

Abstract
Background
Epithelial sodium channel (ENaC) is an important regulator of airway surface liquid volume; ENaC is hyperactivated in cystic fibrosis (CF). ENaC inhibition is a potential therapeutic target for CF. Here, we report in vitro and in vivo results for BI 1265162, an inhaled ENaC inhibitor currently in Phase II clinical development, administered via the Respimat® Soft Mist™ inhaler.
Methods
In vitro inhibition of sodium ion (Na+) transport by BI 1265162 was tested in mouse renal collecting duct cells (M1) and human bronchial epithelial cells (NCI-H441); inhibition of water transport was measured using M1 cells. In vivo inhibition of liquid absorption from rat airway epithelium and acceleration of mucociliary clearance (MCC) in sheep lungs were assessed. Fully differentiated normal and CF human epithelium was used to measure the effect of BI 1265162 with or without ivacaftor and lumacaftor on water transport and MCC.
Results
BI 1265162 dose-dependently inhibited Na+ transport and decreased water resorption in cell line models. BI 1265162 reduced liquid absorption in rat lungs and increased MCC in sheep. No effects on renal function were seen in the animal models. BI 1265162 alone and in combination with CF transmembrane conductance regulator (CFTR) modulators decreased water transport and increased MCC in both normal and CF airway human epithelial models and also increased the effects of CFTR modulators in CF epithelium to reach the effect size seen in healthy epithelium with ivacaftor/lumacaftor alone.
Conclusion
These results demonstrate the potential of BI 1265162 as a mutation agnostic, ENaC-inhibitor-based therapy for CF.
Short abstract
ENaC inhibition is a potential strategy for a mutation-agnostic therapy in CF. In preclinical studies, BI 1265162 is a potent ENaC inhibitor, alone and in synergy with CFTR modulators, supporting Phase I clinical development. https://bit.ly/3mCeWE9
Introduction
Cystic fibrosis (CF) is an autosomal recessive genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene [1], which facilitates chloride secretion across apical cell membranes. CFTR and the epithelial sodium channel (ENaC) together maintain a finely tuned homeostatic mechanism to keep airway surfaces hydrated [2, 3]. Sufficient airway surface hydration facilitates mucociliary clearance (MCC), which is needed for a normal lung environment [4, 5]. In CF airways, the marked reduction of functional CFTR leads to reduction in airway surface liquid (ASL) height, dehydration of airway surfaces and sticky mucus, leading to impaired MCC, mucous plugging, neutrophilic infiltration and chronic bacterial infection [4–6]. This can cause airway obstruction/destruction, and ultimately respiratory failure and death [7].
Previous pulmonary treatments of CF have targeted downstream effects of the disease, such as mucous plugging and infection, as well as providing supportive care. More recent corrective therapy options target the CFTR defects [8–10]. However, with over 2000 identified mutations in the CFTR gene [11] that could lead to either complete absence of the CFTR protein or a malformed CFTR protein [12], targeting CFTR is currently not an option for all patients with CF [7]. At least 10% of patients with CF are unable to receive CFTR modulator therapy due to unsuitable genotype [13]. In addition, despite the best available therapy targeting specific genotypes of CF, patients continue to have exacerbations, with room for improvement in lung function [10, 14], and inflammation is not always reversible with current therapies [15]. Therapies that work on pathways independent of CFTR correction (additive) or synergistic to CFTR modulation would provide additional therapeutic benefit. A combination of multiple therapies will optimally improve quality of life and best prolong patient survival [5].
An ion-transporting pathway that could be targeted to ameliorate the impact of CF is the ENaC pathway. ENaC facilitates sodium ion (Na+) and, as a consequence, water resorption from the apical surface [16] and plays an important role in ASL volume regulation and effective MCC [17]. Hyperactivation of ENaC is known in CF, potentially as a consequence of both an increase in channel number and an increase in open probability of the channel [18]. This leads to increased absorbance of Na+ followed by water resorption from the apical surface, causing reduced ASL height, dehydrated mucus and compressed cilia, subsequently resulting in poor MCC [16, 19]. It is thought that the CFTR protein may play a role in ENaC modulation; however, the nature of this interaction is not fully understood [5, 20].
ENaC inhibition provides a mutation-agnostic therapeutic approach (i.e. independent of the type of underlying CFTR mutation) that can improve ASL height, mucus hydration and MCC in airways [5, 21]. This is predicted to be synergistic or at least additive with CFTR modulators [5]. Unfortunately, development of ENaC inhibitors so far has not translated into clinical success [16], likely due to poor potency, inadequate dosing and/or deposition by inhalation, induced hyperkalaemia, short study duration, non-study-related exacerbations or poor endpoint sensitivity.
