

Summary
Mitochondria constantly adapt to the metabolic needs of a cell. This mitochondrial plasticity is critical to T cells, which modulate metabolism depending on antigen-driven signals and environment. We show here that de novo synthesis of the mitochondrial membrane-specific lipid cardiolipin maintains CD8+ T cell function. T cells deficient for the cardiolipin-synthesizing enzyme PTPMT1 had reduced cardiolipin and responded poorly to antigen because basal cardiolipin levels were required for activation. However, neither de novo cardiolipin synthesis, nor its Tafazzin-dependent remodeling, was needed for T cell activation. In contrast, PTPMT1-dependent cardiolipin synthesis was vital when mitochondrial fitness was required, most notably during memory T cell differentiation or nutrient stress. We also found CD8+ T cell defects in a small cohort of patients with Barth syndrome, where TAFAZZIN is mutated, and in a Tafazzin-deficient mouse model. Thus, the dynamic regulation of a single mitochondrial lipid is crucial for CD8+ T cell immunity.
Keywords: cardiolipin, mitochodria, immunometabolism, PTPMT1, Tafazzin, Barth Syndrome, immune memory, CD8 T cells
Graphical Abstract
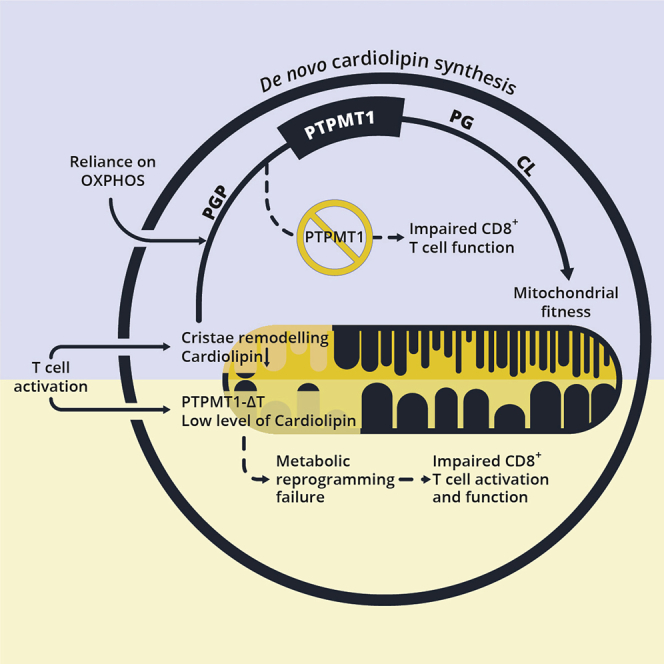
Highlights
-
•
Cardiolipin is essential for in vivo and in vitro CD8+ T cell responses
-
•
Active cardiolipin synthesis and remodeling occurs during T cell differentiation
-
•
Cardiolipin synthesis supports CD8+ TM cell development, metabolism, and function
-
•
T cell defects are evident in TAZ KO mice and in Barth syndrome patients
Corrado et al. show that the mitochondrial membrane-specific lipid cardiolipin is required for the metabolic plasticity that is essential for effective CD8+ T cell function. Cardiolipin-deficient CD8+ T cells fail to respond to pathogens and are not able to adapt to nutrient stress.
Introduction
Upon activation, CD8+ T cells shift from a quiescent to a highly active metabolic state where both glycolysis and OXPHOS increase (Bailis et al., 2019; Buck et al., 2017; Chang et al., 2013; Geltink et al., 2018; Klein Geltink et al., 2017; Sena et al., 2013). During this phase, mitochondria generate signals, such as reactive oxygen species (ROS) or metabolites, required to drive T cell differentiation (Bailis et al., 2019; Sena et al., 2013). Although effector T (TE) cells increase aerobic glycolysis during in vitro culture, they display greater rates of oxidative phosphorylation (OXPHOS) in vivo (Ma et al., 2019). When the infection or cancer has been eradicated, the majority of TE cells die, leaving behind a small number of long-lived memory T (TM) cells that confer long-term immune protection. TM cells engage catabolic pathways like fatty acid oxidation (FAO) to fuel mitochondrial respiration (Pearce et al., 2009; van der Windt et al., 2012). Overall, cellular metabolic changes supported by mitochondria are integral to a functional T cell response.
Mitochondria continuously undergo fusion and fission (Chan, 2020; Pernas and Scorrano, 2016), and Optic atrophy 1 (OPA1), a protein critical for mitochondrial shape and metabolism (Cipolat et al., 2004; Cogliati et al., 2013; Frezza et al., 2006), is required to generate TM cells (Buck et al., 2016). Mitochondria also differ at the sub-organellar level, where different properties and lipid compositions exist between the inner (IMM) and outer mitochondrial membrane (OMM) (Frey and Mannella, 2000). The IMM is organized in discrete invaginations called cristae, where electron transport chain (ETC) complexes are located (Mannella et al., 2001; Wolf et al., 2019). Cardiolipin (CL) is exclusively synthesized and localized in the IMM and accounts for 15%–20% of the total phospholipid mass (Dudek, 2017). CL is a four-acyl chain lipid with a small negative glycerol polar head responsible for the negative curvature of the cristae. Here, CL binds to ETC complexes, making respiration more efficient and reducing ROS (Paradies et al., 2014), and also modulates substrate carrier activity and protein import (Paradies et al., 2019). When mislocalized to the OMM, CL recruits caspases, promoting apoptosis or triggering an inflammatory response (Gonzalvez et al., 2008; Iyer et al., 2013). In addition to its role in heart and muscle (Dudek et al., 2019), CL synthesis is essential for systemic energy homeostasis to prevent insulin resistance (Sustarsic et al., 2018).
CL composition varies greatly in both acyl chain length and saturation, with the tetra-linoleic form (CL 72:8) being the most abundant species (Minkler and Hoppel, 2010). CL localization at the site of OXPHOS makes it susceptible to oxidation by cytochrome c (Kagan et al., 2005). When oxidation occurs, phospholipases remove the altered acyl chain, generating an intermediate monolysocardiolipin (MLCL), which is then “remodeled” into mature CL by the enzyme TAFAZZIN (Hsu et al., 2013; Schlame, 2013). Mutations in TAFAZZIN result in reduced CL content in all cells (Schlame et al., 2003) and are responsible for Barth syndrome, an X-linked recessive human disease characterized by dilated cardiomyopathy, muscle weakness, and fatigue (Barth et al., 1983; Bione et al., 1996; Clarke et al., 2013). Neutropenia and susceptibility to infections have been reported in 90% of Barth syndrome patients, but mechanistic analyses into the impaired immunity is lacking (Steward et al., 2019). Clinically relevant total lymphopenia has only been described in one Barth syndrome patient, where it was a prelude to development of non-Epstein Barr virus (EBV)-associated T cell non-Hodgkin lymphoma following cardiac transplantation (Ronghe et al., 2001).
Our previous work has shown how changes in mitochondrial shape, cristae morphology, and function directly impact CD8+ T cell activation, differentiation, and functional TM cell development (Buck et al., 2016; Klein Geltink et al., 2017; van der Windt et al., 2012). The role of CL presence, de novo synthesis, or remodeling in the assumption of distinct metabolic programs and mitochondrial function in T cells has not been investigated. Given these collective observations, and the pivotal function of CL in regulating OXPHOS and cristae structure, we set out to investigate the role of CL in the CD8+ T cell response.
Results
De Novo Synthesis of Cardiolipin Is a Hallmark of CD8+ T Cells with High-Reserve Respiratory Capacity
We investigated how CL was modulated in CD8+ T cell culture settings that invoked higher spare respiratory capacity (SRC), a measure of a cell’s ability to make extra ATP from OXPHOS upon increased energy demand (Nicholls, 2009). CD28 co-stimulation during activation promotes SRC in TM cells generated in vitro with IL-15 (Figure 1A) and is also required for the generation of functional TM cells in vivo (Klein Geltink et al., 2017). We analyzed lipids from interleukin (IL)-15 TM cells activated with αCD3 or with αCD3/αCD28 and observed higher CL72:8 and CL72:6 (Figure 1B) as well as total CL content (Figure 1C) in cells activated with CD28 co-stimulation. Nutrient restriction also induces CD28-dependent mitochondrial elongation in CD8+ T cells (Klein Geltink et al., 2017) and the assembly of ETC supercomplexes in order to sustain viability and protect from cell death (Gomes et al., 2011). Thus, we generated in vitro IL-2 TE cells and cultured them in 1 mM versus 10 mM glucose for 20 h before analyzing lipid composition. Culturing IL-2 TE cells in 1 mM glucose for 20 h generated higher SRC (Figure 1D) (Klein Geltink et al., 2017). Lipidomics analysis revealed that various CL species were significantly enriched in 1 mM glucose (Figure 1E), increasing overall CL content (Figure 1F). We obtained similar results culturing IL-2 CD8+ TE cells in a medium containing 10 mM galactose (instead of glucose) to force mitochondrial respiration (Chang et al., 2013) (data not shown). Of note, the metabolic rewiring that led to increased CL content (Figures 1B–1F) occurred without an increase in mitochondrial mass measured by TOM20 and MnSOD, ETC protein levels (Figures 1G, 1H, S1A, and S1B), MitoTracker Green dye accumulation (Figures 1I and 1J), and mtDNA/nuclear DNA (nDNA) ratio (Figures S1C and S1D). De novo CL synthesis in the 1 mM condition was needed to generate higher SRC and protect cells from starvation-induced death as these cells lost SRC (Figure 1K) and viability (Figure 1L) when treated with the PTPMT1-specific inhibitor alexidine dihydrochloride (AD).
Figure 1.

De Novo Cardiolipin Synthesis Is a Hallmark of CD8+ T Cells with a High-Reserve Respiratory Capacity
(A) OCR of mouse WT CD8+ T cells activated with αCD3 only or αCD3/αCD28 and differentiated in IL-15 TM cells for 6 days. OCR analysis at baseline and after exposure to αCD3/αCD28 coated beads, Oligomycin (Oligo), FCCP, and Rotenone/Antimycin (Rot/Ant).
(B) Lipids extracted from cells activated and differentiated as in (A). Data show Log2FC and p value calculated with ANOVA test.
(C) Total CL content in cells treated and analyzed as in (B).
(D) OCR of mouse WT CD8+ T cells activated with αCD3/αCD28 + IL-2 and cultured in complete medium until day 3, then cultured for 24 h in either complete medium (10mM glucose) or glucose restricted medium (1mM glucose) for 24 h. Normalized OCR at baseline and after (Oligo) FCCP and Rot/Ant injections.
(E) Lipids extracted from cells activated and differentiated as in (F). Data show Log2FC and p value calculated with ANOVA test.
(F) Total CL content in cells treated and analyzed as in (F).
(G) Immunoblot analysis of cells activated and differentiated as in (A).
(H) Immunoblot analysis of cells treated as in (D).
(I) MitoTracker Green staining in cells activated and differentiated as in (A).
(J) MitoTracker Green staining in cells activated and differentiated as in (D).
(K) SRC of CD8+ T cells activated as in (D) and cultured ± AD during 20 h of glucose restriction.
(L) Survival of CD8+ T cells activated as in (D) and cultured ± AD during 20 h of glucose restriction.
(M) OCR of mouse WT CD8+ T cells activated with αCD3/αCD28 + IL-2 and cultured in complete medium until day 3, then for additional three days in IL-2 (IL-2 TE) or IL-15 (IL-15 TM).
(N) Lipids extracted from WT CD8+ T cells differentiated as in (M). Data show Log2FC and p value calculated with ANOVA test.
(O) Total CL content from IL-2 TE and IL-15 TM CD8+ T cells cultured as in (M).
(P) Lipids extracted from TEM (CD44hiCD62Llo) and TCM (CD44hiCD62Lhi) cells isolated ex vivo from C57BL/6 mice. Data show Log2FC and p value calculated with ANOVA test from 9 animals per group.
(Q) Total CL content from TEM and TCM cells isolated as in (P). Dots represent individual mice.
(R) CD8+ T cells were activated with αCD3/αCD28 + IL-2 for 3 days and then differentiated into IL-15 TM cells in presence of 13C-glucose. At indicated time points lipids were extracted and 13C-glucose derived carbons traced into CL 72:8.
