

Abstract
Objective:
To explore the role of alpha-synuclein (αSyn) oligomers and neurofilament light chain (NFL) in cerebrospinal fluid (CSF) as markers of early multiple system atrophy (MSA) and to contrast findings with Lewy body synucleinopathies.
Methods:
In a discovery cohort of well-characterized early MSA patients (n = 24) and matched healthy controls (CON, n = 14), we utilized enzyme-linked immunosorbent assay to measure NFL and protein misfolding cyclic amplification (PMCA) to detect αSyn oligomers in CSF. We confirmed findings in a separate prospectively enrolled cohort of patients with early MSA (n = 38), Parkinson disease (PD, n = 16), and dementia with Lewy bodies (DLB, n = 13), and CON subjects (n = 15).
Results:
In the discovery cohort, NFL was markedly elevated in MSA patients, with perfect separation from CON. αSyn-PMCA was nonreactive in all CON, whereas all MSA samples were positive. In the confirmatory cohort, NFL again perfectly separated MSA from CON, and was significantly lower in PD and DLB compared to MSA. PMCA was again nonreactive in all CON, and positive in all but 2 MSA cases. All PD and all but 2 DLB samples were also positive for αSyn aggregates but with markedly different reaction kinetics from MSA; aggregation occurred later, but maximum fluorescence was higher, allowing for perfect separation of reactive samples between MSA and Lewy body synucleinopathies.
Interpretation:
NFL and αSyn oligomers in CSF faithfully differentiate early MSA not only from CON but also from Lewy body synucleinopathies. The findings support the role of these markers as diagnostic biomarkers, and have important implications for understanding pathophysiologic mechanisms underlying the synucleinopathies.
Introduction
Parkinson disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) comprise a group of neurodegenerative disorders that share the pathophysiological mechanism of abnormal aggregation of alpha-synuclein (αSyn) as neuronal or glial inclusions, and are therefore referred to as the “synucleinopathies.” MSA is rare, rapidly progressive, and universally fatal. Its clinical presentation is characterized by motor dysfunction that may predominantly involve parkinsonism (MSA-P) or cerebellar impairment (MSA-C), along with prominent autonomic failure.1 In contrast, PD is much more common, generally associated with a more benign clinical course, and besides the typical features of parkinsonism is also often accompanied by sleep disorders, cognitive decline, and typically milder autonomic dysfunction.2,3 DLB is the second most common form of dementia and characterized by cognitive decline with prominent fluctuations, visual hallucinations, rapid eye movement sleep behavior disorder, and parkinsonism, as well as typically moderate autonomic dysfunction.4,5
Although in our experience MSA can usually be differentiated from PD and DLB with the help of standardized autonomic testing, that differentiation can be difficult at early disease stages or when the presentation is atypical.6,7 Misdiagnosis is common among community neurologists, even movement disorder specialists. A recent large referral autopsy series of patients with an antemortem clinical diagnosis of MSA revealed diagnostic accuracy of only 62%, which is in line with previous reports.8–11 Most commonly mistaken for MSA was Lewy body disease, followed by progressive supranuclear palsy and PD.9
These reports highlight the dire need for reliable disease biomarkers to solidify a clinical diagnosis of MSA. MSA results in structural and signal abnormalities of selected regions on magnetic resonance imaging (MRI), but characteristic findings typically only occur at later disease stages.1,12 Efforts at quantitative MRI approaches, including morphometric and diffusivity measures, have shown promising results but are still far from entering clinical practice.13,14
Neurofilament light chain (NFL) in the spinal fluid provides an index of central axonal degeneration and has been reported to be a valuable marker in a number of neurologic conditions associated with rapid disease progression or considerable neuronal injury.15–17 Although not disease-specific, given the tempo of disease progression and neuronal degeneration in MSA compared to other synucleinopathies, it comes as no surprise that NFL has been reported to be increased in both MSA-C and MSA-P and to possibly distinguish MSA from PD.16,18,19
Another marker of particular recent interest is the misfolded, aggregated form of αSyn in the spinal fluid of patients with synucleinopathies. We recently showed not only that these abnormal αSyn aggregates can be reliably detected in the cerebrospinal fluid (CSF) of patients with MSA and PD using a protein misfolding cyclic amplification (PMCA) assay, but also that the product of the αSyn-PMCA assay was biochemically and biophysically distinct between MSA and PD, allowing for clear differentiation of CSF from patients with MSA and PD.20,21
With this study, we therefore sought to confirm the value of NFL and αSyn oligomers in the CSF as diagnostic biomarkers differentiating MSA from the Lewy body synucleinopathies, utilizing prospectively collected CSF samples of clinically well-characterized and longitudinally followed patients with early MSA, PD, and DLB, and healthy controls.
Subjects and Methods
Study Design
In a discovery phase, we prospectively collected CSF from well-characterized patients with early MSA and healthy controls. In a subsequent confirmation phase, we again prospectively collected CSF from well-characterized patients with early MSA and healthy controls, in addition to patients with PD and DLB for comparison of selected CSF biomarkers.
Participants
Subjects were enrolled as a part of a prospective, longitudinal study of synucleinopathies (MSA, PD, and controls; Mayo Longitudinal Synucleinopathy Biomarker Study, NS092625) and prospective studies of dementia (DLB; Mayo Alzheimer’s Disease Research Center, AG016574 and Longitudinal Imaging Bio-markers of Disease Progression in DLB, NS100620). Only spinal fluid samples collected at baseline were utilized for the data presented in this article.
Patients with MSA received a diagnosis of MSA-C or MSA-P by a Mayo Clinic movement disorder specialist. All patients had autonomic function testing to support the diagnosis. To be enrolled, patients were required to fulfill consensus criteria for possible or probable MSA and to have a score ≤ 17 (omitting the erectile dysfunction score) on part one of the Unified Multiple System Atrophy Rating Scale (UMSARS) to ensure enrollment at an early disease stage.1,22
Patients with PD were diagnosed by a Mayo Clinic movement disorder specialist and had to be at Hoehn and Yahr stage 2 to 3 to qualify for the study. Patients with DLB were diagnosed by a Mayo Clinic behavioral neurologist based on published criteria and were within 4 years from DLB diagnosis.4 Healthy controls were without evidence of neurologic disease or autonomic dysfunction.
