

Abstract
Rationale:
Fibroblast growth factor homologous factors (FHFs) are key regulators of sodium channel inactivation. Mutations in these critical proteins have been implicated in human diseases including Brugada syndrome, idiopathic ventricular arrhythmias, and epileptic encephalopathy. The underlying ionic mechanisms by which reduced sodium channel availability in Fhf2 knockout mice predisposes to abnormal excitability at the tissue level are not well defined.
Objective:
Using animal models and theoretical multicellular linear strands, we examined how FHF2 orchestrates the interdependency of sodium, calcium, and gap junctional conductances to safeguard cardiac conduction.
Methods and Results:
Fhf2KO mice were challenged by reducing calcium conductance using verapamil or by reducing gap junctional conductance using carbenoxolone or by backcrossing into a connexin 43 heterozygous (Cx43+/−) background. All conditions produced conduction block in Fhf2KO mice, with Fhf2WT showing normal impulse propagation. To explore the ionic mechanisms of block in Fhf2KO hearts, multicellular linear strand models incorporating FHF2-deficient sodium channel inactivation properties were constructed and faithfully recapitulated conduction abnormalities seen in mutant hearts. The mechanisms of conduction block in mutant strands with reduced calcium conductance or gap junction uncoupling are very different. Enhanced sodium channel inactivation due to FHF2 deficiency shifts dependence onto calcium current to sustain electrotonic driving force, axial current flow, and action potential generation from cell-to-cell. In the setting of gap junction uncoupling, slower charging time from upstream cells conspires with accelerated sodium channel inactivation in mutant strands to prevent sufficient downstream cell charging for action potential propagation.
Conclusions:
FHF2-dependent effects on sodium channel inactivation ensure adequate sodium current reserve to safeguard against numerous threats to reliable cardiac impulse propagation.
Subject Terms: Arrhythmias, Electrophysiology, Ion Channels/Membrane Transport, Sudden Cardiac Death, Ventricular Fibrillation
Keywords: Fhf2, Fgf13, fibroblast growth factor, impulse propagation, Shaw–Rudy, sodium channel, arrhythmia, Brugada syndrome, conduction, modeling
Graphical Abstract
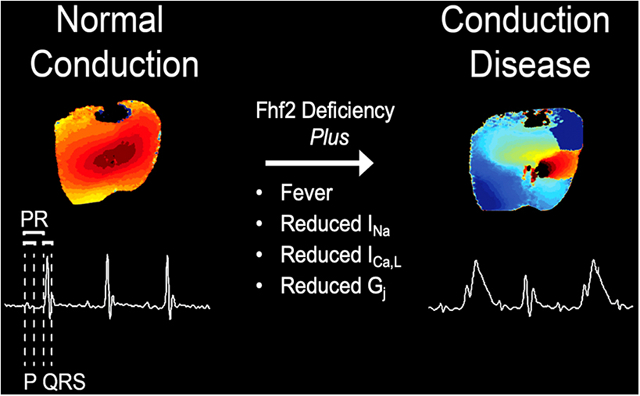
INTRODUCTION
Cardiac impulse propagation reflects a complex interplay between action potential (AP) generation by excitable cells and passive conduction of the excitation wave front from cell-to-cell. A reduction in either membrane excitability or gap junction coupling can adversely affect impulse propagation by decreasing conduction velocity (CV) or causing conduction block, which increases vulnerability to brady and tachyarrhythmias. Although the fundamental importance of reliable impulse propagation is indisputable, the factors that influence conduction safety in myocardial tissue have not been rigorously tested. It is well-accepted that the cardiac sodium current (INa) is the principal determinant of membrane excitability, and therefore conduction, in atrial, Purkinje, and ventricular myocytes. What is less certain, however, is how cardiac conduction is maintained when sodium channel (NaV) availability is reduced in vivo. A major limitation has been the lack of an animal model with sufficiently low membrane excitability in which the multiplicity of factors that influence impulse propagation can be studied. We1 and others2 have recently identified fibroblast growth factor homologous factor 2 (FHF2), also known as fibroblast growth factor 13 (FGF13), as a critical member of the voltage-gated NaV complex that optimizes cardiac excitability and impulse propagation. In this study, we used the Fhf2 mouse model to study the limits of conduction behavior and generate FHF2-aware theoretical linear strands to investigate the ionic basis of conduction failure.
FHF2 belongs to a family of FHFs that bind to the cytoplasmic tails of voltage-gated sodium channels and modulates channel gating properties and trafficking3. Underscoring their importance as regulators of NaV function, mutations in FHFs have been implicated in heritable diseases of excitability, including Brugada syndrome4, atrial and ventricular arrhythmias with sudden cardiac death5, neonatal onset epileptic encephalopathy6 and febrile-onset seizures7. Fhf2 knockout mice (Fhf2KO mice) exhibit marked conduction abnormalities when challenged with the sodium channel blocking drug flecainide or with temperature elevation. At higher temperatures, Fhf2KO mice display a constellation of reversible electrocardiographic (ECG) findings including conduction slowing with eventual conduction block and ECG changes suggestive of Brugada syndrome1, an arrhythmic condition associated with sodium channel loss-of-function8–10. Fhf2KO ventricular myocytes have reduced excitability at higher temperature, which is attributable in part to a hyperpolarizing shift of steady-state inactivation of INa and an accelerated rate of closed-state and open-state NaV inactivation.
Similar to our findings, Wang and colleagues2 reported that their cardiac conditional Fhf2KO mice have conduction defects that become fully apparent with flecainide treatment. In addition, flecainide triggered ventricular arrhythmias in mutant animals. FHF2-deficient cardiomyocytes exhibit altered action potential characteristics consistent with reduced sodium channel availability, including slower maximal upstroke velocity of phase 0 ([dV/dt]max) and reduced action potential amplitude (APA). Defects in the sodium current include a reduction in INa density by ~25% and hyperpolarization of V1/2 of steady state inactivation.
In addition to cardiac conduction defects, FHF2-deficient mice also exhibit temperature sensitive neuronal phenotypes. Fhf2 heterozygous female mice exhibit hyperthermia-induced seizures likely due to impaired excitability of inhibitory neurons of the hippocampus7. Loss of Fhf2 also causes impaired heat nociception by diminishing NaV1.7 current density resulting in reduced excitability and conduction of dorsal root ganglia neurons11, 12 (Marra and M. Goldfarb, unpublished data). The collection of cardiac and neuronal phenotypes of Fhf2KO mice suggest that FHF proteins may have evolved, in part, to antagonize temperature-dependent acceleration of NaV inactivation rates and safeguard excitable tissues such as the heart and brain during transient febrile episodes.
The severe reduction in INa availability in Fhf2KO cardiomyocytes allows us to study the various factors that contribute to conduction behavior in vivo. Shaw and Rudy first utilized the theoretical multicellular fiber to study the impact of reduced membrane excitability or gap junction uncoupling on conduction parameters, including CV, [dV/dt]max, and safety factor (SF)13. SF is a dimensionless parameter calculated as a ratio of charge generated by excitation divided by charge required for depolarization, with SF<1 indicating conduction failure. The Shaw–Rudy model predicts that ICa,L and gap junctional conductance play a larger role in safe impulse propagation when membrane excitability is reduced. Using the Fhf2 mouse model, we validate the observations of Shaw and Rudy in a biological context. We also generate FHF2 aware linear strand models that identify distinct mechanisms whereby ICa,L and Gj contribute to conduction safety.
METHODS
The authors declare that all supporting data are available within the article (and its online supplementary files).
Cardiomyocyte computational model files are available for download at the ModelDB portal (https://senselab.med.yale.edu/modeldb/). All other data and materials, including mouse lines, are available upon corresponding author request.
See Supplemental Material for details on the parameters for the computational model and other experimental procedures.
RESULTS
Fhf2KO mice exhibit increased sensitivity to calcium channel blockade.
To investigate whether ICa,L contributes to conduction safety in the Fhf2KO heart, we challenged Fhf2WT and Fhf2KO mice with the L-type calcium channel blocker, verapamil (15mg/kg)22 and measured conduction parameters using surface ECG. Fhf2KO mice were highly sensitive to verapamil treatment, developing significant conduction abnormalities on ECG. Fhf2KO mice showed uniform lethality due to ventricular asystole with verapamil treatment (Figure 1A,B). Electrocardiographic features included QRS interval prolongation, which is suggestive of conduction block in the His-Purkinje system, and atrioventricular block that ultimately progressed to ventricular asystole (Figure 1B, Online Figure I and Online Table I). In contrast, Fhf2WT mice tolerated verapamil treatment without developing QRS interval prolongation or 2nd or 3rd degree atrioventricular block (Figure 1A,B).
Figure 1. Pharmacological L-type calcium channel blockade produces conduction defects in Fhf2KO hearts.
