GAPDH was used as a loading control.
E. Schematic representation of 3’UTR regions of AURKA and KLF4 showing potential miR-363 binding site/sites.
F. Luciferase reporter activity assays with reporter constructs carrying cloned 3’UTR binding sites (wt/mutant) of AURKA (upper panels) and KLF4 (lower panels) in anti-miR-CON/anti-miR-363 transfected C42B and LNCaP cells. Luciferase activities (Firefly/renilla) were calculated relative to corresponding control 3’UTR constructs and plotted.
G-H. Correlation of miR-363 with AURKA in; G. SU2C dataset (33) and H. Taylor et al dataset (30) of prostate adenocarcinomas.
I. ‘Phenocopy’ experiment in LNCaP (left) and C4B cells (right) transiently transfected with control/ AURKA ORF constructs for 72 hours followed by in vitro transwell invasion assay.
J-K. ‘Rescue’ experiment in LNCaP cells wherein cells were transiently transfected with anti-miR-CON or anti-miR-363 construct combined with control (CON siRNA) or AURKA siRNA. After 72 hours, cells were harvested and subjected to functional assays. J. Western blot analyses of AURKA in transfectants. GAPDH was used as a loading control. K. In vitro transwell invasion assay in transfected cells. *P<0.05.
Upregulation of miR-301a and miR-375 drive PCa NED states
To confirm NGS data, we performed real time PCR analyses of miR-301a-3p and miR-375 in CRPC-NE (LuCaP 49, 145.1 and 145.2) vs CRPC-Adeno PDX models (LuCaP 70, 78, 81 and 92) (27) (Fig. 8A) that confirmed specific overexpression of these miRNAs in CRPC-NE. Expression analyses in prostate cell lines (Fig. 8B) showed that as compared to BPH1 cells (34) and PCa cell lines PC3 and LNCaP-AR, LNCaP-AR-ENZR and NCI-H660 cells show significantly increased expression of these miRNAs pointing to their potential roles in NEPC. To examine their functional roles, we transfected miR-375/miR-301a/control miRNA mimics transiently in LNCaP and C42B cells followed by functional assays. Following transfections, real time PCR analyses confirmed miR-375/miR-301a overexpression in miR-375/miR-301a-mimic transfected cells as compared to control cells (Fig. 8C). Ectopic expression of these miRNAs in these cell lines led to increased invasiveness and migratory ability of PCa cells as compared to corresponding