Subjects were excluded if they were pregnant or breastfeeding, scored 24 points or less on the Mini-Mental Status Examination (except for DLB patients), had a clinically significant or unstable medical or surgical condition that might preclude safe completion of the study or might affect the results of the study, or had taken any investigational products within 60 days prior to baseline.
Standard Protocol Approvals, Registrations, and Patient Consent
The study was approved by the Mayo Clinic Institutional Review Board, and written informed consent was obtained from all participants (or their proxies).
Clinical and Laboratory Study Assessments
A medical and neurologic history was obtained from all subjects, and all underwent a comprehensive general and neurologic examination. Medications that could potentially bias evaluations were held for 5 half-lives prior to neurologic assessments, autonomic testing, and spinal fluid collection. Neurologic impairment and deficits were quantified in MSA patients using the UMSARS (consisting of part I, which quantifies patients’ symptoms and function, and part II, which quantifies findings on neurologic examination).22 All subjects underwent MRI of the head, and all except for DLB patients underwent autonomic function testing including autonomic reflex screen and thermoregulatory sweat test. To quantify autonomic deficits, the Composite Autonomic Severity Score (CASS), a validated instrument to quantify the overall severity and distribution of autonomic failure based on standardized autonomic testing, was derived.23 Autonomic symptoms were assessed using the Composite Autonomic Severity Scale, COMPASS-select.24
CSF Collection and Analysis
After completion of clinical, laboratory, and imaging assessments, CSF was collected via routine spinal tap. A lumbar spinal needle was placed in the subarachnoid space via a standard posterior, intervertebral approach between lumbar levels 2 and 5; the specific level was determined individually for each patient based on anatomical considerations.
After collection of 5ml of CSF for safety assessments (cell count, protein), 10ml of CSF was collected into separate tubes for biomarker studies. CSF collection for the discovery cohort took place between May 1, 2013 and June 24, 2015, and for the confirmatory cohort between March 22, 2016 and August 9, 2019. CSF was placed on ice immediately after collection and hand-delivered to the laboratory for further processing. After centrifuging at 10,000rpm for 10 minutes at 4°C to remove any potential blood contamination, CSF was aliquoted into cryotubes and transferred to a −80°C freezer until the day needed for the biomarker assays. Assays were performed with the technologist blinded to clinical information.
NFL was measured using the UmanDiagnostics (Umeå, Sweden) enzyme-linked immunosorbent assay kit according to the manufacturer’s instructions. Standards and test samples were measured in duplicate, and the average of the 2 measurements was used for quantitative analyses. Samples were diluted 1:2 with kit diluent. Absorbance (450nm) was measured using an Omega Fluorescence Base microplate reader (BMG, Ortenberg, Germany). NFL levels were quantified using a third-order polynomial fit and measured in pg/ml.
αSyn oligomers were measured using PMCA, taking advantage of the seeding-nucleation process of αSyn aggregation, where misfolded oligomers seed the polymerization of monomeric protein. In principal, using a cyclical process, monomeric protein in excess is combined with the study sample containing minute amounts of misfolded oligomers and incubated to induce growth of polymers. The sample is then subjected to a mechanical force to break down the polymers, multiplying the number of seeds, resulting in an exponential increase in the number of seeds.
Monomeric protein was purified and characterized as previously described.21 In brief, a bacterial plasmid pET-21b carrying the coding DNA sequence for human αSyn containing 6xHis-tag at the C terminus was overexpressed in BL21(DE3) pLysS Escherichia coli cells (Invitrogen, Carlsbad, CA) at 25°C using 0.1mM isopropyl β-D-thiogalactoside for 6 hours. The bacterial pellets were lysed in 300mM sodium chloride (NaCl), 50mM sodium dihydrogen phosphate (pH 8.0), 1mM phenylmethylsulfonyl fluoride, 0.1mM tris-(2-carboxyethyl) phosphine, and 1mg/mL lysozyme followed by sonication on ice. After sonication, the solution was centrifuged at 12,000 × g for 15 minutes at 4°C, followed by 100,000 × g for 30 minutes at 4°C. Supernatant was filtered through a 0.45μm filter and loaded on to a nickel-affinity column (Nickel Sepharose Fast Flow, GE Healthcare, Chicago, IL). Bound proteins were eluted with a buffer containing 250mM imidazole. The fractions containing αSyn were pooled and dialyzed overnight at 4°C against 1X phosphate-buffered saline, pH 7.4. To remove any preformed seed or aggregate, protein solution containing αSyn was filtered through a 100kDa cutoff filter (Amicon Ultra, Millipore, Billerica, MA). Small aliquots (500μl/tube) of αSyn at a concentration of 5 to 6mg/ml were prepared in sterile Protein LoBind Tubes (Eppendorf, Hamburg, Germany), snap-frozen in liquid nitrogen, and stored at −80°C until use. The purity of αSyn preparation was evaluated by silver staining. Before experiment, an aliquot of αSyn was thawed in a cold-water bath.
αSyn monomers at a concentration of 1mg/mL in 100mM piperazine-N,N′-bis(ethanesulfonic acid) at pH 6.5 in 500mM NaCl were placed in black 96-well plates in the presence of 5μM concentration of thioflavin T (ThT) at a final volume of 200μl. Forty microliters of CSF sample was added to each well. Samples were then subjected to cyclic agitation for 1 minute at 500rpm followed by 29 minutes without shaking at 37°C. The increase in ThT fluorescence over time was monitored at excitation of 435nm and emission of 485nm daily for 15 days using a microplate spectrofluorometer (Gemini EM, Molecular Devices, Sunnyvale, CA). Each sample was run in duplicate, and the average of the maximum ThT fluorescence (arbitrary units [AU]) was measured during the 15 days of the assay for each sample used for quantitative analyses.
Statistical Analysis
Descriptive statistics were used to describe demographic, clinical, and autonomic characteristics by disease group, including mean, standard deviation, median, interquartile range, frequencies, and percentages as appropriate.
