

The ECM is an essential component of the tumour microenvironment and plays important functions during metastatic colonisation. Depletion of the matricellular protein TGFBI in MMTV‐PyMT mice normalises the vasculature and reduces hypoxia. As a result, Tgfbi loss leads to a dramatic decrease in CSC content and lung metastasis, unveiling its potential as a novel target in breast cancer.
Keywords: breast cancer, cancer stem cells, hypoxia, metastasis, TGFBI
Abstract
Breast cancer metastasis is a complex process that depends not only on intrinsic characteristics of metastatic stem cells, but also on the particular microenvironment that supports their growth and modulates the plasticity of the system. In search for microenvironmental factors supporting cancer stem cell (CSC) growth and tumour progression to metastasis, we here investigated the role of the matricellular protein transforming growth factor beta induced (TGFBI) in breast cancer. We crossed the MMTV‐PyMT model of mammary gland tumorigenesis with a Tgfbi Δ/Δ mouse and studied the CSC content of the tumours. We performed RNAseq on wt and ko tumours, and analysed the tumour vasculature and the immune compartment by IHC and FACS. The source of TGFBI expression was determined by qPCR and by bone marrow transplantation experiments. Finally, we performed in silico analyses using the METABRIC cohort to assess the potential prognostic value of TGFBI. We observed that deletion of Tgfbi led to a dramatic decrease in CSC content and lung metastasis. Our results show that lack of TGFBI resulted in tumour vessel normalisation, with improved vessel perfusion and decreased hypoxia, a major factor controlling CSCs and metastasis. Furthermore, human data mining in a cohort of breast cancer patients showed that higher expression of TGFBI correlates with poor prognosis and is associated with the more aggressive subtypes of breast cancer. Overall, these data reveal a novel biological mechanism controlling metastasis that could potentially be exploited to improve the efficacy and delivery of chemotherapeutic agents in breast cancer.
Abbreviations
- CSC
cancer stem cell
- ECM
extracellular matrix
- EMT
epithelial‐to‐mesenchymal transition
- FACS
fluorescence‐activated cell sorting
- TGFBI
transforming growth factor beta induced
1. Introduction
Many cancers are organised as a hierarchy in which the so‐called cancer stem cells (CSC) can give rise to both CSCs and a differentiated progeny [1]. CSCs sustain tumour growth, and subsets of CSCs are responsible for metastatic colonisation [2, 3, 4, 5, 6], which is especially relevant since in most solid tumours, metastasis represents the late steps of tumour progression and is the main cause of death by cancer. The degree of plasticity of the CSC pool is a reflection of both intrinsic cellular properties and external signals derived from the tumour microenvironment [1]. Indeed, we and others have shown that specific components of the tumour microenvironment, in particular extracellular matrix (ECM) proteins, have essential functions during metastatic colonisation [4, 7, 8].
The ECM is a highly dynamic and complex network of biochemically discrete elements such as proteins, glycoproteins, polysaccharides and proteoglycans. Besides its fundamental role in maintaining tissue morphology, the ECM is known to control mechanotransduction, to modulate tumour angiogenesis and to provide a suitable niche for CSCs [9, 10]. Due to its glycosylated and charged nature, the ECM can bind many secreted growth factors and molecules [11], thus regulating their distribution and availability and therefore influencing signalling activity. The role of the ECM in promoting tumour angiogenesis is well established, with many ECM fragments having angiostatic or angiogenic properties [12]. Importantly, aberrant tumour angiogenesis is typically associated with hypoxia, a process that has been linked to the activation of a number of signalling pathways that govern CSC maintenance and expansion [13, 14]. The above‐mentioned ability of the ECM to generate localised sources of certain molecules or growth factors, its composition, its topography and its role in tumour angiogenesis are all factors that will determine the properties of the CSC niche and therefore the potential of a given tumour to colonise secondary organs.
Here, we aimed at clarifying the niche‐supporting role of the ECM protein transforming growth factor beta induced (TGFBI) in breast cancer. TGFBI is a conserved, fasciclin family, matricellular protein that has important functions during development, including in tissue branching morphogenesis and mesoderm differentiation in vertebrates, and somitogenesis in zebrafish [15, 16, 17, 18]. Our findings reveal that TGFBI plays an important role in tumour angiogenesis, thereby affecting tumour hypoxia and immune cell infiltration, which then ultimately generates a permissive microenvironment for CSCs and metastasis.
2. Methods
2.1. Antibodies and reagents
CD90.1 (HIS51), CD24 (M1/69), eBioscience (San Diego, CA, USA); TruStain FcX (CD16/32, clone 93), Ter119, CD8a (53‐6.7), CD11b (M1/70), CD31 (MEC13/3), CD44 (IM7), CD45 (30‐F11), F4/80 (BM8), BioLegend (San Diego, CA, USA); CD11b, Abcam (Cambridge, UK); mouse TGFBI, Merck‐Millipore (Burlington, MA, USA); human TGFBI, Thermo Scientific (Waltham, MA, USA); VINCULIN, Santa Cruz Biotechnology (Dallas, TX, USA); α‐SMA (1A4), CD31 (SP164), Sigma‐Aldrich (St. Louis, MO, USA); VIMENTIN, Lifespan Biosciences (Seattle, WA, USA); AldeFluor Assay, STEMCELL Technologies (Vancouver, Canada). All plasmids were produced by classical molecular cloning, and the Tgfbi sgRNAs were cloned into the lentiCRISPRv2 lentiviral vector as described previously [19]. The lentiCRISPRv2 was a gift from Feng Zhang (Addgene plasmid #52961). The sgRNA and Cas9 nuclease containing lentiviruses were used to infect C(3)TAg cells at low MOI and select them with 1 μg·mL−1 puromycin.
2.2. Cell culture
Mouse tumour tissue was dissociated mechanically, followed by an incubation with 1 : 66 Liberase TH (Roche, Basel, Switzerland) and DNAse (10 mg·mL−1) at 37 °C for 1 h. Cells were then washed twice in 2 mm EDTA in PBS and once in PBS and then plated in collagen‐coated plates (HBSS, BSA 100 mg·mL−1, HEPES 1 m pH 6.5 and bovine collagen biomatrix by Cell Systems). Cells were grown in DMEM:F12 (PAN Biotech, Aidenbach, Germany) supplemented with 2% FBS, 1% penicillin/streptomycin 20 ng·mL−1 EGF (Invitrogen, Carlsbad, CA, USA) and 10 μg·mL−1 insulin (Invitrogen) and let attach overnight. Human cell lines and 4T1 cells were obtained from the ATCC and grown as recommended. For TGFBI treatments, MMTV‐PyMT;Tgfbi Δ/Δ cells were grown on collagen‐coated plates and incubated with either 100 ng·mL−1 or 2 μg·mL−1 of exogenous TGFBI protein (a kind gift from Prof. J. Huelsken) for 72 h.