(S) CD8+ T cells were activated with αCD3/αCD28 + IL-2 for 3 days and then differentiated into IL-15 TM cells for 72 h in the presence of 13C-glucose or 13C-linoleic acid ± CL synthesis inhibitor (AD). 13C-glucose and 13C-linoleic acid derived carbons traced into CL 72:8.
(T) CD8+ T cells were differentiated as in (S). Also shown is the percentage of isotopologue distribution of 13C-glucose and 13C-linoleic acid-derived carbons traced into CL 72:8.
Data shown as mean ± SEM of ≥3 independent experiments unless indicated. Statistical comparisons for two groups calculated by unpaired two-tailed Student’s t test or ANOVA test, where indicated. Comparison among multiple groups (in [R] and [S]) calculated by One-Way ANOVA and correcting for multiple comparison using Tukey test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S1.
In vitro generated IL-15 TM cells are characterized by higher SRC (Figure 1M) as well as by elongated mitochondria together with greater mitochondrial mass (Buck et al., 2016; van der Windt et al., 2012). These cells also displayed a selective enrichment in CL (Figures 1N, 1O, and S1E), accompanied by higher expression of PTPMT1 and CRLS1 (Figure S1F), as compared to what was seen in IL-2 TE cells. Although there was up to a 1.5-/2-fold difference in mitochondrial mass observed between IL-2 TE and IL-15 TM cells (considering mtDNA/nDNA ratio or TFAM expression; van der Windt et al., 2012), the CL amount increased almost three times, suggesting a specific enrichment in CL in the mitochondria of IL-15 TM cells. In vitro culture and in vivo differentiation of T cells could be different (Ma et al., 2019). Therefore, we sorted CD8+ T effector memory (TEM CD62LloCD44hi) and CD8+ T central memory (TCM CD62LhiCD44hi) cells from the same animal and analyzed their lipid composition. Lipidomics analysis showed CL 72:8 among the most significantly enriched lipids (Figures 1P and S1G) and almost exclusively responsible for the increase in total CL mass in CD8+ TCM cells (Figure 1Q). Notably, human (h) CD8+CD45RO+ TEM cells also had a higher basal and maximal respiration rate than did hCD8+CD45RA+ naive T (TN) cells (Figure S1H). They also exhibited, although with higher variability, a trend toward the enrichment of specific CL species (Figure S1I) and higher total CL content (Figure S1J), without major changes in mitochondrial mass (Figure S1K). Taken together these data suggest that T cells, upon stimuli that provoke mitochondrial respiration (e.g., CD28 co-stimulation, glucose restriction, galactose culture, or IL-15 supported TM cell development), rewire metabolism to increase mitochondrial efficiency through de novo cardiolipin synthesis before, or independently from, mitochondrial biogenesis.
Substrate Utilization Reveals Active De Novo Cardiolipin Synthesis and Remodeling during TM Cell Differentiation
To gain insight into the metabolic rewiring responsible for the increase in CL content in IL-15 TM cells, we performed 13C-glucose and 13C-linoleic acid tracing to measure incorporation of labeled carbons into CL. The first was used to measure whether the classic energetic substrate of IL-15 TM cells was used for de novo CL synthesis (O’Sullivan et al., 2014), the latter to investigate CL remodeling by the acyltransferase TAFAZZIN, whose main substrate is linoleic acid. Given the large number of carbons in a CL molecule (81 for a CL 72:8), it was not surprising to measure a natural occurrence of a few 13C atoms in a non-exogenously labeled molecule (Figures 1R and S1L, untreated samples, 0 ≤ M ≤ 3). Thus, we considered isotopologues with a mass of M≥ +4 as a cut-off for our analysis (dashed line in Figures S1L and S1M). In IL-15 wild-type (WT) CD8+ TM cells cultured with 13C-glucose, almost all the newly synthesized CL contained glucose-derived carbons (Figures 1R and S1L). We next treated cells cultured in IL-15 plus 13C-glucose or 13C-linoleic acid for 72 h with or without AD to inhibit de novo CL synthesis during TM differentiation. The data showed that both glucose- and linoleic-acid-derived 13C accumulated efficiently in newly synthesized CL (Figures 1S and S1M) and revealed the differential molecular origin of carbon incorporation (Figures 1S, 1T, S1M, and S1N). When normalized to total CL, the percentage of M≥ +4 CL was reduced by AD only in cells cultured in 13C-glucose (Figure 1T), suggesting that glucose-derived carbons are incorporated through PTPMT1-dependent de novo CL synthesis, whereas linoleic acid molecules are incorporated into CL through active remodeling of CL (substitution of CL acyl chains), a process not affected by AD during TM generation (Figure S1N).
Cardiolipin Is Essential for In Vivo and In Vitro CD8+ T Cell Responses
CL is synthesized in a two-step reaction in which phosphatidylglycerophosphate (PGP) is dephosphorylated by the mitochondrial phosphatase PTPMT1 into phosphatidylglycerol (PG) then fused to another diacylglycerol molecule (DAG) to generate CL by CRLS1(Zhang et al., 2011) (Figure 2A). To investigate the physiological relevance of CL in T cells, we crossed mice carrying floxed alleles of Ptpmt1 with mice expressing CD4-Cre to selectively ablate Ptpmt1 in T cells (Ptpmt1flox/floxCD4-Cre− mice are referred to as PTPMT1 WT, Ptpmt1flox/floxCD4-Cre+ as PTPMT1-ΔT) (Figure 2B). Lower amount of total CL (Figure 2C) and a reduction in CL 72:8 and CL 72:6 species (Figure S2A) confirmed the effective inhibition of CL synthesis in Ptpmt1−/− CD8+ T cells. CD8+ TN cells from PTPMT1-ΔT mice showed no major differences in expression of subunits of ETC complexes, TOM20, or OPA1 in comparison with PTPMT1 WT cells (Figure S2B). PTPMT1-ΔT mice had grossly normal thymic development (Figure S2C) but peripheral lymphopenia with altered frequencies and numbers of TCRβ CD8+ and CD4+ (Figures 2D, S2D, and S2E) and TCRγδ CD8+ and CD4+ T cells (Figures S2F and S2G) in multiple organs.
Figure 2.

Cardiolipin Is Necessary for In Vivo and In Vitro CD8+ T Cell Responses
(A) Schematic of the cardiolipin synthesis pathway.
(B) PTPMT1 expression in CD8+ T cells from PTPMT1 WT (Ptpmt1flox/floxCD4-Cre−/−) and PTPMT1-ΔT (Ptpmt1flox/floxCD4-Cre+/−) mice.
(C) Cardiolipin content in freshly isolated Ptpmt1 WT and Ptpmt1−/− CD8+ T cells.
(D) Percentage of CD8+ T cells in spleens from PTPMT1 WT and PTPMT1-ΔT mice. Dots represent individual mice.
(E) PTPMT1 WT and PTPMT1-ΔT mice infected i.v. with 1 × 106 colony-forming units (CFU) LmOVA ΔActa and challenged i.v. with 5 × 107 CFU LmOVA 21 days after primary infection. Blood analyzed at indicated time.
(F) Percentage of CD8+ T cells in PTPMT1 WT and PTPMT1-ΔT mice infected as in (E). Dots represent individual mice.
(G) Percentage of CD8+ Tet+ T cells in PTPMT1 WT and PTPMT1-ΔT mice infected as in (E). Dots represent individual mice.
(H) Bacterial burden shown as CFU per μg of liver of PTPMT1 WT and PTPMT1-ΔT mice 7 days post-LmOVA ΔActa challenge. Dots represent individual mice.
(I) TN (CD62LhiCD44lo), TE (CD62LloCD44hi), and TM (CD62LhiCD44hi) CD8+ T cells from PTPMT1 WT and PTPMT1-ΔT mice 7 days after primary infection. Dots represent individual mice.
(J) EEC, SLEC, MPEC, and DPEC CD8+ T cells from PTPMT1 WT and PTPMT1-ΔT mice 7 days after primary infection according to KLRG1 and CD127 expression. Dots represent individual mice.
(K) OCR/ECAR ratio of WT and Ptpmt1−/− CD8+ T cells analyzed 7 days after secondary infection. Dots represent individual mice.
(L) Ptpmt1 WT and Ptpmt1−/− CD8+ T cells stimulated with PMA/Iono for 4 h. Also shown is the percentage of CD8+ T cells producing IFN-γ and IFN-γ MFI of cytokine-producing cells.
(M) Cytoplasmic calcium levels at baseline and after αCD3 crosslinking via IgG, tapsigargin (TG), and ionomycin (iono) in Ptpmt1 WT and Ptpmt1−/− CD8+ T cells. Representative of 3 experiments.
(N) CD5, CD44, CD25, and CD69 expression in Ptpmt1 WT and Ptpmt1−/− CD8+ T cells 24 h after activation with αCD3/αCD28 + IL-2.
(O) Survival of Ptpmt1 WT and Ptpmt1−/− CD8+ T cells 24 h after activation.
(P) Cell cycle analysis assessed by FxCycle + Ki67 staining of Ptpmt1 WT and Ptpmt1−/− CD8+ T cells 24 h and 48 h after activation with αCD3/αCD28 + IL-2.
(Q) Active proliferation assessed by Ki67 expression in Ptpmt1 WT and Ptpmt1−/− CD8+ T cells 24 h and 48 h after activation with αCD3/αCD28 + IL-2.
(R) Cell proliferation assessed by CTV dilution of Ptpmt1 WT and Ptpmt1−/− CD8+ T cells 72 h after activation with αCD3/αCD28 + IL-2. Histogram representative of 4 independent experiments.
Data shown as mean ± SEM of ≥3 independent experiments unless indicated. Statistical comparisons for 2 groups calculated by unpaired two-tailed Student’s t test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S2.
To assess the effect of CL deficiency during CD8+ T cell responses in vivo, we infected PTPMT1 WT and PTPMT-ΔT mice with a sublethal dose of attenuated L. monocytogenes expressing Ova (LmOVA ΔActa) and then followed the total, as well as the Ova-specific CD8+ T cell response, for 3 weeks before challenging the mice with a higher dose of bacteria to evaluate recall responses and protective immunity. Although PTPMT1 WT mice mounted a primary CD8+ T cell response that peaked 7 days post-infection and an enhanced secondary response after challenge, PTPMT-ΔT mice failed to do so, mounting only a very limited Ova-specific response after secondary immunization (Figures 2E–2G). This defect resulted in the persistence of bacteria in the liver of PTPMT1-ΔT mice after secondary challenge (Figure 2H). A significantly higher proportion of TN (CD62LhiCD44lo) compared to TE (CD62LloCD44hi) cells (Figures 2I) as well as a higher proportion of early TE cells (EECs, KLRG1loCD127lo) compared to short-lived TE cells (SLECs, KLRG1hiCD127lo) and memory precursor TE cells (MPECs, KLRG1loCD127hi) (Figure 2J) was present in PTPMT1-ΔT mice 7 days post-infection, suggesting impaired T cell activation. Consistent with the role of CL in regulating cristae organization and OXPHOS efficiency, Ptpmt1−/− CD8+ T cells isolated 7 days post-challenge exhibited a lower OCR/ECAR (oxygen consumption rate/extracellular acidification rate) ratio relative to WT counterparts (Figure 2K).
The failure of Ptpmt1−/− CD8+ T cells to expand upon infection could be caused by defective T cell activation, a proliferative defect due to impaired mitochondrial biogenesis needed to keep up with cell division or increased cell death. We also found impaired interferon (IFN)-γ production in freshly isolated Ptpmt1−/− CD8+ T cells (Figure 2L). This defect was not due to differential cytoplasmic calcium handling, which was grossly similar between Ptpmt1 WT and Ptpmt1−/− CD8+ T cells upon T cell receptor (TCR) activation (Figure 2M). CD69, CD44, and CD25 expression 24 h after αCD3/αCD28 activation was higher in Ptpmt1−/− CD8+ T cells (Figure 2N), corroborating the observation that the signals that drive early T cell activation were normal, at least by surface-marker expression. At the same time point, there was no sign of increased TCR stimulation-dependent apoptosis (Figure 2O). Nevertheless, CL-deficient CD8+ T cells displayed a marked defect in cell cycle entry and progression at 24 h and 48 h post-activation measured by DNA content (Figure 2P) and Ki67 expression (Figure 2Q), resulting in impaired proliferation 72 h post-activation (Figure 2R). Comprehensively, these data suggest that CL is required for T cell cytokine production, cell cycle entry, and proliferation upon in vitro and in vivo antigen challenge.