Given non-normal distribution in some groups, group differences in the spinal fluid markers were compared using Kruskal–Wallis test for comparing multiple groups with pairwise post hoc comparisons using Dunn test. Mann–Whitney U test was used for a priori comparisons of only 2 groups. Receiver operating characteristic (ROC) curves and optimal cutoff values were derived for differentiating MSA samples from controls and Lewy body synucleinopathies for both markers. All statistical tests were 2-sided, and p values < 0.05 were considered statistically significant. Analysis was performed using SPSS (SPSS Statistics 25, IBM, Armonk, NY).
Results
Participants
A total of 120 subjects were included in this study. The discovery cohort consisted of 24 subjects with MSA (19 MSA-C) and 14 control (CON) subjects. In the confirmatory cohort, 38 subjects with MSA, 15 CON, 16 PD, and 13 DLB were included.
Demographic, clinical, and autonomic characteristics of the combined groups are summarized in the Table 1. Patients with PD and DLB had longer disease duration and were slightly older than MSA and CON subjects. There was even gender distribution in the CON group, whereas there was male predominance in the MSA, and particularly PD and DLB groups. Both autonomic deficits and autonomic symptom burden were notably higher in MSA patients compared to PD and CON subjects, as expected.
TABLE. 1.
Demographic, Clinical, and Autonomic Characteristics by Disease Group
| Category | MSA | CON | PD | DLB |
|---|---|---|---|---|
| Age, yr | 59.2 ± 6.8 | 58.5 ± 7.4 | 65.9 ± 6.0 | 66.8 ± 8.3 |
| Sex, M (%) | 43 (69) | 14 (48) | 15 (94) | 13 (100) |
| Disease duration, yr (IQR) | 3.2 (2.4–4.4) | — | 7.5 (5.4–10.8) | 5.1 (4.2–8.5) |
| MSA-C (%) | 44 (71) | — | — | — |
| UMSARS I | 13.9 ± 3.7 | — | — | — |
| UMSARS II | 17.1 ± 6.0 | — | — | — |
| UMSARS total | 31.0 ± 9.1 | — | — | — |
| COMPASS-select | 33.2 ± 17.4 | 2.4 ± 2.3 | 12.3 ± 10.8 | — |
| CASS | 6.1 ± 1.8 | 1.1 ± 1.3 | 2.3 ± 2.0 | — |
| TST, % anhidrosis (IQR) | 53.6 (8.7–86.0) | 2.1 (0.5–15.7) | 2.5 (1.4–4.5) | — |
Normally distributed numeric variables are shown as mean and standard deviation, not normally distributed variables as median and IQR. Frequencies are reported as number and category percentage.
CASS = Composite Autonomic Severity Score; COMPASS = Composite Autonomic Severity Scale; CON = control; DLB = dementia with Lewy bodies; IQR = interquartile range; M = male; MSA = multiple system atrophy; MSA-C = MSA with cerebellar impairment; PD = Parkinson disease; TST = thermoregulatory sweat test; UMSARS = Unified Multiple System Atrophy Rating Scale.
Ten patients with a clinical diagnosis of MSA came to autopsy, and all were confirmed as MSA pathologically.
CSF Markers: NFL
In the discovery cohort, NFL in the CSF was markedly elevated in all MSA patients, resulting in perfect separation from the CON group (Fig 1, left panel, p < 0.001, Mann–Whitney). This finding was confirmed with the confirmatory cohort (see Fig 1, right panel, p < 0.001, Dunn). NFL was also significantly higher in MSA when compared to PD and DLB samples with still near-perfect separation, whereas there was no significant difference between PD, DLB, and CON groups (see Fig 1, right panel, MSA vs PD and MSA vs DLB, both p < 0.001, Dunn). There was no difference between MSA-P and MSA-C (p = 0.47, Mann–Whitney).
FIGURE 1:
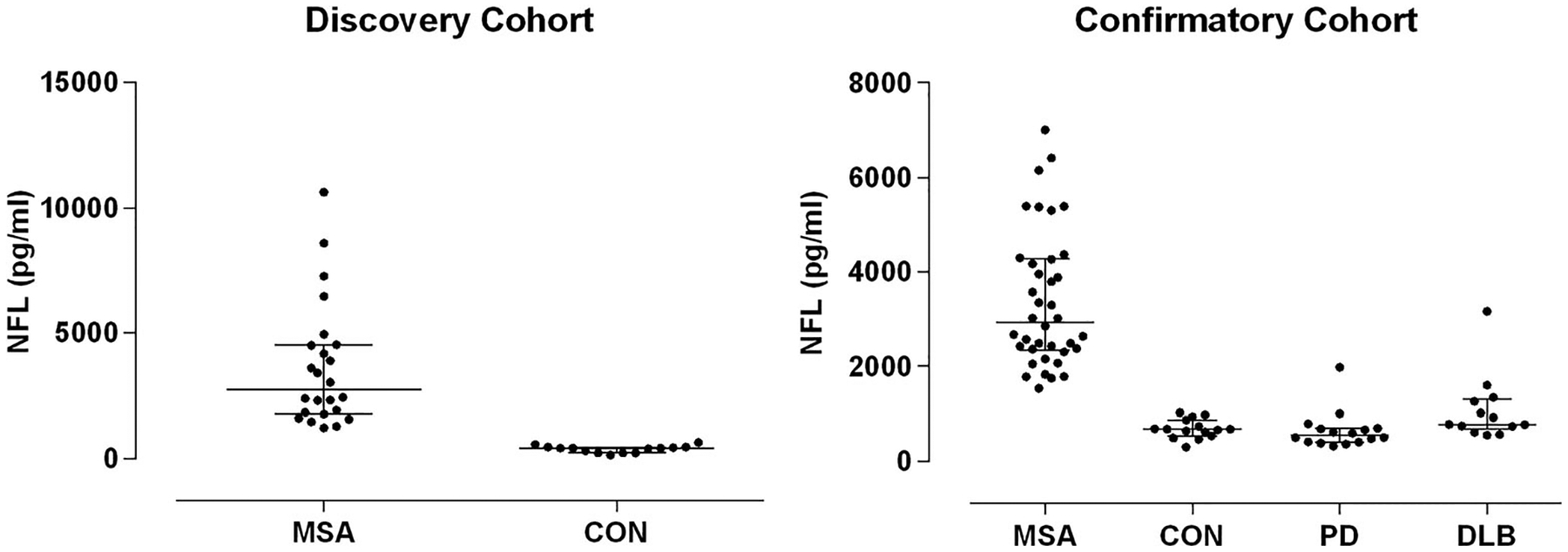
Neurofilament light chain (NFL) in cerebrospinal fluid. Discovery cohort (left) with multiple system atrophy (MSA) and control (CON) subjects, and confirmatory cohort (right) with MSA, CON, Parkinson disease (PD), and dementia with Lewy bodies (DLB) subjects are shown. There was perfect separation of MSA from CON subjects in both cohorts. There was also excellent separation of MSA and PD/DLB subjects. Values in CON subjects ranged from 153 to 1,024pg/ml.