BI 1265162 is an inhaled ENaC inhibitor that is expected to provide a mutation-agnostic approach for treating CF. It is administered via the Respimat® Soft Mist™ inhaler, which is a handheld, propellant-free device that produces a slow-moving, long-lasting mist that enables medication to reach deep into the lungs [22]. BI 1265162 is currently in Phase II clinical development. Here, we report the results from in vitro experiments and animal models designed to establish the clinical potential of BI 1265162 for the treatment of CF.
Methods
A brief summary of methods is given below. For detailed methodology, see the supplementary material.
In vitro inhibition of Na+ transport
The inhibition of Na+ transport by BI 1265162 was tested in a mouse renal collecting duct cell line (M1) and a human bronchial epithelial cell line (NCI-H441).
M1 and NCI-H441 cells were cultivated on polyester Transwell® filters to electrically tight monolayers. ENaC expression was then upregulated with dexamethasone to achieve sufficient short circuit current (ISC) [23]. The apical bath solution was removed to create an air–liquid interface, and filters containing cell layers were inserted into an Ussing chamber. BI 1265162 was administered at final concentrations of 10−10, 10−9, 10−8, 10−7 and 10−6 M in a stepwise manner to the apical solution of each filter. At the end of each experiment, ISC was measured by inhibiting Na+ transport with 3 µM amiloride (maximum inhibition), as previously described [24, 25]. Differences in ISC after addition of test compounds were compared against maximum inhibition, and the inhibition concentration that reduced the effect by 50% (IC50) was calculated. M1 experiments were repeated four times and H441 experiments were repeated twice (n=5 for each experiment).
In vitro inhibition of water resorption
ENaC-mediated water transport with and without BI 1265162 was measured in electrically tight monolayers of M1 cells using the Transwell® system (E-figure 1).
After culture, the apical liquid volume from the M1 cell layers was carefully aspirated and replaced with either 0.9% saline control or 0.9% saline containing BI 1265162. For determination of volume of water on the apical side, tritiated water was then added and mixed with the apical volume of the Transwell®, and water resorption measured by scintigraphy using a radiometric dilution assay. Experiments were carried out in duplicate.
In vivo inhibition of liquid absorption
Efficacy of BI 1265162 on inhibition of liquid absorption from airway epithelium was determined in male Wistar rats.
Male Wistar rats were anaesthetised with isoflurane prior to intratracheal instillation of test compound or vehicle. Ringer Lactate Solution pH 5 alone or with increasing concentrations (0.03, 0.3, 3 and 10 μg·kg−1) of BI 1265162 was instilled into the lungs. Three hours after instillation, renal effects of BI 1265162 were determined by measuring serum aldosterone by ELISA. The animals were then sacrificed under anaesthesia, lungs were removed, non-pulmonary tissue was dissected and the weight of the lung was determined. Inhibition of liquid transport was calculated from lung weights in the treated group versus lung weights of a negative control group, and an untreated control group was used as reference. Experiments were carried out on three separate occasions.
Acceleration of MCC in a sheep model
Stimulation of MCC by BI 1265162 was tested in a sheep model of whole-lung clearance measuring the retention of a radioactive tracer.
Adult ewes (n=2 per dose) were anaesthetised by local application of lidocaine prior to nasal intubation via bronchoscope, then administered doses (0.1, 1.0 and 10 μg·kg−1) of BI 1265162 or vehicle (deionised water) using an AirLife™ nebuliser. The effects of BI 1265162 or vehicle on MCC were measured by administration of aerosolised technetium-labelled sulfur colloid (99mTc-SC) followed by γ scintigraphy as previously described [26]. Serial images were obtained over a 2-h period, and counts were obtained from the right lung.
Blood was collected and plasma potassium levels were determined to assess renal effects of BI 1265162.
In vitro effect on water transport and MCC in CF donor cells
The effect of BI 1265162 (0.1 and 10 μM), with or without ivacaftor (IVA; CFTR corrector; 0.1 µM), lumacaftor (LUM; CFTR potentiator; 3 µM) and isoproterenol (ISO; a non-selective β-adrenoreceptor agonist that upregulates CFTR expression [27]; 100 µM) on water transport, MCC and ciliary beat frequency (CBF) was evaluated using MucilAir™ (Epithelix Sàrl (Suisse), Plan-Les-Ouates, Geneva, Switzerland; a pseudostratified, fully differentiated 3D model of human epithelium on semi-porous membrane inserts) consisting of either primary human bronchial cells isolated from a donor without CF or from a donor with ΔF508 homozygous CF.
Water transport was measured on Days 2 and 5 of the study by weighing the semi-porous membrane inserts containing the cells. MCC was measured on Day 7 by video-tracking the movements of polystyrene microbeads added to the apical surface of MucilAir™.