controls (Fig. 8D). Importantly, miR-375 and miR-301a could drive induction of NE genes ENO2, SYP and CHGA as analyzed by real time PCR and/or Western blotting (Fig. 8E, 8G, 8H). We further probed the signaling molecules that may mediate induction of tumor aggressiveness and NE genes. Interestingly, we found that miR-375 overexpression induced Akt and Src expression in PCa cell lines as compared to controls (Fig. 8F). miR-301a was found to repress AR expression (Fig. 8H).
Fig. 8. Upregulation of miR-375 and miR-301a drive neuroendocrine differentiation states in prostate cancer.
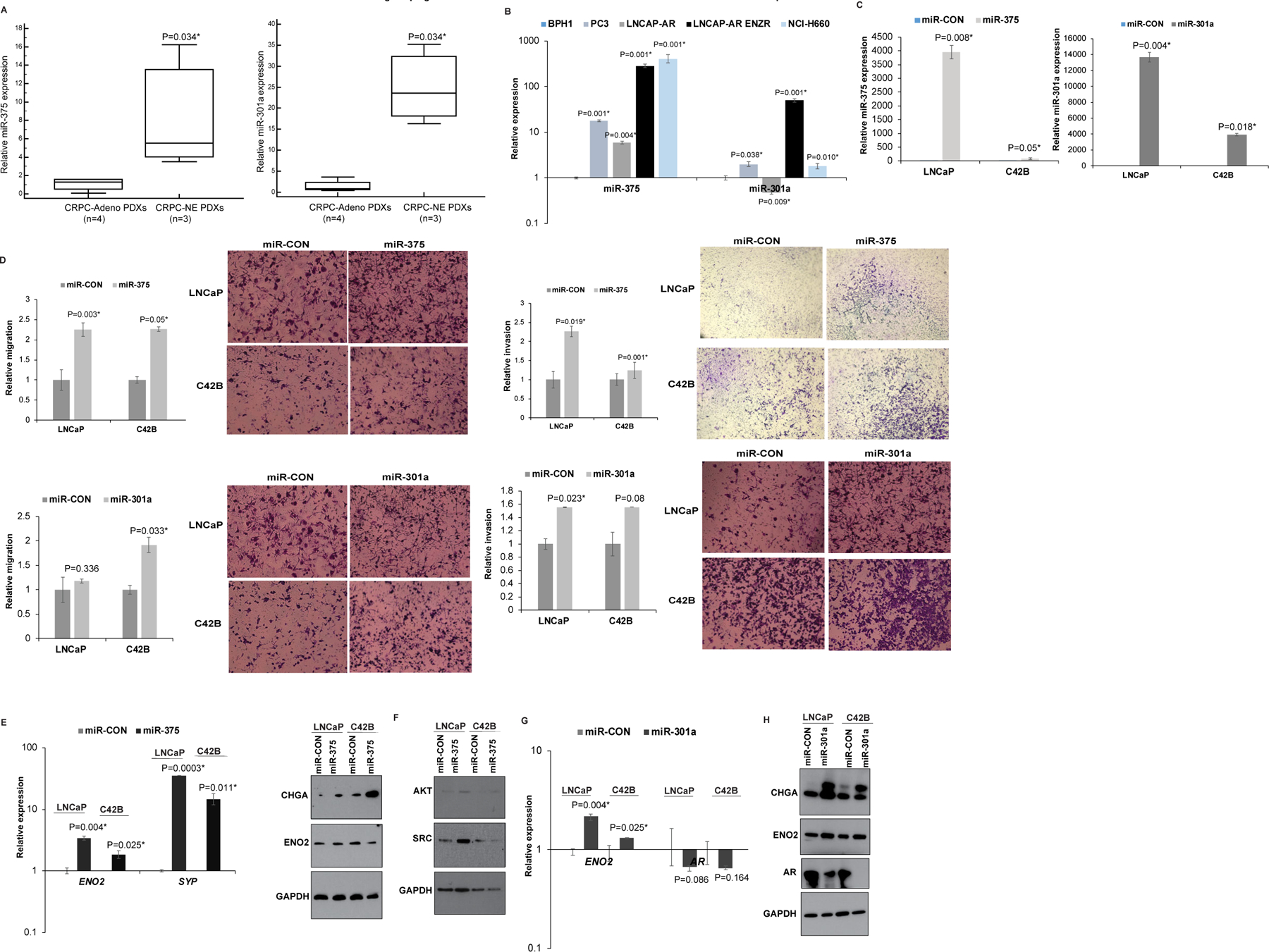
A. Real time PCR analyses of miR-375 (left panel) and miR-301a (right panel) in CRPC-NE (LuCaP 49, 145.1 and 145.2) vs CRPC-Adeno PDX models (LuCaP 70, 78, 81 and 92). RNU48 was used an endogenous control.
B. Real time PCR analyses of miR-375 (left panel) and miR-301a (right panel) in BPH1 and PCa cell lines (PC3, LNCaP, LNCaP-AR, LNCaP-AR-Enzalutamide resistant, NCI-H660). RNU48 was used as an endogenous control.
C. LNCaP and C42B cells were transinetly transfected with control/miR-375/miR-301a mimics for 72 hours followed by functional assays. Real time PCR analyses of miR-375 and miR-301a expression in control/miR-375 transfected or control/miR-301a transfected LNCaP and C42B cells. RNU48 was used an endogenous control.
D. In vitro migration (left panels) and invasion assays (right panels) in control/miR-375 (upper panels) and control/miR-301a-transfected (lower panels) LNCaP and C42B cells.
E. Left panel: Real time PCR analyses of ENO2 and SYP expression in miR-CON/miR-375 transfected LNCaP and C42B cells. GAPDH was used as an endogenous control. Right panel: Western blot analyses of CHGA and ENO2 in miR-CON/miR-375 transfected LNCaP and C42B cells. GAPDH was used as a loading control.
F. Western blot analyses of Akt and Src in miR-CON/miR-375 transfected LNCaP and C42B cells. GAPDH was used as a loading control.
G. Real time PCR analyses of ENO2 and AR expression in miR-CON/miR-301a transfected LNCaP and C42B cells. GAPDH was used as an endogenous control.
H. Western blot analyses of CHGA, ENO2 and AR in miR-CON/miR-301a transfected LNCaP and C42B cells. GAPDH was used as a loading control.