CSF Markers: αSyn Oligomers
In the discovery cohort, there was no change in ThT fluorescence during the PMCA assay in any of the CON samples, whereas ThT fluorescence increased in all MSA samples, with the resulting maximum ThT fluorescence during the assay markedly elevated in all MSA compared to CON samples, resulting in even stronger group separation than for NFL (Fig 2, left panel, p < 0.001, Mann–Whitney). This finding was reproduced in the confirmatory cohort with clean separation of CON from MSA samples with the exception of 2 MSA samples that did not result in aggregation and remained at background ThT fluorescence for the duration of the assay (see Fig 2, middle panel, p = 0.002, Dunn); both were MSA-C, and there were no apparent clinical features that would distinguish those from the other cases. PD and DLB samples resulted in even greater maximum ThT fluorescence than MSA samples, with the exception of 2 samples in the DLB group that did not result in aggregation (see Fig 2, right panel). Nonreactive cases were again clinically indistinct. Apart from the nonreactive samples, there was perfect separation of PD and DLB not only from CON, but also MSA samples (PD vs MSA and CON, and DLB vs MSA and CON, both p < 0.001, Dunn). Maximum ThT fluorescence was not significantly different between PD and DLB samples. There was also no difference between MSA-P and MSA-C (p = 0.96, Mann–Whitney).
FIGURE 2:
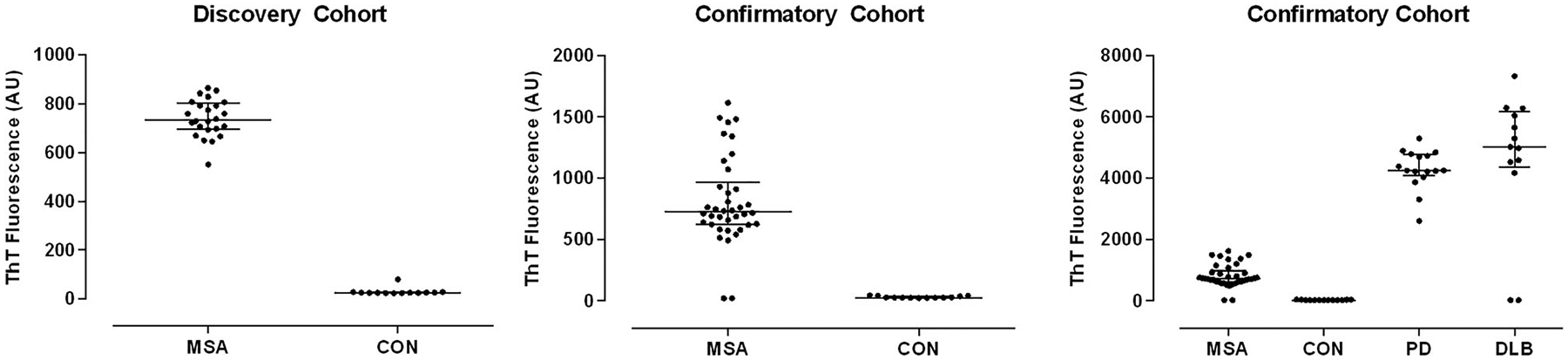
Alpha-synuclein oligomers in cerebrospinal fluid by protein misfolding cyclic amplification. Discovery cohort (left) with MSA and CON subjects, confirmatory cohort with multiple system atrophy (MSA) and control (CON) subjects to illustrate comparable separation (middle), and confirmatory cohort with MSA, CON, Parkinson disease (PD), and dementia with Lewy bodies (DLB) subjects (right) are shown. Apart from very few nonreactive samples in the disease cohorts, there was perfect separation of MSA from both CON and PD/DLB subjects. AU = arbitrary units; ThT = thioflavin T.
Further analysis of the reaction kinetics of the PMCA assay demonstrated notable differences beyond the magnitude of the reaction. The start of polymerization and formation of a reaction plateau occurred invariably earlier in MSA samples, both occurring in less than half the time compared to PD and DLB samples (Fig 3).