Results
BI 1265162 inhibits Na+ transport in vitro
Stepwise increasing doses of BI 1265162 (10−10–10−6 M) in the Ussing chamber assay resulted in a dose-dependent decrease in amiloride-sensitive ISC, reflecting Na+ transport inhibition in both cell lines (mean IC50 with M1 and NCI-H441 cells: 3×10−9 M and 8×10−9 M, respectively). Full inhibition was reached at the highest concentration (figure 1). For amiloride, IC50 was 2.10×10−7 M in M1 cells and 2.38×10−7 M in NCI-H441 cells.
FIGURE 1.
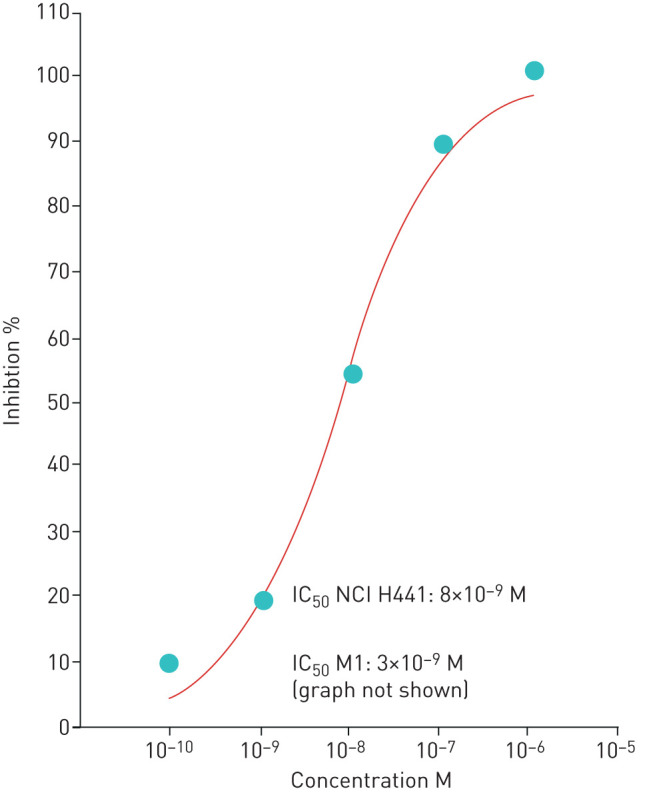
Concentration–response curves and IC50 values for BI 1265162 in NCI-H441 cells. Inhibition of Na+ transport by BI 1265162 was tested in M1 and NCI-H441 cells. Cells were cultivated on polyester Transwell® filters to electrically tight monolayers. Filters containing cell layers were inserted into a Ussing chamber and short-circuit current (ISC) measured at increasing BI 1265162 final concentrations. ISC was compared against maximum inhibition with amiloride and IC50 calculated. IC50: inhibition concentration that reduced the effect by 50%; M1: mouse renal collecting duct cell line.
BI 1265162 inhibits water resorption in vitro
The M1 cells seeded in the Transwell® insert formed a monolayer and transported Na+ via ENaC from the applied saline out of the apical compartment leading to water resorption. The activity representing 0% transport was calculated to be 3763 disintegrations per minute (dpm) for experiment 1 and 2660 dpm for experiment 2. The mean cell control activity was measured to be 4401 dpm and 3205 for experiments 1 and 2, respectively, resulting in a delta activity corresponding to 100% transport of 638 dpm and 545 dpm and a 1% activity of 6.38 dpm and 5.45 dpm, respectively. Addition of 3 μM BI 1265162 resulted in an inhibition of ENaC-mediated water resorption of 76% (mean of both experiments) compared with saline control (experiment 1: saline, 638 dpm; BI 1265162, 174 dpm (73% decrease); experiment 2: saline, 545 dpm; BI 1265162, 117 dpm (78% decrease). Variation between experiments was 5%) (figure 2).
FIGURE 2.
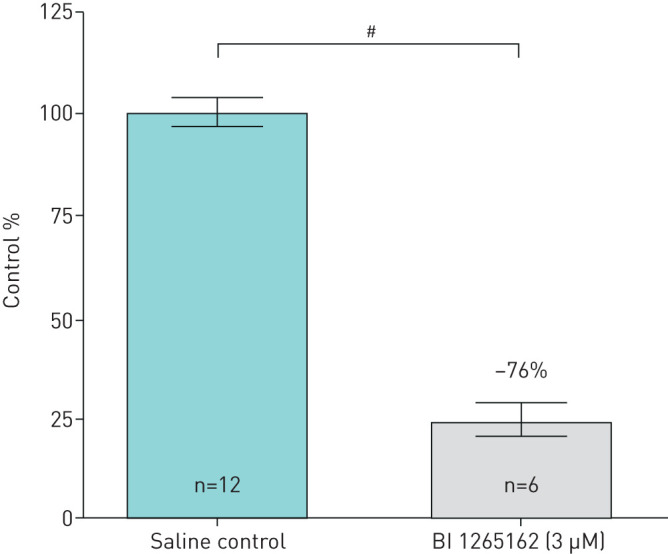
Inhibition of water resorption by BI 1265162 in M1 cell line. Cells were cultivated on polyester Transwell® filters to electrically tight monolayers. Apical liquid volume replaced with either saline control or saline containing 3 µM BI 1265162. Tritiated water was then added and mixed with the apical volume of the Transwell®, and water resorption measured by scintigraphy using a radiometric dilution assay. #: p<0.0001, unpaired t-test.