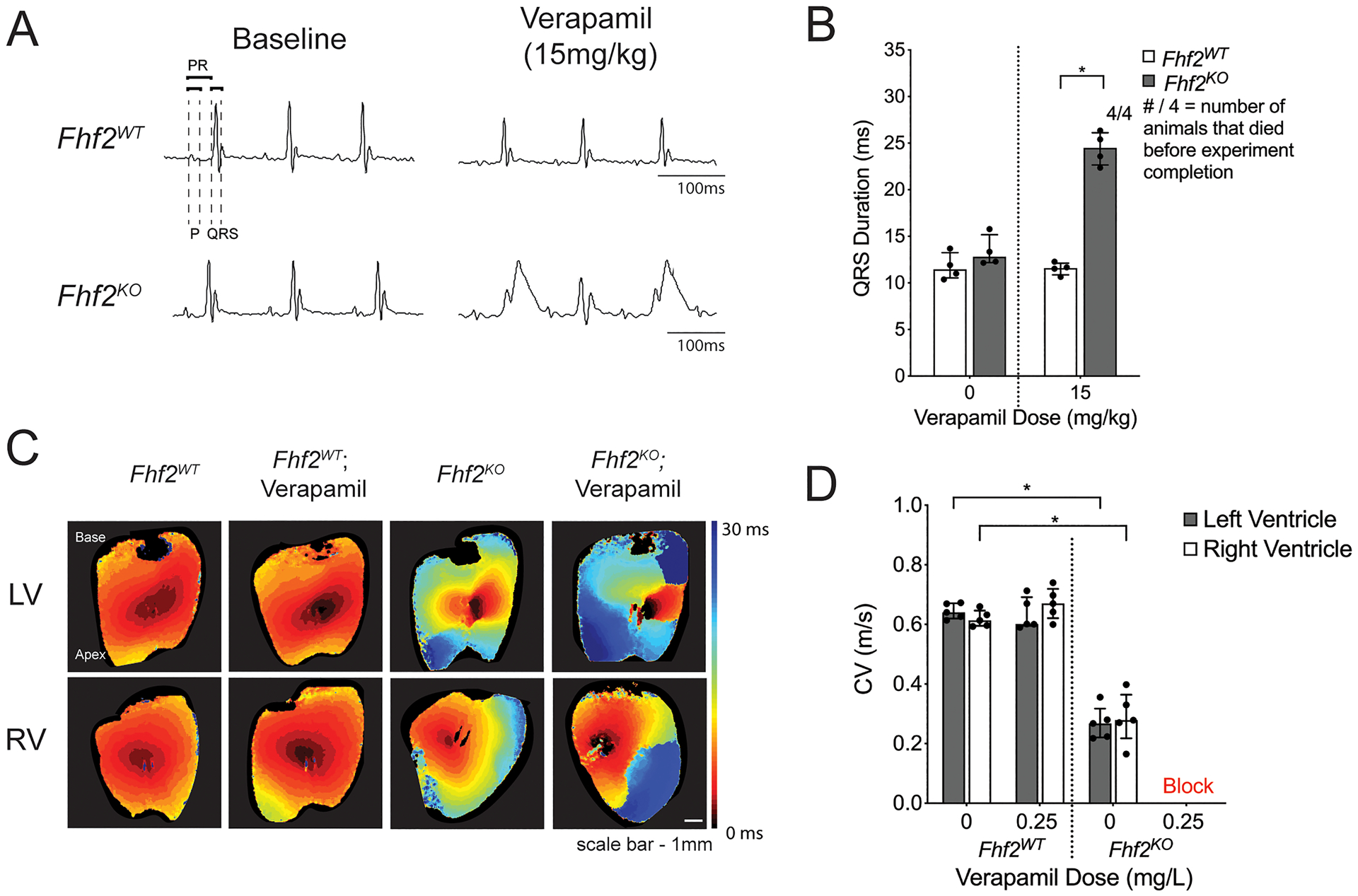
(A,B) In vivo experiments with calcium channel blocker verapamil. (A) Representative surface ECG tracings of Fhf2WT (n=4) and Fhf2KO (n=4) mice subjected to verapamil (0mg/kg or 15mg/kg) via intraperitoneal (IP) injection and observed over 30 minutes. (B) QRS duration plotted against verapamil dose at 0mg/kg or 15mg/kg. QRS measured at 30 minutes or at last measurable time point prior to the onset of complete atrioventricular block. Fhf2WT treated with verapamil displayed no changes in QRS duration, and all mice survived verapamil treatment. Fhf2KO treated with verapamil experienced QRS prolongation and complete atrioventricular block. None of the Fhf2KO mice survived verapamil treatment. (C) Representative epicardial left and right ventricular activation maps. Multiple regions of conduction block were observed in Fhf2KO hearts (n=5) after treatment with verapamil. Fhf2WT hearts (n=5) did not show any change in CV after treatment. Hearts were paced at 100 milliseconds basic cycle length. Isochronal maps were generated based on the time to action potential peak. Isochrones are drawn 5 milliseconds apart. Time-scale index shown in sidebar. (D) Quantification of left and right ventricular epicardial conduction velocity before and after treatment with verapamil. Multiple regions of block precluded quantitative epicardial CV measurements in Fhf2KO hearts treated with verapamil. Data represent median ± interquartile range. *significant p values < 0.05, Wilcoxon signed-rank test was performed for comparisons between paired samples; Mann–Whitney U-test used for comparison of independent variables. See Online Table VIII for individual p-values.
As surface ECG is a poor reflection of ventricular myocardial conduction velocity, optical mapping of Fhf2WT and Fhf2KO hearts using voltage-sensitive dye was performed to examine epicardial impulse propagation and CV at 37°C with and without verapamil treatment (0.25mg/L)18 (Figure 1C,D and Online Table II). Activation maps of the anterior left ventricular (LV) and right ventricular (RV) walls were obtained at a pacing cycle length of 100ms. Fhf2WT hearts displayed normal impulse propagation and normal epicardial CV irrespective of verapamil treatment. Verapamil did not affect transverse (CVmin) or longitudinal (CVmax) conduction in Fhf2WT hearts (Online Table III). Fhf2KO hearts without verapamil displayed reduced conduction velocity, as previously reported1. CV was reduced in both transverse and longitudinal axes in Fhf2KO hearts, although transverse conduction was disproportionately affected (Fhf2WT CVmin = 0.42 m/s [0.38, 0.45] versus Fhf2KO CVmin = 0.14 m/s [0.13, 0.14]; p=0.004 and Fhf2WT CVmax = 0.76 m/s [0.66, 0.87] versus Fhf2KO CVmax = 0.42 m/s [0.35, 0.42]; p=0.004; Online Table III). As a result, the anisotropic ratio (CVmax/CVmin) was significantly increased in Fhf2KO hearts (1.78 [1.73, 1.91] in Fhf2WT hearts versus 3.29 [3.11, 3.54] in Fhf2KO hearts; p=0.004; Online Table III). Fhf2KO hearts treated with verapamil developed conduction block during epicardial ventricular pacing. Epicardial CV was not calculated in verapamil treated Fhf2KO hearts as conduction block precludes accurate calculation in anisotropically conducting tissues.
We next generated action potential duration (APD) maps of intact Fhf2WT and Fhf2KO hearts paced at 100ms (Online Figure IIA–C). Unlike a previous report2 that suggests that APD is prolonged in dissociated ventricular myocytes from cardiac-specific Fhf2 knockout mice, we show that APD 75 and 90 are not significantly different between Fhf2WT and Fhf2KO LV and RV. APD50 is significantly shorter in the RV of Fhf2KO compared to Fhf2WT hearts. (Online Figure IID).
Given a prior report of FHF dependence of voltage-gated calcium current density23, we empirically measured calcium currents in Fhf2WT and Fhf2KO ventricular myocytes (Figure 2). Across a broad voltage spectrum, calcium current activation (Figure 2A), steady-state inactivation (Figure 2B), rate of inactivation (Table 1), and peak (Figure 2A; WT pA/pF = 5.38; KO pA/pF = 5.94) were indistinguishable in Fhf2WT versus Fhf2KO cells. Therefore, the increased dependency of cardiac conduction on ICa,L in mutant hearts is not due to an FHF2-dependent defect of the calcium current. To rule out the possibility that delayed recovery from inactivation of the fast sodium channel might contribute to conduction block in Fhf2KO hearts while pacing at 100ms, whole cell patch clamp recordings performed at −88mV showed recovery from inactivation in Fhf2KO cells was more rapid than that of Fhf2WT cells (Online Figure III).
Figure 2. Measured calcium currents are similar in Fhf2WT and Fhf2KO cardiomyocytes.