FIGURE 3:
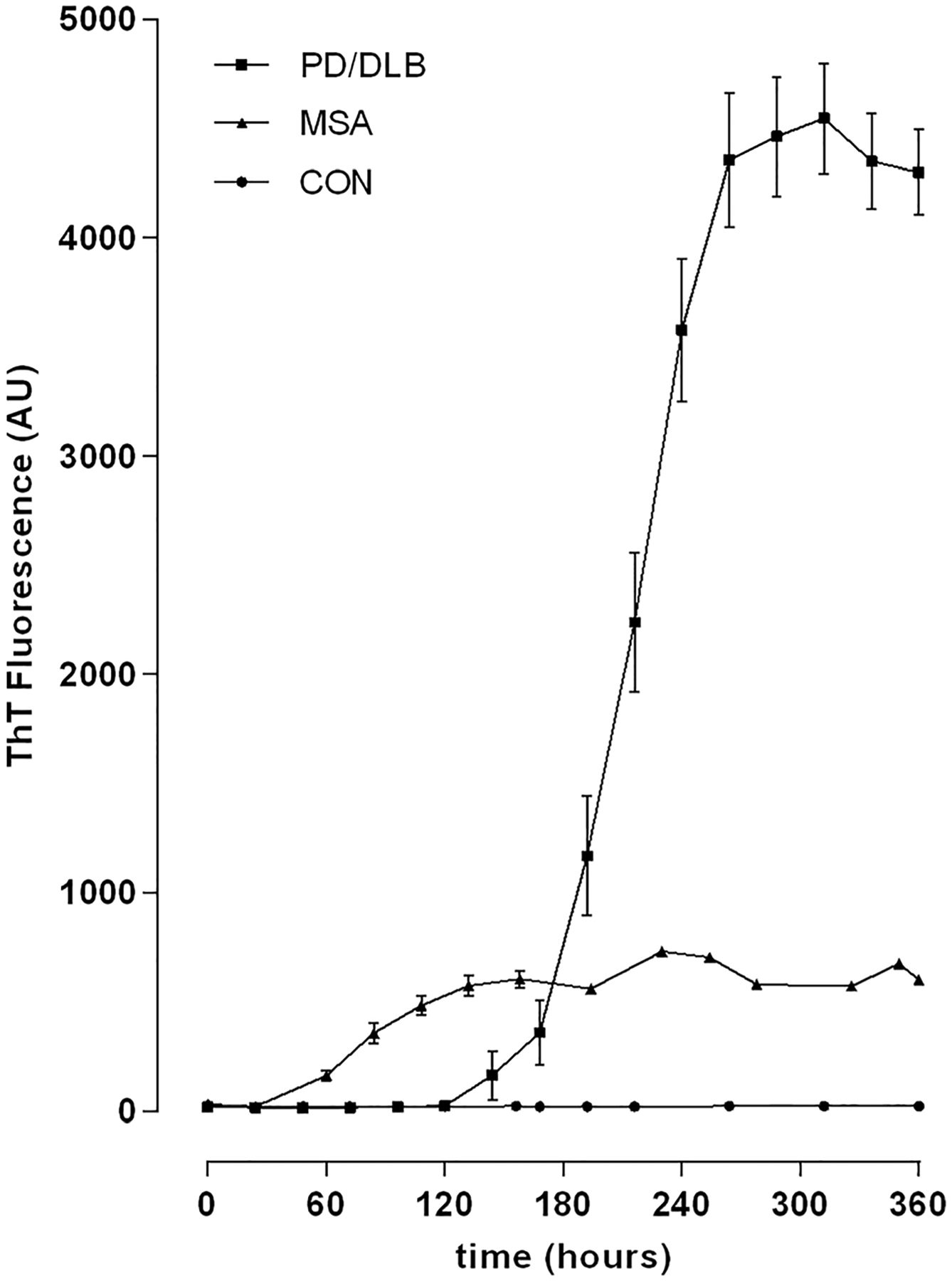
Protein misfolding cyclic amplification (PMCA) assay kinetics. Mean and standard error of the mean are shown at all measurement timepoints during the PMCA assay in multiple system atrophy (MSA), control (CON), and combined Parkinson disease (PD)/dementia with Lewy bodies (DLB) cohorts. Note nonreactive assay in CON samples, early start of polymerization and early, lower plateau in MSA samples, and late start of polymerization and formation of a late plateau at high polymerization levels in PD and DLB samples. AU = arbitrary units; ThT = thioflavin T.
ROC Analysis
For the purposes of ROC analysis, we combined samples from discovery and confirmatory cohorts. We also combined the 2 Lewy body synucleinopathy cohorts (PD and DLB), given almost identical findings for both CSF markers.
For differentiating MSA from CON using NFL, we found not surprisingly a perfect ROC curve, with an area under the curve of 1.0 (Fig 4A). Any cutoff value between 1,024 and 1,225pg/ml would result in 100% sensitivity and 100% specificity in differentiating MSA from CON; given the low variability of values in CON samples, a cutoff value of 1,100pg/ml is suggested. The differentiation of MSA from CON using αSyn oligomers resulted in a similarly impressive ROC area under the curve of 0.97, imperfect only because of the 2 nonreactive MSA samples (see Fig 4B). Any maximum ThT fluorescence cutoff value between 80 and 491AU would result in 97% sensitivity and 100% specificity in differentiating MSA from CON; given the very low variability of values in CON samples, a cutoff value of 150AU is suggested.
FIGURE 4:
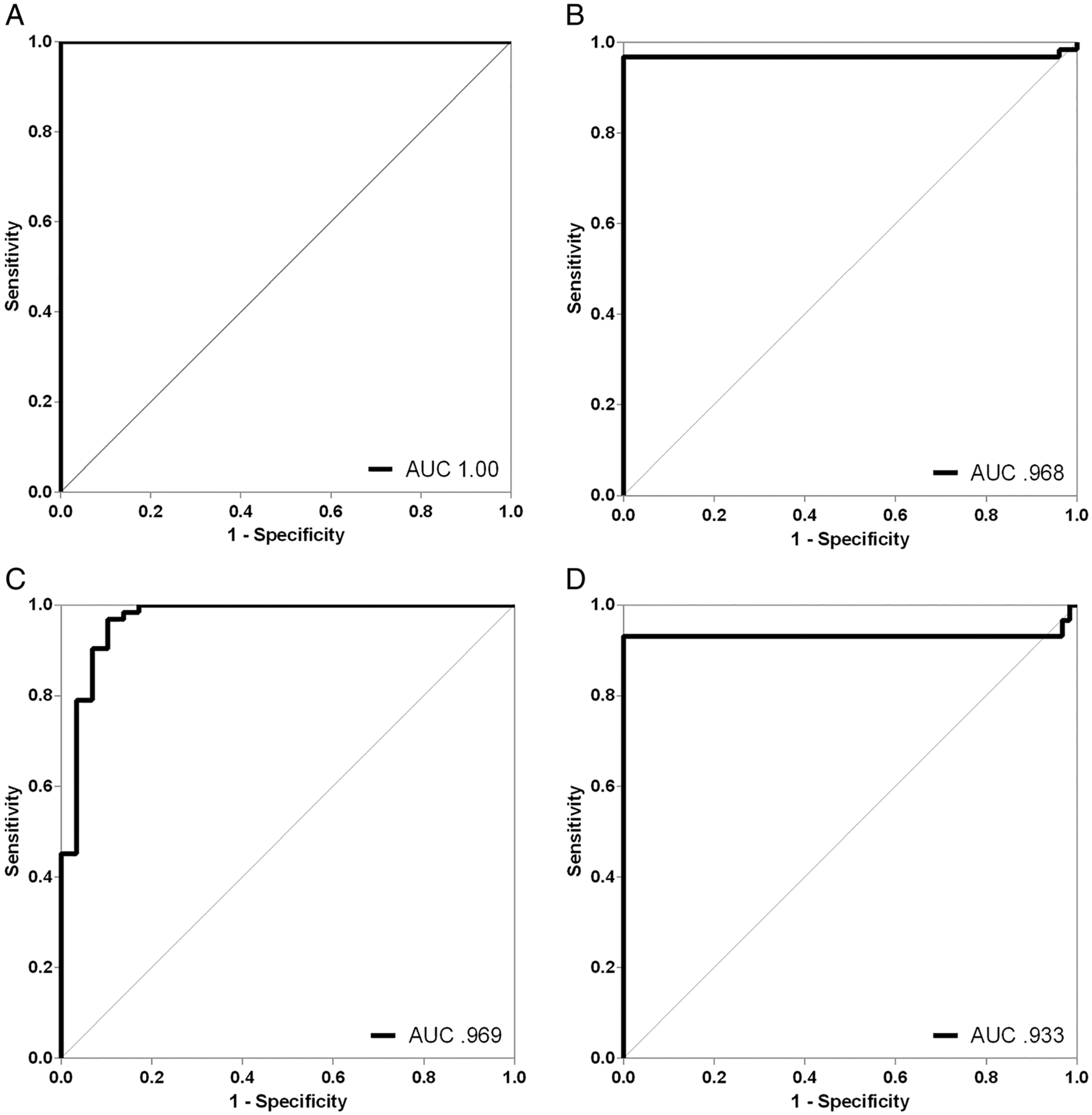
Receiver operating characteristic (ROC) curves. ROC curves for neurofilament light chain (NFL; A) and maximum thioflavin T (ThT) fluorescence during protein misfolding cyclic amplification (PMCA; B) differentiating multiple system atrophy (MSA) from control subjects, and ROC curves for NFL (C) and maximum ThT fluorescence during PMCA (D) differentiating MSA from Parkinson disease/dementia with Lewy bodies are shown. Both cerebrospinal fluid markers provided excellent separation of cohorts. AUC = area under the curve.
When combining both markers and cutoffs, requiring an NFL value > 1,100pg/ml AND ThT fluorescence > 150AU for differentiating MSA from CON would result in the same slightly imperfect sensitivity and perfect specificity as seen with ThT fluorescence alone, and requiring NFL > 1,100pg/ml OR ThT fluorescence > 150AU would result in the same perfect sensitivity and specificity as with NFL alone.
For differentiating MSA from PD/DLB using NFL, we found again a very high ROC area under the curve of 0.97, reflecting only minimal overlap between the groups (see Fig 4C). The best cutoff value was 1,400pg/ml, resulting in 97% sensitivity and 90% specificity in separating MSA from PD/DLB. The differentiation of MSA from PD/DLB using αSyn oligomers resulted in an ROC area under the curve of 0.93, imperfect only because of the 2 nonreactive samples in either group (see Fig 4D). Any maximum ThT fluorescence cutoff value between 1,615 and 2,605AU would provide 100% sensitivity and 93% specificity in separating MSA from PD/DLB. Given lower variability of values in MSA samples, a cutoff value of 2,000AU is suggested.
When combining both markers and cutoffs, requiring an NFL value > 1,400pg/ml and ThT fluorescence < 2,000AU for differentiating MSA from PD/DLB would result in 97% sensitivity and 100% specificity, whereas requiring an NFL value > 1,400pg/ml OR ThT fluorescence < 2,000AU for differentiating MSA from PD/DLB would result in 100% sensitivity, but specificity would decrease to 83%.
Discussion
An early and accurate clinical diagnosis of MSA has been and remains challenging. This is evidenced not only by the high rate of misdiagnosis in autopsy series, but also by the considerable delay between symptom onset and diagnosis.9,25 This delay is documented in several large natural history studies, which all report median survival from first symptom to death of 8 to 10 years, but median survival from time of diagnosis of 3 years or less, suggesting that the average patient has symptoms for >5 years before the diagnosis is made.25–27