BI 1265162 inhibits liquid absorption in a rat model
Treatment with BI 1265162 showed a reduction of airway fluid absorption in rat lungs. BI 1265162 doses of 10, 3, 0.3 and 0.03 μg·kg−1 showed an inhibition of 32.9%, 31.3%, 26.2% and 5.9% after 3 h, respectively (figure 3). BI 1265162 reduced the liquid absorption from rat lungs with a dose required to reach 50% (ED50), 70% (ED70) and 90% (ED90) of the compound effect of 0.09, 0.18 and 0.54 μg·kg−1, respectively. Compared with placebo, mean change in aldosterone was 18%, 18%, 45% and –18% for 10, 3, 0.3 and 0.03 μg·kg−1 of BI 1265162, respectively, without dose dependency. No dose-dependent effect on serum aldosterone level was observed in the study.
FIGURE 3.
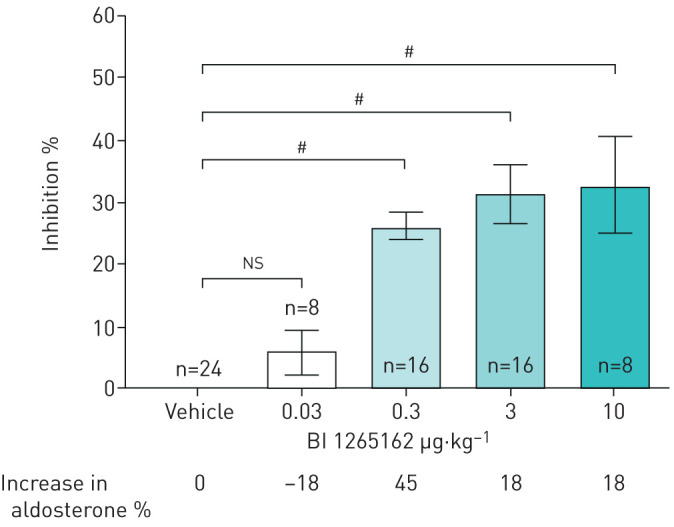
Inhibition of lung fluid absorption by BI 1265162 in rats. Ringer lactate solution pH 5 alone or with increasing concentrations of BI 1265162 was instilled into the lungs of male Wistar rats. Three hours after instillation, animals were sacrificed and the weight of the lungs was determined. Inhibition of liquid transport was calculated from lung weights in the treated group versus lung weights of a negative control group, and an untreated control group was used as reference. Data are presented as mean±sem. #: p<0.0001, one-way ANOVA, Dunnett's multiple comparison test.
BI 1265162 accelerates MCC in a sheep model without affecting plasma potassium
In the sheep model of whole-lung clearance, BI 1265162 led to a reduction in the retention of radioactive tracer over 2 h, indicating a higher MCC. The mean differences from vehicle for 1 and 10 μg·kg−1 of BI 1265162 were –12.6% and –19.3%, respectively (p<0.05; figure 4a). ED50 to accelerate MCC in sheep was estimated to be 1 μg·kg−1; ED90 was calculated to be 2.3 μg·kg−1. There were no increases in plasma K+ over a 24-h period (figure 4b).
FIGURE 4.

Effect of BI 1265162 on a) retention of tracer in lung of sheep and b) K+ concentration in sheep; n=2 per dose. Adult ewes were administered varying doses of BI 1265162 or vehicle using an AirLife™ nebuliser. Mucociliary clearance was measured by administration of aerosolised technetium-labelled sulfur colloid followed by γ scintigraphy. Serial images were obtained over a 2-h period, and counts were obtained from the right lung. Blood was collected and plasma potassium was determined to assess renal effects of BI 1265162. Data are presented as mean (range). #p=0.0027 and 0.0003 at 2 h after radiolabel administration, vehicle versus 1 μg·kg−1 and 10 μg·kg−1, respectively; one-way ANOVA.