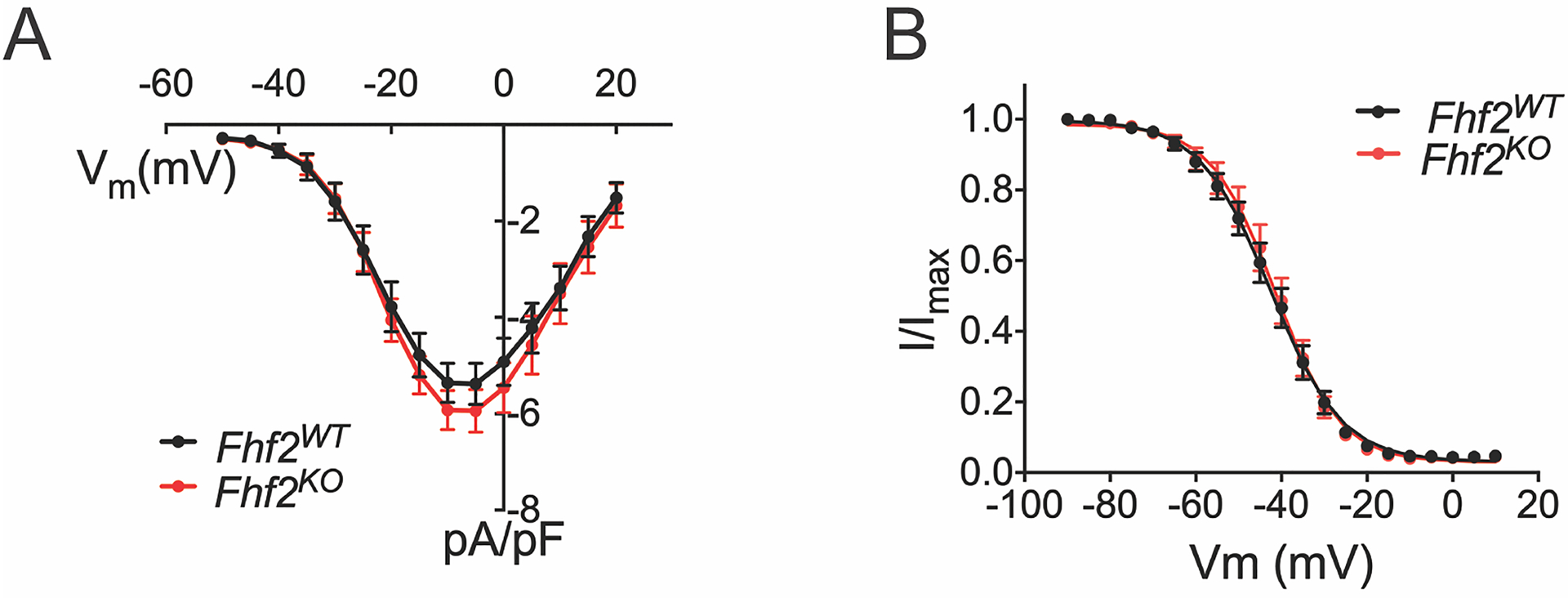
(A) Calcium current density as a function of voltage. 200 ms voltage pulses were applied from holding Vm −90 mV to between −50 mV and +30mV in 5mV steps. Fhf2WT and Fhf2KO cardiomyocytes do not differ in peak calcium current density or voltage dependence of channel activation. (B) Cardiomyocyte voltage-gated calcium channel steady-state inactivation. Available ICa at 25°C after 500 ms conditioning at voltages spanning −130 to −20 mV is expressed as fraction of maximal ICa. Voltage dependence of inactivation does not significantly differ between Fhf2WT and Fhf2KO cardiomyocytes. (n=3 mice in each cohort, 10 cells/mouse). Data presented as mean ± standard error of the mean. Two-sample Kolmogorov-Smirnov test was used to compare current behavior between genotypes. Online Table VIII for individual p-values.
Table 1: Calcium channel inactivation rates.
The rate of ICa decline at −20mV, −10mV, or 0mV was fitted to a two-component exponential decay. The fast and slow time constants and the fraction of current associated with each decay component do not significantly differ between Fhf2WT and Fhf2KO cardiomyocytes at any tested voltage. (n=2 mice in each cohort, N=8 Fhf2WT cells, N=9 Fhf2KO cells). Data presented as mean ± standard error of the mean. Mann–Whitney U-test used for comparison of independent variables. See Online Table VIII for individual p-values.
| WT (N=8) | KO (N=9) | WT (N=8) | KO (N=9) | WT (N=8) | KO (N=9) | WT (N=8) | KO (N=9) | |
|---|---|---|---|---|---|---|---|---|
| Voltage (mV) | Tau1 Fraction | Tau1 Fraction | Tau1 (ms) | Tau1 (ms) | Tau2 Fraction | Tau2 Fraction | Tau2 (ms) | Tau2 (ms) |
| −20 | 0.53 +/− 0.04 | 0.49 +/− 0.03 | 111 +/− 12 | 114 +/− 12 | 0.48 +/− 0.04 | 0.53 +/− 0.04 | 19.2 +/− 1.7 | 15.8 +/− 2.1 |
| −10 | 0.50 +/− 0.05 | 0.44 +/− 0.05 | 77 +/− 5 | 73 +/− 4 | 0.50 +/− 0.05 | 0.56 +/− 0.06 | 17.9 +/− 1.9 | 14.7 +/− 1.5 |
| 0 | 0.52 +/− 0.06 | 0.45 +/− 0.05 | 73 +/− 5 | 72 +/− 6 | 0.48 +/− 0.06 | 0.55 +/− 0.05 | 20.5 +/− 1.9 | 17.7 +/− 1.8 |
Fhf2KO mice display increased sensitivity to reduced gap junctional conductance.
We next explored whether FHF2 deficiency predisposes to conduction block under conditions of gap junction uncoupling. Fhf2KO and Fhf2WT mice were exposed to the uncoupling agent carbenoxolone (CBX)24, 25 and conduction parameters were measured by surface ECG. Fhf2KO mice developed progressive QRS interval prolongation with CBX treatment in a dose-dependent fashion (Figure 3A,B and Online Table IV). In contrast, Fhf2WT mice were resistant to the maximum tested dose of CBX (120mg/kg). Importantly, loss of FHF2 did not affect Cx43 expression levels or localization to intercalated discs (Online Figure IV). We next mated Fhf2KO mice into a cardiomyocyte-specific Cx43 heterozygous (Cx43 cHet) background. Fhf2KO;Cx43 cHet mice displayed prolonged QRS interval duration compared to Fhf2WT, Cx43 cHet, and Fhf2KO mice, although significance was only present with Cx43 cHet mice (Online Figure V). Sodium current density and voltage dependence of inactivation were indistinguishable between Fhf2WT versus Cx43 cHet ventricular myocytes and Fhf2KO versus Fhf2KO;Cx43 cHet ventricular myocytes (Online Figure VI). Fhf2KO;Cx43 cHet mice were more sensitive to CBX treatment compared to Fhf2KO mice (Figure 3A,B). While Fhf2KO mice tolerated CBX up to 120mg/kg, the maximum tolerated dose for Fhf2KO;Cx43 cHet mice was 90mg/kg (Figure 3B and Online Table IV). At CBX 120mg/kg, all Fhf2KO;Cx43 cHet mice developed marked conduction abnormalities with uniform progression to complete heart block and ventricular asystole.
Figure 3. Fhf2KO hearts have increased sensitivity to reduced gap junctional conductance.
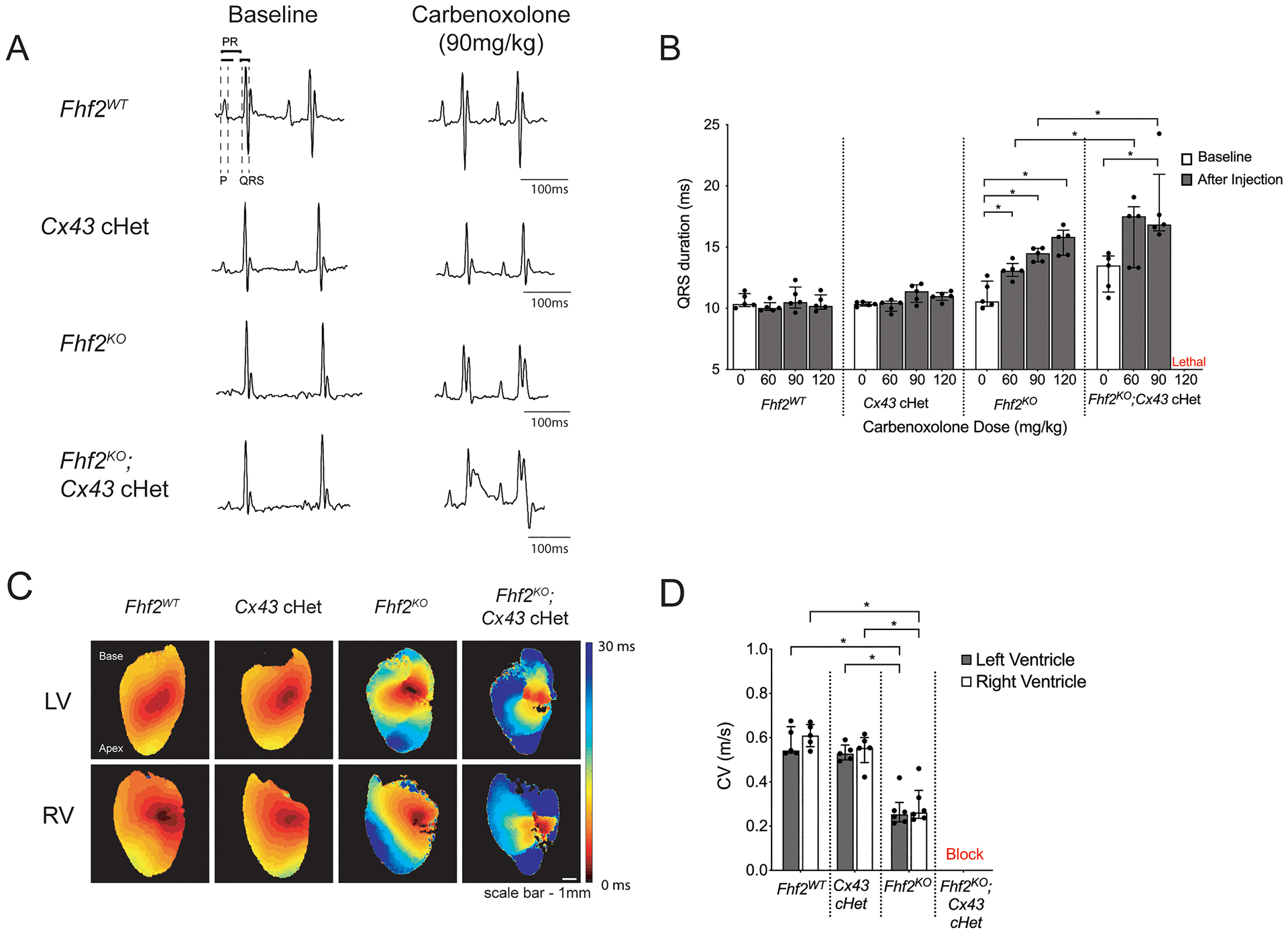
(A,B) In vivo experiments with reduced gap junctional conductance using carbenoxolone or by mating into a cardiomyocyte-specific Cx43 heterozygous (Cx43 cHet) background. (A) Representative surface ECG tracings of Fhf2WT, Cx43 cHet, Fhf2KO, and Fhf2KO;Cx43 cHet mice subjected to carbenoxolone. (B) QRS duration plotted against varying doses of carbenoxolone (60mg/kg, 90mg/kg, and 120mg/kg) via intraperitoneal (IP) injection and observed over 30 minutes (n=5 mice per cohort). (C) Representative epicardial left and right ventricular activation maps. Multiple regions of conduction block were observed in Fhf2KO; Cx43 cHet hearts during epicardial pacing. Hearts were paced at 100 milliseconds basic cycle length. Isochrones are drawn 5 milliseconds apart (n=5 mice per cohort). (D) Quantification of left and right ventricular epicardial conduction velocity. Multiple regions of block precluded quantitative epicardial CV measurements in Fhf2KO; Cx43 cHet hearts. Data represent median ± interquartile range. *significant p values < 0.05, Wilcoxon signed-rank test was performed for comparisons between paired samples; Mann–Whitney U-test used for comparison of independent variables; Kruskal-Wallis test for more than two groups followed by Benjamin-Hochberg post-hoc test for pairwise comparisons. See Online Table VIII for individual p-values.