2.3. Tumour sphere assays
MMTV‐PyMT and C(3)TAg sphere cultures were prepared from fresh tumours. Tumour cells were obtained by tissue dissociation and plated on collagen‐coated plates overnight, trypsinised the next day, and plated in 150 μL of sphere media (DMEM/F12 with B27, 20 ng·mL−1 EGF, 20 ng·mL−1 FGF, 4 μg·mL−1 heparin, 1% penicillin/streptomycin) into 96‐well low attachment plates (Corning, NY, USA) at 1 × 104 cells per well and at least three wells per tumour. For all other models, we plated 103 cells per well. Spheres were counted after one week (7–10 days).
2.4. FACS analysis
For FACSorting experiments, tumour cells were obtained by enzymatic disaggregation as described above. Cells were then washed twice with PBS, strained through 70 μm nylon mesh strainers, stained with the appropriate antibodies for 30 min at 4 °C and sorted using either a FACSAria, a FACSAria III (BD Biosciences, Franklin Lakes, NJ, USA), or a MoFlo Astrios (Beckman Coulter, Brea, CA, USA). For FACS analysis, tumour cells were obtained by enzymatic disaggregation or trypsinised, washed and stained with the appropriate antibodies for 30 min at 4 °C. DAPI or 7‐AAD was used to discard dead cells. ALDH activity was tested using the AldeFluor assay kit (STEMCELL Technologies) as per the manufacturer's protocol. Briefly, cells were incubated with either the AldeFluor reagent alone or together with the inhibitor diethylaminobenzaldehyde (DEAB) for 30 min at 37 °C. Cells were then centrifuged, washed and immunophenotyped when required. Fluorescence was analysed using either a Cyan ADP (Dako‐Agilent, Santa Clara, CA, USA) or a MACSQuant (Miltenyi Biotec, Bergisch Gladbach, Germany) instrument. Data were processed and analysed using flowjo (Becton Dickinson, Franklin Lakes, NJ, USA).
2.5. MACS
For MACSorting experiments, tumour cells were obtained by enzymatic disaggregation, washed twice with PBS and strained through 70 μm nylon mesh strainers. MACS was then performed using the EasySep APC Positive Selection Kit (STEMCELL Technologies) following the manufacturer's instructions of. Briefly, FcR blocker was added to the sample and then incubated with a CD11b APC‐conjugated antibody. Next, the sample was incubated with the selection cocktail, followed by an incubation with immunomagnetic beads. CD11b+ cells were then isolated using an EasySep magnet (STEMCELL Technologies).
2.6. Mouse work
MMTV‐PyMT (FVB) mice were breed and housed in ventilated cages in the OHB mouse husbandry of the University of Fribourg. The Tgfbi ∆/∆ mouse was a kind gift by E. Wagner. For PyMT and C(3)TAg tumour cell transplantation to the 4th mammary fat pad or tail vein injection experiments, we used NODSCID‐Il2rg and immunocompetent FVB mice. The experiments involving 4T1 cell injections were done in immunocompetent BALB/c mice. Human breast cancer cells were injected into the 4th mammary fat pad or via tail vein in NODSCID‐Il2rg mice. Cells were trypsinised, resuspended in complete media and centrifuged at 370 g. They were washed twice in PBS, counted and resuspended in PBS at the desired concentration for injection in PBS or Matrigel : PBS (1 : 3). For bone marrow transplantation experiments, female mice were anaesthetised with injection anaesthesia. Once the mice were asleep, they were placed on a warming pad, given eye moistening drops while limbs were fixed to a board with tape in the 50 cm stage. Mice were then irradiated at 220 kV, 20 mA and Al filtering using a dedicated X‐ray unit (Precision X‐Ray X‐RAD‐225) for a total dose of 13 Gy. Once finished, the mice were then transferred in their cage under IR light and observed till they recovered. In a period ranging from 6 to 48 h, the irradiated mice (recipients) were injected i.v. > 3 × 106 bone marrow cells from donor mice. All the experiments involving mice were carried out in accordance with the Swiss Animal Welfare Regulations and were previously approved by the Cantonal Veterinary Service of the Canton Fribourg (2015_20_FR and 2017_26_FR).
2.7. Tail vein injections
Mice were warmed by placing the cage under an IR light bulb. One mouse at a time was placed in a tube rodent holder for tail vein injection with the tail outside of the tube. The tail was cleaned with 70% ethanol. IR light bulb was placed above the tail to cause the veins to dilate. MMTV‐PyMT tumour cells (5 × 105 per mouse) were resuspended in PBS and injected very slowly in a 100 μL volume into one of the two tail veins using an insulin syringe (26G needle). The spot of injection was then compressed with a tissue to make sure the tail was not bleeding. Mice were returned to the cage and kept for observation for 15 min. Metastatic foci in the lungs were counted after 3–5 weeks using a Leica M125 stereomicroscope (Wetzlar,Germany).
2.8. Vasculature analysis and immunostainings
Immunostaining was performed on 4 μm thick paraffin sections using antigen retrieval for 20 min in boiling 10 mm citric acid, pH6.0. After blocking, we incubated the sections with the indicated antibodies overnight at 4 °C. We used secondary fluorescently labelled antibodies Alexa Fluor 488, 568 and 647 (Molecular Probes, Invitrogen), Cy3 (Jackson ImmunoResearch, West Grove, PA, USA) or HRP‐conjugated secondary antibodies. The breast cancer tissue array containing information on the molecular subtype was purchased from US Biomax (Derwood, MD, USA). The intensity of the IHC was evaluated independently by both authors and graded 1 to 4. To study hypoxia, we used pimonidazole (Hypoxyprobes). 60 mg·kg−1 pimonidazole was injected i.p. and was left to circulate for 1 h. For perfusion experiments, mice were given an intravenous injection of 100 μg of fluorescein isothiocyanate‐labelled tomato lectin (Lycopersicon esculentum; Vector Laboratories, Burlingame, CA, USA). After 10 min, the tissues were fixed with 4% PFA, and tumours were frozen in Tissue‐Tek optimum cutting temperature compound (Sakura, Torrance, CA, USA). To study vessel leakiness, mice were given an intravenous injection of 1 mg of 70‐kDa fluorescein isothiocyanate–dextran (Sigma). After 10 min, the tissues were fixed with 4% PFA, and tumours were frozen in Tissue‐Tek optimum cutting temperature compound. Mice were anaesthetised with 100 mg·kg−1 of ketamine and 10 mg·kg−1 of xylazine before injection of the reagents. Fluorescent images were taken with an automated upright microscope system DM5500 (Leica) or a LSM700 upright or inverted confocal microscope (Zeiss, Oberkochen, Germany). Light images were taken with an AX70 widefield microscope (Olympus, Shinjuku, Tokyo, Japan).
2.9. Western blot
Protein was extracted with complete RIPA buffer [20 mm Tris/HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% NP‐40, 1% sodium deoxycholate, 2.5 mm sodium pyrophosphate, 1 mm β‐glycerophosphate, 1 mm Na3VO4, 1 μg·mL−1 leupeptin; Cell Signaling], separated by electrophoresis, transferred to PVDF membranes (Millipore, Burlington, Massachusetts, USA), blocked with 5% BSA (Carl Roth, Karlsruhe, Germany) in 0.1% Tween 20 containing Tris‐buffered saline (TBST) and incubated overnight with primary antibodies. Immunoreactive bands were visualised using HRP‐conjugated secondary antibodies (Cell Signaling, Danvers, MA, USA, and Dako).