BI 1265162 attenuates water transport and improves MCC in normal and CF tissues
BI 1265162 dose-dependently induced increases of the inserts’ weight, showing reduced water transport from the apical to basolateral compartment in both normal and CF epithelial cells, either alone, in combination with ISO or IVA/LUM, or both. This indicates clearly that BI 1265162 acts on the liquid homeostasis of airways epithelia. The effect was preserved at Day 5 (figure 5a). In normal epithelium, MCC was increased by ISO or with a combination of BI 1265162 with other treatments, but not by BI 1265162 alone. In CF epithelium, MCC was almost 0 µm·s−1 with just vehicle added. Addition of BI 1265162 increased MCC dose-dependently, reaching levels seen in normal epithelium. Addition of IVA/LUM increased MCC alone or together with ISO to ∼15 µm·s−1 and ∼50 µm·s−1, respectively (∼50% of the respective values derived from normal epithelium with the same treatments). Addition of BI 1265162 led to a nearly complete restoration of MCC in CF epithelium also treated with ISO or IVA/LUM (figure 5b).
FIGURE 5.

Effect of BI 1265162 on a) water transport and b) MCC in normal and CF MucilAirTM cultures. The effect of BI 1265162 with or without CFTR modulators and isoproterenol (an upregulator of CFTR expression) on water transport and MCC was evaluated in a pseudostratified, fully differentiated 3D model of human epithelium (n=5 cultures per treatment) employing bronchial cells from a CF and a non-CF donor. Water transport was measured on Days 2 and 5 of the study by weighing the membrane inserts containing the cells. MCC was measured on Day 7 by video-tracking the movements of polystyrene microbeads added to the apical surface of MucilAir™. Error bars show standard error of mean. MCC: mucociliary clearance; CF: cystic fibrosis; CFTR: cystic fibrosis transmembrane conductance regulator; ISO: isoproterenol; IVA/LUM: ivacaftor/lumacaftor. #: MucilAir™ culture medium + penicillin/streptomycin + 0.1% dimethyl sulfoxide. *: p<0.05; **: p<0.01; ***: p<0.001; ¶: p<0.0001 vehicle versus BI 1265162.
Discussion
A comprehensive preclinical analysis demonstrates that BI 1265162 is an effective ENaC inhibitor. BI 1265162 dose-dependently inhibited Na+ transport and increased water resorption in cell line models. This was further confirmed in animal models, with reduced liquid absorption in rat lungs and increased MCC in sheep. No amiloride-like effects on renal function were demonstrated in the animal models. When tested on normal and CF airway human epithelial models, BI 1265162 alone and in combination with CFTR modulators decreased water transport and increased MCC. BI 1265162 increased the effects of CFTR modulators IVA/LUM to reach the effect size seen in healthy epithelium with IVA/LUM alone.
CFTR has a putative role in ENaC regulation [16, 17]. In healthy airway epithelium, the transepithelial potential difference generated by active ENaC-mediated Na+ absorption is the driving force for chloride secretion and water absorption. In CF airway epithelium, ENaC is hyperactivated but CFTR function is defective, which leads to an imbalanced secretion and absorption of ions and fluid [17]. This gives rise to the hypothesis that CFTR is not only an ion channel, but also a regulator of ENaC. Hyperactivation of ENaC entails increased channel opening time and/or modulation of the total number of channels in the epithelium [18, 28], leading to increased Na+ and water resorption [17]. In addition, other extrinsic factors found at increased levels in CF, such as proteases (e.g. neutrophil elastase, cathepsin and some released by Gram-negative bacteria), or ENaC-specific mutations in atypical CF can also activate ENaC [5]. Mutations in ENaC leading to reduced activity in patients who are also homozygous for the F508del mutation in CFTR have been associated with a long-term non-progressive CF phenotype, further implicating ENaC as a potential modifier of CF [29].
Despite this, and promising preclinical results, most ENaC inhibitors to date have been unsuccessful in clinical settings [30]. Target engagement of the inhibitor in the kidney, leading to hyperkalaemia, and unfavourable pharmacokinetics and pharmacodynamics of the test compound, are two common reasons [5]. It is also not clear whether ENaC inhibitors have been dosed adequately in previous studies [31].
The airway fluid absorption model in rats was used to calculate the BI 1265162 dose in humans. Unlike MCC, which is influenced by several other factors (e.g. CBF [32]), fluid absorption in the lung depends mainly on the activity of ENaC [17]. The MCC model is appropriate for the assessment of activity rather than comparison across compounds or dose calculations. In the rat model, BI 1265162 was intratracheally inserted into the lungs, which ensured complete deposition of the test compound into the rat airways, whereas in the sheep model, this could not be ensured as the animals were nebulised. The ED70 of BI 1265162 was determined to be 0.18 μg·kg−1 bodyweight in the rat. Extrapolated to a typical human of 75 kg, and with inhalation as a means of delivery, the expected dose to inhibit about 95% of ENaC in the airways is 200 µg.
BI 1265162 had no relevant effect on rat serum aldosterone at the doses tested in the study, suggesting that kidney function was not affected. Similarly, in sheep, BI 1265162 administered via inhalation increased whole-lung MCC without any relevant changes in plasma potassium after 24 h, indicating no inhibition of kidney ENaC at the doses required for lung effects.
We have shown that BI 1265162 is highly potent in inhibiting Na+ transport in a dose-dependent manner with IC50 of 3×10−9 M in the M1 mouse cell line and 8×10−9 M in the NCI-H441 human bronchial epithelial cell line. IC50 of amiloride in our study was 2.10×10−7 M and 2.38×10−7 M in M1 cells and NCI-H441 cells, respectively, indicating a 30- to 70-fold higher potency of BI 1265162 compared with amiloride. Na+ inhibition by BI 1265162 reduced water transport by 76% in M1 cells compared with saline control.
MCC regulation is a complex, multi-component system involving at least three major processes: ion and water transport; mucin synthesis and secretion; and CBF and ciliary coordination [32]. Via inhibition of ENaC, BI 1265162 increased MCC by 19% in the sheep model compared with vehicle. In a study in adult female sheep by Åstrand et al. [33], nebulised isotonic (0.9%) saline did not increase MCC. In a similar study by Coote et al. [26], hypertonic (7%) saline had only limited efficacy. Both nebulised isotonic and hypertonic saline are commonly used by CF patients to promote hydration of the airways. In our study, BI 1265162 significantly increased MCC in female adult sheep, thereby demonstrating the potential of BI 1265162 as an inhaled therapy with superior efficacy in promoting MCC. BI 1265162-mediated improvement in MCC was also seen in human CF epithelium culture. The increase in MCC indicates restoration of the depleted fluid layer on the apical surface of the CF epithelia.
The MucilAir™ system used in this study is a 3D model of human epithelium reconstituted from primary human bronchial cells. It demonstrates the full functionality of the epithelial tissue when cultured on a semi-porous membrane and therefore provides a suitable model to test ENaC inhibitors in healthy and CF cells. Using this system, we found that in cells from a CF donor treated with IVA/LUM (an approved drug combination for treatment of CF), maximal MCC was 68% lower than that of a healthy donor. Concomitant BI 1265162 treatment restored MCC to the same levels as healthy epithelium, indicating a possible synergistic effect between ENaC inhibition and CFTR modulation. It has been hypothesised that ion channels such as CFTR are passive and only move Cl− according to the electrochemical gradient. Inhibition of ENaC can cause apical membrane hyperpolarisation, thereby having a CFTR-activating effect, creating an electrical driving force to induce Cl− secretion into ASL [5]. In vitro studies have reported decreased activity of ENaC with IVA, indicating an inhibitory effect of the drug on ENaC and possible synergism between CFTR modulation and ENaC inhibition [34]. Further work is needed to elaborate the effects of ENaC inhibition on CFTR channels in CF, either rescued by CFTR modulators or not. In addition, investigating the effects of ENaC inhibition on other mutant CFTR channels (e.g. G551D), either rescued or non-rescued, in human airway epithelial cells would be an interesting future study.
This study has some limitations. Cytotoxic effects of BI 1265162 at the concentrations assessed were not directly investigated in experiments measuring blockade of sodium absorption in M1 and H441 cell lines. However, cytotoxicity has been assessed during independent experiments using a human bronchial epithelial cell line, a human monocytic cell line and rat hepatocytes, with EC50s >800 µM. The resorption of an instilled liquid load from rat lung has been used as readout for ENaC modulators previously [21]. We employed this method in examining the effect of BI 1265162 on airway fluid absorption in our rat model; however, we measured lung weight to determine fluid resorption, whereas the previously published study employed magnetic resonance imaging. In our sheep model of MCC, experiments were carried out in two sheep per BI 1265162 dose and per vehicle; in previous studies of ENaC inhibitors in sheep, numbers of animals per treatment group ranged from two to five [18, 26, 33, 35]. Rat and sheep models do not exhibit airway mucous plugging and structural lung damage common in patients with CF, and instilled or inhaled ENaC inhibitor doses demonstrating positive results in these models may not achieve similar results in CF patients. However, these models are established, validated and widely used in studies of CF therapies. Finally, our experiments investigating the effect of BI 1265162 on fluid transport and MCC in non-CF and CF air–liquid interface cultures are based on bronchial cells provided by single donors. In addition, we did not analyse apical fluid electrolyte composition, and the determination of fluid transport by weighing Transwell® inserts in these experiments, as compared with measurements of fluid height, is an uncommon methodology.