Optical mapping of Fhf2WT, Cx43 cHet, Fhf2KO, and Fhf2KO;Cx43 cHet hearts was performed to evaluate differences in epicardial impulse propagation and CV at 37°C. Activation maps of the anterior LV and RV walls were obtained at a pacing cycle length of 100ms (Figure 3C,D and Online Table V). Consistent with previously published work1, 20, epicardial CV from Cx43 cHet hearts were not significantly different from Fhf2WT controls, whereas Fhf2KO hearts had markedly slower CV compared to Fhf2WT and Cx43 cHet hearts (0.24 m/sec [0.22, 0.27] vs. 0.54 m/sec [0.53, 0.62], Fhf2KO vs Fhf2WT LV, respectively P < 0.05; 0.24 m/sec [0.22, 0.27] vs. 0.53 m/sec [0.51,0.54], Fhf2KO vs Cx43 cHet LV, respectively P < 0.05). Fhf2KO;Cx43 cHet hearts were markedly abnormal and displayed areas of conduction block, precluding epicardial CV measurements.
Computational models of impulse propagation using Fhf2WT and Fhf2KO cardiomyocytes.
To study the ionic mechanisms of conduction disease in Fhf2KO hearts and probe the interdependencies between INa, ICa,L, and Gj, we generated FHF2-aware computational linear strand models. Our previously described models of Fhf2WT and Fhf2KO ventricular myocytes incorporated sodium conductance Markov models that gave the same peak amplitude I-NaT at 25°C but with different rate constants for inactivation from closed and open states, consistent with empirical voltage clamp recordings of I-NaV that showed a 10 mV hyperpolarizing shift in V1/2 inactivation in KO cells1. Inactivation rates are accelerated with increasing temperature independent of genotype, such that Fhf2 deletion and elevated temperature combine to give even faster channel inactivation from closed and open states, which is reflected in our cardiomyocyte models. Based on our calcium current recordings, we designed an equivalent voltage-gated calcium conductance for both Fhf2WT and Fhf2KO ventricular myocyte models (Online Figure VII).
Single-cell cardiomyocyte model cells were assembled into 11-millimeter linear strands by coupling 111 cells end-to-end with a gap junctional conductance (Figure 4A). Gj was set to 772.8 nS at 37°C based prior empirical measurement of Gj between murine ventricular myocytes at room temperature26 and the temperature Q10 scaling factor of 1.4327. Using this value of Gj, our modeled wild-type action potential conduction velocity at 37°C is similar to that determined by voltage optical mapping on epicardially paced ventricular myocardium1.
Figure 4. Computational models of Fhf2WT and Fhf2KO cardiomyocyte strands.
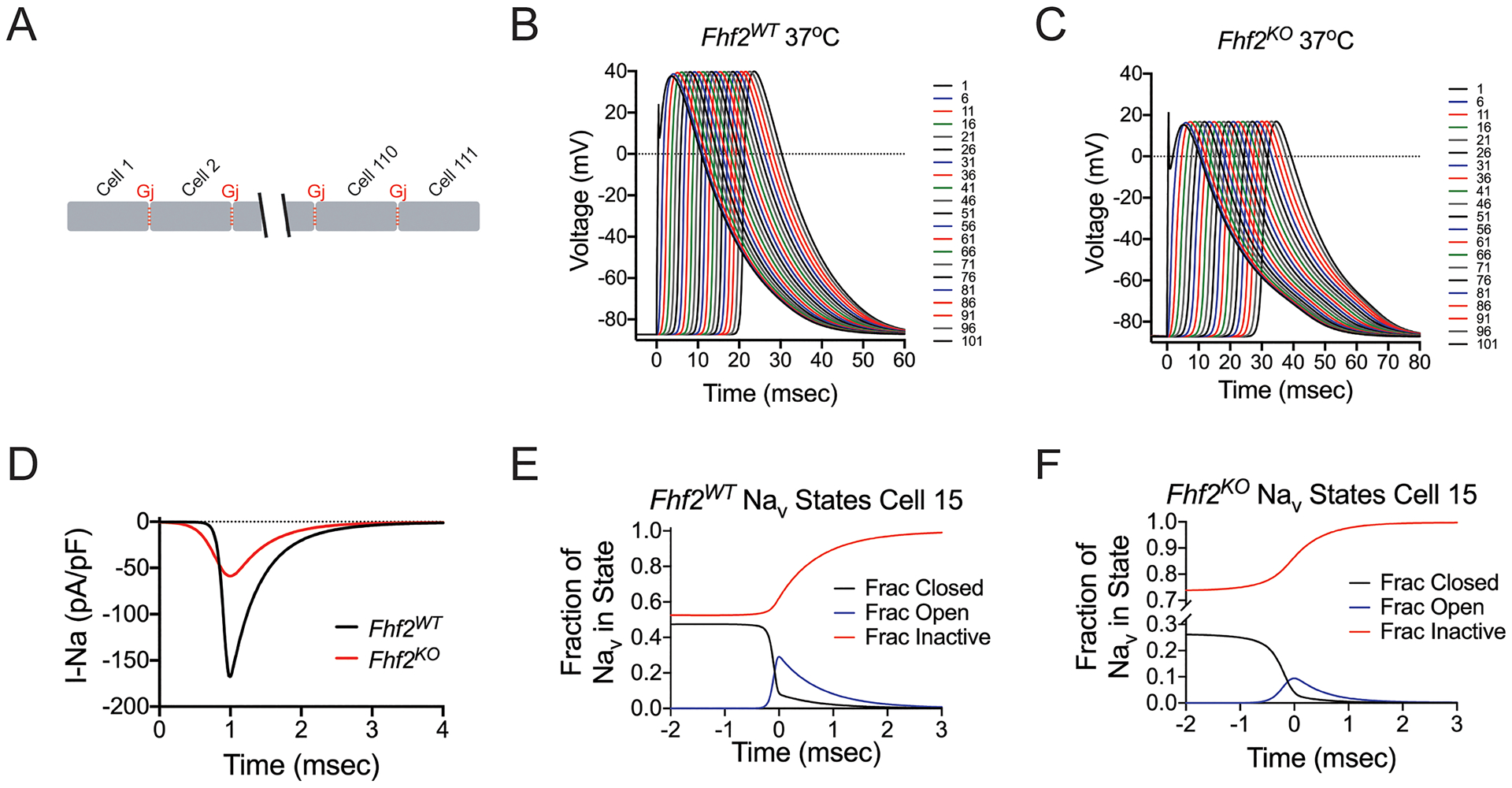
(A) Strand model designed to include 111 cells, each measuring 100 microns aligned end-to-end, connected by gap junctions (Gj = 772.8 nS at 37°C). (B) Simulation of action potential (AP) propagation through strand. 0.5msec current injection into the first cell results in AP propagation through entire Fhf2WT strand. The AP propagates with conduction velocity (CV) = 0.475 mm/msec. (C) Similar injection of current into the Fhf2KO strand results in slowed AP propagation (CV = 0.339 mm/msec) with decreased AP amplitude in all cells. (D) Sodium current influx in cell 50 as a function of time. Sodium current influx is reduced in the Fhf2KO strand, despite the same density of sodium conductance (gNaV). (E) In cell 15 of the Fhf2WT strand, sodium channel availability is approximately 50% at resting potential with peak NaV activation peaking much earlier than NaV inactivation. (F) In cell 15 of the Fhf2KO strand, reduced sodium current is the consequence of approximately 74% NaV steady-state inactivation at the resting potential, further NaV closed state inactivation during the voltage rise preceding peak NaV activation, and faster decay of the current due to faster NaV open state inactivation.