Unlike CD8+ IL-2 TE Cells, CD8+ IL-15 TM Cells Require PTPMT1 for Survival and Cytokine Production
To assess whether the differences in activation and proliferation (Figures 2L–2R) would affect in vitro IL-2 TE or IL-15 TM cell differentiation, we activated Ptpmt1 WT and Ptpmt1−/− CD8+ T cells for 3 days and then differentially cultured them in IL-2 or IL-15 to generate TE-like cells or TM-like cells, respectively. Ptpmt1−/− IL-15 TM cells showed a strong defect in survival (Figure S2H) and lower CD62L expression (Figure S2I) and IFN-γ production upon restimulation (Figure S2J) than did Ptpmt1 WT IL-15 TM cells. Ptpmt1−/− IL-2 TE cell properties were largely unaffected with the exception of the persistent proliferative defect (Figures S2H–S2K). Lower mitochondrial membrane potential was observed both in IL-2 TE and IL-15 TM cells (Figure S2L, upper panel), whereas mitochondrial mass was unaltered (Figure S2L, lower panel). The metabolic deficiencies in Ptpmt1−/− IL-15 TM cells were further highlighted by the absence of SRC (Figure S2M) and defective maximal glycolytic rate (Figure S2N).
Metabolic Reprogramming upon CD8+ T Cell Activation Requires Cardiolipin, but Not Its De Novo Synthesis
We found that CL is necessary for CD8+ T cell responses both in vitro and in vivo (Figure 2). Yet, whether CD8+ T cells needed de novo CL synthesis early during activation was not clear. We activated WT CD8+ T cells in the presence of the CL synthesis inhibitor AD during the first 48 h of stimulation, then washed out AD and differentiated the cells in IL-15 (Figure S3A). Lipidomics analysis of cells 48 h after activation, at the moment of AD wash out, showed no difference in total CL content with untreated cells (Figure S3B), although some changes were observed in the CL species profile (Figure S3C). Cells from the same experiment analyzed 3 days post-IL-15 TM differentiation showed no defects in basal or maximal respiration (Figure S3D) or IFN-γ production (Figure S3E). Then, we compared CL content of unstimulated cells with cells at different time points after αCD3/αCD28 stimulation (Figure 3A). Total CL content remained stable in the first 6 h after stimulation before strongly decreasing at later time points (Figure 3B). Of note, this drop occurred before the first cell division and without mitochondrial content dilution (Figure S3F). The drop in total CL was also paralleled by changes in acyl-chain composition (Figure 3C). CL 72:6 started to increase 24 h post-activation (Figure 3C). This observation would suggest that CL 72:6 could be the first CL species to be generated when the process of CL synthesis is re-activated, followed by CL 70:8, CL 70:6, and CL 72:9, whose increased levels are blunted by AD treatment during 48-h activation (Figures 3C and S3C). Overall, these results suggest that de novo synthesis of CL is not necessary for T cell priming and that its inhibition during early activation does not affect in vitro TM cell differentiation later on.
Figure 3.

Metabolic Reprogramming upon CD8+ T Cell Activation Requires Cardiolipin, but Not Its De Novo Synthesis
(A) Schematic of CD8+ T cells activated with αCD3/αCD28 +IL-2. At indicated time points cell pellets were collected for lipidomics analysis.
(B) Total CL amount in CD8+ T cells treated as in (A)
(C) Cardiolipin species in CD8+ T cells treated as in (A).
(D) Schematic of WT CD8+ T cells activated with αCD3/αCD28 +IL-2 with ± AD.
(E) Cytokine production of WT CD8+ T cells activated as in (D) analyzed at indicated time points.
(F) Schematic of WT and Ptpmt1−/− CD8+ T cells activated with αCD3/αCD28 + IL-2.
(G) Cytokine production of WT and Ptpmt1−/− CD8+ T cells activated as in (F) analyzed at indicated time points.
(H) WT CD8+ T cells activated with αCD3/αCD28 + IL-2 in a medium containing 10mM 13C-glucose ± AD. Twenty-four h after activation cell pellets were collected for metabolomics analysis.
(I) WT and Ptpmt1−/− CD8+ T cells activated with αCD3/αCD28 + IL-2 in a medium containing 10mM 13C-glucose. Twenty-four h after activation cell pellets were collected for metabolomics analysis.
(J) 13C-glucose derived carbons incorporated into indicated metabolites in WT CD8+ T cells treated as in (H). Data represent mean ± SEM of 3 samples per genotype.
(K) 13C-glucose derived carbons incorporated into indicated metabolites in Ptpmt1 WT and Ptpmt1−/− CD8+ T cells treated as in (I). Data represent mean ± SEM of 3 samples per genotype.
(L) Schematic of 13C-glucose tracing experiment results in WT CD8+ T cells ± AD, Ptpmt1 WT, and Ptpmt1−/− CD8+ T cells.
Data shown as mean ± SEM of ≥3 independent experiments unless indicated. Statistical comparisons for 2 groups calculated by unpaired two-tailed Student’s t test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S3.
Even though de novo synthesis of CL is not necessary for T cell activation the presence of a threshold level of CL is required for in vivo and in vitro immune responses (Figure 2). Considering the metabolic reprogramming that characterizes CD8+ T cells upon activation (Buck et al., 2017), we hypothesized that during antigen stimulation CL might maintain efficient mitochondrial metabolism, thus promoting proper T cell activation. To test this hypothesis, we performed experiments analyzing cytokine production and metabolic properties of WT CD8+ T cells cultured with or without AD during activation, as well as of Ptpmt1 WT and Ptpmt1−/− CD8+ T cells. WT cells were fully equipped with CL, and only de novo synthesis was pharmacologically inhibited. In Ptpmt1−/− CD8+ T cells, as a result of lacking PTPMT1 and de novo CL synthesis, the cells were CL deficient. Cytokine production was tested at different time points post-activation (6 h, 24 h, and 48 h) (Figure 3D). Although pharmacological inhibition of PTPMT1 by AD did not affect cytokine production (Figures 3D and 3E), Ptpmt1−/− CD8+ T cells showed a marked reduction in IFN-γ and tumor necrosis factor (TNF) production with unaltered IL-2 synthesis (Figures 3F and 3G). Then, we traced 13C-glucose during the first 24 h post-αCD3/αCD28 stimulation in WT CD8+ T cells cultured with or without AD and in Ptpmt1 WT and Ptpmt1−/− CD8+ T cells (Figures 3H and 3I). Both WT CD8+ T cells incubated with or without AD (Figures 3J and S1G–S1I) and Ptpmt1 WT and Ptpmt1−/− T cells (Figures 3K and S3L–S3N) acquired glucose equally and then metabolized it to pyruvate and lactate to the same extent. However, although pyruvate entered the tricarboxylic acid (TCA) cycle and 13C carbons were incorporated into TCA cycle intermediates in both in WT CD8+ T cells incubated with or without AD (Figures 3J, S1J, and S1K) as well as in Ptpmt1 WT control CD8+ T cells (Figures 3K, S3O, and S3P), it was preferentially shunted to alanine with decreased incorporation into TCA cycle intermediates in Ptpmt1−/− T cells (Figure 3L). Consistent with the role of PTPMT1 in modulating SDH function (Nath et al., 2015), AD-treated WT cells had lower levels of 13C-glucose-derived carbon incorporation into fumarate (Figure 3J). These results highlight that CD8+ T cells require basal levels of CL, but not its de novo synthesis, to allow metabolic activation upon TCR triggering.
Cardiolipin Synthesis Supports CD8+ TM Cell Development, Metabolism, and Function
PTPMT1-ΔT mice allowed us to investigate the role of de novo CL synthesis in T cell responses in vivo. However, this mouse model produced T cells deficient for Ptpmt1 from embryonic development and therefore did not allow us to test the role of de novo CL synthesis exclusively in TM development. We tackled this question from multiple angles by using pharmacological inhibition (Figure 4A), inducible genetic deletion (Figures 4E), and a CRISPR approach (Figure 4I). Inhibition of PTPMT1 with AD during IL-15 TM differentiation (Figure 4A) reduced total CL content (Figure 4B) by additive reduction of the most abundant CL species in CD8+ T cells (Figure S4A). AD worked selectively on CL, because variations in other lipids were limited (Figure S4A). CL synthesis is a slow process, and IL-15 TM cells did not show alterations in OCR upon direct AD treatment (Figure S4B). Mitochondrial mass (Figure S4C, left panel) or ETC complex levels were not altered (Figure S4C, right panel), although AD treatment induced accumulation of short forms of OPA1 (Figure S4C, right panel), commonly observed during mitochondrial fission (Ban et al., 2017; Cipolat et al., 2006; Cogliati et al., 2013). We previously described how TM cells have tight cristae with closely associated ETC complexes, which renders their mitochondrial membranes less susceptible to digitonin disruption than those of TE cells (Buck et al., 2016). Hypothesizing that CL might play a key role in this process, we performed a similar experiment where untreated or AD-treated IL-15 TM cells were incubated with increasing concentrations of digitonin followed by separation of the crude membrane-bound fraction from solubilized proteins by centrifugation. We found a greater proportion of ETC complex subunits in the soluble fraction of AD-treated cells (Figure S4D), indicating that ETC complexes were more loosely embedded in the IMM or that the protein/lipid ratio was altered, because the membranes were more susceptible to digitonin disruption when CL synthesis was inhibited. Electron micrographs of untreated and AD-treated IL-15 TM cells further confirmed cristae structure alterations with increased maximal cristae width (Figure S4E), and live-cell imaging revealed mitochondrial fragmentation (Figure S4F) upon CL synthesis inhibition. In vitro IL-15 TM rely on OXPHOS to meet their energetic needs (Buck et al., 2017). In line with this, only IL-15 TM cells exhibited a survival defect when CL synthesis was inhibited (Figure S4G). Furthermore, AD treatment impaired CD62L expression (Figure S4H), SRC (Figure 4C), and IFN-γ production (Figure 4D) in IL-15 TM cells.
Figure 4.

Cardiolipin Synthesis Supports CD8+ TM Cell Development, Metabolism, and Function
(A) Schematic of CD8+ T cells activated with αCD3/αCD28 + IL-2 for 48 h and then cultured ± AD for 72 h during IL-15 TM cell development.
(B) Quantification of total CL content in IL-15 TM cells cultured as in (A).
(C) OCR of IL-15 TM cells cultured as in (A) at baseline and after exposure to αCD3/αCD28 coated beads, Oligo, FCCP, and Rot/Ant.
(D) IL-15 TM cells were cultured as in (A) and then restimulated with αCD3/αCD28 for 20 h. Percentage of producing IFN-γ cells, and IFN-γ MFI are shown.
(E) Schematic of the activation of CD8+ T cells isolated from Ptpmt1flox/floxErt2-Cre−/− (PTPMT1 iWT) Ptpmt1flox/floxErt2-Cre+/− (PTPMT1 iΔT) mice and 4-OHT treatment.
(F) CL content in Ptpmt1 iWT and Ptpmt1 iΔT CD8+ T cells cultured as in (E).
(G) OCR of Ptpmt1 iWT and Ptpmt1 iΔT CD8+ T cells cultured as in (E) at baseline and after exposure to PMA/Ionomycin, Oligo, FCCP, and Rot/Ant.
(H) Ptpmt1 iWT and Ptpmt1 iΔT CD8+ T cells cultured as in (E) and stimulated with αCD3/αCD28 for 20 h. Percentage of producing IFN-γ cells and IFN-γ MFI are shown.
(I) Schematic of WT CD8+ T cells activated with αCD3/αCD28 + IL-2 for 48 h, cultured additional 24 h in IL-2 before performing Ctrl/Ptpmt1 CRISPR. 24 h after CRISPR, equal number of CRISPR Ctrl/Ptpmt1 KO cells were transferred in congenic recipient mice and donor cell frequencies followed for 21 days before LmOVA WT infection.