Conclusion
BI 1265162 shows promising results as an inhibitor of ENaC, both in vitro and in vivo. The compound effectively inhibited Na+ transport, leading to a reduction in water resorption in cell lines and rat models, and accelerated MCC in a sheep model without an effect on renal function. In normal and CF human epithelial models, BI 1265162 decreased apical to basal water transport, and increased MCC alone and in combination with CFTR modulation. BI 1265162 allowed IVA/LUM in CF epithelium to reach MCC levels seen in normal epithelium, suggesting a possible synergistic effect. Taken together, these preclinical results demonstrate the potential of BI 1265162 as an ENaC inhibitor-based therapy for respiratory effects of CF. The benefit could lie with treatment with ENaC inhibition alone or in combination, additively, with CFTR modulators, and be mutation agnostic.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00429-2020.SUPPLEMENT (412.6KB, pdf)
Acknowledgements
The authors would like to thank Kerstin Benediktus, Madlen Hahn, Gabriele Göggerle, Julia Wolf, Almut Schüle, Mathilde Borsch and Angelika Hoffmann (all of Boehringer Ingelheim International GmbH, Biberach, Germany) for their excellent technical assistance during the conduct of these studies. Findings from the studies were presented at the European Cystic Fibrosis Conference, June 5–8, 2019, Liverpool, UK.
Footnotes
This article has supplementary material available from openres.ersjournals.com.
Conflict of interest: P. Nickolaus is an employee of Boehringer Ingelheim International GmbH.
Conflict of interest: B. Jung was an employee of Boehringer Ingelheim at the time of the study.
Conflict of interest: J. Sabater reports grants from Boehringer Ingelheim during the conduct of the study.
Conflict of interest: S. Constant reports grants from Boehringer Ingelheim during the conduct of the study.
Conflict of interest: A. Gupta is an employee of Boehringer Ingelheim International GmbH.
Support statement: This work was supported by Boehringer Ingelheim International GmbH. Medical writing assistance, in the form of the preparation and revision of the manuscript, was supported financially by Boehringer Ingelheim, and provided by Lee Kempster and Ishmam Nawar of MediTech Media (London, UK) under the authors’ conceptual direction and based on feedback from the authors. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Ratjen F, Bell SC, Rowe SM, et al. Cystic fibrosis. Nat Rev Dis Primers 2015; 1: 15010. doi: 10.1038/nrdp.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest 2002; 109: 571–577. doi: 10.1172/JCI0215217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mall M, Bleich M, Greger R, et al. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest 1998; 102: 15–21. doi: 10.1172/JCI2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Althaus M. ENaC inhibitors and airway re-hydration in cystic fibrosis: state of the art. Curr Mol Pharmacol 2013; 6: 3–12. doi: 10.2174/18744672112059990025 [DOI] [PubMed] [Google Scholar]
- 5.Moore PJ, Tarran R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin Ther Targets 2018; 22: 687–701. doi: 10.1080/14728222.2018.1501361 [DOI] [PubMed] [Google Scholar]
- 6.Hill DB, Long RF, Kissner WJ, et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur Respir J 2018; 52: 1801297. doi: 10.1183/13993003.01297-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponzano S, Nigrelli G, Fregonese L, et al. A European regulatory perspective on cystic fibrosis: current treatments, trends in drug development and translational challenges for CFTR modulators. Eur Respir Rev 2018; 27: 170124. doi: 10.1183/16000617.0124-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edmondson C, Davies JC. Current and future treatment options for cystic fibrosis lung disease: latest evidence and clinical implications. Ther Adv Chronic Dis 2016; 7: 170–183. doi: 10.1177/2040622316641352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019; 394: 1940–1948. doi: 10.1016/S0140-6736(19)32597-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019; 381: 1809–1819. doi: 10.1056/NEJMoa1908639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cystic Fibrosis Mutation Database www.genet.sickkids.on.ca/Home.html Date last updated April 25, 2011. Date last accessed: November 4, 2019.
- 12.Cystic Fibrosis Foundation www.cff.org/What-is-CF/Genetics/Types-of-CFTR-Mutations/ Types of CFTR mutations. Date last accessed: November 4, 2019.