Simulation of action potential conduction through Fhf2WT and Fhf2KO cardiomyocyte model strands at a resting potential of −87 mV was initiated by in silico injection of current for 0.5 msec into the first cell of the strand, using sufficient current to drive maximal activation of sodium channels in that cell. For simulations run at 37°C, both Fhf2WT and Fhf2KO strands faithfully conduct an action potential throughout (Figure 4B,C). The action potential conducts more slowly and has lower amplitude in the mutant strand in comparison to the wild-type (Online Table VI; Fhf2KO CV= 0.339 m/sec, Vpeak = 16.8 mV vs. Fhf2WT CV= 0.475 m/sec, Vpeak= 39.7 mV) due to reduced total sodium influx for each cell in the Fhf2KO vs. Fhf2WT strands (Figure 4D). Reduced sodium current in the Fhf2KO strand in comparison to the Fhf2WT strand is the combined consequences of greater NaV steady-state inactivation at the resting potential, further NaV closed state inactivation during the voltage rise preceding peak NaV activation, and faster rate of NaV open state inactivation (Figure 4E,F, Online Table VII). Calcium currents during action potential propagation were similar in both wild-type and mutant strands (Online Figure VIII), reflecting the identical calcium channel formulations in the two strands.
The Fhf2KO strand model recapitulates increased sensitivity to sodium channel blockade and temperature elevation demonstrated in knockout mice.
We previously reported that Fhf2KO hearts have an increased propensity for conduction block when challenged with the sodium channel blocker flecainide or with hyperthermic stress1. Similar to our in vivo findings, the Fhf2KO strand is far more sensitive to either of these parameter perturbations compared to the Fhf2WT strand (Online Table VI). Conduction block in the strand was defined as simulation in which a portion of the strand failed to depolarize or only weakly and passively depolarized without sodium channel activation. To simulate sodium channel blockade, NaV conductance (gNaV) was reduced in a graded fashion in Fhf2WT and Fhf2KO models (Figure 5A,B). The mutant strand undergoes block when gNaV is reduced by as little as 32%. In contrast, the wild-type strand tolerates a 78% reduction in sodium conductance, which leads to diminished AP amplitude and slowed conduction velocity, but no block is observed. Block occurs at 79% reduction in gNaV (data not shown). Consistent with reduced sodium channel availability as the mechanism of conduction block in the Fhf2KO strand, the maximum upstroke velocity of the action potential ([dV/dt]max) is severely reduced at baseline, and similar to the Fhf2WT strand, decreases monotonically as gNaV is reduced (Figure 5C). SF13 and CV behave similarly to the [dV/dt]max curve when plotted against percentage of gNaV (Figure 5D,E). The Fhf2KO strand also recapitulates the response of mutant hearts to hyperthermic stress, with CV slowing upon temperature elevation1 prior to conduction block when simulation temperature reaches 41°C, while the Fhf2WT strand tolerates temperatures up to and above 45°C with accelerating CV (Figure 5F–H).
Figure 5. Fhf2KO strands are susceptible to conduction failure with reduction in gNaV and elevated temperatures.
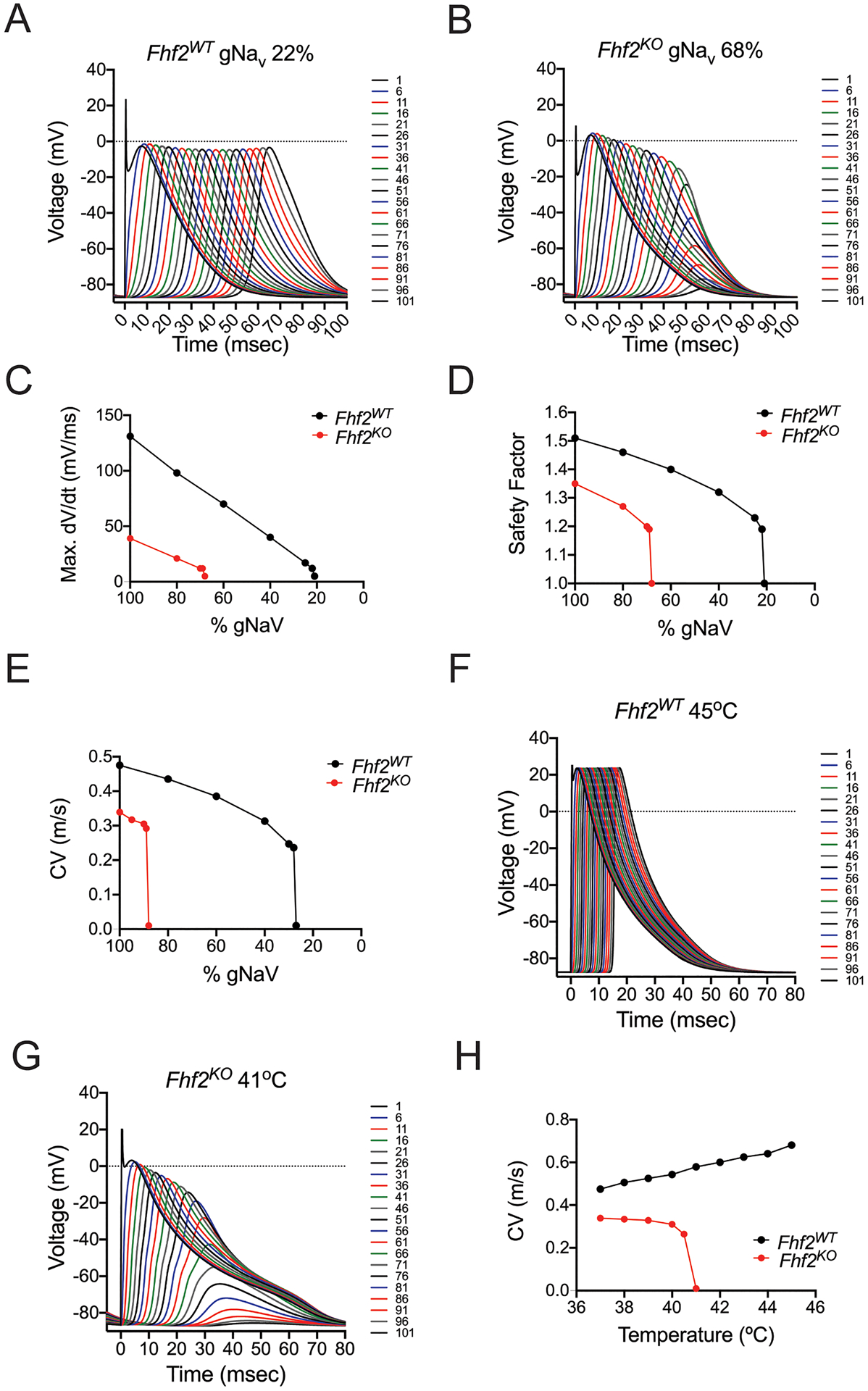
(A) In Fhf2WT strand, a 78% reduction in sodium conductance leads to diminished action potential (AP) amplitude and slowed conduction velocity, but no block is observed. (B) In Fhf2KO strand, a 32% reduction in sodium conductance leads to progressive decay in AP amplitude, and conduction block. (C) [dV/dt]max for Fhf2WT and Fhf2KO strands plotted versus reductions in gNaV. For each strand, [dV/dt]max declines as gNaV decreases until point of failure. (D) Safety factor for Fhf2WT and Fhf2KO strands plotted versus reductions in gNaV. SF declines as gNaV decreases until point of failure. (E) Conduction velocity for Fhf2WT and Fhf2KO strands plotted versus reductions in gNaV. For each strand, CV declines as gNaV decreases until point of failure. (F) The Fhf2WT strand faithfully conducts action potentials when temperature is increased to 45°C. (G) The Fhf2KO strand fails to conduct at 41°C due to faster rates of open and closed state inactivation. (H) Conduction velocity for Fhf2WT and Fhf2KO strands plotted versus rising temperature. CV increases with temperature in the Fhf2WT strand, while decreasing in the Fhf2KO strand until failure temperature.
The Fhf2KO strand model is dependent on calcium current for safe impulse propagation.
We next explored the mechanism underlying verapamil-induced conduction block in Fhf2KO hearts. Action potential conduction through the Fhf2KO cardiomyocyte strand model is characterized by reduced sodium current flux and a greater percentage of the total inward current carried by calcium ions. Therefore, we suspected that mutant strand models would show enhanced susceptibility to conduction block in response to conditions that reduce calcium conductance. Consistent with this hypothesis, the wild-type strand faithfully conducted without calcium conductance but the Fhf2KO strand did not (Figure 6A, B, Online Table VI). The mutant strand underwent progressive decay of the action potential amplitude followed by block if the calcium conductance was reduced to 54% its initial density (Figure 6B). In the absence of FHF2, the contribution of ICa,L to action potential amplitude is essential to augment electrotonic driving force between the upstream excited cell and downstream cells. To determine the durability of the ICa,L contribution to action potential regeneration in the Fhf2KO strand, we simulated a condition where gCaV was set to zero starting from cell 51 and beyond. Under these conditions, the decline of INa was not seen until cell 52, suggesting that ICa contributes to forward currents to ensure sufficient NaV activation in several cells downstream (Figure 6C). The Fhf2WT strand displayed virtually normal NaV activation throughout the fiber despite the abrupt loss of ICa (Figure 6C).