DISCUSSION
Currently, the clinical management of therapy-induced NEPC is challenging as clinicians do not have effective ways to definitively diagnose this variant. Though current NEPC markers SYP, NSE, CHGA and CD56 (7, 8) augment the sensitivity of detecting NED based on morphologic criteria alone, these markers lack specificity and are unable to discriminate between CRPC and NEPC (9). Here, we report for the first time, a robust ‘miRNA- based classifier’, translatable to the clinic, for effective diagnosis of treatment-induced NED in CRPC patients. Importantly, this classifier could not only identify NE states but also have the ability to accurately distinguish between ‘adenocarcinoma’ and ‘NE’ states as confirmed by its application to a validation cohort of mCRPC patients and TCGA primary PCa cohort (29) and Taylor et al (30) cohort of primary and metastatic adenocarcinomas, cross-validating its robustness. Though several groups have recently reported ‘gene expression based molecular classifiers’ for NEPC (9, 13, 35, 36), our ‘miRNA classifier’ is clearly advantageous over other molecular classifiers considering the small size of miRNAs, their resistance to endogenous RNase activity contributing to their stability in formalin-fixed tissues and ease of detection. To our knowledge, our study is the first to report a ‘miRNA based classifier’.
Though several recent large scale genomic studies identified key genomic/epigenomic alterations driving therapy-induced NED (9, 13, 18), the molecular pathogenesis is incompletely understood contributing to late diagnosis, poor prognosis and lack of effective therapies. Dual inactivation of RB1 and p53 along with NMYC overexpression, amplifications of AURKA and EZH2 (9, 13, 15) appear to be critical for transitioning to NEPC (13–15, 17). RB1 acts by restricting expression of E2F1, a TF that regulates cell-cycle G1–S transition. RB1 loss releases E2F1 restriction, promoting NEPC as E2F1 directly regulates expression of CHGA, SYP and NSE (37). We demonstrate here for the first time that NEPC is accompanied by key miRNA alterations, including downregulation of miR-106a~363 cluster, −148a-3p, −203a-3p and upregulation of miR-375 and miR-301a. In view of distinct miRNA expression patterns in CRPC-Adeno vs CRPC-NE states, tumors could be stratified into ‘Adeno’ and ‘NE’ states leading to generation of a ‘miRNA-based classifier’ to distinguish between these two states. Importantly, predominant features of the classifier included miRs-301a-3p, −375 and miR-106a~363 cluster. Validation of the classifier in an independent cohort preserved these top miRNA alterations, suggesting a key role of these miRNA alterations in driving PCa NED. In view of these data, we propose that these miRNA alterations are required in conjunction with key genomic alterations, such as RB1/TP53 loss (13–15, 17) to drive tumor plasticity of advanced PCa to NEPC. IPA analyses with potential miRNA targets and functional studies with miR-106a~363, miR-375 and miR-301a strongly support this notion. Interestingly, miR-106a~363 inhibition or miR-301a/miR-375 overexpression were sufficient to induce neuronal genes, leading to more aggressive phenotypes suggesting that tumor plasticity in advanced PCa is driven likely by multiple miRNA alterations. Importantly, miR-106a~363 was found to pleiotropically regulate multiple NEPC drivers, including N-Myc, E2F1, STAT3 and AURKA. While AURKA has been shown to stabilize N-Myc (14, 15, 25), N-Myc has been shown to co-bind with E2F1 to drive transcription of CHGA, SYP and ENO2 (37). Similarly, STAT3 has been implicated in IL6-induced NED (9). The effects of miR-106a~363 cluster or miR-363 alone on PCa invasiveness were ‘phenocopied’ by MYCN, E2F1 and AURKA constructs, respectively that were rendered insensitive to miRNA inhibition. miR-106a~363 family is located on chromosome Xq26.2, a region reported to be associated with CRPC-NE specific copy number alterations (8, 9). Further, AURKA inhibition in miR-363 inhibited cells could restore the invasive potential of LNCaP cells, consolidating a critical role of AURKA in miR-363-mediated role in PCa invasiveness. In view of these studies and our results, we propose that miR-106a~363 cluster downregulation may constitute a ‘cardinal NE alteration’ that drives NEPC via promoting multiple ‘oncogenic’ events. We propose restoration of this miRNA cluster as a novel targeted therapeutic modality for NEPC.
Analyses for potential target genes for other dysregulated miRNAs suggest that these miRNAs potentially influence crucial signaling axes that drive NEPC including glucocorticoid receptor, p53, PTEN/PI3K/Akt, cell cycle G1/S checkpoint regulation, ERK/MAPK, TGFβ and Wnt/β-cat signaling. Importantly, we found that miR-375 expression upregulates Akt and Src signaling signaling. PI3K/Akt signaling has been shown to promote development of NEPC. Active myristoylated AKT1 coupled with MYCN expression in normal prostate basal cells have been demonstrated to lead to the development of invasive, metastatic CRPC with NE marker expression (38). Further, Src signaling has been shown to interplay with Akt to regulate prostate cancer progression (39). Previous studies have shown an oncogenic role of miR-375 in PCa and a mediator of docetaxel resistance via its ability to suppress targets such as YAP1 and SEC23A (40, 41).
Importantly, IPA analyses showed that 32/70 gene ‘NE signature’ generated by Beltran et al. (13), are potentially targeted by NE- dysregulated miRNAs including RB1 (13, 14), NMYC (15, 42) and EZH2. Similarly, a set of identified dysregulated miRs (let-7a-5p, miR-92a, −421, miR-28) potentially target AR signaling. Though NEPC is recognized to be AR indifferent, the expression of AR and AR-V7 (43, 44), has been reported to be varied (8, 9, 13, 14). Considering predominant loss of AR expression upon NEPC induction, we expect miRNAs directly targeting AR to be upregulated and miRNAs induced by androgens and/or AR to show attenuated expression. In agreement, we found a predominant downregulation of miR-148a and miR-203 upon NED induction that can be attributed to AR signaling axis inhibition as these miRNAs have been shown to possess androgen response elements (AREs) within their cis-regulatory regions (45–48). In addition, miR-301a-3p was found to be upregulated and constituted the most important feature of ‘miRNA classifier’. miR-301a has been previously reported to be oncogenic via its ability to regulate E-cadherin, control EMT (49) and has been reported to be associated with PCa recurrence and metastasis (49–51). Our data suggest that miR-301a overexpression leads to induction of NE states coupled with AR repression. Future studies are warranted to mechanistically dissect the roles of other observed miRNA alterations and to validate their potential targets.
In conclusion, our study defines important molecular alterations in miRNAs that cause lineage switching in advanced PCa. We propose that these key alterations are required in addition to characteristic genomic changes to drive PCa NED, in concert with the notion that multiple hits are required for complete lineage switching in PCa (12). Our study is clinically significant with potential translational implications as we expect that our ‘miRNA predictor’ can help stratify mCRPC patients into those with/without NED and guide treatment decisions. Further, our data with miR-106a~363 cluster and other NEPC promoting miRNA alterations can be potentially exploited for deriving effective targeted NEPC therapies.
MATERIALS AND METHODS
Cell lines and cell culture
Immortalized non-transformed prostate epithelial cell line (BPH1) (34) and PCa cell lines LNCaP, C42B, NCI-H660 (CRL-5813) (28) were obtained from the American Type Culture Collection (ATCC) and cultured under recommended conditions (detailed in supplemental methods). All cell lines were maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C. Prostate cell lines were authenticated by DNA short-tandem repeat analysis. All experiments with cell lines were performed within 6 months of their procurement/resuscitation. All cell lines were tested and found negative for mycoplasma.
Clinical Samples
The study was conducted in accordance with ethical guidelines of US Common Rule and was approved by the institutional committee on human research. Written informed consent was obtained from all patients. FFPE tissues with corresponding clinical information were procured from Prostate Cancer Biorepository Network (PCBN). Cohort 1+2 included human CRPC clinical samples with adenocarcinoma features (CRPC-adeno, mCRPC patients with no evidence of NED) (n=21+18) vs those with neuroendocrine features (CRPC-NE, metastatic AR- patients with therapy-induced NED) (n=6+2) (Table S1). Follow up information included prior therapies for all the clinical samples. RNA from PDX models with CRPC-Adenocarcinoma characteristics (LuCaP 70, 78, 81 and 92) vs those with CRPC-NE alterations (LuCaP 49, 145.1 and 145.2) (27) were procured through PCBN.
Small RNA sequencing
Using 0.2μg of purified total RNA, libraries were generated using an Illumina® TruSeq® small RNA library prep kit (cat no. RS-200–0012) as per manufacturer’s instructions. Set A indices 1–12 and set B indices 13–24 were employed to generate cDNA libraries. Index libraries were equally pooled and sequenced on Illumina NextSeq 500 platform at the institutional molecular core facility using an Illumina NextSeq® 500/550 Mid Output Kit v2 (150 cycles) as per manufacturer’s instructions. Sequencing reads were adapter trimmed and analyzed by BaseSpace Small RNA app (Illumina).
Statistics
All quantified data represents an average of triplicate samples (technical replicates) except sequencing data for clinical samples. Data are represented as mean ± S.E.M or as indicated. Each experiment (except sequencing) was performed at least twice or thrice. Statistical analyses were performed using MedCalc version 10.3.2 or R package (https://www.r-project.org/). Results were considered statistically significant at P ≤ 0.05.
Supplementary Material
Acknowledgements
We sincerely thank Dr. Paul Jedlicka at University of Colorado, Denver for miR-106a~363 sponge and control constructs and Dr. Felix Feng at UCSF for LNCaP-AR-Enzalutamide resistant cell line. This work is supported by the US Army Medical Research Acquisition Activity (USAMRAA) through the Idea Development Award under Award No. W81XWH-18-1-0303 and W81XWH-18-2-0013. Additionally, supported by Award no. W81XWH-18-2-0015, W81XWH-18-2-0016, W81XWH-18-2-0017, W81XWH-18-2-0018, and W81XWH-18-2-0019 Prostate Cancer Biorepository Network (PCBN). Funding support by the National Cancer Institute at the National Institutes of Health (Grant Number RO1CA177984) is also acknowledged. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense or U.S. Army.
Footnotes
Competing interests: The authors declare no competing interests.
REFERENCES
- 1.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15(15):4792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24(18):1967–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hussain M, Fizazi K, Saad F, Rathenborg P, Shore N, Ferreira U, et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N Engl J Med. 2018;378(26):2465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussain M, Saad F, Sternberg CN. Enzalutamide in Castration-Resistant Prostate Cancer. N Engl J Med. 2018;379(14):1381. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97. [DOI] [PubMed] [Google Scholar]
- 6.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aggarwal R, Zhang T, Small EJ, Armstrong AJ. Neuroendocrine prostate cancer: subtypes, biology, and clinical outcomes. J Natl Compr Canc Netw. 2014;12(5):719–26. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal RR, Small EJ. Small-cell/neuroendocrine prostate cancer: a growing threat? Oncology (Williston Park). 2014;28(10):838–40. [PubMed] [Google Scholar]
- 9.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol. 2018;36(24):2492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu CD, Choo R, Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Front Oncol. 2015;5:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki T, Komiya A, Suzuki H, Shimbo M, Ueda T, Akakura K, et al. Changes in chromogranin a serum levels during endocrine therapy in metastatic prostate cancer patients. Eur Urol. 2005;48(2):224–9; discussion 9–30. [DOI] [PubMed] [Google Scholar]
- 12.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest. 2019;129: 4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell. 2016;30(4):563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24(6):820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maina PK, Shao P, Liu Q, Fazli L, Tyler S, Nasir M, et al. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget. 2016;7(46):75585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017;7(1):54–71. [DOI] [PubMed] [Google Scholar]
- 19.Bhagirath D, Yang TL, Tabatabai ZL, Majid S, Dahiya R, Tanaka Y, et al. BRN4 is a novel driver of neuroendocrine differentiation in castration-resistant prostate cancer and is selectively released in extracellular vesicles with BRN2. Clin Cancer Res. 2019;25:6532–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang Q, Li L, Xie H, He D, Chen J, Song W, et al. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (NE) differentiation via altering the infiltrated mast cells --> androgen receptor (AR) --> miRNA32 signals. Mol Oncol. 2015;9(7):1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dankert JT, Wiesehofer M, Czyrnik ED, Singer BB, von Ostau N, Wennemuth G. The deregulation of miR-17/CCND1 axis during neuroendocrine transdifferentiation of LNCaP prostate cancer cells. PLoS One. 2018;13(7):e0200472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding M, Lin B, Li T, Liu Y, Li Y, Zhou X, et al. A dual yet opposite growth-regulating function of miR-204 and its target XRN1 in prostate adenocarcinoma cells and neuroendocrine-like prostate cancer cells. Oncotarget. 2015;6(10):7686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Sun Y, Chen X, Squires J, Nowroozizadeh B, Liang C, et al. p53 Mutation Directs AURKA Overexpression via miR-25 and FBXW7 in Prostatic Small Cell Neuroendocrine Carcinoma. Mol Cancer Res. 2015;13(3):584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nam RK, Benatar T, Amemiya Y, Wallis CJD, Romero JM, Tsagaris M, et al. MicroRNA-652 induces NED in LNCaP and EMT in PC3 prostate cancer cells. Oncotarget. 2018;9(27):19159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell. 2016;29(4):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res. 2019;25(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate. 2017;77(6):654–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai SL, Brauch H, Knutsen T, Johnson BE, Nau MM, Mitsudomi T, et al. Molecular genetic characterization of neuroendocrine lung cancer cell lines. Anticancer Res. 1995;15(2):225–32. [PubMed] [Google Scholar]
- 29.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dylla L, Jedlicka P. Growth-promoting role of the miR-106a~363 cluster in Ewing sarcoma. PLoS One. 2013;8(4):e63032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4(9):721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;162(2):454. [DOI] [PubMed] [Google Scholar]
- 34.Hayward SW, Dahiya R, Cunha GR, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In vitro cellular & developmental biology Animal. 1995;31(1):14–24. [DOI] [PubMed] [Google Scholar]
- 35.Alshalalfa M, Liu Y, Wyatt AW, Gibb EA, Tsai HK, Erho N, et al. Characterization of transcriptomic signature of primary prostate cancer analogous to prostatic small cell neuroendocrine carcinoma. Int J Cancer. 2019;145(12):3453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng S, Yu X. Bioinformatics analyses of publicly available NEPCa datasets. Am J Clin Exp Urol. 2019;7(5):327–40. [PMC free article] [PubMed] [Google Scholar]
- 37.Liu B, Li L, Yang G, Geng C, Luo Y, Wu W, et al. PARP Inhibition Suppresses GR-MYCN-CDK5-RB1-E2F1 Signaling and Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2019;25(22):6839–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018;362(6410):91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta K, Gupta S. Neuroendocrine differentiation in prostate cancer: key epigenetic players. Transl Cancer Res. 2017;6(Suppl 1):S104–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selth LA, Das R, Townley SL, Coutinho I, Hanson AR, Centenera MM, et al. A ZEB1-miR-375-YAP1 pathway regulates epithelial plasticity in prostate cancer. Oncogene. 2017;36(1):24–34. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Lieberman R, Pan J, Zhang Q, Du M, Zhang P, et al. miR-375 induces docetaxel resistance in prostate cancer by targeting SEC23A and YAP1. Mol Cancer. 2016;15(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J Clin Invest. 2019;129:3924–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol. 2017;35(19):2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kojima S, Goto Y, Naya Y. The roles of microRNAs in the progression of castration-resistant prostate cancer. J Hum Genet. 2017;62(1):25–31. [DOI] [PubMed] [Google Scholar]
- 46.Thieu W, Tilki D, de Vere White R, Evans CP. The role of microRNA in castration-resistant prostate cancer. Urol Oncol. 2014;32(5):517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandes R, Hickey T, Tilley WD, Selth LA. Interplay between the androgen receptor signalling axis and microRNAs in prostate cancer. Endocr Relat Cancer. 2019;26:R237–R257. [DOI] [PubMed] [Google Scholar]
- 48.Takayama KI, Misawa A, Inoue S. Significance of microRNAs in Androgen Signaling and Prostate Cancer Progression. Cancers. 2017;9:102 (1-16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nam RK, Benatar T, Wallis CJ, Amemiya Y, Yang W, Garbens A, et al. MiR-301a regulates E-cadherin expression and is predictive of prostate cancer recurrence. Prostate. 2016;76(10):869–84. [DOI] [PubMed] [Google Scholar]
- 50.Damodaran C, Das TP, Papu John AM, Suman S, Kolluru V, Morris TJ, et al. miR-301a expression: A prognostic marker for prostate cancer. Urol Oncol. 2016;34(8):336e13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nam RK, Amemiya Y, Benatar T, Wallis CJ, Stojcic-Bendavid J, Bacopulos S, et al. Identification and Validation of a Five MicroRNA Signature Predictive of Prostate Cancer Recurrence and Metastasis: A Cohort Study. J Cancer. 2015;6(11):1160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.