- 13.Couroux P, Farias P, Rizvi L, et al. First clinical trials of novel ENaC targeting therapy, SPX-101, in healthy volunteers and adults with cystic fibrosis. Pulm Pharmacol Ther 2019; 58: 101819. doi: 10.1016/j.pupt.2019.101819 [DOI] [PubMed] [Google Scholar]
- 14.Skilton M, Krishan A, Patel S, et al. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 2019; 1: CD009841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khoury O, Barrios C, Ortega V, et al. Immunomodulatory cell therapy to target cystic fibrosis inflammation. Am J Respir Cell Mol Biol 2018; 58: 12–20. doi: 10.1165/rcmb.2017-0160TR [DOI] [PubMed] [Google Scholar]
- 16.Shei R-J, Peabody JE, Kaza N, et al. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol 2018; 43: 152–165. doi: 10.1016/j.coph.2018.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mall MA, Galietta LJV. Targeting ion channels in cystic fibrosis. J Cyst Fibros 2015; 14: 561–570. doi: 10.1016/j.jcf.2015.06.002 [DOI] [PubMed] [Google Scholar]
- 18.Scott DW, Walker MP, Sesma J, et al. SPX-101 is a novel epithelial sodium channel–targeted therapeutic for cystic fibrosis that restores mucus transport. Am J Respir Crit Care Med 2017; 196: 734–744. doi: 10.1164/rccm.201612-2445OC [DOI] [PubMed] [Google Scholar]
- 19.Clunes MT, Boucher RC. Cystic fibrosis: the mechanisms of pathogenesis of an inherited lung disorder. Drug Discov Today Dis Mech 2007; 4: 63–72. doi: 10.1016/j.ddmec.2007.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst 2009; 5: 123–127. doi: 10.1039/B810471A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blé FX, Cannet C, Collingwood S, et al. ENaC-mediated effects assessed by MRI in a rat model of hypertonic saline-induced lung hydration. Br J Pharmacol 2010; 160: 1008–1015. doi: 10.1111/j.1476-5381.2010.00747.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhand R, Eicher J, Hansel M, et al. Improving usability and maintaining performance: human-factor and aerosol-performance studies evaluating the new reusable Respimat inhaler. Int J Chron Obstruct Pulmon Dis 2019; 14: 509–523. doi: 10.2147/COPD.S190639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Güney Ş, Schuler A, Ott A, et al. Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am J Physiol Lung Cell Mol Physiol 2007; 293: L1332–L1338. doi: 10.1152/ajplung.00338.2006 [DOI] [PubMed] [Google Scholar]
- 24.Gondzik V, Awayda MS. Methods for stable recording of short-circuit current in a Na+-transporting epithelium. Am J Physiol Cell Physiol 2011; 301: C162–C170. doi: 10.1152/ajpcell.00459.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blouquit S, Morel H, Hinnrasky J, et al. Characterization of ion and fluid transport in human bronchioles. Am J Respir Cell Mol Biol 2002; 27: 503–510. doi: 10.1165/rcmb.4869 [DOI] [PubMed] [Google Scholar]
- 26.Coote KJ, Paisley D, Czarnecki S, et al. NVP-QBE 170: an inhaled blocker of the epithelial sodium channel with a reduced potential to induce hyperkalaemia. Br J Pharmacol 2015; 172: 2814–2826. doi: 10.1111/bph.13075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taouil K, Hinnrasky J, Hologne C, et al. Stimulation of β2-adrenergic receptor increases cystic fibrosis transmembrane conductance regulator expression in human airway epithelial cells through a cAMP/protein kinase A-independent pathway. J Biol Chem 2003; 278: 17320–17327. doi: 10.1074/jbc.M212227200 [DOI] [PubMed] [Google Scholar]
- 28.Thibodeau PH, Butterworth MB. Proteases, cystic fibrosis and the epithelial sodium channel (ENaC). Cell Tissue Res 2013; 351: 309–323. doi: 10.1007/s00441-012-1439-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agrawal PB, Wang R, Li HL, et al. The epithelial sodium channel is a modifier of the long-term nonprogressive phenotype associated with F508del CFTR mutations. Am J Respir Cell Mol Biol 2017; 57: 711–720. doi: 10.1165/rcmb.2017-0166OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cystic Fibrosis Foundation Drug development pipeline www.cff.org/Trials/pipeline Date last accessed: January 8, 2020.
- 31.Danahay H, McCarthy C, Abraham W, et al. 210 – Pharmacokinetic and pharmacodynamic profile of a novel inhaled enac blocker, ETD001. In: 32nd Annual North American Cystic Fibrosis Conference (NACFC), October 18–20, 2018, Denver, CO, USA, 2018.
- 32.Sears PR, Yin W-N, Ostrowski LE. Continuous mucociliary transport by primary human airway epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol 2015; 309: L99–L108. doi: 10.1152/ajplung.00024.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Åstrand AB, Hemmerling M, Root J, et al. Linking increased airway hydration, ciliary beating, and mucociliary clearance through ENaC inhibition. Am J Physiol Lung Cell Mol Physiol 2015; 308: L22–L32. doi: 10.1152/ajplung.00163.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cholon DM, Esther CR Jr, Gentzsch M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Rev Precis Med Drug Dev 2016; 1: 235–243. doi: 10.1080/23808993.2016.1175299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terryah ST, Fellner RC, Ahmad S, et al. Evaluation of a SPLUNC1-derived peptide for the treatment of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol 2018; 314: L192–L205. doi: 10.1152/ajplung.00546.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00429-2020.SUPPLEMENT (412.6KB, pdf)