Figure 6. Fhf2KO computational strand has increased dependence on calcium currents and gap junctional conductance.
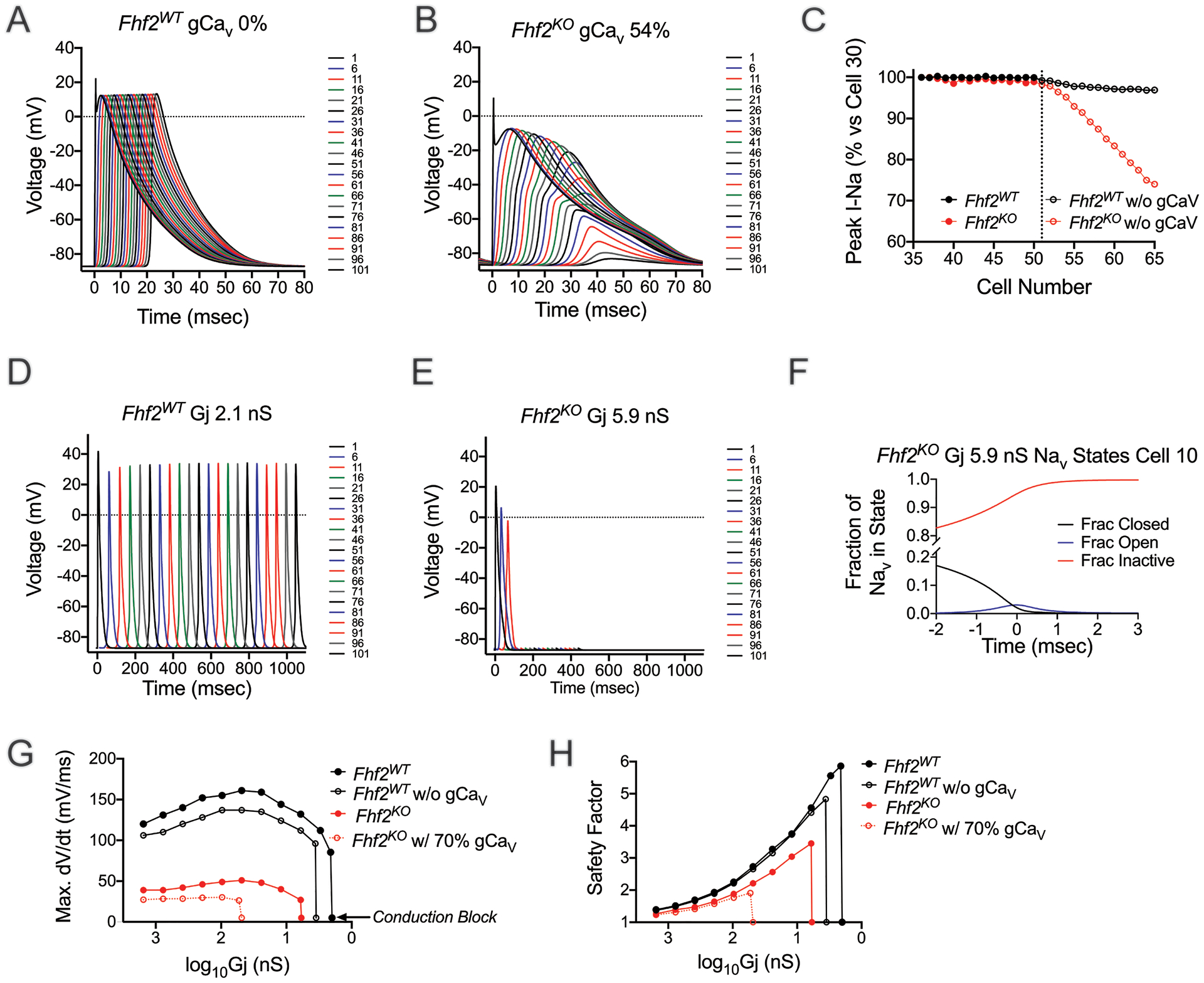
(A) Fhf2WT strand model shows no dependence on calcium currents for the propagation of the AP. (B) Reduction of calcium currents to 54% leads to progressive decay of the AP and block in Fhf2KO strand. Simulated data are consistent with in vivo administration of verapamil. (C) In the Fhf2KO strand, when calcium current is set to zero at and beyond cell 51, not until cell 52 is a decrease in peak sodium current noted, with a steady decline thereafter until impulse propagation failure. In contrast, the Fhf2WT strand is virtually unaffected by reductions in calcium currents. (D) In the Fhf2WT strand, reduction in gap junctional conduction to 2.1 nS dramatically slows conduction, but does not lead to block. Block occurs at Gj of 2.0 nS (data not shown). (E) Reduction of Gj to 5.9 nS in the Fhf2KO strand leads to block. (F) In the Fhf2KO strand with reduced junctional conductance as in panel E, poor charging from upstream cells results in greater NaV channel inactivation and diminished INa. (G,H) Fhf2KO strand demonstrates increased reliance of ICa,L and Gj for safe conduction. (G) A biphasic relationship exists between maximum upstroke velocity ([dV/dt]max) and junctional currents in both Fhf2KO and Fhf2WT strands, with Fhf2KO strand showing markedly reduced maximum upstroke velocity and increased dependence on Gj at baseline conditions. Elimination of calcium currents in Fhf2WT model only slightly reduces [dV/dt]max at all Gj values and has virtually no impact on the threshold value of Gj causing block. By contrast, reduction of calcium conductance in the Fhf2KO model causes a far greater left shift in the threshold value of Gj inducing block. (H) Safety factor rises as junctional conductance decreases until nearing uncoupling values that cause conduction block. Reduction in calcium currents has only minimal effect on SF in both Fhf2WT and Fhf2KO strands at Gj values sufficient for safe conduction, while more dramatically impacting the Gj for conduction block in the Fhf2KO strand.
The Fhf2KO strand model demonstrates an increased sensitivity to gap junctional uncoupling.
Based on sodium channel availability at resting potential and more rapid open and closed state NaV inactivation prior to peak activation, we predicted that Fhf2KO models would have a greater sensitivity to junctional uncoupling. Indeed, the Fhf2KO strand model recapitulates the enhanced sensitivity to junctional uncoupling seen in vivo (Online Table VI). The Fhf2KO strand undergoes conduction block at Gj = 5.9 nS, while the Fhf2WT strand still conducts at Gj 2.1 nS (Figure 6D,E). The accelerated closed state NaV inactivation in the mutant strand enhanced its sensitivity to junctional uncoupling. The slow charging process from upstream cells due to junctional uncoupling provided for greater NaV inactivation and markedly reduced INa availability (Figure 6F).
We next examined [dV/dt]max and SF when plotted against Gj (Figure 6G,H). The Fhf2WT strand displays a rise in [dV/dt]max and SF as Gj is reduced, which reflects preservation of charge in the upstream cell and slower subthreshold depolarization in the downstream cell as coupling between cells decreases. As cells reach near isolated conditions, [dV/dt]max and SF decline as the upstream cell fails to charge the neighboring downstream cell. These relationships also hold true for the mutant strand, although the curves initiate at a lower starting point and fail at significantly higher Gj.
To investigate the role of gCaV in Fhf2WT and Fhf2KO strands for conduction safety when Gj is reduced, we plotted [dV/dt]max and SF against Gj with 0% gCaV in the Fhf2WT strand and 70% gCaV in the Fhf2KO strand. In the absence of gCaV, the profile of [dV/dt]max and SF vs. Gj in the Fhf2WT strand is minimally affected (Figure 6G,H), as INa is the principal component driving upstroke velocity and SF of the action potential. ICa makes a small contribution to depolarizing current in the Fhf2WT strand, such that gCaV removal slightly left-shifts the Gj point at which junctional uncoupling causes failure. In the Fhf2KO strand, ICa makes a greater contribution to the overall depolarizing current, markedly increasing sensitivity to junctional uncoupling. A 30% reduction in gCaV left-shifts the Gj point at which junctional uncoupling causes failure of [dV/dt]max and SF by more than 8-fold (Figure 6G,H).
The increased sensitivities of the Fhf2KO strand are due to steady state and dynamic reductions to sodium conductance availability.
In order to better understand the individual factors leading to conduction failure in Fhf2KO mice, a third model was created, termed: Fhf2WTNaVHYPO. Fhf2KO mice possess reduced NaV availability as a consequence of a hyperpolarizing shift in NaV steady state inactivation and an increased rate of channel inactivation1. This third Fhf2WTNaVHYPO model was designed by reducing the sodium conductance density of the Fhf2WT model to 49% such that the Fhf2WTNaVHYPO and Fhf2KO models generate the same peak sodium current upon step depolarization from the −87 mV resting potential (Online Table VII). Simulations were run to tease out if the dramatic phenotypes observed in vivo were a consequence of the reduced sodium currents alone, or some additional factor, specifically the gating properties of the channel.