(J) Percentage of CD8+ CD45.2+ Tet+ CRISPR Ctrl/Ptpmt1 KO cells treated as in (I), shown as mean ± SEM.
(K) Bacterial burden shown as CFU per μg of liver isolated from mice treated as in (I) 3 days post-LmOVA challenge. Dots represent individual mice.
(L) Schematic of WT CD8+ T cells activated and cultured in IL-2 in the presence of BSA-conjugated 50 μM PG(18:1)2 or 50 μM PG(18:2)2 for 48 h.
(M) Quantification of CL species in IL-2 TE CD8+ T cell cultured ± PG(18:2)2 for 48 h.
(N) Maximal cristae width measured in IL-2 TE CD8+ T cell cultured as in (M).
(O) SRC of IL-2 TE CD8+ T cells cultured as in (M) stimulated with αCD3/αCD28 coated beads.
(P) Cytokine production (normalized to untreated cells) measured in IL-2 TE CD8+ T cells activated as in (M) and stimulated for 20 h with αCD3/αCD28 + IL-2.
(Q) Schematic of adoptive transfer strategy of OT-I IL-2 CD8+ TE cells—pre-treated as in (L)— into congenic recipient mice. Twenty-one days post-transfer, mice were infected i.v. with 1 × 106 CFU of LmOVA ΔActa.
(R) Survival of donor cells 21 days post-transfer, expressed as number of donor cells in the spleen. Dots represent individual mice.
(S) Percentage of circulating donor cells 21 days post-transfer. Dot represent individual mice.
(T) Number of donor cells recovered from spleens of recipient mice 5 days after infection with LmOVA ΔActa. Dots represent individual mice.
Data shown as mean ± SEM of ≥3 independent experiments unless indicated. Statistical comparisons for 2 groups calculated by unpaired two-tailed Student’s t test, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S4.
Although our data showed that AD selectively inhibited CL synthesis (Figures 4B and S4A) to rule out the possibility of off-target effects of the drug we crossed a tamoxifen (4-OHT) inducible (Ert2-CRE) mouse to the Ptpmt1flox/flox mouse to generate a strain (Ptpmt1 iΔT) whose cells could be inducibly deleted of Ptpmt1 upon exposure to 4-OHT in vitro. To avoid any effect on activation and early differentiation, we induced Ptpmt1 deletion in vitro only during TM cell differentiation in IL-15 (Figures 4E and S4I), without a reduction in Crls1 and Tafazzin (Taz) expression levels (Figure S4I). Ptpmt1 iΔT cells recapitulated results from AD-treated cells showing lower CL content than in Ptpmt1flox/floxErt2-Cre− (Ptpmt1 iWT) T cells subjected to the same 4-OHT treatment (Figure 4F). IL-15 Ptpmt1 iΔT cells also showed a survival defect and a lower CD62L expression (Figures S4J), SRC (Figure 4G), and IFN-γ production (Figure 4H) than did Ptpmt1 iWT T cells (Figure S4K).
Finally, to test the role of PTPMT1 in vivo during TM cell generation, we used a CRISPR approach. We activated OT-I cells in vitro and deleted Ptpmt1 only at the peak of effector phase (3 days post-activation), which allowed normal T cell activation and expansion prior to deleting Ptpmt1. After an additional 24 h, we transferred an equal number of CRISPR Ctrl and CRISPR Ptpmt1 knockout (KO) cells into congenic mice and tracked donor cells for 21 days before LmOVA infection (Figure 4I). CRISPR Ptpmt1 KO cells exhibited lower survival in vivo during the TE cell contraction phase (Figure 4J). Strikingly, when mice were then infected with LmOVA, only CRISPR Ctrl cells expanded, whereas CRISPR Ptpmt1 KO cells failed to do so (Figure 4J). As a result, only CRISPR Ctrl cells were able to efficiently clear bacteria from the liver of infected mice (Figure 4K). In summary, PTPMT1 and CL synthesis support CD8+ TM cell generation both in vitro and in vivo.
Enhancing Cardiolipin Synthesis Promotes the Generation of Memory-like CD8+ T Cells
T cells with elongated mitochondria and tight cristae are more fit in adoptive cellular immunotherapy models in mice (Buck et al., 2016). We asked whether increasing CL content, which could promote cristae tightness, would also have a similar effect on T cell fitness. To increase CL content we supplemented IL-2 TE and IL-15 TM cells with different forms of the CL precursor phosphatidylglycerol (PG), i.e., PG(18:2)2 and PG(18:1 Δ9-cis)2, for 48 h (Figure 4L). We found that, without affecting the overall lipidome (Figure S4L), only PG(18:2)2 increased total CL content as well as CL 72:6 and CL 72:8 in both IL-2 TE and IL-15 TM cells (Figures 4M and S4M–S4O). PG(18:1)2 did not increase total CL or the main CL forms containing oleic acid CL 72:4, Cl 72:5, and CL 70:4 (data not shown). The higher CL content observed in IL-2 TE cells, although not sufficient to counteract mitochondrial fragmentation (Figure S4P), tightened cristae (Figures 4N and S4Q) and resulted in higher SRC (Figure 4O), and increased IFN-γ, TNF production (Figure 4P), and expression of CD25 and CD62L (Figure S4R). The further increase in CL content in IL-15 TM cells mediated by PG(18:2)2 (Figure S4M) did not induce changes in surface marker expression (Figure S4R), SRC (Figure S4S), or cytokine production (Figure S4T), suggesting the existence of a maximal threshold for CL content to modulate T cell function. To investigate whether these observations held true in vivo, we adoptively transferred untreated and PG(18:2)2-treated OT-I IL-2 TE cells into congenic mice and tracked cell survival and responses against LmOVA ΔActa infection (Figure 4Q). There were no differences in the number of donor cells in the spleen (Figure 4R), with only a trend toward more circulating PG(18:2)2-treated cells 21 days post-cell transfer (Figure 4S), suggesting no difference in long-term survival between the two populations. However, 5 days after LmOVA ΔActa infection, a greater number of PG(18:2)2-treated cells were recovered in the spleen of infected mice (Figure 4T). These data raise the possibility that higher CL content protects cristae structure upon restimulation, limiting cytochrome c release and activation-induced cell death (AICD) (Corrado et al., 2016). Of note, PG(18:2)2 supplementation in the context of 4-OHT-induced deletion of Ptpmt1 in IL-15 TM cells (Figure S4U) did not rescue CL content (Figure S4V), survival (Figure S4W), or cytokine production (Figure S4X), likely reflecting inefficient incorporation of external PG into CL in the absence of PTPMT1 (Serricchio et al., 2018). Collectively, these data show that enhancing CL content in vitro and in vivo generates T cells that are metabolically and functionally fit.
CD8+ T Cell Defects Observed in TAZ KO Mice Are Not Intrinsic to CD8+ T Cells
Given the functions of CL in CD8+ T cell biology outlined here, we set out to investigate the role of Tafazzin in T cells in a mouse model of Barth syndrome and in a small cohort of Barth syndrome patients. We analyzed CL content in CD8+ T cells isolated from Tafazzin KO mice (full-body deletion) (Figure 5A) at 20 weeks of age, when cardiomyopathy and muscle weakness start to develop (Cadalbert et al., 2015). CL deficiency was not only limited to muscles but also observed in CD8+ T cells of Tafazzin KO mice (Figures 5B and 5C) together with a slight but significant reduction in CD62LhiCD44hi TCM cells (Figure 5D). Upon in vitro stimulation, Tafazzin KO CD8+ T cells produced less IFN-γ (Figures 5E–5G) and proliferated less efficiently (Figure 5H). In addition, IL-15 TM CD8+ Tafazzin KO cells showed an accumulation of short forms of OPA1, a reduction in CIII protein levels (Figure S5A), and altered expression of CD25 and CD62L (Figure S5B) in comparison with IL-2 TE cells.
Figure 5.

CD8+ T Cell Defects n Tafazzin-Deficient Mice and in a Small Cohort of Barth Syndrome Patients
(A) Immunoblot analysis of cell extracts from freshly isolated WT and Tafazzin KO CD8+ T cells.
(B) Lipids extracted from freshly isolated CD8+ T cells from 20-week old WT and Tafazzin KO. Data show Log2FC and p value calculated with ANOVA test.
(C) Total CL amount in freshly isolated CD8+ T cells from 20-week old WT and Tafazzin KO mice. Dots represent individual mice.
(D) TN (CD62LhiCD44lo), TEM (CD62LloCD44hi), and TCM (CD62LhiCD44hi) CD8+ T cells from 20-week--old WT and Tafazzin KO mice. Dots represent individual mice.
(E–G) Representative flow cytometry analysis (E), percentage of IFNγ producing CD8+ T cells (F), and IFN-γ MFI of CD8+ IFN-γ+ T cells (G) of freshly isolated CD8+ T cells from 20-weeks old WT and Tafazzin KO mice stimulated for 4 h with PMA/iono. Dots represents individual mice.
(H) Cell proliferation of CD8+ T cells from 20-week-old WT and Tafazzin KO mice 48 h after activation with αCD3/αCD28 + IL-2. Dots represent individual mice.
(I) Monolysocardiolipin/cardiolipin (18:2)4 ratio in healthy donors and Barth syndrome patients. Dots represent individual donors.
(J) Representative flow cytometry analysis and quantification of the percentage of CD8+ T cells in hPBMCs from healthy donors and Barth syndrome patients. Dots represent individual donors.
(K) Representative flow cytometry analysis and quantification of IFN-γ production of CD8+ T cells from healthy donors and Barth syndrome patients activated with PMA/iono for 5 h. Dots represent individual donors.
Data shown as mean ± SEM of ≥3 independent experiments unless indicated. Statistical comparisons for two groups calculated by unpaired two-tailed Student’s t test or Anova test, where indicated, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figure S5.
To understand whether the T cell impairment observed in mice with total body deletion of Tafazzin was CD8+ T cell intrinsic, we generated Taz CRISPR KO OT-I cells and investigated, in vitro and in vivo, the result of TAZ acute deletion on T cell activation, expansion, and response to infection (Figure S5C). The absence of TAFAZZIN (Figures S5D and S5E) did not change the overall lipid profile (Figure S5F), CL amount (Figure S5G), or composition (Figure S5H) comparable to those of Ctrl CRISPR T cells 6 h post-activation in vitro, the same time point when we observed alterations in CLs in WT cells (Figure 3C), as well as the activation-induced expression of CD5, CD25, CD44, and CD69 (Figure S5I). Thus, TAFAZZIN was dispensable for CD8+ T cell activation. To translate these observations in vivo, we adoptively transferred Ctrl and Taz CRISPR KO OT-I cells into recipient mice and followed their response to LmOVA infection (Figure S5J). Despite Taz CRISPR KO CD8+ T cells showing a defective switch from EEC to SLEC (Figures S5K and S5L) (both after primary and secondary challenge), similar to what observed in PTPMT1-ΔT mice (Figure 2J), CD62L and CD44 expression was similar (Figure S5M). Moreover, Taz CRISPR KO cells responded to antigen stimulation similarly to Ctrl cells and generated TM cells that efficiently expanded upon secondary infection (Figure S5J). As per TAZ function, 7 days post-secondary infection-sorted CD8+ OT-I Taz CRISPR KO OT-I cells showed a reduced level of CL in comparison with WT cells (Figures S5N and S5O). Nevertheless, even increasing the window between primary and secondary LmOVA challenge from 3 weeks to 2 months did not alter CD8+ T cell function but confirmed defects in the EEC to SLEC transition (Figures S5Q–S5T). In both experiments, Taz CRISPR KO CD8+ T cells cleared bacteria from the liver of infected mice (Figures S5P and S5U). Overall, these data suggest that the defects observed in the T cell compartment of TAZ KO mice either are not CD8+ T cell intrinsic or might derive from hematopoietic lineage developmental alterations.