2.10. Lentiviral production
Lentiviral particles were produced in HEK293T cells by calcium phosphate precipitation. Briefly, 2 h prior to transfection, 11 × 106 HEK293T cells/15 cm dish were incubated with DMEM + 10%FBS supplemented with 25 μm chloroquine (Sigma). Lentivirus was produced by cotransfection of HEK293T cells with the vectors of interest together with the pCMV‐dR8.74 and the pMD2G (VSVG). Next day, media was removed and replaced with fresh media containing 3 mm caffeine (Sigma). On the third day, the supernatant containing the viral particles was collected, ultracentrifuged for 2.5 h at 20 000 r.p.m. to concentrate the lentiviruses and used to infect cells or aliquoted and stored at −80 °C.
2.11. Real‐time PCR
RNA was prepared using the mini or micro RNA kit (Qiagen, Hilden, Germany) as per the manufacturer's instructions. cDNAs were generated using oligo‐T priming and the M‐MLV Reverse Transcriptase RNase H (–) Point Mutant (Promega, Madison, WI, USA). qPCR was performed in a StepOnePlus thermocycler (Applied Biosystems, Foster City, CA, USA) using the SYBR green PCR Master Mix (Kapa) and following the manufacturer's instructions. A list of primers used is shown in Table S1.
2.12. RNA sequencing
The RNA sequencing experiments were performed in the Swiss Integrative Center for Human Health (SICHH). All samples were first tested for integrity on a Fragment Analyser (ATTI) with the (DNF‐471) Standard Sensitivity RNA Analysis Kit (15 nt). Sequencing libraries were prepared and pooled using the TruSeq® Stranded mRNA Library Prep kit (Illumina, San Diego, CA, USA) as instructed by the manufacturer. The sequencing was performed on a NextSeq 500 sequencer (Illumina), using the NextSeq 500/550 HT reagent kit v2 as instructed by the manufacturer. cutadapt version 1.16 was used to remove the adapter sequences from the reads and for quality trimming. All paired‐end reads were discarded if one of the reads was shorter than 20 bases.
The reads were then aligned against the mouse genome (assembly GRCm38/mm10) using star version 2.6.0a [20]. We acknowledge our use of the gene set enrichment analysis, gsea software and Molecular Database (MSigDB) [21].
2.13. Cibersort
cibersort is an algorithm designed to estimate the cell composition of a complex sample [22]. METABRIC data analyses were performed with 1000 permutations, the default signature matrix for immune cell subtypes (LM22), default statistical parameters and enabled quantile normalisation. Samples were filtered for P ≤ 0.05. The analysis of MMTV‐PyMT samples was performed using the signature matrix ImmuCC [23], with 1000 permutations and disabled quantile normalisation.
2.14. Statistics
The results were analysed using the graphpad prism software (San Diego, CA, USA). Means were compared with two‐tailed unpaired Student's t‐test. In case groups would not pass normality test (assessed using D'Agostino‐Pearson's omnibus normality test), samples were analysed with Mann–Whitney's nonparametric test. When comparing more than two variables, we performed one‐way analysis of variance. To isolate differences between groups, we performed the LSD test. In case groups would not pass normality, samples were analysed using the Kruskal–Wallis test. The METABRIC dataset [24], including clinical data and normalised gene expression, was retrieved through cbioportal [25]. Survival analyses were performed using the Mantel–Cox and the Gehan–Breslow–Wilcoxon tests. Cutpoints for survival analyses were determined using the application Evaluate cutpoints [26]. P‐values are indicated for each experiment. Limiting dilution assay data were analysed using ELDA (extreme limiting dilution assay) [27]. Experiments were done at least in triplicate. Error bars indicate standard deviation unless stated otherwise. Significant differences between experimental groups are indicated with asterisks as follows: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
3. Results
3.1. TGFBI affects tumour‐initiating potential and metastasis in breast cancer
We first analysed MMTV‐PyMT mammary gland tumours for the expression of TGFBI. As shown in Fig. 1A, immunostainings revealed that TGFBI is mainly expressed in the stromal compartment, close to regions containing vimentin‐positive cells and CD90+ cells [4]. Since it has been shown that TGFBI can bind and recruit macrophages through the integrin ITGAM (CD11b) [28], we performed costainings and showed that indeed TGFBI is found in close contact to CD11b+ cells (Fig. 1A). To identify and quantify the cellular sources of TGFBI, we next isolated by FACS and MACS different cell populations from these tumours and analyse them by qPCR. We found that CD11b+ cells expressed the highest levels of Tgfbi, followed by CD24+CD90+ CSCs and CD24−CD90+ stromal cells (Fig. 1B). Of note, up to 90% of CD11b+ MACSorted cells were also F4/80+, indicating that they are macrophages (Fig. S1a). The pattern of expression is similar in other murine breast cancer models (Fig. S1b). To understand the biological effects that TGFBI exerts on CSCs, we crossed a Tgfbi knockout mouse with the MMTV‐PyMT model (Fig. S1c–e). We first tested the ability of these tumours to form mammospheres and observed that MMTV‐PyMT;Tgfbi Δ/Δ tumours possess less sphere‐forming cells (Fig. 1C). We had previously shown that MMTV‐PyMT derived spheres are formed by tumour‐initiating ALDHhigh cells and contain a mixture of cell types [6]. Indeed, FACS analyses revealed that MMTV‐PyMT;Tgfbi Δ/Δ tumours have reduced numbers of ALDHhigh cells and express lower levels of Aldh1a3 (Fig. 1D and Fig. S1f,g). Accordingly, tumour cells derived from TGFBI‐deficient mice have 37 times less tumour‐initiating capacity in limiting dilution assays (Fig. 1E). In addition, MMTV‐PyMT;Tgfbi Δ/Δ tumours also showed a significant reduction in metastatic Lin−CD24+CD90+ cells (Fig. 1F) and consequently seeded less lung metastases (Fig. 1G). These results were confirmed by lung metastasis assays, which demonstrated that the metastatic capacity of the tumour cells is determined by their origin, regardless of the genotype of the host in which they were injected (Fig. 1H). However, sphere, tumour and metastasis formation were not significantly decreased or increased by either knocking out or overexpressing Tgfbi in breast cancer cells (Fig. S2). These results indicate that TGFBI‐driven microenvironmental changes in the primary tumour influence the CSC phenotype. Taken together, these data suggest that TGFBI in the ECM is an important driver of tumour‐ and metastasis‐initiating capacity in breast cancer.
Fig. 1.