The Fhf2WTNaVHYPO strand conducts safely at 37°C with AP amplitude and CV less seriously impaired than in the Fhf2KO strand (Online Table VI). In comparison to the Fhf2KO strand, safe conduction through the Fhf2WTNaVHYPO strand is less sensitive to uncoupling (failure at Gj= 2.9 nS; Online Table VI) or reduction in gNaV density (failure upon reduction to 42%; Online Table VI). Furthermore, the Fhf2WTNaVHYPO strand safely conducts when temperature is raised to 45°C or when gCaV is eliminated (Online Table VI). These findings support the contention that accelerated sodium channel inactivation in the absence of FHF is an important contributor to the multitude of conduction block sensitivities in the Fhf2KO heart.
DISCUSSION
Here we identify FHF2 as an essential component of the cardiac sodium channel complex that ensures INa availability is sufficient for optimal conduction performance in the murine heart. FHF2-dependent effects on cardiac sodium channel inactivation provide adequate depolarization and kinetic reserve of the sodium current to withstand various threats to safe conduction. We show using both in vivo and in silico approaches that FHF2 deficiency increases vulnerability to propagation failure when challenged with temperature elevation or reductions in sodium, calcium, or junctional conductance. These results not only validate the findings of the original Shaw–Rudy model but also identify FHFs as the principal modulator of INa availability that ensures propagation safety.
Although it is unknown precisely when FHFs first appeared in the cardiovascular or nervous systems on an evolutionary timescale, FHFs may have conferred a survival advantage in two ways: 1) to enable INa dynamics to be modulated rapidly and in a cell-type specific manner to optimize signal processing in neural networks28, 29 and 2) to ensure sufficient depolarization and kinetic reserve of INa to protect the heart and nervous system against fever, an essential response to infection. In the heart, Fhf2KO mice develop profound conduction abnormalities, Brugada-like ECG changes, ventricular contractile dyssynchrony30, and asystole with temperature elevation1. In the nervous system, Fhf2KO mice develop hyperthermia-induced seizures attributable to impaired excitability of inhibitory hippocampal neurons7 as well as defects in heat nociception caused by an inability to sustain action potential firing and conduction in dorsal root ganglia neurons in response to noxious heat stimuli11, 12 (C.Marra and M. Goldfarb, unpublished data).
The FHF2-aware strand models recapitulate the temperature-dependent propagation failure seen in mutant mice. Conduction defects in the mutant strand is due to a combination of 74% steady-state NaV inactivation at the resting potential and accelerated closed-state and open-state NaV inactivation that markedly reduces INa. Diminished INa leaves the mutant strand highly vulnerable to reductions in sodium conductance or to temperature elevation. Temperature elevation causes an enhancement of inactivation rate that synergizes with FHF2 loss-of-function effects on NaV inactivation to severely suppress INa.
Our data also demonstrate another critical function of FHF2 in the heart, to decrease the dependency of impulse propagation on ICa,L or junctional conductance. Fhf2KO hearts readily develop conduction block when challenged with either calcium channel blockade or junctional uncoupling. In the absence of FHF2, ICa,L plays an important role in augmenting depolarization reserve by contributing to action potential amplitude to sustain electronic driving force and axial current flow. The mechanism of conduction block in Fhf2KO hearts during junctional uncoupling differs from calcium dependent effects. As the rate of passive charging of downstream cells during the subthreshold depolarization phase in the strand is a function of Gj, uncoupling conspires with more rapid acceleration of NaV inactivation to markedly diminish sodium channel availability in the mutant strand. We show that conduction fails at a ~3-fold higher level of Gj in the Fhf2KO strand compared to the Fhf2WT strand. Therefore, FHF2 provides sufficient INa kinetic reserve by slowing sodium channel closed state inactivation rates, which increases tolerance to transient reductions in gap junctional conductance. By maximizing sodium channel availability, FHF2 ensures that depolarization and kinetic reserve are sufficient so that impulse propagation is dependent on INa and not ICa,L and less sensitive to reduced Gj. This configuration also allows for functionally distinct roles between different ionic currents, where INa is dedicated to conduction and ICa,L is dedicated to intracellular signaling and myocardial contraction31.
The full manifestation of conduction defects in Fhf2KO mice cannot be explained simply by a reduction in sodium conductance density alone. It is rather the constellation of inactivation defects, including the left shift in steady state inactivation and the accelerated rate of open and closed state inactivation, that yields a myocardial substrate that is highly prone to conduction failure. Whereas the Fhf2KO strand developed conduction block when challenged with temperature elevation or reduced gNaV, gCaV, or Gj, the Fhf2WTNaVHYPO strand, which models an equivalent reduction in sodium conductance density to the Fhf2KO strand but with wildtype gating parameters, behaves differently when challenged. Fhf2WTNaVHYPO strand is more resistant to reductions in gNaV or Gj than the Fhf2KO strand and did not block with temperature elevation or elimination of gCaV. Additionally, conduction failure is not a function of delayed recovery from inactivation or prolonged refractoriness in FHF2 deficient hearts.
The disproportionate effect of FHF2 loss-of-function on transverse CV is an intriguing observation. We have previously shown that sodium current behavior in adult cardiac ventricular myocytes differs as a function of subcellular localization30. Peak INa density was larger when measured at the intercalated disc versus at the lateral membrane. Furthermore, a positive shift in steady-state activation, a negative shift in steady-state inactivation, and a slower recovery from inactivation were observed in INa at the lateral membrane. Based on these observations, the majority of sodium channels in the lateral membrane of ventricular myocytes would be predicted to be inactivated at resting potential. Consequently, transverse conduction may be especially vulnerable to the added insult of FHF2 deficiency. Measurements of subcellular sodium current behavior in FHF2KO ventricular myocytes and incorporation of these data into two dimensional models will be required to test this hypothesis. Moreover, such an approach may provide additional insight into the mechanisms of arrhythmogenesis in the Fhf2 knockout mice, which develop spontaneous ventricular arrhythmias when challenged with flecainide2.
Our study identifies FHF2 as the major component of the cardiac sodium channel macromolecular complex that ensures adequate depolarization and kinetic reserve of INa for optimal conduction performance in the murine heart. Remarkably, we show that FHF2 is singularly sufficient to protect against conduction block due to hyperthermic stress and in response to reductions in ICa,L or Gj. It will be of interest to determine how cardiovascular diseases, such as myocardial ischemia or heart failure, influence FHF expression levels or interactions with other components of the cardiac sodium channel complex. Furthermore, given that FHF2 is an X-linked gene, examination of sex-based differences is in order, and will be the focus of ongoing work. Another important question is whether FHF1, the dominant family member expressed in human ventricular myocytes, behaves in a similar manner to FHF2. This has become all the more important as FHF1 (aka FGF12) was reported to be a candidate Brugada syndrome locus4. A missense mutation in FGF12 (Q7R-FGF12) decreased binding to the NaV1.5 C terminus, reducing sodium current density and sodium channel availability. Incorporation of these FHF1-dependent changes on the sodium current into our theoretical multicellular fiber will be an important tool for unraveling disease pathogenesis in these and related patients, and may also be of use in formulating mutation and mechanism-specific therapies. Lastly, our results underscore the importance of NaV inactivation properties in maintaining normal cardiac excitation and conduction and suggest that targeting inactivation may prove a fruitful strategy for enhancing depolarization and kinetic reserve to preserve safe conduction.
Supplementary Material
Animals (in vivo studies)
| Species | Vendor or Source | Background Strain | Sex | Persistent ID / URL |
|---|---|---|---|---|
| Fhf2KO mice | In house generated | 129/svPas | Male/Female | MGI:4434341; Park et al., Nat Com 2016 |
| Cx43flox/+; mice (Gja1tm1Gfi) | In house generated | C57Bl/6J | Male/Female | MGI:3051881; Gutstein et al., Circ Res 2001 |
| α-MHC-Cre mice (Tg(Myh6-cre)2182Mds) | Jackson Labs | FVB/N | Male/Female | MGI:2386742; Agah et al., JCI 1997; https://www.jax.org/strain/011038 |
| Fhf2KO; Cx43flox/+; α-MHC-Cre mice | In house generated | Backcrossed more than four generations on the 129/svPas background | Male/Female | This study |
Antibodies
| Target antigen (antimouse if not indicated otherwise) | Host Species | Vendor or Source Catalog # | Working concentration | Persistent ID / URL |
|---|---|---|---|---|
| Primary Antibodies: | ||||
| CX43 | Rabbit | Sigma Aldrich C6219 | 1:100 | https://www.sigmaaldrich.com/catalog/nroduct/sigma/c6219 |
| N-Cadherin | Mouse | BD Biosciences, 610921 | 1:100 | https://www.bdbiosciences.com/us/annlications/research/stem-cell-research/cancer-research/human/nurified-mouse-anti-n-cadherin-32n-cadherin/n/610921 |
| Secondary Antibodies: | ||||
| Donkey anti-Rabbit, Alexa Fluor 594 | Donkey | Invitrogen, A32754 | 1:400 | https://www.thermofisher.com/antibody/nroduct/Donkey-anti-Rabbit-IgG-H-L-Highly-Cross-Adsorbed-Secondary-Antibody-Polyclonal/A32754 |
| Donkey anti-Mouse IgG, Alexa Fluor 488 | Donkey | Invitrogen, A21202 | 1:400 | https://www.thermofisher.com/antibody/nroduct/Donkey-anti-Mouse-IgG-H-L-Highly-Cross-Adsorbed-Secondary-Antibody-Polyclonal/A-21202 |
DNA/cDNA Clones
| Not applicable |
Cultured Cells
| Not applicable |
Data & Code Availability
| Cardiomyocyte computational model files are available for download at the ModelDB portal: (https://senselab.med.yale.edu/modeldb/) |
other
NOVELTY AND SIGNIFICANCE.