T Cell Defects in a Small Cohort of Barth Syndrome Patients
We analyzed peripheral blood mononuclear cells (PBMCs) from 5 patients under the care of the NHS Barth syndrome Service (Barth syndrome group average age 13 years, min 6 years, max 28 years; healthy donor group average age 28 years, min 22 years, max 39 years) with pathology confirmed both by MLCL/CL(18:2)4 ratio (Figure 5I) and TAFAZZIN sequencing (data not shown). Patients that had undergone cardiac transplantation, and hence were on immunosuppressive drugs, were excluded from the analysis. We found that patients had significantly lower relative frequencies of CD8+ T cells in PBMCs than did healthy donors (Figure 5J), with no apparent differences in the CD4+ T cell compartment (Figure S5V). Although no differences were detected in the expression of naive/memory CD45RA and CD45RO (Figure S5W), nor in surface markers of different CD8+ TM subpopulations (Figure S5X), CD8+ T cells from Barth syndrome patients showed a trend toward impaired IFN-γ production after stimulation (Figure 5K). Overall, our analysis suggests a possible CD8+ T cell impairment in patients affected by Barth syndrome.
Discussion
Although metabolic reprogramming during the adaptive immune response has been widely explored in recent years, the role of mitochondrial lipid composition and, in particular, CL in this scenario remained uninvestigated. Our study shows that the dynamic regulation of synthesis and remodeling of a single lipid confers to T cell mitochondria the plasticity necessary for adapting to the different metabolic and functional requirements during all phases of the T cell response—from activation, to expansion, contraction, and eventually long-term immune protection.
In the absence of CL, PTPMT1-ΔT mice mounted a defective CD8+ T cell response upon infection. We found that de novo CL synthesis was not necessary for T cell activation, but the presence of a sufficient initial pool of CL in the mitochondrial membrane was required. Indeed, ex vivo-activated Ptpmt1−/− CD8+ T cells were defective in cytokine production and metabolic adaptation even before a proliferative defect emerged. During activation the CL profile is modulated in human T cells (Mürke et al., 2016), whereas in murine CD4+ T cells stomatin-like protein 2 (SLP2) organizes CL in lipid rafts (Christie et al., 2012). Given the interaction of CL with different subunits of the ETC, and the burst in ROS during T cell activation (Dudek, 2017; Sena et al., 2013), it is tempting to speculate that CL modifications/oxidations are required for the metabolic shifts that occur during early T cell activation and that Ptpmt1 ablation might alter ROS production upon TCR triggering. It also remains to be determined whether during antigen stimulation ROS causes CL oxidation to instruct mitochondrial structural and metabolic reprograming or whether ROS acts in a CL-independent manner.
Our lab previously showed that Opa1-deficient T cells are able to form TE cells after primary infection with a later failure in TM cell generation (Buck et al., 2016). Here, we characterize how Ptpmt1−/− T cells have a broader defective phenotype, failing to activate properly and expand into TE cells. Thus, whereas OPA1 and CL might in part functionally share the role of maintaining cristae structure, the different nature of a protein versus a lipid, a specific signaling role for CL during T cell activation, and a more structural requirement for CL synthesis for mitochondrial biogenesis during T cell expansion might contribute to the differential cellular behavior of the two models.
When autophagy is triggered by low nutrient availability, mitochondria elongate to sustain viability and avoid starvation-induced cell death (Buck et al., 2016; Gomes et al., 2011). This change is accompanied by an increase in the number of cristae, assembly of respiratory chain supercomplexes, and in the dimerization and activity of the ATP synthase (Balsa et al., 2019; Gomes et al., 2011). Here we show that upon glucose restriction, cellular metabolism rapidly responds by synthesizing CL, independently from changes in mitochondrial mass, thus increasing SRC and limiting apoptosis. Of note, both these pro-survival features are lost if de novo CL synthesis is inhibited during glucose restriction. CL binds selectively but transiently to the ATP synthase, affecting its supramolecular organization (Acehan et al., 2011; Duncan et al., 2016; Mehdipour and Hummer, 2016), as well as to complex I (Jussupow et al., 2019) and complex II (Schwall et al., 2012), modulating their stability and function. Of note, in our experiments the subunit levels of complex I and complex II (NDUFB8 and SDHB, respectively) are modulated together with CL during glucose restriction, or upon CD28 co-stimulation, which also results in increased OXPHOS efficiency and higher CL content.
Safeguarding mitochondrial integrity and metabolism is instrumental in generating long-lived and fit TM cells (Buck et al., 2016). Indeed, mitochondrial fusion-fission cycles allow the segregation and elimination of dysfunctional mitochondria through autophagy (Pickles et al., 2018). Induction of autophagy sustains TM cell development, whereas its inhibition combined with mitochondrial dysfunction contributes to AICD of short-lived TE cells (Corrado et al., 2016; Puleston et al., 2014; Xu et al., 2014). We show that de novo CL synthesis is required to increase mitochondrial metabolic capacity and allow AICD resistance during TM cell differentiation. Thus, increasing CL content might improve adoptive cellular immunotherapy, where a lack of T cell persistence after transfer remains an issue (Restifo et al., 2012; Tschumi et al., 2018).
Mutations in TAFAZZIN responsible for Barth syndrome alter CL remodeling and reduce total CL content over time due to the inability of cells to regenerate mature CL from MLCL (Schlame et al., 2003). In the mouse gut, intraepithelial lymphocytes (IELs) maintain a controlled activation state that relies on mitochondria through a TAFAZZIN-dependent CL remodeling mechanism (Konjar et al., 2018). Although able to migrate to the site of infection and produce cytokines, bone marrow chimera-derived Tafazzin-deficient IELs are not able to restrict E. vermiformis infection (Konjar et al., 2018). Here, we reinforce these observations in a total body Tafazzin KO mouse model, resembling more closely the human pathology where TAFAZZIN inactivating mutations are not confined to the hematopoietic compartment. In particular, experiments in TAZ KO mice suggested that reduced number and impaired function of CD8+ T cells follow long-term systemic CL deficiency. In addition, our CRISPR data support the idea that the T cell functional defects observed in bone marrow chimera Tafazzin-deficient mice, as well as in total body TAZ KO mice, might not be CD8+ T cell intrinsic or might derive from hematopoietic lineage developmental alterations.
Cardiomyopathy and neutropenia are life-threatening features of Barth syndrome. Some Barth syndrome patients require cardiac transplantation followed by lifelong immunosuppressive drug treatment (Mangat et al., 2007). Our analysis of hPBMCs from a limited number of Barth syndrome patients suggests the possibility of a relative lymphopenia and functional impairment in the CD8+ T cell compartment. However, more detailed clinical studies to assess the absolute number, frequency, and function of lymphocyte subsets in a larger number of patients are required. Less efficient responses to T cell-mediated vaccines or lower susceptibility to acute organ rejection after heart transplant in Barth syndrome patients compared to non-affected age-matched individuals might be expected if our hypothesis holds true. In our view it is possible that the adaptive immune phenotype in these patients might exacerbate the impact of neutropenia in the susceptibility to infections by a combination of the impaired activation, proliferation, and differentiation of TM cells.
In conclusion, we describe the pivotal role of the dynamic regulation of CL synthesis in modulating CD8+ T cell function during the immune response, from activation to TM cell differentiation. Absence of CL due to impaired synthesis (PTPMT1-ΔT) or remodeling (Tafazzin KO) results in compromised CD8+ T cell function, although our data suggest the latter is not CD8+ T cell intrinsic. Moreover, we propose modulation of CL content and/or composition as a strategy to improve adoptive cell immunotherapy. Lastly, our findings suggest a potentially unrecognized T cell impairment in Barth syndrome patients, broadening the spectrum of pathologic features of Barth syndrome and prompting a further evaluation of immune function in patients and in clinical trials targeting CL modifications.
Limitations of study
Although our study shows that cardiolipin synthesis is regulated during CD8+ T cell responses to infection, it addresses neither the role of different CL species nor the regulation of acyl chain modifications. It also remains to be determined the role of cardiolipin oxidation and its interplay with ROS production upon CD8+ T cell activation. Lastly, analysis of T cell function in larger cohort of Barth syndrome patients and epidemiological studies on susceptibility to acute organ rejection after heart transplant are necessary to further corroborate our observations.
STAR★Methods
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD3, inVivoMAb | Bio Cell | Cat#BE0002; RRID: AB_1107630 |
| anti-mouse CD28, inVivoMab | Bio Cell | Cat#BE0015; RRID: AB_1107628 |
| BV421 anti-mouse CD8a | Biolegend | Cat#100738; RRID: AB_11204079 |
| T-select I-Ab OVA Tetramer-PE | MBL | Cat#TS-M710-1 |
| FITC anti-mouse CD45.2 | Biolegend | Cat#109806; RRID: AB_313443 |
| PE anti-mouse CD62L | Biolegend | Cat#104408; RRID: AB_313095 |
| APC-Cy7 anti-mouse CD44 | Biolegend | Cat#103028; RRID: AB_830785 |
| PerCP/Cyanine5.5 anti-mouse/human KLRG1 | Biolegend | Cat#138418; RRID: AB_2563015 |
| AF647 anti-mouse CD127 (IL-7Rα) | Biolegend | Cat#135020; RRID: AB_1937209 |
| AF488 anti-mouse IFNg | Biolegend | Cat#505813; RRID: AB_493312 |
| APC anti-mouse CD5 | Biolegend | Cat#100626; RRID: AB_2563929 |
| AF647 anti-mouse CD25 | Biolegend | Cat#102020; RRID: AB_493458 |
| PE anti-mouse CD69 | Biolegend | Cat#104508; RRID: AB_313111 |
| PE-Cy7 anti-Ki67 | Biolegend | Cat#652426; RRID: AB_2632694 |
| AF647 anti-mouse TNF-alpha | Biolegend | Cat#506314; RRID: AB_493330 |
| PE anti-mouse IL-2 | Biolegend | Cat#503808; RRID: AB_315302 |
| APC-Cy7 anti-human CD14 | Biolegend | Cat#301819; RRID: AB_493694 |
| APC-Cy7 anti-human CD16 | Biolegend | Cat#302017; RRID: AB_314217 |
| APC-Cy7 anti-human CD19 | Biolegend | Cat#302217; RRID: AB_314247 |
| APC-Cy7 anti-human CD8 | Biolegend | Cat#300926; RRID: AB_10613636 |
| APC-Cy7 anti-human CD25 | Biolegend | Cat#302613; RRID: AB_314283 |
| BV711 anti-human CD45RO | Biolegend | Cat#304235; RRID: AB_11218600 |
| FITC anti-human IFNg | Biolegend | Cat#502507; RRID: AB_315232 |
| BV421 anti-human CD4 | Biolegend | Cat#357423; RRID: AB_2721518 |
| PE anti-human hCD45RO | Biolegend | Cat#304206; RRID: AB_314422 |