Such delay in diagnosis is to some extent understandable in that making a diagnosis of a disabling and invariably fatal disease warrants a sensible degree of certainty. This approach is also reflected in the current consensus criteria for MSA, which have improved the certitude of diagnosis but at the price of deferring diagnosis until an advanced stage of disease.1 Based on these criteria, a diagnosis of “definitive” MSA can only be made postmortem. “Probable” MSA defines the highest level of diagnostic certainty achievable during life and requires strict clinical criteria to be met. A diagnosis of “possible” MSA still requires the presence of clinical criteria but can be aided by findings on MRI, positron emission tomography, or single photon emission computed tomography.1
Making an accurate diagnosis of MSA early in the disease course is not only important for patients and their families to learn about and better understand the cause of patients’ symptoms and impairments and the significant impact on life expectancy; it is also critically important considering that several promising treatment trials aiming at disease modification in MSA have failed, and there is reasonable concern that these trials may have failed because enrolled patients were at a disease stage that was too advanced for disease-modifying agents to be efficacious.28–31 With a number of new treatment strategies in the pipeline, the need for reliable biomarkers of early disease has become even more critical.32,33
This study was designed to systematically explore and confirm the role of 2 promising CSF markers, NFL and αSyn oligomers, as diagnostic biomarkers of early MSA. Expanding upon our previous work, we aimed with this study for the first time to establish the clinical value of these markers in separate, prospective cohorts of carefully phenotyped patients with early disease and at matched stages of disease, contrasting findings in early MSA with those in CON subjects, as well as early PD and DLB. In an analysis blinded to clinical data, we found that both markers provided clean separation of MSA patients not only from CON subjects, but also from patients with Lewy body synucleinopathies, that is, PD and DLB, who were studied at a stage of comparable disease severity. NFL values > 1,100pg/ml allowed for perfect separation of MSA patients from CON subjects, and values > 1,400pg/ml provided excellent separation of MSA from PD and DLB (97% sensitivity, 90% specificity). NFL was not elevated in the CSF of the vast majority of PD and DLB cases, apart from a few outliers, consistent with previous studies.16–18 αSyn oligomers measured utilizing PMCA were not detectable in any of our CON samples, but in all but 2 MSA and in all but 2 PD/DLB cases. We confirmed our previously reported highly different αSyn reaction kinetics between MSA and PD/DLB samples, which resulted in perfect separation between these disorders with the exception of the 4 nonreactive samples.20 The cutoff values of 150AU for separating MSA from CON samples and of 2,000AU for separating MSA from PD/DLB samples define 3 ranges: (1) nonreactive or normal range (<150AU), (2) MSA range (150–2,000AU), and (3) PD/DLB range (>2,000AU). Due to the nonreactive disease samples, the assay provided “only” 97% sensitivity but 100% specificity in separating MSA cases from CON subjects, and 100% sensitivity but 93% specificity separating MSA from PD/DLB cases. Combining the 2 assays and requiring both NFL and αSyn oligomers to be “positive” for MSA would provide a test with 97% sensitivity and 100% specificity in separating MSA from PD/DLB. This remarkable separation of early MSA cases not only from normal controls, but also from the Lewy body synucleinopathies achieved using 2 independent CSF markers confirms these markers as valuable disease biomarkers that achieve better diagnostic certitude than any previously described clinical, autonomic, biofluid, or imaging marker. Combining these markers as a dual biofluid marker for MSA may add little additional sensitivity or specificity, but would be expected to add additional confirmatory value, particularly if the results of one of the markers is borderline or the assay is nonreactive.