What Is Known?
Cardiac sodium channels are the principal determinant of conduction behavior in the heart.
Fibroblast growth factor homologous factors (FHFs) are important modulators of sodium channels that have been implicated in heart rhythm and seizure disorders.
Fhf2 knockout (Fhf2KO) mice exhibit defects in sodium channel inactivation that give rise to severe conduction defects in the heart.
What New Information Does This Article Contribute?
Changes in sodium channel inactivation due to Fhf2 deficiency increases the dependency of cardiac conduction onto auxiliary factors, including calcium and gap junctional conductances.
FHF2 aware computational models faithfully recapitulate conduction abnormalities seen in mutant hearts and elucidate distinct mechanisms by which sodium, calcium, and gap junctional conductances contribute to conduction in the absence of FHF2.
FHFs are key regulators of sodium channel (NaV) behavior. NaV and FHF mutations have been implicated in human heart rhythm (ie. Brugada syndrome) and seizure (ie. epileptic encephalopathy) disorders. Fhf2KO mice exhibit cardiac conduction disease and arrhythmias when challenged with temperature elevation and sodium channel blockade. Studies at the cellular level have identified that FHF2 modulates NaV inactivation properties to optimize channel availability and membrane excitability. What remains unresolved is how altered NaV gating in Fhf2KO hearts influences conduction properties at the tissue level. Here we show that FHF2KO hearts are vulnerable to conduction block when challenged with a broad range of stressors, including reduced calcium or gap junctional conductance. Using FHF2 aware theoretical linear strands, we define the underlying ionic mechanisms of conduction failure, which differs depending on the pathological insult. Our findings underscore the importance of NaV inactivation properties in maintaining normal cardiac excitation and conduction and suggest that targeting FHF2-dependent NaV inactivation may prove a fruitful strategy for preserving safe conduction in the heart.
ACKNOWLEDGEMENTS
We thank Fang-Yu Liu and Jie Zhang for their technical assistance.
SOURCES OF FUNDING
Supported by National Institutes of Health grants (R01HL142498 to G.I.F. and T32 GM066704 and F31 HL132438 to A.S.) and a Fondation Leducq Transatlantic Network of Excellence Award (G.I.F. and D.S.P.)
Nonstandard Abbreviations and Acronyms:
- FHF2
fibroblast growth factor homologous factor 2
- FGF13
fibroblast growth factor 13
- CV
conduction velocity
- INa
sodium current
- ICa, L
L-type calcium current
- Gj
junctional conductance
- [dV/dt]max
maximum upstroke velocity of the action potential
- SF
safety factor
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
ONLINE MATERIALS AND METHODS
REFERENCES
- 1.Park DS, Shekhar A, Marra C, Lin X, Vasquez C, Solinas S, Kelley K, Morley G, Goldfarb M and Fishman GI. Fhf2 gene deletion causes temperature-sensitive cardiac conduction failure. Nat Commun. 2016;7:12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Tang H, Wei EQ, Wang Z, Yang J, Yang R, Wang S, Zhang Y, Pitt GS, Zhang H and Wang C. Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J Mol Cell Cardiol. 2017;104:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldfarb M Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci. 2012;69:1067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ and Pitt GS. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm. 2013;10:1886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, Higgins RS, Janssen PM, Fedorov VV, Weiss R, Salazar C, Hund TJ, Pitt GS and Mohler PJ. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A. 2015;112:12528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siekierska A, Isrie M, Liu Y, Scheldeman C, Vanthillo N, Lagae L, de Witte PA, Van Esch H, Goldfarb M and Buyse GM. Gain-of-function FHF1 mutation causes early-onset epileptic encephalopathy with cerebellar atrophy. Neurology. 2016;86:2162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puranam RS, He XP, Yao L, Le T, Jang W, Rehder CW, Lewis DV and McNamara JO. Disruption of Fgf13 causes synaptic excitatory-inhibitory imbalance and genetic epilepsy and febrile seizures plus. J Neurosci. 2015;35:8866–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brugada J, Brugada P and Brugada R. The syndrome of right bundle branch block ST segment elevation in V1 to V3 and sudden death--the Brugada syndrome. Europace. 1999;1:156–66. [DOI] [PubMed] [Google Scholar]
- 9.Brugada P and Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–6. [DOI] [PubMed] [Google Scholar]
- 10.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA and Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. [DOI] [PubMed] [Google Scholar]
- 11.Yang L, Dong F, Yang Q, Yang PF, Wu R, Wu QF, Wu D, Li CL, Zhong YQ, Lu YJ, Cheng X, Xu FQ, Chen L, Bao L and Zhang X. FGF13 Selectively Regulates Heat Nociception by Interacting with Nav1.7. Neuron. 2017;93:806–821 e9. [DOI] [PubMed] [Google Scholar]
- 12.Effraim PR, Huang J, Lampert A, Stamboulian S, Zhao P, Black JA, Dib-Hajj SD and Waxman SG. Fibroblast growth factor homologous factor 2 (FGF-13) associates with Nav1.7 in DRG neurons and alters its current properties in an isoform-dependent manner. Neurobiol Pain. 2019;6:100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw RM and Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–41. [DOI] [PubMed] [Google Scholar]
- 14.Hines ML and Carnevale NT. NEURON: a tool for neuroscientists. Neuroscientist. 2001;7:123–35. [DOI] [PubMed] [Google Scholar]
- 15.Bondarenko VE, Szigeti GP, Bett GC, Kim SJ and Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1378–403. [DOI] [PubMed] [Google Scholar]
- 16.Cohen SD, Hindmarsh AC and Dubois PF. CVODE, a stiff/nonstiff ODE solver in C Computers in Physics. 1996;10:138–143. [Google Scholar]
- 17.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H and Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aydin O, Becker R, Kraft P, Voss F, Koch M, Kelemen K, Katus HA and Bauer A. Effects of protein kinase C activation on cardiac repolarization and arrhythmogenesis in Langendorff-perfused rabbit hearts. Europace. 2007;9:1094–8. [DOI] [PubMed] [Google Scholar]
- 19.Gloschat C, Aras K, Gupta S, Faye NR, Zhang H, Syunyaev RA, Pryamonosov RA, Rogers J, Kay MW and Efimov IR. RHYTHM: An Open Source Imaging Toolkit for Cardiac Panoramic Optical Mapping. Sci Rep. 2018;8:2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M and Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–75. [DOI] [PubMed] [Google Scholar]
- 21.Lang D, Glukhov AV, Efimova T and Efimov IR. Role of Pyk2 in cardiac arrhythmogenesis. Am J Physiol Heart Circ Physiol. 2011;301:H975–83. [DOI] [PubMed] [Google Scholar]
- 22.Choi JS and Li X. The effect of verapamil on the pharmacokinetics of paclitaxel in rats. Eur J Pharm Sci. 2005;24:95–100. [DOI] [PubMed] [Google Scholar]
- 23.Hennessey JA, Wei EQ and Pitt GS. Fibroblast growth factor homologous factors modulate cardiac calcium channels. Circ Res. 2013;113:381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosseinzadeh H and Nassiri Asl M. Anticonvulsant, sedative and muscle relaxant effects of carbenoxolone in mice. BMC Pharmacol. 2003;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashizume M, Miyazaki T, Sakimura K, Watanabe M, Kitamura K and Kano M. Disruption of cerebellar microzonal organization in GluD2 (GluRdelta2) knockout mouse. Front Neural Circuits. 2013;7:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao JA, Gutstein DE, Liu F, Fishman GI and Wit AL. Cell coupling between ventricular myocyte pairs from connexin43-deficient murine hearts. Circ Res. 2003;93:736–43. [DOI] [PubMed] [Google Scholar]
- 27.Bukauskas FF and Weingart R. Temperature dependence of gap junction properties in neonatal rat heart cells. Pflugers Arch. 1993;423:133–9. [DOI] [PubMed] [Google Scholar]
- 28.Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ, Copeland NG, Jenkins NA and Nathans J. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc Natl Acad Sci U S A. 1996;93:9850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dover K, Marra C, Solinas S, Popovic M, Subramaniyam S, Zecevic D, D’Angelo E and Goldfarb M. FHF-independent conduction of action potentials along the leak-resistant cerebellar granule cell axon. Nat Commun. 2016;7:12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shekhar A, Aristizabal O, Fishman GI, Phoon CKL and Ketterling JA. Characterization of vortex flow in a mouse model of ventricular dyssynchrony by plane-wave ultrasound using hexplex processing. IEEE Trans Ultrason Ferroelectr Freq Control. 2020;PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liebeskind BJ, Hillis DM and Zakon HH. Evolution of sodium channels predates the origin of nervous systems in animals. Proc Natl Acad Sci U S A. 2011;108:9154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.