| APC anti-human CD45RA | Biolegend | Cat#304112; RRID: AB_314416 |
| AF488 anti-human CD62L | Biolegend | Cat#304816; RRID: AB_528857 |
| BV785 anti-human CD197 | Biolegend | Cat#353229; RRID: AB_2561371 |
| PE-Cy7 anti-humna CD95 | Biolegend | Cat#305621; RRID: AB_2100370 |
| BV711 anti-human CD28 | Biolegend | Cat#302947; RRID: AB_2616856 |
| PE-Cy7 anti-human CD4 | Biolegend | Cat#357409; RRID: AB_2565661 |
| BV711 anti-human CD25 | Biolegend | Cat#302635; RRID: AB_11219793 |
| AF647 anti-human IFNg | Biolegend | Cat#502516; RRID: AB_493031 |
| APC-Cy7 anti-human CD56 | Biolegend | Cat#318331; RRID: AB_10898118 |
| BV605 anti-mouse TCRb | Biolegend | Cat#109241; RRID: AB_2629563 |
| APC anti-mouse TCR gammadelta | Biolegend | Cat#118115; RRID: AB_1731824 |
| PE-Cy7 anti-mouse CD8b | Biolegend | Cat#140416; RRID: AB_2564385 |
| OPA1 | BD Biosciences | Cat#612606; RRID: AB_399888 |
| ACONITASE | Abcam | Cat#ab126595; RRID: AB_11130595 |
| SDHA | Cell Signaling | Cat#11998S; RRID: AB_2750900 |
| IDH | Cell Signaling | Cat#8137S; RRID: AB_10950504 |
| SCS | Cell Signaling | Cat#5557S; RRID: AB_10691593 |
| CPT1a | Proteintech | Cat#15184-1-AP; RRID: AB_2084676 |
| TUBULIN | Cell Signaling | Cat#2144S; RRID: AB_2210548 |
| CALNEXIN | Santa Cruz | Cat#sc-23954; RRID: AB_626783 |
| GAPDH | Cell Signaling | Cat#5174P; RRID: AB_10622025 |
| TAFAZZIN | Santa Cruz | Cat#sc-365810; RRID: AB_10842049 |
| CRLS1 | Abcam | Cat#ab156882 |
| TOM20 | Cell Signaling | Cat#42406; RRID: AB_2687663 |
| MnSOD | Enzo LifeScience | Cat#ADI-SOD-110-F; RRID: AB_2750987 |
| OXPHOS cocktail | Abcam | Cat#ab110413; RRID: AB_2629281 |
| ACTIN | Cell Signaling | Cat#4970; RRID: AB_2223172 |
| PTPMT1 | Sigma | Cat#HPA043932; RRID: AB_2678738 |
| Bacterial and Virus Strains | ||
| LmOVA ΔActa | N/A | N/A |
| LmOVA WT | N/A | N/A |
| Biological Samples | ||
| Healthy control blood | UniKlinik Freiburg | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator | StemCell Technologies, Inc. | Cat#10990 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| Mitotracker green FM | Thermofisher Scientific | Cat#M7514 |
| TMRM | Thermofisher Scientific | Cat#T668 |
| D-GLUCOSE-13C6, 99 ATOM % 13C, 399% | Sigma | Cat#389374 |
| LINOLEIC ACID-13C18, 99 ATOM % 13C, 97%+ | Sigma | Cat#605735 |
| Alexidine dihydrochloride | Sigma | Cat#A8986 |
| PMA | Sigma | Cat#P1585 |
| Ionomycin calcium salt | Sigma | Cat#I3909 |
| Anti-Rat IgG (Fc specific)-Biotin antibody | Sigma | Cat#SAB3700543 |
| Indo-1 AM | Thermofisher Scientific | Cat#I1223 |
| Thapsigarargin | Tocris | Cat#1138 |
| Fx cycle | Thermofisher Scientific | Cat#R37166 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Thermofisher Scientifi | Cat#L34957 |
| LIVE/DEAD™ Fixable IR Dead Cell Stain Kit | Thermofisher Scientific | Cat#L34975 |
| Cell Trace Violet Proliferation Kit | Thermofisher Scientific | Cat#C34557 |
| Recombinant human IL-2 | Peprotech | Cat#200-02 |
| Recombinant murine IL-15 | Peprotech | Cat#210-15 |
| PG(18:1)2 | Avanti Lipids | Cat#840475 |
| PG(18:2)2 | Avanti Lipids | Cat#840485 |
| Oligomycin | Sigma | Cat#1404-19-9 |
| FCCP | Sigma | Cat#370-86-5 |
| Tamoxifen | Sigma | Cat#T5648 |
| Rotenone | Sigma | Cat#83-79-4 |
| Antimycin A | Sigma | Cat#1397-94-0 |
| Alt-R® S.p. Cas9 Nuclease V3 | IDT | Cat#1081059 |
| Brefeldin A solution | BioLegend | Cat#BLD-420601 |
| Brain Heart infusion Broth (BHI) | Sigma | Cat#53286 |
| Cysclosporine H | Sigma | Cat#SML1575 |
| 7-AAD Viability Staining Solution | BioLegend | Cat#420404 |
| Critical Commercial Assays | ||
| EasySep Mouse CD8+ TCell Isolation Kit | StemCell Technologies, Inc. | Cat#19853 |
| EasySep Human CD45RO+ CD8+ T Cell Isolation Kit | StemCell Technologies, Inc. | Cat#19159 |
| EasySep Human CD45RA+ CD8+ T Cell Isolation Kit | StemCell Technologies, Inc. | Cat#19258 |
| High Capacity cDNA Reverse Transcription Kit | Thermofisher Scientific | Cat#4368814 |
| iTaq Universa Probes Supermix | Bio-Rad | Cat#1725131 |
| Pierce BCA protein assay kit | Thermofisher Scientific | Cat#23225 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Jackson laboratories | RRID: IMSR_JAX:000664 |
| PTPMT1 floxed mice | Jackson laboratories | RRID: IMSR_JAX:020775 |
| PhAM mice | Jackson laboratories | RRID: IMSR_JAX:018397 |
| OT-I mice | Jackson laboratories | RRID: IMSR_JAX:003831 |
| CD45.1 congenic mice | Jackson laboratories | RRID: IMSR_JAX:002014 |
| TAZ KO | Dr. Douglas Strathdee | N/A |
| Oligonucleotides | ||
| PTPMT1 sgRNA1 | IDT | Mm.Cas9.PTPMT1.1.AA |
| PTPMT1 sgRNA2 | IDT | Mm.Cas9.PTPMT1.1.AB |
| PTPMT1 sgRNA3 | IDT | Mm.Cas9.PTPMT1.1.AC |
| TAZ sgRNA1 | IDT | Mm.Cas9.TAZ.1.AA |
| TAZ sgRNA2 | IDT | Mm.Cas9.TAZ.1.AB |
| TAZ sgRNA3 | IDT | Mm.Cas9.TAZ.1.AC |
| TaqMan Gene expression Assay, Assay ID: Mm00504978_m1 TAZ FAM-MGB | Applied Biosystem | Cat#4448892 |
| TaqMan Gene expression Assay, Assay ID: Mm04225274_s1 Gene Symbol: ND1 FAM-MGB | Applied Biosystem | Cat#4331182 |
| TaqMan Gene Expression Assay, Assay ID: Mm03024075_m1 Gene: Hprt, FAM-MGB | Applied Biosystem | Cat#4351368 |
| Recombinant DNA | ||
| Plasmid: pMAX-GFP (control vector in nucleofector kit) | LONZA | N/A |
| Software and Algorithms | ||
| Wave software version 2.4 | Agilent | http://www.agilent.com/cs/ContentServer?c=Page&pagename=Sapphire/Page/HomePage |
| FlowJO | FlowJO | https://www.flowjo.com |
| Graphpad Prism 7 (seahorse and statistical analysis) | Graphpad software | http://www.graphpad.com |
| ImageJ | NIH | N/A |
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erika L. Pearce (pearce@ie-freiburg.mpg.de)
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
This study did not generate datasets
Experimental Model and Subject Details
Cell Culture and Drug Treatment
Mouse Cells
CD8+ T cells were isolated from spleens of 8-10 week-old mice using the naive or bulk CD8+ T cell kit (Stem Cell technologies, Cat# 19858, 19853) according to the manufacturers protocol. Both male and female mice were used and within experiments mice were age and sex matched. Sample size is indicated in the figure legends. Isolated T cells were activated using plate bound αCD3 (5 μg/mL) (anti-mouse CD3, inVIVOMAb Cat# BE0002), in 1640 media supplemented with 10% fetal calf serum, 4mM L-glutamine, 1% penicillin/streptomycin, 100 U/mL hrIL-2 (Peprotech), 55 μM beta-mercaptoethanol, and ± 0.5 μg/mL soluble αCD28 (anti-mouse CD28, inVivoMab Cat# BE0015) under 5% CO2, atmospheric oxygen, at 37°C in a humidified incubator. TE and TM cell differentiation was performed as previously described (Buck et al., 2016). Briefly, media changes were performed from day 2 post activation and then daily up to day 6. For TE cell differentiation cells were incubated in 100 U/mL hrIL-2 (Peprotech), for TM cells in 100U/mL mIL-15 (Peprotech). Where indicated cells were treated with vehicle control (DMSO or EtOH)) or 1 μM Alexidine dihydrochloride (AD, Sigma, A8986), 50 μM PG(18:2)2 (Cat# 840485), PG(18:1 Δ9-cis)2 (Cat# 840475), 5nM 4-OHT. For in vitro survival assays, cells were activated for 3 days as described, then cultured in either IL-2 at 5x104 cells/mL, or in IL-15 at 1x105 cells/mL in 96 well round bottom plates. Survival was analyzed by 7-AAD exclusion using flow cytometry.
Human Cells
Fresh buffy coats from healthy donors were kindly provided by the Institute for Transfusion Medicine and Gene Therapy, Medical Center, University of Freiburg under approval by the ethical committee of the University of Freiburg. PBMCs from healthy donors and Barth Syndrome patients were collected at University of Bristol in accordance with the Helsinki Declaration with approval from the UK NHS Research Ethics committee (permit number 09/H0202/52). Age and sex of healthy donors provided by the Institute for Transfusion Medicine and Gene Therapy, Medical Center, University of Freiburg were blinded to the authors. Barth syndrome patients were males (Barth syndrome group average age 13 years, min 6 years, max 28 years; healthy donor group average age 28 years, min 22 years, max 39 years). Informed consent was provided by all patients, or by their parents, in the case of children. Human CD45RA+ and CD45RO+ CD8+ T cells were isolated using CD8+ T cell kits (Stem Cell technologies, Cat# 19258, 19159). Researchers were blinded to the identity of the donors, and age or sex matching was not performed. Sample size is indicated in the figure legends.
Mice and Immunizations
C57BL/6J (RRID: IMSR_JAX:000664), PhAM (RRID: IMSR_JAX:018397), major histocompatibility complex (MHC) class I-restricted OVA specific TCR OT-I transgenic mice (RRID: IMSR_JAX:003831), and CD45.1 congenic (RRID: IMSR_JAX:002014) mouse strains were purchased from The Jackson Laboratory. PTPMT1 floxed (RRID: IMSR_JAX:020775) mice were a kind gift form Jack E. Dixon (University of California, San Diego, CA). TAZ KO mice were generated and maintained at the University of Glasgow by D. Strathdee under license approved by University of Glasgow Ethical Review Process and the UK Animal Procedures Committee London. PTPMT1 floxed mice were crossed to CD4-Cre or tamoxifen inducible ERT2-Cre mice. Experimental studies including mice of the PTPMT1 strain were approved by the Regierungspräsidium Freiburg. All mice were maintained in C56Bl6/J background and were the results in in-house matings. In all experiments including WT and KO mice, WT littermates from same breedings were used as controls. All mice, apart from TAZ KO, were maintained at the Max Planck Institute of Immunobiology and Epigenetics and cared for according to the Institutional Animal Use and Care Guidelines. Briefly, mice were maintained in IVC Tecniplast green line cages with max five mice per cage, light cycle was 14/10 h, temperature 20-24°C, humidity 45%–64%. Ssniff MZ-Ereich chow (Cat# V1185-300) was provided ad libitum. Health monitoring program followed FELASA recommendations (Laboratory Animals 2014, Vol. 48(178-192). Briefly, germ free NMRI mice are used as bedding sentinels receiving dirty bedding from up to 100 other cages weekly for three months before being tested for all pathogens and opportunists recommended in the guidelines. Within experiments, age and sex matched mice were injected intravenously (i.v.) as indicated with a sublethal dose of 1x106 colony forming units (CFU) of recombinant L. monocytogenes expressing OVA deleted for actA (LmOVA ΔActa) for primary immunizations and challenged with 5x107 CFU for secondary immunizations. Where indicated, 3x104 CFU of L. monocytogenes expressing OVA WT were used for primary infection and 1x106 for secondary immunization.
All mice were used for experiments between 8-10 weeks of age apart from TAZ KO mice that were 20 weeks old (time in which pathology develops) and animals used for TEM and TCM cell sorting that were 20-month old (in order to obtain a sufficient proportion of both TEM and TCM cells from the same animal, 20% and 40% respectively (in 2-month old mice TEM cells constituted only 5% of total CD8+ T cells). For in vivo experiments male mice were used, for in vitro studies both sexes were used interchangeably (no influence or association of sex on the results was observed).