NFL is considered a marker of axonal degeneration that has been reported to be elevated in CSF and plasma in many neurodegenerative, neuroinflammatory, and neurotraumatic conditions.17,34,35 Although not disease-specific, in neurodegenerative disorders it likely reflects the tempo and distribution of neuronal degeneration, and has previously been described not only to be greatly increased in MSA but to have value in distinguishing MSA from PD and DLB.15,16,18,19 Our findings confirm the previous reports and extend previous observations to patients at an early disease stage, when disease biomarkers are particularly helpful. Despite our focus on early disease, our data show even cleaner separation between groups than previous reports; this may relate to the standardization and detail of clinical and autonomic phenotyping of our patients, who were prospectively enrolled and followed longitudinally.
A few previous studies on αSyn levels in the CSF of patients with synucleinopathies have reported decreased total αSyn levels in both MSA and PD, although the separation from controls was suboptimal.36–38 In contrast, in a study utilizing postmortem CSF and immunoassays, Foulds and colleagues report increased total αSyn levels, more prominently increased αSyn oligomers, and particularly phosphorylated αSyn oligomers in MSA compared to controls, PD, and DLB patients.39 αSyn oligomers are of particular interest in the synucleinopathies, as they represent the misfolded, aggregated, and neurotoxic protein.40,41 Among approaches to detect αSyn oligomers, the PMCA approach has evolved as one of the most promising techniques.20,21 This technique is particularly intriguing also because it mimics in vitro the process of seeding and progressive misfolding of αSyn that has recently been recognized to underlie the propagation and progressive spread of misfolded protein from cell to cell.42–44 PMCA has been reported to identify patients with PD with high sensitivity and specificity, and we could recently demonstrate that αSyn aggregates associated with PD and MSA correspond to different conformational strains of α-synuclein utilizing this technique.20,21 In that study, we also showed that the maximum ThT fluorescence during PMCA was significantly greater in CSF samples from PD patients than in samples from patients with MSA derived from different cohorts and centers. Our prospective data presented here reproduce these earlier findings with similar if not stronger separation of early MSA not only from PD, but also DLB patients. We could furthermore reproduce the markedly different reaction kinetics with start of polymerization and formation of a reaction plateau occurring invariably earlier, and the maximum fluorescence being invariably lower in MSA samples than in PD and DLB samples. This observation supports the reported biological differences in αSyn of MSA and Lewy body synucleinopathies and is in line with the concept of different self-perpetuating conformational strains that perpetuate misfolding of normal monomer according to a specific template.20,43
The strengths of the study presented here include the focus on early disease, when diagnostic biomarkers are of greatest clinical utility; the prospective collection of CSF samples in both discovery and confirmatory cohorts; all patients in this study being longitudinally followed, ensuring a disease course consistent with the diagnosis at the time of CSF collection; the meticulous clinical, autonomic, and imaging phenotyping of all subjects; and the standardized, rigorously careful handling of samples in this single-center study. Weaknesses include differences in gender distribution and mild differences in age between the groups.
In conclusion, our findings confirm and establish 2 CSF markers, NFL and αSyn oligomers via PMCA, as robust diagnostic biomarkers that confidently distinguish early MSA from healthy controls on the one hand, and MSA from Lewy body synucleinopathies on the other hand. These markers, alone and in combination, possess diagnostic value that may be superior to any other currently available clinical, laboratory, or imaging marker, and appear ready for clinical implementation. Applying these biomarkers may prove particularly valuable in (1) aiding the clinical diagnosis at early disease stages when MSA is considered, but diagnostic criteria are not or only partially met; (2) the workup of parkinsonism, particularly when atypical features are present; and (3) future clinical trials in synucleinopathies, where enhanced diagnostic certainty may be particularly valuable. The PMCA findings furthermore highlight important biological differences of αSyn in MSA and Lewy body synucleinopathies, aiding our understanding of the pathophysiology of these disorders. Applying these CSF markers to even earlier, prodromal disease stages would be a logical next step.
Acknowledgments
This publication was made possible by the National Institute of Neurological Disorders and Stroke (NINDS) (R01NS092625, K23NS075141), the National Center for Advancing Translational Sciences (NCATS) (UL1TR 000135), the National Institute on Aging (NIA) (P50AG 016574, U01NS100620, R01AG055053, R01AG061069), the US Food and Drug Administration (FDA; R01FD 004789), grants from the Michael J. Fox Foundation for PD, Cure MSA Foundation, Sturm Foundation, Mayo Clinic Dorothy and Harry T. Mangurian Jr Lewy Body Dementia Program, and Little Family Foundation, and Mayo funds. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH or FDA.
Footnotes
Potential Conflicts of Interest
C.S. is a Founder, Chief Scientific Officer, and Member of the Board of Directors of Amprion, a biotechnology company that focuses on the commercial use of PMCA (real-time quaking-induced conversion) for high-sensitivity detection of misfolded protein aggregates. The remaining authors have nothing to report.
References
- 1.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kowal SL, Dall TM, Chakrabarti R, et al. The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 2013;28:311–318. [DOI] [PubMed] [Google Scholar]
- 3.Marras C, Beck JC, Bower JH, et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thaisetthawatkul P, Boeve BF, Benarroch EE, et al. Autonomic dysfunction in dementia with Lewy bodies. Neurology 2004;62:1804–1809. [DOI] [PubMed] [Google Scholar]
- 6.Figueroa JJ, Singer W, Parsaik A, et al. Multiple system atrophy: prognostic indicators of survival. Mov Disord 2014;29:1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iodice V, Lipp A, Ahlskog JE, et al. Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 2012;83:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125:861–870. [DOI] [PubMed] [Google Scholar]
- 9.Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osaki Y, Wenning GK, Daniel SE, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002;59:1486–1491. [DOI] [PubMed] [Google Scholar]
- 11.Wenning GK, Tison F, Ben Shlomo Y, et al. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133–147. [DOI] [PubMed] [Google Scholar]
- 12.Meijer FJ, Aerts MB, Abdo WF, et al. Contribution of routine brain MRI to the differential diagnosis of parkinsonism: a 3-year prospective follow-up study. J Neurol 2012;259:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huppertz HJ, Moller L, Sudmeyer M, et al. Differentiation of neurodegenerative Parkinsonian syndromes by volumetric magnetic resonance imaging analysis and support vector machine classification. Mov Disord 2016;31:1506–1517. [DOI] [PubMed] [Google Scholar]
- 14.Worker A, Blain C, Jarosz J, et al. Diffusion tensor imaging of Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy: a tract-based spatial statistics study. PLoS One 2014;9:e112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdo WF, van de Warrenburg BP, Munneke M, et al. CSF analysis differentiates multiple-system atrophy from idiopathic late-onset cerebellar ataxia. Neurology 2006;67:474–479. [DOI] [PubMed] [Google Scholar]
- 16.Hall S, Ohrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or Parkinsonian disorders. Arch Neurol 2012;69:1445–1452. [DOI] [PubMed] [Google Scholar]
- 17.Wang SY, Chen W, Xu W, et al. Neurofilament light chain in cerebrospinal fluid and blood as a biomarker for neurodegenerative diseases: a systematic review and meta-analysis. J Alzheimers Dis 2019;72:1353–1361. [DOI] [PubMed] [Google Scholar]
- 18.Abdo WF, Bloem BR, Van Geel WJ, et al. CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson’s disease. Neurobiol Aging 2007;28:742–747. [DOI] [PubMed] [Google Scholar]
- 19.Abdo WF, van de Warrenburg BP, Kremer HP, et al. CSF biomarker profiles do not differentiate between the cerebellar and Parkinsonian phenotypes of multiple system atrophy. Parkinsonism Relat Disord 2007;13:480–482. [DOI] [PubMed] [Google Scholar]
- 20.Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020;578:273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 2017;74:163–172. [DOI] [PubMed] [Google Scholar]
- 22.Wenning GK, Tison F, Seppi K, et al. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19:1391–1402. [DOI] [PubMed] [Google Scholar]
- 23.Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc 1993;68:748–752. [DOI] [PubMed] [Google Scholar]
- 24.Lipp A, Sandroni P, Ahlskog JE, et al. Prospective differentiation of multiple system atrophy from Parkinson’s disease, with and without autonomic failure. Arch Neurol 2009;66:742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon EA, Suarez MD, Ahlskog JE, et al. Survival in multiple system atrophy—insights from a large retrospective cohort. Ann Neurol 2014;76:S39 (Abstract). [Google Scholar]
- 26.Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14:710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12:264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levin J, Maass S, Schuberth M, et al. Safety and efficacy of epigallocatechin gallate in multiple system atrophy (PROMESA): a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2019;18:724–735. [DOI] [PubMed] [Google Scholar]
- 29.Low PA, Robertson D, Gilman S, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poewe W, Seppi K, Fitzer-Attas CJ, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 2015;14:145–152. [DOI] [PubMed] [Google Scholar]
- 31.Singer W, Low PA. Optimizing clinical trial design for multiple system atrophy: lessons from the rifampicin study. Clin Auton Res 2015;25:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palma JA, Kaufmann H. Novel therapeutic approaches in multiple system atrophy. Clin Auton Res 2015;25:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singer W, Dietz AB, Zeller AD, et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology 2019;93:e77–e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bagnato S, Grimaldi LME, Di Raimondo G, et al. Prolonged cerebrospinal fluid neurofilament light chain increase in patients with post-traumatic disorders of consciousness. J Neurotrauma 2017;34:2475–2479. [DOI] [PubMed] [Google Scholar]
- 35.Varhaug KN, Torkildsen O, Myhr KM, Vedeler CA. Neurofilament light chain as a biomarker in multiple sclerosis. Front Neurol 2019;10:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 2011;69:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tokuda T, Qureshi MM, Ardah MT, et al. Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 2010;75:1766–1772. [DOI] [PubMed] [Google Scholar]
- 38.Tokuda T, Salem SA, Allsop D, et al. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun 2006;349:162–166. [DOI] [PubMed] [Google Scholar]
- 39.Foulds P, Mann DM, Allsop D. Phosphorylated alpha-synuclein as a potential biomarker for Parkinson’s disease and related disorders. Expert Rev Mol Diagn 2012;12:115–117. [DOI] [PubMed] [Google Scholar]
- 40.Outeiro TF, Putcha P, Tetzlaff JE, et al. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 2008;3:e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tetzlaff JE, Putcha P, Outeiro TF, et al. CHIP targets toxic alpha-synuclein oligomers for degradation. J Biol Chem 2008;283:17962–17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 2015;112:E5308–E–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 2018;21:1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woerman AL, Stohr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A 2015;112:E4949–E4958. [DOI] [PMC free article] [PubMed] [Google Scholar]