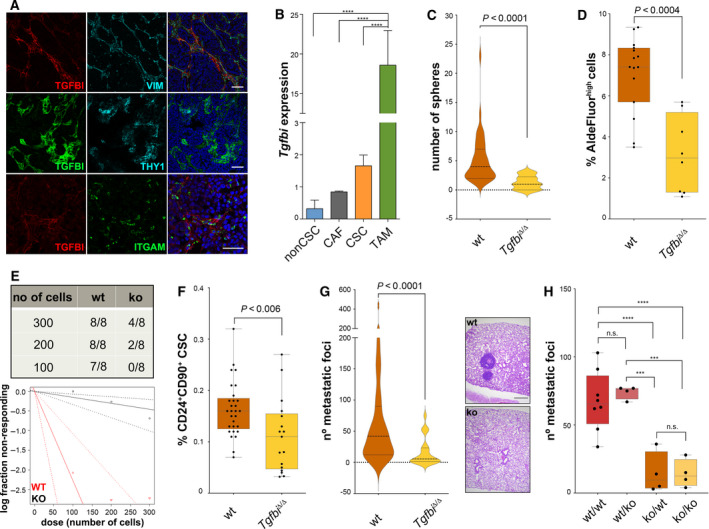
TGFBI and CSC potential. (A) Immunofluorescent staining for TGFBI, VIM, THY1 (CD90) and ITGAM (CD11b) in MMTV‐PyMT tumour sections. Scale bars, 50 μm. (B) qPCR analyses on FACS‐sorted Lin−CD24+CD90− (nonCSC, n = 4), Lin−CD24−CD90+ (CAF, n = 2), and Lin−CD24+CD90+ (CSC, n = 4), or MACS‐sorted CD11b+ cells (TAM, n = 4) from fresh MMTV‐PyMT tumours showed differences in Tgfbi expression. Data were analysed using one‐way ANOVA followed by Fisher's LSD test, and are presented as mean and SD (n = 4 independent tumours); Actb was used as a housekeeping gene. (C) Tumour cells were obtained from fresh MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours, grown overnight in collagen‐coated plates, and seeded as spheres (104 cells/well). Spheres were counted 10 days later. Data were analysed by Mann–Whitney test and are presented as a violin plot showing the median and the quartiles (n = 4 wt, n = 2 ko independent tumours). (D) Cells from fresh MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours were analysed by FACS using the AldeFluor assay. Data were analysed by Mann–Whitney test (n = 15 wt, n = 8 ko). (E) MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours were digested; tumour cells were counted and injected orthotopically in limiting dilution assays in FVB/N mice. The presence or absence of tumours was evaluated for a maximum of 3 months after injection. Data were analysed using ELDA (P = 1.61e−08). (F) Cells from fresh MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours were analysed by FACS for their expression of CD24 and CD90. Data were analysed by Mann–Whitney test (n = 29 wt, n = 17 ko). (G) Number of metastatic foci in the lungs of MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ mice. Data were analysed by Mann–Whitney test and are presented as a violin plot showing the median and the quartiles (n = 55 wt, n = 17 ko). Representative haematoxylin–eosin staining of MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ lungs (scale bar 500 μm). (H) Number of metastatic foci in the lungs of wt and ko FVB/N mice injected with 5 × 105 MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumour cells. Data were analysed using one‐way ANOVA followed by Fisher's LSD test (n = 8 wt/wt, n = 4 wt/ko, n = 4 ko/wt, n = 4 ko/ko). CSC, cancer stem cell; CAF, cancer‐associated fibroblast; TAM, tumour‐associated macrophage. ***P < 0.001; ****P < 0.0001; n.s., not significant.
3.2. TGFBI depletion normalises the tumour vasculature and reduces tumour hypoxia
To gain a better understanding of the pathways affected by the deletion of TGFBI, we performed RNA sequencing of wt and ko tumours. GSEA analyses showed that both angiogenesis and hypoxia were significantly reduced in ko tumours (Fig. 2A, and Figs S3 and S4). In view of these results, we next aimed at understanding whether depletion of TGFBI impacts the tumour vasculature functionality. We first determined vascular perfusion and leakiness performing tomato lectin and 70 kDa FITC‐labelled dextran intravenous injections. The former stains all vessels with an active perfusion and the latter seeps out of the vasculature in ruptured vessels. Our results indicate that TGFBI depletion leads to increased perfusion (Fig. 2B) and reduced vascular leakage (Fig. 2C). Interestingly, we observed a positive correlation of Tgfbi with regulator of G protein signalling 5 (Rgs5), a gene whose expression has been linked to the formation of aberrant vasculature in tumours (Fig. 2D and Fig. S5) [29]. Furthermore, TGFBI loss led to increased and more structured pericyte coverage (Fig. 2E). As a consequence of improved perfusion, Tgfbi ko tumours exhibit less hypoxic areas (Fig. 2F) and consequently, decreased features of epithelial‐to‐mesenchymal (EMT) transition (Fig. S6a). To uncouple potential direct effects of TGFBI on cancer cells from those on the tumour vasculature, we added exogenous TGFBI to MMTV‐PyMT;Tgfbi Δ/Δ cells in culture. However, adding TGFBI did not trigger EMT (Fig. S6b), which again indicates that decreased CSC numbers and metastases are mainly the result of TGFBI's microenvironmental effects. In silico analyses using the METABRIC cohort confirmed that the expression of TGFBI in human breast tumours positively correlates with the expression of many hypoxia‐related genes (Fig. S7). Likewise, Tgfbi overexpression in breast cancer cells increased tumour hypoxia (Fig. 2G). Overall, these results indicate that TGFBI promotes aberrant angiogenesis and increases tumour hypoxia in breast cancer.
Fig. 2.
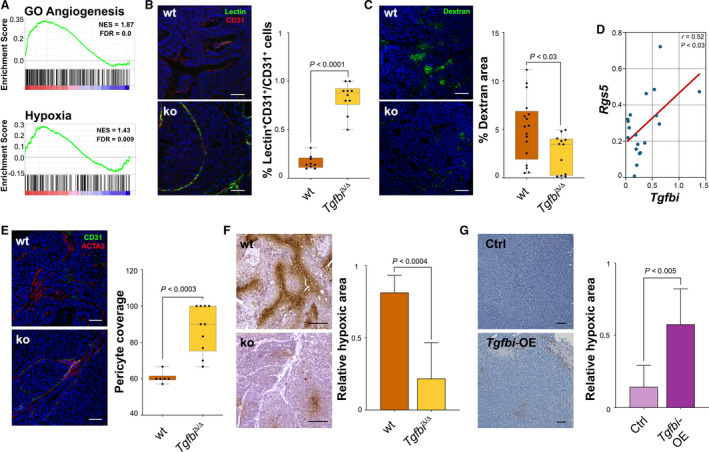
TGFBI depletion normalises the tumour vasculature. (A) Enrichment score analyses of RNAseq data obtained from MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours (n = 5) for GO angiogenesis and hallmark hypoxia. (B) MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ mice were injected via tail vein with 100 μg of tomato lectin, and tumours were embedded in OCT and costained with CD31 (scale bar 100 μm). Data were analysed by unpaired t‐test. (C) MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ mice were injected via tail vein with 50 μg of 70 kDa FITC‐labelled dextran, and tumours were embedded in OCT (scale bar 100 μm). Data were analysed by unpaired t‐test. (D) Tgfbi and Rgs5 expression correlation in MMTV‐PyMT tumours. The expression of both genes was determined by qPCR. The correlation was estimated by Pearson's r coefficient (n = 19). Rplp0 was used as a housekeeping gene. (E) Immunofluorescent staining of MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours with CD31 and ACTA2. Data were analysed by unpaired t‐test. (F) PIMO staining of MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours and quantification of stained area (scale bar 100 μm). Data were analysed by unpaired t‐test on two independent tumours, and are presented as mean and SD. (G) PIMO staining of MDA‐MB‐453 control and Tgfbi‐overexpressing tumours and quantification of stained area (scale bar 100 μm). Data were analysed by unpaired t‐test, and are presented as mean and SD (n = 3 ctrl tumours, n = 4 overexpression tumours). PIMO, pimonidazole.
3.3. TGFBI levels determine tumour hypoxia
In order to evaluate the effects of TGFBI deletion in the tumour immune landscape, we first used the cibersort software [22, 23]. The analysis of our tumour samples revealed significant differences in the myeloid and the T‐cell compartments (Fig. 3A), with Tgfbi Δ/Δ tumours having less myeloid cells and more T lymphocytes. These findings were confirmed by immunophenotyping the MMTV‐PyMT tumours by FACS (Fig. 3B,C). Since we had previously found that in MMTV‐PyMT tumours macrophages secreted high levels of TGFBI, we argued that it should be possible to induce tumour hypoxia by transferring wt macrophages into lethally irradiated mice bearing Tgfbi Δ/Δ tumours. Therefore, we performed wt and ko bone marrow transplants (BMT) into wt mice, which were afterwards orthotopically injected with Tgfbi Δ/Δ tumour cells (Fig. 3D). Tgfbi‐deficient tumours grown in wt mice transplanted with ko bone marrow cells showed a decreased number of CSCs (Fig. 3E). In addition, these tumours were less hypoxic and expressed lower levels of Rgs5 (Fig. 3F,G). These results confirm that CD11b+ cells are a main source of TGFBI in MMTV‐PyMT tumours, and they indicate that TGFBI levels associate with tumour angiogenesis and hypoxia.
Fig. 3.

TGFBI levels determine tumour hypoxia. (A) cibersort analysis of MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ. (B) The presence of CD45+CD11b+ cells was determined by FACS in fresh MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours. Data were analysed by unpaired t‐test (n = 12 wt, n = 10 ko). (C) FACS analysis of CD8+ T cells in fresh MMTV‐PyMT;Tgfbi +/+ and MMTV‐PyMT;Tgfbi Δ/Δ tumours. Data were analysed by Mann–Whitney's test (n = 19 wt, n = 20 ko). (D) Bone marrow transplantation experimental scheme. GFP+ bone marrows from Tgfbi +/+ or Tgfbi Δ/Δ mice were transplanted into lethally irradiated wild‐type hosts. The resultant chimeras were orthotopically injected with MMTV‐PyMT;Tgfbi Δ/Δ tumour cells. The content of CD24+CD90+ (E) in these tumours was analysed by FACS. Data were analysed by unpaired t‐test, and are presented as mean and SD (n = 5 wt, n = 4 ko). (F) PIMO staining in Tgfbi +/+ and Tgfbi Δ/Δ bone marrow transplanted mice tumours and quantification of stained area (scale bar 100 μm). Data were analysed by unpaired t‐test, and are presented as mean and SD (n = 3). (G) Tgfbi, Rgs5, and Hif1α qPCR on pulverised tumour material from Tgfbi Δ/Δ chimeras. Data were analysed by unpaired t‐test, and are presented as mean and SD (n = 5). Rplp0 was used as a housekeeping gene. BM, bone marrow; BMT, bone marrow transplantation; PIMO, pimonidazole. *P < 0.05; ***P < 0.001; n.s., not significant.
3.4. TGFBI is associated with poor prognosis
In order to understand the relevance of TGFBI in human breast cancer, we performed in silico analysis using the METABRIC cohort [30] and found that TGFBI predicts poor prognosis (Fig. 4A). Interestingly, in patients undergoing chemotherapy, low TGFBI expression levels predict better prognosis (Fig. S8), supporting our experimental data on vessel normalisation, which had previously been linked to enhanced therapeutic efficacy [31, 32]. Moreover, its expression increases as a function of the Nottingham prognostic index (NPI, Fig. 4B), especially between patients with NPI ≤ 3.4 and NPI > 3.4. We next compared the expression of TGFBI in the different molecular subtypes of the METABRIC database and found that basal and claudin‐low tumours, which are known to contain high percentages of CSCs [33, 34], have the highest expression levels (Fig. 4C). We further confirmed these results by IHC on a panel of human breast cancer tissue samples. Our results indicate that basal tumours show higher TGFBI staining scores compared to luminal A tumours (Fig. 4D,E). We then estimated the abundance of different immune cell types in tumours expressing high or low levels of TGFBI in the METABRIC cohort using cibersort. As shown in Fig. 4F, the estimated relative content of myeloid cells in general, and M2 macrophages in particular, positively associates with TGFBI expression, while that of CD8+ T cells is negatively linked with TGFBI expression. Likewise, when every subtype was subdivided into high and low expressers, we found that TGFBI high tumours within the same molecular subtype contain more myeloid cells and M2 macrophages, but less CD8+ T cells (Fig. 4F), which agrees with our experimental data. Furthermore, GSEA analyses confirmed that TGFBI high tumours display molecular signatures of increased angiogenesis, hypoxia and EMT (Fig. S9). Taken together, these results indicate that TGFBI expression predicts tumour aggressiveness and poorer outcomes in human breast cancer, and it is associated with higher macrophage infiltration and angiogenesis.
Fig. 4.
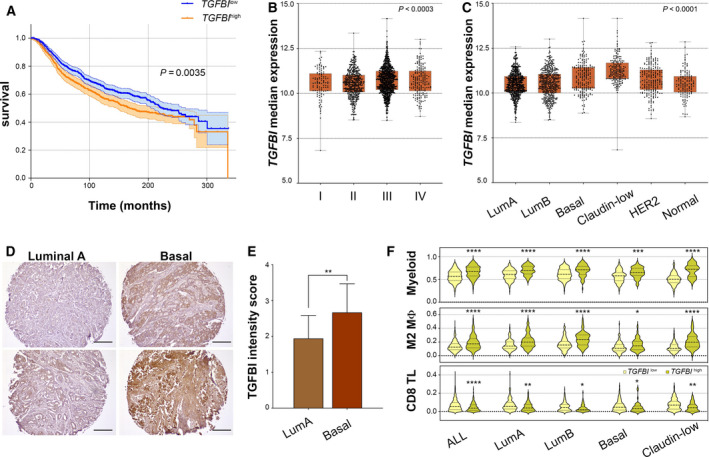
TGFBI is associated with poor prognosis. (A) Survival curves of breast cancer patients in the METABRIC cohort classified according to the expression of TGFBI (n high = 620; n low = 803). Patients were stratified using the application Evaluate cutpoints, and the survival curves were compared using the log‐rank (Mantel–Cox) test. Only ‘living’ and ‘died of disease’ patients were included in the analysis. (B) TGFBI expression in the METABRIC cohort classified according to the Nottingham prognostic index (n I = 108, n II = 452, n III = 1064, n IV = 191). Results were analysed using the Kruskal–Wallis test. (C) TGFBI expression of breast cancer patients in the METABRIC cohort classified according to the molecular subtype (n LumA = 679, n LumB = 461, n Basal = 199, n Claudin‐low = 199, n HER2 = 220, n Normal = 140). Results were analysed using the Kruskal–Wallis test followed by Dunn's multiple comparison test (LumA vs. LumB n.s., LumA vs. Basal P < 0.0001, LumA vs. Claudin‐low P < 0.0001, LumA vs. HER2 P < 0.0003, LumA vs. Normal n.s., LumB vs. Basal P < 0.0001, LumB vs. Claudin‐low P < 0.0001, LumB vs. HER2 P < 0.02, LumB vs. Normal n.s., Basal vs. Claudin‐low P < 0.0001, Basal vs. HER2 n.s., Basal vs. Normal P < 0.002, Claudin‐low vs. HER2 P < 0.0001, Claudin‐low vs. Normal P < 0.0001, HER2 vs. Normal n.s.) (D) Representative immunohistochemistry stainings for TGFBI in luminal A and basal‐like human breast tumours. (E) TGFBI IHC scoring according to the staining's intensity. Data was analysed using an unpaired t‐test, and are presented as mean and SD (n = 18 luminal A tumours, n = 22 basal‐like tumours). (F) Relative myeloid cell, M2 macrophage, and CD8 T‐cell content of TGFBI l ow (< 75th percentile) and TGFBI high tumours (≥ 75th percentile) in METABRIC tumours unclassified and classified by molecular subtype. The relative cell content estimation was calculated with cibersort. The data were analysed using either unpaired Student's t‐tests or Mann–Whitney's nonparametric tests, and they are presented as violin plots showing the median and the quartiles. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; n.s., not significant.
4. Discussion
Aberrant angiogenesis and hypoxia influence each other by creating a positive forward loop that promotes tumour progression. Increased hypoxia exerts severe metabolic constraints on tumour cells and is a major driver that maintains and expands CSCs [13, 35, 36, 37]. Despite its promising potential, antiangiogenic therapies have failed to produce consistent, long‐lasting effects in mouse models and human patients [38]. In fact, reducing the tumour vasculature inevitably results in increased hypoxia, which in turn may increase CSC expansion and tumour chemoresistance [39]. Tumour vessel normalisation has several potential advantages, which include the improvement of drug delivery and the reduction of metastatic spread through the generation of a proper, well‐organised layer of pericytes around the vessels [31, 32, 40, 41, 42]. However, the molecular mechanisms controlling this reversal of the structure and function of the tumour vasculature are yet unclear. In the present study, we show that deletion of the matricellular protein TGFBI is sufficient to normalise the tumour vasculature and mitigate hypoxia. The role of TGFBI in cancer is still controversial and seems to be context‐dependent. For instance, TGFBI has been suggested to have tumour suppressive activities in mesothelioma, breast, and lung cancer cells [43, 44]. In addition, Zhang et al. [45] reported that homozygous null deletions of Tgfbi in mice result in increased frequency of spontaneous tumours and increased predisposition to cancer induction. Conversely, in colon cancer, TGFBI has been shown to favour extravasation, which in turn promotes metastasis [46]. In melanoma cells, TGFBI plays an anti‐adhesive role and its knockdown decreases tumour growth and invasion [47]. Likewise, in prostate cancer, TGFBI contributes to tumour progression [48]. Finally, recent work by Costanza et al. [49] showed that TGFBI predicts poor prognosis in pancreatic adenocarcinoma patients and promotes pancreatic cancer cell migration. Our results in breast cancer also suggest a tumour‐promoting role of TGFBI, but they indicate that modulating TGFBI in breast cancer cells does not directly impact the CSC phenotype and therefore the metastatic potential of tumour cells. However, using a Tgfbi straight knockout model, we found that the overall expression levels of TGFBI in a given tumour will determine the extent of hypoxia, EMT, and ultimately the CSC content and metastatic potential of the tumour. The differences observed between our study and previously published data might be due to methodological differences, in particular the fact that we focused on the role of TGFBI in the tumour microenvironment using a knockout mouse [43, 44]. We found that in the MMTV‐PyMT model, TGFBI is expressed mostly by macrophages, mesenchymal CSCs and stromal cells. Interestingly, Martinez et al. compared the transcriptional profiles of M1 vs M2 macrophages and found that TGFBI is expressed at higher levels in the latter [50], which are known to contribute to tumour angiogenesis [51]. Indeed, our analyses reveal that there is a positive association between TGFBI expression and M2 macrophage content in human breast tumours. Moreover, the levels of expression of TGFBI in human breast tumours significantly correlate with their molecular subtype. Interestingly, those subtypes enriched in mesenchymal CSCs [52] express higher levels of TGFBI, which is in agreement with our experimental findings. The mechanism by which TGFBI regulates tumour hypoxia in breast cancer and whether this effect is direct or indirect remains unclear. A number of nonexclusive factors may explain the effects that we observed. For instance, TGFBI is known to interact not only with several integrins [28, 46, 53, 54, 55, 56, 57, 58], but also with other components of the ECM, such as collagens, fibronectin, laminin, periostin and proteoglycans [59, 60, 61, 62, 63]. The reorganisation of the extracellular matrix and the initiation of integrin and focal adhesion kinase (FAK) signalling have direct implications for the regulation of angiogenesis and hypoxia [12, 64]. However, further work is needed to explore the interactions of TGFBI with endothelial cells and its potential role in macrophage biology and vascular maturation.
5. Conclusions
In summary, our study reveals a new biological role for the matricellular protein TGFBI in breast cancer. Our results indicate that TGFBI, which we found secreted by macrophages, mesenchymal tumour cells and CAFs, is a crucial player regulating breast tumour‐ and metastasis‐initiating potential through the modulation of the tumour microenvironment and hypoxia, and emphasise the importance of the extracellular matrix on breast cancer progression and metastasis. Taken together, our findings suggest that TGFBI may be used as a prognostic factor in breast cancer and open potential new opportunities for combinatorial therapies.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AS‐M conceptualized the study. AS‐M and FF contributed to methodology. FF and AS‐M involved in investigation. FF and AS‐M performed formal analysis. AS‐M provided resources. AS‐M and FF wrote the original draft of the manuscript. AS‐M and FF wrote, reviewed and edited the manuscript. AS‐M acquired funding. AS‐M supervised the study. All authors read and approved the final manuscript.
Supporting information
Fig. S1. CD11b+ MACS, TGFBI IHC, Tgfbi knockout mouse model and ALDH in MMTV‐PyMT;Tgfbi Δ/Δ tumours.
Fig. S2. TGFBI genetic modulation in C(3)TAg and MDA‐MB‐453 cells.
Fig. S3. Heatmap of angiogenesis and hypoxia pathway genes regulated upon Tgfbi deletion.
Fig. S4. List of differentially expressed genes in MMTV‐PyMT;Tgfbi Δ/Δ tumours.
Fig. S5. Correlation between Tgfbi and Rgs5 expression in 4T1 and MDA‐MB‐453 tumours.
Fig. S6. TGFBI and EMT.
Fig. S7. Correlation between TGFBI and hypoxia‐related genes in the METABRIC cohort.
Fig. S8. Survival curves for METABRIC patients who underwent chemotherapy stratified for TGFBI expression.
Fig. S9. GSEA analyses in the METABRIC cohort.
Table S1. List of primers used for qPCR.
Acknowledgements
The authors are grateful to Vanina Lauper, Emanuel Lubart, Marta Valeri‐Sala, Pierre Dessen and Oriana Coquoz for excellent technical assistance, and to Prof Joerg Huelsken and Megan Young for critical revision of the manuscript. The TGFBI knockout mouse was a kind gift by Dr Erwin Wagner. The authors are deeply grateful to the Swiss Cancer League and the Swiss National Science Foundation (SNSF) for supporting this project. FF was supported by a Swiss Cancer League grant to AS‐M (KLS‐4121‐02‐2017). AS‐M was supported with a SNSF Ambizione grant (PZ00P3_154751).
Data accessibility
All data generated and analysed during this study are included in this manuscript and its supplementary files.
References
- 1. Batlle E & Clevers H (2017) Cancer stem cells revisited. Nat Med 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- 2. Reya T, Morrison SJ, Clarke MF & Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. [DOI] [PubMed] [Google Scholar]
- 3. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ & Heeschen C (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1, 313–323. [DOI] [PubMed] [Google Scholar]
- 4. Malanchi I, Santamaria‐Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF & Huelsken J (2012) Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89. [DOI] [PubMed] [Google Scholar]
- 5. Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, Ng L, Cheung LW, Lan XR, Lan HY et al (2010) A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 6, 603–615. [DOI] [PubMed] [Google Scholar]
- 6. Fico F, Bousquenaud M, Ruegg C & Santamaria‐Martinez A (2019) Breast cancer stem cells with tumor‐ versus metastasis‐initiating capacities are modulated by TGFBR1 inhibition. Stem Cell Reports 13, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB et al (2011) VEGF‐A and Tenascin‐C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA 108, 16002–16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova‐Todorova K, Brogi E & Massague J (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nallanthighal S, Heiserman JP & Cheon DJ (2019) The role of the extracellular matrix in cancer stemness. Front Cell Dev Biol 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu P, Weaver VM & Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonnans C, Chou J & Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15, 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mongiat M, Andreuzzi E, Tarticchio G & Paulitti A (2016) Extracellular matrix, a hard player in angiogenesis. Int J Mol Sci 17, 1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carnero A & Lleonart M (2016) The hypoxic microenvironment: a determinant of cancer stem cell evolution. BioEssays 38 (Suppl 1), S65–S74. [DOI] [PubMed] [Google Scholar]
- 14. Peng G & Liu Y (2015) Hypoxia‐inducible factors in cancer stem cells and inflammation. Trends Pharmacol Sci 36, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirate Y, Okamoto H & Yamasu K (2003) Structure of the zebrafish fasciclin I‐related extracellular matrix protein (betaig‐h3) and its characteristic expression during embryogenesis. Gene Expr Patterns 3, 331–336. [DOI] [PubMed] [Google Scholar]
- 16. Kim HR & Ingham PW (2009) The extracellular matrix protein TGFBI promotes myofibril bundling and muscle fibre growth in the zebrafish embryo. Dev Dyn 238, 56–65. [DOI] [PubMed] [Google Scholar]
- 17. Schorderet DF, Menasche M, Morand S, Bonnel S, Buchillier V, Marchant D, Auderset K, Bonny C, Abitbol M & Munier FL (2000) Genomic characterization and embryonic expression of the mouse Bigh3 (Tgfbi) gene. Biochem Biophys Res Commun 274, 267–274. [DOI] [PubMed] [Google Scholar]
- 18. Schwab K, Patterson LT, Aronow BJ, Luckas R, Liang HC & Potter SS (2003) A catalogue of gene expression in the developing kidney. Kidney Int 64, 1588–1604. [DOI] [PubMed] [Google Scholar]
- 19. Sanjana NE, Shalem O & Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M & Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M & Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen Z, Huang A, Sun J, Jiang T, Qin FX & Wu A (2017) Inference of immune cell composition on the expression profiles of mouse tissue. Sci Rep 7, 40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ et al (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ogluszka M, Orzechowska M, Jedroszka D, Witas P & Bednarek AK (2019) Evaluate cutpoints: adaptable continuous data distribution system for determining survival in Kaplan‐Meier estimator. Comput Methods Programs Biomed 177, 133–139. [DOI] [PubMed] [Google Scholar]
- 27. Hu Y & Smyth GK (2009) ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- 28. Kim HJ & Kim IS (2008) Transforming growth factor‐beta‐induced gene product, as a novel ligand of integrin alphaMbeta2, promotes monocytes adhesion, migration and chemotaxis. Int J Biochem Cell Biol 40, 991–1004. [DOI] [PubMed] [Google Scholar]
- 29. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Grone HJ, Hammerling GJ et al (2008) Vascular normalization in Rgs5‐deficient tumours promotes immune destruction. Nature 453, 410–414. [DOI] [PubMed] [Google Scholar]
- 30. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, Chaturvedi P, Hol J, Thienpont B, Teuwen LA et al (2016) Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30, 968–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park JS, Kim IK, Han S, Park I, Kim C, Bae J, Oh SJ, Lee S, Kim JH, Woo DC et al (2016) Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 30, 953–967. [DOI] [PubMed] [Google Scholar]
- 33. Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI et al (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor‐initiating features. Proc Natl Acad Sci USA 106, 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X & Perou CM (2010) Phenotypic and molecular characterization of the claudin‐low intrinsic subtype of breast cancer. Breast Cancer Res 12, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwab LP, Peacock DL, Majumdar D, Ingels JF, Jensen LC, Smith KD, Cushing RC & Seagroves TN (2012) Hypoxia‐inducible factor 1alpha promotes primary tumor growth and tumor‐initiating cell activity in breast cancer. Breast Cancer Res 14, R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiang L, Gilkes DM, Hu H, Takano N, Luo W, Lu H, Bullen JW, Samanta D, Liang H & Semenza GL (2014) Hypoxia‐inducible factor 1 mediates TAZ expression and nuclear localization to induce the breast cancer stem cell phenotype. Oncotarget 5, 12509–12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim H, Lin Q, Glazer PM & Yun Z (2018) The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res 20, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bergers G & Hanahan D (2008) Modes of resistance to anti‐angiogenic therapy. Nat Rev Cancer 8, 592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG & Wicha MS (2012) Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA 109, 2784–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jain RK (2014) Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P & Hammerling GJ (2015) Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol 16, 609–617. [DOI] [PubMed] [Google Scholar]
- 42. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, Sheng K, Dobrolecki LE, Zhang X, Putluri N et al (2017) Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li B, Wen G, Zhao Y, Tong J & Hei TK (2012) The role of TGFBI in mesothelioma and breast cancer: association with tumor suppression. BMC Cancer 12, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen G, Partridge MA, Li B, Hong M, Liao W, Cheng SK, Zhao Y, Calaf GM, Liu T, Zhou J et al (2011) TGFBI expression reduces in vitro and in vivo metastatic potential of lung and breast tumor cells. Cancer Lett 308, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Wen G, Shao G, Wang C, Lin C, Fang H, Balajee AS, Bhagat G, Hei TK & Zhao Y (2009) TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res 69, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ et al (2008) Extracellular matrix protein betaig‐h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev 22, 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nummela P, Lammi J, Soikkeli J, Saksela O, Laakkonen P & Holtta E (2012) Transforming growth factor beta‐induced (TGFBI) is an anti‐adhesive protein regulating the invasive growth of melanoma cells. Am J Pathol 180, 1663–1674. [DOI] [PubMed] [Google Scholar]
- 48. Chen WY, Tsai YC, Yeh HL, Suau F, Jiang KC, Shao AN, Huang J & Liu YN (2017) Loss of SPDEF and gain of TGFBI activity after androgen deprivation therapy promote EMT and bone metastasis of prostate cancer. Sci Signal 10, eaam6826. [DOI] [PubMed] [Google Scholar]
- 49. Costanza B, Rademaker G, Tiamiou A, De Tullio P, Leenders J, Blomme A, Bellier J, Bianchi E, Turtoi A, Delvenne P et al (2019) Transforming growth factor beta‐induced, an extracellular matrix interacting protein, enhances glycolysis and promotes pancreatic cancer cell migration. Int J Cancer 145, 1570–1584. [DOI] [PubMed] [Google Scholar]
- 50. Martinez FO, Gordon S, Locati M & Mantovani A (2006) Transcriptional profiling of the human monocyte‐to‐macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303–7311. [DOI] [PubMed] [Google Scholar]
- 51. De Palma M, Biziato D & Petrova TV (2017) Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 17, 457–474. [DOI] [PubMed] [Google Scholar]
- 52. Brooks MD, Burness ML & Wicha MS (2015) Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 17, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ahmed AA, Mills AD, Ibrahim AE, Temple J, Blenkiron C, Vias M, Massie CE, Iyer NG, McGeoch A, Crawford R et al (2007) The extracellular matrix protein TGFBI induces microtubule stabilization and sensitizes ovarian cancers to paclitaxel. Cancer Cell 12, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim JE, Kim SJ, Lee BH, Park RW, Kim KS & Kim IS (2000) Identification of motifs for cell adhesion within the repeated domains of transforming growth factor‐beta‐induced gene, beta ig‐h3. J Biol Chem 275, 30907–30915. [DOI] [PubMed] [Google Scholar]
- 55. Nam EJ, Sa KH, You DW, Cho JH, Seo JS, Han SW, Park JY, Kim SI, Kyung HS, Kim IS et al (2006) Up‐regulated transforming growth factor beta‐inducible gene h3 in rheumatoid arthritis mediates adhesion and migration of synoviocytes through alpha v beta3 integrin: regulation by cytokines. Arthritis Rheum 54, 2734–2744. [DOI] [PubMed] [Google Scholar]
- 56. Nam JO, Kim JE, Jeong HW, Lee SJ, Lee BH, Choi JY, Park RW, Park JY & Kim IS (2003) Identification of the alphavbeta3 integrin‐interacting motif of betaig‐h3 and its anti‐angiogenic effect. J Biol Chem 278, 25902–25909. [DOI] [PubMed] [Google Scholar]
- 57. Ohno S, Noshiro M, Makihira S, Kawamoto T, Shen M, Yan W, Kawashima‐Ohya Y, Fujimoto K, Tanne K & Kato Y (1999) RGD‐CAP ((beta)ig‐h3) enhances the spreading of chondrocytes and fibroblasts via integrin alpha(1)beta(1). Biochim Biophys Acta 1451, 196–205. [DOI] [PubMed] [Google Scholar]
- 58. Thapa N, Kang KB & Kim IS (2005) Beta ig‐h3 mediates osteoblast adhesion and inhibits differentiation. Bone 36, 232–242. [DOI] [PubMed] [Google Scholar]
- 59. Billings PC, Whitbeck JC, Adams CS, Abrams WR, Cohen AJ, Engelsberg BN, Howard PS & Rosenbloom J (2002) The transforming growth factor‐beta‐inducible matrix protein (beta)ig‐h3 interacts with fibronectin. J Biol Chem 277, 28003–28009. [DOI] [PubMed] [Google Scholar]
- 60. Hashimoto K, Noshiro M, Ohno S, Kawamoto T, Satakeda H, Akagawa Y, Nakashima K, Okimura A, Ishida H, Okamoto T et al (1997) Characterization of a cartilage‐derived 66‐kDa protein (RGD‐CAP/beta ig‐h3) that binds to collagen. Biochim Biophys Acta 1355, 303–314. [DOI] [PubMed] [Google Scholar]
- 61. Kim BY, Olzmann JA, Choi SI, Ahn SY, Kim TI, Cho HS, Suh H & Kim EK (2009) Corneal dystrophy‐associated R124H mutation disrupts TGFBI interaction with Periostin and causes mislocalization to the lysosome. J Biol Chem 284, 19580–19591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim JE, Park RW, Choi JY, Bae YC, Kim KS, Joo CK & Kim IS (2002) Molecular properties of wild‐type and mutant betaIG‐H3 proteins. Invest Ophthalmol Vis Sci 43, 656–661. [PubMed] [Google Scholar]
- 63. Reinboth B, Thomas J, Hanssen E & Gibson MA (2006) Beta ig‐h3 interacts directly with biglycan and decorin, promotes collagen VI aggregation, and participates in ternary complexing with these macromolecules. J Biol Chem 281, 7816–7824. [DOI] [PubMed] [Google Scholar]
- 64. Lechertier T & Hodivala‐Dilke K (2012) Focal adhesion kinase and tumour angiogenesis. J Pathol 226, 404–412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CD11b+ MACS, TGFBI IHC, Tgfbi knockout mouse model and ALDH in MMTV‐PyMT;Tgfbi Δ/Δ tumours.
Fig. S2. TGFBI genetic modulation in C(3)TAg and MDA‐MB‐453 cells.
Fig. S3. Heatmap of angiogenesis and hypoxia pathway genes regulated upon Tgfbi deletion.
Fig. S4. List of differentially expressed genes in MMTV‐PyMT;Tgfbi Δ/Δ tumours.
Fig. S5. Correlation between Tgfbi and Rgs5 expression in 4T1 and MDA‐MB‐453 tumours.
Fig. S6. TGFBI and EMT.
Fig. S7. Correlation between TGFBI and hypoxia‐related genes in the METABRIC cohort.
Fig. S8. Survival curves for METABRIC patients who underwent chemotherapy stratified for TGFBI expression.
Fig. S9. GSEA analyses in the METABRIC cohort.
Table S1. List of primers used for qPCR.
Data Availability Statement
All data generated and analysed during this study are included in this manuscript and its supplementary files.