Methods Details
Bacterial Burden
Liver biopsies were weighed and homogenized in 1ml of PBS. 10 μL per serial dilution of the original homogenate was plated on a Brain Heart infusion Broth (BHI, Sigma Cat# 53286) agar and incubated over night at 37°C. One day later the number of colonies were counted and normalized to the weight of the liver biopsy.
Adoptive Transfers
For in vivo TM cell experiments, £1x104 OT-I+ CD8+ T cells/mouse from donor splenocytes were transferred intravenously (i.v.) into congenic recipient mice. Blood or spleens were collected at indicated time points and analyzed by flow cytometry. For in vivo survival experiments, 1-2x106 treated IL-2 TE cells (6 days post activation) cells/mouse were injected i.v. into naive congenic CD45.1 mice. Cells were recovered at indicated time points from the spleen or lymph nodes and analyzed by flow cytometry.
Flow Cytometry
Fluorochrome-conjugated monoclonal antibodies were purchased from eBioscience, BD PharMingen, or Biolegend. Staining was performed in 1% FBS/PBS for 30 min on ice, dead cells were excluded with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo scientific). OVA-specific CD8+ T cells from spleen, lymph node, or blood were quantified after red blood cell lysis by direct staining with H2-KbOVA257-264 (KbOVA) MHC-peptide tetramers. For intracellular cytokine staining cells were reactivated with αCD3/αCD28 overnight or 50 ng/mL phorbol 12-myristate 13-acetate (PMA) + 500 ng/mL ionomycin (all Sigma) for 4 h and cultured in the presence of Brefeldin A for 4 h prior to fixation using Cytofix Cytoperm (BDBioscience). MitoTracker and TMRM staining were performed according to the manufacturer’s instructions (Life Technologies) using respectively 10nM and 2nM of Mitotracker Green and TMRM in presence of 0.5 μM of Cysclosporine H. Cells were collected on Fortessa flow cytometers (BD Biosceince) and analyzed using FlowJo software (FlowJo).
Confocal and Electron Microscopy Imaging
Spinning disk confocal and EM imaging was performed as described previously (Buck et al., 2016). Cells were activated as indicated and transferred to glass bottom dishes (MatTek) coated with fibronectin(Sigma) in complete medium containing IL-2 or IL-15 using a Zeiss spinning disk confocal with an Evolve (EMCCD) camera. Cells were kept in a humidified incubation chamber at 37°C with 5% CO2 during image collection. Confocal imaging was analyzed using Imaris imaging software. For Electron Microscope imaging 2 × 106 T cells were fixed in 2.5% glutaraldehyde in 100 mM sodium cocodylate, washed in cocodylate buffer. After dehydration samples were embedded in Eponate 12 resin (Ted Pella) and sections were cut. Images were acquired using a FEI Tecnai 12 Transmission electron microscope equipped with a TIETZ digital camera. Cristae width was measured using ImageJ software and averaged over 50 independent images, acquisition of EM micrographs and measurements of max cristae width displayed were performed using ImageJ software (NIH). Brightness and contrast were adjusted in ImageJ software (NIH)
Metabolic Phenotyping
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured in XF media (non-buffered RPMI 1640 containing 25 mM glucose, 2mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to 1 μM oligomycin, 1.5 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 100 nM rotenone + 1 μM antimycin A, or 50 ng/mL phorbol 12-myristate 13-acetate (PMA) + 500 ng/mL ionomycin (all Sigma) using a 96 well XF or XFe Extracellular Flux Analyzer (EFA) (Seahorse Bioscience). 2 x105 T cells per well (33 wells per sample) were spun onto poly-D-lysine coated seahorse 96 well plates and preincubated at 37°C for a minimum of 45 min in the absence of CO2. For activation of T cells, αCD3 ± CD28 coated beads were used at 1:1 ratio of beads:T cell Thermofisher, Cat# 11452D). For metabolic tracing naive (TN) cells (Figure 4) were isolated and activated as described above in glucose-free media supplemented with 11 mM D-[U13C] glucose or 11mM unlabeled D-glucose for 24 h. Cells were washed with ice cold 0.9% w/v NaCl buffer and metabolites were extracted three times with cold methanol (80%) and analyzed by GCMS. For glucose and palmitate tracing into CL, T cells were cultured in IL-2 or IL-15 condition in presence of 11 mM D-[U13C] glucose or 20 μM [U13C]-Palmitate for 72 h. Medium was changed daily. Cell pellets for lipid extraction were collected at indicated time points.
Lipid Extraction and Quantification
Lipids were extracted using a biphasic Methyl tert-butyl ether (MTBE) extraction protocol (adapted from Matyash et al., 2008). In brief, cells were resuspended in 100uL cold PBS in glass vials. Cold methanol (750uL), MTBE (2mL), and water (625uL) were added sequentially with vortexing. Samples were centrifuged to separate phases, and the upper organic phase was dried using a Genevac EZ2 speed vac. Samples were resuspended in 2:1:1 isopropanol:acetonitrile:water prior to analysis. LC-MS was carried out using an Agilent Zorbax Eclipse Plus C18 column using an Agilent 1290 Infinity II UHPLC in line with an Agilent 6495 Triple Quad QQQ-MS. Lipids were identified by fragmentation and retention time, and were quantified using Agilent Mass Hunter software.
Glucose and Linoleic Acid Tracing into Cardiolipin
Label tracing by LC-MS was carried out using an Agilent 1290 Infinity II UHPLC inline with a Bruker Impact II QTOF-MS operating in negative ion mode. Scan range was from 50 to 1600 Da. Mass calibration was performed at the beginning of each run. LC separation was on a Zorbax Eclipse plus C18 column (100 × 2 mm, 1.8 μm particles) using a solvent gradient of 70% buffer A (10 mM ammonium formiate in 60:40 acetonitrile:water) to 97% buffer B (10 mM ammonium formiate in 90:10 2-propanol:acetonitrile). Flow rate was from 400 μL/min, autosampler temperature was 5°C and injection volume was 2 μL. Data processing including correction for natural isotope abundance was carried out by an in-house R script.
Glucose Tracing into Central Carbon Metabolites
Dried metabolite extracts were resuspended in pyridine and derivatized with methoxyamine (sc-263468 Santa Cruz Bio) for 60 min at 37°C and subsequently with N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamid, with 1% tert-utyldimethylchlorosilane (375934 Sigma-Aldrich) for 30 min at 80°C. Isotopomer distributions were measured using a DB5-MS GC column in a 7890 GC system (Agilent Technologies) combined with a 5977 MS system (Agilent Technologies). Data processing, including correction for natural isotope abundance was performed by an in-house R script.
RT-PCR
RNA isolations were done by using the RNeasy kit (QIAGEN) and single-strand cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). All RT-PCR were performed using iTaq Universa Probes Supermix (Biorad) and Taqman primers (Thermo Fisher Sceintific) using an Applied Biosystems 7000 sequence detection system. The expression levels of mRNA were normalized to the expression of a housekeeping gene (HPRT ot ActB).
mtDNA/nDNA Quantification
Genomic DNA was extracted with QIAGEN QIAamp DNA Micro Kit. DNA was quantified, diluted to 10ng/μl and 50 μg of DNA was used for qPCR analysis of mtDNA (ND1) and nuclear DNA transcripts (Hprt).
Calcium Flux
CD8+ T cells were isolated using CD8+ T cell kit (Stem Cell technologies, Cat# 19853). 3∗10ˆ6 cells were loaded 30min at 37°C in humidified incubator with 2uM Indo-AM dissolved in complete medium + 0,5 μM CsH. Then, cells were washed and resuspended in 300 μL of complete medium supplemented with 0,5 μM CsH and 5 μg/mL anti-mouse CD3 (inVivoMAb Cat# BE0002) 30min at 4°C in the dark. At cytofluorimeter indoviolet and indoblue were acquired in linear scale. Tube was placed in 37°C temperature-controlled tube holder immediately before acquisition and signal was acquired as follows: 30 s basal, 6min after addition of Anti-Rat IgG (Fc-specific)biotin crosslinking antibody (Sigma, SAB3700543, diluted 1:50 directly in the acquisition tube), 2 min after adding tapsigargin (final concentration 100nM) to measure endoplasmic reticulum calcium stores depletion, 1min after adding Ionomycin (final concentration 500 ng/mL) to evaluate maximal calcium flux.
Cell Cycle Analysis
At indicated time points after activation CD8+ T cells were stained with NearIR Live dead staining (Thermofisher) 10min RT in the dark, fixed in fixative buffer from eBioscience Foxp3 / Transcription Factor Staining Buffer Set (00-5523-00) for 60min at RT. After a wash in perm buffer, cells were stained with PE-Cy7 Anti-Ki67 antibody in perm buffer 45min 4°C in the dark. After additional wash cells were resuspended in FxCycle (Thermofisher R37166) diluted 1:4000 in PBS immediately before flow cytometry acquisition.
Western Blotting
For western blot analysis, cells were washed with ice cold PBS and lysed in 1 x Cell Signaling lysis buffer (20 mM Tris-HCL [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1mM Na3VO4, 1 μg/mL leupeptin), supplemented with 1mM PMSF for 30min followed by centrifugation at 20.000 x g for 10 min at 4°C. Cleared protein lysate were quantified using Pierce BCA protein assay kit (Cat#23225) according to manufacturer’s instruction. Cleared protein extracts were denatured with LDS loading buffer for 10 min at 70°C and loaded onto precast 4% to 12% bis-tris protein gels. Proteins were transferred onto nitrocellulose membranes using the iBLOT2 system following the manufacturers protocols. Membranes were blocked with 5% w/v milk and 0.1% Tween-20 in TBS and incubated with the appropriate antibodies in 5% w/v BSA in TBS with 0.1% Tween-20 overnight at 4°C. All primary antibody incubations were followed by incubation with secondary HRP-conjugated antibody (Pierce) in 5% milk and 0.1% Tween-20 in TBS and visualized using SuperSignal West Pico or Femto Chemiluminescent Substrate (Pierce) on Biomax MR Film (Kodak). Optical density of the signals on the film was quantified using grayscale measurements in ImageJ software (NIH) and converted to fold change, normalized to the loading control.
Generation of CRISPR KO T Cells
CD8+ T cells were electroporated using Amaxa electroporation system with 60 nmol of sgRNA-CAS9 (Alt-R® S.p. Cas9 Nuclease V3) complexes (sgRNAs: Mm.Cas9.TAZ.1AA, Mm.Cas9.TAZ.1AB, Mm.Cas9.TAZ.1C targeting TAZ, Mm.Cas9.PTPMT1.1.AA, Mm.Cas9.PTPMT1.1.AB, Mm.Cas9.PTPMT1.1.AC targeting PTPMT1) following manufacturer’s guidelines. Naive CD8+ T cells were cultured for 24 h in IL-7 before being activated with αCD3/αCD28 + IL-2.
Quantification and Statistical Analysis
Flow cytometry data were analyzed using FlowJo 10 (BD Biosciences). Statistical Analyses were performed using Prism 7 software (GraphPad) and results are represented as Mean ± SEM, unless otherwise indicated. Comparisons for two groups were calculated using unpaired two-tailed Student’s t tests. Comparisons of more than two groups were calculated using one-way ANOVA with Tukey multiple comparison tests. We observed normal distribution and no difference in variance between groups in individual comparisons. Selection of sample size was based on extensive experience with metabolic assays. The sample size for in vivo experiments was based on previous experience with infection experiments. The number of independent experiments performed, and the p values for each experiment are reported in the corresponding figure legends. For both in vitro and in vivo experiments, no initial exclusion criteria were used and no animals or replicates were excluded from the study.
Acknowledgments
We thank members of the Pearce labs for support and helpful discussions; Johan Friden for assistance with the graphical abstract; the Electron Microscopy Laboratory at the University of Padova; and the Metabolomics, Imaging, FACS facilities at the MPI-IE for technical support. This work was supported by the Max Planck Society. M.C. was supported by an Alexander von Humboldt Fellowship. D.S. and E.A. were supported by Cancer Research UK grant A31287. B.A. and C.G.S. were supported by Barth Syndrome Foundation.
Author Contributions
M.C. and E.L.P. designed research. M.C., J.H.E., M.V., B.A., C.G.S., D.S., E.T., D.O., E.J.P., and E.L.P. provided conceptual input. M.C., J.H.E., M.V., L.J.F., D.E.S., M.J., F.B., M.S., E.A., M.A., A.Q., J.D.C., T.C., K.M.G., A.M.K., R.K., A.E.P., and R.K.G. performed experiments and analyzed data. M.C. and E.L.P. wrote the manuscript.
Declaration of Interests
E.L.P. is a SAB member of Immunomet, and E.L.P. and E.J.P. are founders of Rheos Medicines. E.L.P. is Advisory Board member of Cell and Cell Metabolism.
Published: December 1, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.11.003.
Supplemental Information
References
- Acehan D., Malhotra A., Xu Y., Ren M., Stokes D.L., Schlame M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 2011;100:2184–2192. doi: 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W., Shyer J.A., Zhao J., Canaveras J.C.G., Al Khazal F.J., Qu R., Steach H.R., Bielecki P., Khan O., Jackson R. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature. 2019;571:403–407. doi: 10.1038/s41586-019-1311-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa E., Soustek M.S., Thomas A., Cogliati S., García-Poyatos C., Martín-García E., Jedrychowski M., Gygi S.P., Enriquez J.A., Puigserver P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol. Cell. 2019;74:877–890.e6, e876. doi: 10.1016/j.molcel.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T., Ishihara T., Kohno H., Saita S., Ichimura A., Maenaka K., Oka T., Mihara K., Ishihara N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017;19:856–863. doi: 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- Barth P.G., Scholte H.R., Berden J.A., Van der Klei-Van Moorsel J.M., Luyt-Houwen I.E., Van ’t Veer-Korthof E.T., Van der Harten J.J., Sobotka-Plojhar M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983;62:327–355. doi: 10.1016/0022-510x(83)90209-5. [DOI] [PubMed] [Google Scholar]
- Bione S., D’Adamo P., Maestrini E., Gedeon A.K., Bolhuis P.A., Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996;12:385–389. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- Buck M.D., O’Sullivan D., Klein Geltink R.I., Curtis J.D., Chang C.H., Sanin D.E., Qiu J., Kretz O., Braas D., van der Windt G.J. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M.D., Sowell R.T., Kaech S.M., Pearce E.L. Metabolic Instruction of Immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadalbert L.C., Ghaffar F.N., Stevenson D., Bryson S., Vaz F.M., Gottlieb E., Strathdee D. Mouse Tafazzin Is Required for Male Germ Cell Meiosis and Spermatogenesis. PLoS One. 2015;10:e0131066. doi: 10.1371/journal.pone.0131066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020;15:235–259. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- Chang C.H., Curtis J.D., Maggi L.B., Jr., Faubert B., Villarino A.V., O’Sullivan D., Huang S.C., van der Windt G.J., Blagih J., Qiu J. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie D.A., Mitsopoulos P., Blagih J., Dunn S.D., St-Pierre J., Jones R.G., Hatch G.M., Madrenas J. Stomatin-like protein 2 deficiency in T cells is associated with altered mitochondrial respiration and defective CD4+ T cell responses. J. Immunol. 2012;189:4349–4360. doi: 10.4049/jimmunol.1103829. [DOI] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Rudka T., Hartmann D., Costa V., Serneels L., Craessaerts K., Metzger K., Frezza C., Annaert W., D’Adamio L. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Clarke S.L., Bowron A., Gonzalez I.L., Groves S.J., Newbury-Ecob R., Clayton N., Martin R.P., Tsai-Goodman B., Garratt V., Ashworth M. Barth syndrome. Orphanet J. Rare Dis. 2013;8:23. doi: 10.1186/1750-1172-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M., Mariotti F.R., Trapani L., Taraborrelli L., Nazio F., Cianfanelli V., Soriano M.E., Schrepfer E., Cecconi F., Scorrano L., Campello S. Macroautophagy inhibition maintains fragmented mitochondria to foster T cell receptor-dependent apoptosis. EMBO J. 2016;35:1793–1809. doi: 10.15252/embj.201593727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017;5:90. doi: 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J., Hartmann M., Rehling P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:810–821. doi: 10.1016/j.bbadis.2018.08.025. [DOI] [PubMed] [Google Scholar]
- Duncan A.L., Robinson A.J., Walker J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. USA. 2016;113:8687–8692. doi: 10.1073/pnas.1608396113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey T.G., Mannella C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000;25:319–324. doi: 10.1016/s0968-0004(00)01609-1. [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Geltink R.I.K., Kyle R.L., Pearce E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018;36:461–488. doi: 10.1146/annurev-immunol-042617-053019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes L.C., Di Benedetto G., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvez F., Schug Z.T., Houtkooper R.H., MacKenzie E.D., Brooks D.G., Wanders R.J., Petit P.X., Vaz F.M., Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y.H., Dumlao D.S., Cao J., Dennis E.A. Assessing phospholipase A2 activity toward cardiolipin by mass spectrometry. PLoS One. 2013;8:e59267. doi: 10.1371/journal.pone.0059267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S.S., He Q., Janczy J.R., Elliott E.I., Zhong Z., Olivier A.K., Sadler J.J., Knepper-Adrian V., Han R., Qiao L. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussupow A., Di Luca A., Kaila V.R.I. How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 2019;5:v1850. doi: 10.1126/sciadv.aav1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan V.E., Tyurin V.A., Jiang J., Tyurina Y.Y., Ritov V.B., Amoscato A.A., Osipov A.N., Belikova N.A., Kapralov A.A., Kini V. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- Klein Geltink R.I., O’Sullivan D., Corrado M., Bremser A., Buck M.D., Buescher J.M., Firat E., Zhu X., Niedermann G., Caputa G. Mitochondrial Priming by CD28. Cell. 2017;171:385–397.e11. doi: 10.1016/j.cell.2017.08.018. e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konjar Š., Frising U.C., Ferreira C., Hinterleitner R., Mayassi T., Zhang Q., Blankenhaus B., Haberman N., Loo Y., Guedes J. Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci. Immunol. 2018;3:3. doi: 10.1126/sciimmunol.aan2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E.H., Verway M.J., Johnson R.M., Roy D.G., Steadman M., Hayes S., Williams K.S., Sheldon R.D., Samborska B., Kosinski P.A. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity. 2019;51:856–870.e5. doi: 10.1016/j.immuni.2019.09.003. e855. [DOI] [PubMed] [Google Scholar]
- Mangat J., Lunnon-Wood T., Rees P., Elliott M., Burch M. Successful cardiac transplantation in Barth syndrome--single-centre experience of four patients. Pediatr. Transplant. 2007;11:327–331. doi: 10.1111/j.1399-3046.2006.00629.x. [DOI] [PubMed] [Google Scholar]
- Mannella C.A., Pfeiffer D.R., Bradshaw P.C., Moraru I.I., Slepchenko B., Loew L.M., Hsieh C.E., Buttle K., Marko M. Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life. 2001;52:93–100. doi: 10.1080/15216540152845885. [DOI] [PubMed] [Google Scholar]
- Matyash V., Liebisch G., Kurzchalia T.V., Shevchenko A., Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res. 2008;49:1137–1146. doi: 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdipour A.R., Hummer G. Cardiolipin puts the seal on ATP synthase. Proc. Natl. Acad. Sci. USA. 2016;113:8568–8570. doi: 10.1073/pnas.1609806113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkler P.E., Hoppel C.L. Separation and characterization of cardiolipin molecular species by reverse-phase ion pair high-performance liquid chromatography-mass spectrometry. J. Lipid Res. 2010;51:856–865. doi: 10.1194/jlr.D002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mürke E., Stoll S., Lendeckel U., Reinhold D., Schild L. The mitochondrial phospholipid cardiolipin is involved in the regulation of T-cell proliferation. Biochim. Biophys. Acta. 2016;1861(8 Pt A):748–754. doi: 10.1016/j.bbalip.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Nath A.K., Ryu J.H., Jin Y.N., Roberts L.D., Dejam A., Gerszten R.E., Peterson R.T. PTPMT1 Inhibition Lowers Glucose through Succinate Dehydrogenase Phosphorylation. Cell Rep. 2015;10:694–701. doi: 10.1016/j.celrep.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D.G. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem. Soc. Trans. 2009;37:1385–1388. doi: 10.1042/BST0371385. [DOI] [PubMed] [Google Scholar]
- O’Sullivan D., van der Windt G.J., Huang S.C., Curtis J.D., Chang C.H., Buck M.D., Qiu J., Smith A.M., Lam W.Y., DiPlato L.M. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41:75–88. doi: 10.1016/j.immuni.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G., Paradies V., Ruggiero F.M., Petrosillo G. Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox Signal. 2014;20:1925–1953. doi: 10.1089/ars.2013.5280. [DOI] [PubMed] [Google Scholar]
- Paradies G., Paradies V., Ruggiero F.M., Petrosillo G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells. 2019;8:8. doi: 10.3390/cells8070728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce E.L., Walsh M.C., Cejas P.J., Harms G.M., Shen H., Wang L.S., Jones R.G., Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L., Scorrano L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016;78:505–531. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- Pickles S., Vigié P., Youle R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleston D.J., Zhang H., Powell T.J., Lipina E., Sims S., Panse I., Watson A.S., Cerundolo V., Townsend A.R., Klenerman P., Simon A.K. Autophagy is a critical regulator of memory CD8(+) T cell formation. eLife. 2014;3:3. doi: 10.7554/eLife.03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo N.P., Dudley M.E., Rosenberg S.A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronghe M.D., Foot A.B., Martin R., Ashworth M., Steward C.G. Non-Epstein-Barr virus-associated T-cell lymphoma following cardiac transplantation for Barth syndrome. Acta Paediatr. 2001;90:584–586. [PubMed] [Google Scholar]
- Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta. 2013;1831:582–588. doi: 10.1016/j.bbalip.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Schlame M., Kelley R.I., Feigenbaum A., Towbin J.A., Heerdt P.M., Schieble T., Wanders R.J., DiMauro S., Blanck T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- Schwall C.T., Greenwood V.L., Alder N.N. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta. 2012;1817:1588–1596. doi: 10.1016/j.bbabio.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Sena L.A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D.A., Wang C.R., Schumacker P.T., Licht J.D., Perlman H. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serricchio M., Vissa A., Kim P.K., Yip C.M., McQuibban G.A. Cardiolipin synthesizing enzymes form a complex that interacts with cardiolipin-dependent membrane organizing proteins. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2018;1863:447–457. doi: 10.1016/j.bbalip.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Steward C.G., Groves S.J., Taylor C.T., Maisenbacher M.K., Versluys B., Newbury-Ecob R.A., Ozsahin H., Damin M.K., Bowen V.M., McCurdy K.R. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr. Opin. Hematol. 2019;26:6–15. doi: 10.1097/MOH.0000000000000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sustarsic E.G., Ma T., Lynes M.D., Larsen M., Karavaeva I., Havelund J.F., Nielsen C.H., Jedrychowski M.P., Moreno-Torres M., Lundh M. Cardiolipin Synthesis in Brown and Beige Fat Mitochondria Is Essential for Systemic Energy Homeostasis. Cell Metab. 2018;28:159–174.e11, e111. doi: 10.1016/j.cmet.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschumi B.O., Dumauthioz N., Marti B., Zhang L., Lanitis E., Irving M., Schneider P., Mach J.P., Coukos G., Romero P., Donda A. CART cells are prone to Fas- and DR5-mediated cell death. J. Immunother. Cancer. 2018;6:71. doi: 10.1186/s40425-018-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt G.J., Everts B., Chang C.H., Curtis J.D., Freitas T.C., Amiel E., Pearce E.J., Pearce E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D.M., Segawa M., Kondadi A.K., Anand R., Bailey S.T., Reichert A.S., van der Bliek A.M., Shackelford D.B., Liesa M., Shirihai O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019;38:e101056. doi: 10.15252/embj.2018101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Araki K., Li S., Han J.H., Ye L., Tan W.G., Konieczny B.T., Bruinsma M.W., Martinez J., Pearce E.L. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 2014;15:1152–1161. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Guan Z., Murphy A.N., Wiley S.E., Perkins G.A., Worby C.A., Engel J.L., Heacock P., Nguyen O.K., Wang J.H. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets